

Abstract
Background:
Hydrogen sulfide (H2S) exerts mitochondria-specific actions that include the preservation of oxidative phosphorylation, biogenesis, and ATP synthesis, while inhibiting cell death. 3-mercaptopyruvate sulfurtransferase (3-MST) is a mitochondrial H2S producing enzyme whose functions in the cardiovascular disease are not fully understood. In the current study, we investigated the effects of global 3-MST deficiency in the setting of pressure overload-induced heart failure.
Methods:
Human myocardial samples obtained from heart failure patients undergoing cardiac surgeries were probed for 3-MST protein expression. 3-MST knockout mice and C57BL/6J wild-type mice were subjected to transverse aortic constriction (TAC) to induce pressure-overload HFrEF. Cardiac structure and function, vascular reactivity, exercise performance, mitochondrial respiration and ATP synthesis efficiency were assessed. Additionally, untargeted metabolomics were utilized to identify key pathways altered by 3-MST deficiency.
Results:
Myocardial 3-MST was significantly reduced in heart failure patients compared to non-failing controls. 3-MST KO mice exhibited increased accumulation of branched-chain amino acids (BCAA) in the myocardium, which was associated with reduced mitochondrial respiration and ATP synthesis, exacerbated cardiac and vascular dysfunction, and worsened exercise performance following TAC. Restoring myocardial BCAA catabolism with 3,6-dichlorobenzo1[b]thiophene-2-carboxylaic acid (BT2) and administration of a potent H2S donor JK-1 ameliorates the detrimental effects of 3-MST deficiency in HFrEF.
Conclusions:
Our data suggest that 3-MST derived mitochondrial H2S may play a regulatory role in BCAA catabolism and mediate critical cardiovascular protection in heart failure.
Keywords: Hydrogen sulfide, Mitochondrial respiration, Heart Failure with reduced ejection fraction, Branched chain amino acids
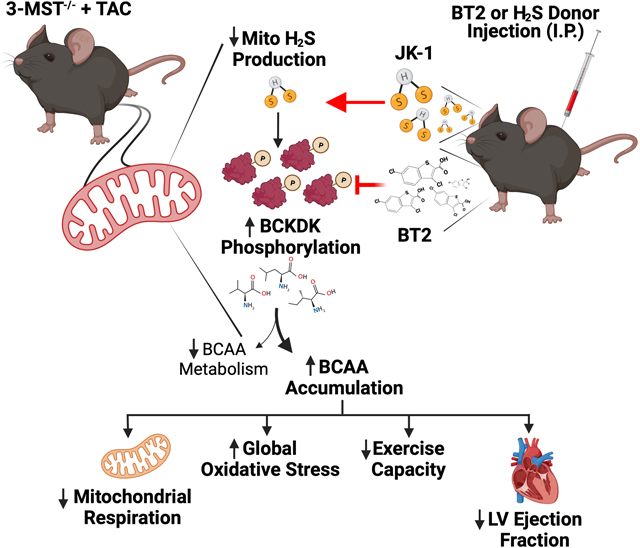
Graphical Abstract
INTRODUCTION
Cardiovascular disease, including heart failure (HF), is one of the leading causes of death and a growing economic problem world-wide1. Despite considerable progress in developing therapeutic options for HF patients, the 5-year mortality remains at 50%1. Additionally, the economic burden resulting from heart failure-related costs is projected to increase to $69.8 billion in the US and $108 billion worldwide by 20302. Today, there is no approved treatment to stimulate repair, regeneration and recovery of heart function after HF ensues. Therefore, new therapeutic measures are urgently needed.
Defects in branched-chain amino acids (BCAA) catabolism including leucine, isoleucine, and valine are associated with the pathogenesis and progression of multiple diseases3. Genetic deficiency of branched-chain alpha-keto acid dehydrogenase (BCKD), the rate-limiting enzyme that governs BCAA catabolism in mitochondria, leads to Maple Syrup Urine disease in human3. Abnormal levels of BCAA or BCAA metabolites such as branched chain keto acids (BCKA) in plasma or tissues have also been linked to Huntington’s disease, pancreatic cancer, diabetes and obesity3–7. More recently, Sun et al. discovered that BCAA catabolic defect in the myocardium promotes the progression of heart failure with reduced ejection fraction (HFrEF), while resetting the BCAA catabolic balance with 3,6-dichlorobenzo1[b]thiophene-2-carboxylaic acid (BT2), a branched-chain alpha-keto acid dehydrogenase kinase (BCKDK) inhibitor, preserved cardiac function in a pressure overload HFrEF model8, 9. However, despite recent efforts, the regulation of BCAA pathways remains poorly understood, hindering the development of targeted therapies8, 10, 11.
Hydrogen sulfide (H2S), produced endogenously by three enzymes, cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (3-MST), is an important physiological signaling molecule that modulates cardiovascular homeostasis12, 13. H2S exerts several protective actions in the mitochondria, including preservation of mitochondrial respiration, biogenesis, and ATP synthesis while inhibiting apoptosis and promoting cellular survival. 3-MST is the only endogenous H2S producing enzyme that localized to mitochondria, where it uses 3-mercaptopyruvate as a substrate to produce H2S, persulfides and polysulfides14–16. The intra-mitochondrial production of H2S and other sulfur species mediated by 3-MST have been shown to maintain mitochondrial electron flow and support cellular bioenergetics17–21. Additionally, it was previously reported that genetic deletion of 3-MST leads to reduced levels of cellular antioxidants and elevated basal levels of cardiac reactive oxygen species22. Taken together, these data suggest a critical role of 3-MST is maintaining mitochondrial function, cellular bioenergetics, and cardiovascular homeostasis. However, whether alterations in 3-MST and its intra-mitochondrial production of H2S and other sulfur species contributes to chronic diseases states such as heart failure has yet to be investigated.
In the current study, building on our preliminary discovery of reduced 3-MST expression in human myocardial samples obtained from patients with HFrEF, we investigated the roles of 3-MST in the setting of pressure overload induced HFrEF. A genetic mouse model lacking 3-MST was subjected to TAC and evaluated for changes in cardiac and vascular function, exercise tolerance, and mitochondrial morphology and function. Additionally, to better delineate the crosstalk between 3-MST - intramitochondrial H2S system and other vital metabolic pathways, we performed untargeted metabolomics in the heart to identify key metabolites affected by 3-MST deletion.
METHODS
Detailed Materials and Methods and Major Resources Table are available in the Online Supplemental Material. The data that support the findings of this study, experimental materials, and analytic methods are available from the corresponding author upon reasonable request.
RESULTS
3-MST expression is down-regulated in patients with end-stage heart failure.
Myocardial samples were obtained from non-failing donor hearts (n=10) and patients with ischemic cardiomyopathy induced end-stage HF (n=20) undergoing heart transplantation or mechanical circulatory support device implantation. Demographic information and clinical features of the HF patients are described in Table 1. Notably, patients with end-stage HF suffered from severe cardiac remodeling and dysfunction, evidenced by significantly larger left ventricular end-diastolic diameter (LVEDD) and reduced LVEF (Table 1). Further, cardiac index was significantly lower while circulating BNP levels were significantly higher in failing hearts than normal ranges (Table 1). Western blotting demonstrated that 3-MST expression declined in the HF group while CBS and CSE levels remained unaltered as compared to donor controls (Figure 1A and B, Figure S1), which was associated with significant reduction in myocardial sulfane sulfur levels (Figure 1C). However, the level of free hydrogen sulfide, the most labile sulfur species, was not significantly different between non-failing controls and heart failure (Figure 1D).
Table 1.
Patient Information
| HF Cohort (n=20) | |
|---|---|
| Age | 59 ± 10 |
|
| |
| Race | |
| African-American | 7 (35%) |
| Caucasian | 13 (65%) |
|
| |
| Sex | |
| Male | 15 (80%) |
| Female | 5 (20%) |
|
| |
| Hypertension | 17 (85%) |
|
| |
| Diabetes | 7 (35%) |
|
| |
| Inotrope Use | 16 (70%) |
|
| |
| LV Ejection Fraction (%) | 19 ± 7 |
|
| |
| LV End-Diastolic Diameter (cm) | 6.8 ± 1.1 |
|
| |
| Right Atrial Pressure (mmHg) | 13 ± 6 |
|
| |
| Precapillary Wedge Pressure (mmHg) | 28 ± 6 |
|
| |
| Fick Cardiac Index (L/min/m 2 ) | 1.9 ± 0.4 |
|
| |
| Cardiac Power Output (Watts) | 0.7 ± 0.2 |
|
| |
| GFR (mL/min) | 59 ± 26 |
|
| |
| BNP (pg/mL) | 1187 ± 1119 |
Myocardial samples were obtained from non-failing donor hearts and failing hearts from patients undergoing heart transplantation or mechanical circulatory support device implantation. The demographic information and clinical features of these patients are described. Categorial variables are expressed as numbers and percentages. Continuous variables are expressed as mean ± SD.
Figure 1: 3-MST and H2S/Sulfane Sulfur Levels in Human Non-Failing Control Hearts and Failing Hearts.

(A) Representative Western blots of human 3-MST, (B) Quantification of the Western blots images, (C) sulfane sulfur, and (D) free H2S levels in human myocardial samples obtained from non-failing vs. failing hearts. Circles inside bars indicate samples size. Data were analyzed with student unpaired 2-tailed t test and presented as mean ± SD.
Genetic deletion of 3-MST exacerbated the severity of pressure overload heart failure.
To determine whether global deletion of 3-MST impacts the severity of heart failure following pressure overload, WT and 3-MST KO mice underwent TAC were followed for 12 weeks (Figure 2). Prior to TAC surgery, LVEF (Figure 3B), as measured by echocardiography, was similar between WT and 3-MST KO mice, indicating that loss of 3-MST does not impact normal cardiac function. At 3 weeks following TAC surgery however, LVEF was significantly reduced in 3-MST KO mice compared to WT mice. Over the course of the 12-week study, LVEF was significantly reduced in 3-MST KO mice compared to WT mice. Similar to that observed with LVEF, deletion of 3-MST had no effect on LVEDD at baseline prior to TAC surgery (Figure 3A). Ventricular diastolic dilatation progressed at a slower rate than that observed with LVEF as there was a significant increase in LVEDD only at the later 9- and 12-week timepoints.
Figure 2: Study Timeline of Murine Model of Pressure Overload Heart Failure.

Echocardiography was performed prior to the induction of HF by TAC procedure then once every 3 weeks for a duration of 12 weeks. Invasive hemodynamic, vascular reactivity, exercise capacity, mitochondrial function assessments, metabolomic analysis, and other molecular determination were performed at 12 weeks post TAC.
Figure 3: Exacerbated Pressure Overload HF in 3-MST KO Mice.

(A) LV end-diastolic diameter (LVEDD) and (B) LV ejection fraction (LVEF) throughout the 12 weeks study for 3-MST KO and wildtype (WT) control mice. (C) LV end-diastolic pressure (LVEDP), (D) LV relaxation constant Tau, (E) circulating B-type natriuretic peptide (BNP), (F) aortic vascular reactivity to acetylcholine (Ach), (G) aortic vascular reactivity to sodium nitroprusside (SNP), and (H) treadmill running duration in 3-MST KO and control mice at 12 weeks post TAC. Circles inside bars indicates samples size. Data in (A), (B), (F) and (G) were analyzed with ordinary 2-way ANOVA; data in other panels were analyzed with student unpaired 2-tailed t test. Data are presented as mean ± SD.
The detrimental impact of global deletion of 3-MST on LV diastolic function was also corroborated by measurements of LV hemodynamics. At the 12-week timepoint, LV end-diastolic pressure was significantly elevated in 3-MST KO mice as compared to WT controls (Figure 3C). Furthermore, the LV diastolic relaxation time constant, Tau, was significantly higher in 3-MST KO mice (Figure 3D). In addition to increased LV chamber diameter and intracardiac filling pressures at end-diastole, we observed significantly higher circulating BNP levels in mice lacking 3-MST (Figure 3E). These data strongly suggest that the absence of 3-MST exacerbates pressure overload induced cardiac remodeling and failure.
To investigate the vascular effects of 3-MST deletion in HFrEF, we studied the relaxation responses of isolated thoracic aorta segments to acetylcholine and sodium nitroprusside ex vivo. 12-week post-TAC 3-MST KO mice exhibited impaired endothelial cell-dependent vasorelaxation responses to ACh as compared to WT controls (Figure 3F). Endothelial-independent vasorelaxation responses of aortic vascular rings to SNP were also perturbed in 3-MST KO mice compared to WT control (Figure 3G). In addition, at 12 weeks post-TAC, 3-MST KO mice displayed exercise intolerance as there was a significant reduction in treadmill running duration compared to WT controls (Figure 3H). Impaired arterial vasorelaxation and reduced exercise capacity are commonly observed in HF patients and contribute to decreased quality of life. We determined that systemic genetic deficiency of 3-MST further diminishes vascular function and exercise tolerance in the setting of HFrEF.
3-MST deficiency results in reduced mitochondrial H2S production, which is associated with elevated oxidative stress, altered mitochondrial content, structure, and function.
Since 3-MST resides primarily in mitochondria, we first measure the H2S production with isolated cardiac mitochondrial proteins. Following TAC, we observed diminished mitochondrial H2S producing ability in 3-MST KO mice, with reduced sulfane sulfur and increased levels of circulating 8-isoprostane, a marker for oxidative stress (Figures 4A to C). It is well-established that alterations in mitochondrial content, structure and function occur in response to heart failure26; therefore, we investigated whether the absence of 3-MST alters mitochondrial content and function in TAC-induced HFrEF. Analysis of transmission electron microscopy images of the LV of 3-MST KO mice subjected to TAC-induced HFrEF revealed extensive mitochondrial damage and disorganization that was more severe than observed in hearts from WT mice subjected to TAC HFrEF (Figure 4D). Following 12 weeks of TAC-induced heart failure, genetic deletion of 3-MST resulted in a reduction myocardial mitochondrial number compared to WT hearts (Figure 4E). Structural abnormalities were observed in remnant mitochondria from 3-MST KO hearts as exhibited by increased circularity compared to WT hearts (Figure 4F). Subsequent analysis revealed that the altered mitochondrial content observed in failing 3-MST KO hearts was likely a result of impaired mitochondrial biogenesis. An array of key enzymes regulating mitochondrial biogenesis, mitochondrial fusion and fission, as well as mitophagy were evaluated. We observed significantly reduction in relative expression of genes regulating mitochondrial biogenesis (i.e., atp5b, tfam, and mt-co1), while there was no difference in genes involved in mitochondrial fission/fusion and mitophagy (i.e., Mfn1, Mfn2, Fis1, PINK1, and Parkin) (Figure 4G).
Figure 4: Mitochondrial Morphology, Respiration and ATP Synthesis Efficiency in 3-MST KO Mice after TAC.

(A) Mitochondrial H2S generation capacity using 3-mercaptopyruvate as substrate in Wild-type and 3-MST KO mice at baseline and 12 weeks post TAC. Circulating (B) Sulfane Sulfur and (C) 8-isoprostane in wildtype or 3-MST KO mice at 12 weeks post TAC. (D) Representative transmission electron microscopy microphotography of cardiac mitochondria from 3-MST KO + HF vs. WT + HF mice. Scale bar at the bottom right corner represents 1 μm. (E) Number of mitochondria per area and (F) circularity of the mitochondria quantified. (G) Relative fold changes in gene expression of atp5b, tfam, mt-co1, Mfn1, Mfn2, Fis1, PINK1, and Parkin. Respiration using (H) pyruvate or (I) carnitine as substrate and (J) ATP synthesis efficiency in mitochondria isolated from wildtype or 3-MST KO hearts. Respiration using (K) pyruvate or (L) carnitine as substrate and (M) ATP synthesis efficiency in mitochondria isolated from wildtype or 3-MST KO skeletal muscle. Circles inside bars indicates samples size. Data in (A), (E), and (F) were analyzed with Mann-Whitney test; data in other panels were analyzed with student unpaired 2-tailed t test. Data are presented as mean ± SD.
To determine if global deletion of 3-MST impacts mitochondrial respiration, oxygen consumption rate was evaluated under various conditions in mitochondria isolated from the hearts or skeletal muscle of WT and 3-MST KO mice at baseline or at 12 weeks post-TAC. At baseline, there was a modest yet significant reduction in state 3 pyruvate and palmitoyl-L-carnitine mediated respiration observed in the 3-MST KO hearts. However, only pyruvate mediated state 3 respiration was significantly suppressed in 3-MST KO skeletal muscle. No difference was observed in ATP synthesis efficiency in both cardiac and skeletal muscle tissues (Figure S2). Following 12 weeks of TAC-induced HFrEF, global deletion of 3-MST resulted in significantly reduced mitochondrial state 3 pyruvate respiration and palmitoyl-L-carnitine respiration of both cardiac and skeletal muscle compared to WT controls (Figures 4H, I, K, L). Subsequent determination of post-oligomycin pyruvate respiration returned to similar levels in cardiac (Figure 4H) and skeletal muscle (Figure 4K) mitochondria from WT and 3-MST KO mice. While there was no difference in palmitoyl-L-carnitine respiration between WT and 3-MST KO skeletal muscle, 3-MST KO cardiac mitochondria exhibited significant reduction in post-oligomycin respiration mediated by palmitoyl-L-carnitine as compared to WT controls (Figure 4I). Furthermore, independent of the specific metabolic substrate, metabolic efficiency of ATP synthesis (ATP/O) was significantly impaired in 3-MST KO hearts (Figure 4J) and skeletal muscle (Figure 4M) compared to WT controls following TAC.
3-MST deficiency is associated with BCAA catabolic defects.
Given the reduction in mitochondrial number, alteration in morphology, impaired ATP synthesis, and decreased mitochondrial respiration observed in 3-MST KO hearts, we sought to investigate the impact of 3-MST deficiency on key metabolic pathways. Untargeted metabolomic analysis was performed to identify metabolic changes that are unique or in common between WT and 3-MST KO hearts following 12 weeks of TAC. We identified a total of 15,922 m/z features (6197 in positive mode and 6725 in negative mode). After statistical analysis of these features, we identified 85 metabolites that were different between the WT Sham and WT TAC hearts and 105 metabolites different between the 3-MST Sham and 3-MST TAC hearts (unadjusted p values < 0.05; Tables S1–2). This list was further narrowed by identifying those metabolites with an adjusted p value < 0.1 (Tables S3–5). In WT hearts subjected to 12 weeks of TAC a total of 17 metabolites (9 up and 8 down) were significantly changed compared to Sham-TAC WT hearts (Figure 5A). In contrast, 12 weeks of TAC significantly changed 51 metabolites (33 up and 1 down) in the hearts of 3-MST KO mice (Figure 5B). With changes in only 4 metabolites (1 up and 3 down) found common to both WT and 3-MST KO hearts (Figure 5C), suggesting that the lack of 3-MST expression contributes to greater metabolic pathway alterations in the failing myocardium.
Figure 5: Metabolomic Analysis of Wildtype and 3-MST KO Hearts.

(A) Z-score plot analysis of metabolic changes unique to WT hearts following TAC. (B) Z-score plot analysis of metabolic changes unique to 3-MST KO hearts following TAC. Red arrows represent the significantly elevated levels of BCAA while blue arrows indicate the significant reduction in the downstream metabolites of BCAA. (C) Z-score plot analysis of metabolic changes common to WT and 3-MST KO hearts following TAC. In each plot, the data are shown as standard deviation from the mean of the respective shams. Each dot represents a single metabolite in 1 sample. n=5 per group. (D) Dotplot showing the top 25 enriched metabolite sets for WT hearts following TAC. (E) Dotplot showing the top 25 enriched metabolite sets for 3-MST KO hearts following TAC. For these analyses, metabolites showing significant changes were analyzed using MetaboAnalyst metabolite set enrichment analysis.
For WT hearts, the top enriched metabolite groups were related to fatty acid biosynthesis, sphingolipid metabolism and glycolysis (Figure 5D). For 3-MST KO hearts, the top enriched metabolite sets were related to BCAA degradation and the citric acid cycle (Figure 5E). Further evaluation revealed that the BCAA, valine, leucine, and isoleucine, were significantly elevated in 3-MST KO following TAC (red arrows; Figure 5B), whereas downstream metabolites that follow the catabolism of BCAA, such as succinate (succinic acid), isovalerylcarnitine and 3-hydroxyisobutyrate were significantly decreased (blue arrows, Figure 5B). Taken together, this suggests that that cardiac BCAA catabolism is impaired and results in an accumulation of BCAA in 3-MST KO hearts following TAC. Importantly, impaired BCAA catabolism resulting in pathological amino acid accumulation has previously been shown to exacerbate heart failure severity4, 7–8.
Inhibition of BCKDK resets the BCAA catabolic balance and ameliorates the severity of HFrEF in 3-MST KO mice.
Given that global deletion of 3-MST was associated with dysregulation of amino acid metabolism and increased BCAA levels in failing hearts, we investigated whether resetting BCAA catabolism could improve cardiac function and mitochondrial respiration in 3-MST KO hearts subjected to TAC-induced heart failure. 3-MST knockout mice were treated with the BCKDK inhibitor, 3,6-dichlorobensothiophene-2-carboxylic acid (BT2), or vehicle daily over the course of the 12-week study. Despite no difference was observed in the protein expression of key regulators of BCAA catabolism such as BCKDK, PP2Cm, and BCKDH between WT and 3-MST KO mice (Figure S3), daily administration of BT2 effectively inhibited BCKDK activity as evidenced by reduced levels of phosphorylated BCKDH (Figure 6A, B) and was accompanied by significant reductions in the myocardial levels of leucine, isoleucine, and valine (Figure 6C).
Figure 6: Reduced BCAA Accumulation and Attenuated Severity of Pressure Overload HF in BT2 Treated 3-MST KO Mice.

(A) Representative Western blots of branched-chain ketoacid dehydrogenase kinase (BCKDK) and phosphorylated BCKDK, (B) quantified pBCKDK/BCKDK ratio, and (C) targeted metabolomic analysis of BCAA in vehicle treated vs. ,6-dichlorobenzo1[b]thiophene-2-carboxylaic acid (BT2) treated 3-MST KO mice. (D) LV end-diastolic diameter (LVEDD) and (E) LV ejection fraction (LVEF) throughout the 12 weeks study for vehicle or BT2 treated mice. (F) LV end-diastolic pressure (LVEDP), (G) LV relaxation constant Tau, (H) circulating B-type natriuretic peptide (BNP), in vehicle vs. BT2 treated 3-MST KO mice at 12 weeks post TAC. Circles inside bars indicates samples size. Data in (B) and (C) were analyzed with Mann-Whitney test; data in (D) and (E) were analyzed with ordinary 2-way ANOVA; data in (F) to (H) were analyzed with student unpaired 2-tailed t test. Data are presented as mean ± SD.
With respect to cardiac structure, inhibition of BCKDK activity by BT2 in 3-MST KO heart failure mice significantly attenuated LV chamber dilation at 9- and 12-weeks post-TAC (Figure 6D). Starting at 3 weeks following TAC surgery, there was significant preservation of LVEF that was sustained over the 12-week study in 3-MST KO mice treated with BT2 compared to vehicle control (Figure 6E). Furthermore, BT2 treatment in 3-MST KO mice resulted in improved LV hemodynamics as demonstrated by reductions in LVEDP (Figure 6F) and Tau (Figure 6G), as compared to vehicle control. Moreover, circulating BNP levels were significantly reduced in 3-MST KO heart failure mice treated with BT2 (Figure 6H), providing additional evidence for the pathological actions of impaired BCAA metabolism in the absence of 3-MST. Interestingly, the beneficial effects of BT2 treatment in adverse cardiac remodeling and functions in failing 3-MST KO hearts were comparable to those seen in WT controls (Figures S4A to C).
Given the global deletion of 3-MST in the 3-MST KO heart failure mice, the effect of BCKDK activity inhibition with BT2 beyond those observed in the heart was investigated. Preservation of endothelial-dependent (Figure 7A) and endothelial-independent relaxation (Figure 7B) were observed in 3-MST KO heart failure mice treated with BT2 compared to vehicle control. Additionally, exercise intolerance was attenuated in 3-MST KO heart failure mice treated with BT2 as evidenced by increased treadmill running duration (Figure 7C). However, BT2 treatment improved the exercise capacity to a greater extent in WT mice than in 3-MST mice following TAC (Figure S4D).
Figure 7: Ameliorated Vascular Dysfunction, Exercise Intolerance, and Mitochondrial Dysfunction in BT2 Treated 3-MST KO Mice after TAC.

(A) Aortic vascular reactivity to acetylcholine (Ach), (B) aortic vascular reactivity to sodium nitroprusside (SNP), (C) treadmill running duration in 3-MST KO mice treated with Vehicle or BT2. Respiration using (D) pyruvate or (E) carnitine as substrate and (F) ATP synthesis efficiency in mitochondria isolated from Vehicle or BT2 treated 3-MST KO hearts. Respiration using (G) pyruvate or (H) carnitine as substrate and (I) ATP synthesis efficiency in mitochondria isolated from from Vehicle or BT2 treated 3-MST KO skeletal muscle. Circles inside bars indicates samples size. Data in (A) and (B) were analyzed with ordinary 2-way ANOVA; data in other panels were analyzed with student unpaired 2-tailed t test. Data are presented as mean ± SD.
Inhibition of BCKDK improves mitochondrial oxidative phosphorylation in 3-MST KO mice in pressure overload HFrEF.
Our data indicate that the severity of cardiac and vascular dysfunction resulting from 3-MST deficiency is attenuated following inhibition of BCKDK activity and normalization of BCAA catabolism in the failing heart. Both BCKDK and 3-MST are primarily localized in the mitochondria and it is appreciated that mitochondria play a central role in cellular function and survival in heart failure. Therefore, we sought to investigate the effects of resetting BCAA catabolic balance on mitochondrial respiration and ATP biogenesis efficiency in cardiac (Figure 7D–F) and skeletal (Figure 7G–I) tissue mitochondria. There were no differences in basal respiration in (pyruvate or palmitoyl-L-carnitine as substrates) mitochondria isolated from BT2- or vehicle treated 3-MST KO mouse hearts (Figure 7D, E) or skeletal muscle (Figure 7G, H). Compared to vehicle control, BT2 treatment significantly improved state 3 respiration of cardiac and skeletal muscle mitochondrial isolates. Interestingly, BT2 treatment resulted in a modest yet significant increase in state 4 respiration in cardiac mitochondria when using pyruvate as a substrate (Figure 7D). Furthermore, independent of the specific metabolic substrate, pyruvate and palmitoyl-L-carnitine substrates, metabolic efficiency of ATP synthesis (ATP/O) was significantly improved in BT2 treated 3-MST KO mouse hearts (Figure 7F) and skeletal muscle (Figure 7I) compared to vehicle 3-MST KO mouse controls.
Hydrogen sulfide therapy reverses HFrEF in 3-MST KO mice.
Given that 3-MST synthesizes H2S in the mitochondria, we sought to evaluate the effects of H2S supplementation in the setting of TAC-induced HFrEF in 3-MST KO mice compared to WT. We utilized a well-characterized H2S donor, JK-1, that has previously been shown to attenuate myocardial ischemia-reperfusion injury and HFrEF 23, 24. Data from these experiments are summarized in Figure S5. Administration of JK-1 (200 μg/Kg/day) starting at the time of TAC surgery significantly preserved LV ejection fraction and attenuated cardiac dilatation (Figures S5A and B). H2S therapy significantly reduced LV end-diastolic pressure and TAU (Figures S5C and D) while improving exercise performance (Figure S5E) and aortic vascular reactivity to acetylcholine and sodium nitroprusside (Figures S5F and G).
DISCUSSION
Of the three endogenous hydrogen sulfide generating enzymes, 3-MST is the most abundant although its function and importance in cardiovascular system remains largely unknown16, 25. In this study we observed significantly reduced protein expression of 3-MST while CSE and CBS remained unaltered in myocardial samples obtained from end-stage heart failure patients requiring heart transplantation or mechanical circulatory support device implantation. Additionally, sulfane sulfur levels were significantly lower in end-stage heart failure myocardial tissue further confirming that there is a reduction in 3-MST governed H2S/sulfur species production in heart failure patients. However, the reduction in myocardial sulfane sulfur may be attributed to not only a reduction in 3-MST expression but also to the dysregulation of CSE and CBS enzymatic activity. Similarly, in murine model of HFrEF induced by TAC, we previously reported that myocardial protein expression of 3-MST was significantly reduced, which was associated with significantly lower myocardial and circulating H2S and sulfane sulfur levels26. Interestingly, in this murine model of HFrEF, CBS also remained unchanged but CSE was upregulated, possibly through a protective compensatory mechanism26.
In order to study the impact of genetic deficiency of 3-MST in the setting of HFrEF, we employed global 3-MST knockout mice. We recently characterized 3-MST KO mice under baseline conditions. We observed that circulating and myocardial tissue H2S levels were similar to wild-type controls while CSE and CBS expression remains unchanged22. We also reported normal left ventricular structure and function in global 3-MST KO mice22. Deletion of 3-MST was associated with increased myocardial reactive oxygen species and reduced antioxidants resulting in “preconditioning” of the myocardium against myocardial ischemia and reperfusion injury22. While deletion of 3-MST might be cardioprotective in the acute setting, we hypothesized, that similar to heart failure patients with reduced 3-MST expression, the reduced baseline cardiac mitochondrial function and early accumulation of reactive oxygen species seen in 3-MST KO mice may predispose the heart to more chronic insults, and the absence of 3-MST in the setting of pressure overload would exacerbate heart failure pathophysiology. Indeed, we observed significantly compromised cardiac structure and function in mice that lack 3-MST when compared to the wild-type controls. This pathological response is evidenced by exacerbated LV chamber dilation at 9- and 12- weeks post TAC, as well as reduced LV ejection fraction at 3-, 6-, 9-, and 12-weeks post-TAC, respectively. Additionally, invasive hemodynamic measurements revealed significantly elevated LVEDP and relaxation constant tau in 3-MST KO mice when compared to wildtype control mice. Global deletion of 3-MST in failing hearts resulted in not only profound cardiac remodeling and dysfunction, but severely compromised vascular reactivity and exercise performance.
3-MST has been detected in both the cytoplasm and in the mitochondria16, 25. However, because the concentration of cysteine is much higher in the mitochondria, as compared to the cytoplasm, it is plausible that most of the H2S generated by 3-MST occurs in mitochondria14, 27. Furthermore, given that H2S is a very powerful modulator of mitochondrial respiration and function, it is likely that 3-MST exerts its most critical physiological actions within the mitochondria20, 21, 28. It has been previously reported that therapeutic levels of H2S exerts mitochondria-specific protective effects including preservation of mitochondrial respiration and biogenesis, as well as improving ATP synthesis efficiency under stress conditions20, 21, 28. Together with the previous findings indicating 3-MST is the only intra-mitochondrial-expressed H2S producing enzyme that supports mitochondrial electron flow and bioenergetics16, 19, we hypothesized 3-MST deficiency would lead to impaired mitochondrial content, morphology and functions after TAC. 3-MST KO hearts displayed significantly reduced mitochondrial H2S production, which was associated with increased circulating levels of oxidative stress marker 8-isoprostane. Moreover, Transmission electron microscopic images revealed a disorganized pattern of mitochondrial ultrastructure, along with reduced mitochondrial density and increased mitochondrial swollenness in 3-MST KO hearts. We believed that the changes in mitochondrial content seen in failing 3-MST KO hearts were resulted from reduction in mitochondrial biogenesis, consistent with a previous report28. We observed significantly reduced expression of genes regulating mitochondrial biogenesis, namely tfam, atp5b, and mt-co1. In contrast, there was no significant difference in expression of genes regulating mitochondrial fusion/fission and mitophagy. Further evaluation with Clark-type oxygen electrodes demonstrated that 3-MST deletion resulted in impaired mitochondrial respiration and reduced ATP synthesis efficiency using pyruvate or carnitine as substrates at 12 weeks after TAC as compared to wild-type control mice. Notably, we observed similar degrees of mitochondrial dysfunction in mitochondria isolated from either cardiac or skeletal muscle tissue of 3-MST KO mice indicating a global effect of 3-MST on mitochondrial function that is not restricted to the heart. Interestingly, we observed a significant reduction in skeletal muscle mitochondrial respiration in wild-type mice after TAC when compared to healthy wild-type mice (data not shown), indicating that mitochondrial dysfunction in distal organs such as skeletal muscle is involved in the pathophysiology of HFrEF, which may contribute to the profound exercise intolerance seen in patients. Taken together, these data strongly suggest that 3-MST plays important roles in maintaining proper mitochondrial morphology and function during the pathogenesis and progression of pressure overload HFrEF.
To further define the mechanisms by which 3-MST deficiency exacerbates pressure overload HF, un-biased metabolomic analysis of myocardial tissue samples was performed. We discovered that 3-MST deletion significantly downregulated the catabolism of all three BCAA including leucine, isoleucine, and valine following TAC, indicating a potential regulatory role by 3-MST in BCAA catabolism in the setting of pressure overload HF. BCAA are essential amino acids that serve as building blocks for peptides and proteins29. It has been well-established that dysregulations in BCAA catabolism and pathological accumulation of BCAA/BCKA play a causal role in the onset of obesity and diabetes3, 5–7, 30. More recently, amino acids metabolism, specifically BCAA catabolism, has emerged as a one of the key metabolic components that contribute to the pathogenesis and progression of HFrEF in addition to the extensively-studied fatty acid and glucose metabolism4, 8, 10, 11. BCAA catabolism are catalyzed by a host of enzymes including branched chain aminotransferase (BCAT), BCKDs, BCKD kinases as well as protein phosphatase 2Cm (PP2Cm) which are located within the mitochondria4, 8, 10. It was recently shown that genetic defects such as BCKD phosphatase deletion that reduced BCAA catabolism led to accelerated progression of pressure overload HF while resetting the BCAA catabolic balance through inhibiting BCKD kinase by BT2 or overexpressing PP2Cm rescued such phenotype8, 31. To understand the relations between 3-MST deficiency and BCAA catabolic defects seen in 3-MST KO mice, we administered BT2 to 3-MST KO mice following TAC and investigated the effects of BT2 treatment on cardiac and vascular function, exercise performance, and mitochondrial respiration as well as ATP synthesis. Our data indicate that BT2 treatment effectively attenuated the phosphorylated BCKD kinase ratio, reduced the accumulation of all three BCAA, and attenuated the severity of pressure overload HFrEF in 3-MST deficient mice, as evidenced by preserved cardiac structure and function, lower LVEDP and Tau, as well as improved vascular reactivity and exercise performance. BT2 treatment in 3-MST deficient mice also significantly improved cardiac and skeletal muscle mitochondrial respiration and ATP synthesis efficiency. Taken together, these data provide the proof-of-concept evidences suggesting 3-MST plays an upstream regulatory role in BCAA catabolism and 3-MST deficiency leads to dampened BCAA degradation, which ultimately contributes to worse HFrEF. Additionally, we observed significantly attenuated HFrEF severity in BT2 treated 3-MST KO mice when compared to WT mice following TAC. In contrast, when compared to BT2-treated WT mice, there was no difference observed in BT2 treated 3-MST KO mice in cardiac remodeling and functions following TAC. These data indicate that BCAA catabolic defect may be a major pathogenic factor downstream of 3-MST deficiency. However, BT2 treatment in WT mice resulted in greater preservation of exercise capacity compared to 3-MST KO mice following TAC, suggesting that 3-MST deficiency impairs endurance exercise tolerance through additional mechanisms, possibly by affecting fatty acid metabolism and energy expenditure, as indicated in unbiased metabolomic analysis. Further, we were also able to halt the rapid progression of HFrEF in 3-MST KO with H2S supplementation. Treatment with a potent H2S donor JK-1 effectively attenuated the adverse cardiac remodeling, preserved the cardiac functions, and improved vascular reactivity as well as exercise capacity in 3-MST KO mice following TAC. These data provide additional insights into possible mechanisms by which 3-MST might regulate mitochondrial function, BCAA metabolism, and protect against heart failure. Clearly, additional experiments utilizing additional approaches such as mitochondrial-targeted H2S donors and confirmation of the effects of H2S on BCAA metabolism and mitochondrial function are required to fully define the actions of 3-MST in the mitochondria.
Interestingly, no difference was observed in phosphorylated BCKD kinase/total BCKD kinase ratio when compared 3-MST KO heart versus WT control hearts following TAC, indicating that 3-MST modulates BCAA catabolism pathway through a non-canonical mechanism. 3-MST has been shown to express and produce H2S and other sulfur species such as persulfides and polysulfides14, 15. Notably, BCAA catabolic enzymes all locate in mitochondria, and are rich in thiol-containing amino acid residues such as cysteine that are highly susceptible to oxidative modification by H2S or other reactive sulfur species produced by 3-MST3, 10. Future studies are warranted to investigate whether 3-MST regulates the activity of key BCAA catabolic enzymes via direct post-translation modification by 3-MST-produced intra-mitochondrial H2S/sulfur species.
In summary, this is the first study addressing the role of 3-MST and its intra-mitochondrial H2S/sulfur species production in the setting of pressure overload induced heart failure. Our results provide evidence that 3-MST deficiency resulted in impaired mitochondrial ultrastructure, respiration, and ATP synthesis efficiency, which might ultimately exacerbate the severity of HF. Moreover, we demonstrated that under the stress of pressure overload, 3-MST deficiency was associated with significant accumulation of BCAA in the failing heart, while resetting the BCAA catabolic balance ameliorates the detrimental effects of 3-MST deficiency, suggesting a regulatory role of 3-MST on BCAA catabolism. Modulation of 3-MST and intra-mitochondrial H2S/sulfur species may represent a novel therapeutic strategy for the treatment of heart failure and other diseases that involve BCAA catabolic defects.
Supplementary Material
Novelty and Significance.
What is Known?
Mitochondrial H2S/Sulfur species produced by 3-MST (3-mercaptopyruate sulfurtransferase) play important roles in cardiovascular homeostasis.
Under physiological conditions, 3-MST facilitates and maintains normal mitochondrial function.
What New Information Does This Article Contribute?
Myocardial 3-MST protein expression is reduced in patients with severe end-stage HFrEF (Heart failure with reduced ejection fraction).
Genetic deficiency of 3-MST resulted in increased cardiac remodeling, impaired vascular reactivity, and reduced mitochondrial respiration in murine pressure overload-induced HFrEF.
Unbiased metabolomic revealed impaired catabolism of myocardial BCAAs (branched-chain amino acids), and restoration of BCAA catabolism reversed the cardiac and vascular dysfunction in 3-MST deficient mice following pressure overload induced HFrEF.
Building on the observation of reduced myocardial 3-MST protein expression in patients with severe end-stage HFrEF, we studied the roles of 3-MST in HFrEF utilizing a global 3-MST deficient mouse model. Our data demonstrates 3-MST deficiency led to exacerbated LV (left ventricular) chamber dilation, accelerated decline in LV ejection fraction, elevated LV filling pressure, reduced vascular reactivity, impaired mitochondrial function, as well as severe exercise intolerance in pressure overload-induced HFrEF. Unbiased metabolomic profiling of 3-MST deficient hearts demonstrated enhanced accumulation of BCAA and reduction of their downstream metabolites and this defect was corrected by pharmacological restoration (i.e., BT2) of BCAA catabolism. Furthermore, H2S (hydrogen sulfide) supplementation with a potent H2S donor (i.e., JK-1) reversed the pathological effects of pressure overload HFrEF. These data suggest a key regulatory role of 3-MST derived H2S/sulfur species on mitochondrial BCAA metabolism in the setting of HFrEF. Ongoing studies are aimed at investigating the post-translational modifications of 3-MST derived H2S/sulfur species on key mitochondrial enzymes involved in BCAA metabolism.
STUDY LIMITATIONS.
Human myocardial samples were obtained from heart failure patients suffering from ischemic cardiomyopathy, which is most commonly caused by coronary artery disease. Despite that 85% of the current patient cohort had hypertension, the cardiac remodeling, left ventricular enlargement and chamber dilatation seen in these patients were largely due to the formation of plaque in coronary blood vessels, which results in a decreased capacity of the coronary arteries to supply oxygen to the myocardium. The TAC heart failure model used in this study is one of chronic pressure overload with a pathological cardiac remodeling process that also results in LV enlargement and dilatation, albeit one that may differ from that of ischemic heart disease. Additionally, most of the currently available H2S donors release H2S spontaneously in an uncontrolled manner, resulting in increased H2S bioavailability globally while it remains difficult to target H2S delivery to a specific tissue or organelle. This approach provided limited knowledge on the specific effects of mitochondria-derived H2S and sulfur species from 3-MST. Design and development of mitochondrial-targeted H2S prodrugs or H2S scavengers whose actions are catalyzed restrictively by local mitochondrial enzymes are in need for future studies. In addition, tissue targeted 3-MST knock mice are required to more fully understand the role of 3-MST and mitochondrial H2S in various organs during cardiovascular diseases.
SOURCES OF FUNDING
This work was supported by Grants from NIH National Heart, Lung, and Blood Institute (R01 HL146098, R01 HL146514, R01 HL137711) to DJL, (R01 HL151398) to DJL and MX, (R01 HL133167, R35 HL155651) to FNS, American Heart Association Postdoctoral Grant (20POST3520075) to ZL, and (18POST34020143) to TES.
Nonstandard Abbreviations and Acronyms
- 3-MST
3-mercaptopyruvate sulfurtransferase
- Ach
acetylcholine
- BCAA
branched-chain amino acids
- BCKD
branched-chain alpha-keto acid dehydrogenase
- BCKDK
branched-chain alpha-keto acid dehydrogenase kinase
- BNP
B-type natriuretic peptide
- BT2
3,6-dichlorobenzo1[b]thiophene-2-carboxylaic acid
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase
- EDD
end-diastolic dimension
- EDP
end-diastolic pressure
- EF
ejection fraction
- HF
heart failure
- HFrEF
heart failure with reduced ejection fraction
- H2S
hydrogen sulfide
- LV
left ventricle
- SNP
sodium nitroprusside
Footnotes
CONFLICT OF INTEREST DISCLOSURES
Dr. David Lefer serves as scientific consultant for Sulfagenix Inc, a company focusing on developing H2S-based therapy for clinical use. Dr. Lefer also has stock in both NovoMedix and SAJE Pharma, biotech companies that are developing novel therapeutics for cardiovascular diseases. Other authors declare no conflict of interests.
References
- 1.Bozkurt B, Hershberger RE, Butler J, Grady KL, Heidenreich PA, Isler ML, Kirklin JK and Weintraub WS. 2021 ACC/AHA Key Data Elements and Definitions for Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Clinical Data Standards for Heart Failure). Circ Cardiovasc Qual Outcomes. 2021;14:e000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG, American Heart Association Advocacy Coordinating C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention and Stroke C. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nie C, He T, Zhang W, Zhang G and Ma X. Branched Chain Amino Acids: Beyond Nutrition Metabolism. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun H and Wang Y. Branched chain amino acid metabolic reprogramming in heart failure. Biochim Biophys Acta. 2016;1862:2270–2275. [DOI] [PubMed] [Google Scholar]
- 5.Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, Kim E, Ghosh CC, Parikh SM, Jiang A, Chu Q, Forman DE, Lecker SH, Krishnaiah S, Rabinowitz JD, Weljie AM, Baur JA, Kasper DL and Arany Z. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016;22:421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloomgarden Z. Diabetes and branched-chain amino acids: What is the link? J Diabetes. 2018;10:350–352. [DOI] [PubMed] [Google Scholar]
- 7.Zhou M, Shao J, Wu CY, Shu L, Dong W, Liu Y, Chen M, Wynn RM, Wang J, Wang J, Gui WJ, Qi X, Lusis AJ, Li Z, Wang W, Ning G, Yang X, Chuang DT, Wang Y and Sun H. Targeting BCAA Catabolism to Treat Obesity-Associated Insulin Resistance. Diabetes. 2019;68:1730–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK and Wang Y. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation. 2016;133:2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tso SC, Gui WJ, Wu CY, Chuang JL, Qi X, Skvora KJ, Dork K, Wallace AL, Morlock LK, Lee BH, Hutson SM, Strom SC, Williams NS, Tambar UK, Wynn RM and Chuang DT. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain alpha-ketoacid dehydrogenase kinase. J Biol Chem. 2014;289:20583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang Y, Zhou M, Sun H and Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. 2011;90:220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen M, Gao C, Yu J, Ren S, Wang M, Wynn RM, Chuang DT, Wang Y and Sun H. Therapeutic Effect of Targeting Branched-Chain Amino Acid Catabolic Flux in Pressure-Overload Induced Heart Failure. J Am Heart Assoc. 2019;8:e011625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polhemus DJ and Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res. 2014;114:730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Polhemus DJ and Lefer DJ. Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ Res. 2018;123:590–600. [DOI] [PubMed] [Google Scholar]
- 14.Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K and Kimura H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–14. [DOI] [PubMed] [Google Scholar]
- 15.Pedre B and Dick TP. 3-Mercaptopyruvate sulfurtransferase: an enzyme at the crossroads of sulfane sulfur trafficking. Biol Chem. 2021;402:223–237. [DOI] [PubMed] [Google Scholar]
- 16.Nagahara N, Ito T, Kitamura H and Nishino T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem Cell Biol. 1998;110:243–50. [DOI] [PubMed] [Google Scholar]
- 17.Abdollahi Govar A, Toro G, Szaniszlo P, Pavlidou A, Bibli SI, Thanki K, Resto VA, Chao C, Hellmich MR, Szabo C, Papapetropoulos A and Modis K. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. Br J Pharmacol. 2020;177:866–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, Chen S, Wen JY and Chen ZW. 3-Mercaptopyruvate sulfurtransferase/hydrogen sulfide protects cerebral endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury via mitoprotection and inhibition of the RhoA/ROCK pathway. Am J Physiol Cell Physiol. 2020;319:C720–C733. [DOI] [PubMed] [Google Scholar]
- 19.Modis K, Coletta C, Erdelyi K, Papapetropoulos A and Szabo C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013;27:601–11. [DOI] [PubMed] [Google Scholar]
- 20.Nagahara N. Multiple role of 3-mercaptopyruvate sulfurtransferase: antioxidative function, H2 S and polysulfide production and possible SOx production. Br J Pharmacol. 2018;175:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow CW and Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peleli M, Bibli SI, Li Z, Chatzianastasiou A, Varela A, Katsouda A, Zukunft S, Bucci M, Vellecco V, Davos CH, Nagahara N, Cirino G, Fleming I, Lefer DJ and Papapetropoulos A. Cardiovascular phenotype of mice lacking 3-mercaptopyruvate sulfurtransferase. Biochem Pharmacol. 2020;176:113833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang J, Li Z, Organ CL, Park CM, Yang CT, Pacheco A, Wang D, Lefer DJ and Xian M. pH-Controlled Hydrogen Sulfide Release for Myocardial Ischemia-Reperfusion Injury. J Am Chem Soc. 2016;138:6336–9. [DOI] [PubMed] [Google Scholar]
- 24.Li Z, Organ CL, Kang J, Polhemus DJ, Trivedi RK, Sharp TE 3rd, Jenkins JS, Tao YX, Xian M and Lefer DJ. Hydrogen Sulfide Attenuates Renin Angiotensin and Aldosterone Pathological Signaling to Preserve Kidney Function and Improve Exercise Tolerance in Heart Failure. JACC Basic Transl Sci. 2018;3:796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomita M, Nagahara N and Ito T. Expression of 3-Mercaptopyruvate Sulfurtransferase in the Mouse. Molecules. 2016;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr., Gojon G Jr., Wang R, Karusula N, Nicholson CK, Calvert JW and Lefer DJ H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura H. Hydrogen sulfide and polysulfides as signaling molecules. Proc Jpn Acad Ser B Phys Biol Sci. 2015;91:131–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimizu Y, Polavarapu R, Eskla KL, Nicholson CK, Koczor CA, Wang R, Lewis W, Shiva S, Lefer DJ and Calvert JW. Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J Mol Cell Cardiol. 2018;116:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neinast M, Murashige D and Arany Z. Branched Chain Amino Acids. Annu Rev Physiol. 2019;81:139–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, Al Batran R, Pherwani S, Ho KL, Boisvenue J, Karwi QG, Altamimi T, Wishart DS, Dyck JRB, Ussher JR, Oudit GY and Lopaschuk GD. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol. 2019;18:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lian K, Guo X, Wang Q, Liu Y, Wang RT, Gao C, Li CY, Li CX and Tao L. PP2Cm overexpression alleviates MI/R injury mediated by a BCAA catabolism defect and oxidative stress in diabetic mice. Eur J Pharmacol. 2020;866:172796. [DOI] [PubMed] [Google Scholar]
- 32.Nagahara N, Nagano M, Ito T, Shimamura K, Akimoto T and Suzuki H. Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: a model for human mercaptolactate-cysteine disulfiduria. Sci Rep. 2013;3:1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luczak ED and Leinwand LA. Sex-based cardiac physiology. Annu Rev Physiol. 2009;71:1–18. [DOI] [PubMed] [Google Scholar]
- 34.Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, Tsai EJ, Molkentin JD, Chen X, Madesh M, Houser SR and Elrod JW. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature. 2017;545:93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Polhemus D, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, Lefer DJ and Calvert JW. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail. 2013;6:1077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R and Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc Natl Acad Sci U S A. 2014;111:3182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia HJ, Li Z, Sharp TE, Polhemus DJ, Carnal J, Moles KH, Tao YX, Elrod J, Pfeilschifter J, Beck KF and Lefer DJ. Endothelial Cell Cystathionine gamma-Lyase Expression Level Modulates Exercise Capacity, Vascular Function, and Myocardial Ischemia Reperfusion Injury. Journal of the American Heart Association. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu KH, Nellis M, Uppal K, Ma C, Tran V, Liang Y, Walker DI and Jones DP. Reference Standardization for Quantification and Harmonization of Large-Scale Metabolomics. Anal Chem. 2020;92:8836–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu KH, Owens JA, Saeedi B, Cohen CE, Bellissimo MP, Naudin C, Darby T, Druzak S, Maner-Smith K, Orr M, Hu X, Fernandes J, Camacho MC, Hunter-Chang S, VanInsberghe D, Ma C, Ganesh T, Yeligar SM, Uppal K, Go YM, Alvarez JA, Vos MB, Ziegler TR, Woodworth MH, Kraft CS, Jones RM, Ortlund E, Neish AS and Jones DP. Microbial metabolite delta-valerobetaine is a diet-dependent obesogen. Nat Metab. 2021;3:1694–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu T, Park Y, Johnson JM and Jones DP. apLCMS--adaptive processing of high-resolution LC/MS data. Bioinformatics. 2009;25:1930–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uppal K, Soltow QA, Strobel FH, Pittard WS, Gernert KM, Yu T and Jones DP. xMSanalyzer: automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinformatics. 2013;14:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W and Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith MR, Jarrell ZR, Orr M, Liu KH, Go YM and Jones DP. Metabolome-wide association study of flavorant vanillin exposure in bronchial epithelial cells reveals disease-related perturbations in metabolism. Environ Int. 2021;147:106323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carlson NS, Frediani JK, Corwin EJ, Dunlop A and Jones D. Metabolic Pathways Associated With Term Labor Induction Course in African American Women. Biol Res Nurs. 2020;22:157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uppal K, Walker DI and Jones DP. xMSannotator: An R Package for Network-Based Annotation of High-Resolution Metabolomics Data. Anal Chem. 2017;89:1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.