

Abstract
Biogenic volatile organic compounds (BVOCs) affect climate via changes to aerosols, aerosol-cloud interactions (ACI), ozone and methane. BVOCs exhibit dependence on climate (causing a feedback) and land use but there remains uncertainty in their net climatic impact. One factor is the description of BVOC chemistry. Here, using the earth-system model UKESM1, we quantify chemistry’s influence by comparing the response to doubling BVOC emissions in the pre-industrial with standard and state-of-science chemistry. The net forcing (feedback) is positive: ozone and methane increases and ACI changes outweigh enhanced aerosol scattering. Contrary to prior studies, the ACI response is driven by cloud droplet number concentration (CDNC) reductions from suppression of gas-phase SO2 oxidation. With state-of-science chemistry the feedback is 43% smaller as lower oxidant depletion yields smaller methane increases and CDNC decreases. This illustrates chemistry’s significant influence on BVOC’s climatic impact and the more complex pathways by which BVOCs influence climate than currently recognised.
Subject terms: Atmospheric chemistry, Climate and Earth system modelling
The modelling of BVOC chemistry strongly affects how doubling of BVOC emissions affects climate. Lower oxidant depletion with state-of-science chemistry leads to 43% smaller positive forcing from smaller methane increases and cloud albedo decreases.
Introduction
Atmospheric composition, and its response to a perturbation, plays a key role in climate1. Tropospheric chemistry in current state-of-the-art climate models used in the 6th Coupled Model Intercomparison Project (CMIP6) is highly parameterised in terms of reactions, emissions, aerosol chemistry and gas-aerosol coupling and there remains considerable uncertainty in the modelling of chemistry in the lower atmosphere.
In a climatic context this uncertainty is important because tropospheric chemistry is a major factor in determining the atmosphere’s oxidative capacity. Oxidants control the lifetimes of methane (CH4), and thus its efficacy as a greenhouse gas (GHG), and a huge range of reactive gases, including volatile organic compounds (VOCs). Oxidation of VOCs in the presence of nitrogen oxides can produce ozone (O3), another GHG. Unlike CH4, well-mixed in the troposphere, O3 is spatially heterogeneous. O3’s potency as a GHG is much greater in the cold upper troposphere2 and thus dependent on dispersion of O3-precursors. Oxidants also influence aerosol processes, termed aerosol-oxidant coupling, through the oxidation of sulfur dioxide (SO2) to sulfate aerosol and of VOCs to low volatility species which can contribute to secondary organic aerosol (SOA). Aerosols influence climate directly by scattering or absorbing solar radiation and indirectly by affecting cloud properties3. Oxidants control where the key reactions for aerosol production occur and therefore influence the resulting aerosol’s lifetime, effect on cloud properties, and consequently their climatic impact4,5.
Biogenic volatile organic compounds (BVOCs) play a central role in these chemistry-climate interactions by influencing oxidant concentrations, via direct reaction and secondary production from oxidation products6, and providing condensable material for SOA (e.g.,7). However, BVOC emissions (EBVOC) depend strongly on climate themselves, leading to a BVOC-climate feedback (BCF). Determining the sign and magnitude of this feedback is important for predicting future climate change (e.g.,8).
EBVOC, especially isoprene (the most widely emitted BVOC9), are strongly dependent on atmospheric conditions and land use. Rising CO2 inhibits isoprene production10 but also drives increased vegetation-mass via fertilisation11. Higher temperatures also increase emissions of isoprene and monoterpenes12. Perturbations to aerosols and clouds change photosynthetically active radiation (PAR) and precipitation, also influencing emissions13,14. Simulated isoprene emissions exhibit increases from the present day to 2100, albeit with significant variation between both models and future climate scenarios15. Proposed re/afforestation policies would likely drive even greater increases in BVOC emissions.
The climatic impact of BVOCs has been studied with varying degrees of sophistication over the last two decades. Most studies predict that increases to SOA following enhanced EBVOC would cause a negative radiative forcing (RF) (via increased aerosol scattering and cloud albedo (e.g.,16,17)), constituting a negative feedback. When changes to gas phase chemistry are considered, increases to CH4 lifetime and O3 cause a positive RF, although the extent to which this opposes the negative RF from aerosols is uncertain. Ref. 18 found the negative RF from aerosols still outweighed the positive forcing from O3 and CH4 while19, using a different model, found the opposite. AerChemMIP also revealed significant inter-model variation in the response to 2xEBVOC (Fig. S1) with UKESM1 and GISS predicting a positive forcing while GFDL and CESM2 a negative forcing20. The impact of oxidant changes on sulfate aerosol from increased EBVOC has not previously been examined in detail and is a key factor in this work.
Thus, the uncertainty in BVOCs’ climatic impact depends on the uncertainty in multiple chemical and physical processes governing the net radiative forcing. Several studies have investigated how modelling aerosol processes (principally nucleation, condensation and growth) can affect BVOCs’ climatic impact21–23. By contrast, there has been no rigorous assessment of the influence the description of BVOCs’ chemistry, and the effects to oxidants, has on the climatic impact of BVOCs despite recent advancements in the understanding of this chemistry24,25. For isoprene this centres on reactions of the peroxy radical formed by reaction with OH (ISOPOO) (Fig. 1). Some ISOPOO isomers can undergo intramolecular hydrogen shifts (H-shifts) which produce species which regenerate OH, termed HOx-recycling. These reactions, along with natural emissions of NOx from soil, increase simulated OH in environments with high isoprene emissions and low anthropogenic and biomass burning emissions of NOx, helping to reconcile the persistent model low biases for OH against observations26,27. We expect the smaller depletion of OH by isoprene with this chemistry to have ramifications for the atmospheric and radiative response to an EBVOC perturbation via changes to CH4, O3, aerosol and cloud properties.
Fig. 1. Mechanistic differences between Strat-Trop (ST) and CRI-Strat 2 (CS2) for the key processes of isoprene oxidation by OH and oxidation of monoterpenes (represented by α-pinene and β-pinene in CS2).

Processes in black are featured in ST and CS2 while processes in red are only in CS2. RO2, RCHO and ROOH refer to peroxy radicals, carbonyl and hydroperoxides respectively.
In this study we assess how the description of BVOC chemistry affects the simulated climatic impact of BVOCs. We compare the change to the atmosphere’s composition and energy balance, specifically the RF, following a doubling of EBVOC in the preindustrial atmosphere (PI) with two chemical mechanisms. We use Strat-Trop (ST)28, the standard mechanism in UKESM1 and practical for long climate studies, and CRI-Strat 2 (CS2)29. CS2 includes a much more comprehensive description of tropospheric chemistry including ISOPOO H-shifts and a more complete treatment of monoterpene oxidation, with both important for oxidant production. These differences are described in Fig. 1 and Methods. Following 2xEBVOC we find the positive RF from changes to O3, CH4 and ACI outweighs the negative RF from aerosol scattering but the net RF is 43% smaller with CS2 due to a smaller depletion in oxidants. This highlights the multiple pathways by which chemistry, oxidants and aerosols interact to affect radiatively-active atmospheric components and thus demonstrates the importance of uncertainty in BVOC chemistry.
Results and discussion
Figure 2 shows the RF, radiative efficiency and feedback factor from changes in O3 (SARFO3), CH4, aerosol scattering (IRFDRE) and the interactions of clouds with radiation, termed the cloud radiative effect (CRE) (Methods). Mechanism acronyms ST or CS2 refer to a particular detail of the mechanism (e.g. the OH + CH4 rate constant in ST). Individual runs are denoted with the mechanism acronym and subscript (e.g. STcon, ST2x for the control run and run with doubled BVOC (2xEBVOC) respectively). STΔ and CS2Δ refer to the change between the control and 2xEBVOC simulations for a given parameter (e.g., the change in O3 in STΔ refers to the change in O3 for ST2x - STcon) (Methods). Both mechanisms simulate a net positive radiative forcing (and therefore a positive feedback), but the forcing in CS2Δ (168 ± 33 mWm−2) is 43% smaller than STΔ (298 ± 37 mWm−2). This is driven by smaller positive forcings from CH4 (−45 mWm−2; −16%) and CRE (−92 mWm−2; −50%) in CS2Δ compared to STΔ which, along with the 8% smaller SARFO3 (−8 mWm−2), outweigh the 7% (16 mWm−2) stronger negative IRFDRE in STΔ. The negative IRFDRE and positive CH4 and SARFO3 forcings following an EBVOC increase are qualitatively in agreement with prior studies (e.g.,19,20), but the positive CRE contrasts with most studies18,23: both simulated negative CRE with increased EBVOC. The key processes controlling these forcings and the factors driving the mechanistic differences are now reviewed.
Fig. 2. Radiative forcing, radiative efficiency (ϕ) and feedback factor (α) from a doubling of BVOC emissions in a pre-industrial (PI) atmosphere.
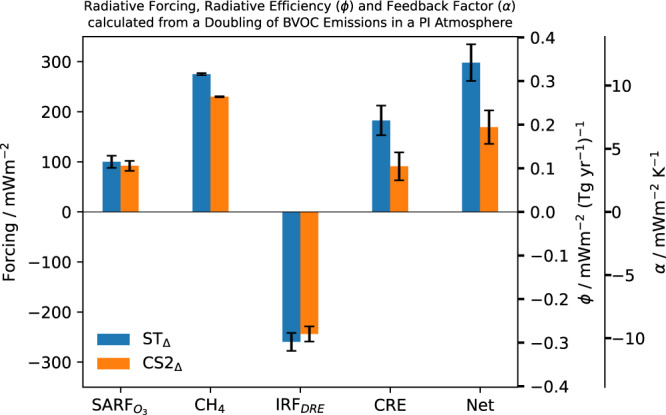
We show the individual forcing components from changes to O3 (SARFO3), CH4, the aerosol direct radiative effect (IRFDRE) and aerosol-cloud interactions (CRE) and their combined totals (Net) for Strat-Trop (STΔ) and CRI-Strat 2 (CS2Δ). The left axis shows the radiative forcing, the inner right axis the radiative efficiency and the outer right axis the feedback factor. Error bars show the standard error.
The hydroxyl radical & methane
The larger positive CH4 forcing in STΔ than CS2Δ can be understood with reference to changes in the OH concentration. 2xEBVOC depletes OH throughout the troposphere in both ST2X and CS22X but the larger relative reduction in STΔ of −31% (cf. −24% in CS2Δ) is one of the fundamental causes of the different climatic responses between the chemical mechanisms. In the lowest 5 km, OH decreases by >65% (>55%) and >50% (>35%) over Amazonia and central Africa respectively in STΔ (CS2Δ), two of the regions with greatest BVOC emissions (Fig. 3a, b). In the lowest ~1 km CS2’s enhanced HOx-recycling from isoprene is particularly influential while in the lower tropical FT (~1–5 km) the mechanistic differences come from a greater increase in OH production from HO2 + NO and hydroperoxide (ROOH) photolysis, primarily coming from the ROOH derived from -pinene and -pinene (omitted in ST), in CS2Δ.
Fig. 3. OH, CH4 oxidation and O3 radiative forcing.

Percentage change in OH in lowest 5 km for (a) Strat-Trop (STΔ) and (b) CRI-Strat 2 (CS2Δ). Zonal mean change in CH4 oxidation flux for (c) STΔ and (d) CS2Δ - STΔ. Forcing from O3 changes (SARFO3) for (e) STΔ and (f) CS2Δ, values in title show global mean forcing.
CS2 produces higher yields of the major hydroperoxide (H2O2) than ST from the ozonolysis of isoprene (38.5% vs. 9%). The consideration of monoterpene chemistry in CS2, in contrast to ST, also leads to higher production of H2O2 (18% direct yield vs. zero in ST) as well as HO2-precursors (e.g., HCHO). Thus, 2xEBVOC produces a greater increase in H2O2 and HO2 in CS2Δ than STΔ, driving a greater increase in secondary OH production in the lower FT (Fig. S2a, b).
As the major tropospheric sink for CH4, the decrease in OH following 2xEBVOC leads to reductions in CH4 oxidation and increases in simulated CH4 concentration. The reduction in oxidation flux is greatest in the warm tropical lower troposphere (Fig. 3c) given the large OH reduction and strong positive temperature dependence of OH + CH4. The larger reduction of OH in STΔ leads to a larger decrease in CH4 oxidation flux (Fig. 3d), corresponding to larger increases in CH4 concentration (STΔ 276 ppbv vs. CS2Δ 223 ppbv) and forcing (STΔ 275 mWm−2 vs. CS2Δ 230 mWm−2) (Methods).
Ozone
The forcing from O3 changes is dictated by the partitioning of nitrogen between reactive NOx and reservoir species (predominantly peroxyacetylnitrate (PAN) and nitric acid (HONO2)), the availability of peroxy radical (RO2) precursors and the location of O3 production: the radiative efficiency of O3 (forcing per unit change in concentration) is greater around the tropical tropopause than in the lower troposphere2.
2xEBVOC reduces PBL O3 over the major biogenic emission regions via O3’s direct reaction with BVOCs. PAN formation also increases but mechanistic differences mean PAN has a ~35% longer lifetime in the warm PBL in ST than CS2. This leads to greater vertical transport of PAN into the FT where the lower temperature increases PAN’s lifetime (from ~1 h in the PBL to ~2 days in the FT).
The increase in PAN in the middle troposphere in both mechanisms leads to lower NOx throughout the region and a reduction in O3, greater in STΔ. However, around the tropical tropopause, increases in HO2, driven by the photolysis of carbonyls (RCHO) such as HCHO produced from BVOC oxidation products, result in increased O3 via the reaction of HO2 + NO and subsequent NO2 photolysis. The increase in HO2, and thus O3, is greater in STΔ since the greater reduction of OH leads to greater vertical transport of these HO2 precursors, allowing them to reach the region with maximum O3 radiative efficiency. By contrast, HO2 production from carbonyl photolysis increases by more in CS2Δ in the lower and middle tropical troposphere where O3’s radiative efficiency is lower (Fig. 3e, f).
The result is an 8% smaller forcing in CS2Δ (92 ± 9 mWm−2) compared to STΔ (100 ± 10 mWm−2) despite CS2Δ producing a 20% greater increase in tropospheric O3 burden; highlighting the influence of O3 precursor-transport and thus oxidant concentrations.
Aerosol scattering (IRFDRE)
The increase in EBVOC not only increases the fuel for SOA production (and thus burden), but also, via oxidant depletion, influences the location of SOA production. The reduction in OH (Fig. 3a, b) increases BVOC lifetimes meaning SOA-precursors are formed at higher altitude, further from EBVOC sources, and the resulting SOA has a longer lifetime and greater climatic impact. The greater OH reduction in STΔ yields greater increases in isoprene lifetime (8.2 h (66%) vs. 3.7 h (61%)) and thus transport away from source than in CS2Δ. Accordingly, SOA burden (lifetime) increases by 121% (12%) in STΔ compared to 114% (7%) in CS2Δ. The greater vertical transport of SOA-precursors in STΔ also means SOA concentrations increases by more in the FT in STΔ and in the PBL for CS2Δ (Fig. 4a) while column SOA increases are greater over EBVOC source regions in CS2Δ and over more remote regions, particularly the central Atlantic, in STΔ (Fig. 4b).
Fig. 4. Secondary organic aerosol (SOA) changes, aerosol scattering forcing, cloud droplet number concentration changes and associated cloud forcing.

Difference in (a) zonal mean SOA increase and (b) accumulation mode SOA column increase between CRI-Strat 2 (CS2Δ) and Strat-Trop (STΔ). Radiative forcing from the change in the aerosol direct radiative effect (IRFDRE) for (c) STΔ and (d) CS2Δ. Percentage change in vertically averaged cloud droplet number concentration (CDNC) concentration (e) STΔ and (f) CS2Δ and radiative forcing due to change in CDNC (ERFCDNC) for (g) ST∆ and (h) CS2∆ calculated using the offline approach of34 (Methods). Values in titles are global mean forcing (c, d) and CDNC change (e, f). Non-hatched regions in (c–h) show areas where the change is statistically significant (95% confidence).
STΔ and CS2Δ also differ in how the extra SOA-precursors alter the SOA size and number distribution. The greater increase of precursors within the PBL in CS2Δ results in a larger increase in condensation to accumulation mode particles than in STΔ. Conversely, the greater transport of precursors into the FT in STΔ means condensation flux to the Aitken mode increases by a greater extent. In turn this yields a greater increase in accumulation number concentration in STΔ, via growth of Aitken particles to accumulation mode size, over much of Amazonia, central Africa and the central Atlantic.
The differences in SOA dispersion and accumulation mode number concentration between STΔ and CS2Δ have direct consequences for the spatial changes in aerosol scattering and the attendant forcing. The statistically significant (95% confidence) IRFDRE is slightly stronger over Amazonia and central Africa in CS2Δ but noticeably stronger over the central Atlantic in STΔ (Fig. 4c, d), correlating well with the difference in SOA column and aerosol number concentration. The IRFDRE is the single largest forcing component and the greater dispersion of additional SOA in STΔ leads to a 7% stronger forcing (−260 vs. −244 mWm−2). Similarly30, found that following a doubling of SOA, greater transport of SOA in the EC-Earth model, compared to NorESM and ECCHAM, led to a stronger IRFDRE.
Cloud forcing (CRE)
In STΔ and CS2Δ SOA increases drive higher cloud droplet number concentration (CDNC) over Amazonia and over the central Atlantic (Fig. 4e, f) (downwind of central Africa) following the spatial change of SOA accumulation mode aerosol, although much of the CDNC increase is not statistically significant (95% confidence).
However, statistically significant decreases in CDNC occur over large areas of the south Atlantic, south Pacific and Southern Ocean (Fig. 4e, f), regions downwind of the Amazon31 and with high stratocumulus coverage32. This is driven by sulfate aerosol changes, not SOA, and the response in STΔ is much stronger than in CS2Δ (Fig. 4e, f). Co-located increases in cloud droplet effective radius are also simulated and, for a given cloud liquid water content, such changes reduce cloud albedo33. Accordingly, both mechanisms simulate positive global SW CRE (STΔ 222 mWm−2, CS2Δ 137 mWm−2). Offline calculations isolating the impact of CDNC changes (34, Methods) also find reductions in outgoing SW radiative flux (i.e., positive forcings) over the south Atlantic and Southern Ocean (Fig. 4g, h).
The CDNC decreases are driven by reduction in gas phase oxidation of SO2 by OH to form H2SO4 which in turn nucleates new aerosol particles. The suppression of H2SO4 production is greater in STΔ (3.0 vs. 2.0 Tg yr−1) due to the greater reduction in OH. The reduction in H2SO4 production and thus new particle nucleation (25 Gg yr−1 STΔ vs. 14 Gg yr−1 CS2Δ) (Fig. S3) leads to compensatory increases in aqueous phase SO2 oxidation by H2O2, which only adds mass to existing particles predominantly in the accumulation mode (whereas nucleation adds to aerosol mass and number). The increase in aqueous SO2 oxidation is reinforced by the increases in H2O2 from the additional BVOC loading.
The net effect is a shift in the aerosol size distribution to fewer, larger particles. Accordingly, STΔ exhibits a 26% decrease in Aitken mode SO4 burden compared to 21% in CS2Δ, particularly downwind of the major biogenic emission regions (e.g., south Atlantic from Amazonia), and a more widespread decrease in Aitken mode number concentrations. Larger Aitken mode particles can activate to cloud condensation nuclei (CCN) in remote regions30 and so their decrease reduces CDNC concentrations. The LW component of the CRE is small and very similar between the mechanisms but the net CRE of 183 mWm−2 in STΔ compared to 91 mWm−2 in CS2Δ constitutes the largest difference between the mechanisms among the forcing components.
The central role of oxidants
Figure 5 contrasts the feedback loops which arise when model simulations consider the impact of BVOC emissions (a) solely from aerosol changes and (b) when chemistry and oxidants change as well. The latter yields a more complex response and highlights the central role of oxidants in influencing not only the forcing from gas phase composition changes but from aerosol and cloud property changes too.
Fig. 5. Atmospheric composition response and BVOC feedback from an increase in CO2 driving an increase in BVOC emissions (EBVOC).

In (a) only aerosols are considered yielding a negative feedback (adapted from23) whereas in (b) chemistry and oxidants are also allowed to respond, leading to a more complex response. Aerosol optical depth (AOD) is a measure of aerosol scattering while IRFDRE and CRE correspond to the forcing from changes to the aerosol direct radiative effect and aerosol cloud interactions respectively. Dashed lines in (b) show important oxidant-driven responses including reduced OH driving (A) increased secondary organic aerosol (SOA) lifetime and climatic impact, (B) greater vertical transport of O3 precursors and thus O3 forcing (SARFO3), (C) increased CH4 lifetime () and climatic impact and (D, E) reduction in gas phase SO2 oxidation with attendant decreases in H2SO4, new particle formation, cloud condensation nuclei (CCN), cloud droplet number concentration (CDNC) and cloud albedo. The strength of the feedback for each loop is shown by the feedback factor (see Methods) for the Strat-Trop mechanism () and CRI-Strat 2 mechanism () in parentheses.
The most noticeable difference between these paradigms is the sign of the CRE which is negative in (a) but positive in (b). This illustrates the subtle difference between the IRFDRE and CRE from a perturbation to EBVOC with interactive oxidants (as done here) and the IRFDRE and CRE from a perturbation to SOA or EBVOC with prescribed oxidants (e.g.,22,23 respectively). In the latter case, the only way an EBVOC or SOA increase can impact ACI is via changes to SOA which typically results in a negative CRE (although not always, cf. EC-Earth in30) by providing additional condensable mass which grows aerosol particles to sizes where they can act as cloud condensation nuclei and therefore increase CDNC, with a minor contribution from enhanced aerosol nucleation. This tends to make BVOCs appear strong cooling agents. When simulating a doubling of CO2, ref. 23 found the total negative aerosol forcing (direct and CRE) from the accompanying EBVOC increase offset 13% of the positive forcing from CO2. This substantial offsetting arose from a very strong positive dependence of EBVOC on temperature (highest among AerChemMIP models) causing a large increase in SOA which yielded strong negative forcings, particularly from CRE, with no concomitant oxidant-driven forcing from changes to sulfate aerosol, O3 and CH4.
By contrast, the use of interactive oxidants here not only results in radiatively-important changes to O3 and CH4 but also changes in SOA transport (affecting IRFDRE) and significant perturbations to sulfate aerosol via reduction in gas phase SO2 oxidation. This reduces new particle formation and CDNC, yielding a positive CRE which outweighs the impact of increased SOA and leads to the opposite conclusion to23.
The link between oxidants, CDNC and CRE has also been simulated in35 where OH-suppression from increases to CH4 concentration yielded CDNC reductions and a positive CRE. However, the impact of increased H2O2 (substantial from BVOC increases but less so from CH4 increases) favouring aqueous phase SO2 oxidation further highlights the wider range of pathways via which BVOCs can affect climate.
Fully understanding the climatic impact of BVOC emissions requires capturing as many of these oxidant-influenced interactions as possible. This is particularly important in the context of nature-based climate policies since incorrectly diagnosing their effects on climate could lead to implementation of ineffective or even counterproductive policies.
Wider context
Changes to CO2 concentration, climate (temperature, flooding, droughts) and land use policies, including well-intentioned efforts to promote biodiversity and mitigate climate change by increasing CO2 sequestration (via re/afforestation or energy crops), will affect future BVOC emissions in a complex manner. Understanding how these changes will influence climate change is therefore critical for reducing uncertainty in future climate projections and ensuring that such mitigation policies are beneficial and not counterproductive.
While most prior work on the climatic impact of BVOCs has focused on the impact to aerosols and the accompanying uncertainty in BVOC-aerosol parameterisations, this work demonstrates the important coupling between aerosols, chemistry and oxidants. The necessity of using interactive (rather than prescribed) oxidants in the context of the BVOC feedback has already been demonstrated by the radiatively-important changes to O3 and CH4 (e.g.,20). By comparing the response to an EBVOC increase with two interactive chemical mechanisms, this study progresses beyond prior studies by identifying the wider reach of oxidants as they impact not only the forcing from gas phase composition changes but also the forcing from aerosol and cloud property changes; previously overlooked interactions. The strong dependence of the BVOC feedback on oxidants, and therefore the chemical mechanism, demonstrates the importance of accurately representing tropospheric chemistry for determining the influence of BVOCs on climate.
Improving the understanding of the pristine PI atmosphere is important given the large degree of uncertainty in the period and the associated consequences for radiative forcing from the PI to the present day36. The use of the PI highlights the importance of simulated chemistry to understanding this period and its response to perturbations. It also allows this study to serve as a baseline for future work since the critical role of oxidants and sulphate aerosol identified here means the background atmospheric composition, particularly species which affect atmospheric oxidising capacity and background aerosol (e.g., NOx and SOx which are higher in the present day than the PI), will be influential in determining how changes to BVOC emissions will affect O3, CH4, aerosol burdens and CDNC and thus the magnitude of the opposing radiative effects which ultimately determine the climatic impact. Improvements to the description of SOA formation beyond the current fixed yield, condensation-driven approach include the adoption of more realistic processes including dimer formation from terpenes (e.g.,37), the reactive uptake of isoprene epoxy-diols (IEPOX)38 and SOA formation in aqueous aerosol and cloud droplets which is believed to be comparable to gas phase SOA formation in some circumstances (e.g.,39). These updates may alter, to varying extents, the DRE and ACI response to a BVOC emission perturbation, thus warranting further work. The response of the DRE and ACI will be influenced by background atmospheric composition and the requirement for multiple oxidation steps for SOA-precursor formation will alter (and indeed likely accentuate) the effect of oxidants on SOA dispersion and lifetime while the complex role of NOx in IEPOX and dimer formation and the influence of aerosol composition (e.g., acidity) on IEPOX reactive uptake will drive a greater dependence on NOx and SOx and the wider background atmospheric composition.
The wide-ranging influence of oxidants and chemistry identified in this study, and the attendant dependence on atmospheric chemical composition, means a doubling of BVOC emissions in a present-day or future climate is likely to have a different climatic impact to that simulated here. Such experiments would provide further information regarding the sensitivity of BVOC’s climatic impact to background atmospheric conditions and make for interesting follow up studies. When assessing the future climatic impact of a re/afforestation policy the application of the radiative efficiency or feedback factor determined using the doubling of emissions in a PI atmosphere following the CMIP6 convention may not suitable. Instead, contemporaneous background atmospheric composition must be used with the processes highlighted in this study providing a framework for such research.
Doubling BVOC emissions represents a substantial perturbation and extrapolation of this study’s results to different emission scalings (e.g., 50% increase) should be performed with care since different components of the model’s response are likely to scale with emissions with varying degrees of linearity. For example, the current use of a fixed SOA yield means the modelled IRFDRE may scale quite linearly with emissions while the non-linearity of Ox-NOx-VOC chemistry (e.g.,40) means changes to OH, and thus to CH4 forcing, are likely to be less linear. The complexity of the interactions and role of background atmospheric composition mean the extent of linearity can only truly be determined by further experiments.
Increasing emissions of BVOCs leads to a cascade of chemical and climatic impacts in the Earth system by driving complex changes in the distribution of oxidants with concomitant effects on the burden and lifetime of radiatively important gases, aerosols and cloud properties. Overall, we find, in a PI climate, a doubling of EBVOC in UKESM1 leads to increases in O3 and CH4 and decreases to CDNC/cloud albedo through a reduction in gas phase SO2 oxidation. In ST, the combined positive forcing from these changes outweighs the negative forcing arising from the scattering of radiation from enhanced SOA, yielding a positive feedback. However, when a state-of-the-science chemistry scheme (CS2), featuring recent developments in isoprene chemistry, is used the net positive feedback is 43% smaller. The central driver of this difference is a smaller reduction in oxidants and attendant smaller increases in CH4 and smaller decreases in gas phase SO2 oxidation, CDNC and cloud albedo. The smaller oxidant depletion also limits the transport of O3-precursors up to the upper-troposphere, where O3 is most potent as a GHG, yielding a smaller positive forcing despite a greater increase in tropospheric O3 burden. The wide-scale transport of SOA from the enhanced EBVOC is lower in CS2 following the lower oxidant depletion, yielding a smaller negative aerosol and cloud forcing, but this effect is outweighed by the diminished positive forcings from CH4, cloud albedo and O3. Thus, we demonstrate the important coupling between aerosols, chemistry, and oxidants in determining the climatic impact of BVOC emissions.
Methods
Model runs
All model runs were performed for 45 years (15 years spin up, 30 years analysis) with pre-industrial timeslice conditions using the UKESM1-AMIP setup at a horizontal resolution of 1.25° × 1.875° with 85 vertical levels up to 85 km41. All simulations had fully interactive stratospheric and tropospheric chemistry, including interactive oxidants, using either the Strat-Trop (ST) mechanism28 or the CRI-STRAT 2 (CS2) mechanism29. The simulations used the GLOMAP‐mode aerosol scheme which simulates sulfate (SO4), sea‐salt (SS), black carbon (BC), primary organic aerosol (POA), secondary organic aerosol (SOA) and dust but not nitrate aerosol42,43. In this setup, the model tracks the mass concentration of each mode present in each component (e.g. SO4 nucleation mode) and the total particle number concentration for the nucleation, Aitken (soluble and insoluble), accumulation and coarse modes.
Emissions of well‐mixed greenhouse gases (WMGHGs), such as methane (CH4) and CO2, were not simulated; rather, prescribed lower boundary conditions at PI levels were applied for CO2 (284 ppm), CH4 (808 ppb) and N2O (273 ppb), consistent with control runs of UKESM1’s contributions to AerChemMIP44.
The setup of these runs followed the AerChemMIP protocol44 to allow calculation of the ERF. Fields for SSTs, SI, ocean biogeochemistry (DMS and chlorophyll) and land cover were taken from monthly mean climatologies derived from 30 years of output of the UKESM1 fully-coupled pre-industrial control experiment (piControl) discussed in45. Timeslice PI anthropogenic and biomass burning emissions were taken from the CEDS dataset46,47 respectively. While the atmosphere-only setup with fixed SSTs does constrain the wider Earth system response (for example aerosol-driven changes to PAR cannot change land cover via fertilisation of additional vegetation), it does reduce the noise which would occur with a coupled ocean. Importantly it also allows this study’s results to be directly comparable to other studies such as the emission perturbation runs in AerChemMIP. The use of ERF, as opposed to other definitions of radiative forcing such as instantaneous radiative forcing, allows the inclusion of stratospheric temperature adjustments but also rapid adjustments in the troposphere including temperature, water vapour, clouds, and land surface temperature35.
All terrestrial biogenic emissions, except isoprene and MT, were based on 2001–2010 climatologies from Model of Emissions of Gases and Aerosols from Nature under the Monitoring Atmospheric Composition and Climate project (MEGAN-MACC) version 2.148. Oceanic emissions were from the POET 1990 dataset49. Oceanic DMS emissions were calculated from seawater DMS concentrations45 which were prescribed from the fully coupled UKESM1 PI control run.
As in the UKESM1 runs for AerChemMIP, isoprene and MT emissions were calculated using the iBVOC emissions system50 which calculates the emissions interactively based on temperature, CO2, plant functional type and photosynthetic activity. The use of iBVOC allows for a more faithful estimate of pre-industrial emissions of biogenic species compared to using present-day emissions inventories such as MEGAN-MACC9 since iBVOC considers the PI land use and atmospheric conditions such as lower CO2. In the CS2 runs, the MT emissions calculated by the iBVOC system were split into α-pinene and β-pinene in a 2:1 ratio as in previous studies using the CRI mechanisms29,51.
Chemical mechanisms
The scale of tropospheric chemistry (~19,000 reactions for organic species alone in the near-explicit Master Chemical Mechanism (MCM)52); prevents explicit simulation and necessitates the use of condensed mechanisms which reduce complexity by lumping chemical species together and considering only the most important reactions.
The Strat-Trop (ST) and CRI-Strat 2 (CS2) chemical mechanisms are described in detail in28,29 respectively with a full description of every tropospheric chemistry reaction in CS2 also available at http://cri.york.ac.uk/home.htt (last accessed 5th June 2022). ST considers 73 species and 305 reactions while CS2 has 228 species and 766 reactions with the bulk of the added complexity coming from a wider range of organic species (Tables 2 and S0151). ST does not feature the CS2 species C2H2, C2H4, C3H6, C2H5OH, C2H5CHO and methyl ethyl ketone but does add their emissions to species it does consider (e.g., emissions of C2H4 are included in C2H6 in ST). Some species are omitted entirely by ST and are only included in CS2. These are butane, butene, benzene, toluene, oxylene, formic acid and ethanoic acid (Table 351).
CS2 is based on the tropospheric chemistry scheme CRI v2.253 which is traceable to the latest version of the MCM (v3.3.1) and conserves its ozone forming potential.
As illustrated in Fig. 1, a major difference between the mechanisms is the inclusion of the H-shift pathways of ISOPOO (C5H9O3). ST features isoprene chemistry from54 where ISOPOO forms the isoprene hydroperoxide (ISOPOOH) via reaction with HO2 and methacrolein the major product from reaction of ISOPOO with NO, NO3 and other peroxy radicals (RO2). By contrast, CS2 also features the 1,4- and 1,6-H-shift reactions of ISOPOO. CS2 also simulates organonitrate formation from a wide range of RO2 whereas ST uses the methyl nitrate (CH3ONO2), and isoprene nitrate (C5H9NO3) and nitrooxy aldehyde (C2H3NO4) to represent all organonitrates. CS2 simulates 50–100% higher OH concentration in terrestrial tropical regions than ST, improving model performance for OH, isoprene and monoterpenes29,53. CS2 is comparable to other more advanced chemical mechanisms such as the CalTech reduced isoprene scheme55 but the effect of this chemistry on the climatic impact of BVOCs has not been assessed. Isoprene oxidation also produces the chemically-inert species, Sec_OrgISOP (Fig. 1), which condenses onto aerosol.
For monoterpenes ST features a single tracer (MT) whose oxidation by O3, OH and NO3 produces only a chemically-inert species, Sec_OrgMT (Fig. 1), which condenses onto aerosol or nucleates new aerosol with sulfuric acid. This lack of further chemistry means MT only acts as an oxidant sink rather than behaving as reactive organic carbon (ROC)56. In CS2, monoterpene chemistry features oxidation of α-pinene and β-pinene (a sink of oxidants) which produces both Sec_OrgMT and other chemically active products. These oxidation products undergo further chemical reactions57 (Fig. 1). The transport of these oxidation products can lead to the regeneration of O3 and OH away from emission sources, offsetting some of the oxidant depletion from initial oxidation of the monoterpenes, with associated effects on CH4 and aerosol.
In the PI, CS2 simulates an extra ~5 TgC yr−1 of ROC emissions than ST due to the wider range of emitted VOCs considered by CS251. In addition, CS2 features an extra ~120 TgC yr−1 of reactive organic carbon produced in the atmosphere in the form of 1st generation oxidation products from monoterpenes since monoterpene oxidation in ST does not produce any chemically-active species (Fig. 1). Prior mechanistic analysis has identified this additional ROC to lead to lower surface OH but greater OH in the tropical lower free troposphere (FT)51.
UKESM performance using ST and CS2 was evaluated against present day observational data of BVOCs and other important chemical species from surface sites, flight campaigns and satellites with a full description in29. Relative to ST, CS2 reduced the model’s high isoprene and monoterpene bias at the surface by increasing the local OH concentration. CS2 also yielded substantial improvements in isoprene column over Amazonia, Africa and southeast Asia.
The rate constant for the reaction of MT + NO3 in ST was corrected from the erroneously high expression of 1.19 × 10−12e925/T to 1.19 × 10−12e490/T, bringing it into line with the IUPAC preferred value (https://iupac-aeris.ipsl.fr/htdocs/datasheets/pdf/NO3_VOC9_NO3_apinene.pdf, last accessed 14th September 2021) for α-pinene on which the ST tracer MT is based. This results in a reduction in the rate constant of ~80%, but as NO3 is a minor sink for monoterpenes, this change does not have a huge impact on aerosol formation.
SOA scheme improvements
The UKESM1 contributions to AerChemMIP (which also used the Strat-Trop chemical mechanism) simulated SOA production only from monoterpene oxidation with a doubled molar yield of 26% (28.6% mass yield) to account for the lack of SOA production principally from isoprene but also other VOCs43. However, as a greater fraction of monoterpenes are produced in high latitude forests compared to isoprene9, this approach skewed SOA production to higher latitudes with implications for SOA lifetime and climatic impact. Nucleation of new particles from the clustering of oxidised organic species and sulfuric acid was also omitted in the UKESM1 simulations for AerChemMIP.
In this current study, the description of SOA formation was improved from that used by UKESM in AerChemMIP to include SOA production from isoprene as well as monoterpenes and aerosol nucleation in the boundary layer from Sec_OrgMT and H2SO4. Inert SOA-precursors were produced from monoterpenes (Sec_OrgMT) at the original molar yield of 13% (14.3% mass yield) (Eq. (1)) and isoprene (Sec_OrgIsop 3% molar yield; 3.3% mass yield) (Eq. (2)). SOA-precursors from both monoterpenes and isoprene could condense onto existing aerosol while nucleation of new particles via the clustering of H2SO4 and Sec_OrgMT was also simulated following the scheme of58 (Eq. (3)) but constrained to the model boundary layer. The inclusion of isoprene SOA and boundary layer nucleation (BLN) represent improvements over the standard UKESM1 model setup used for AerChemMIP (e.g.,20). The formation of SOA was the same in ST and CS2.
| 1 |
| 2 |
| 3 |
The change in SOA precursor yields leads to total organic aerosol (primary + secondary) burdens which are 9% and 17% higher in the STcon and ST2x simulations in this study compared to the corresponding PI control and 2xEBVOC UKESM1 simulations in AerChemMIP.
Forcing definitions
For each mechanism pair, the ERF is defined as the difference in TOA net radiative flux (Eq. (4))
| 4 |
Following the approach of35,59, the ERF can be decomposed in aerosol direct radiative effects () (Eq. (5)), aerosol‐cloud effects (CRE) (Eq. (6)), and clear‐sky effects (CS) (Eq. (7)).
| 5 |
| 6 |
| 7 |
is the net flux excluding scattering and absorption by aerosols, and, is the flux excluding scattering and absorption by aerosols and clouds. Thus, the IRFDRE corresponds to the difference in net TOA radiative flux due solely to the scattering and absorption of aerosols (changes to land surface albedo are negligible due to prescribed land use) while the CRE reflects changes to cloud forcing via aerosol indirect effects. The clear sky forcing corresponds to change due to the absorption and emission of radiation by gas phase species.
The prescribed surface concentration of CH4 in the model setup significantly constrains the response of CH4 concentrations to oxidant perturbations and thus the radiative effect. However, the change in CH4 concentration which would have occurred had surface CH4 concentration not been constrained can be diagnosed (Eq. (8)).
| 8 |
Where is the CH4 concentration, is the methane lifetime and is the feedback of methane on its own lifetime60 taken as 1.28 for the pre-industrial period35. The forcing due to the change in CH4 concentration was then calculated using the approach in61 using the baseline concentrations of CH4 and N2O of 808 ppb and 273 ppb respectively. Following20, this forcing was then scaled by 1.52 to account for the additional chemical production of ozone and stratospheric water vapour.
Unlike methane, O3 concentrations can respond to changes in EBVOC, and the resulting forcing is included in the clear sky forcing component, . The forcing from ozone changes was isolated using the radiative kernel from62 as in20 which yielded the stratospheric-temperature adjusted radiative forcing (SARFO3).
Offline CDNC forcing calculation
Offline radiative flux calculations were performed to calculate the forcing due to changes in CDNC alone (ERFCDNC). Monthly mean values were used for all variables for these calculations. This followed the technique described in34,63 for TOA fluxes and used CDNC, total cloud fraction (fc, calculated using maximum random overlap), in-cloud (as opposed to all-sky) liquid water path (LWPic), SW clear-sky upwelling flux at TOA (), SW downwelling flux at TOA () and the surface albedo (Asurf) as inputs. The approach used here differs slightly to those studies due to the inclusion here of from the model for the clear-sky regions rather than assuming a constant transmissivity. A transmissivity of 0.89 was use above cloud. Multiple scattering between the surface and cloud was also included here following64. Asurf was calculated by dividing the upwelling clear-sky SW surface fluxes by the corresponding downwelling fluxes. LWPin-cloud is the LWP from the cloudy regions only and was calculated by dividing the all-sky LWP data (as output by the model) by fc (e.g., as in65).
ERFCDNC was calculated firstly by using the control (con) values as a baseline for the SW TOA flux (FSW) calculation and then calculating the difference between this and an Fsw value calculated using the 2× BVOC (2×) values for CDNC and control values for everything else (Eq. (9)).
| 9 |
Then the 2xBVOC run was used as a baseline and the CDNC from the control substituted in Eq. (10).
| 10 |
An overall value for ERFCDNC was calculated as the average of ERFCDNC,con base and ERFCDNC, 2x base.
Feedback factor
For a given forcing , the resultant change to TOA radiative imbalance, , can be expressed by where is the climate feedback parameter and represents the rate of change of the TOA radiative imbalance with respect to the global mean change in surface temperature, . can be decomposed into individual feedback terms, , arising from changes to different climate variables, (Eq. (11)).
| 11 |
In this study the climate variable of interest is . The corresponding feedback factor, , can be considered as the forcing arising from the change in EBVOC in response to a temperature change (Eq. 12).
| 12 |
Where is the radiative efficiency per unit change in emissions (i.e., the change in TOA radiative imbalance per unit change in emissions with typical units of Wm−2 (Tg yr−1)−1) and is the change in EBVOC with climate (Tg yr−1 K−1).
is calculated by dividing the radiative forcing diagnosed from the timeslice model simulation pairs (STcon & ST2x, CS2con & CS22x) by the change in emissions.
is diagnosed from a pair of timeslice model simulations: the piControl which simulates an 1850s atmosphere and the abrupt-4xCO2 which is initialised from the piControl before atmospheric CO2 concentrations are instantly quadrupled. These simulations were run for 150 years as part of the AerChemMIP project44 and the changes in temperature and emissions were calculated from the mean of years 121–150. The change in EBVOC per unit temperature change was then calculated.
Nomenclature
The terms used to represent the response of an atmospheric parameter for a given mechanism are defined in Eqs. (13) and (14) while Eq. (15) shows the difference between two responses.
| 13 |
| 14 |
| 15 |
Supplementary information
Acknowledgements
Vice-Chancellor’s Award from the Cambridge Trust (J.W.). NERC through the University of Cambridge ESS-DT (Y.M.S.). NERC PROMOTE (grant no. NE/P016383/1) (S.A.N., A.T.A.). Support from NERC and NCAS through the ACSIS project (N.L.A. and A.T.A.). National Environmental Research Council (NERC) national capability grant for The North Atlantic Climate System Integrated Study (ACSIS) program (grant NE/N018001/1) via NCAS, and by the ADVANCE (Aerosol-cloud-climate interactions derived from Degassing VolcANiC Eruptions; NE/T006897/1) program (D.P.G.). This work used Monsoon2, a collaborative high-performance computing facility funded by the Met Office and the Natural Environment Research Council. This work used JASMIN, the UK collaborative data analysis facility.
Author contributions
J.W. set up and executed the simulations with help from N.L.A., S.A.N., Y.M.S. and C.E.S. J.W. analysed the output with input from A.T.A., S.A.N., Y.M.S., P.T.G. and D.P.G. D.P.G. performed the offline CDNC forcing calculations.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Data availability
The UKESM1 data generated in this study have been deposited in the University of Cambridge Apollo database 10.17863/CAM.83526. The data are available for all users.
Code availability
Due to intellectual property right restrictions, we cannot provide either the source code or documentation papers for the UM. The Met Office United Model is available for use under licence. A number of research organisations and national meteorological services use the UM in collaboration with the UK Met Office to undertake atmospheric process research, produce forecasts, develop the UM code, and build and evaluate Earth system models. For further information on how to apply for a licence, see https://www.metoffice.gov.uk/research/approach/modelling-systems/unified-model (last access: 18 July 2022).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-34944-9.
References
- 1.Naik, V. et al. Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change. Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R. & Zhou, B. (eds.)]. (Cambridge University Press, 2021).
- 2.Lacis AA, Wuebbles DJ, Logan JA. Radiative forcing of climate by changes in the vertical distribution of ozone. J. Geophys. Res.: Atmospheres. 1990;95:9971–9981. doi: 10.1029/JD095iD07p09971. [DOI] [Google Scholar]
- 3.Boucher, O. et al. Clouds and Aerosols. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. [Stocker, T. F., Qin, D., Plattner, G. -K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V. & Midgley, P. M. (eds.)]. (Cambridge University Press, 2013) pp. 571–658.
- 4.Karset IH, et al. Strong impacts on aerosol indirect effects from historical oxidant changes. Atmos. Chem. Phys. 2018;18:7669–7690. doi: 10.5194/acp-18-7669-2018. [DOI] [Google Scholar]
- 5.Tilmes S, et al. Climate forcing and trends of organic aerosols in the Community Earth System Model (CESM2) J. Adv. Modeling Earth Syst. 2019;11:4323–4351. doi: 10.1029/2019MS001827. [DOI] [Google Scholar]
- 6.Archibald, A. T. et al. Impacts of HOx regeneration and recycling in the oxidation of isoprene: Consequences for the composition of past, present and future atmospheres. Geophys. Res. Lett.38, (2011).
- 7.Shrivastava M, et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophysics. 2017;55:509–559. doi: 10.1002/2016RG000540. [DOI] [Google Scholar]
- 8.Forster, P. et al. Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change. [Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R. & Zhou, B. (eds.)]. (Cambridge University Press, 2013).
- 9.Sindelarova K, et al. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years. Atmos. Chem. Phys. 2014;14:9317–9341. doi: 10.5194/acp-14-9317-2014. [DOI] [Google Scholar]
- 10.Yáñez-Serrano AM, et al. Dynamics of volatile organic compounds in a western Mediterranean oak forest. Atmos. Environ. 2021;257:118447. doi: 10.1016/j.atmosenv.2021.118447. [DOI] [Google Scholar]
- 11.Zhu Z, et al. Greening of the Earth and its drivers. Nat. Clim. Change. 2016;6:791–795. doi: 10.1038/nclimate3004. [DOI] [Google Scholar]
- 12.Fares, S., Mahmood, T., Liu, S., Loreto, F. & Centritto, M. Influence of growth temperature and measuring temperature on isoprene emission diffusive limitations of photosynthesis and respiration in hybrid poplars. Atmos. Environ.45, 155–161 (2011).
- 13.van Meeningen Y, et al. Isoprenoid emission variation of Norway spruce across a European latitudinal transect. Atmos. Environ. 2017;170:45–57. doi: 10.1016/j.atmosenv.2017.09.045. [DOI] [Google Scholar]
- 14.Rap A, et al. Enhanced global primary production by biogenic aerosol via diffuse radiation fertilization. Nat. Geosci. 2018;11:640–644. doi: 10.1038/s41561-018-0208-3. [DOI] [Google Scholar]
- 15.Cao Y, et al. Ensemble projection of global isoprene emissions by the end of 21st century using CMIP6 models. Atmos. Environ. 2021;267:118766. doi: 10.1016/j.atmosenv.2021.118766. [DOI] [Google Scholar]
- 16.Carslaw KS, et al. A review of natural aerosol interactions and feedbacks within the Earth system. Atmos. Chem. Phys. 2010;10:1701–1737. doi: 10.5194/acp-10-1701-2010. [DOI] [Google Scholar]
- 17.Scott CE, et al. Substantial large-scale feedbacks between natural aerosols and climate. Nat. Geosci. 2018;11:44–48. doi: 10.1038/s41561-017-0020-5. [DOI] [Google Scholar]
- 18.Scott CE, et al. Impact on short-lived climate forcers increases projected warming due to deforestation. Nat. Commun. 2018;9:1–9. doi: 10.1038/s41467-017-02412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Unger N. On the role of plant volatiles in anthropogenic global climate change. Geophys. Res. Lett. 2014;41:8563–8569. doi: 10.1002/2014GL061616. [DOI] [Google Scholar]
- 20.Thornhill G, et al. Climate-driven chemistry and aerosol feedbacks in CMIP6 Earth system models. Atmos. Chem. Phys. 2021;21:1105–1126. doi: 10.5194/acp-21-1105-2021. [DOI] [Google Scholar]
- 21.Makkonen R, et al. BVOC-aerosol-climate interactions in the global aerosol-climate model ECHAM5. 5-HAM2. Atmos. Chem. Phys. 2012;12:10077–10096. doi: 10.5194/acp-12-10077-2012. [DOI] [Google Scholar]
- 22.Scott CE, et al. The direct and indirect radiative effects of biogenic secondary organic aerosol. Atmos. Chem. Phys. 2014;14:447–470. doi: 10.5194/acp-14-447-2014. [DOI] [Google Scholar]
- 23.Sporre MK, et al. BVOC–aerosol–climate feedbacks investigated using NorESM. Atmos. Chem. Phys. 2019;19:4763–4782. doi: 10.5194/acp-19-4763-2019. [DOI] [Google Scholar]
- 24.Peeters J, et al. HOx radical regeneration in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2009;11:5935–5939. doi: 10.1039/b908511d. [DOI] [PubMed] [Google Scholar]
- 25.Wennberg PO, et al. Gas-phase reactions of isoprene and its major oxidation products. Chem. Rev. 2018;118:3337–3390. doi: 10.1021/acs.chemrev.7b00439. [DOI] [PubMed] [Google Scholar]
- 26.Khan MAH, et al. Changes to simulated global atmospheric composition resulting from recent revisions to isoprene oxidation chemistry. Atmos. Environ. 2021;244:117914. doi: 10.1016/j.atmosenv.2020.117914. [DOI] [Google Scholar]
- 27.Shrivastava M, et al. Urban pollution greatly enhances formation of natural aerosols over the Amazon rainforest. Nat. Commun. 2019;10:1–12. doi: 10.1038/s41467-019-08909-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Archibald AT, et al. Description and evaluation of the UKCA stratosphere–troposphere chemistry scheme (StratTrop vn 1.0) implemented in UKESM1. Geoscientific Model Dev. 2020;13:1223–1266. doi: 10.5194/gmd-13-1223-2020. [DOI] [Google Scholar]
- 29.Weber J, et al. Improvements to the representation of BVOC chemistry–climate interactions in UKCA (v11. 5) with the CRI-Strat 2 mechanism: incorporation and evaluation. Geoscientific Model Dev. 2021;14:5239–5268. doi: 10.5194/gmd-14-5239-2021. [DOI] [Google Scholar]
- 30.Sporre MK, et al. Large difference in aerosol radiative effects from BVOC-SOA treatment in three Earth system models. Atmos. Chem. Phys. 2020;20:8953–8973. doi: 10.5194/acp-20-8953-2020. [DOI] [Google Scholar]
- 31.Putnam, W. & Sokolowsky, E. NASA’s Goddard Space Flight Center/Global Modeling and Assimilation Office. https://svs.gsfc.nasa.gov/30637, accessed 18th March 2022 (2014).
- 32.Wood R. Stratocumulus clouds. Monthly Weather Rev. 2012;140:2373–2423. doi: 10.1175/MWR-D-11-00121.1. [DOI] [Google Scholar]
- 33.Twomey, S. Pollution and the planetary albedo. Atmos. Environ. (1967)8, 1251–1256 (1974).
- 34.Grosvenor DP, Carslaw KS. The decomposition of cloud–aerosol forcing in the UK Earth System Model (UKESM1) Atmos. Chem. Phys. 2020;20:15681–15724. doi: 10.5194/acp-20-15681-2020. [DOI] [Google Scholar]
- 35.O’Connor FM, et al. Assessment of pre-industrial to present-day anthropogenic climate forcing in UKESM1. Atmos. Chem. Phys. 2021;21:1211–1243. doi: 10.5194/acp-21-1211-2021. [DOI] [Google Scholar]
- 36.Carslaw KS, et al. Large contribution of natural aerosols to uncertainty in indirect forcing. Nature. 2013;503:67–71. doi: 10.1038/nature12674. [DOI] [PubMed] [Google Scholar]
- 37.Weber JM, et al. CRI-HOM: A novel chemical mechanism for simulating highly oxygenated organic molecules (HOMs) in global chemistry–aerosol–climate models. Atmos. Chem. Phys. 2020;20:10889–10910. doi: 10.5194/acp-20-10889-2020. [DOI] [Google Scholar]
- 38.Gaston CL, et al. Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles. Environ. Sci. Technol. 2014;48:11178–11186. doi: 10.1021/es5034266. [DOI] [PubMed] [Google Scholar]
- 39.Ervens B, et al. Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies. Atmos. Chem. Phys. 2011;11:11069–11102. doi: 10.5194/acp-11-11069-2011. [DOI] [Google Scholar]
- 40.Jenkin ME, Clemitshaw KC. Ozone and other secondary photochemical pollutants: chemical processes governing their formation in the planetary boundary layer. Atmos. Environ. 2000;34:2499–2527. doi: 10.1016/S1352-2310(99)00478-1. [DOI] [Google Scholar]
- 41.Walters D, et al. The Met Office Unified Model global atmosphere 7.0/7.1 and JULES global land 7.0 configurations. Geoscientific Model Dev. 2019;12:1909–1963. doi: 10.5194/gmd-12-1909-2019. [DOI] [Google Scholar]
- 42.Mann GW, et al. Description and evaluation of GLOMAP-mode: A modal global aerosol microphysics model for the UKCA composition-climate model. Geoscientific Model Dev. 2010;3:519–551. doi: 10.5194/gmd-3-519-2010. [DOI] [Google Scholar]
- 43.Mulcahy JP, et al. Description and evaluation of aerosol in UKESM1 and HadGEM3-GC3. 1 CMIP6 historical simulations. Geoscientific Model Dev. 2020;13:6383–6423. doi: 10.5194/gmd-13-6383-2020. [DOI] [Google Scholar]
- 44.Collins WJ, et al. AerChemMIP: quantifying the effects of chemistry and aerosols in CMIP6. Geoscientific Model Dev. 2017;10:585–607. doi: 10.5194/gmd-10-585-2017. [DOI] [Google Scholar]
- 45.Sellar AA, et al. UKESM1: Description and evaluation of the UK Earth System Model. J. Adv. Modeling Earth Syst. 2019;11:4513–4558. doi: 10.1029/2019MS001739. [DOI] [Google Scholar]
- 46.Hoesly RM, et al. Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS) Geoscientific Model Dev. 2018;11:369–408. doi: 10.5194/gmd-11-369-2018. [DOI] [Google Scholar]
- 47.Van Marle M, et al. Historic global biomass burning emissions for CMIP6 (BB4CMIP) based on merging satellite observations with proxies and fire models (1750–2015) Geoscientific Model Dev. 2017;10:3329–3357. doi: 10.5194/gmd-10-3329-2017. [DOI] [Google Scholar]
- 48.Guenther AB, et al. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2. 1): an extended and updated framework for modeling biogenic emissions. Geoscientific Model Dev. 2012;5:1471–1492. doi: 10.5194/gmd-5-1471-2012. [DOI] [Google Scholar]
- 49.Olivier, J. G. J. et al: Present and future surface emissions of atmospheric compounds, POET Report #3, EU project EVK2-1999-00011, available at: http://www.aero.jussieu.fr/projet/ACCENT/Documents/del2_final.doc (2003)
- 50.Pacifico F, et al. Evaluation of a photosynthesis-based biogenic isoprene emission scheme in JULES and simulation of isoprene emissions under present-day climate conditions. Atmos. Chem. Phys. 2011;11:4371–4389. doi: 10.5194/acp-11-4371-2011. [DOI] [Google Scholar]
- 51.Archer‐Nicholls S, et al. The Common Representative Intermediates Mechanism version 2 in the United Kingdom Chemistry and Aerosols Model. J. Adv. Modeling Earth Syst. 2021;13:e2020MS002420. [Google Scholar]
- 52.Jenkin ME, Young JC, Rickard AR. The MCM v3. 3.1 degradation scheme for isoprene. Atmos. Chem. Phys. 2015;15:11433–11459. doi: 10.5194/acp-15-11433-2015. [DOI] [Google Scholar]
- 53.Jenkin ME, et al. The CRI v2. 2 reduced degradation scheme for isoprene. Atmos. Environ. 2019;212:172–182. doi: 10.1016/j.atmosenv.2019.05.055. [DOI] [Google Scholar]
- 54.Pöschl U, et al. Development and intercomparison of condensed isoprene oxidation mechanisms for global atmospheric modeling. J. Atmos. Chemistry37. 2000;1:29–52. doi: 10.1023/A:1006391009798. [DOI] [Google Scholar]
- 55.Bates KH, Jacob DJ. A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol. Atmos. Chem. Phys. 2019;19:9613–9640. doi: 10.5194/acp-19-9613-2019. [DOI] [Google Scholar]
- 56.Heald CL, Kroll JH. The fuel of atmospheric chemistry: Toward a complete description of reactive organic carbon. Sci. Adv. 2020;6:eaay8967. doi: 10.1126/sciadv.aay8967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jenkin ME. Modelling the formation and composition of secondary organic aerosol from α- and β-pinene ozonolysis using MCM v3. Atmos. Chem. Phys. 2004;4:1741–1757. doi: 10.5194/acp-4-1741-2004. [DOI] [Google Scholar]
- 58.Metzger S, et al. Gas/aerosol partitioning: 1. A computationally efficient model. J. Geophys. Res.: Atmospheres. 2002;107:ACH-16. [Google Scholar]
- 59.Ghan SJ. Estimating aerosol effects on cloud radiative forcing. Atmos. Chem. Phys. 2013;13:9971–9974. doi: 10.5194/acp-13-9971-2013. [DOI] [Google Scholar]
- 60.Fiore, A. M. et al. Multimodel estimates of intercontinental source‐receptor relationships for ozone pollution. J. Geophys. Res.: Atmos. 114.D4 (2009).
- 61.Etminan M, et al. Radiative forcing of carbon dioxide, methane, and nitrous oxide: a significant revision of the methane radiative forcing. Geophys. Res. Lett. 2016;43:12–614. doi: 10.1002/2016GL071930. [DOI] [Google Scholar]
- 62.Skeie RB, et al. Historical total ozone radiative forcing derived from CMIP6 simulations. Npj Clim. Atmos. Sci. 2020;3:1–10. doi: 10.1038/s41612-020-00131-0. [DOI] [Google Scholar]
- 63.Grosvenor DP, et al. The relative importance of macrophysical and cloud albedo changes for aerosol-induced radiative effects in closed-cell stratocumulus: insight from the modelling of a case study. Atmos. Chem. Phys. 2017;17:5155–5183. doi: 10.5194/acp-17-5155-2017. [DOI] [Google Scholar]
- 64.Seinfeld, J. H. & Pandis, S. N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change. 2nd Edition, (John Wiley & Sons, New York, 2006).
- 65.Seethala, C. & Horváth, A. Global assessment of AMSR‐E and MODIS cloud liquid water path retrievals in warm oceanic clouds. J. Geophys. Res. Atmos. 115.D13 (2010). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The UKESM1 data generated in this study have been deposited in the University of Cambridge Apollo database 10.17863/CAM.83526. The data are available for all users.
Due to intellectual property right restrictions, we cannot provide either the source code or documentation papers for the UM. The Met Office United Model is available for use under licence. A number of research organisations and national meteorological services use the UM in collaboration with the UK Met Office to undertake atmospheric process research, produce forecasts, develop the UM code, and build and evaluate Earth system models. For further information on how to apply for a licence, see https://www.metoffice.gov.uk/research/approach/modelling-systems/unified-model (last access: 18 July 2022).