

Abstract
Background
Acute COVID‐19–related myocardial, pulmonary, and vascular pathology and how these relate to each other remain unclear. To our knowledge, no studies have used complementary imaging techniques, including molecular imaging, to elucidate this. We used multimodality imaging and biochemical sampling in vivo to identify the pathobiology of acute COVID‐19. Specifically, we investigated the presence of myocardial inflammation and its association with coronary artery disease, systemic vasculitis, and pneumonitis.
Methods and Results
Consecutive patients presenting with acute COVID‐19 were prospectively recruited during hospital admission in this cross‐sectional study. Imaging involved computed tomography coronary angiography (identified coronary disease), cardiac 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose positron emission tomography/computed tomography (identified vascular, cardiac, and pulmonary inflammatory cell infiltration), and cardiac magnetic resonance (identified myocardial disease) alongside biomarker sampling. Of 33 patients (median age 51 years, 94% men), 24 (73%) had respiratory symptoms, with the remainder having nonspecific viral symptoms. A total of 9 patients (35%, n=9/25) had cardiac magnetic resonance–defined myocarditis. Of these patients, 53% (n=5/8) had myocardial inflammatory cell infiltration. A total of 2 patients (5%) had elevated troponin levels. Cardiac troponin concentrations were not significantly higher in patients with and without myocarditis (8.4 ng/L [interquartile range, IQR: 4.0–55.3] versus 3.5 ng/L [IQR: 2.5–5.5]; P=0.07) or myocardial cell infiltration (4.4 ng/L [IQR: 3.4–8.3] versus 3.5 ng/L [IQR: 2.8–7.2]; P=0.89). No patients had obstructive coronary artery disease or vasculitis. Pulmonary inflammation and consolidation (percentage of total lung volume) was 17% (IQR: 5%–31%) and 11% (IQR: 7%–18%), respectively. Neither were associated with the presence of myocarditis.
Conclusions
Myocarditis was present in a third patients with acute COVID‐19, and the majority had inflammatory cell infiltration. Pneumonitis was ubiquitous, but this inflammation was not associated with myocarditis. The mechanism of cardiac pathology is nonischemic and not attributable to a vasculitic process.
Registration
URL: https://www.isrctn.com; Unique identifier: ISRCTN12154994.
Keywords: CMR, COVID‐19, FDG‐PET, myocarditis, pneumonitis
Subject Categories: Computerized Tomography (CT), Magnetic Resonance Imaging (MRI), Nuclear Cardiology and PET, Inflammatory Heart Disease, Vascular Disease
Nonstandard Abbreviations and Acronyms
- 18F‐FDG
2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose
- CTCA
computed tomography coronary angiography
- hs‐cTnI
high‐sensitivity cardiac troponin I
- LGE
late gadolinium enhancement
- T1
longitudinal relaxation time
- T2
horizontal relaxation time
- TBR
target‐to‐background ratio
Clinical Perspective.
What Is New?
This is a prospective multimodality imaging study of acute COVID‐19 in an unselected population presenting to hospital.
Cardiac magnetic resonance, cardiac and vascular molecular positron emission tomography imaging, and computed tomography coronary angiography in conjunction with biochemical biomarkers were used to comprehensively phenotype patients with acute COVID‐19.
What Are the Clinical Implications?
Rates of myocarditis were high in an unselected population of acute COVID‐19 and may occur in the absence of biochemical markers of injury.
Cardiac involvement in COVID‐19 may not be appreciated clinically without imaging and can occur in the absence of severe pulmonary involvement.
Vasculitis or coronary artery thrombosis are not the cause of myocardial injury.
COVID‐19 has been mostly associated with pulmonary injury, but its association with cardiac and vascular pathobiology remains poorly understood. 1 , 2 , 3 Patients with cardiac involvement are at a higher risk of mortality, with 8% to 28% of patients showing biochemical evidence of myocardial injury. 4
2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose (18F‐FDG) positron emission tomography (PET)/computed tomography (CT) can identify cellular inflammation in pulmonary, cardiac, and vascular tissues, but prospective studies in COVID‐19 remain limited. 5 , 6 , 7 Although cardiac magnetic resonance (CMR) 1 , 2 , 3 and chest CT imaging in COVID‐19 have been conducted,8 these have been limited to the recovery phase and restricted to a single modality. As such, these studies were unable to differentiate ischemic from nonischemic cardiac pathology. A multisystem inflammatory syndrome in children with myocarditis and cardiac impairment as hallmarks of the presentation has been described. 9 Whether similar mechanisms of cardiac and vascular injuries occur in adults with acute COVID‐19 remains unknown. Finally, it is unknown if myocarditis can develop with only minimal pulmonary involvement.
Using CMR, CT coronary angiography (CTCA),10 and 18F‐FDG‐PET/CT 5 , 6 , 7 imaging during acute COVID‐19 infection, we investigated in vivo pathobiology of the myocardium, arterial vasculature, and pulmonary parenchyma. We hypothesized that myocardial or pulmomary inflammation and injury could be described by CMR and 18F‐FDG‐PET/CT, the presence of vascular inflammation identified by 18F‐FDG‐PET/CT, and the contribution by coronary artery disease shown by CTCA. We investigated the relationship between imaging findings and biomarkers as well as any association between pulmonary and cardiac pathology.
Methods
Study Design and Population
The data that support the findings of this study are available from the corresponding author upon reasonable request; however, any data that would allow possible identification of anonymized research patients will not be made available.
Participants hospitalized with COVID‐19 at the Aga Khan University Hospital in Nairobi, Kenya, were recruited in this single‐center exploratory observational study (Figure 1). Inclusion criteria were patients aged >18 years diagnosed with COVID‐19 (positive on polymerase chain reaction testing) on presentation to the hospital. The full study methodology with imaging techniques and protocols has been published. 11 Briefly, participants were recruited on admission to hospital. Following informed consent, blood draws were taken. Patients then underwent multimodality imaging as described in the Image Acquisition and Assessment section. The study complies with the Declaration of Helsinki with study approval from the Aga Khan University Nairobi Institutional Ethics Review Committee (reference: 2020/IERC‐74 [v2]).
Figure 1. Study design.
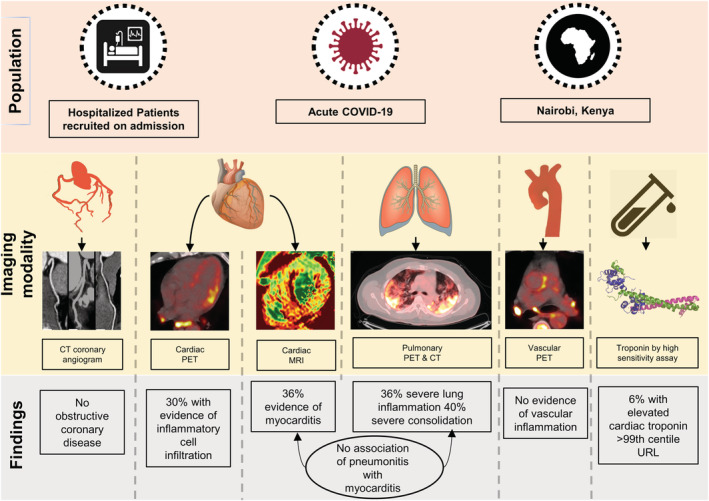
Patients with acute COVID‐19 were scanned on hospital admission. Cardiac magnetic resonance revealed myocarditis in 1 in 3 patients using the most stringest diagnostic criteria. Myocardial inflammatory cell infiltration identified by 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose PET/CT was present in 30% of all patients, and in the majority of patients with cardiac magnetic resonance–defined myocarditis. No patient had significant coronary artery disease on CT coronary angiography scanning. No patient had vasculitis. Although significant pulmonary inflammation and consolidation was common, it was not associated with the presence of myocarditis. Troponin testing did not identify patients with imaging evidence of myocardial edema or inflammatory cell infiltration. CT indicates computed tomography; MRI, magnetic resonance imaging; PET, positron emission tomography; and URL, upper reference limit.
Exclusion criteria were contraindication to CMR, known previous myocardial pathology, and those with severe symptoms requiring noninvasive or invasive ventilation. Patients underwent multimodality imaging and serological testing (1 sample on admission) for high‐sensitivity cardiac troponin I (hs‐cTnI; Siemens Healthineers; normal, <2.5 pg/mL), NT‐proBNP (N‐terminal pro–brain natriuretic peptide; Siemens Atellica Solution; normal, <300 pg/mL), CRP (C‐reactive protein; Siemens Atellica Solution; precision levels ≤0.3 mg/L), and viral load 12 (using cycle threshold; RealStar SARS‐CoV‐2 RT‐PCR Kit, Altona Diagnostics—limit of detection at 625 copies/mL). We additionally identified a small prospective control population of individuals who had no symptoms and had COVID‐19 excluded by polymerase chain reaction. This control group underwent the complete study protocol. In addition, for vascular analysis, we identified an age‐ and sex‐matched historical control population who had previously undergone 18F‐FDG‐PET/CT for another indication. This historical control group had no other pathology, for example, had undergone 18F‐FDG‐PET/CT for follow‐up of a benign pulmonary nodule.
Image Acquisition and Assessment
Participants underwent simultaneous CTCA and thoracic 18F‐FDG‐PET/CT (GE Discovery MI series PET/CT scanner) following admission, followed by CMR (Ingenia, Philips Healthcare as described previously; Data S1, 11 ).
Atherosclerotic Disease by CTCA
CTCA scanning was ECG gated and performed in diastole during a single breath hold with prospective ECG gating, detector collimation 64×0.625 mm, tube voltage 120 kV, and window of acquisition 70% to 90% (or wider if necessary because of heart rate). Tube current varied depending on body mass index using a prespecified manufacturer protocol. After the acquisition of scout images, CTCA was performed with iodinated contrast (Ultravist 370 mg/mL) in a biphasic injection protocol. Image acquisition was triggered by contrast enhancement of 100 HU in the ascending aorta. Presence of coronary artery disease in each major coronary artery and the main side branches were classified as potentially obstructive (>50% stenosis) or nonobstructive.
Myocardial Disease by CMR
CMR was performed using a 3 Tesla system (Ingenia, Philips Healthcare). Ejection fraction (EF) and regional wall motion abnormalities (by cine balanced steady‐state free precession sequence), myocardial fibrosis, edema, and presence of infarction in the left and right ventricles by late gadolinium enhancement (LGE) were determined as previously described (phase‐sensitive inversion recovery 5 minutes after administration of 0.1 mmol/kg gadolinium‐based contrast agent). 11 The anatomical 17‐segment model was used to derive T1, T2, and extracellular volume values for each segment excluding the apex. 13 Before gadolinium administration, native T1 and T2 maps were acquired at the base, mid‐ventricle, and apex. Postcontrast T1 mapping was repeated in an identical manner to precontrast T1 mapping 12 minutes after gadolinium injection.
T1 mapping was acquired using a modified look locker sequence using 10 images. Imaging parameters were the following: field of view, 300 mm; slice thickness, 10 mm; flip angle, 20°; repetition time, 2.26 ms; echo time, 1.03 ms; matrix, 256×256; 2.5 pixels/mm; trigger delay end diastole; and inversion times ranging from 137 to 5272 ms.
T2 mapping was performed using a multi‐echo gradient‐spin‐echo sequence on the same ventricular slices as T1 mapping. Repetition time was 1 RR interval. A total of 9 echoes were acquired using echo time 6 to 88 ms and echo train length 27. Slice thickness was 10 mm; matrix, 300×300 pixels; 1.4 pixels/mm; and field of view, 288×288 mm.
Myocardial, Vascular, and Pulmonary Pathology by 18F‐FDG‐PET/CT
We assessed myocardial inflammation as previously described. 5 Participants underwent imaging after a high‐fat, low‐carbohydrate meal for 24 hours with an 18‐hour fast to reduce physiologic myocardial 18F‐FDG uptake. 6 , 14 The PET imaging was performed 60 to 90 minutes after administration of 10 to 15 mCi of 18F‐FDG. The carotid arteries were the superior aspect of imaging, and the entire thoracic aorta was covered using 3‐minute different bed positions with additional dedicated 10‐minute cardiac acquisition. CT images were obtained immediately after PET scan acquisition. A low‐dose CT using 100 to 120 kVp and 30 to 50 mAs (automatic exposure control system) was performed immediately after the PET emission scan. Images were reconstructed using ordered subset expectation maximization. The PET images were attenuation corrected using the CT data and fused with CT for anatomical registration. CT and 18F‐FDG‐PET scan images were coregistered, and analysis was performed using the 17‐segment anatomical framework. 13 Myocardial uptake was scored based on a visual scale. Patients with focal or diffuse uptake were identified as having acute myocardial inflammation. 5
Semiquantative vascular inflammation on 18F‐FDG‐PET/CT for the aorta was assessed by the American Society of Nuclear Cardiologists visual grading criteria. 15 Quantitative assessment was also undertaken on large vessel inflammation. 6 A maximum arterial standardized uptake value was derived in serial axial measurements across the ascending, arch, and descending aorta. The target‐to‐background ratio (TBR) for each aortic region was calculated by averaging the ratio of the maximum arterial standardized uptake value to the mean venous standardized uptake value for each segment. A total of 21 age‐ and sex‐matched historical controls who had previously undergone clinical 18F‐FDG‐PET/CT scans for other indications (eg, investigation of pulmonary nodules and reported as normal) and 5 healthy active controls were also scanned.
For pulmonary analysis, chest CT and 18F‐FDG‐PET/CT images were analyzed separately for lung consolidation and inflammation, respectively. The 3‐dimensional lung contours were generated and linked to the coregistered PET and CT images. Thresholds, for pathology, were determined at 3 pooled SDs above the population means. Control patients were used to define thresholds to delineate consolidation on CT (by lung density in Hounsfield units) and inflammation on 18F‐FDG‐PET (by standardized uptake value). The volumes of consolidated lung and inflamed lung were presented as percentages of total lung volume.
Statistical Analysis
Baseline clinical and imaging data were expressed as the median (interquatile range) for continuous data and categorical data as proportions. Clinical and imaging data were presented by tertile of cardiac troponin (a priori analysis), presence of myocarditis on CMR, myocardial cell infiltration on PET, and degree of pulmonary inflammation/consolidation. A priori hypothesis testing was carried out across categorical and continuous covariates by tertile of cardiac troponin. 11 Exploratory hypothesis testing was further conducted when comparing clinical and imaging parameters by myocarditis and myocardial cell infiltration status. A priori comparisons of covariate values by categories of troponin levels were performed using the tableone package in R (https://cran.r‐project.org/web/packages/tableone/vignettes/introduction.html). This included the Fisher exact test for categorical variables and the Kruskal–Wallis test for continuous variables. All other hypothesis testing reported in the Results section was considered exploratory. Hypothesis testing for troponin values by cardiac pathology (presence of myocarditis on CMR or myocardial cell infiltration on cardiac PET) was done on nontransformed data using a nonparametric test. A P value of <0.05 was considered statistically significant. No correction for multiple testing was done. Analysis was done in R (version 4.0.3; http://www.R‐project.org/).
Results
Study Population
Of 64 consecutive patients with acute COVID‐19, 33 were recruited (median age, 51 years [interquartile range, IQR: 34–55], 31 [94%] men, and 31 (94%) Black men from Kenya [Table 1, Table S1]). Of the patients, 13 declined to participate and 18 had exclusion criteria. A total of 24 (73%) patients were hospitalized because of respiratory symptoms of cough with or without shortness of breath in the context of COVID‐19. The remaining patients had nonspecific viral symptoms (fever, myalgia, arthralgia fatigue, diarrhea, nausea, or vomiting). No patients had been vaccinated. A total of 29 patients underwent cardiac 18F‐FDG‐PET/CT, 26 underwent CTCA, and 26 underwent CMR scanning (Figure S1). CTCA and 18F‐FDG‐PET/CT scans were performed at a median time of 4 days after presentation (IQR: 2–9 days). CMR scans were performed at a median time of 10 days (IQR: 5–20 days). A total of 6 patients who were COVID‐19 negative were recruited as controls.
Table 1.
Baseline Characteristics of Patients With Acute COVID‐19 (N=33)
| Patients | |
|---|---|
| Demographics and past medical history | |
| Age, y | 51 [34–56] |
| Current/exsmokers | 6 (18.2) |
| Diabetes | 10 (31) |
| Hypertension | 11 (33) |
| HIV | 4 (13) |
| Clinical assessments | |
| Symptom duration, days | 4 [2–7] |
| Systolic BP, mm Hg | 127 [120–136] |
| Diastolic BP, mm Hg | 78 [70–85] |
| Heart rate, bpm | 88 [80–92] |
| COVID‐19 treatments | |
| Oxygen requirement | 19 (58) |
| Remdesevir | 4 (13) |
| Dexamethasone | 15 (47) |
| SARS‐CoV‐2 PCR (cycle threshold) | 25 [20–29] |
| Laboratory measurements | |
| Creatinine, μmol/L | 97 [60–108] |
| White cell count, ×109/L | 6 [5–9] |
| D‐dimer, mcg/mL | 0.66 [0.37–1.09] |
| C‐reactive protein, mg/L | 55 [25–101] |
| Procalcitonin, ng/mL | 0.07 [0.04–0.12] |
| NT‐proBNP, pg/mL | 35 [28–151] |
| Troponin, ng/L | 3.88 [2.76–7.18] |
Data are provided as number (percentage) or median [interquartile range]. BP indicates blood pressure; NT‐proBNP, N‐terminal pro–brain natriuretic peptide; and PCR, polymerase chain reaction.
The prevalence of biochemical evidence of myocardial injury (hs‐cTnI >99th centile upper reference limit) was 5% (n=2/31). Tertiles of hs‐cTnI levels only correlated with CRP (22 mg/L [IQR: 12–32] versus 85 [IQR: 50–100] versus 153 [IQR: 59–194]; P=0.001) (Table S2). A total of 25 patients underwent assessment of viral load by cycle threshold testing (7 high, 19 medium, 5 low) (Table S3). There was no association of viral load by cycle threshold (25 [IQR: 25–28] versus 27 [IQR: 22–29]; P=0.57), CRP (34 mg/L [IQR: 13–75] versus 45 mg/L [IQR: 30–101]; P=0.47), NT‐proBNP (35 pg/mL [IQR: 9–252] versus 35 pg/mL [IQR: 28–58]; P=0.89), or procalcitonin (0.04 ng/mL [IQR: 0.02–0.08] versus 0.11 ng/mL [IQR: 0.05–0.12]; P=0.17) levels comparing patients with and without myocarditis. There was a numerical but nonsignificant trend toward a lower duration of symptoms (6.5 days [IQR: 5‐7] versus 4 days [IQR: 3–7] versus 3 days [IQR: 2–5.5]; P=0.23) with increasing tertile of cardiac troponin.
CMR Imaging
A total of 26 patients underwent CMR scanning. All scans were of adequate quality for volume and wall motion analysis. One scan was of insufficient quality for T1‐mapping analysis, 1 was of insufficient quality for T2 analysis, and 1 scan was inadequate for LGE analysis. Myocarditis status was therefore available in 25 patients using the specific 2018 Lake Louise Criteria. 16
In the patient population, the median left ventricle EF was 51% (IQR: 57–57), and right ventricle EF was 55% (IQR: 48–50). Median global native T1 was 1275 ms (IQR: 1250–1317), global extracellular volume was 25% (IQR: 24–28), and global T2 was 51 ms (IQR: 47–54). A total of 9 patients (35%, n=9/25) had LGE. Of these, 2 (22%) had subendocardial LGE, and 8 (89%) had mid‐wall or epicardial LGE. Of the 9 patients with LGE, 7 (78%) also had evidence of active myocardial edema by T2 value.
A total of 9 (35%, n=9/25) patients had evidence of active myocarditis by the most specific 2018 Lake Louise criteria (Table 2, Figure 2). Of these patients, 6 (67%, n=6/9) had evidence of LGE, with 4 in a myocarditis pattern (mid‐wall), 1 with subendocardial LGE, and 1 with both. A total of 13 patients (50%, n=13/25) had evidence of myocarditis by the sensitive criteria (Table S4).
Table 2.
CMR Imaging Results Stratified by the Specific 2018 Lake Louis II Diagnosis of Myocarditis: Cardiac 18F‐FDG‐PET/CT Evidence of Myocardial Inflammatory Cell Infiltration
| Patients | Myocarditis (CMR) | No myocarditis (CMR) | Inflammatory cell infiltration (cardiac PET) | No inflammatory cell infiltration (cardiac PET) | |
|---|---|---|---|---|---|
| No. | 26* | 9 | 16 | 7 | 16 |
| LV ejection fraction, % | 61 [57–67] | 59 [56–62] | 64 [59–68] | 62 [60–63] | 60 [56–69] |
| LV EDVi, mL/m2 | 65 [62–72] | 65 [60–73] | 66 [62–72] | 64 [56–75] | 66 [62–72] |
| LV ESVi, mL/m2 | 25 [21–29] | 25 [21–32] | 22 [21–27] | 2 [20–32] | 26 [21–30] |
| LV SVi, mL/m2 | 42 [34–48] | 38 [30–44] | 44 [38–49] | 38 [31–42] | 44 [36–50] |
| RV ejection fraction, % | 56 [48–60] | 50 [47–58] | 58 [54–60] | 52 [48–56] | 59 [52–61] |
| T1–maximum, ms | 1356 [1304–1412] | 1342 [1301–1403] | 1372 [1322–1414] | 1317 [1297–1405] | 1380 [1335–1424] |
| T2–maximum, ms | 62 [57–68] | 68 [67–70] | 58 [56–62] | 66 [64–69] | 60 [56–63] |
| ECV, maximum % | 31 [28–34] | 34 [31–36] | 29 [27–32] | 33 [31–35] | 30 [27–34] |
| LGE, % | 9 (35) | 6 (67) | 2 (12) | 6 (86) | 3 (19) |
| Subendocardial LGE, % | 2 (8) | 2 (22) | 0 (0) | 2 (29) | 0 (0) |
| Mid‐wall, % | 8 (31) | 5 (56) | 2 (12) | 5 (71) | 3 (19) |
| Myocarditis‐specific Lake Louis criteria, CMR | 9 (36) | 5 (71) | 3 (20) | ||
| Myocardial inflammatory cell infiltration, PET | 7 (30) | 5 (63) | 2 (14) | ||
| Pulmonary inflammation, percentage of lung | 17 [3–29] | 16 [2–30] | 17 [10–31] | 7 [2–20] | 22 [13–38] |
| Pulmonary consolidation, percentage of lung | 12 [7–18] | 10 [8–16] | 13 [7–18] | 9 [8–12] | 16 [7–19] |
Data are provided as number, number (percentage), or median [interquartile range]. 18F‐FDG indicates 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose; CMR, cardiac magnetic resonance; CT, computed tomography; ECV, extracellular volume; EDVi, indexed end‐diastolic volume; ESVi, indexed end‐systolic volume; LGE, late gadolinium enhancement; LV, left ventricle; PET, positron emission tomography; RV, right ventricle; SVi, systolic volume indexed; T1, longitudinal relaxation time; and T2, horizontal relaxation time.
Denominators differ for each modality because not all scans were performed/diagnostic on every patient.
Figure 2. Severe myocarditis with minimal lung injury.
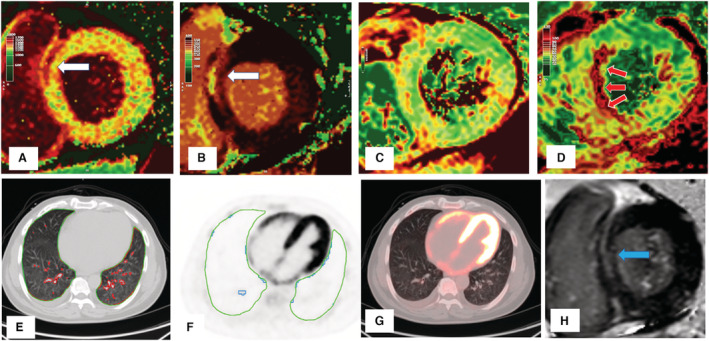
There is mid‐wall injury at the basal myocardium in the septum (white arrows) shown by cardiac magnetic resonance (CMR) (A) native T1, (B) postcontrast T1, and late gadolinium enhancement (H; blue arrow). There is no increase in T2 values in this basal region (C), but there is gross increase in mid‐ventricular septal T2 (D; red arrows), indicating edema remote to prior myocardial fibrosis. There was minimal lung consolidation (E; red contours) or inflammation (F; blue contours). There is diffuse biventricular 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose uptake (significantly higher than in the liver) (G). The patient had severe left and right ventricle impairment with elevated high‐sensitivity cardiac troponin I (110 ng/L) and NT‐proBNP (N‐terminal pro–brain natriuretic peptide; 7140 pg/mL) but low CRP (C‐reactive protein; 10 mg/L).
Cardiac troponin concentrations were numerically higher in patients with myocarditis compared with those without (8.4 [IQR: 4.0–55.3] versus 3.5 [IQR: 2.5–5.5]; P=0.07) (Table S5). No differences in viral load (25 [IQR: 20–28] versus 27 [IQR: 22–29]; P=0.70), left ventricle diastolic volume (55 mL/m2 [IQR: 50–73] versus 55 [IQR: 52–72]; P=0.84), or left ventricle EF (59% [IQR: 55–52] versus 54 [IQR: 59–58]; P=0.23) were found in patients with and without myocarditis (Table 1, Table S1, and Table S2).
Computed Tomography Coronary Angiography
A total of 25 patients underwent CTCA, and all had sufficient image quality. No patients had significant obstructive coronary artery disease (lumen stenosis>50%; Figure 3).
Figure 3. Focal inferolateral myocarditis with no atherosclerotic disease.
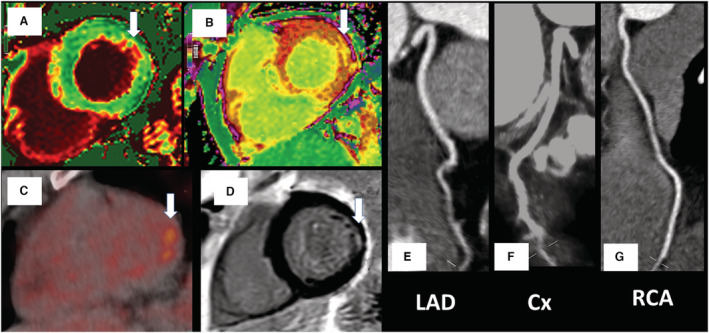
Changes (white arrows) in the native and postcontrast cardiac magnetic resonance (CMR) T1 values (A and B), 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose positron emission tomography focal uptake (C), and subendocardial fibrosis on CMR late gadolinium enhancement (D). There was no significant coronary artery disease on computed tomography coronary angiography (E through G). Biochemical cardiac and inflammatory markers were low (high‐sensitivity cardiac troponin I, 2.72 ng/L; NT‐proBNP [N‐terminal pro–brain natriuretic peptide], <35 pg/mL; CRP [C‐reactive protein], 4 mg/L). Cx indicates left circumflex coronary artery; LAD, left anterior descending coronary artery; and RCA, right coronary artery.
Positron Emission Tomography/Computed Tomography
Vascular Inflammation
Arterial inflammation in the ascending aorta by TBR was 1.97±0.35 (Figure S2 and Table S6) and similar to historical or active controls (1.92±0.32 and 2.03±0.05; P=0.74). There was no significant regional variation in TBR values in different aortic segments (Table S7). No patients fulfilled the visual criteria for inflammation in the aorta or carotids (Figure S3). There was no correlation with aortic fluorodeoxyglucose uptake (TBR) and CRP, hs‐cTnI, or viral load (Table S8). Ascending aorta TBR was similar in patients with and without CMR myocarditis (1.93±0.18 versus 2.00±0.44; P=0.55) and with and without myocardial inflammatory cell infiltration on 18F‐FDG‐PET (1.97±0.17 versus 1.91±0.21; P=0.47).
Myocardial Inflammatory Cell Infiltration
A total of 2 patients were not adequately fasted for cardiac analysis. Of the remaining patients, 8 (30%, n=8/27) had evidence of myocardial inflammatory cell infiltration. Of these patients, 3 had focal uptake (Figure 3), 4 had focal on diffuse (Figure 4), and 1 had diffuse (Figure 2).
Figure 4. Cardiac and pulmonary 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose positron emission tomography/CT imaging in 2 patients showing discordance between pulmonary and myocardial involvement.
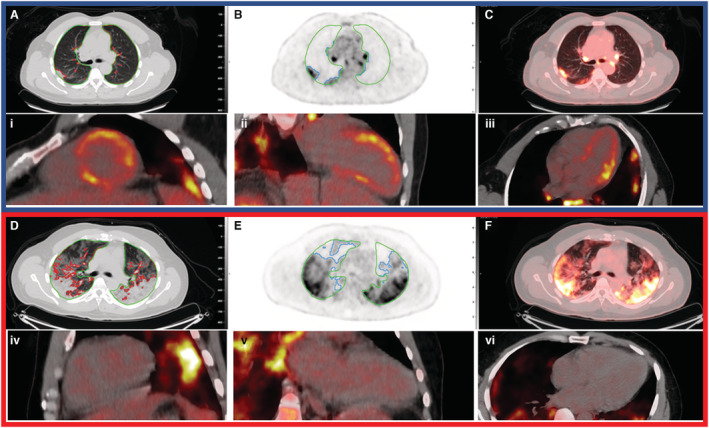
Top panel (blue outline) represents a patient with significant myocardial inflammatory cell infiltration with some pulmonary involvement—17% lung consolidation and 29% inflammation. Cardiac inflammatory cell infiltration (focal on diffuse bright spots in lateral anterior and septal walls). Bottom panel (red outline) represents another patient with no myocardial involvement but with significant lung consolidation (35%) and inflammation (54%). Lung consolidation on computed tomography (CT; A and D; red contours) and lung inflammation (B and E; blue contours) are shown. Green contours indicate lung parenchyma. Fused image (C and F) showing lung inflammation with heat maps on CT. Cardiac 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose positron emission tomography shows inflammatory cell infiltration in the short axis (i and iv), 2‐chamber (ii and v), and 4‐chamber views (iii and vi).
A total of 22 patients had both CMR and 18F‐FDG‐PET/CT. Of these, 8 patients had CMR‐defined myocarditis by using the specific 2018 Lake Louise criteria, and 5/8 (53%) also had simultaneous evidence of inflammatory cell infiltration. Similarly, of the 8 patients who had evidence of inflammatory cell infiltration by 18F‐FDG‐PET/CT, 5/8 (53%) had myocardial edema on CMR. A total of 12 patients (55%, n=12/22) had no evidence of myocarditis or cellular uptake on CMR or 18F‐FDG‐PET/CT.
Pulmonary Injury
Of the patients, 29 with acute COVID‐19 infection and 5 controls underwent pulmonary 18F‐FDG‐PET/CT. Overall, 25 patients had both CMR and pulmonary 18F‐FDG‐PET/CT performed.
In patients with acute COVID‐19, the median amount of inflammation (based on 18F‐FDG‐PET) and consolidation (based on CT) as a percentage of total lung volume was 17% (IQR: 5%–31%) and 11% (IQR: 7%–18%), respectively. In controls, there was 0.18% (IQR: 0.15%–0.57%) inflammation and 3.0% (IQR: 2.7%–3.1%) consolidation.
When categorizing patients who underwent both CMR and pulmonary 18F‐FDG‐PET/CT into tertiles, 7 of 25 patients had 0% to 5%, 9 of 25 had 5% to 25%, and 9 of 25 had >25% inflammation of the total lung volume (Table S7). Similarly, 5 of 25 patients had 0% to 7%, 10 of 25 had 7% to 15%, and 10 of 25 had >15% consolidation of total lung volume (Table S9).
The degree of lung inflammation (15% [IQR: 2%–30%] versus 17% [IQR: 10%–31%]; P=0.95) or consolidation (10% [IQR: 8%–15%] versus 13% [IQR: 7%–18%]; P=0.85) was comparable in patients with and without myocarditis (Figure 4). Similarly, the degree of lung inflammation (7% [IQR: 2%–20%] versus 22% [IQR: 13%–38%]; P=0.11) or consolidation (9% [IQR: 8%–12%] versus 15% [IQR: 7%–19%]; P=0.23) was comparable in patients with and without myocardial inflammatory cell infiltration.
There was no association between the presence of myocarditis and the degree of lung injury. Of patients with CMR‐based myocarditis, 3 of 9 (37.5%) had severe pulmonary inflammation compared with 5 of 17 (35.3%) without myocarditis (P=1.0). Patients with myocardial inflammatory cell infiltration, compared with those without, had numerically lower pulmonary inflammatory involvement (5% [IQR: 2%–17%] versus 24% [IQR: 13%–38%]; P=0.05). There was no correlation between severity of lung inflammation (55% [IQR: 52%–50%] versus 54% [IQR: 45%–59%]; P=0.57) or consolidation (57% [IQR: 51%–50%] versus 54% [IQR: 49%–59%]; P=0.52) with right ventricle EF. Similarly, no correlation was seen for severity of lung inflammation (81 ms/m2 [IQR: 71–85] versus 71 [55–79]; P=0.35) or consolidation (79 ms/m2 [IQR: 70–85] versus 74 [IQR: 55–82]; P=0.52) with indexed right ventricle diastolic volumes (Table 3).
Table 3.
CMR Imaging Results Stratified by 18F‐FDG‐PET/CT Evidence of Pulmonary Inflammation or Consolidation
| Patients | <25% Pulmonary inflammation (PET) | ≥25% Pulmonary inflammation (PET) | <15% Pulmonary consolidation (CT) | ≥15% Pulmonary consolidation (CT) | |
|---|---|---|---|---|---|
| No. | 26* | 9 | 16 | 10 | 15 |
| LV ejection fraction, % | 61 [57–67] | 57 [55–62] | 64 [60–68] | 58 [54–64] | 64 [60–68] |
| LV EDVi, mL/m2 | 65 [62–72] | 66 [60–73] | 65 [64–72] | 64 [60–72] | 65 [64–76] |
| LV ESVi, mL/m2 | 25 [21–29] | 26 [21–32] | 22 [21–27] | 26 [22–30] | 22 [20–28] |
| LV SVi, mL/m2 | 42 [34–48] | 41 [32–46] | 44 [37–49] | 38 [32–48] | 44 [38–48] |
| RV ejection fraction, % | 56 [48–60] | 54 [45–59] | 56 [52–60] | 54 [49–59] | 57 [51–60] |
| T1–maximum, ms | 1356 [1304–1412] | 1407 [1369–1415] | 1321 [1298–1400] | 1376 [1337–1412] | 1334 [1299–1424] |
| T2–maximum, ms | 62 [57–68] | 62 [57–67] | 62 [56–67] | 60 [57–67] | 62 [58–67] |
| ECV, maximum % | 31 [28–34] | 32 [26–34] | 30 [29–34] | 30 [26–34] | 31 [29–35] |
| LGE, % | 9 (35) | 3 (33) | 6 (38) | 4 (40) | 5 (33) |
| Subendocardial LGE, % | 2 (8) | 0 (0) | 2 (12) | 0 (0) | 2 (13) |
| Mid‐wall LGE, % | 8 (31) | 3 (33) | 5 (31) | 4 (40) | 4 (27) |
| Myocarditis‐specific Lake Louis criteria, CMR | 9 (36) | 3 (33) | 5 (33) | 3 (33) | 5 (33) |
| Myocardial inflammatory cell infiltration, PET | 7 (30) | 1 (11) | 6 (43) | 1 (11) | 6 (43) |
| Pulmonary inflammation, percentage of lung | 17 [3–29] | 38 [29–42] | 10 [1–17] | 35 [29–41] | 7 [1–15] |
| Pulmonary consolidation, percentage of lung | 12 [7–18] | 18 [17–26] | 8 [6–13] | 18 [18–25] | 8 [5–11] |
Data are provided as number, number (percentage), or median [interquartile range]. 18F‐FDG indicates 2‐deoxy‐2‐[fluorine‐18]fluoro‐D‐glucose; CMR, cardiac magnetic resonance; CT, computed tomography; ECV, extracellular volume; EDVi, end‐diastolic volume indexed; ESVi, end‐systolic volume indexed; IQR, interquartile range; LGE, late gadolinium enhancement; LV, left ventricle; PET, positron emission tomography; RV, right ventricle; SVi, systolic volume indexed; T1, longitudinal relaxation time; and T2, horizontal relaxation time.
Denominators differ for each modality because not all scans were performed/diagnostic on every patient.
Discussion
To our knowledge, this is the first study to systematically use molecular imaging alongside anatomical and functional modalities to explore cardiovascular and pulmonary pathobiology in acute COVID‐19 infection. We make some important observations. First, rates of myocarditis (by CMR criteria) and myocardial inflammatory cell infiltration (by 18F‐FDG‐PET/CT imaging) were significant at 35% and 30%, respectively. Second, the median burden of lung inflammation and consolidation was quantified at 17% and 11% of total lung volume, respectively. Lung involvement, both inflammation and consolidation, did not correlate with the presence of myocarditis or myocardial inflammatory cell infiltration. Third, vasculitis was not present in acute COVID‐19. Finally, biochemical evidence of myocardial injury was not common with only 2 patients with acute COVID‐19 showing elevated troponin levels.
Our rates of myocarditis, despite recruiting patients with acute COVID‐19, were lower than previously reported 1 but similar to other recent studies. 2 , 3 This in part reflects our choice of using the more specific 2018 Lake Louise criteria to define CMR‐based myocarditis. Indeed, the prevalence of myocarditis rose from 1 in 3 to 1 in 2 when applying the most sensitive criteria as in previous studies. 1 Using 18F‐FDG‐PET/CT imaging, myocardial inflammatory cell infiltration was present in 1 in 3 cases. Surprisingly, neither the presence of myocarditis nor myocardial cell infiltration was associated with biochemical evidence of cardiac injury. Myocarditis may not always result in cell necrosis and troponin release. 17 , 18 Furthermore, troponin release may be dynamic 19 , 20 and may not be appreciated on single‐point blood sampling on hospital admission. Alternatively, troponin release may occur weeks after initial presentation with myocarditis. 19 , 20 Finally, studies on myocarditis have generally been restricted to patients with troponin elevations in whom significant coronary disease has been excluded. 21 In contrast, our study involved cardiac imaging of an unselected population with an acute viral infection, regardless of troponin concentration.
Although CMR‐based tissue characterization can indicate myocardial edema, molecular imaging with 18F‐FDG‐PET/CT reflects myocardial cellular infiltration—a better indicator of an acute inflammatory process. 22 , 23 Of those patients who had CMR‐defined myocarditis, only 53% had an inflammatory cell presence. This suggests that acute myocardial inflammation may have either occurred before presentation or edema is not always attributed to direct cellular infiltration. SARS‐CoV‐2 infection is present in the myocardium in the majority of individuals dying from COVID‐19. 23 Furthermore, in vitro studies have shown SARS‐CoV‐2 cytopathic infection of cardiac myocytes with macrophage and lymphocytic infiltration. 1 , 23 , 24 However, a cytokine storm has also been implicated in COVID‐19 infection. 25 , 26 This process occurs sometime after viral inoculation and may also result in cardiac pathology without the presence of SARS‐CoV‐2 in the myocardium. 27 In this case, systemic cytokines may also cause systemic capillary leak (with resultant edema) without cellular infiltration of all tissues. 28 Therefore, cardiac injury may result from a dual‐injury process: initially from viral infection followed by a subsequent cardiac insult from a systemic inflammatory response. In keeping with this, we demonstrated that some patients had evidence of prior myocardial fibrosis without associated edema but then also had active edema without fibrosis in other regions (Figure 2).
Although the pathogenesis of hypercoagulability in COVID‐19 remains unclear, vascular thrombosis has been described in hospitalized patients. 29 Endothelial injury and vascular inflammation have been postulated to play a central role. 30 , 31 In contrast, our study did not find any supporting evidence of arterial inflammation in acute COVID‐19. We further found no evidence of coronary thrombosis to explain the myocardial pathology observed (Figure 3). A previous study demonstrated coronary artery obstruction and ischemic injury patterns on CMR; however, the study population was restricted to those with troponin elevations. 3 As such, we can conclude that the mechanism of cardiac pathology in acute COVID‐19 is unlikely to have occurred secondary to coronary atherosclerosis, and the reported high prevalence of vascular thrombosis is not attributed to an arterial vasculitic process. 29
Macrophages and monocytes are known to be involved in the pathogenesis of acute respiratory distress syndrome, and there is growing evidence of their involvement in COVID‐19–related pulmonary injury. 32 We showed that the degree of pneumonitis, by 18F‐FDG‐PET/CT, was variable, correlated with the degree of lung consolidation but was not associated with presence of myocarditis. This suggests that myocarditis can occur in patients with minimal lung involvement.
Our study has some limitations. First, although achieving comprehensive phenotyping, this was an observational study in a small COVID‐19 population. Almost half of the patients received either dexamethasone or remdesivir, which may have supressed the inflammatory response and underestimated myocardial inflammation. Scanning, however, was performed early in the clinical course. Second, our assessment of vasculitis was based on 18F‐FDG‐PET/CT uptake in the large vessels. Vascular inflammation in the smaller vessels, because of limited spatial resolution, may be undetected. However, if vascular inflammation was secondary to a systemic cytokine storm or immune response, it would have been expected that this would have been reflected in the aorta and the medium‐sized carotids. Third, we excluded patients with severe COVID‐19 who were unable to tolerate imaging, limiting the generalizability of our findings in this population. Finally, we did not perform cardiac biopsy. Although this is the gold standard for the diagnosis of myocarditis, we performed deep phenotyping using 3 different imaging modalities. The combination of myocardial inflammatory cell identification by 18F‐FDG‐PET and myocarditis detection by CMR (using the strictest criteria) make our findings robust.
In conclusion, with the use of multimodality imaging in acute COVID‐19 infection we make several observations. Myocarditis was present in 1 in 3 patients, and the majority of these patients had evidence of inflammatory cell infiltration by cardiac 18F‐FDG‐PET/CT. Pneumonitis was ubiquitous in acute COVID‐19, but this inflammation was not associated with CMR myocarditis. The mechanism of cardiac pathology in acute COVID‐19 is nonischemic, and vascular thrombosis in acute COVID‐19 is not attributable to a vasculitic process that involves large‐ or medium‐sized vessels.
Sources of Funding
This work was supported by the British Heart Foundation (FS/19/17/34172) and the Global Challenges Research Fund (COV_03).
Disclosures
None.
Supporting information
Acknowledgments
We thank Rehema Sunday and Norah Matheka for coordinating the study.
Author contributions: Alam and Shah conceived the study, constructed the study design, wrote the study and imaging protocols, ran the study, and wrote the first draft of the paper; Vinayak, Nganga, Gitau, Makhdomi, and Ombati were responsible for setting up imaging protocols and performing imaging; Shah, Kimeu, Gachoka, Obino, Jeilan, Riunga, Adam, and Chung were responsible for recruitment, data collection, and application of study design; Doolub and Joshi performed vascular positron emission tomography (PET) analysis; Lee performed computed tomography coronary angiography analysis; Vesselle, Horn, and Bowen performed pulmonary PET analysis; Alam performed cardiac magnetic resonance analysis; Shah and Gitau performed cardiac PET analysis. All authors made critical revisions of the manuscript.
Preprint posted on MedRxiv March 22, 2022. doi: https://doi.org/10.1101/2022.03.21.22272698.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.122.026399
For Sources of Funding and Disclosures, see page 11.
References
- 1. Puntmann VO, Carerj ML, Wieters I, Fahim M, Arendt C, Hoffmann J, Shchendrygina A, Escher F, Vasa‐Nicotera M, Zeiher AM, et al. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID‐19). JAMA Cardiol. 2020;5:1265–1273. doi: 10.1001/jamacardio.2020.3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang H, Li R, Zhou Z, Jiang H, Yan Z, Tao X, Li H, Xu L. Cardiac involvement in COVID‐19 patients: mid‐term follow up by cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2021;23:14. doi: 10.1186/s12968-021-00710-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kotecha T, Knight DS, Razvi Y, Kumar K, Vimalesvaran K, Thornton G, Patel R, Chacko L, Brown JT, Coyle C, et al. Patterns of myocardial injury in recovered troponin‐positive COVID‐19 patients assessed by cardiovascular magnetic resonance. Eur Heart J. 2021;42:1866–1878. doi: 10.1093/eurheartj/ehab075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, et al. Clinical course and risk factors for mortality of adult inpatients with COVID‐19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nensa F, Kloth J, Tezgah E, Poeppel TD, Heusch P, Goebel J, Nassenstein K, Schlosser T. Feasibility of FDG‐PET in myocarditis: comparison to CMR using integrated PET/MRI. J Nucl Cardiol. 2018;25:785–794. doi: 10.1007/s12350-016-0616-y [DOI] [PubMed] [Google Scholar]
- 6. Subramanian S, Tawakol A, Burdo TH, Abbara S, Wei J, Vijayakumar J, Corsini E, Abdelbaky A, Zanni MV, Hoffmann U, et al. Arterial inflammation in patients with HIV. JAMA. 2012;308:379–386. doi: 10.1001/jama.2012.6698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jonsson CB, Camp JV, Wu A, Zheng H, Kraenzle JL, Biller AE, Vanover CD, Chu YK, Ng CK, Proctor M, et al. Molecular imaging reveals a progressive pulmonary inflammation in lower airways in ferrets infected with 2009 H1N1 pandemic influenza virus. PLoS One. 2012;7:e40094. doi: 10.1371/journal.pone.0040094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Afshar‐Oromieh A, Prosch H, Schaefer‐Prokop C, Bohn KP, Alberts I, Mingels C, Thurnher M, Cumming P, Shi K, Peters A, et al. A comprehensive review of imaging findings in COVID‐19—status in early 2021. Eur J Nucl Med Mol Imaging. 2021;48:2500–2524. doi: 10.1007/s00259-021-05375-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Consiglio CR, Cotugno N, Sardh F, Pou C, Amodio D, Rodriguez L, Tan Z, Zicari S, Ruggiero A, Pascucci GR, et al. The immunology of multisystem inflammatory syndrome in children with COVID‐19. Cell. 2020;183:968–981.e7. doi: 10.1016/j.cell.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aldrovandi A, Cademartiri F, Arduini D, Lina D, Ugo F, Maffei E, Menozzi A, Martini C, Palumbo A, Bontardelli F, et al. Computed tomography coronary angiography in patients with acute myocardial infarction without significant coronary stenosis. Circulation. 2012;126:3000–3007. doi: 10.1161/CIRCULATIONAHA.112.117598 [DOI] [PubMed] [Google Scholar]
- 11. Alam SR, Shah ASV, Ombati KO, Nganga E, Gitau S, Makhdomi K, Chung MH, Vinayak S. CardiOvaScular mechanisms in Covid‐19: methodology of a prospective observational multimodality imaging study (COSMIC‐19 study). BMC Cardiovasc Disord. 2021;21:234. doi: 10.1186/s12872-021-02027-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kampf G, Lemmen S, Suchomel M. Ct values and infectivity of SARS‐CoV‐2 on surfaces. Lancet Infect Dis. 2021;21:e141. doi: 10.1016/S1473-3099(20)30883-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, Verani MS, et al. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart. A statement for healthcare professionals from the cardiac imaging Committee of the Council on clinical cardiology of the American Heart Association. Circulation. 2002;105:539–542. doi: 10.1161/hc0402.102975 [DOI] [PubMed] [Google Scholar]
- 14. Yarasheski KE, Laciny E, Overton ET, Reeds DN, Harrod M, Baldwin S, Davila‐Roman VG. 18FDG PET‐CT imaging detects arterial inflammation and early atherosclerosis in HIV‐infected adults with cardiovascular disease risk factors. J Inflamm (Lond). 2012;9:26. doi: 10.1186/1476-9255-9-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Slart R, Writing G, Reviewer G, Members of EC, Members of EI, Inflammation, Members of Committees SC, Members of Council PETIG, Members of A, Coordinator EC . FDG‐PET/CT(a) imaging in large vessel vasculitis and polymyalgia rheumatica: joint procedural recommendation of the EANM, SNMMI, and the PET interest group (PIG), and endorsed by the ASNC. Eur J Nucl Med Mol Imaging. 2018;45:1250–1269. doi: 10.1007/s00259-018-3973-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferreira VM, Schulz‐Menger J, Holmvang G, Kramer CM, Carbone I, Sechtem U, Kindermann I, Gutberlet M, Cooper LT, Liu P, et al. Cardiovascular magnetic resonance in nonischemic myocardial inflammation: expert recommendations. J Am Coll Cardiol. 2018;72:3158–3176. doi: 10.1016/j.jacc.2018.09.072 [DOI] [PubMed] [Google Scholar]
- 17. Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation. 1997;95:163–168. [PubMed] [Google Scholar]
- 18. Lauer B, Niederau C, Kuhl U, Schannwell M, Pauschinger M, Strauer BE, Schultheiss HP. Cardiac troponin T in patients with clinically suspected myocarditis. J Am Coll Cardiol. 1997;30:1354–1359. doi: 10.1016/s0735-1097(97)00317-3 [DOI] [PubMed] [Google Scholar]
- 19. Galea N, Marchitelli L, Pambianchi G, Catapano F, Cundari G, Birtolo LI, Maestrini V, Mancone M, Fedele F, Catalano C, et al. T2‐mapping increase is the prevalent imaging biomarker of myocardial involvement in active COVID‐19: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2021;23:68. doi: 10.1186/s12968-021-00764-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blanco‐Dominguez R, Sanchez‐Diaz R, de la Fuente H, Jimenez‐Borreguero LJ, Matesanz‐Marin A, Relano M, Jimenez‐Alejandre R, Linillos‐Pradillo B, Tsilingiri K, Martin‐Mariscal ML, et al. A novel circulating MicroRNA for the detection of acute myocarditis. N Engl J Med. 2021;384:2014–2027. doi: 10.1056/NEJMoa2003608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vago H, Szabo L, Dohy Z, Czimbalmos C, Toth A, Suhai FI, Barczi G, Gyarmathy VA, Becker D, Merkely B. Early cardiac magnetic resonance imaging in troponin‐positive acute chest pain and non‐obstructed coronary arteries. Heart. 2020;106:992–1000. doi: 10.1136/heartjnl-2019-316295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dall'Armellina E, Karia N, Lindsay AC, Karamitsos TD, Ferreira V, Robson MD, Kellman P, Francis JM, Forfar C, Prendergast BD, et al. Dynamic changes of edema and late gadolinium enhancement after acute myocardial infarction and their relationship to functional recovery and salvage index. Circ Cardiovasc Imaging. 2011;4:228–236. doi: 10.1161/CIRCIMAGING.111.963421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bearse M, Hung YP, Krauson AJ, Bonanno L, Boyraz B, Harris CK, Helland TL, Hilburn CF, Hutchison B, Jobbagy S, et al. Factors associated with myocardial SARS‐CoV‐2 infection, myocarditis, and cardiac inflammation in patients with COVID‐19. Mod Pathol. 2021;34:1345–1357. doi: 10.1038/s41379-021-00790-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perez‐Bermejo JA, Kang S, Rockwood SJ, Simoneau CR, Joy DA, Silva AC, Ramadoss GN, Flanigan WR, Fozouni P, Li H, et al. SARS‐CoV‐2 infection of human iPSC‐derived cardiac cells reflects cytopathic features in hearts of patients with COVID‐19. Sci Transl Med. 2021;13. doi: 10.1126/scitranslmed.abf7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moore JB, June CH. Cytokine release syndrome in severe COVID‐19. Science. 2020;368:473–474. doi: 10.1126/science.abb8925 [DOI] [PubMed] [Google Scholar]
- 26. Group RC , Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, Linsell L, Staplin N, Brightling C, Ustianowski A, et al. Dexamethasone in hospitalized patients with Covid‐19. N Engl J Med. 2021;384:693–704. doi: 10.1056/NEJMoa2021436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, Law KI, Tang BS, Hon TY, Chan CS, et al. Clinical progression and viral load in a community outbreak of coronavirus‐associated SARS pneumonia: a prospective study. Lancet. 2003;361:1767–1772. doi: 10.1016/s0140-6736(03)13412-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuribayashi S, Sakoda Y, Kawasaki T, Tanaka T, Yamamoto N, Okamatsu M, Isoda N, Tsuda Y, Sunden Y, Umemura T, et al. Excessive cytokine response to rapid proliferation of highly pathogenic avian influenza viruses leads to fatal systemic capillary leakage in chickens. PLoS One. 2013;8:e68375. doi: 10.1371/journal.pone.0068375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bilaloglu S, Aphinyanaphongs Y, Jones S, Iturrate E, Hochman J, Berger JS. Thrombosis in hospitalized patients with COVID‐19 in a New York City health system. JAMA. 2020;324:799–801. doi: 10.1001/jama.2020.13372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lowenstein CJ, Solomon SD. Severe COVID‐19 is a microvascular disease. Circulation. 2020;142:1609–1611. doi: 10.1161/CIRCULATIONAHA.120.050354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Libby P, Luscher T. COVID‐19 is, in the end, an endothelial disease. Eur Heart J. 2020;41:3038–3044. doi: 10.1093/eurheartj/ehaa623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kosyreva A, Dzhalilova D, Lokhonina A, Vishnyakova P, Fatkhudinov T. The role of macrophages in the pathogenesis of SARS‐CoV‐2‐associated acute respiratory distress syndrome. Front Immunol. 2021;12:682871. doi: 10.3389/fimmu.2021.682871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roy C, Slimani A, de Meester C, Amzulescu M, Pasquet A, Vancraeynest D, Vanoverschelde JL, Pouleur AC, Gerber BL. Age and sex corrected normal reference values of T1, T2 T2* and ECV in healthy subjects at 3T CMR. J Cardiovasc Magn Reson. 2017;19:72. doi: 10.1186/s12968-017-0371-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suinesiaputra A, Bluemke DA, Cowan BR, Friedrich MG, Kramer CM, Kwong R, Plein S, Schulz‐Menger J, Westenberg JJ, Young AA, et al. Quantification of LV function and mass by cardiovascular magnetic resonance: multi‐center variability and consensus contours. J Cardiovasc Magn Reson. 2015;17:63. doi: 10.1186/s12968-015-0170-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ferreira VM, Piechnik SK, Dall'Armellina E, Karamitsos TD, Francis JM, Choudhury RP, Friedrich MG, Robson MD, Neubauer S. Non‐contrast T1‐mapping detects acute myocardial edema with high diagnostic accuracy: a comparison to T2‐weighted cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2012;14:42. doi: 10.1186/1532-429X-14-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haaf P, Garg P, Messroghli DR, Broadbent DA, Greenwood JP, Plein S. Cardiac T1 mapping and extracellular volume (ECV) in clinical practice: a comprehensive review. J Cardiovasc Magn Reson. 2016;18:89. doi: 10.1186/s12968-016-0308-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baessler B, Schaarschmidt F, Treutlein M, Stehning C, Schnackenburg B, Michels G, Maintz D, Bunck AC. Re‐evaluation of a novel approach for quantitative myocardial oedema detection by analysing tissue inhomogeneity in acute myocarditis using T2‐mapping. Eur Radiol. 2017;27:5169–5178. doi: 10.1007/s00330-017-4894-9 [DOI] [PubMed] [Google Scholar]
- 38. Kim RJ, Fieno DS, Parrish TB, Harris K, Chen EL, Simonetti O, Bundy J, Finn JP, Klocke FJ, Judd RM. Relationship of MRI delayed contrast enhancement to irreversible injury, infarct age, and contractile function. Circulation. 1999;100:1992–2002. doi: 10.1161/01.cir.100.19.1992 [DOI] [PubMed] [Google Scholar]
- 39. Rudd JH, Myers KS, Bansilal S, Machac J, Rafique A, Farkouh M, Fuster V, Fayad ZA. (18)fluorodeoxyglucose positron emission tomography imaging of atherosclerotic plaque inflammation is highly reproducible: implications for atherosclerosis therapy trials. J Am Coll Cardiol. 2007;50:892–896. doi: 10.1016/j.jacc.2007.05.024 [DOI] [PubMed] [Google Scholar]
- 40. Gropler RJ, Siegel BA, Lee KJ, Moerlein SM, Perry DJ, Bergmann SR, Geltman EM. Nonuniformity in myocardial accumulation of fluorine‐18‐fluorodeoxyglucose in normal fasted humans. J Nucl Med. 1990;31:1749–1756. [PubMed] [Google Scholar]
- 41. Bartlett ML, Bacharach SL, Voipio‐Pulkki LM, Dilsizian V. Artifactual inhomogeneities in myocardial PET and SPECT scans in normal subjects. J Nucl Med. 1995;36:188–195. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.