

Abstract
Cancer stem cells (CSCs) serve an essential role in failure of conventional antitumor therapy. In breast cancer, CD24−/low/CD44+ phenotype and high aldehyde dehydrogenase activity are associated with CSC subtypes. Furthermore, CD24−/low/CD44+ pattern is also characteristic of mesenchymal cells generated by epithelial-mesenchymal transition (EMT). CD24 is a surface marker expressed in numerous types of tumor, however, its biological functions and role in cancer progression and treatment resistance remain poorly documented. Loss of CD24 expression in breast cancer cells is associated with radiation resistance and control of oxidative stress. Reactive oxygen species (ROS) mediate the effects of anticancer drugs as well as ionizing radiation; therefore, the present study investigated if CD24 mediates radiation- and chemo-resistance of breast cancer cells. Using a HMLE breast cancer cell model, CD24 expression has been artificially modulated and it was observed that loss of CD24 expression induced stemness properties associated with acquisition of a hybrid E/M phenotype. CD24−/low cells were more radiation- and chemo-resistant than CD24+ cells. The resistance was associated with lower levels of ROS; CD24 controlled ROS levels via regulation of mitochondrial function independently of antioxidant activity. Together, these results suggested a key role of CD24 in de-differentiation of breast cancer cells and promoting acquisition of therapeutic resistance properties.
Keywords: breast cancer, CD24, cancer stem cells, epithelial- mesenchymal transition, resistance
Introduction
Tumor heterogeneity is a key characteristic of cancer and the presence of cancer stem cells (CSCs) has become increasingly associated with treatment failure and tumor progression/relapse (1,2). CSCs are operationally defined as a subset of cancer cells that i) is capable of self-renewal, ii) has tumor-initiating ability and iii) is resistant to ionizing radiation and chemotherapy (3). CSCs can be identified by various markers. CSCs from human breast tumors, characterized as having a CD24−/low/CD44+ phenotype, were first identified for their tumorigenic potential in xeno-transplanted immune-deficient mice (4). Additional markers are associated with CSC characteristics and high aldehyde dehydrogenase activity (ALDH+), a marker of normal and malignant human mammary stem cells, is a predictor of poor clinical outcome in breast cancer (5). The subpopulations detected by these markers only partially overlap, indicating that different lineages of CSCs may coexist within the same tumor (6).
In addition, evidence connects epithelial-mesenchymal transition (EMT) with the acquisition of stem cell properties (7). Many dysregulated pathways in breast CSCs [Notch, hedgehog, Wingless (Wnt), transforming growth factor β (TGFβ) and NFκB], via activation of mesenchymal transcription factors (Twist, Snail and Zeb), induce EMT (8). Most mesenchymal (M) cells generated by EMT acquire a CD24−/low/CD44+ pattern (7,9,10). On the other hand, studies have shown the existence of epithelial (E)-like breast CSCs with a CD24+/CD44low phenotype and high ALDH activity (5,11). Finally, several transition states occurring during EMT have been identified and a hybrid E/M phenotype is associated with increased tumor stemness (12,13). In breast cancer, CSCs are endowed with plasticity that enables them to reverse transition between E and M states and increased metastatic or tumorigenic potential and poor clinical prognosis have been associated with CSCs in a hybrid E/M state (12,14).
By definition, breast CSCs are relatively resistant to traditional cancer therapies, including chemotherapy and radiation therapy (15) and this resistance has been observed for CD24−/low/CD44+ M CSCs but also for ALDH+ E CSCs (16–19). Different mechanisms have been proposed to explain the genotoxic stress survival of CSCs (20), including cell quiescence, increased ability to repair DNA damage, increased antiapoptotic defense, dysregulation of autophagy, metabolic changes and resistance to reactive oxygen species (ROS); many of these pathways are mediated by redox imbalance (21).
Our previous study showed that high-dose irradiation (IR) of breast cancer cell lines leads to transitory selection of a CD24−/low subpopulation independently of CD44 expression (22) and that loss of CD24 expression promotes radiation resistance association with decreased ROS levels (23). Lower intrinsic levels of ROS, associated with radioresistance, are also characteristic of CD24−/low/CD44+ M breast CSCs (10,16,24).
CD24 is a small cell surface protein molecule frequently overexpressed in human cancer, especially breast cancer (25,26). CD24 is hypothesized to function as an adhesion molecule but due to variable glycosylation pattern it acts as a versatile ligand with diverse physiological functions (26), making its mechanisms complex to understand. Despite CD24 being a marker strongly associated with EMT in breast cancer, its biological functions and role in cancer progression and treatment resistance remain poorly documented. Moreover, clinical studies on the association between CD24 expression and breast tumor progression are conflicting, especially due to the poor specificity of CD24 antibodies used in these studies (27,28).
ROS mediate the effects of anticancer drugs as well as ionizing radiation (29–32), and our previous study observed an association between CD24 expression and ROS levels (10); therefore it was hypothesized CD24 may serve a role in the radio- and chemo-resistance of breast cancer cells.
The present study used the non-tumorigenic HMLE cell model developed by Mani in R. Weinberg's team (7) to study EMT in breast cancer cells, with the main advantage to study E and M cells in the same genetic background (CD24−/low/CD44+ M-HMLE cells are obtained after induction of EMT by TGFβ in parental CD24+/CD44low E-HMLE). Here, we investigated if CD24 could be defined as an actor of both radiation- and chemo-resistance of breast cancer cells.
Materials and methods
Cell culture
All cell lines were cultivated in Heraeus Thermo Scientific BBD6220 incubator at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Presence of mycoplasma was regularly tested with the MycoAlert™ Mycoplasma detection kit from Lonza Group, Ltd. Tumor breast cancer cell lines T47D and MCF7 were from the American Type Culture Collection and Human Mammary Epithelial HMLE cell line was kindly provided by Professor Robert A. Weinberg (Whitehead Institute). HMLE cells come from Human Mammary Epithelial Cells (HMECs) immortalized by human telomerase reverse transcriptase (hTERT) and transformed by large T antigen (SV40) (33). E and M HMLE phenotypes were previously characterized by Mani in R. Weinberg's team (7) and our group (10). All cell lines were grown in adherent conditions in cell culture media supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Sigma-Aldrich; Merck KGaA) and 1 mM antibiotic-antimycotic (Invitrogen; Thermo Fisher Scientific, Inc.) For T47D and MCF7, Dulbecco's Modified Eagle's Medium (DMEM), high glucose, GlutaMAX™ Supplement, pyruvate (Gibco; Thermo Fisher Scientific, Inc.) was used. HMLE cells were cultured in DMEM/F-12 Nutrient Mixture, GlutaMAX Supplement (Gibco; Thermo Fisher Scientific, Inc.) with 10 ng/ml human epidermal growth factor, 0.5 µg/ml hydrocortisone and 10 µg/ml insulin (all Sigma-Aldrich; Merck KGaA). Medium for transfected cell lines was supplemented with 0.4 puromycin and 16.0 µg/ml blasticidin (both Gibco; Thermo Fisher Scientific, Inc.) during the transfectant selection phase and 0.2 puromycin and 8.0 µg/ml blasticidin for routine cell culture. Mesenchymal M HMLE cells were induced and FACS-sorted after a 10 day treatment with 2.5 ng/ml recombinant TGFβ1 (Thermo Fisher Scientific, Inc.). Cell proliferation and survival analysis were performed in ≥3 experiments, by scoring cells with a TC20 automated cell counter (Bio-Rad Laboratories, Inc.). Mammosphere formation assay was performed as described by Lombardo et al (34) (Appendix S1).
Chemicals, reagents and antibodies
5-Fluorouracil (5FU), cisplatin and paclitaxel (Taxol) were from Sigma-Aldrich (Merck KGaA). 5FU and paclitaxel were diluted in DMSO to concentration stock of 400 and 1 mM, respectively. Cisplatin was diluted with sodium chloride to a concentration stock of 1 mM. Each drug was used at different concentrations depending on the experiments. Antibodies against CD24 (clone ML5, Cat no: 555428), CD44 (clone no. G44-26, Cat. no: 555478) and isotypic controls were from BD Biosciences.
Plasmids and transfection
To stably knock down expression of CD24 in MCF-7, T47D and HMLE cells, replicative short hairpin (shRNA)-expressing vectors [puromycin-resistant pEBV-small interfering (si)RNA] that impose a strong and stable gene silencing in human cells, even after several months in culture, were used (plasmid backbone: pEBVsiRNA, patent n°WO2006085016, CEA). siRNA design and cloning in pEBVsiRNA vectors and establishment of stable knockdown and control clones were performed as previously described (35). To design shRNA sequences, DSIR program developed by Vert et al (36) was used. Forward shCD24 sequence (pBD2506 plasmid): 5′-GATCCCGCCAAGAAACGTCTTCTAAATTCAAGAGATTTAGAAGACGTTTCTTGGTTTTTTGGAAA-3′; Reverse shCD24 sequence: 5′-AGCTTTTCCAAAAAACCAAGAAACGTCTTCTAAATCTCTTGAATTTAGAAGACGTTTCTTGGCGG-3′. Control cells carried the pBD650 plasmid which expressed an inefficient shRNA validated on more than 200 genes. Forward shCTL sequence: 5′-GATCCCGAATTGCGGCGAGCAGTAATTCAAGAGATTACCTGCTCGCCGCCAATTCTTTTTGGAAA-3′; Reverse shCTL sequence: 5′-AGCTTTTCCAAAAAAGTCAAGAAGCATTAGAAGATCTCTTGAATCTTCTAATGCTTCTTGACGG-3′.
To express CD24 into HMLE cells already depleted for CD24, open reading frame (ORF) of human CD24 was amplified from an IMAGE clone (ID_5591617; Thermo Fisher Scientific, Inc.) with the following primers: 5′-ATGGGCAGAGCAATGGTGGCCAGGCTC-3′ and 5′-TTAAGAGTAGAGATGCAGAAGAGAGAG-3′ and introduced into a blasticidin-resistant pEBV plasmid downstream of a CAG promoter (pBD2915; CEA).
After one day of seeding (density: 50%) into 6-well plate; MCF-7, T47D and HMLE cells were transfected with jetPRIME® reagent (Polyplus-transfection SA) according to the manufacturer's recommendations. Following 24 h incubation, cells were trypsinized and seeded (density: 25%) in culture medium supplemented with puromycin alone or with blasticidin. Experiments were performed either on the whole transfected population or on selected clones.
IR
For all experiments except clonogenic assay, cells were plated at least 24 h prior to IR. On day 0, γ-IR of cells were performed on a GSR D1 irradiator (Gamma Medical Service GmbH; Appendix S1). Studied cells were irradiated at 2, 4, 6 and 10 Gy and control cells were submitted to sham IR.
Aldefluor assay
The Aldefluor kit (Stemcell Technologies, Inc.) was used to analyze the population with high ALDH enzymatic activity according to the manufacturer's intructions. HMLE cells were incubated in the Aldefluor assay buffer containing ALDH substrate (BODIPY aminoacetaldehyde (BAAA); 1 µmol/l/1×106 cells) and incubated for 40 min at 37°C. As negative control, for each sample of cells, an aliquot was treated with 50 mmol/l diethylaminobenzaldehyde, a specific ALDH inhibitor.
Clonogenic assay
Sub-confluent HMLE cells were trypsinized using TrypLE express solution (Thermo Fisher Scientific, Inc.). Live cells were counted using an automated cell counter (TC20; Bio-Rad Laboratories, Inc.) and trypan blue exclusion. In the IR group, cells were immediately irradiated in suspension to generate a dose curve of 0, 2, 4 and 6 Gy and colony-forming assay was performed immediately following IR by plating cells in 60-mm Petri dishes, in triplicate.
In the drug treatment group, following trypsinization and counting, cells were plated in 60-mm diameter Petri dishes, in triplicate. At 6 h after plating, drug treatment (4 to 16 µM exposition to 5FU, 2 to 6 µM exposition to Cisplatin and 1 to 3 nM exposition to Paclitaxel) was performed for three days, then cells were washed (PBS) and incubated for 7 days with fresh medium.
Number of cells seeded increased with radiation dose or drug concentration but was identical for each cell line tested. After 7 days, cells were fixed at room temperature for 30 min in 4% paraformaldehyde, washed (PBS) and stained at room temperature for at least 2 h in methylene blue/30% methanol. Colonies containing >50 cells were manually counted. The surviving fraction at each radiation dose was normalized to that of the non-irradiated sample and points were fitted using an exponential tendency curve. At least three independent experiments were performed.
Cell staining
CSC marker labeling and analysis were performed as described by Bensimon et al (22,23). Intracellular concentrations of ROS prooxidants were determined using dihydroethidium (DHE) probe (Invitrogen; Thermo Fisher Scientific, Inc.). Adherent HMLE cells were incubated with 10 µM DHE for 30 min at 37°C, then washed with PBS, trypsinized and immediately analyzed by flow cytometry. Intracellular concentrations of mitochondrial ROS were determined using MitoSOX™ Red Mitochondrial Superoxide Indicator (Invitrogen; Thermo Fisher Scientific, Inc.). Adherent cells were incubated with 5 µM MitoSOX Red for 10 min at 37°C, washed with PBS, trypsinized and immediately analyzed by flow cytometry. Mitochondrial mass was analyzed using MitoTracker™ Green (MTG) probe (Invitrogen; Thermo Fisher Scientific, Inc.). Following soft trypsinization, cells were loaded with 200 nM MTG and incubated for 20 min at 37°C, then immediately analyzed by flow cytometry. Mitochondrial membrane potential was quantified using tetramethylrhodamine, ethyl ester, perchlorate (TMRE) probe (Invitrogen; Thermo Fisher Scientific, Inc.). Following soft trypsinization, cells were loaded with 10 nM TMRE and incubated for 20 min at 37°C, then immediately analyzed by flow cytometry.
Flow cytometry
Cells were analyzed on a SORP LSR-II analyzer (configuration: 488, 561, 405, 355 and 635 nm; BD Biosciences) or BD FACSCalibur (configuration: 488 and 635 nm; BD Biosciences). Data were analyzed with FlowJo v10.7.1 (Tree Star). Cells were sorted on a BD Influx sorter (BD Biosciences; configuration: 488, 561, 405, 355 and 635 nm).
RNA extraction and reverse transcription-quantitative (RT-q)PCR
Total RNA was extracted from frozen MCF-7, T47D and HMLE cell pellets (−80°C) with Total RNA purification kit (Norgen Biotek Corp.), according to the manufacturer's instructions. DNAse treatment was performed using TURBO DNA-free kit (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions. cDNA synthesis was performed with the SuperScript™ VILO™ cDNA Synthesis kit (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. RT-qPCR was performed with ABI Prism 7300 detection apparatus (Applied Biosystems; Thermo Fisher Scientific, Inc.) using Taqman Universal Master Mix according to the manufacturer's recommendations. Cq value was determined with Sequence Detection System software. All primers were from Applied Biosystems (Thermo Fisher Scientific, Inc.; Appendix S1). Levels of gene expression were determined using GENORM software and normalized using GAPDH and RPLPO.
Cell migration analysis
Cells were plated on 24-well glass bottom dishes at 4,000 cells/well. Live-cell imaging was performed using Plan APO 20× DIC objective (Numerical aperture: 0.7) on a Nikon A1R confocal laser scanning microscope system attached to an inverted ECLIPSE Ti (Nikon Corporation) thermostated at 37°C under a 5% CO2 atmosphere. Mosaic images were recorded every 15 min for 5 h. Tracking of individual cells was performed using the MtrackJ plugin in ImageJ software (37). Dynamic parameters such as migration velocity and mean square displacement (MSD) were calculated with the open-source computer program DiPer (38).
Statistical analysis
All statistical tests were performed using GraphPad Prism 8 (GraphPad Software, Inc.). ANOVA or Kruskal-Wallis tests followed by a suitable multiple comparisons correction test and Mann-Whitney tests were used. Each test used is specified in the legend of the figures. Data are presented as the mean ± SD of independent experiments (minimum three, depending of the experiments). P<0.05 was considered to indicate a statistically significant difference.
Results
Low CD24 expression defines a radio- and chemo-resistant subpopulation of breast cancer cells
To investigate the role of CD24 in resistance to γ-IR and widely used anticancer drugs, the present study investigated CD24 expression in epithelial HMLE cells following high dose IR and treatment with high concentrations of three drugs with different mechanisms of action: 5FU, cisplatin and paclitaxel. CD24 expression was analyzed by flow cytometry and CD24−/low subpopulation was defined as the 10% of cells expressing the lowest fluorescence in untreated cells.
Following IR, dose-dependent cell death was observed, which is a delayed apoptotic process (10) (Fig. 1A). IR selectively enriches the CD24−/low cells subpopulation (Fig. 1A). Similarly, chronic exposure to increased concentrations of anticancer drugs led to dose-dependent cell death (Fig. 1B-D). For the three drugs used, a clear increase in the percentage of CD24−/low cells was observed following chronic exposure. It was next investigated whether CD24−/low cells were more radio- and chemo-resistant than CD24+ cells. These populations were isolated by flow cytometry (Fig. 1E) and studied separately. Membrane expression levels of CD24 observed by FACS were associated with CD24 mRNA expression in CD24−/low and CD24+ cells (Fig. 1E). Following 6 and 10 Gy IR, death rate was significantly lower in CD24−/low cells than in CD24+ cells (Fig. 1F). In the same way, following treatment with high doses of anticancer drugs, death rate was significantly decreased in CD24−/low cells compared with CD24+ cells. Therefore, CD24−/low cells exhibited an enhanced ability to survive following IR and chronic exposure to chemotherapeutic agents. Long-term culture of sorted CD24−/low HMLE cells showed the appearance of CD24+ cells after six days and after 21 days, the heterogeneity of CD24 levels in parental population was observed (Fig. S1).
Figure 1.

Low CD24 expression defines a radio- and chemo-resistant subpopulation of HMLE cells. CD24−/low subpopulation was defined as the 10% of cells expressing the lowest fluorescence in control cells. (A) Percentage of dead and CD24−/low HMLE cells at five days following high dose irradiation (4–10 Gy). Percentage of dead and CD24−/low HMLE cells following three day exposure to high concentrations of (B) 5-fluorouracil (5FU), (C) cisplatin and (D) paclitaxel. Data are presented as the mean ± SD of 3 independent experiments. Significant differences (compared with untreated cells) were analyzed by one-way ANOVA followed by Dunnett's multiple comparisons correction test. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. ns, not significant. (E) CD24−/low and CD24+ HMLE cell subpopulations were analyzed by flow cytometry following CD24/CD44 staining. CD24 expression was analyzed by reverse transcription-quantitative PCR. (F) Percentage of CD24−/low and CD24+ dead cells five days after 4 and 6 Gy irradiation and following three day exposure to 400 µM 5FU, 15 µM cisplatin and 10 nM paclitaxel. Data are presented as the mean ± SD of 3 independent experiments. Significant differences (compared with untreated cells) were analyzed by 2 way ANOVA followed by Sidak's multiple comparisons correction test. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. ns, not significant.
Taken together, these results indicated that the CD24−/low cell subpopulation was more radio- and chemo-resistant than CD24+ cells. Hence, IR or drug exposure led to transient enrichment of the CD24−/low cell subpopulation in the whole cell culture.
Loss of CD24 expression does not modify overall E features of HMLE cells but induces hybrid E/M state
To determine whether CD24 mediated radio- and chemo-resistance of breast cancer cells, E HMLE cells were transfected with p-EBV-plasmid expressing siRNA against CD24 (E_CD24−). As control, the parental CD24+/CD44low HMLE cells were transfected with p-EBV vector expressing inefficient shRNA sequence (E_vec). Flow cytometry confirmed the loss of surface expression of CD24 in E_CD24− cells compared with E cells (Fig. 2A). A purified population of M HMLE cells was obtained following FACS analysis of CD24−/low/CD44+ cells induced by prolonged exposure to TGFβ1 (10). To discard potential off-target effects of the siRNA, CD24-complemented E cells (E_CD24−c) were generated by transfecting E_CD24− cells with a p-EBV-plasmid coding for CD24 ORF. High levels of CD24 were observed at the membrane of E_CD24−c cells (Fig. 2A). Modulation of CD24 expression levels was further validated by RT-qPCR analysis, showing a strong downregulation in E_CD24− cells compared with E or E_vec cells (Fig. S2).
Figure 2.

Characterization of E, M and E-transfected HMLE cells. (A) Flow cytometry characterization following CD24/CD44 labelling of parental E cells, E cells transfected with control p-EBV vector (E_vec) and p-EBV-plasmid expressing a CD24 small interfering RNA (E_CD24−), E_CD24− cells transfected with p-EBV-plasmid expressing CD24 mRNA (E_CD24−c) and M cells obtained following FACS analysis of CD24−/low/CD44+ cells induced by prolonged exposure to TGFβ1. (B) Phase-contrast images of E, E_vec, E_CD24−, E_CD24−c and M cells. Analysis by reverse transcription-quantitative PCR of relative expression of mRNAs encoding (C) EMT-associated factors (CD44, ESA, E-cad, N-cad and Fn1), (D) primary transcription factors of EMT (Twist-1, Twist-2, Snail-1, Snail-2, Zeb-1 and Zeb-2) and (E) other factors involved in EMT (Vim, Krt14, DTP63a and OVOL2) in E_CD24-, E_CD24-c and M compared with E cells. Results were normalized to expression levels in E cells. Data are presented as the mean ± SD of 3 independent experiments. Significant differences (compared with E cells) were analyzed by one-way ANOVA followed by Dunnett's multiple comparisons correction test. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. ns, not significant; E, epithelial; M, mesenchymal; EMT, E-M transition; ESA, epithelial cell adhesion molecule; Cad, cadherin; Fn, fibronectin; Zeb, zinc finger E-box binding homeobox; Vim, vimentin; Krt, keratin; OVOL2, ovo-like zinc finger 2.
The effect of CD24 expression on cellular phenotypes classically associated with EMT in breast cancer was assessed. E_CD24− cells kept the cuboidal-like E morphology of the parental E cells and formed cobblestone cell islands (Fig. 2B). This cobblestone morphology was different from that observed for fibroblast-like M cells. To confirm that silencing of CD24 did not modify E characteristics of transfected E HMLE cells, the expression of EMT-associated genes was measured by RT-qPCR (Fig. 2C). The results were normalized to expression levels in E cells. ESA (Ep-CAM) and E-cadherin, detectable at low levels in M cells compared with E cells were not affected by altered expression of CD24. In the same way, CD44, N-cadherin and fibronectin mRNAs expression levels were markedly increased in M cells but not in transfected E cells. The expression of mRNAs encoding the primary transcription factors driving EMT was also studied [Twist-1, Twist-2, Snail-1, Snail-2, zinc finger E-box binding homeobox (Zeb)-1 and Zeb-2; Fig. 2D]. Expression of these mRNAs was increased in M cells but not in E transfected cells with the exception of Snail-1, which was upregulated when CD24 expression was decreased. However, expression of other markers suggested that E_CD24− cells were not in a completely E state (Fig. 2E). Keratin 14, a classical E marker in breast cancer (39), was downregulated in E_CD24− and M cells compared with E cells. Vimentin was weakly upregulated and E transcription factors ovo-like zinc finger (OVOL)2 and ΔNp63α, the predominant p63 isoform in mammary E cells (40) were weakly expressed in E_CD24− cells. Because recent studies have suggested that NRF2 activates partial EMT and is maximally present in a hybrid E/M phenotype (41,42), NRF2 expression was investigated. Upregulation of NRF2 was observed in M cells but not in E_CD24- cells (data not shown). Therefore, in the present model, overexpression of NRF2 was not associated with hybrid E/M state.
To ensure these results were not specific to the HMLE cells model, the T47D epithelial breast cell line was transfected with the p-EBV-plasmid expressing CD24 siRNA to obtain a low CD24 expression (T47D_CD24− cells). T47D_CD24− cells kept the cuboidal-like E morphology of parental cells and formed cobblestone cell islands, but upregulation of Vimentin and downregulation of DNp63a expression was also observed, indicating that cells were not in a completely E state (Fig. S3).
Therefore, these results indicated that loss of CD24 expression does not modify E characteristics of breast E cells but induces an intermediate hybrid E/M state.
Stemness properties and migration potential are associated with hybrid E/M phenotype of E_CD24− HMLE cells
Stemness properties acquisition during EMT is associated with the appearance of cells in an intermediate state, often characterized as E-like CSCs displaying increased ALDH1 activity (11). The present study investigated whether ALDH1 activity was associated with CD24 expression in HMLE cells (Figs. 3A and S4). A total of 15–20% of parental E cells were ALDH+; this subpopulation reached 40% in E_CD24− cells and re-expression of CD24 (E_CD24−c cells) abolished this increase. ALDH+ subpopulation was absent in CD24−/low/CD44+ M cells.
Figure 3.

Stemness properties and migration potential of E_CD24− HMLE cells. (A) Percentage of Aldefluor-positive subpopulation defined by Aldefluor assay. (B) Mammosphere formation efficiency. Data are presented as the mean ± SD of 3 independent experiments. Significant differences were analyzed by ANOVA, followed by Sidak's for (A) and Dunnett's multiple comparisons correction test for (B). **P<0.01, ****P<0.0001. Cell migration potential analysis using DiPer program. (C) Visualization of cell trajectory. (D) Measurement of cell surface area using MSD analysis. Data are presented as the mean ± SD of 3 independent experiments. ns, not significant; E, epithelial; M, mesenchymal; MSD, mean square displacement.
Another feature of breast CSCs is their potential to form spheres (mammospheres) (7). There was a significant increase in mammosphere formation efficiency (MFE) of CD24−/low/CD44+ M cells compared with E cells (Fig. 3B). The MFE was also significantly increased in E_CD24− cells compared with parental E cells and re-expression of CD24 (E_CD24−c cells) abolished this increase.
CSCs, as well as hybrid and M tumor cells, are associated with motility and tumor propagating characteristics (12); therefore, migration potential of HMLE cells was investigated. MSD analysis indicated that M cells exhibited a markedly higher migratory potential than E cells (Fig. 3C and D). In the same way, E_CD24− cells presented significantly increased migratory potential than E cells; this increase was abolished following re-expression of CD24 (E_CD24−c cells).
Altogether, these results indicated that E_CD24− cells in a hybrid E/M state were associated with the acquisition of stemness properties. Thus, CD24 knockdown may influence stemness properties, leading to ALDH+ E-like CSCs rather than CD24−/low/CD44+ M-like CSCs.
Loss of CD24 expression alone promotes radio- and chemo-resistance of E HMLE cells
To determine effects of CD24 on radiation and drug sensitivity, HMLE cells were irradiated at 10 Gy or treated with high concentration 5FU, cisplatin and paclitaxel. Delayed cell death was observed between E and M cells 6 days after IR (Fig. 4A). In CD24-/low/CD44+ M cells, cell death was significantly delayed and remained lower than in E (or E_vec) cells. Cell death rate after IR was significantly decreased in E_CD24− cells compared with parental E cells and re-expression of CD24 (E_CD24−c cells) abolished this resistance, indicating that CD24 controls radiation response. In the same way, loss of CD24 expression induced resistance to chronic exposure of anticancer drugs, similar to that observed in M cells (Fig. 4B). When CD24 was re-expressed, E_CD24−c cells became drug sensitive, cell death was restored to a similar level as that in parental cells. These data indicated that E_CD24− cells exhibited an enhanced ability to survive following IR and chronic treatment with anticancer drugs.
Figure 4.

Decreased CD24 expression enhances radio- and chemo-resistance in HMLE.E cells. (A) Time course of death of 10 Gy-irradiated cells. Data are presented as the mean ± SD of 3 independent experiments. (B) Percentage of dead cells following three day exposure to 400 µM 5FU, 15 µM cisplatin and 10 nM paclitaxel. Data are presented as the mean ± SD of 4–12 independent experiments. Significant differences (compared with E cells) were analyzed by one-way ANOVA with Kruskal-Wallis followed by Dunn's multiple comparisons correction test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (C) Clonogenic cell survival curves following 2–6 Gy irradiation, 4–16 µM 5FU, 2–6 µM cisplatin and 1–3 nM paclitaxel treatment. ns, non-significant; 5FU, 5-fluorouracil; E, epithelial; M, mesenchymal.
To ensure that these results were not specific to the HMLE cell model, E breast cell lines MCF7 and T47D were also transfected with the p-EBV-plasmid expressing a CD24 siRNA to obtain two lines with a low CD24 expression (MCF7_CD24− and T47D_CD24− cells). Forced extinction of CD24 expression alone promoted resistance of MCF7_CD24− and T47D_CD24− cells against chronic exposure to high dose 5FU and cisplatin (Fig. S5).
It was next investigated whether differences in cell death rate following IR or drug treatment between CD24− and CD24+ cells had an impact on long-term survival, as measured by clonogenic assay. Following IR (Fig. 4C), surviving fractions at 2, 4 and 6 Gy were significantly higher in E_CD24− than in control E/E_vec cells and similar to those observed in M cells. E_CD24−c cells exhibited decreased of cloning efficiency, similar to that in parental E cells. Following drug treatment, similar results were observed: Surviving fractions for all doses of drug tested were higher in E_CD24− and M cells than in parental E and E_CD24− cells. These results indicate that E_CD24− and M cells exhibited greater clonogenic capacity than CD24+ E cells.
Taken together, these data indicated that CD24 controlled the response to radiation and chemotherapeutic drugs.
Decreased CD24 expression decreases intracellular ROS concentration
As ROS production is reported to be an essential inducer of apoptosis (43), it was investigated whether changes in ROS levels were associated with radio- and chemo-sensitivity in HMLE cells. First, the intracellular concentrations of ROS were measured using DHE staining. Flow cytometry analysis indicated that M cells contained significantly lower concentrations of ROS than E/E_vec cells (Fig. 5A). E_CD24− cells also displayed lower DHE staining than E control cells and re-expression of CD24 abolished this decreased staining. Because mitochondria are the primary source of ROS in cancer cells, a similar experiment was performed using Mitosox-Red, a selective probe for mitochondrial superoxide (Fig. 5B). Same results were observed: a lower mitochondrial ROS level in M cells than in their parental counterparts, a decrease of mitochondrial ROS in CD24 knockdown cells and a return to the level observed in E cells when CD24 was re-expressed.
Figure 5.

CD24 downregulation decreases total and mitochondrial ROS levels. (A) Cellular ROS levels were assessed by DHE and (B) mitochondrial ROS levels were assessed by Mitosox-Red probe. ROS levels were studied in untreated control cells and three days following 10 Gy irradiation and exposure to 400 µM 5FU. Data are presented as the mean ± SD of 4–10 independent experiments. Significant differences were analyzed by one-way ANOVA with Kruskal-Wallis followed by Dunn's multiple comparisons correction test. *P<0.05, ***P<0.001. ns, non-significant; ROS, reactive oxygen species; 5FU, 5-fluorouracil; E, epithelial; M, mesenchymal.
The effect of CD24 expression on ROS levels 3 days following IR or 5FU treatment was investigated. ROS levels increased in all cell lines tested (Fig. 5A and B), but a lower concentration of ROS was maintained in E_CD24− and M cells compared with parental E or E_CD24−c cells. Similar results were obtained with both DHE and Mitosox-Red.
Taken together, these data indicated that CD24 downregulation led to decreased basal levels of total and mitochondrial ROS. Following IR or drug treatment, there was an increase in ROS levels but these remained lower in E_CD24− cells than in parental E cells, consistent with the rate of cell death observed in different cell lines. Therefore, CD24 expression may affect radio- and chemo-resistance by controlling ROS levels.
CD24 controls ROS levels via regulation of mitochondrial function independently of antioxidant activity
Lower ROS levels are commonly ascribed to the CSC phenotype in breast tumors and are associated with enhanced ROS scavengers and/or decreased mitochondrial mass (44). First, it was determined whether ROS modulation was associated with differential regulation of oxidative stress-associated genes. Our previous study showed that HMLE.M cells exhibit higher antioxidant activity than HMLE.E cells and characterized genes involved in ROS metabolism (10). Thus, RT-qPCR was performed to assess expression of four key genes in this model: Superoxide dismutase 2, heme oxygenase 1, glutathione-sulfide reductase and thioredoxin reductase 1 (Fig. 6A). The expression of all these genes was increased in M cells, but not modified in transfected E cells. Lower ROS levels observed in E_CD24− cells were not associated with high intracellular levels of radical scavengers.
Figure 6.

CD24 downregulation has no impact on ROS scavengers but decreases mitochondrial mass and membrane potential. (A) Analysis by reverse transcription-quantitative PCR of the relative expression of mRNAs encoding stress-associated factors involved in ROS metabolism in E_CD24-, E_CD24-c and M compared with E cells. For each gene, expression in E cells was normalized to 1 and ratio of relative mRNA level of E to E_CD24-, E_CD24-c and M cells is presented. Data are presented as the mean ± SD of 3 independent experiments. Significant differences (compared with E cells) were analyzed by one-way ANOVA followed by Dunnett's multiple comparisons correction test. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. ns: not significant. Mitochondrial (B) mass was assessed by Mitotracker Green probe and (C) membrane potential was assessed using TMRE staining. Mitochondrial mass and membrane potential were studied in untreated cells and at three days following 10 Gy irradiation and exposure to 400 µM 5FU. Data are presented as the mean ± SD of 4–10 independent experiments. Significant differences were analyzed by one-way ANOVA with Kruskal-Wallis followed by Dunn's multiple comparisons correction test. *P<0.05, **P<0.01, ***P<0.001. ns, non-significant. SOD, superoxide dismutase 2; HMOX1, heme oxygenase 1; GSR, glutathione-sulfide reductase; TXNDR1, thioredoxin reductase 1; TMRE, tetramethylrhodamine, ethyl ester; E, epithelial; M, mesenchymal; 5FU, 5-fluorouracil; ROS, reactive oxygen species.
It was next studied if modulation of CD24 expression affects mitochondrial function. Mitochondrial mass was analyzed using MTG, a mitochondrial selective probe, and mitochondrial membrane potential was quantified using TMRE staining. Flow cytometry analysis indicated that mitochondrial mass was significantly lower in M and E_CD24− cells than in parental E and E_CD24−c cells (Fig. 6B). In the same way, mitochondrial membrane potential decreased in M and in E_CD24− cells compared with E cells, and a return to the level observed in E cells was showed in E_CD24−c cells (Fig. 6C). The TMRE/MTG ratio was similar for all the cell lines analyzed (Fig. S6), indicating that mitochondrial membrane potential observed in the cell lines was associated with mitochondrial mass. Then, similar experiments were performed following IR or drug treatment (Fig. 6B and C). Mitochondrial mass and membrane potential were lower in E_CD24−cells (with the exception of TMRE staining following 5FU treatment: a small but not significant decrease is observed) compared with parental E cells and E_CD24−c cells.
Taken together, these data indicated that lower ROS levels observed in CD24−/low cells were associated with lower mitochondrial membrane potential and mass independently of intrinsic antioxidant activity. Markedly decreased mitochondrial ROS production may be a key event in radio- and chemo-resistance controlled by CD24 expression.
Discussion
The present data demonstrated that intrinsic radio- and chemo-resistance of breast cancer cells was directly associated with membrane expression level of CD24 and that CD24 may be considered not only as a marker but also as a mediator of this resistance. In our model, as for the mesenchymal phenotype (45), the CD24−/low phenotype of epithelial HMLE cells is reversible (46,47). CD24 expression exhibits dynamic regulation (48) as its expression is reversible and under epigenetic control. Expression of CD24 was heterogenous in the HMLE cell model and is frequently observed in breast cancer cell lines and tumor tissue (49).
Radio- and chemo-resistance of breast cancer cells has been highlighted using IR or different classes of chemotherapeutic drugs (50). The present study used high doses of IR (4–10 Gy) and chronic exposure with high concentration of drugs from three categories of antitumor agents: 5FU, an antimetabolite, cisplatin, an alkylating agent, and paclitaxel, a mitotic inhibitor. In parental HMLE.E cells dose-dependent death was observed following IR and drug treatment; this cell death is primarily associated with late apoptosis (9,51–54). Cell death was concomitant with a clear increase in percentage of CD24−/low surviving cells. This enrichment in CD24−/low cells may be the consequence of selection (22) and associated with induction of an active EMT program (55). CD24−/low cells exhibited greater ability to survive than CD24+ cells following IR or antitumor drug treatment. Altogether, the present results demonstrated an association between CD24−/low expression and treatment resistance, independent of the mechanism of CD24−/low cell enrichment.
Failure of conventional treatments is commonly associated with CSC survival (15), suggesting that E_CD24− cells have acquired stemness properties. In the past 20 years, numerous studies have associated loss of CD24 expression with EMT. The widely used CD24−/low/CD44+ marker allows characterization of M cells enriched in CSC properties compared with CD24+/CD44low E cells (4,7,10). However, in the present study, E_CD24− cells retained the primary characteristics of E cells: Cuboidal-like E morphology and formation of cobblestone cell islands. Furthermore, the expression of the primary EMT-associated genes (ESA, E-cadherin, CD44, N-cadherin and fibronectin) and 5 out of 6 transcription factors driving EMT (Twist-1, Twist-2, Snail-2, Zeb-1 and Zeb-2) were not modified. Therefore, treatment resistance in E_CD24− cells was not associated with acquisition of M characteristics.
The development of biomarkers to identify breast CSCs demonstrated that cells with high ALDH activity also exhibit stemness properties; radio- and chemo-resistance have been also associated with ALDH+ breast cancer cells (15–18). Loss of CD24 expression may promote stemness characteristics, allowing genotoxic stress survival of E_CD24− cells and radio- and chemo-resistance of E_CD24− cells are associated with ALDH activity. Moreover, the association between stem cell features and E_CD24− cells was supported by the increased potential to form mammospheres and migratory potential observed for these cells.
The hypothesis that only ‘full’ EMT is associated with increased stemness was challenged by later studies demonstrating the existence of E-like breast CSCs (11,13,14). These E CSCs with a CD24+/CD44low phenotype are characterized by high ALDH activity (11). E-M plasticity is a spectrum of transitory cell states, and tumor cells with the highest stem cell capabilities reside in a hybrid E/M state, associated with increased tumor propagating potential (13,14,39,56).
In the present study, when overall E characteristics of HMLE cells were maintained, deregulation of other genes associated with transition states occurring during EMT was also observed: Snail-1, Vimentin, Keratin 14, ΔNp63α and OVOL2. The strong downregulation of Keratin 14 expression (E marker) and the low but significant overexpression of Vimentin (M marker) suggested that E_CD24− cells acquired few M characteristics. Among the primary EMT transcription factors, only Snail1 was strongly overexpressed in E_CD24− cells. Snail1 expression has been implicated in reprogramming somatic cells to pluripotency (57,58) and Snail1 maintains stem-like properties, chemoresistance and ALDH activity in mouse breast cancer cells (59). Kroger et al (14) showed that both in vitro and in vivo, breast cancer cells reside stably and with low plasticity in a highly tumorigenic hybrid E/M state, which is driven primarily by Snail1 and canonical Wnt signaling. The E transcription factors ΔNp63α and OVOL2 were weakly downregulated in E_CD24− cells but at a lower level than in M cells. Suppression of ΔNp63α has been implicated in EMT induction in mammary E cells (40). OVOL2 is a key regulator of E-M plasticity as well as CSCs (13). Expression of OVOL2 induces mesenchymal-to-epithelial transition (MET), antagonizes TGF-β signaling and has been implicated in inducing mammary E cells to enter an intermediate E/M state (60–62). Moreover, OVOL2 and ΔNp63α may serve a key role in partial retention of E traits associated with collective migration by clusters of circulating tumor cells and high tumor-initiation potential (63,64). NRF2 signaling may be involved in CSC-like properties of several types of cancer cell (65). High NRF2 levels activate partial EMT (41), mediate cancer stem cell-like properties of ALDH+ cells (42) and contribute to radio-resistance (18). In the present model, NRF2 overexpression was associated with M phenotype but not intermediate E/M state, indicating that NRF2 pathway was not modulated by CD24 expression and was not involved in radio- and chemo-resistance of E_CD24− cells.
E_CD24− cells exhibit significantly increased migratory potential compared to E cells, which is strongly associated with the increased tumor propagating potential observed in breast cancer cells in a hybrid E/M state (12). Altogether, our results reinforced the hypothesis that the hybrid E/M state of E_CD24− cells leads to acquisition of stemness and maintenance of stem cell properties is independent of phenotypic plasticity (66,67). The present results are in agreement with the cooperation metastasis model proposed by Grosse-Wilde et al (68), where metastasis-initiating cells are in a mixed E/M state and may originate from cells in the E state.
The model used in the present study has the advantage of using cells with the same genetic background and plasticity between E and M allows the characterization of the alterations induced by regulation of CD24 expression in E cells and a fine mapping in the E/M hybrid state of E_CD24− cells. This model not only allows generalizability of our previous observations (23) but facilitates understanding of the sequence of events and consequences of downregulation of CD24.
The intrinsic radio- and chemo-resistance of CSCs is associated with many mechanisms, often mediated by redox imbalance and ROS control (21). Moreover, ROS production is reported to be an essential regulator or inducer of apoptosis in cancer cells and increased intracellular ROS levels mediate cell death induced by ionizing radiation (29) as well as anticancer drugs (30–32). Increased ROS scavengers are associated with lower ROS levels observed in CSCs (10,24), but few papers also report that CSCs that have undergone EMT show decreased mitochondrial mass and membrane potential, consume less oxygen per cell and produce markedly lower levels of ROS (69,70). The present results demonstrated that CD24 downregulation led to decreased basal levels of total and mitochondrial ROS via regulation of mitochondrial function. An association between mitochondrial suppression and metabolic reprogramming has been proposed and lower mitochondrial levels and activity reflect the metabolic switch that has been reported during EMT (71,72). Moreover, Snail may serve a central role in this process (13,57–59). Altogether, the present results showed an association between CD24 expression and therapeutic resistance. In our study, we observe that in breast cancer cell culture, CD24 expression is heterogeneous and CD24−/low subpopulation is selected following radiation and drug treatment. Artificial modulation of CD24 expression shows that radio- and chemo-sensitivity are directly controlled by CD24, and that loss of CD24 expression in E HMLE cells increase the presence of ALDH+ E-like CSCs, in a hybrid E/M state. In CD24−/low/CD44+ CSCs, the resistance properties are strongly associated to a low ROS level, but for the two subtypes of CSCs the mechanisms leading to decrease ROS level are different. For ALDH+ E_CD24− cells, we observed decreased mitochondrial mass and membrane potential, while for CD24−/low/CD44+ M cells, decrease ROS level is under the control of both enhanced ROS scavenger and decrease of mitochondrial mass and membrane potential. These results are summarized in a model in Fig. 7.
Figure 7.
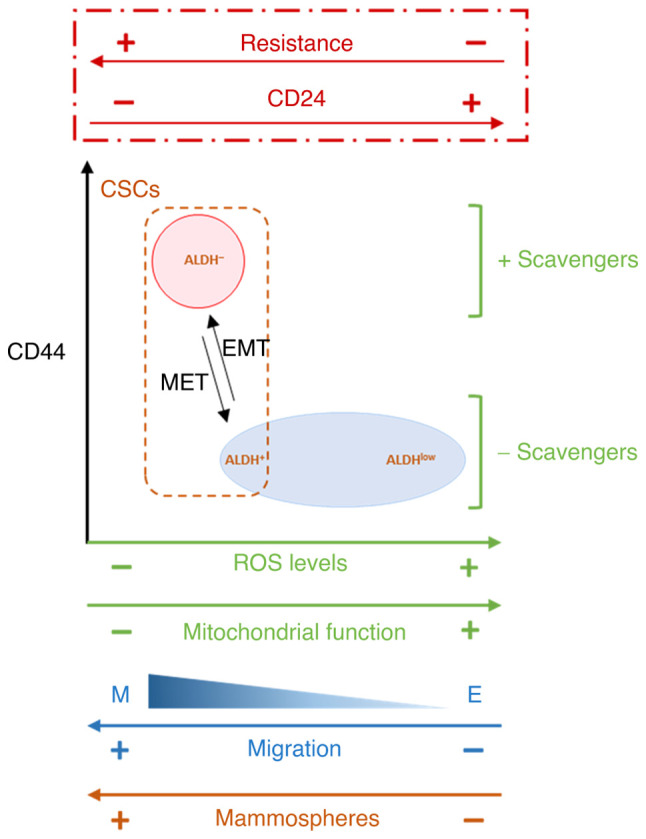
Proposed model of the association between CD24 expression and EMT, stemness properties, ROS levels and scavengers and mitochondrial function. In HMLE cells cultures, CD24 expression is heterogeneous and CD24−/low subpopulation is selected following radiation and drug treatment. Artificial modulation of CD24 expression shows that radio- and chemo-sensitivity are directly controlled by CD24, and that loss of CD24 expression in E HMLE cells increase the presence of ALDH+ E-like CSCs, in a hybrid E/M state, in association with decreased mitochondrial mass and membrane potential. EMT, epithelial-mesenchymal transition; ROS, reactive oxygen species; CSC, cancer stem cell; ALDH, aldehyde dehydrogenase; MET, mesenchymal-to-epithelial transition.
In the past decade, few papers have studied the role of CD24 in human tumors; when different functions of CD24 have been proposed, its role in cancer progression and treatment resistance remain poorly documented and the signaling downstream of CD24 has not been clearly elucidated (26,73). The intrinsic chemo-resistance of CD24−/low cells may depend on the type of drug. Deng et al (74) reported that cells with CD24-knockdown are more sensitive to docetaxel, while CD24-overexpressing cells are more sensitive to doxorubicin in triple-negative breast cancer. Therefore, CD24 may be a promising biomarker candidate to guide chemotherapy. Finally, the heterogeneity of breast cancer and molecular subtypes should be considered (75). In breast tumors, high CD24 expression is frequently associated with a terminally differentiated, luminal phenotype, while most basal-like tumors are classified as CD24−/low. In parallel, studies have shown that CD44/CD24 and ALDH1 are expressed differentially in different subtypes of breast cancer (49). ALDH+ cells are more common in HER2-overexpressing and basal/E breast cancer, while CD24−/low/CD44+ phenotype is associated with basal-like breast cancer (49,76). Moreover, only a fraction of CD24−/low/CD44+ breast cancer cells are ALDH+, these cells being more tumorigenic in athymic mice (5,11).
In conclusion, the present study suggested that CD24 expression may be a key factor in transition between different subtypes of breast CSCs and that loss of CD24 expression was associated with de-differentiation. De-differentiation is associated with invasion potential and metastasis and promotes resistance to a wide spectrum of chemotherapy drugs and radiation exposure (77).
Supplementary Material
Acknowledgements
We would like to thank Professor Robert A. Weinberg (Whitehead Institute, Cambridge, MA, USA), for the donation of HMLE cells.
Funding Statement
The present study was supported by Electricité de France and by the Transverse Division n. 4 (Radiobiology) of the French Alternative Energies and Atomic Energy Commission (Segment n. 4 Radiobiologie).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
IB, AC, FB, DB and JL designed the study and wrote the manuscript. IB, CL, ND, TK, EB, DB CS and JL performed the experiments. DB, TK and FB contributed reagents/analytic tools. IB, SC, AC and JL analyzed data. IB, EB, FB, AC and JL edited the paper. EB and AC confirm the authenticity of all the raw data. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer. 2012;12:323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 2.Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 3.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–1134. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 4.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang-Verslues WW, Kuo WH, Chang PH, Pan CC, Wang HH, Tsai ST, Jeng YM, Shew JY, Kung JT, Chen CH, et al. Multiple lineages of human breast cancer stem/progenitor cells identified by profiling with stem cell markers. PLoS One. 2009;4:e8377. doi: 10.1371/journal.pone.0008377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang R, Tu J, Liu S. Novel molecular regulators of breast cancer stem cell plasticity and heterogeneity. Semin Cancer Biol. 2022;82:11–25. doi: 10.1016/j.semcancer.2021.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Konge J, Leteurtre F, Goislard M, Biard D, Morel-Altmeyer S, Vaurijoux A, Gruel G, Chevillard S, Lebeau J. Breast cancer stem cell-like cells generated during TGFβ-induced EMT are radioresistant. Oncotarget. 2018;9:23519–23531. doi: 10.18632/oncotarget.25240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2013;2:78–91. doi: 10.1016/j.stemcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. 2019;29:212–226. doi: 10.1016/j.tcb.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Pasani S, Sahoo S, Jolly MK. Hybrid E/M phenotype(s) and stemness: A mechanistic connection embedded in network topology. J Clin Med. 2020;10:60. doi: 10.3390/jcm10010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, Thiru P, Bierie B, Ye X, Burge CB, Weinberg RA. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci USA. 2019;116:7353–7362. doi: 10.1073/pnas.1812876116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015;21:1301–1310. doi: 10.2174/1381612821666141211120604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips TM, McBride WH, Pajonk F. The response of CD24(−/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 18.Kamble D, Mahajan M, Dhat R, Sitasawad S. Keap1-Nrf2 pathway regulates ALDH and contributes to radioresistance in breast cancer stem cells. Cells. 2021;10:83. doi: 10.3390/cells10010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y, Noguchi S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-base chemotherapy for breast cancers. Clin Cancer Res. 2009;15:4234–4241. doi: 10.1158/1078-0432.CCR-08-1479. [DOI] [PubMed] [Google Scholar]
- 20.Palomeras S, Ruiz-Martínez S, Puig T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules. 2018;23:2193. doi: 10.3390/molecules23092193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.García-Heredia JM, Carnero A. Role of mitochondria in cancer stem cell resistance. Cells. 2020;9:1693. doi: 10.3390/cells9071693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bensimon J, Altmeyer-Morel S, Benjelloun H, Chevillard S, Lebeau J. CD24(−/low) stem-like breast cancer marker defines the radiation-resistant cells involved in memorization and transmission of radiation-induced genomic instability. Oncogene. 2013;32:251–258. doi: 10.1038/onc.2012.31. [DOI] [PubMed] [Google Scholar]
- 23.Bensimon J, Biard D, Paget V, Goislard M, Morel-Altmeyer S, Konge J, Chevillard S, Lebeau J. Forced extinction of CD24 stem-like breast cancer marker alone promotes radiation resistance through the control of oxidative stress. Mol Carcinog. 2016;55:245–254. doi: 10.1002/mc.22273. [DOI] [PubMed] [Google Scholar]
- 24.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JH, Kim SH, Lee ES, Kim YS. CD24 overexpression in cancer development and progression: A meta-analysis. Oncol Rep. 2009;22:1149–1156. doi: 10.3892/or_00000548. [DOI] [PubMed] [Google Scholar]
- 26.Altevogt P, Sammar M, Hüser L, Kristiansen G. Novel insights into the function of CD24: A driving force in cancer. Int J Cancer. 2021;148:546–559. doi: 10.1002/ijc.33249. [DOI] [PubMed] [Google Scholar]
- 27.Kristiansen G, Machado E, Bretz N, Rupp C, Winzer KJ, König AK, Moldenhauer G, Marmé F, Costa J, Altevogt P. Molecular and clinical dissection of CD24 antibody specificity by a comprehensive comparative analysis. Lab Invest. 2010;90:1102–1116. doi: 10.1038/labinvest.2010.70. [DOI] [PubMed] [Google Scholar]
- 28.Weber E, Lehmann HP, Beck-Sickinger AG, Wawrzynczak EJ, Waibel R, Folkers G, Stahel RA. Antibodies to the protein core of the small cell lung cancer workshop antigen cluster-w4 and to the leucocyte workshop antigen CD24 recognize the same short protein sequence leucine-alanine-proline. Clin Exp Immunol. 1993;93:279–285. doi: 10.1111/j.1365-2249.1993.tb07980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riley PA. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65:27–33. doi: 10.1080/09553009414550041. [DOI] [PubMed] [Google Scholar]
- 30.Mohiuddin M, Kasahara K. Cisplatin activates the growth inhibitory signaling pathways by enhancing the production of reactive oxygen species in non-small cell lung cancer carrying an EGFR exon 19 deletion. Cancer Genomics Proteomics. 2021;18((3 Suppl)):S471–S486. doi: 10.21873/cgp.20273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosca L, Ilari A, Fazi F, Assaraf YG, Colotti G. Taxanes in cancer treatment: Activity, chemoresistance and its overcoming. Drug Resist Updat. 2021;54:100742. doi: 10.1016/j.drup.2020.100742. [DOI] [PubMed] [Google Scholar]
- 32.Pan X, Zhang X, Sun H, Zhang J, Yan M, Zhang H. Autophagy inhibition promotes 5-fluorouraci-induced apoptosis by stimulating ROS formation in human non-small cell lung cancer A549 cells. PLoS One. 2013;8:e56679. doi: 10.1371/journal.pone.0056679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, Popescu NC, Hahn WC, Weinberg RA. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lombardo Y, de Giorgio A, Coombes CR, Stebbing J, Castellano L. Mammosphere formation assay from human breast cancer tissues and cell lines. J Vis Exp. 2015:52671. doi: 10.3791/52671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biard DS, Despras E, Sarasin A, Angulo JF. Development of new EBV-based vectors for stable expression of small interfering RNA to mimick human syndromes: Application to NER gene silencing. Mol Cancer Res. 2005;3:519–529. doi: 10.1158/1541-7786.MCR-05-0044. [DOI] [PubMed] [Google Scholar]
- 36.Vert JP, Foveau N, Lajaunie C, Vandenbrouck Y. An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics. 2006;7:520. doi: 10.1186/1471-2105-7-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- 38.Gorelik R, Gautreau A. Quantitative and unbiased analysis of directional persistence in cell migration. Nat Protoc. 2014;9:1931–1943. doi: 10.1038/nprot.2014.131. [DOI] [PubMed] [Google Scholar]
- 39.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, Van Keymeulen A, Brown D, Moers V, Lemaire S, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556:463–468. doi: 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 40.Yoh KE, Regunath K, Guzman A, Lee SM, Pfister NT, Akanni O, Kaufman LJ, Prives C, Prywes R. Repression of p63 and induction of EMT by mutant Ras in mammary epithelial cells. Proc Natl Acad Sci USA. 2016;113:E6107–E6116. doi: 10.1073/pnas.1613417113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bocci F, Tripathi SC, Vilchez Mercedes SA, George JT, Casabar JP, Wong PK, Hanash SM, Levine H, Onuchic JN, Jolly MK. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr Biol (Camb) 2019;11:251–263. doi: 10.1093/intbio/zyz021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim D, Choi B, Ryoo I, Kwak MK. High NRF2 level mediates cancer stem cell-like properties of aldehyde dehydrogenase (ALDH)-high ovarian cancer cells: Inhibitory role of all-trans retinoic acid in ALDH/NRF2 signaling. Cell Death Dis. 2018;9:896. doi: 10.1038/s41419-018-0903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang JY, Ou-Yang F, Hou MF, Huang HW, Wang HR, Li KT, Fayyaz S, Shu CW, Chang HW. Oxidative stress-modulating drugs have preferential anticancer effects-involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin Cancer Biol. 2019;58:109–117. doi: 10.1016/j.semcancer.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 44.Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:55. doi: 10.1186/s13058-016-0712-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhatia S, Monkman J, Blick T, Pinto C, Waltham M, Nagaraj SH, Thompson EW. Interrogation of phenotypic plasticity between epithelial and mesenchymal states in breast cancer. J Clin Med. 2019;8:893. doi: 10.3390/jcm8060893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 47.Ruscetti M, Dadashian EL, Guo W, Mulholland DJ, Park JW, Tran LM, Kobayashi N, Bianchi-Frias D, Xing Y, Nelson PS, Wu H. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene. 2016;35:3781–3795. doi: 10.1038/onc.2015.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meyer MJ, Fleming JM, Ali MA, Pesesky MW, Ginsburg E, Vonderhaar BK. Dynamic regulation of CD24 and the invasive, CD44posCD24neg phenotype in breast cancer cell lines. Breast Cancer Res. 2009;11:R82. doi: 10.1186/bcr2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, Milanezi F, Schmitt F, Paredes J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64:937–946. doi: 10.1136/jcp.2011.090456. [DOI] [PubMed] [Google Scholar]
- 50.Xia F, Powell SN. The molecular basis of radiosensitivity and chemosensitivity in the treatment of breast cancer. Semin Radiat Oncol. 2002;12:296–304. doi: 10.1053/srao.2002.35250. [DOI] [PubMed] [Google Scholar]
- 51.Luce A, Courtin A, Levalois C, Altmeyer-Morel S, Romeo PH, Chevillard S, Lebeau J. Death receptor pathways mediate targeted and non-targeted effects of ionizing radiations in breast cancer cells. Carcinogenesis. 2009;30:432–439. doi: 10.1093/carcin/bgp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 53.Tchounwou PB, Dasari S, Noubissi FK, Ray P, Kumar S. Advances in our understanding of the molecular mechanisms of action of cisplatin in cancer therapy. J Exp Pharmacol. 2021;13:303–328. doi: 10.2147/JEP.S267383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abu Samaan TM, Samec M, Liskova A, Kubatka P, Büsselberg D. Paclitaxel's mechanistic and clinical effects on breast cancer. Biomolecules. 2019;9:789. doi: 10.3390/biom9120789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity-implications for chemoresistance. Cancer Lett. 2013;341:56–62. doi: 10.1016/j.canlet.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, Liu Donaher J, Reinhardt F, Chaffer CL, Keckesova Z, Weinberg RA. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci USA. 2017;114:E2337–E2346. doi: 10.1073/pnas.1618298114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Unternaehrer JJ, Zhao R, Kim K, Cesana M, Powers JT, Ratanasirintrawoot S, Onder T, Shibue T, Weinberg RA, Daley GQ. The epithelial-mesenchymal transition factor SNAIL paradoxically enhances reprogramming. Stem Cell Reports. 2014;3:691–698. doi: 10.1016/j.stemcr.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gingold JA, Fidalgo M, Guallar D, Lau Z, Sun Z, Zhou H, Faiola F, Huang X, Lee DF, Waghray A, et al. A genome-wide RNAi screen identifies opposing functions of Snai1 and Snai2 on the Nanog dependency in reprogramming. Mol Cell. 2014;56:140–152. doi: 10.1016/j.molcel.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma SY, Park JH, Jung H, Ha SM, Kim Y, Park DH, Lee DH, Lee S, Chu IH, Jung SY, et al. Snail maintains metastatic potential, cancer stem-like properties, and chemoresistance in mesenchymal mouse breast cancer TUBO-P2J cells. Oncol Rep. 2017;38:1867–1876. doi: 10.3892/or.2017.5834. [DOI] [PubMed] [Google Scholar]
- 60.Roca H, Hernandez J, Weidner S, McEachin RC, Fuller D, Sud S, Schumann T, Wilkinson JE, Zaslavsky A, Li H, et al. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS One. 2013;8:e76773. doi: 10.1371/journal.pone.0076773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu RS, Hong JJ, Wu JF, Yan S, Wu D, Liu N, Liu QF, Wu QW, Xie YY, Liu YJ, et al. OVOL2 antagonizes TGF-β signaling to regulate epithelial to mesenchymal transition during mammary tumor metastasis. Oncotarget. 2017;8:39401–39416. doi: 10.18632/oncotarget.17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hong T, Watanabe K, Ta CH, Villarreal-Ponce A, Nie Q, Dai X. An Ovol2-Zeb1 mutual inhibitory circuit governs bidirectional and multi-step transition between epithelial and mesenchymal states. PLoS Comput Biol. 2015;11:e1004569. doi: 10.1371/journal.pcbi.1004569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Westcott JM, Camacho S, Nasir A, Huysman ME, Rahhal R, Dang TT, Riegel AT, Brekken RA, Pearson GW. ΔNp63-regulated epithelial-to-mesenchymal transition state heterogeneity confers a leader-follower relationship that drives collective invasion. Cancer Res. 2020;80:3933–3944. doi: 10.1158/0008-5472.CAN-20-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jolly MK, Boareto M, Debeb BG, Aceto N, Farach-Carson MC, Woodward WA, Levine H. Inflammatory breast cancer: A model for investigating cluster-based dissemination. NPJ Breast Cancer. 2017;3:1–8. doi: 10.1038/s41523-017-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ryoo I, Lee S, Kwak MK. Redox modulating NRF2: A potential mediator of cancer stem cell resistance. Oxid Med Cell Longev. 2016;2016:2428153. doi: 10.1155/2016/2428153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jia D, Tan Y, Liu H, Ooi S, Li L, Wright K, Bennett S, Addison CL, Wang L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget. 2016;7:771–785. doi: 10.18632/oncotarget.5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vipparthi K, Hari K, Chakraborty P, Ghosh S, Patel AK, Ghosh A, Biswas NK, Sharan R, Arun P, Jolly MK, Singh S. Emergence of hybrid states of stem-like cancer cells correlates with poor prognosis in oral cancer. iScience. 2022;25:104317. doi: 10.1016/j.isci.2022.104317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grosse-Wilde A, Kuestner RE, Skelton SM, MacIntosh E, d'Hérouël AF, Ertaylan G, Del Sol A, Skupin A, Huang S. Loss of inter-cellular cooperation by complete epithelial-mesenchymal transition supports favorable outcomes in basal breast cancer patients. Oncotarget. 2018;9:20018–20033. doi: 10.18632/oncotarget.25034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gammon L, Biddle A, Heywood HK, Johannessen AC, Mackenzie IC. Sub-sets of cancer stem cells differ intrinsically in their patterns of oxygen metabolism. PLoS One. 2013;8:e62493. doi: 10.1371/journal.pone.0062493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sciacovelli M, Frezza C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017;284:3132–3144. doi: 10.1111/febs.14090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee SY, Ju MK, Jeon HM, Lee YJ, Kim CH, Park HG, Han SI, Kang HS. Reactive oxygen species induce epithelial-mesenchymal transition, glycolytic switch, and mitochondrial repression through the Dlx-2/Snail signaling pathways in MCF-7 cells. Mol Med Rep. 2019;20:2339–2346. doi: 10.3892/mmr.2019.10466. [DOI] [PubMed] [Google Scholar]
- 72.Jia D, Park JH, Kaur H, Jung KH, Yang S, Tripathi S, Galbraith M, Deng Y, Jolly MK, Kaipparettu BA, et al. Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br J Cancer. 2021;124:1902–1911. doi: 10.1038/s41416-021-01385-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fang X, Zheng P, Tang J, Liu Y. CD24: From A to Z. Cell Mol Immunol. 2010;7:100–103. doi: 10.1038/cmi.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deng X, Apple S, Zhao H, Song J, Lee M, Luo W, Wu X, Chung D, Pietras RJ, Chang HR. CD24 expression and differential resistance to chemotherapy in triple-negative breast cancer. Oncotarget. 2017;8:38294–38308. doi: 10.18632/oncotarget.16203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen Y, Schmidt BUS, Kubitschke H, Morawetz EW, Wolf B, Käs JA, Losert W. Detecting heterogeneity in and between breast cancer cell lines. Cancer Converg. 2020;4:1. doi: 10.1186/s41236-020-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li W, Ma H, Zhang J, Zhu L, Wang C, Yang Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci Rep. 2017;7:13856. doi: 10.1038/s41598-017-14364-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: Driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell. 2019;24:65–78. doi: 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.