

Abstract
Background
Monkeypox Virus (MPV) is the cause of zoonotic disease characterized by skin-eruption with pus cell formation and lymphadenopathy. This virus belongs to the Orthopoxvirus genus with DNA as its genetic material. Previously, this infection was reported from Africa and occasionally from USA and UK. However, recently there is a sudden surge of infection in non-epidemic countries and a new strain of MPVhas been discovered. Therefore it is important to revisit the phylogeny of MPV with the addition of new strains. Recently WHO also stressed the need of developing vaccines for new strains. In this scenario we have two objectives for this study -first, to reveal the exact phylogenetic position of the 2022 strain and second, to identify specific peptides which may be used for vaccine development in the future.
Methods
The phylogenetic analysis was done with the help of Bayesian phylogeny. The dN/dS calculation was performed based on DNA polymerase genes of selected MPV strains. The peptidyl-epitope was searched in MPV2022/2 SLO strain with the help of several algorithms implemented in Allergen FP v.1.0, NetMHCII 2.3, MHCpred and Toxin Pred. The structure prediction of the proteins and peptides was performed through Hpepdock. The quality of the structures was validated through the Ramachandran plot. The molecular dynamics and simulation were performed through Gromacs software. The interaction between peptide and protein was assessed through Ligplot software.
Results
The phylogenetic analysis revealed that the considered 2022 MPVstrains were close to the USA strains. The evolutionary analysis showed the volatile nature of the genome. Moreover, 9-mer peptide sequence was identified as an epitope for vaccine development.
Conclusions
The emergence of more virulent strains in near future may not be ruled out. Immunocompromised patients are more susceptible to this virus hence sub-unit vaccine is a better choice than a recombinant or attenuated vaccine against monkeypox. We have identified a small stretch of specific peptide which may be used for developing a subunit vaccine against this virus.
Keywords: Monkeypox virus, Phylogenomics, Evolution, Epitopes, Molecular docking and dynamics
1. Introduction
Poxviruses are generally brick or oval-shaped viruses with sizeable double-stranded DNA as their genomes. They are well-known throughout the globe as the causative agent of pox, a disease which is chiefly marked by the skin and mucous membrane eruption with lesion formation, development of skin nodules, and disseminated rash (Giulio and Eckburg, 2004). This disease generally occurs in mammals including humans. Different poxviruses cause different diseases. For instance, Smallpox is caused by the Variola virus which belongs to the Orthopoxvirus genus. Other members of this genus are the Vaccinia virus, Cowpox virus, and Monkeypox Virus (MPV) which have the capacity to infect humans. In 1980, the World Health Organization (WHO) declared that smallpox has been eradicated from the World (Au et al., 2022). However, within that period, another Orthopox virus came into the limelight, the Monkeypox virus. Infrequent cases were reported here and there in Africa, the USA, and the UK until 2022 when a sudden outbreak ensued in the USA and spread to neighboring countries and continents. Monkeypox was first reported in 1970 in a group of monkeys that were kept for research purposes, hence the causative agent was named MonkeyPoxVirus (MPV) (Cho and Wenner, 1973). The first report on human infection of this virus came from the Democratic Republic of Congo (DRC) in 1970 and 2003, for the first time this infection was reported from the USA i.e. outside Africa (Giulio and Eckburg, 2004).
The primary reservoir of this virus is still unknown but the African rodents and primates harbor this virus and transmit them to humans, establishing it as a zoonotic (disease that has jumped from a non-human animal to a human) disease (Cho and Wenner, 1973). However, human-to-human transmission happens principally through respiratory droplets and skin lesions contact. People infected with the MPVexperience febrile prodrome with widespread vesiculopustular rash in palms and soles (Cho and Wenner, 1973). Another distinguishing feature of this virus is lymphadenopathy (a disease affecting the lymph nodes) (https://wwwnc.cdc.gov/travel/yellowbook/2020/travel-related-infectious-diseases/smallpox-and-other-orthopoxvirus-associated-infections).
Monkeypox was previously reported to be endemic to tropical forest regions of West and Central Africa, mostly the Congo Basin (Velavan and Meyer, 2022). Refugees and immigrants leaving DRC might be one of the reasons for exposing countries outside Africa to this virus. Travelers returning from Africa may also expose people of their own country to this virus. For example, in 2018, both UK and Israel reported some imported cases of the MPV from travelers returning from African countries. It has been reported that imported rodents from Africa were the sole source of the MPVoutbreak in the USA in 2003.
Although the main source of MPVis the wild African primates, scientists have proved that in due course this virus has changed its genome, and now two distinct clades are available – the Congo Basin clade or central African clade and the West-African clades (the USA strains) (Likos et al., 2005, Shanbehzadeh et al., 2022). The Congo basin stains are more pathogenic with higher morbidity and transmission rate than the USA strains (Likos et al., 2005). However, it is still not determined whether the 2022 strains are the Congo-Basin strains or the West-African strains.
Vaccination with the live vaccinia virus can afford long-lasting protection against MPVsince this virus is an orthologue of Variola virus (smallpox-causing virus). Previous reports have shown that vaccinia-specific B-cell response is sufficient and essential for protection against the MPV (Sánchez-Sampedro et al., 2015). Antibody-mediated depletion of B-cells but not T-cells (both CD4+ and CD8+) gives vaccine-induced protection from the MPV(Sánchez-Sampedro et al., 2015). However, after the eradication of smallpox from the human population, vaccination against smallpox has been closed. This has made today’s generation more exposed to monkeypox attacks (Sánchez-Sampedro et al., 2015). Moreover, the countless intake of corticosteroids during the Covid-19 situation has made us immunocompromised (Kruh and Foster, 2012), a condition favorable for MPVattack. All this situation along with the fact that any viral genome is always prone to mutation has made us think to develop a vaccine against this virus specifically. Since in immunocompromised patients, epitope-based subunit vaccines work relatively better, we tried to identify a specific peptide-epitope from one of the 2022 MPVstrains for vaccine development in near future.
2. Materials and methods
2.1. Retrieval of sequences and phylogeny
The complete genome and proteome sequences of the MPV and other available Orthopox viruses were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/). There were a total of 100 MPV genomes available before the 2022 strains appeared. Among them, 33 were found to be unverified and were not considered for further analysis. Four whole-genome sequences of MPV_2022 strains were available while doing this study. Thus a total of 71 complete valid MPV genomes and proteomes were present in NCBI. However, these were not 2022 strains. When we initially started the work only a few 2022 MPV strains were available. Among them, we used Monkeypox virus isolate 2022/2 SLO (ON631241.1) since its sequence was complete and fully annotated. Along with MPV, we considered other Orthopox viruses (Vaccinia virus, Buffalopox virus, Racoonpox virus, and Variola virus) whose whole genome and proteome sequences are available in NCBI. These were used as Outgroups in the phylogenetic analysis. We have used the Bayesian phylogeny tree for this study. The DNA polymerase protein sequences of the considered MPV strains were aligned through Clustal Omega and the alignment file in Nexus format was fed into Mr. Bayes software for the analysis.
2.2. Evolutionary analysis
We exploited the ratio of non-synonymous substitution rate per synonymous site (dN) and synonymous substitution rate per synonymous site (dS) for predicting the evolutionary rate (ω). The idea here was, that ω value less than 1 indicates positive Darwinian selection pressure, and ω more than one signifies purifying selection (Roy et al., 2015). This analysis was performed using the PAML package (Yang, 2007) which uses the aligned sequence to calculate the substitution rate per synonymous sites. The dN/dS values range from 0 to 1. We have used the DNA polymerase genes of the selected strains for this analysis and the analysis was done via Ka/Ks Calculation tool (http://services.cbu.uib.no/tools/kaks).
2.3. Prediction of allergic proteins
The viral proteome contains some proteins which may cause allergies in our body. Those proteins should not be considered a candidate for epitopes. Hence, we identified the allergen and non-allergen proteins from the MPV isolate 2022/2 SLO. We used this particular strain since the annotated protein files were available for this strain during this study. Before identifying any protein as an allergen we should consider the ‘five E-descriptors’ of the protein (Venkatarajan et al., 2001). These five ‘E’ are- E1-hydrophobicity of the amino acids, E2- the size of the protein, E3- the propensity of helix formation of the amino acids, E4- a relative abundance of the amino acids, and E5- whether the protein is dominated by the β-strand forming propensity. An auto-cross covariance (ACC) transformation should also be used so that the length of the proteins becomes equal (Wold et al., 1993). ACC is a tool to transform a protein sequence into a vector of fixed length. This method is now successfully used for protein interaction study as well as for the classification of protein families. Each amino acid residue has some specific physical and chemical properties like hydrophobicity, hydrophilicity, polarity, polarizability, normalized van der Waals volume, sequence profile, etc (Wold et al., 1993). Hence, a sequence made up of amino acids can be depicted as a numeric matrix. ACC can correlate either the same or two different properties along the protein sequence and can transform the matrix into a vector of fixed length (Wold et al., 1993). Allergen FP v.1.0 is a server that uses all these aforementioned criteria for the prediction of a protein as an allergen (https://ddg-pharmfac.net/AllergenFP/method.html). Moreover, this server predicts Tanimoto coefficients for all protein pairs in a multiple-protein set.
A Tanimoto coefficient refers to a score or metric to measure the identity level between two different sets (Godden et al., 2000). There are a number of thresholds available for similarity searching. The higher the threshold level the more the similarity between the query and the target sequence. To predict the similarities between the query and the target sequences. Tanimoto coefficient plays a vital role. Fingerprints of both the query and the target sequences are used. Here fingerprints are the compiled predefined structural fragments or features present in a structure. Each feature present in a structure is represented by ‘ON’ or 1 (one bit). Tanimoto coefficient can be calculated by the following formula:
NA is the number of features ‘ON’ structure A and NB is the number of features ‘ON’ structure B and NA&B are the features ‘ON’ in both structure A and B (common features in both A and B) (Godden et al., 2000). Allergen-FP v.1.0 was used for this analysis. The non-allergen proteins were further considered for the T-cells’ epitope prediction.
2.4. Prediction of T cells’ epitope
Vaccine development in the post-genomic era requires in silico screening of the genomic information so that the most effective antigen can be predicted. This approach has its advantage. For instance, this does not require growing the microorganism in lab conditions; this is less time-consuming and cost-effective (Doytchinova and Flower, 2007). However, there are some problems. The in silico vaccine development strategies often depend upon the sequence alignment algorithm and in some cases, significant protein similarity is not found among the protein sequences even if their tertiary structure and functionality are similar (Doytchinova and Flower, 2007). Hence the antigenicity of a protein must be identified in a subtle and recondite manner that is not totally dependent only on sequence alignment. To overcome this sequence alignment dependability a new approach has been proposed which uses auto-cross covariance (ACC) transformation of the protein sequences into undeviating vectors with properties of principal amino acids (Doytchinova and Flower, 2007). Vaxijen server is one such server that predicts the antigenicity of the proteins based on the ACC transformation. The non-allergen proteins were fed into the Vaxijen server for predicting the antigenicity of those proteins. A cut-off score was set to 0.9 for this analysis. The epitopes should always be antigenic to trigger our immune system. Thus, proteins with high antigenic properties were fed into NetMHCII 2.3 (Hoof et al., 2009) for identifying the specific peptide parts which can interact with the HLA allele. Human leukocyte antigens (HLA) are genes of major histocompatibility complex (MHC) among humans which are associated with the recognition of self and non-self-antigens. HLA plays an immense role in the immune system. The subunit vaccine which is a cost-effective alternative to the high-cost vaccine development program is largely based on peptide-MHC interaction. Accordingly, we have used HLA for the molecular docking study. MHCpred (Guan et al., 2003) was used to further validate the interaction between the identified epitopes and HLA alleles.
2.5. Toxicity prediction of epitopes
There are several membrane-associated peptides with amphipathic structures which are hemolytic and thus are toxic to the human body. The predicted epitopes were further investigated for their probability of being suitable for epitope-based subunit vaccine generation and should not be toxic to the human system. The major function of those epitopes is just to trigger the immune system, however; they should not do any harm to the cellular mechanism of the recipients’ body. Hence those peptides were examined for toxicity through Toxin Pred (Kaushik, 2020). Non-toxic peptides were considered for further analysis.
2.6. Molecular modeling and docking of HLA alleles and epitopes
Protein-peptide interaction is important for cellular functionality and prediction of the protein-peptide interaction is crucial for understanding the molecular mechanism of biological processes and the development of peptide drugs. The interaction between the HLA proteins and the identified peptides (which can be used as T cells’ epitopes) was studied through ‘HpepDock’ (Zhou et al., 2018). HpepDock server is a tool for peptide-protein blind docking by fast modeling of the peptidyl conformations along with a global sampling of the orientations used in binding. We have provided the protein sequences of the HLA alleles obtained from NCBI (HLA-DP- NP_002112.3; HLA-DQ- NP_002113.2; HLA-DR- NP_061984.2) and the peptide sequences of the epitopes. HpepDock provided the structures of both the protein and the peptides. The tertiary structures of the HLA proteins were validated through Ramachandran plots. We used Ramachandran Plot Server (https://zlab.umassmed.edu/bu/rama/) by Zlab for this analysis. Those 3D structures were used to identify the specific interacting amino acids through Lig-plot (https://www.ebi.ac.uk/thornton-srv/software/LIGPLOT/) (Wallace et al., 1995) software and then a site-specific docking was performed through the AutoDock Vina software (Trott and Olson, 2010) to further support the results from HpepDock server. The results were visualized through Pymol (https://pymol.org/2/) (Yuan et al., 2017) and the interacting amino acids were visualized via LigPlot. A diagrammatic flow chart of the strategies used in this study has been given in Fig. 1 .
Fig. 1.
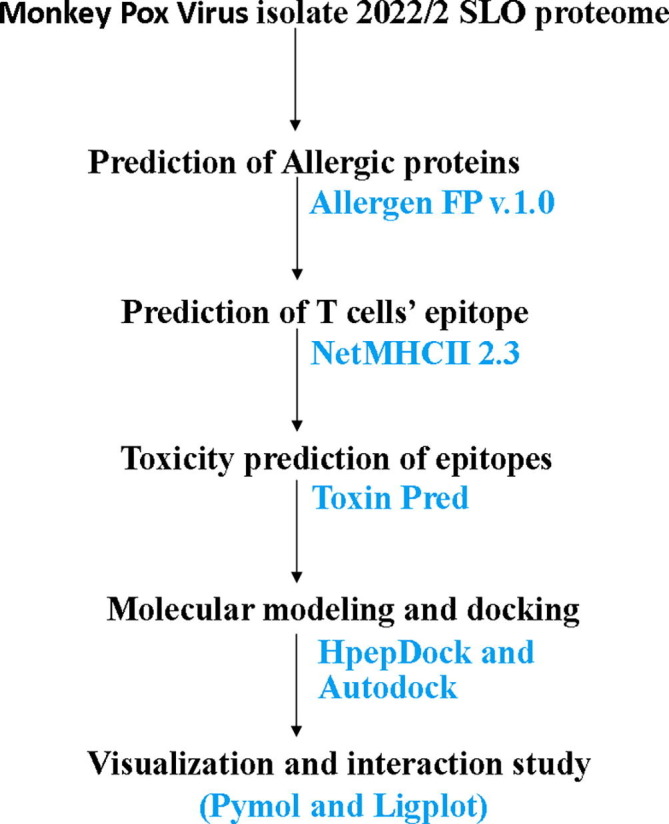
A flow chart showing the strategy used to identify the peptide for possible epitope vaccine formation.
3. Results
3.1. Phylogenomics analysis
The linear double stranded DNA genome of MPV 2022/2 SLO strain has been provided in Fig. 2 . Two distinct clades were resolved by Bayesian phylogeny (Fig. 2). One clade contains Zaire, Boende, Cameroon, Congo, Sudan, Ikubai, and Gabon strains. The other clades included USA strains along with Nigeria-SE, and Cote strains. The 2022/2 SLO was in the second clade i.e. with the USA strains. The Buffalopox, Vaccinia, Variola, and Racoonpox viruses were presented as outgroup. The linear double-stranded DNA of 2022/2 SLO strain has been diagrammatically represented in Fig. 3 .
Fig. 2.

The linear double-stranded DNA genome of Monkey Pox Virus isolate 2022/2 SLO genome.
Fig. 3.

Bayesian Phylogeny analysis of selected Monkey Pox Virus strains.
3.2. Evolutionary analysis
The evolutionary analysis revealed that MPV strains are under positive Darwinian selection and are govered by constraints. The results related to this analysis have been given in Supplementary File 1. Purifying selection reduces genetic diversity by modulating both the sites which are under direct selection as well as the neutral sites. The purifying (negative) selection hinders the spread of deleterious allels in the population whereas the Darwinian (positive) selections are known to promote the spread of beneficial alleles through the population. Although smallpox vaccines can protect us from this viral infection, we thought it would be better if an MPV-specific vaccine is developed. In this scenario, in silico prediction of epitopes for subunit vaccine development comes in handy due to its energy economic and time-saving approach. Hence the next phase of our study focused on the prediction of a peptide that may be utilized as a T cell epitope for the development of the MPV-specific vaccine.
3.3. Allergen and Non-allergen protein in MPV
Allergens are the type of antigens producing vigorous immune response so that the immune system can fight against a specific threat that is otherwise harmless to the body. These antigens can induce type-1 hypersensitivity and thus can produce allergic reactions in the body. These allergens are not a good candidate for epitope-based vaccine development. Hence we avoided all the allergens and used only non-allergens for further studies.
A total of 54 proteins were predicted to be allergen proteins and they were mostly TNF-alpha-receptor-like proteins, bifunctional zinc finger-like protein/E3 ubiquitin ligase, interleukin-18-binding proteins, alpha-amanitin target protein, 36 kDa major membrane protein F5, assembly protein G7, myristyl protein, RNA polymerase-associated transcription-specificity factor RAP94, scaffold protein D13, major core protein 4a precursor, 6 kDa intracellular viral protein, ser/thr protein kinase-like protein, interleukin-1-binding protein, ankyrin-like protein, Kelch repeat, and BTB domain-containing protein 2, TNF-alpha-receptor-like protein and some glycoproteins. The rest of the proteins were non-allergens which were further used for epitope identification.
3.4. Identification of T cells’ epitope
The Vaxijen server predicted two non-allergen proteins (protein_id URK21097.1 and URK21207.1) to be probable antigenic with a cut-off value of more than 0.9. Among them, URK21097.1 protein contained a specific peptide sequence -‘DIKRRYRHTIESVYF’. The core part of the peptide was found to be a 9-mer sequence ‘YRHTIESVY’. This core peptide part showed strong binding interaction with HLA-DP, DQ, and DR with 189.3 nM affinity. However, the URK21207.1 protein identified a total of 43 peptides with 10 different 9-mer core peptide sequences. All of them showed strong binding with the three HLA alleles (DP, DQ, and DR). The affinity ranges from 7.3 to 309.6 nM. All of them were non-toxic. Thus, a total of 11 peptide cores were predicted as T cells’ epitopes which showed strong binding with HLA alleles. This result was also validated through the data obtained from the MHCpred server which also indicated that the ‘YRHTIESVY’ peptide sequence was the most suitable candidate for epitope among the predicted ones.
3.5. Molecular docking study
The Ramachandran plot validated the quality of the HLA structures. Most of the amino acids were found to be in the most preferable regions (green cross) of the plots and very few were in the preferable regions (orange dots) (Fig. 4 ). Molecular docking was performed to visualize the interaction between the HLA alleles and the predicted epitope peptides. The best result was obtained for ‘YRHTIESVY’ against HLA-DP, DQ, and DR. The docking scores were −9.2 kcal/mol, −8.7 kcal/mol, and −9.8 kcal/mol for HLA-DP, DQ, and DR respectively. From the Ligplot analysis, it was revealed that hydrogen bonds and hydrophobic interactions were mainly associated with the peptide-protein interaction. Asp54 of HLA-DR, Glu51, Ser 165 of HLA-DQ, Gln42, Asn109, Cys44, Ala46, Tyr45, and Gly152 of HLA-DP were mainly associated with the hydrogen bond formation. Along with them, several other amino acids were forming the hydrophobic interactions in all three HLA-peptide interactions as shown in (Fig. 5 ).
Fig. 4.

Ramachandran plots for HLA-DP, DQ and DR tertiary structures. The most preferred amino acid residues are shown in green crosses and the preferred residues are in orange triangles.
Fig. 5.

The HLA alleles and peptidyl epitope interaction. The interacting amino acids are shown through Ligplot software.
4. Discussion
Monkeypox virus, a DNA virus is causing recurrent infection in several countries of the Western world. The reason behind this sudden outbreak is still elusive. Since we haven’t overcome the Covid-19 pandemic situation till now, these new viral cases are creating havoc and concern among the population (Rudan, 2022). During Covid, we became dependent on steroids to protect ourselves from the pandemic. Prednisone-like corticosteroids became our first and last hope to fight against Covid-19, until recently. These corticosteroids are broadly immune-suppressant since they act upon the whole body and try to lower inflammation and other related symptoms (Kruh and Foster, 2012). This is due to their unusual mechanism of action. They act by imitating the naturally produced corticosteroids (from the adrenal gland) in the body. As a result, the activity of the adrenal gland is reduced. Although it relieves pain and inflammation throughout the body, however, this reduced adrenal activity and abridged adrenal secretion ultimately leave us immune-compromised, a condition favorable for MPV attack (Kruh and Foster, 2012). Another major aspect of the recent MPV outbreaks is the lack of a vaccine against this DNA virus. Previously, the smallpox vaccine cured MPV infection (Yang, 2022). Since smallpox has been eradicated from the population and vaccination against smallpox has also been stopped for a few generations, most young adults in today’s population do not have specific antibodies in their system against MPV (Yang, 2022). The shortage of the smallpox vaccine in the market has made this situation worst. The 2022/2 SLO was closely related to the USA strains. Furthermore, the evolutionary analysis revealed that MPV strains are under positive Darwinian selection and are governed by constraints. Purifying selection reduces genetic diversity by modulating both the sites which are under direct selection as well as the neutral sites. The purifying (negative) selection hinders the spread of deleterious alleles in the population whereas the Darwinian (positive) selection is known to promote the spread of beneficial alleles through the population. Therefore it would be wise enough to be prepared with a vaccine against MPV before such a situation arise. Recently, the WHO has recognized the need for a specific vaccine against the Monkeypox virus.
As previously mentioned MPV attack is more prevalent among immunocompromised patients (Adalja and Inglesby, 2022) and live/attenuated/inactivated virus vaccines are also reported to have lots of disadvantages in such patients (Sirohi et al., 2022), epitope-based subunit vaccines seem to be the best option against MPV. Those vaccines are also easy to produce and have a broad safety profile. In this study, we tried to identify the most potent peptide from the MPV 2022 strain which can be further utilized for epitope-based vaccine production against MPV. Previous studies have reported major criteria to establish a peptide as an epitope for vaccine generation. These are, peptides should be non-allergen, antigenic, non-toxic, and should strongly interact with the MLC alleles. All those criteria were studied thoroughly and we could identify a short 9-mer peptide sequence ‘YRHTIESVY’ as a potent epitope for MPV vaccine generation. Human leukocyte antigens (HLA) are genes of major histocompatibility complex (MHC) among humans which are associated with the recognition of self and non-self-antigens. HLA plays an immense role in the immune system. The subunit vaccine which is a cost-effective alternative to the expensive vaccine development program is largely based on peptide-MHC interaction. The molecular docking analysis revealed a strong interaction between this peptide and the HLA-DP, DQ, and DR alleles. The molecular dynamics study revealed no significant (or major) alteration in the protein backbone movement thus, indicating the potency of this peptide as a T-cells-epitope for vaccine generation.
5. Conclusion
MPV is a DNA virus that originated in Central Africa and is transmitted across the globe. Two specific clades are there- the DRC clade from Central- Africa and another one is Western-African clade. We have determined that the 2022 strains of this virus are close to Western African and may not be that deadly. The evolutionary analysis revealed the presence of both positive Darwinian selection and purifying pressure on the considered 2022 strains of the Monkeypox virus. This also underlines the possibility of the emergence of more devastating MPV strains in near future. Hence vaccine development against this virus is the need of the hour.
For developing a subunit vaccine, the prediction of a non-allergen, antigenic and non-toxic epitope from the viral proteome is an excellent approach that can be done in a less time-consuming and economic manner. In this scenario, we came up with the idea of identifying some MPV-specific peptides which may be used as epitopes for sub-unit vaccine generation. We have also identified a specific peptide sequence ‘YRHTIESVY’ in MPV strain 2022/2 SLO. This peptide was found to be non-allergen, antigenic, and non-toxic. The molecular docking and dynamics study also validated the potency of this peptide to be used in the subunit vaccine generation against MPV. Vaccine manufacturers may consider this study for developing vaccines against the MPV_2022 strains.
Authors contribution
IS, MAA, and AS jointly conceived the idea. IS, GS, MAA, RMA, and JL performed the bioinformatic analysis. AS, MAA, and IS prepared the manuscript. All authors have agreed to the final version of the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
AS acknowledges Biswas Bangla Genome Center, University of North Bengal. IS acknowledges the Bioinformatics facility center, University of North Bengal. AS and IS acknowledge Sutapa Dutta for her scientific suggestions. The authors would like to extend their sincere appreciation to the Researchers Supporting Project number (RSP-2021/306), King Saud University, Riyadh, Saudi Arabia.
Footnotes
Peer review under responsibility of King Saud University.
Supplementary material to this article can be found online at https://doi.org/10.1016/j.jksus.2022.102458.
Appendix A. Supplementary material
The following are the Supplementary material to this article:
References
- Adalja A., Inglesby T. A novel international monkeypox outbreak. Ann. Intern. Med. 2022;175(8):1175–1176. doi: 10.7326/M22-1581. [DOI] [PubMed] [Google Scholar]
- Au N.H., Portillo M.T., Marwah A., Thomas-Bachli A., Demarsh P.A., Khan K., Bogoch I.I. Potential for monkeypox exportation from west and Central Africa through global travel networks. J. Travel Med. 2022 doi: 10.1093/jtm/taac072. [DOI] [PubMed] [Google Scholar]
- Cho C.T., Wenner H.A. Monkeypox virus. Bacteriol. Rev. 1973;37(1):1–8. doi: 10.1128/br.37.1.1-18.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giulio D.B., Eckburg P.B. Human monkeypox: an emerging zoonosis. Lancet Infect. Dis. 2004;4(1):15–25. doi: 10.1016/S1473-3099(03)00856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doytchinova I.A., Flower D.R. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007;8(1):1–7. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godden J.W., Xue L., Bajorath J. Combinatorial preferences affect molecular similarity/diversity calculations using binary fingerprints and Tanimoto coefficients. J. Chem. Inf. Model. 2000;40(1):163–166. doi: 10.1021/ci990316u. [DOI] [PubMed] [Google Scholar]
- Guan P., Doytchinova I.A., Zygouri C., Flower D.R. MHCPred: a server for quantitative prediction of peptide–MHC binding. Nucleic Acids Res. 2003 Jul 1;31(13):3621–3624. doi: 10.1093/nar/gkg510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoof I., Peters B., Sidney J., Pedersen L.E., Sette A., Lund O., Buus S., Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009 Jan;61(1):1–3. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik V. In silico identification of epitope-based peptide vaccine for Nipah virus. Int. J. Pept. Res. Ther. 2020;26(2):1147–1153. [Google Scholar]
- Kruh J., Foster C.S. Corticosteroid-sparing agents: conventional systemic immunosuppressants. Dev. Ophthalmol. 2012;51:29–46. doi: 10.1159/000336185. [DOI] [PubMed] [Google Scholar]
- Likos A.M., Sammons S.A., Olson V.A., Frace A.M., Li Y., Olsen-Rasmussen M., Davidson W., Galloway R., Khristova M.L., Reynolds M.G., Zhao H. A tale of two clades: monkeypox viruses. J. Gen. Virol. 2005;86(10):2661–2672. doi: 10.1099/vir.0.81215-0. [DOI] [PubMed] [Google Scholar]
- Roy A., Mukhopadhyay S., Sarkar I., Sen A. Comparative investigation of the various determinants that influence the codon and amino acid usage patterns in the genus Bifidobacterium. World J. Microbiol. Biotechnol. 2015;31(6):959–981. doi: 10.1007/s11274-015-1850-1. [DOI] [PubMed] [Google Scholar]
- Rudan I. The COVID-19 pandemic: SARS-CoV-2, childhood hepatitis and monkeypox raise five new questions for the global health research community. J. Glob. Health. 2022:22. doi: 10.7189/jogh.12.01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Sampedro L., Perdiguero B., Mejías-Pérez E., García-Arriaza J., Di Pilato M., Esteban M. The evolution of poxvirus vaccines. Viruses. 2015;7(4):1726–1803. doi: 10.3390/v7041726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbehzadeh M., Nopour R., Kazemi-Arpanahi H. Kazemi-Arpanahi HDesigning a standardized framework for data integration between zoonotic diseases systems: Towards one health surveillance. Inform. Med. Unlocked. 2022;30:100893. [Google Scholar]
- Sirohi P.R., Gupta J., Somvanshi P., Prajapati V.K., Grover A. Multiple epitope-based vaccine prediction against SARS-CoV-2 spike glycoprotein. J. Biomol. Struct. Dyn. 2022;40(8):3347–3358. doi: 10.1080/07391102.2020.1846626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velavan T.P., Meyer C.G. Monkeypox 2022 outbreak: an update. Trop. Med. Int. Health. 2022;27(7):604–605. doi: 10.1111/tmi.13785. [DOI] [PubMed] [Google Scholar]
- Venkatarajan M.S., Braun W. New quantitative descriptors of amino acids based on multidimensional scaling of a large number of physical–chemical properties. J. Mol. Model. 2001;7(12):445–453. [Google Scholar]
- Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Wold S., Jonsson J., Sjörström M., Sandberg M., Rännar S. DNA and peptide sequences and chemical processes multivariately modelled by principal component analysis and partial least-squares projections to latent structures. Anal. Chim. Acta. 1993;277(2):239–253. [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z. Monkeypox: a potential global threat? J. Med. Virol. 2022;94(9):4034–4036. doi: 10.1002/jmv.27884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Chan H.S., Hu Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017;7(2):e1298. [Google Scholar]
- Zhou P, Jin B, Li H, Huang S.Y. HPEPDOCK: a web server for blind peptide–protein docking based on a hierarchical algorithm. Nucleic acids research. 2018;46(W1):W443–W450. doi: 10.1093/nar/gky357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.