

Abstract
INTRODUCTION:
As knowledge about neurological examination findings in autosomal dominant Alzheimer disease (ADAD) is incomplete, we aimed to determine the frequency and significance of neurological examination findings in ADAD.
METHODS:
Frequencies of neurological examination findings were compared between symptomatic mutation carriers and non mutation carriers from the DIAN to define Alzheimer disease neurological examination findings. Alzheimer disease neurological examination findings were analyzed regarding frequency, association with and predictive value regarding cognitive decline, and association with brain atrophy in symptomatic mutation carriers.
RESULTS:
Alzheimer disease neurological examination findings included abnormal deep tendon reflexes, gait disturbance, pathological cranial nerve examination findings, tremor, abnormal finger to nose and heel to shin testing, and compromised motor strength. Frequency of Alzheimer disease neurological examination findings was 65.1%. Cross-sectionally, mutation carriers with Alzheimer disease neurological examination findings showed a more than two-fold faster cognitive decline and had greater parieto-temporal including hippocampal atrophy. Longitudinally, Alzheimer disease neurological examination findings predicted a significantly greater decline over time.
DISCUSSION:
ADAD features a distinct pattern of neurological examination findings that is useful to estimate prognosis and may inform clinical care and therapeutic trial designs.
Keywords: Alzheimer disease, Autosomal dominant Alzheimer disease, neurological examination, neurological examination findings, predictive value, prognosis, differential diagnosis
1. Introduction
The neurological examination has formed the base for evaluation of neurological patients for over a century.1 It is highly standardized and the attribution of pathological findings to distinct brain regions is well established.2 The neurological examination guides the process of diagnostic investigations and informs treatment decisions in a noninvasive as well as time and cost effective manner.3 Physical examination, in combination with medical history, determined the correct diagnosis in approximately 40% of patients without any further diagnostic procedures in outpatient cohorts.4,5
Autosomal dominant Alzheimer disease (ADAD) is a rare monogenic form of Alzheimer disease.6 ADAD shows comprehensive overlap with sporadic AD. With respect to clinical manifestation both ADAD and sporadic AD exhibit typical amnestic and atypical non-amnestic cognitive presentations7–10 and non-cognitive clinical symptoms such as motor symptoms, seizures and myoclonus8,9,11–14. Neuropsychological characteristics include memory disturbance, visuospatial deficits, executive dysfunction and in later stages generalized cognitive decline in both AD variants7,10. ADAD and sporadic AD share biomarker changes proposed by the amyloid hypothesis6,15,16. Neuropathological findings in both AD forms comprise amyloid-β plaques and tau tangles with higher burden including higher Braak scores in ADAD. Lewy body disease was reported in about 30–50% in ADAD and sporadic AD. Cerebral amyloid angiopathy is common in both disorders with a higher severity in some ADAD mutations17–19. Non AD co-pathologies such as TDP-43 pathology, argyrophilic grain disease, hippocampal sclerosis and infarcts are much more common in sporadic AD18. ADAD and sporadic AD differ in the mean age at clinical onset, since ADAD starts on average in the mid 40s and sporadic AD in the 70s.20 As a result, individuals with ADAD usually lack the age-related comorbidities commonly seen in sporadic AD, for example peripheral neuropathy, orthopedic problems, falls and consecutive traumatic brain injury, and the aforementioned neuropathological non AD co-pathologies including infarcts18,21–23. Since neurological examination can be substantially influenced by age and age-related comorbidities,24 ADAD provides an opportunity to determine an AD-specific pattern of neurological examination findings.
We hypothesized that ADAD holds a distinct pattern of neurological examination findings that may inform cognitive prognosis and clinical decision making. Therefore, the aims of this study were to 1) determine the frequency of neurological examination findings in ADAD, 2) reveal a potential change in frequency over the disease course, 3) test the capacity of neurological examination findings to distinguish between mutation carriers (MC) and mutation non carriers (non MC) among mildly cognitive impaired individuals at risk, 4) analyze associations between neurological examinations findings in ADAD and both cognitive performance and brain atrophy as assessed by MRI, and 5) investigate the possibility to predict cognitive decline over time based on neurological examination findings.
2. Methods
2.1. Participants
We analyzed data from the observational study of the Dominantly Inherited Alzheimer Network (DIAN) that aims to investigate the clinical and biomarker course in individuals at risk for or with ADAD over time. That is, the DIAN observational study includes data from asymptomatic and symptomatic mutation carriers (MC) for ADAD and mutation negative family members of ADAD mutation carriers (non MC). For this study, all patients with early-onset Alzheimer disease from the DIAN observational study at the time of data freeze 14 (n=118) were evaluated. As it is a prerequisite for entering the DIAN study to be member of a family with a known ADAD mutation, no individuals with early-onset Alzheimer disease without ADAD mutations were included. Hence, all of the early-onset Alzheimer disease patients studied here carried a mutation in either PSEN1, APP or PSEN2. Data were gathered at 17 study sites around the world (USA, UK, Australia, Japan, South Korea, Argentina, Spain, and Germany) between January 2008 and February 2020. Clinical data of the DIAN study participants were collected using the Uniform Data Set version 2 from the National Alzheimer’s Coordinating Center (NACC-UDS2).25 Clinical raters were blinded to the mutation status of the participants. The protocol of the DIAN observational study was approved by the respective institutional review boards of the study sites. The DIAN study is conducted in accordance with the declaration of Helsinki. Each study participant provided written informed consent.
2.2. Genetic analyses
For identification of mutations in PSEN1, PSEN2, and APP the respective exons were amplified using polymerase chain reaction, followed by Sanger sequencing.6
2.3. Neurological Examination
The DIAN observational study includes a comprehensive, structured neurological examination that is completed by a trained clinical rater at each visit. The neurological examination is subdivided into 12 domains including visual impairment, auditory impairment, tremor, consciousness, cranial nerve examination findings, motor strength, finger to nose testing, heel to shin testing, sensory testing, deep tendon reflexes, plantar reflexes, gait, and other findings. Each item is rated either as absent vs. present or normal vs. abnormal depending on whether the respective domain label represents a pathological condition. For each rating as abnormal or present in the case of pathological conditions the study clinician may provide further details.
2.4. Definition and classification of symptomatic ADAD
In accordance with previous studies from the DIAN,6,9,16 the CDR global score26 was used to classify an individual as symptomatic (CDR global>0) or asymptomatic (CDR global=0). For investigating the pattern of neurological examination findings across the course of ADAD, we stratified symptomatic MC by CDR global scores (groups for CDR global 0.5, 1, and 2 or 3). As numbers in the groups with CDR 2 (n=7) and CDR 3 (n=5) were small, these groups were taken together to form a group of MC with CDR scores of >1.
2.5. Determination of Alzheimer disease neurological examination findings and group stratification procedures
The frequency of findings in the single subscale components of the neurological examination was compared between symptomatic MC and non MC. As both groups were relatively young (46.1 and 38.2 years, respectively) and difference in age was only 7.9 years, we did not perform a statistical age matching that can cause bias itself.27 For those neurological examination subscale findings that occurred more frequently in symptomatic MC than in non MC we introduced the term Alzheimer disease neurological examination findings (AD-NEF). Then, symptomatic MC were stratified by the presence of at least one AD-NEF into symptomatic MC with AD-NEF and symptomatic MC without AD-NEF. The latter stratification was done to form a cross-sectional population, i.e. by the use of data from baseline visits, and a longitudinal population that included only symptomatic MC with at least the baseline visit and one follow-up visit (Figure 1).
Figure 1:
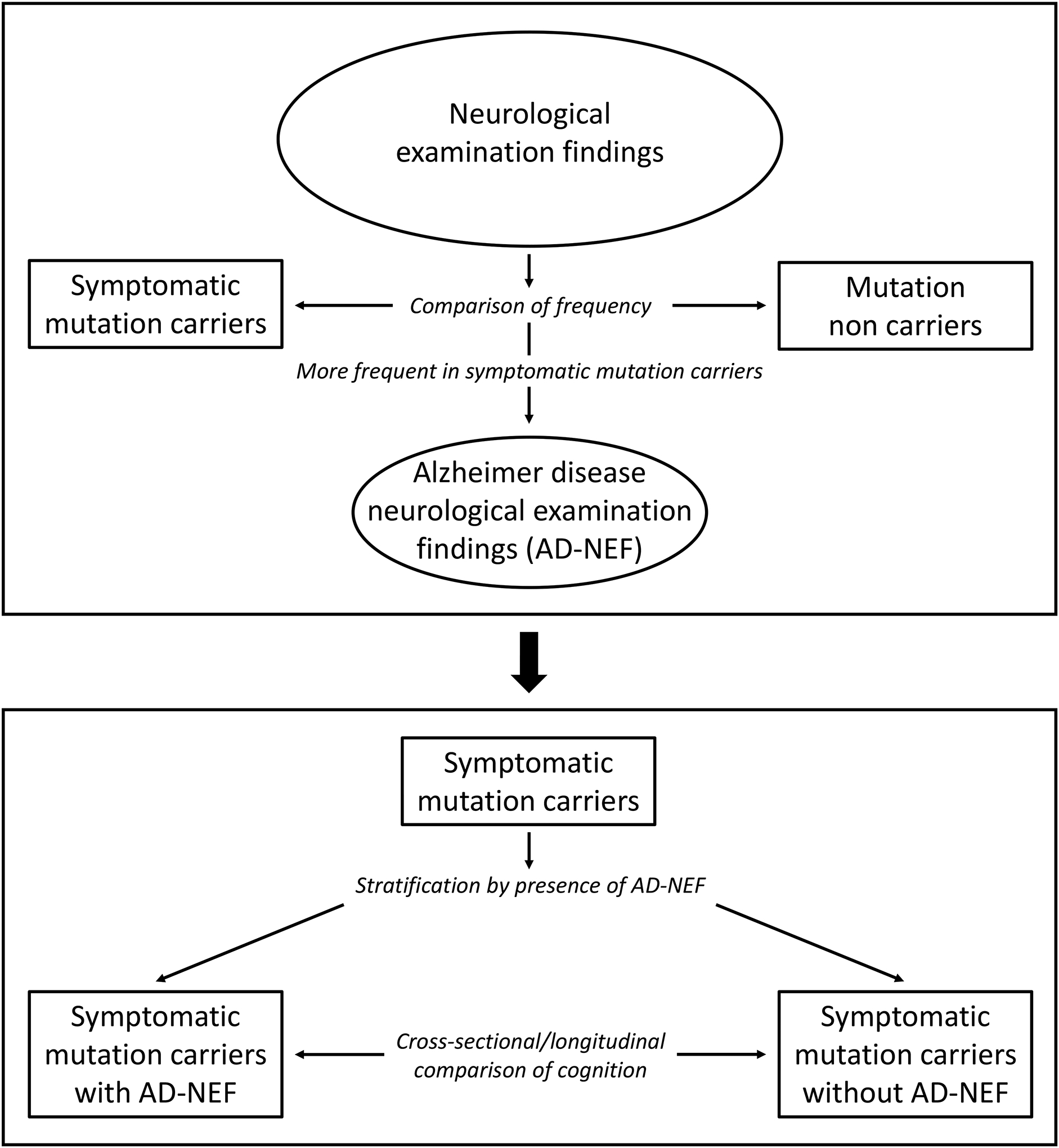
Flow chart depicting the processes to determine Alzheimer disease neurological examinations findings (AD-NEF), of group stratifications, and of analyses performed in this study.
2.6. Calculation of disease duration
If a participant is rated as symptomatic in the DIAN observational study, the rating clinician determines the age at symptom onset by exploring the earliest progressive symptom from a predefined list of symptoms. Disease duration was calculated as the difference between the age of a participant at the time of evaluation minus her/his age at symptom onset.
2.7. Relevant comorbidities
The data set was screened for relevant comorbidities that can influence neurological examination findings. Two participants had a history of stroke, one in the symptomatic MC group and one in the non MC group (0.8 vs. 0.5 %, p=1). Three participants had a history of traumatic brain injury, one in the symptomatic MC group and two in the non MC group (0.8 vs. 0.9 %, p=1). The one symptomatic mutation carrier with a history of stroke was also part of the longitudinal dataset, in the symptomatic MC without AD-NEF group. The one symptomatic MC with traumatic brain injury was not part of the longitudinal dataset. These participants were included in the analyses, as it was not determinable if these comorbidities actually affected neurological examination findings, were very rare, were equally distributed between groups and in case of stroke may be a consequence of AD-associated cerebral amyloid angiopathy.
2.8. Magnetic resonance imaging
Structural MRI included a 3D-MPRAGE sequence on 3T scanners with 1.1×1.1×1.2mm voxel resolution. For the current analysis, we used FreeSurfer-processed (Version 6) region of interest (ROI) data (i.e. cortical thickness and subcortical volumes) in Desikan-Killiany Atlas space,28 provided by the DIAN neuroimaging core.
2.9. Statistical analysis
2.9.1. Baseline comparisons
Baseline parameters were compared using Student’s t-test for continuous variables and chi-squared test or Fisher’s exact test for categorical variables, where appropriate.
2.9.2. Frequencies of neurological examination findings
We used chi-squared test or Fisher’s exact test, as appropriate, for comparison of frequencies of neurological examination findings between groups. False discovery rate correction (via Benjamini-Hochberg method) was used to account for multiple comparisons.
2.9.3. Cross-sectional analyses
To analyze the association between the presence of AD-NEF and cognition over time, we fitted a linear mixed effects model including random intercepts with the main effects disease duration and presence/absence of AD-NEF and a disease duration*presence/absence of AD-NEF interaction term using CDR – Sum of Boxes (CDR-SB) as the outcome measure. The CDR-SB score ranges from 0 to 18 with higher values indicating worse cognitive performance. CDR-SB was chosen based on its advantages as an outcome parameter including a comprehensive assessment of cognitive performance and an almost linear decline in AD.29
In symptomatic MC exploratory cross-sectional structural MRI analyses were performed to determine whether presence of AD-NEF was associated with increased grey matter atrophy determined via analyses of cortical thickness and subcortical volumes, using analyses of covariance (ANCOVA) controlling for disease duration and global Pittsburgh compound B – positron emission tomography standardized uptake value ratio (PiB-PET SUVR). Details with respect to the acquisition of PiB-PET in the DIAN observational study were described before.6
Additionally, exploratory analyses were performed to determine whether the single AD-NEF, ataxia or saccadic smooth pursuit eye movement were associated with specific patterns of grey matter atrophy determined via analyses of cortical thickness and subcortical volumes, using analyses of covariance (ANCOVA) controlling for disease duration, PiB-PET SUVR and for all other AD-NEF. As an indicator for ataxia a pathological finding in either finger to nose or heel to shin testing was used.
2.9.4. Longitudinal analyses
For investigation of a longitudinal association between AD-NEF and cognitive decline over time, that is the rate of change in CDR-SB, a linear mixed effects model that included disease duration and presence/absence of AD-NEF at each visit as the main effects and a disease duration*presence/absence of AD-NEF interaction term with CDR-SB as the outcome parameter was used. To investigate the predictive capacity of AD-NEF regarding a future cognitive decline over time, a linear mixed effects model that included disease duration and presence/absence of AD-NEF at baseline as the main effects and a disease duration*presence/absence of AD-NEF at baseline interaction term with CDR-SB as the outcome parameter was used. The models included random slopes for each individual with variance components as the covariance matrix across random effects. For selection of the best fitting model the goodness-of-fit Akaike information criterion was used for the linear mixed effects models in this study. Linear mixed effects models were chosen for analyses because of several benefits including the ability to increase statistical power and to deal with unequal numbers of measurements or intervals.30
Missing data were considered missing at random. All tests were performed two-sided. P values less than 0.05 were considered statistically significant. IBM SPSS Statistics Version 25 and R statistical software (Version 3.6.1) were used for statistical analyses.
2.10. Role of the funding source
The funding source had no role in study design, in the collection, analysis, and interpretation of data, in the writing of the report, and in the decision to submit the paper for publication.
3. Results
3.1. AD-NEF
Baseline data regarding AD-NEF were available for 118 symptomatic MC and 211 non MC. Clinical and genetic parameters at baseline are listed and compared between groups in Table 1.
Table 1:
Comparison of baseline characteristics between symptomatic mutation carriers and non mutation carriers. Description: p values below 0.05 are italicized.
| Symptomatic mutation carriers (n=118, 35.9%) | Non mutation carriers (n=211, 64.1%) | p value | |
|---|---|---|---|
| Age (years), mean (SD) | 46.1 (10.0) | 38.2 (11.4) | <0.001 |
| Sex (female), n (%) | 56 (47.5) | 87 (41.2) | 0.27 |
| Education (years), mean (SD) | 13.5 (3.4) | 14.7 (2.9) | 0.001 |
| At least one NEF, n (%) | 69 (65.1) | 47 (25.3) | <0.001 |
| Age at onset (years), mean (SD) | 42.6 (8.8) | na | na |
| Disease duration (years), mean (SD) | 3.7 (2.9) | na | na |
| CDR global, n (%) | 0.5, 78 (66.1) : 1, 28 (23.7) : 2, 7 (5.9) : 3, 5 (4.2) | 0, 196 (92.9) : 0.5, 15 (7.1) | <0.001 |
| CDR-SB, mean (SD) | 3.8 (4.0) | 0.07 (0.27) | <0.001 |
| MMSE, mean (SD) | 22.5 (7.0) | 29.0 (1.3) | <0.001 |
| Mutated gene, n (%) | PSEN1, 97 (82.2) : APP, 19 (16.1) : PSEN2, 2 (1.7) | na | na |
| APOE ε4 carrier, n (%) | 34 (29.3) | 59 (29.1) | 0.96 |
Abbreviations: SD=standard deviation; NEF=neurological examination finding; CDR=Clinical Dementia Rating; SB=Sum of Boxes; MMSE=mini mental state examination; APOE=gene encoding Apolipoprotein.
An abnormal neurological examination result, defined by the presence of at least one NEF, occurred more frequently in symptomatic MC compared to non MC (65.1 vs. 25.3 %, p<0.001). Symptomatic MC exhibited more frequently abnormal findings in 9 subdomains of the neurological examination. The highest frequency of abnormal findings showed the subdomain deep tendon reflexes (35.9% in symptomatic MC vs. 3.3% in non MC, p<0.001), followed by other findings (22.4 vs. 2.7%, p<0.001), gait (17.8 vs. 2.8%, p<0.001), cranial nerve examination findings (14.4 vs. 4.7%, p=0.002), tremor (12.7 vs. 4.7%, p=0.009), finger to nose testing (11.0 vs. 0%, p<0.001), heel to shin testing (7.7 vs. 0.5%, p=0.001), plantar reflexes (6.8 vs. 1.4%, p=0.02), and motor strength (5.1 vs. 0.9%, p=0.027). Abnormal findings in these 9 subdomains of the neurological examination are referred to as AD-NEF in this article. All of these differences remained statistically significant after correction for multiple comparisons. There were no subdomains in which abnormal neurological examination findings occurred more frequently in non MC than in symptomatic MC (Figure 2). No statistically significant differences in frequencies of AD-NEF were observed between MC with a global CDR score of 0 or in non MC (11.1% vs. 14.6%; p=0.33).
Figure 2:
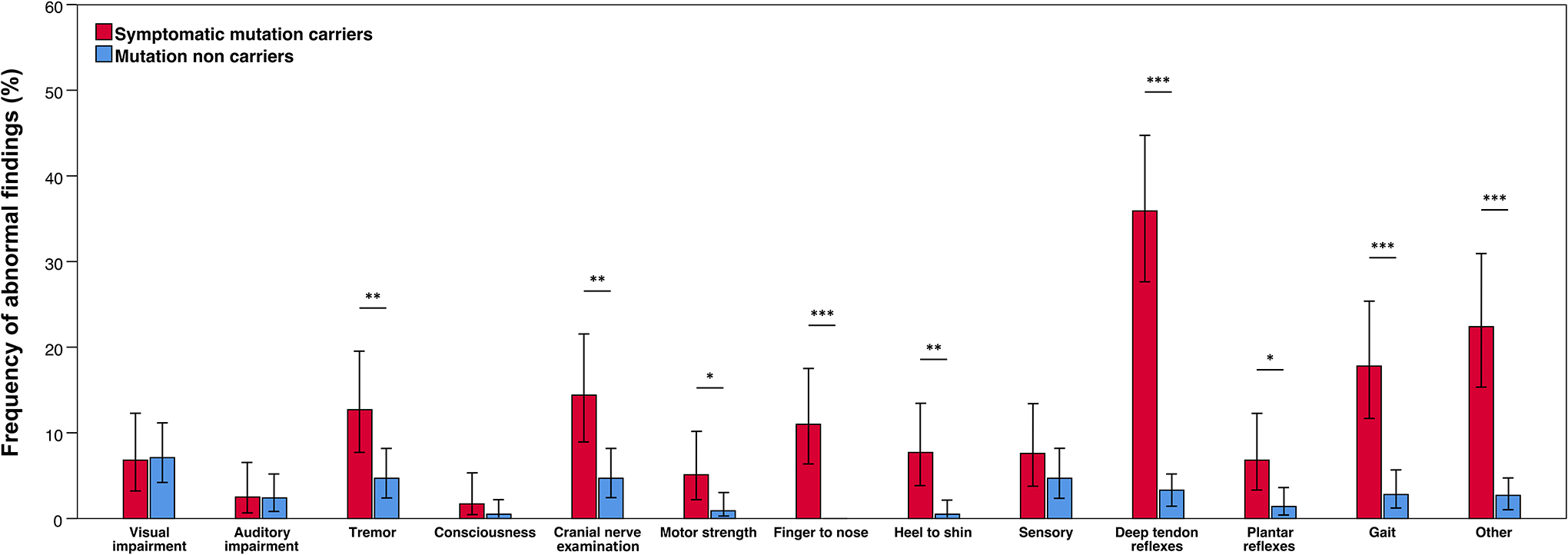
Comparisons of frequencies of neurological examinations findings between symptomatic mutation carriers and mutations non carriers. Description: Tremor, abnormal cranial nerve examination findings, compromised motor strength, abnormal findings on finger to nose testing and heel to shin testing, pathological deep tendon reflexes, abnormal plantar reflexes, gait disturbance, and other findings were more frequent in symptomatic mutations carriers. * p<0.05; ** p<0.01; *** p<0.001. Error bars represent 95% confidence intervals.
For the five most frequent AD-NEF pathological deep tendon reflexes, gait disturbance, abnormal cranial nerve examination findings, tremor, and other findings, specifications provided by the respective clinical raters were available. The most frequent findings within the respective AD-NEF were asymmetrical brisk deep tendon reflexes, reduced arm swing while walking, saccadic smooth pursuit eye movement, postural tremor, and increased muscle tone. Further specifications are depicted in Figure 3.
Figure 3:
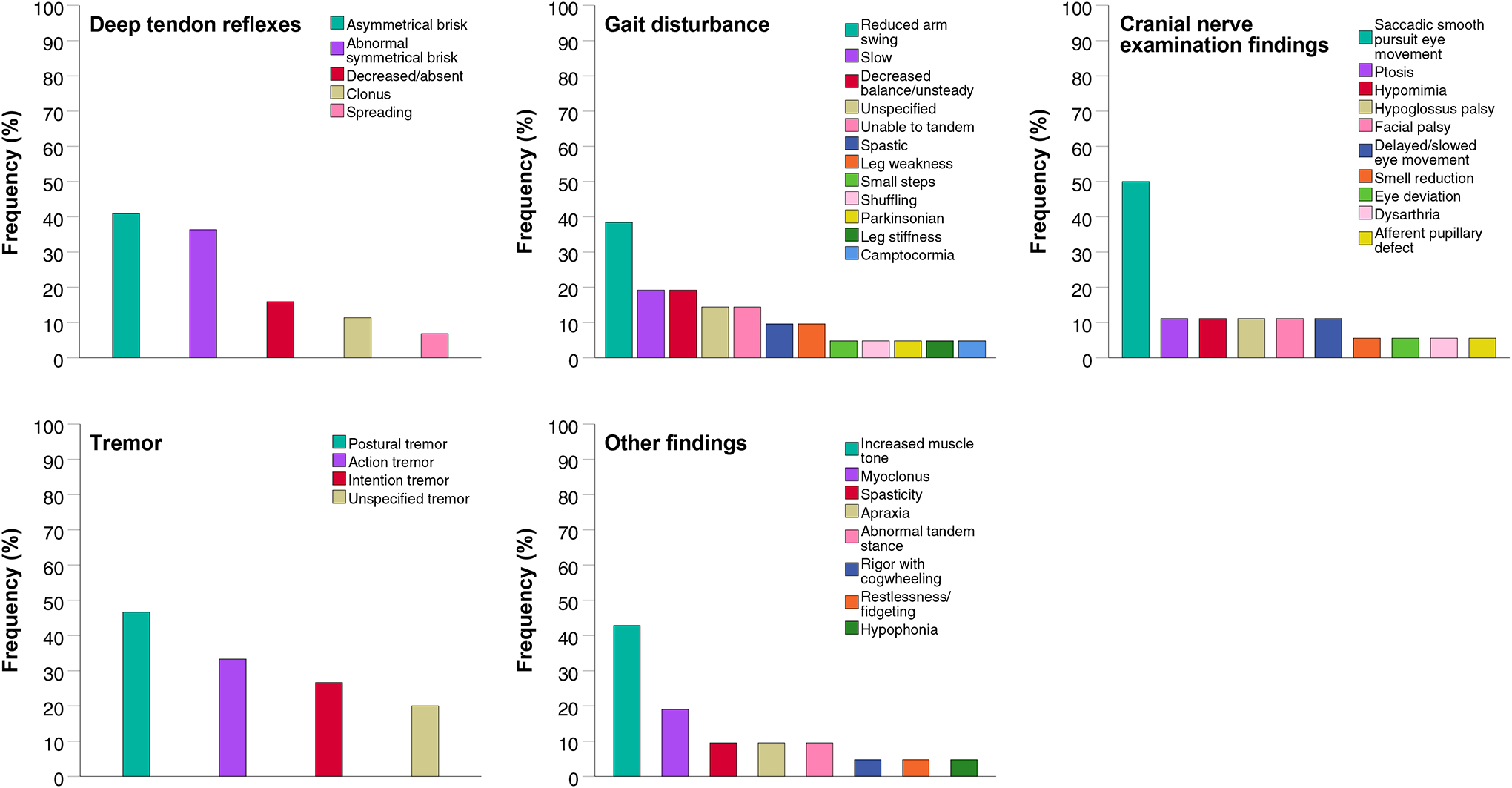
Particular signs and their frequencies within the group of the five most frequent Alzheimer disease neurological examination findings pathological deep tendon reflexes, gait disturbance, abnormal cranial nerve examination findings, tremor, and other findings. Description: The most frequent particular signs of each of the five Alzheimer disease neurological examination findings were asymmetrical brisk deep tendon reflexes, reduced arm swing while walking, saccadic smooth pursuit eye movement, postural tremor, and increased muscle tone.
The ADAD mutation carriers in this study had 46 different mutations (49 PSEN1, 1 PSEN2 and 6 APP mutations). The single AD-NEF were compared regarding their respective frequency between single mutations using chi-squared test and false discovery rate correction (via Benjamini-Hochberg method) to account for multiple comparisons. There was no difference in frequency of any AD-NEF between the single ADAD mutations.
3.2. AD-NEF in symptomatic MC stratified by disease stage
The frequencies of AD-NEF in symptomatic MC were analyzed by disease stage determined by CDR global scores (Figure 4). Frequency of all AD-NEF increased over the whole disease course. In all disease stages abnormal deep tendon reflexes were the most frequent finding among AD-NEF. Frequency was 33.3% at CDR 0.5, remained stable at CDR 1 and rose to 58.3% at CDR>1. Gait disturbance was present in 9.0% at CDR 0.5 and rose steadily to 28.6% at CDR 1 and to 50.0% at CDR>1. Frequency of abnormal cranial nerve examination findings was in the medium range of frequencies across disease stages: 11.5% at CDR 0.5, stayed roughly stable at CDR 1 (10.7%) and increased to 41.7% at CDR>1. Tremor occurred in 9.0% at CDR 0.5, its frequency rose slightly to 14.3% at CDR 1 and then more steeply to 33.3% at CDR>1. Abnormal finger to nose testing was found in a relatively small percentage of 2.6% at CDR 0.5, its frequency increased steeply to 28.6% at CDR 1, and then decreased slightly to 25.0% at CDR>1. Abnormalities in heel to shin testing exhibited the lowest frequency at CDR 0.5 (1.3%), and rose relatively steadily to 17.9% at CDR 1, and to 27.3% at CDR>1, in the medium range of frequencies of AD-NEF in the CDR 1 and CDR>1 groups. Frequencies of abnormal plantar reflexes were in the lower range of frequencies through all disease stages. They were present in 6.4% at CDR 0.5, slightly declined in frequency to 3.6% at CDR 1 and rose relatively steeply to 16.7% at CDR>1. Also in the lower frequency range through all disease stages were abnormalities in motor strength. They occurred in 3.8% at CDR 0.5 and increased slightly to 7.1% at CDR 1 and to 8.3% at CDR>1.
Figure 4:
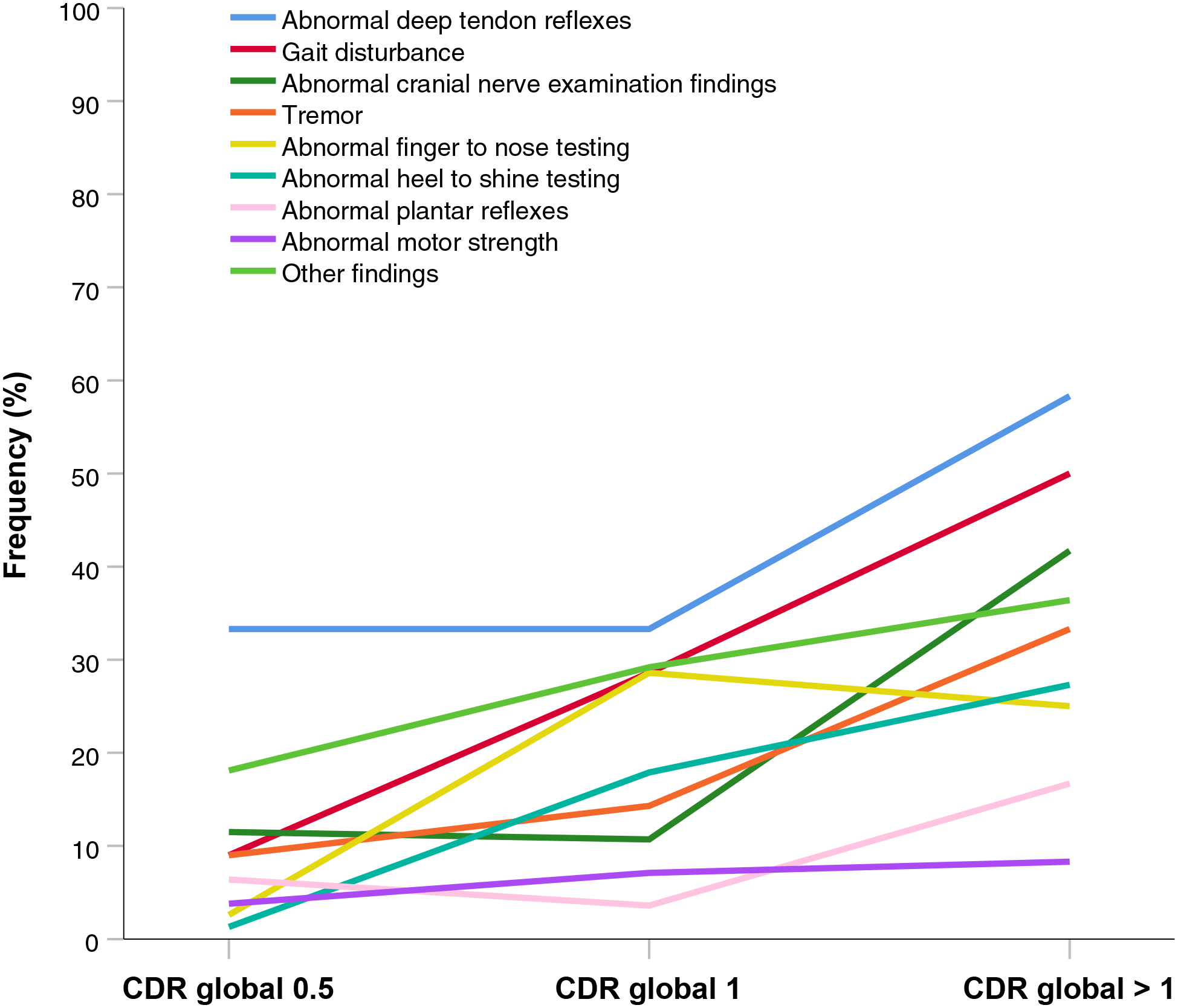
Frequencies of Alzheimer disease neurological examination findings stratified by global CDR scores. Description: All Alzheimer disease neurological examination findings increased in frequency with CDR global stages. Abnormal deep tendon reflexes were the most frequent finding in all disease phases. Gait disturbance exhibited the steepest increase in frequency with autosomal dominant Alzheimer disease progression. Abbreviations: CDR=Clinical Dementia Rating.
3.3. Association between AD-NEF and cognition
To analyze associations between AD-NEF and cognitive performance, symptomatic MC were stratified in groups by the presence (n=64) or absence (n=42) of AD-NEF. Baseline clinical and genetic parameters are shown in Table 2. Symptomatic MC with AD-NEF exhibited a worse cognitive performance as assessed by CDR-SB than symptomatic MC without AD-NEF (mean CDR-SB scores: 4.32 vs. 2.59, p=0.007) while being at the same disease phase as determined by disease duration (mean disease duration: 3.9 vs. 3.6 years, p=0.53). A linear mixed effects model revealed a significant effect of the presence of AD-NEF on cognitive performance as measured by CDR-SB towards abnormal over disease duration (disease duration: estimate=0.406, standard error=0.186, p=0.031; disease duration*presence of AD-NEF interaction: estimate=0.572, standard error=0.221, p=0.011). In this cross-sectional model, the decline in CDR-SB per year was 0.41 points in symptomatic MC without AD-NEF and 0.98 points in MC with AD-NEF. Symptomatic MC with AD-NEF declined significantly more, by 0.57 points on CDR-SB per year (Figure 5A).
Table 2:
Comparison of baseline characteristics between symptomatic MC with and without Alzheimer disease neurological examination findings. Description: p values below 0.05 are italicized.
| Symptomatic MC with AD-NEF (n = 64, 60.4%) | Symptomatic MC without AD-NEF (n = 42, 39.6%) | p value | |
|---|---|---|---|
| Age (years), mean (SD) | 45.9 (10.3) | 46.2 (8.7) | 0.90 |
| Sex (female), n (%) | 30 (46.9) | 25 (59.5) | 0.20 |
| Education (years), mean (SD) | 12.9 (3.8) | 14.0 (2.6) | 0.12 |
| Age at onset (years), mean (SD) | 42.0 (8.9) | 43.1 (8.1) | 0.54 |
| Disease duration (years), mean (SD) | 3.98 (3.18) | 3.46 (2.58) | 0.39 |
| CDR global, n (%) | 0.5, 40 (62.5) : 1, 15 (23.4) : 2, 5 (7.8) : 3, 4 (6.3) | 0.5, 32 (76.2) : 1, 9 (21.4) : 2, 1 (2.4) : 3, 0 (0) | 0.20 |
| CDR-SB, mean (SD) | 4.32 (4.60) | 2.62 (2.10) | 0.012 |
| MMSE, mean (SD) | 21.45 (7.36) | 24.76 (5.22) | 0.008 |
| Mutated gene, n (%) | PSEN1, 52 (81.3) : APP, 11 (17.2) : PSEN2* | PSEN1, 33 (78.6) : APP, 8 (19.0) : PSEN2* | 0.92 |
| APOE ε4 carrier, n (%) | 20 (31.3) | 9 (21.4) | 0.27 |
As there were fewer than 3 PSEN2 mutation carriers in the study, no figures are shown.
Abbreviations: MC=mutation carriers; AD-NEF=Alzheimer disease neurological examination findings; SD=standard deviation; CDR=Clinical Dementia Rating; SB=Sum of Boxes; MMSE=mini mental state examination; APOE=gene encoding Apolipoprotein.
Figure 5:
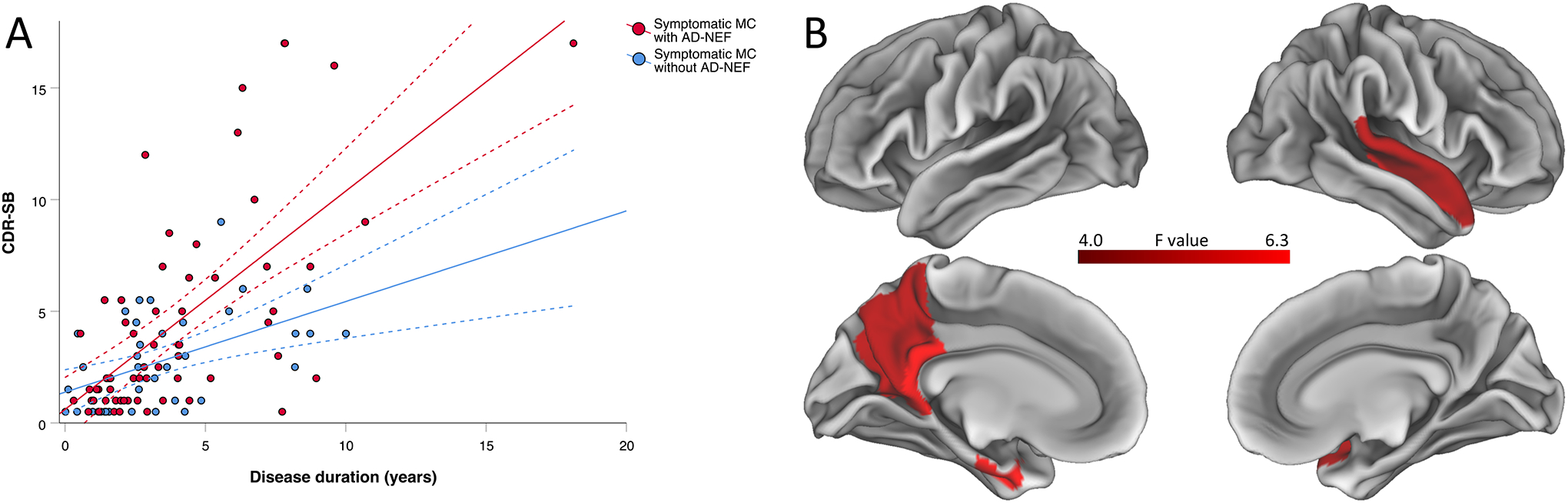
Cross-sectional associations between AD-NEF and (A) cognitive performance and (B) brain atrophy. Description: (A) Grouped scatter plot depicting the cross-sectional relationship between CDR – Sum of Boxes scores and disease duration in symptomatic MC with and without Alzheimer disease neurological examination findings. Symptomatic MC with AD-NEF showed a significantly more pronounced decline in CDR – Sum of Boxes over the disease duration compared to symptomatic MC without AD-NEF. Dashed lines represent 95 % confidence intervals. (B) Differences in brain atrophy between MC with and without AD-NEF. MC with AD-NEF showed a greater atrophy in temporo-parietal brain regions and greater bilateral hippocampal atrophy in an exploratory analysis with an alpha threshold of 0.05. After adjusting for 82 regions of interest using the Bonferroni method (resulting alpha threshold <0.0006), only the left hippocampal volume remained significantly different. Abbreviations: CDR-SB=Clinical Dementia Rating–Sum of Boxes; MC=mutations carriers; AD-NEF=Alzheimer disease neurological examination findings.
3.4. Association between AD-NEF and grey matter atrophy in MC
In symptomatic MC, we found that the presence of AD-NEF was associated with greater grey matter atrophy in the temporo-parietal cortex (left precuneus, left posterior cingulate, left entorhinal cortex, right superior temporal gyrus) and bilateral hippocampus at an exploratory ROI-wise alpha threshold of 0.05, controlling for disease duration and global PiB-PET SUVR (Figure 5B). At a Bonferroni-corrected alpha threshold accounting for 82 ROIs (p<0.0006), only the left hippocampus remained significant.
The results of the exploratory analyses to determine whether the single AD-NEF, ataxia or saccadic smooth pursuit eye movement were associated with specific patterns of grey matter atrophy are summarized in the supplementary figure. In summary, most of the single AD-NEF, ataxia and saccadic smooth pursuit eye movement were associated with a fronto-temporo-parietal pattern of atrophy. There were no significant associations between any AD-NEF, ataxia or saccadic smooth pursuit eye movement and atrophy in any subcortical region. At a Bonferroni-corrected alpha threshold accounting for 82 ROIs (p<0.0006), no brain region remained significant.
3.5. Longitudinal analysis and predictive value of AD-NEF regarding individual rate of cognitive decline
Longitudinal data, i.e. data from the baseline visit and at least one follow-up visit of the same individual, were present for 73 symptomatic MC with a total of 222 visits (≥2 visits: n=73; ≥3 visits: n=39; ≥4 visits: n=21; ≥5 visits: n=12; ≥6 visits: n=3; 7 visits: n=1). Mean number of visits was 3.04 (standard deviation=1.25) and mean follow-up time 2.49 years (standard deviation=1.63; range=0.96–7.03 years). There was a significant difference in slopes as a function of the presence of AD-NEF at each visit and disease duration with CDR-SB as the outcome parameter (disease duration: estimate=0.981, standard error=0.099, p<0.001; disease duration*presence of AD-NEF at each visit interaction: estimate=0.343, standard error=0.136, p=0.012). The rate of yearly decline estimated by the model was 0.98 points on the CDR-SB score in symptomatic MC without AD-NEF compared to 1.32 points in symptomatic MC with AD-NEF. That is, symptomatic mutation carriers with AD-NEF declined significantly more, by 0.34 points per year, than symptomatic mutation carriers without AD-NEF (Figure 6). There was also a significant difference in slopes as a function of the presence of AD-NEF at baseline and disease duration with CDR-SB as the outcome parameter (disease duration: estimate=1.020, standard error=0.120, p<0.001; disease duration*presence of AD-NEF at baseline interaction: estimate=0.494, standard error=0.211, p=0.022). The rate of yearly decline estimated by the model was 1.02 points on the CDR-SB score in symptomatic MC without AD-NEF at baseline compared to 1.51 points in symptomatic MC with AD-NEF at baseline. That is, symptomatic mutation carriers with AD-NEF at baseline showed a significantly increased future cognitive decline, by 0.49 points on CDR-SB per year, than symptomatic mutation carriers without AD-NEF at baseline.
Figure 6:
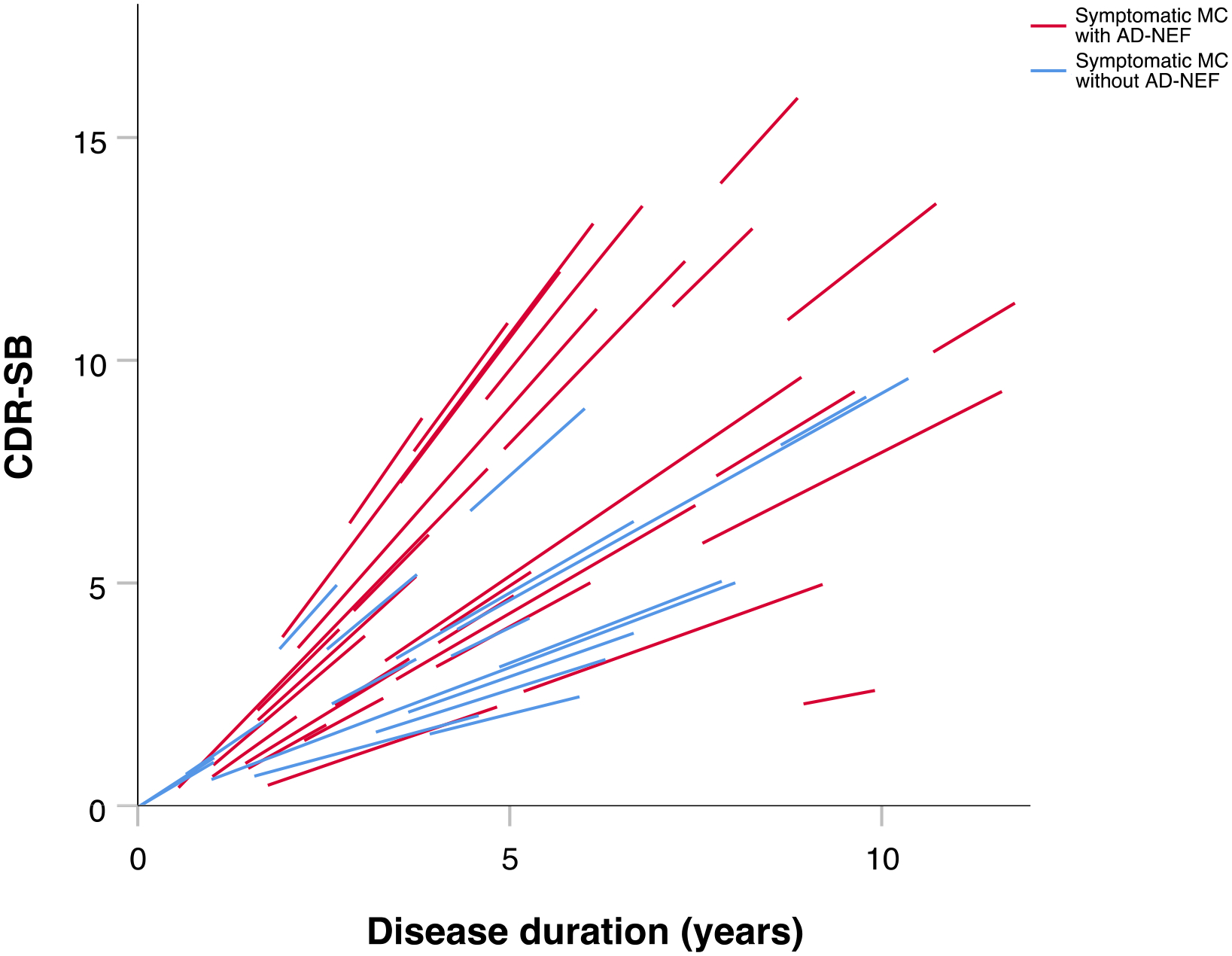
Individual linear estimates of change in Clinical Dementia Rating–Sum of Boxes over time in symptomatic mutation carriers with and without Alzheimer disease neurological examination findings. Description: Individual decline in Clinical Dementia Rating – Sum of Boxes over time was significantly more pronounced in symptomatic mutation carriers with Alzheimer disease neurological examination findings compared to those without. The individual linear changes in Clinical Dementia Rating–Sum of Boxes were predicted by a linear mixed effects model based on longitudinal data, i.e. data from symptomatic mutation carriers with at least the baseline visit and one follow-up visit. Abbreviations: MC=mutations carriers; CDR-SB=Clinical Dementia Rating–Sum of Boxes; AD-NEF=Alzheimer disease neurological examination findings.
3.6. Differential diagnostic significance of AD-NEF
Among individuals at risk for ADAD with a CDR global score of 0.5, i.e. very mild cognitive impairment, AD-NEF were significantly more frequent in MC than in non MC (55.6 vs. 26.7%; p=0.042). The positive predictive value of AD-NEF to predict a MC status was 91%. Sensitivity was 56% and specificity 73%.
4. Discussion
Two recently published studies, one using a European case series and the other comparing DIAN and literature data, recently provided insights about non-amnestic manifestations of ADAD on a symptom and diagnosis level.8,9 Relatively frequent symptoms were seizures, myoclonus, and behavioural or personality changes. Compared to symptoms and diagnoses, findings are less based on inductive generalization and provide the least abstract level of categorization, and therefore may provide more objective information and a high degree of cue validity.31,32 In the current study, a systematic investigation of single neurological findings as subscale components of a structured clinical neurological examination was performed, an approach that has not previously been pursued. Neurological examination findings in ADAD encompass pathological deep tendon reflexes, gait disturbance, cranial nerve examination findings, tremor, abnormal finger to nose and heel to shin test findings, pathological plantar reflexes, as well as compromised motor strength. Neurological examination findings in ADAD were associated with a two-fold faster cognitive decline and ADAD patients with neurological examination findings exhibited a greater parieto-temporal atrophy independent from disease duration. The presence of AD-NEF at baseline predicted an increased rate of future cognitive decline.
Knowledge about these examination findings may help clinicians to corroborate a suspected ADAD diagnosis and to distinguish from differential diagnoses of ADAD. Taking illustratively the five most frequent AD-NEF and their respective most frequent subitem (Figure 3) as the basis, a typical ADAD patient may present with asymmetrical brisk deep tendon reflexes, increased muscle tone, reduced arm swing while walking, saccadic smooth pursuit eye movements, and postural tremor.
A profile of motor symptoms measured by the Unified Parkinson Disease Rating Scale Part III was recently described in ADAD. This profile indicates that bradykinetic symptoms are the primary motor manifestation in ADAD.13 The insights about clinical neurological examination findings of this study may add further to a sharper and more comprehensive clinical picture of ADAD.
The term Alzheimer disease neurological examination findings (AD-NEF) was introduced for those findings that were more frequent in symptomatic mutation carriers than in non mutation carriers. The frequency of AD-NEF increased with disease stage of ADAD. This finding is in accordance with disease phase dependent build-up of non-cognitive symptoms in AD such as for example seizures and motor symptoms.9,13,14
In at risk individuals with mild cognitive symptoms in this study, the presence of AD-NEF was highly indicative for ADAD mutation carrier status. Since the presence of AD-NEF predicts a worse outcome in symptomatic ADAD, identifying this group early might facilitate earlier intervention and perhaps help to provide haste in confirming genetic results. The integration of knowledge of the predictive value of seizures and impaired rapid alternating hand movements regarding mutation carrier status in the cognitively presymptomatic phase of ADAD12,13 and of AD-NEF in cognitively symptomatic at risk persons may help to aid patient evaluation and care throughout disease phases.
In the current study, an association between the presence of AD-NEF and poorer cognitive performance independent from the disease stage was found in ADAD patients. The exploratory MRI-analysis revealed an increased temporo-parietal including hippocampal atrophy in MC with AD-NEF compared to MC without AD-NEF. A similar pattern was seen in a recent study of the spatial distribution of atrophy in ADAD patients.30 Therefore, a potential pathophysiological explanation for the worse cognitive performance associated with AD-NEF may be a greater burden of AD-related atrophy in ADAD patients with AD-NEF independent from the disease stage.
Beyond the cross-sectional association of AD-NEF with poorer cognitive performance in ADAD patients, our intraindividual longitudinal analyses showed an association between the presence of AD-NEF and a significantly higher rate of cognitive decline over time, by approximately 35% per year on CDR-SB. Moreover, the longitudinal analysis showed that the presence of AD-NEF at baseline predicted a significantly higher rate of future decline in CDR-SB, by approximately 50% per year. The predictive capability of AD-NEF offers the opportunity to estimate prognosis and thus may add substance to patient counselling as well as to diagnostic and therapeutic strategies. Taking the stage of very mild dementia (CDR-SB 3.0–4.0) as an assumptive starting point, after 5 years patients without AD-NEF would arrive at the stage of mild dementia (CDR-SB 4.5–9.0), whereas patients with AD-NEF would be at the stage of moderate dementia (CDR-SB 9.5–15.5). After 10 years, ADAD patients without AD-NEF would exhibit moderate dementia and those with AD-NEF would suffer from severe dementia (CDR-SB 16.0–18.0).33 The predictive nature of AD-NEF regarding cognitive decline over time could be explained through a potential capability of the neurological examination to detect subtle and localized AD-associated brain changes that did not yet extend to brain regions that cause cognitive decline when damaged.
Since a population with ADAD formed the basis for the analyses in this study, it is a crucial question how our findings may translate to sporadic AD. In literature, spastic paraparesis is more frequently described in ADAD than in sporadic AD. Nine of the 97 PSEN1 mutation carriers in this study had mutations that were reported to be possibly associated with spastic paraparesis (Val261Phe, Pro264Leu, Leu271Val).34 Only in one of these nine patients a bilateral spastic increase in lower limb tone was reported. Importantly, the higher age and frequency of age related comorbidities in patients with sporadic AD that can cause abnormal neurological examination findings could challenge translatability.24 Exploring for those comorbidities by thorough medical history taking including third-party anamnesis and analyses of medical files may account for these challenges and warrant translatability of the study findings to sporadic AD. However, this requires further study.
In summary, the results of our study may leverage differential diagnostic considerations by revealing neurological examination findings in symptomatic autosomal dominant Alzheimer disease including their stage dependent frequencies. The presence of these findings indicates mutation status in mildly cognitive impaired at risk persons with accuracy. The association of neurological findings typical for ADAD with poor cognitive performance and their predictive value regarding increased cognitive deterioration over time may render the neurological examination suitable to contribute to estimation of prognosis, to improve patient consultation and to inform treatment decisions and future therapeutic trial designs.
Supplementary Material
Research in Context.
Systematic review
A comprehensive literature review in PubMed regarding neurological examination findings (NEF) in Alzheimer disease (AD) including a wide range of neurological manifestations in AD on a symptom and diagnosis level was performed (Pubmed terms: “Alzheimer disease”/“autosomal dominant Alzheimer disease”/“familial Alzheimer disease” AND “neurological examination findings”/“neurological findings”/“neurological symptoms”/“neurological manifestations”).
Interpretation
Neurological examination findings in AD are frequent and indicative of a broader affection of brain areas to those involved not only in cognition but also in motor function. This is associated with a poorer prognosis.
Future directions
The knowledge about the association between the presence of non-cognitive neurological examination findings and a worse cognitive course may inform future therapeutic AD trial designs.
Highlights.
Neurological examination findings in Alzheimer disease (AD-NEF) are frequent
AD-NEF are associated with a two-fold faster cognitive decline
Patients with AD-NEF exhibit greater parieto-temporal including hippocampal atrophy
AD-NEF predict a greater cognitive decline
Acknowledgements
This project was supported by The Dominantly Inherited Alzheimer Network (DIAN, UF1 AG032438) funded by the National Institute on Aging (NIA), the German Center for Neurodegenerative Diseases (DZNE), the NIHR Queen Square Dementia Biomedical Research Centre and the MRC Dementias Platform UK (MR/L023784/1 and MR/009076/1), and AMED JP21dk0207049. This work was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) (FOR2290) and was funded by the Deutsche Forschungsgemeinschaft under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy - ID 390857198). GH acknowledges support by Niedersächsisches Ministerium für Wissenschaft und Kunst, VolkswagenStiftung (Niedersächsisches Vorab), Petermax-Müller Foundation (Etiology and Therapy of Synucleinopathies and Tauopathies). This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families as well as the contributions of the DIAN research and support staff at each of the participating sites.
Funding sources
German Center for Neurodegenerative Diseases (DZNE)
Disclosures
Jonathan Vöglein has nothing to disclose.
Nicolai Franzmeier has received a research grant from Bright Focus Foundation.
John C. Morris has received NIH grants. He has received royalities or licenses for CDR registration. He has received consulting fees from Barcelona BetaBrain Res Center BBRC SAB meeting, Barcelona, Spain; Centre for Brain Research meeting, Bangalore, India. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Montefiore, NY Grand Rounds. He has received support for attending meetings and/or travel from TS Srinavasan 40th Oration, India; World Congress of Neurology; Cure Alzheimer Board meeting; CBR International Advisory Board. He has held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): Cure Alzheimer Board Meeting.
Marianne Dieterich has received grants from the German Research Foundation and the German Foundation for Neurology. She has received support for attending meetings and/or travel: European Academy of Neurology for the annual congress in 2019, to her.
Eric McDade has received grants or contracts from: Institution: National Institute of Health; Janssen; Eli Lilly; Roche. He has received royalties or licenses from UpToDate. He has received personal consulting fees from: Allector- DSMB Eli Lilly- DSMB Alzamend- Scientific Advisory Board Fondation Alzheimer- Scientific Advisor. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Eisai (CME; personal payment). He has received support for attending meetings and/or travel: Personal Support: Fondation Alzheimer Alzheimer Association. He has had any patents planned, issued, or pending: Novel Tau isoforms to predict onset of symptoms and dementia in Alzheimer’s disease. He has participated on a Data Safety Monitoring Board or Advisory Board: see above. He has received equipment, materials, drugs, medical writing, gifts, or other services for medical writing.
Mikael Simons has nothing to disclose.
Oliver Preische has nothing to disclose.
Anna Hofmann has nothing to disclose.
Jason Hassenstab has received several NIH grants, BrightFocus foundation. Payments to institution, personal consulting fees from Roche. He has participated on a Data Safety Monitoring Board or Advisory Board: DSMBs for 2 NIH sponsored studies DSMB for Eisai. payments made to him.
Tammie L. Benzinger has grants to her institution from NIH, personal payments from Biogen, payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events from Biogen and Eisai speakers bureau (payments to her). She has participated on a Data Safety Monitoring Board or Advisory Board: Biogen (payments to her). She has received Precursor for flortaucipir, from Avid Radiopharmacuticals.
Anne Fagan has received many grant from the NIH/NIA paid to her institution. She also received a research grant from Centene, paid to her institution. She is on the scientific advisory boards for Roche Diagnostics/Genentech and also consult for Diadem, DiamiR and Siemens Healthcare Diagnostics Inc. Payments were made to her.
James M. Noble has received grants from NIH (all by his institution): R01AG054536, T35AG044303, UF1AG032438, R21AG065753, P30AG066462, R01NS067443, R01NR017571, R01AG060929. He has any patents planned, issued, or pending: US20190298262A1, does not relate to this manuscript.
Sarah B. Berman has received grants or contracts from Michael J Fox Foundation 18119 – institution.
Neill R. Graff-Radford has nothing to disclose.
Bernardino Ghetti has received NIH grants to Indiana University and two honoraria from University of Utah Northwestern University. He has held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): International Society for Frontotemporal Dementias: Secretary Treasurer.
Martin R. Farlow is a coinventor for US Patent No. 6184435 entitled “Transgenic Mouse Expressing APP770-V717F”. He has received consulting fees from Avanir Biogen Eli Lilly & Company Cognition Therapeutics Longeveron Otsuka Proclara vTv Therapeutics Lexeo Ionis McClena Athira. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: Now Up to Date. He has received payment for expert testimony: confidential. He has participated on a Data Safety Monitoring Board or Advisory Board: All payments made to him: Avanir Biogen Eli Lilly & Company Cognition Therapeutics Longeveron Otsuka Proclara vTv Therapeutics Lexeo Ionis McClena Athira.
Jasmeer P. Chhatwal has received grants or contracts from: NIH R01AG062667 (institution) NIH R01AG071865 (institution) NIH P01AG036694 (institution) Doris Duke Charitable Foundation Career Dev Award (institution). He has received support for attending meetings and/or travel: NIH R01AG062667 (institution) NIH R01AG071865 (institution) NIH P01AG036694 (institution) Doris Duke Charitable Foundation Career Dev Award (institution).
Stephen Salloway has received grants or contracts from Biogen, Institution/Butler Hospital ACTC, Institution/Butler Hospital. He has received consulting fees from Bolden Therapeutics, Ono, Genentech, Biogen, Prothena, Alnylam, ATRI, Roche, Mayo, payments to him. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: Grand Rounds/VA, payment to self CME talk for PlatformQ, payment to self CME talk for PER, payment to self CME Master Class series, Biogen, payment to self. He has received support for attending meetings and/or travel: RSNA symposium, Biogen, payment to self Webinar SNMMI, payment to self Travel costs for AMGEN, GEMVAX and AVID. He has participated on a Data Safety Monitoring Board or Advisory Board: Acumen, payment to self Advisory Board, Genentech, payment to self.
Chengjie Xiong has received grants or contracts from: NIH R01 AG053550 as the PI; and NIH R01 AG067505 as the PI. He has received consulting fees: he provided statistical data analyses for DIADEM, and the fee was paid to him. He participated on a Data Safety Monitoring Board or Advisory Board: he serves on FDA Medical Imaging Drug Advisory Committee. He also serves on the External advisory Committee for University of Wisconsin Alzheimer disease research center.
Celeste M. Karch has nothing to disclose.
Nigel Cairns has nothing to disclose.
Richard J. Perrin has received grants or contracts from: for all grants below, payments made only to institution: R01AG054567 (Benzinger) 09/15/17-06/30/22 P01 AG003991 (Morris) 05/01/19-04/30/24 P30 AG066444 (Morris) 05/01/20-04/30/25 U19AG024904 (Weiner)08/01/16-07/31/22 NCE R01AG068319 (Bateman) 09/15/20-05/31/25 R01 AG053267 (Bateman) 09/01/17-05/31/22 U19 AG032438 (Bateman) 09/15/19-06/30/24 U19 AG032438 (Bateman) 09/15/19-06/30/24 R01 AG052550 (Benzinger) 04/15/18-01/31/23 R01 AG070883 (Kind, Raji) 03/01/21-02/28/26 R01NS097799 (Kotzbauer) 08/01/16-04/30/22 NCE R01NS092865 (Xu) 02/01/16-11/30/21 R01AG054513 (Yablonskiy) 07/01/17-04/30/22 R01 NS075321 (Perlmutter) 05/01/11-04/30/22 NCE APDA (EXTENDED 2019) (Perlmutter) 01/01/99-08/31/21.
Gregory Day has received grants to institution: - K23AG064029 (NIH/NIA) - Chan Zuckerberg Initiative (Neurodegeneration Challenge Network; WU-20-421) - Alzheimer’s Association (LDRFP-21-824473). He has received personal payments: - DynaMED (Topic Editor, Dementia) - Parabon Nanolabs (Consulting for NIA SBAR Grant) Texas Neurological Institute - Continuing Education Company - Barrow Law (medicolegal advice on a case of Wernicke encephalopathy). He has held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): Clinical Director (unpaid): Anti-NMDA Receptor Encephalitis Foundation, Inc.
Ralph Martins has nothing to disclose.
Raquel Sanchez-Valle has receive support for the present manuscript: ISCIII, Spain (grant number PI20/00448 to RSV). She received grants or contracts from ISCIII, Sage Ph and Biogen, payments were made to my institution. She reports personal fees from Wave pharmaceuticals and Ionis Pharmaceuticals for attending Advisory board meetings, personal fees from Roche diagnostics, Janssen and Neuraxpharm for educational activities.
Hiroshi Mori reports a grant for DIAN-J by AMED (Japanese Goverment).
Takeshi Ikeuchi has receive support for the present manuscript: AMED JP21dm0207073 to Takeshi Ikeuchi AMED JP21dk0207057 to Takeshi Ikeuchi. He has received grants or contracts from: AMED JP20dk0207049 to Takeshi Ikeuchi AMED JP20dk0207051 to Takeshi Ikeuchi AMED JP20dk0207045 to Takeshi Ikeuchi. He has received honoraria for lectures from Eisai, Daiichi-Sankyo, Ono, Takeda, and Ajinomoto.
Kazushi Suzuki has nothing to disclose.
Peter R. Schofield has receive support for the present manuscript: US National Institutes of Health, National Institute of Aging. Grant No UF1AG032438 Project Title: “Dominantly Inherited Alzheimer Network” Grant Value: Total value $US 17,935,541 Sydney site value ~$250,000 pa Principal Investigator: Prof Randall Bateman His role is as a Site Investigator. Support to his institution. He has received grants or contracts from: All payments made to my institution. 2019–2020 NSW Health Project Title: “Biospecimen Collection Grant 45 and Up - Bipolar Disorder Substudy” Grant Value $100,000 Chief Investigators: Dr Jan Fullerton, Prof Phil Mitchell, Prof Melissa Green, Prof Peter R Schofield, Dr Claudio Toma. 2020–2024 NHMRC Investigator Grant Leadership 3 Level No 1176716 Project Title: “Improving our Understanding of the Diagnosis, Prevention and Treatment of Bipolar Disorder “ Grant Value: $1,500,000 (2020 $300,000; 2021 $300,000; 2022 $300,000; 2023 $300,000; 2024 $300,000) Chief Investigator: Prof Peter R Schofield 2018–2023 NHMRC NNIDR Boosting Dementia Research Grants Priority Round 3: National Dementia Network Grant 1152623 Project Title: “The Australian Dementia Network (ADNeT): Bringing together Australia’s dementia stakeholders. Grant Value: $18,000,000 (2018 $; 2019 $; 2020 $; 2021 $; 2022 $; 2023 $) Chief Investigators: Prof Chris Rowe, Prof Perminder Sachdev, Prof Sharon Naismith, Prof Michael Breakspear, Prof Henry Brodaty, Prof Kaarin Anstey, Prof Ralph Martins, Dr Stephanie Ward, Prof James Vickers, Prof Colin Masters, Prof Peter R Schofield. 2020–2022 MRFF Mental Health Pharmacogenomics 2020 Grant 1200428 Project Title: A multifaceted approach to the pharmacogenomic signatures of bipolar disorder for improving treatment outcomes. Grant Value: $1,009,768 (2020 $304,492; 2021 $504,434; 2022 $199,942) Chief Investigators: A/Prof Jan Fullerton, Prof Melissa Green, Prof Peter R Schofield, Dr Claudio Toma 2020–2022 Spanish Internationalisation Network I-Link Grant LINKB20329 Project Title: Whole-genome sequencing and pharmacogenomic studies in a large cohort of bipolar patients with electronic administrative health records. Grant Value: Euro24,000 ($40,000) Chief Investigators: Dr Claudio Toma, A/Prof Jan Fullerton, Prof Peter R Schofield. He has held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): Remunerated positions Chief Executive Officer, Neuroscience Research Australia (ABN 94 050 110 346) Company/Association Director Positions Company Director, Neuroscience Research Australia Foundation (ABN 57 008 429 961) Company Director, The Health-Science Alliance (ACN 133 834 376) Company Director and Chief Executive Officer, Schizophrenia Research Institute (ABN 068 846 319) Company Director, Australian Association of Medical Research Institutes (AAMRI) (ABN 12 144 783 728) Company Director, Australian Dementia Network (ADNeT) Ltd (ABN 61 639 095 300) Company Director, StandingTall Pty Ltd (ACN 652 562 751) President, Australasian Neuroscience Society (ICN A842, ABN 68 737 804 032) Other Honorary Positions Member, Steering Committee, Maridulu Budyari Gumal - Sydney Partnership for Health Education, Research and Enterprise (SPHERE) Chair, National Medical Advisory Panel, The Judith Jane Mason & Harold Stannett Williams Memorial Foundation Ambassador, Business Events Sydney.
Colin L. Master has nothing to disclose.
Alison Goate has received NIH grant to support DIAN Institutional grant. She has received grants or contracts from: NIH Rainwater Charitable Foundation Picower Foundation Neurodegeneration COnsortium All were to the institution. She has received royalties or licenses: Taconic - MAPT mutations BOTH Athena Diagnostics - TDP43 mutations BOTH Millipore - licensed cell line – BOTJ. She has received consulting fees from SAB for Genentech - her UK DRI SAB - her SAB for VIB centers in Antwerp and Leuven – her. She has received personal honoraria for presentations at Eisai GSK AbbVie.
Virginia Buckles has received personal consulting fees from Washington University in St. Louis.
Nick Fox: His institution has received payments from Ixico for the use of the Boundary Shift Intergral. He has provided consultancy for Eli Lilly and for Ionis - payments were to his institution. He has participated in advisory boards for Roche and Biogen - payment was to his institution. He has served on a DSMB for Biogen - payment was to him.
Patricio Chrem has nothing to disclose.
Ricardo Allegri has nothing to disclose.
John M. Ringman has received support for the present manuscript: U19AG032438 (U.S. NIH Grant/Contract) - Payments made to his institution for enrollment. He has received grants or contracts from: All payments made to his institution. P01AG052350 (MPIs:Zlokovic/Toga) 09/30/16-05/31/21 0.6 calendar NIH/NIA $12,320,954 Vascular contributions to dementia and genetic risk factors for Alzheimer’s disease This program project proposes to study imaging and molecular biomarkers of neurovascular dysfunction in individuals at genetic risk for AD both familial and sporadic. Role: Co-Investigator U01AG051218 (PI: Ringman) 07/01/15-06/30/20 (NCE) 1.8 calendar NIH/NIA $3,856,728 The structural and functional connectome across Alzheimer Disease subtypes The goal of this project is to test the hypothesis of transynaptic spread of tau in the etiology of AD and to differentiate pathological processes in subtypes of AD and therefore inform approaches to treatment. This study will leverage the relatedness among persons with the A431E PSEN1 and V717I APP mutations to estimate mutation-associated variability in the connectome MRI. Participants are of Mexican Mestizo origin, a population typically under-represented in Alzheimer’s and other neuroscientific research. UH3NS100614 (MPIs: Wang/Ringman/Kashani) 09/30/16-07/31/21 0.8 calendar NIH/NIA $4,842,314 Imaging Cerebral and Retinal Microvasculature in Cerebral Small Vessel Disease In this project we will explore novel MRI and retinal measures of small vessel disease and their relationship to cognitive impairment and decline. We will then contribute the selection of and validation of additional markers of small vessel disease as part of a larger consortium. R01AG025340 (PI:Mather) 09/01/04-02/28/22 0.30 calendar NIH/NIA $5,787,707 Emotion-cognition interactions and aging This competitive renewal application examines the role of the locus coeruleus in attention and memory processes among healthy older adults and those with Alzheimer’s disease. Role: Co-Investigator RF1AG056573 (PI:Shi) 07/01/18-06/30/23 0.4 calendar NIH/NIA $2,457,284 Brainstem connectomes related to Alzheimer’s disease The novel brainstem connectomes developed in this project will provide the enabling technology for the in vivo examination of the relation between brainstem connectivity, tau pathology, and behavior changes in AD. This will greatly improve our understanding of the anatomical basis underlying the development of neurodegeneration and behavior symptoms in AD. These tools will also be highly valuable for studying brainstem changes in other neurological disorders. Role: Co-Investigator R21AG064776 (PI:Shi) 09/01/19-05/31/21 0.4 calendar NIH/NIA $443,940 Tau-associated pathological changes in reward networks for the prediction of neuropsychiatric symptoms in Alzheimer’s disease By studying the impact of tau pathology on reward networks, we can greatly improve our understanding of the biological mechanism underlying the development of neuropsychiatric symptoms in Alzheimer’s disease. This will no doubt help the early diagnosis and management of this important aspect of Alzheimer’s disease. Role: Co-Investigator R01AG062007 (PI:Ringman) 09/01/18-8/31/23 1.8 calendar NIH $4,108,010 Motor, Visual, and Olfactory Changes in Genetic Subtypes of AD Though changes in cognition are the most salient features of Alzheimer’s disease (AD), subtle changes in sensation and motor function occur early in the course of AD and might serve as biomarkers and provide clues as to the cascade of events leading to progressive disability. We will perform comprehensive characterization of motor and sensory changes in autosomal dominant and late-onset sporadic Alzheimer’s Disease to identify biomarkers for diagnosis, prognosis, and therapeutics targets in the management of the disease. R01NS114382 (PI:Wang) 02/01/20-01/31/25 0.30 calendar NIH $1,237,500 BBB Permeability Imaging in CADASIL This US-China Biomedical Research Collaboration project will apply cutting edge MRI technologies in a unique cohort of Chinese patients with genetically defined SVD (NOTCH3 mutation carriers suffering from cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy or CADASIL), to understand the role of blood-brain barrier (BBB) in the initiation and progression of SVD. Role: Co-Investigator P30AG066530 (MPIs:Chui/Toga/Zlokovic) 09/01/20-02/28/2025 1.00 calendar NIH/NIA $16,428,655 Alzheimer Disease Research Center (ADRC) An NIH funded research center that supports basic and clinical AD research through its administrative, clinical, imaging, pathology, education and information transfer cores. Role: Co-Lead, Clinical Core U2CAG057441(MPIs:Gershon/Weintraub) 5/1/20 – 4/30/21 0.3 calendar NIH / NIA $55,559 Advancing Reliable Measurement in Alzheimer’s Disease and cognitive Aging (ARMADA) The purpose of the ARMADA is to validate the NIH Toolbox (English and Spanish version) in existing, well-characterized, ethnically and racially diverse samples of adults ages 65–85 representing the trajectory of cognitive aging and in cognitively normal individuals 86+ years of age. NIHTB measures will be examined in relationship to clinical and biomarker indicators of disease severity and standard measures of cognitive and mental health. Sensitivity in detecting early indicators of cognitive impairment and clinically significant change in disease status will be evaluated. USC will provide Spanish-language NIH Toolbox data from persons of Mexican origin who are either cognitively normal, have mild cognitive impairment Role: PI, USC Subcontract R01AG066711 (MPIs:Jann/Wang) 04/01/20-03/31/25 0.60 calendar NIH $3,269,805 Complexity of FMRI in Alzheimer’s Disease Biomarkers for pre-clinical stages of Alzheimer’s disease (AD) have become increasingly important for the development of preventative interventions. For neuroimaging, positron emission tomography (PET) imaging of amyloid beta (Aβ) and tau protein provide early imaging markers of AD and can track disease progression but are expensive and require the use of radioactive tracers. This project aims to develop and evaluate a noninvasive, quantitative and economical imaging marker of AD based on the complexity or regularity of resting state functional magnetic resonance imaging (fMRI). Role: Co-Investigator R01EB028297(PI:Wang) 04/01/2003/31/21 0.60 calendar NIH $412,500 Multiband ASL for Neurodevelopment Study (Supplement) This project will evaluate cutting edge high-resolution magnetic resonance imaging (MRI) techniques to noninvasively detect cerebral blood flow reductions in elderly humans with mild AD and mild cognitive impairment (MCI). The successful completion of this project will lead to new noninvasive imaging biomarkers for AD and MCI. Role: Co-Investigator 666-2020-06 (PI: Ringman, Co-PI: Lew, M) 10/15/2020-10/15/2021 0.12 calendar CurePSP $30,000 In this study we propose to phenotypically characterize and collect DNA from affected and unaffected members of a family in whom progressive supranuclear palsy appears to be inherited in an autosomal dominant fashion in order to identify the disease-causing gene. Role: PI RF1AG064584 (Shi) 09/01/20-08/31/24 1.20 calendar NIH $2,130,568 Tau-induced connectome imaging markers of Alzheimer’s disease Our project will for the first time provide the comprehensive and in vivo characterization of the fiber pathways affected by tau pathology in Alzheimer’s disease (AD). This systematic investigation is important for improving our understanding of the biological mechanism of AD and provide more targeted imaging makers for the early prediction of AD at the prodromal stage. Role: Co-Investigator. He has received consulting fees from Innosense, LLC, payments made to him. He has participated on a Data Safety Monitoring Board or Advisory Board: Renew, Inc. Payments made to him.
Igor Yakushev has received grants or contracts from: Federal Ministry of Education and Research Germany (BMBF), to his institution. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: Piramal, to him.
He has held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): Board of Directors, Brain Imaging Council, SNMMI.
Christoph Laske has nothing to disclose.
Mathias Jucker has received grants or contracts from: all grants to the instituion (DFG, IMI2, AluCure). He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: Roche (private) Synapsis (private).
Günter Höglinger has nothing to disclose.
Randall J. Bateman has receive support for the present manuscript: National Institute on Aging (NIA). He has received grants or contracts from: Avid Radiopharmaceuticals, Janssen, Eisai, Genentech, Abbvie, Biogen, Centene, United Neuroscience, Eli Lilly & Co, Hoffman-LaRoche. He has equity ownership interest in C2N Diagnostics and receive royalty income based on technology (stable isotope labeling kinetics and blood plasma assay) licensed by Washington University to C2N Diagnostics. He has received consulting fees from Janssen, Eisai, C2N Diagnostics, AC Imunne, Amgen, Hoffman-LaRoche, Pfizer. He has received support for attending meetings and/or travel: AC Immune, Hoffman-LaRoche. He has participated on a Data Safety Monitoring Board or Advisory Board: C2N Diagnostics, Hoffman-LaRoche, Pfizer. He has held stock or stock options in entities related to the current manuscript and/or area of research included in this manuscript or related area of research: C2N Diagnostics- Equity ownership interests.
Adrian Danek has received grants or contracts from: Advocacy for Neuroacanthocytosis Patients -> Institution. He has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events: three hospitals in Switzerland -> him. He has received payment for expert testimony: Munich court of law -> him. He has held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid): Advocacy for Neuroacanthocytosis Patients: unpaid.
Johannes Levin reports speaker fees from Bayer Vital, Biogen and Roche, consulting fees from Axon Neuroscience and Biogen, author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers, non-financial support from Abbvie and compensation for duty as part-time CMO from MODAG, outside the submitted work.
Abbreviations
- AD-NEF
Alzheimer disease neurological examination findings
- MC
mutation carriers
- Non MC
non mutation carriers
- ADAD
autosomal dominant Alzheimer disease
- DIAN
Dominantly Inherited Alzheimer Network
- CDR-SB
Clinical Dementia Rating – Sum of Boxes
References
- 1.Gowers W A manual of diseases of the nervous system. 1888
- 2.Fox MD. Mapping Symptoms to Brain Networks with the Human Connectome. The New England journal of medicine. Dec 6 2018;379(23):2237–2245. doi: 10.1056/NEJMra1706158 [DOI] [PubMed] [Google Scholar]
- 3.Nicholl DJ, Appleton JP. Clinical neurology: why this still matters in the 21st century. J Neurol Neurosurg Psychiatry. 2015;86(2):229–233. doi: 10.1136/jnnp-2013-306881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampton JR, Harrison MJ, Mitchell JR, Prichard JS, Seymour C. Relative contributions of history-taking, physical examination, and laboratory investigation to diagnosis and management of medical outpatients. Br Med J. 1975;2(5969):486–489. doi: 10.1136/bmj.2.5969.486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paley L, Zornitzki T, Cohen J, Friedman J, Kozak N, Schattner A. Utility of Clinical Examination in the Diagnosis of Emergency Department Patients Admitted to the Department of Medicine of an Academic Hospital. Archives of Internal Medicine. 2011;171(15):1393–1400. doi: 10.1001/archinternmed.2011.340 [DOI] [PubMed] [Google Scholar]
- 6.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. The New England journal of medicine. Aug 30 2012;367(9):795–804. doi: 10.1056/NEJMoa1202753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storandt M, Balota DA, Aschenbrenner AJ, Morris JC. Clinical and psychological characteristics of the initial cohort of the Dominantly Inherited Alzheimer Network (DIAN). Neuropsychology. Jan 2014;28(1):19–29. doi: 10.1037/neu0000030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryan NS, Nicholas JM, Weston PSJ, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. The Lancet Neurology. Dec 2016;15(13):1326–1335. doi: 10.1016/s1474-4422(16)30193-4 [DOI] [PubMed] [Google Scholar]
- 9.Tang M, Ryman DC, McDade E, et al. Neurological manifestations of autosomal dominant familial Alzheimer’s disease: a comparison of the published literature with the Dominantly Inherited Alzheimer Network observational study (DIAN-OBS). The Lancet Neurology. Dec 2016;15(13):1317–1325. doi: 10.1016/s1474-4422(16)30229-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol. Oct 2009;66(10):1254–9. doi: 10.1001/archneurol.2009.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauser WA, Morris ML, Heston LL, Anderson VE. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology. Sep 1986;36(9):1226–30. doi: 10.1212/wnl.36.9.1226 [DOI] [PubMed] [Google Scholar]
- 12.Vöglein J, Noachtar S, McDade E, et al. Seizures as an early symptom of autosomal dominant Alzheimer’s disease. Neurobiology of aging. 2019/04/01/ 2019;76:18–23. doi: 10.1016/j.neurobiolaging.2018.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vöglein J, Paumier K, Jucker M, et al. Clinical, pathophysiological and genetic features of motor symptoms in autosomal dominant Alzheimer’s disease. Brain : a journal of neurology. 2019;142(5):1429–1440. doi: 10.1093/brain/awz050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vöglein J, Ricard I, Noachtar S, et al. Seizures in Alzheimer’s disease are highly recurrent and associated with a poor disease course. Journal of Neurology. 2020/10/01 2020;267(10):2941–2948. doi: 10.1007/s00415-020-09937-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR Jr., Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. The Lancet Neurology. Jun 2017;16(6):435–444. doi: 10.1016/s1474-4422(17)30077-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295–e1306. doi: 10.1212/wnl.0000000000006277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bateman RJ, Aisen PS, De Strooper B, et al. Autosomal-dominant Alzheimer’s disease: a review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s research & therapy. Jan 6 2011;3(1):1. doi: 10.1186/alzrt59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cairns NJ, Perrin RJ, Franklin EE, et al. Neuropathologic assessment of participants in two multi-center longitudinal observational studies: the Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology : official journal of the Japanese Society of Neuropathology. Aug 2015;35(4):390–400. doi: 10.1111/neup.12205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ringman JM, Monsell S, Ng DW, et al. Neuropathology of Autosomal Dominant Alzheimer Disease in the National Alzheimer Coordinating Center Database. Journal of neuropathology and experimental neurology. Mar 2016;75(3):284–90. doi: 10.1093/jnen/nlv028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryman DC, Acosta-Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. Jul 15 2014;83(3):253–60. doi: 10.1212/wnl.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanewinckel R, van Oijen M, Ikram MA, van Doorn PA. The epidemiology and risk factors of chronic polyneuropathy. European journal of epidemiology. Jan 2016;31(1):5–20. doi: 10.1007/s10654-015-0094-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tinetti ME, Kumar C. The patient who falls: “It’s always a trade-off”. Jama. Jan 20 2010;303(3):258–66. doi: 10.1001/jama.2009.2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grecula MJ, Caban ME. Common orthopaedic problems in the elderly patient. Journal of the American College of Surgeons. May 2005;200(5):774–83. doi: 10.1016/j.jamcollsurg.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 24.Schott JM. The neurology of ageing: what is normal? Practical neurology. Jun 2017;17(3):172–182. doi: 10.1136/practneurol-2016-001566 [DOI] [PubMed] [Google Scholar]
- 25.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer disease and associated disorders. Oct-Dec 2006;20(4):210–6. doi: 10.1097/01.wad.0000213865.09806.92 [DOI] [PubMed] [Google Scholar]
- 26.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. Nov 1993;43(11):2412–4. [DOI] [PubMed] [Google Scholar]
- 27.Marsh JL, Hutton JL, Binks K. Removal of radiation dose response effects: an example of over-matching. BMJ. 2002;325(7359):327. doi: 10.1136/bmj.325.7359.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage. Jul 1 2006;31(3):968–80. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 29.Cedarbaum JM, Jaros M, Hernandez C, et al. Rationale for use of the Clinical Dementia Rating Sum of Boxes as a primary outcome measure for Alzheimer’s disease clinical trials. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. Feb 2013;9(1 Suppl):S45–55. doi: 10.1016/j.jalz.2011.11.002 [DOI] [PubMed] [Google Scholar]
- 30.Gordon BA, Blazey TM, Su Y, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. The Lancet Neurology. Mar 2018;17(3):241–250. doi: 10.1016/s1474-4422(18)30028-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popper K, Miller D. A proof of the impossibility of inductive probability. Nature. 1983/04/01 1983;302(5910):687–688. doi: 10.1038/302687a0 [DOI] [Google Scholar]
- 32.Rosch E Principles of Categorization. In: Rosch EL BB, ed. Cognition and Categorization. Lawrence Erlbaum Associates; 1978:27–48. [Google Scholar]
- 33.O’Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer’s research consortium study. Arch Neurol. 2008;65(8):1091–1095. doi: 10.1001/archneur.65.8.1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karlstrom H, Brooks WS, Kwok JB, et al. Variable phenotype of Alzheimer’s disease with spastic paraparesis. Journal of neurochemistry. Feb 2008;104(3):573–83. doi: 10.1111/j.1471-4159.2007.05038.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.