

Abstract
Because of its less toxicity and electronic structure analogous to that of lead, tin halide perovskite (THP) is currently one of the most favorable candidates as an active layer for optoelectronic and electric devices such as solar cells, photodiodes, and field‐effect transistors (FETs). Promising photovoltaics and FETs performances have been recently demonstrated because of their desirable electrical and optical properties. Nevertheless, THP's easy oxidation from Sn2+ to Sn4+, easy formation of tin vacancy, uncontrollable film morphology and crystallinity, and interface instability severely impede its widespread application. This review paper aims to provide a basic understanding of THP as a semiconductor by highlighting the physical structure, energy band structure, electrical properties, and doping mechanisms. Additionally, the key chemical instability issues of THPs are discussed, which are identified as the potential bottleneck for further device development. Based on the understanding of the THPs properties, the key recent progress of THP‐based solar cells and FETs is briefly discussed. To conclude, current challenges and perspective opportunities are highlighted.
Keywords: field‐effect transistors, lead‐free perovskites, solar cells, tin halide perovskite
Tin halide perovskite (THP) is a promising material for active layer in optoelectronic and electric devices, due to its less toxicity and analogous electronic structure to that of lead. This review provides a basic understanding of the material, including lattice structure, energy band, doping and chemical instability, and presents key progress and perspectives of THP‐based high performance solar cells and transistors.
1. Introduction
To date, metal halide perovskite (MHP) can be considered as a promising candidate for various next‐generation optoelectronics and electronics, such as solar cells,[ 1 ] field‐effect transistors (FETs),[ 2 ] light‐emitting diodes,[ 3 ] photodetectors,[ 4 ] energy storage,[ 5 ] lasers,[ 6 ] memories,[ 7 ] and piezoelectric applications.[ 8 ] Such prompt progress of MHPs in a broad spectrum of the scientific community is due to not only its impressive optoelectronic properties but also promising cost‐effective, high‐throughput, and low‐temperature manufacturing processes.[ 9 ] The bulky three‐dimensional (3D) form of MHPs comprises of a corner‐sharing BX6 octahedral network with the chemical stoichiometry ABX3, where A is an organic or inorganic monovalent cation, B is a divalent metal ion, and X is a halide anion. The B‐X‐B configuration in a BX6 octahedron has a bonding angle of 180°, inducing a large dispersion of both the conduction and valence bands originating from super degeneracy. This electronic structure implies that MHPs possess intrinsically outstanding and desirable electrical and optoelectronic properties such as high charge‐carrier mobility, tunable bandgaps, large optical absorption coefficient, long exciton diffusion length, and ambipolar charge transport.[ 10 , 11 , 12 , 13 , 14 ] Remarkable performances from MHP‐based FETs and MHP‐based solar cells (PSCs) (power conversion efficiency (PCE) > 25%) imply the potential scopes for developing future functional optoelectronic devices.
Despite these exciting properties and advances, most MHP devices today are manufactured using Pb as the divalent metal in the ABX3 structure, the toxicity issue of Pb remains a long‐lasting debate. The concentration of immediately dangerous to life or health of Pb is 100 mg m−3, which means there is a risk of human exposure to Pb in the fabrication and handling of perovskite devices.[ 15 ] Moreover, our biological systems might be in great jeopardy due to the water‐soluble properties of Pb. Thus, to prevent any perturbing issues related to Pb, the European Union has publicly announced that exposures of up to 1000 ppm are allowed for all electronic devices.[ 15 ] Systematic encapsulation protocol and numerous encapsulation approaches have been given serious consideration to inhibit any possible leakage of Pb; however, it is difficult to ensure how safe it is when it comes to device fabrication as well as long‐term storage. Even with a sophisticated encapsulation procedure, there is no guarantee that potential Pb leakage can be prevented. Therefore, controlling, handling, and securing safety during the manufacturing of any related Pb‐based MHPs devices remain a huge challenge.
Apart from the encapsulation method, recent attention has been devoted to replacing Pb with low toxicity and non‐toxicity cations. Several potential low‐toxic and chemically compatible materials such as Sn,[ 16 ] Bi,[ 17 ] and Ge[ 18 ] have been proposed to replace Pb, not only to reduce its toxicity but also to maintain the unique optoelectronic properties of perovskite. Among these materials, eco‐friendly material Sn has been widely utilized in various promising optoelectronic devices, including solar cells and FETs. Similar to that of Pb, Sn has an inactive outer shell s orbital, which is vital to realizing the distinctive electrical and optical properties of MHP.[ 19 ] The effects of Sn substances may vary, and it is relatively safe for a human when the hydrogen bonds grow longer in Sn substances.[ 20 ] Still, the influences and their adverse effect on human health have raised some eyebrows, there is a big question regarding its use.
The first Sn‐based perovskite (tin halide perovskite; THP) was successfully developed by Fisher et al. in 1974,[ 21 ] followed by a systematic study from Donaldson's research group.[ 22 , 23 ] Later, the first hybrid THP was proposed by Yamada et al.[ 24 , 25 , 26 ] in which Mitzi and colleagues expanded the work and conducted a study in the context of dimensional reduction, and demonstrated the first structural properties under pressure.[ 27 ] These initial discoveries stimulate researchers to devote huge attention to THP‐based devices until now. In 2014, Kanatzidis et al. reported the first Sn‐based perovskite solar cells (TPSCs).[ 28 ] Subsequently, numerous studies have utilized Sn instead of Pb for various type of optoelectronics devices; for instance, the latest breakthrough in TPSCs have witnessed the PCE of 14.8%, whereas the highest hole and electron mobilities of 55 and 2.1 cm2 V−1 s−1 for THP‐based FETs have been demonstrated.[ 20 ] The rapid development in THP‐based optoelectronics devices makes it urgently necessary to review and assess their potential for several optoelectronic and electronic applications. In this regard, earlier published reviews have highlighted the progress and potential of THP's applications with solar cells,[ 29 , 30 , 31 , 32 ] photodetectors,[ 33 ] light‐emitting devices, and radiation detectors.[ 34 ] With intense research for various applications, THP‐based electronic devices show encouraging performances. However, the stability of the THP‐based electronic devices is still poor as compared to the Pb‐based ones. In this article, we aim to provide a comprehensive understanding of THP and highlight the impressive latest results for their functional device applications. First, we discuss the structure, chemistry, and thin‐film formation of THP. Second, we introduce the structure‐electrical property relationship of THP with energy band structure and electrical doping through intrinsic and extrinsic doping. Third, we review the recent progress of TPSCs. In this section, we also discuss the various chemical bonding and chemical structure of THP and their effect on the fabrication of TPSCs. Fourth, we discuss recent progress in THP‐based FETs, including two‐dimensional (2D) perovskite, hybrid 2D/3D perovskite, and 3D perovskite. Finally, we review the prospects for THP‐based PSCs and FETs, aiming to motivate the scientific community and better understand the development of future optoelectronic devices (Scheme 1 ).
Scheme 1.
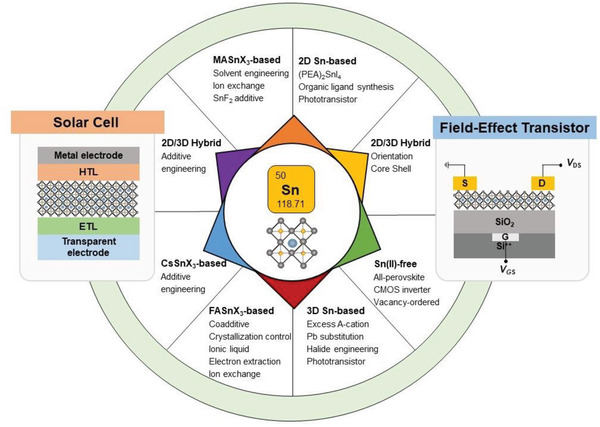
Summary of the THP‐based perovskite materials, and their specific engineering for device performance enhancement of PSCs and FETs.
2. THP
2.1. Structure of THP
THPs consist of ABX3 stoichiometry (Figure 1a). To maintain the neutrality of the structure, the sum of the oxidation states of the A and B cations must be 3+, thus requiring a 3− charge, provided by the three monovalent anions is required. A stoichiometry outside this restriction forms other dimensions of perovskite or non‐perovskite structures. The chemical and structural versatility of perovskites offers many opportunities for research and applications. The inorganic sublattice is composed of [BX6]4− octahedra, where B is a divalent metal such as Pb2+, Sn2+, and Ge2+ or a mixture of monovalent and trivalent metals (e.g., Ag+ and In3+) and X is a halide: I−, Br−, or Cl−. By permutation of the A‐site cations and anions, a huge number of THPs are possible.[ 35 ] Besides the widely used Cs and Rb, 13 protonated amines can be incorporated into the perovskite framework, as A‐site cations and eight anions, to obtain different types of perovskites. Additional structures can also be achieved through doping with various compounds. Nevertheless, to realize stable THPs, much theoretical understanding and fundamental calculation are necessary. At room temperature, the amine cations (e.g., methylammonium (MA), formamidinium (FA)) in the perovskite cavity are generally disordered, and the systems exhibit a remarkable spectrum of ferroelectric and multiferroic characteristics when cooled.[ 36 ] However, to realize an ideal perovskite structure, the cations must be well‐matched with the anions. The Goldschmidt model provides an ideal method to determine the stable perovskite structure.[ 37 ]
| (1) |
where Ra , Rb , and Rx are the ionic radii of the A, B, and X sites, respectively. Hence, the tolerance factor (t) assesses whether the A‐cation can fit within the framework of corner‐sharing octahedra. Experimental data suggest that t values have to be in the range between 0.8 and 1.0 to maintain a 3D perovskite structure. If the t values are within 0.9 to 1.0, an ideal cubic phase is expected. For the largest value of Rb and Rx (R Pb = 1.19 Å, R I = 2.20 Å) within the optimum t limit (0.8 < t < 1), the ionic radius for A‐site cation to form perovskite framework was calculated at 1.60–2.60 Å.[ 38 , 39 ] Hence, MA (2.17 Å) and FA (2.53 Å) are the most suitable organic cations for developing effective perovskite compounds.[ 40 , 41 ] Incorporation of A‐cation beyond the above size limit could reduce the dimensionality of perovskite from 3D to 2D and 1D.[ 42 ] Interestingly, when divalent B‐cation is replaced from large Pb to small Sn cation, the values of t increase and approach toward 1. One of the remarkable aspects making the crystal structures of perovskites versatile is the structural flexibility of organic groups (Figure 1b). A t value close to 1 can be achieved by replacing a larger monovalent A‐cation (FA+) with a smaller cation (Cs+) too. The doping of FASnI3 with Cs could tune the t downward from 1.04 to 1.02.[ 43 , 44 ] THPs have high electrical conductivity (σ) because of the marginal crossing of Sn 5s and Sn 5p bands near the Brillouin zone boundary (due to the large dispersion of Sn 5s hybridized band) point ({½, ½, ½} 2π/a).[ 45 ] Relativistic effects (e.g., spin‐orbital coupling, effective masses of electrons and holes, orbital contraction, or inert pair effect)[ 46 ] are stronger in Pb than Sn, which leads to stabilization of 6s 2 orbital of Pb relative to 5s 2 of Sn.
Figure 1.
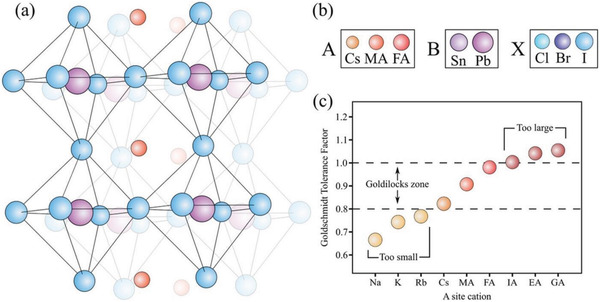
a) ABX3 structure of MHP, b) size of various cations and anions, and c) tolerance factor of MHPs with a variation of cations.Adapted with permission.[ 43 ] Copyright 2017, American Association for the Advancement of Science.
2.1.1. MASnI3
MASnX3 perovskites were studied intensively because of their similarity with MAPbI3. MASnI3 shows a cubic (Pm3m) phase at 295 K; however, a phase transition from cubic to lower symmetry tetragonal (I4/mcm) occurs at 275 K.[ 47 , 48 ] Because the SnI6 octahedra tilt about the vertical axis, the tetragonal structure corresponds to a √2a × √2a × √2a supercell extension of the cubic lattice. By lowering the temperature to 108–114 K, another phase transition from tetragonal to orthorhombic (Pbn21 or Pbnm) can be obtained.[ 49 ] In the adjacent planes, the SnI6 octahedra tilt in the same direction around the c–axis (Figure 2 ). The A‐cation has no rotational flexibility in this arrangement. Similarly, holes become more localized, extending the lifetime of charge carriers.[ 50 ] According to the first principle calculations, this phase has extremely low conductivity,[ 51 ] which makes them an ideal candidate for low‐temperature electronics.
Figure 2.
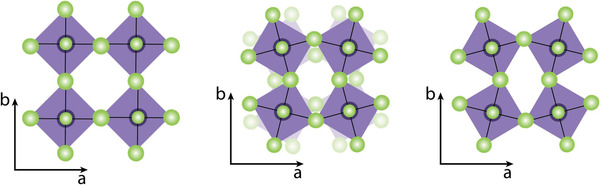
Rotation of adjacent octahedral layers along the c–axis according to crystal phase. a) Cubic, b) tetragonal, and c) orthorhombic phase. a–c) Reproduced under the terms of the Creative Commons CC‐BY license.[ 52 ] Copyright 2022, SciELO Brazil.
2.1.2. FASnX3
At room temperature, MASnI3 and FASnI3 are isostructural. Because their symmetry does not match the Oh site symmetry of the cubic perovskite structure, the FA molecules are orientationally disordered.[ 53 ] Because of the steric effect of the FA cation, the bond length of Sn‐I is 3.158 Å, whereas, in the MASnI3 system, the Sn—I bond length is 3.121 Å. FASnI3 forms a pseudocubic Amm2 crystal structure at 340 K. Interestingly, a phase transition to orthorhombic phase with Imm2 space group can be observed below 180 K for the FASnI3 system.[ 28 ] However, clear conscientious experiments below 100 K to understand the crystal structure of FASnI3 have not been reported yet. Nevertheless, gradual cooling to room temperature revealed some phase transition properties for FASnI3 perovskites. The first phase transition upon cooling from room temperature was observed between 225 K and 250 K in cubic Pm3m to tetragonal P4/mbm, and a further phase transition to orthorhombic Pnma structure occurs between 125 and 150 K was confirmed by Schueller et al.[ 54 ] Another study showed that a phase transition from a cubic room temperature to a tetragonal phase is possible at 255 K and a second tetragonal phase below 155 K.[ 55 ] However, it must be considered that the variation of the crystal phases has been identified with either single crystals or polycrystalline films, which might have an impact. Figure 3 shows the phase transitions of FASnI3. At room temperature, the cubic phase contains rotationally disordered FA molecules. When the temperature is deduced to 255 K, an in‐plane rotation of the SnI6 octahedra occurs. A further deduction of temperature to 155 K depicts the fully ordered FA molecules. At this stage, instead of one, there are two distinct layers of SnI6 octahedra with different rotation angles. The inclusion of a pseudohalide SCN partially with the FASnI3 system can change the orthorhombic phase to monoclinic and triclinic structures.[ 56 ]
Figure 3.
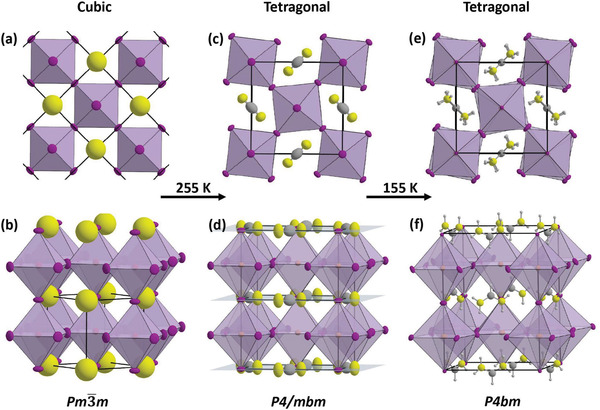
FASnI3 undergoes two phase transitions in the temperature range from 300 to 85 K. a,b) The cubic phase at room temperature contains fully rotationally disordered FA molecules. c,d) The structure becomes tetragonal at 255 K because of an in‐plane rotation of the SnI6 octahedra. The orientation of the FA molecules is two‐fold disordered due to mirror planes (shaded light blue). e,f) Doubling of the c‐lattice parameter occurs at 155 K, as the mirror planes perpendicular to the c–axis are removed and the FA molecules become fully ordered. There are now two distinct layers of SnI6 octahedra with different rotation angles. a–f) Reproduced with permissio.[ 55 ] Copyright 2020, American Chemical Society.
2.1.3. CsSnX3
When compared with MA+ or FA+, Cs+ is smaller in size which leads to rotation and tilt of SnI6 octahedral. This tilt influences the structural stability of the CsSnI3 at room temperature. Interestingly, at a higher temperature (500 K), the CsSnI3 perovskite converts to a black cubic (B‐α) phase (Figure 4a). Upon gradual decrease of temperature to 380 K, the black cubic phase transforms into low symmetry black tetragonal phase (B‐β) (Figure 4b) phase. At 300 K, the phase transition to the black orthorhombic (B‐γ) phase is stable at room temperature.[ 57 ] In this aspect, the B‐γ phase of CsSnI3 perovskite is most suitable for device applications. Nonetheless, studies suggest that exposure to ambient air or organic solvents in this phase tends to transform into a double‐chain structure as shown in Figure 4d. The formation of such a double‐chain structure is susceptible to air and often decomposes to Cs2SnI6, which has poor optoelectric and electric properties making it unsuitable for photovoltaic and FET applications. Kubicki et al. performed a detailed solid‐state nuclear magnetic resonance (ssNMR) study to compare the structures of MASnBr3, FASnBr3, CsSnBr3, MASnI3, FASnI3, and CsSnI3 perovskites.[ 58 ] For every I/Br and Br/Cl mixed ratio, the ssNMR results showed that iodide–bromide and bromide–chloride mixtures can form a stable phase. Iodide–chloride mixes, conversely, produced phase‐segregated mixtures of phases, despite being somewhat miscible.
Figure 4.
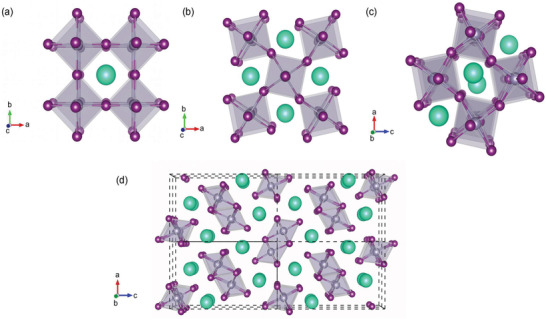
The four polymorphs of CsSnI3. On the top row, the three black perovskite structures are shown: a) cubic B‐α, b) tetragonal B‐β, and c) orthorhombic B‐γ. d) The bottom row shows the yellow crystal where the tin halide octahedral networks are fragmented into 1D chains. In all four images, the green spheres represent the Cs atom, whereas the purple polyhedra represent the octahedral perovskite cage formed by the bonding of the Sn (steel blue) and I (dark purple) atoms. a–d) Reproduced with permission.[ 59 ] Copyright 2015, The American Physical Society.
2.2. Band Structure and Electrical Properties
Sn exhibits a comparable electronic configuration to Pb, as both elements possess s orbital lone‐pairs (Figure 5a).[ 48 , 59 , 60 , 61 , 62 ] Sn and Pb have similar ionic radii and optoelectronic properties, such as direct bandgaps in the visible spectrum and relatively low electron/hole effective mass.[ 63 ] However, THP exhibits a different electronic band structure from that of lead halide perovskites (LHPs). The bandgap of THP is ≈1.4 eV, which is smaller than the bandgap of LHP (≈1.5 eV).[ 64 ] The bandgap of THP approaches Shockley–Queisser optimal bandgap for single‐junction solar cells, and under ideal fabrication conditions, TPSCs are expected to reach a theoretical PCE up to 33.4%. Additionally, the lone‐pair state of Sn 5s is more reactive than Pb 6s because the strong lanthanide shrinkage effect of Pb makes them an inert electron pair.[ 65 ] Because of the absence of this effect, Sn becomes an easily oxidized element with two active Sn 5s electrons (Figure 5b). This active electron pair increases the Lewis acidity of SnI2 over PbI2 and speeds up the reaction of SnI2 with Lewis base starting materials for perovskite such as MAI and FAI. In the following section, the formation of the energy band structure in THPs will be discussed in terms of orbital overlaps and unique properties of Sn.
Figure 5.
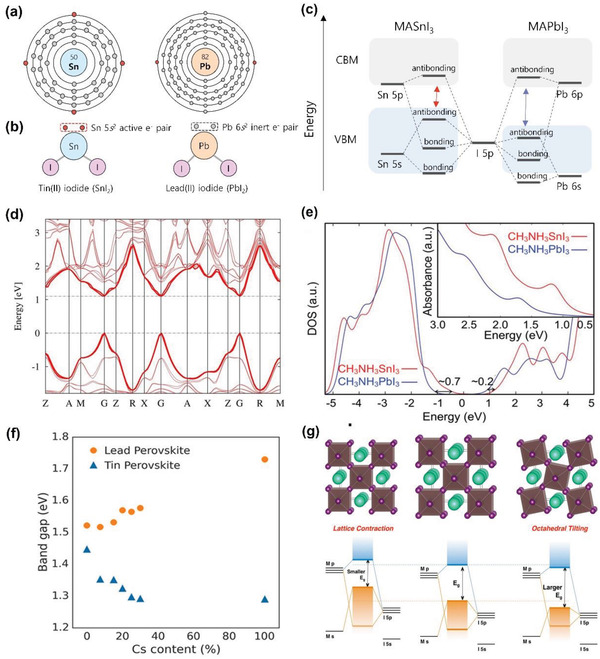
Electronic band structure of THPs. a) Electronic structure of Sn and Pb atoms. b) Schematics of s state valence electrons in SnI2 and PbI2. c) Energy band schematics of MASnI3 and MAPbI3. d) GW + SOC calculated band structure of MASnI3. e) GW + SOC calculated electronic DOSs and optical absorption spectra (inset) of MASnI3 and MAPbI3. d,e) Adapted with permission.[ 48 ] Copyright 2014, Nature Publishing Group. f) Optical bandgaps of THPs and LHPs FA1‐ x Cs x SnI3 (0 ≤ x ≤ 1) as a function of Cs content. g) Perovskite lattice diagrams (top row) and corresponding energy level diagrams (bottom row) of lattice contraction (left panel), normal lattice (center panel), and octahedral tilting (right panel). f,g) Reproduced with permission.[ 66 ] Copyright 2017, American Chemical Society.
2.2.1. Energy Band Structure
The energy band structure is the key to analyzing the optoelectronic and electronic properties of semiconductor materials. Through combined theoretical and experimental approaches, the band structures of THP can be examined. The valence band maximum (VBM) of THP is composed of the antibonding between Sn 5s and I 5p orbitals with its main contribution from I 5p. Conduction band minimum (CBM) is generated from the antibonding of Sn 5p and I 5p orbitals with dominant contribution by Sn 5p orbital (Figure 5c).[ 48 , 60 ] Umari et al. investigated the electronic band structure of MASnI3 by GW + Spin‐orbital coupling (SOC) method in comparison with its Pb counterpart.[ 48 ] The band structure of MASnI3 showed a direct bandgap of 1.1 eV (1.67 eV for MAPbI3 at the same point), comparable with the bandgap of ≈1.3 eV obtained experimentally (Figure 5d).[ 67 , 68 ] MASnI3 exhibited more robust s—p antibonding coupling near the VBM because of the shallower and more active Sn 5s lone‐pair states than those of Pb 6s. Consequently, the VBM of MASnI3 is more dispersive and locates ≈0.7 eV higher than that of MAPbI3 (Figure 5e). The calculated average hole effective mass of MASnI3 (0.13 m0) is also smaller than that of MAPbI3 (0.25 m0). In the case of CBM, the Sn 5p orbital is shallower and less dispersive than Pb 6p orbitals due to much weaker SOC, and therefore, the CBM of MASnI3 is less dispersive and positions at 0.2 eV higher than that of MAPbI3. Thus, the calculated average electron effective mass of MASnI3 (0.28 m0) is larger than that of MAPbI3 (0.19 m0).
The band structure of THP is dependent not only on B‐site cation but also on A‐site cation by modulating its molecular size. Prasanna et al. demonstrated that by reducing the ionic radius of A‐site cation, the bandgap of THP decreases in the following order: FASnI3 (1.41 eV), MASnI3 (1.30 eV), and CsSnI3 (1.25 eV), and a reverse trend in LHPs (Figure 5f).[ 66 ] In the case of LHPs, the reduction of A‐site cation radius causes [PbI6]4− octahedra to tilt. Consequently, the Pb‐I‐Pb angle is reduced, and the orbital overlap between Pb and I is shortened, pushing VBM to a deeper energy level and widening the bandgap. For THPs, because of the smaller size of Sn 2p than that of Pb 2p, [SnI6]4− octahedron simply contracts instead of tilting, pushing the energy band to a shallower level, and ultimately shortens the bandgap (Figure 5g). However, the bandgap tuning with A‐site cation may only be possible in 3D perovskite structures. In 2D perovskites, the electronic state of organic cation is located deep in valence band (VB). Hence, the organic content merely participates in structural stabilization and electrostatic charge balance and does not directly contribute to electronic properties.
THPs have both advantages and disadvantages for optoelectronic applications. For example, THPs exhibit a reduced bandgap and higher absorption coefficient in the visible spectrum than LHPs. Hence, THPs are more suitable for efficient single‐junction solar cells. However, the high energy of Sn 5s 2 states easily breaks the Sn—I bonds and increases the density of Sn vacancies (V Sn). The formation of V Sn and thus intrinsic doping in THPs will be discussed in the next section. In summary, the energy levels of Sn 5s and Sn 5p orbitals and CBM and VBM of THPs are located higher than those of Pb 6s orbitals and LHPs. The higher energy level of Sn 5s than Pb 6s contributes to weaker Sn—I bonding than Pb—I bonding, which increases the density of V Sn in the thin film.[ 69 ] The weak Sn—I bond also facilitates its reaction with oxygen and moisture to generate Sn—O and H—I bonds.[ 70 ] Due to these subtle but critical differences in electronic band structures, THPs and LHPs exhibit different optoelectric properties and ambient stability. THPs show high hole mobility and conductivity due to smaller hole effective mass and low formation energy of V Sn, whereas LHPs show better electron transport and ambient stability as a result of their smaller electron effective mass, deep Pb 6p orbitals, and stronger SOC effect.[ 69 , 71 ] Hence, THPs are more suitable for p‐type electric devices such p‐channel FETs.
Electrical doping can provide additional electrons or holes into the lattice to further tune the Fermi level positions and carrier concentrations. Electrical doping within the lattice can generally be divided into two methods: intrinsic doping and extrinsic doping. Intrinsic doping is induced by self‐generated defects such as cation or anion vacancy within the constituents, whereas extrinsic doping is induced by the introduction of impurity atoms or molecules in or near the crystal lattice. Each method is discussed in the following sections.
2.2.2. Intrinsic Doping
Intrinsic doping in THPs can be induced by defect formation in the ABX3 crystal lattice, such as vacancies (V A, V B, and V X), interstitials (A i, B i, and X i), and substitutions (A B, B A, A X, X A, B X, and X B) (Figure 6a).[ 72 ] Density functional theory (DFT) calculations are often used to predict the range of tuned Fermi level (E F) after defect formation and to calculate the transition level of each defect within the bandgap. For example, Xu et al. demonstrated that V Sn is located closely below the VBM as a shallow trap.[ 73 ] V Sn is the dominant trap with low formation enthalpies, especially under Sn‐poor conditions. Hence, SnI2‐based perovskite films exhibit high hole density above 1017 cm−3 due to easy V Sn formation. The V Sn formation originates from the strong antibonding between Sn 5s‐I 5p: the stronger the antibonding is, the lower the formation energy of V Sn becomes (Figure 6b,c).[ 73 , 74 ] The strength of Sn s‐I p antibonding was measured by crystal orbital Hamilton population (pCOHP) and showed that antibonding strength has a negative correlation with ionic radii of A‐site cation (Figure 6d). The increase in ionic radii of A‐site cation further perturbs the lattice and increases the Sn—I bond length. The optical bandgap is also affected by the extent of orbital overlap between Sn and I ions.[ 60 , 76 ] The large size A‐site cation weakens Sn s—I p antibonding and increases in energy barrier of V Sn formation. For example, DFT calculations performed on ASnI3 (A = Cs, MA, and FA) demonstrated a high level of p‐doping under I‐rich and Sn‐poor conditions.[ 73 , 74 , 77 ] Larger size of FA+ than MA+ resulted in a longer Sn—I bond. Consequently, Sn 5s—I 5p antibonding coupling in FASnI3 was weaker than that of MASnI3, resulting in higher V Sn formation energy for FASnI3 (Figure 6e). Hence, the conductivity of FASnI3 can be regulated from p‐type to intrinsic under Sn‐rich conditions. In comparison, MASnI3 has high conductivity regardless of the condition. Furthermore, Ke et al. used bulky ethylenediammonium cation to modify the physical properties of MASnI3 and FASnI3. [ 78 , 79 ] The high energy state and instability of Sn 5s 2 states facilitated the oxidation of Sn2+ to Sn4+. Therefore, THPs are significantly unstable in ambient air.[ 80 ] The undercoordinated I− ions may result in deep trap states within the bandgap because of the easy loss of Sn2+. Deep trap states are detrimental as non‐radiative carrier recombination centers and result in the loss of photogenerated carriers. Additionally, the deep traps degrade charge transport in FETs and provide large hysteresis in FET operation. To compare the intrinsic properties of THPs, Table 1 lists previously reported carrier concentrations and Hall mobilities.
Figure 6.
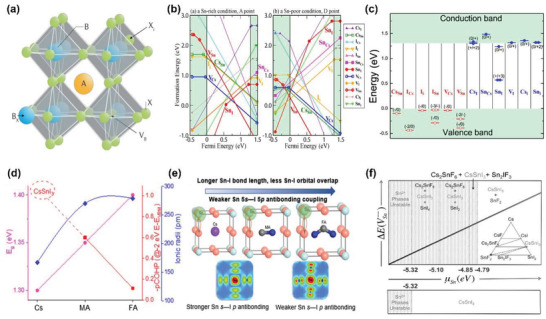
Intrinsic doping. a) Schematic of substitutional (Bx ), vacancy (V B), and interstitial (X i) intrinsic defects in ABX3 perovskite crystal lattice. Reproduced with permission.[ 72 ] Copyright 2021, Nature Publishing Group. b) Calculated defect formation energy as a function of Fermi energy at Sn‐rich condition (left) and Sn‐poor condition (right). c) Calculated transition energy levels for various intrinsic defects in CsSnI3. b,c) Reproduced with permission.[ 73 ] Copyright 2014, American Chemical Society. d) Negative associations between antibonding strength of Sn 5s‐I 5p atomic orbitals (red: measured at −2 eV below VBM via pCOHP method) and size of A‐site cations (blue) or bandgaps of corresponding Sn‐based iodide perovskites. e) Schematics of the bond length impacts on Sn 5s—I 5p antibonding coupling, also regulated by the size of A‐site cation (top), and partial charge densities around VBM level of MASnI3 (bottom, left) and FASnI3 (bottom, right). d,e) Adapted with permission.[ 74 ] Copyright 2017, Royal Society of Chemistry. f) Calculated formation energy of Sn vacancy [ΔE(V Sn)] at a fixed Fermi level, as a function of Sn chemical potential (µ Sn). Reproduced with permission.[ 75 ] Copyright 2014, Wiley‐VCH.
Table 1.
Intrinsic properties of THPs: carrier concentration and Hall mobility
| Material | n [cm−3] | µ Hall (p‐type) [cm2 V−1 s−1] | µ Hall (n‐type) [cm2 V−1 s−1] | T [K] | Process | Measurement | Ref. |
|---|---|---|---|---|---|---|---|
| CsSnI3 | 8.73 × 1014 | 520 | 536 | R.T. | Pressed pellet | Hall measurement | [28] |
| CsSnI3 | 1.00 × 1017 | 585 | R.T. | Pressed powder | Hall measurement | [57] | |
| CsSnI3 | 1.00 × 1019 | 24.5 | R.T. | Thin film | Hall measurement | [90] | |
| CsSnI3/BiI3 | 1.00 × 1017 | 26.9 | R.T. | Thin film | Hall measurement | [90] | |
| CsSnI3 | 3.00 × 1016 | 54 | R.T. | Thin film | Hall measurement | [20] | |
| CsSn x Pb1‐ x I3/SnF2 | 3.00 × 1015 | 486 | R.T. | Thin film | Hall measurement | [20] | |
| Cs2SnI6 | 1.00 × 1014 | 310 | R.T. | Polycrystalline pellet | I–V/Hall measurement | [91] | |
| Cs2SnI6 | 1.50 × 1016 | 79 | R.T. | Thin film | Hall measurement | [92] | |
| Cs2SnI6 | 6.00 × 1016 | 2.9 | R.T. | Thin film | Hall measurement | [93] | |
| Cs2SnI6 | 9.10 × 1018 | 20.2 | R.T. | Nanocrystal solution dropcast | I–V measurement | [94] | |
| MASnI3 | 2.00 × 1019 | 50 | R.T. | Pressed pellet | Hall measurement | [83] | |
| MASnI3 | 9.00 × 1017 | 200 | 250 | As‐grown crystal | Hall measurement | [84] | |
| MASnI3 | 7.94 × 1014 | 322 | R.T. | Pressed pellet | Hall measurement | [28] | |
| MASnI3 | 2.8 × 1017 | 25 | R.T. | Thin film | Hall measurement | [95] | |
| MASn(I/Br/Cl)3 | 2.2 × 1015 | 301 | R.T. | Thin film | Hall measurement | [95] | |
| (4Tm)2FASn2I7 | 5.49 × 1018 | 1.06 | R.T. | Thin film | Hall measurement | [96] | |
| FASnI3/(PEA)2SnI4 | 1.20 × 1016 | 0.21 | R.T. | Thin film | Hall measurement | [97] | |
| HC(NH2)2SnI3 | 8.38 × 1013 | 103 | R.T. | Pressed pellet | Hall measurement | [28] |
n: Carrier concentration, µ Hall: Hall mobility, T: Temperature
The accuracy of DFT calculations should be tested as it depends on several factors such as exchange–correlation functionals, the secondary‐order phase constraints, and the supercell sizes.[ 81 ] Specifically, a large size supercell is necessary to omit defect–defect interaction. Although DFT calculation is an important method to determine the compositional limits of a specific material under thermodynamic equilibrium conditions, it should be verified experimentally. Intrinsic defects can be experimentally confirmed by adjusting the ratio of starting materials in various film deposition methods. The experimental results on THPs are consistent with the theoretical calculations explained above. For example, strong p‐doping is observed in ASnX3 perovskite due to the oxidation of Sn2+ into Sn4+ and easily formed V Sn during synthesis or post‐degradation.[ 47 , 82 , 83 , 84 ] The density of V Sn can be modulated in Sn‐rich conditions, such as the addition of SnF2 in precursor solutions. Although the excess Sn2+ does not incorporate into the crystal lattice, the SnF2 addition can increase the formation energy of V Sn and reduce overall hole density (Figure 6f).[ 75 , 85 , 86 , 87 , 88 , 89 ]
2.2.3. Extrinsic Doping
Extrinsic doping can be achieved by introducing impurity atoms or molecules in or near the perovskite lattice. Figure 7a illustrates the schematic for potential extrinsic dopants in place of each atomic site in ABX3. The introduction of different A‐site cations in ASnX y is discussed in the previous section where the lattice contraction changes Sn—I bond length, the strength of antibonding, and the overall bandgap. The B‐site extrinsic doping in place of Sn2+ is especially important because the inorganic octahedral cage and its network connection theoretically have the most contribution to the electronic properties of organic–inorganic perovskites.[ 60 ] Extrinsic doping of B‐site can be generally divided into two types: homovalent and heterovalent doping. Despite the significant amount of B‐site atomic doping studies for LHPs, doping studies on THPs are still premature.[ 98 , 99 , 100 , 101 ] To select an appropriate atomic substitutional dopant, the ionic size must be suitable to avoid host lattice distortion.
Figure 7.
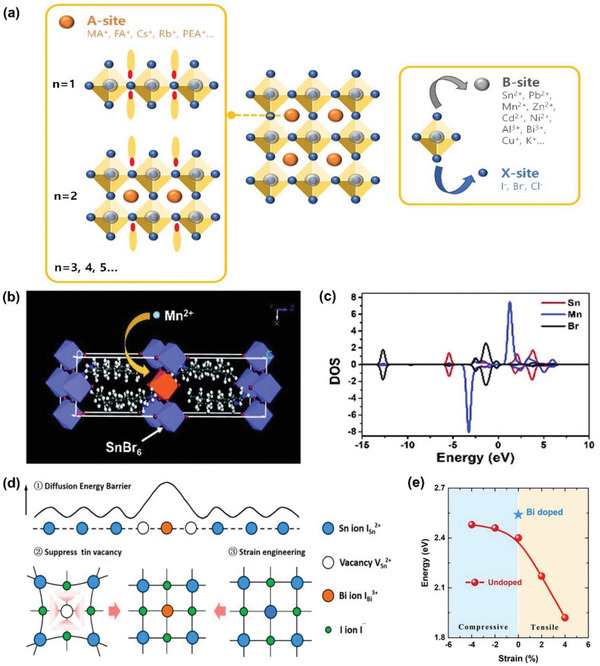
Extrinsic doping. a) Schematics of extrinsic dopants in A‐, B‐, and X‐site of ABX3 crystal structure. b) Schematic illustration of 2D single‐layered (C8H17NH2)2Sn1‐ x Mn x Br4 (x = 0.25). c) Orbital‐projected partial DOS. b,c) Reproduced with permission.[ 102 ] Copyright 2020, Royal Society of Chemistry. d) Schematic representation of CsSnI3 crystal lattice engineering with Bi incorporation: formation of diffusion energy barrier, tin vacancy suppression, and strain engineering. e) Correlation of strain and Bi substitution on the energy barrier for Sn2+ diffusion. d,e) Reproduced with permission.[ 90 ] Copyright 2021, American Chemical Society.
Mn2+ is one of the suitable candidates for homovalent doping of Sn2+. A previous study examined Mn2+ doping into octylammonium tin bromide and resulted in a red shift of PL spectrum, showing orange‐red light (Figure 7b).[ 102 ] A series of measurements showed that Mn2+ atoms were bound to the lattice. X‐ray diffraction peaks showed a positive shift in the (001) signals, indicating Mn alloy formation with Sn.[ 99 ] Additionally, according to the calculated electronic band structure and density of state (DOS), the top of VB is predominately occupied by Br orbitals and the bottom of the CB is dominated by Sn and Mn orbitals (Figure 7c). Mn orbitals are mostly located in CB enabling photogenerated electrons to jump into Mn orbitals for efficient energy transfer. Hence, the close orbital positions of Mn and Sn generate a harmonious yet competitive relationship between self‐trapped exciton emission and Mn d‐d‐transition emission. Another example candidate for homovalent doping of Sn2+ is Zn2+. Bowman et al. observed that slight addition of ZnI2 into the precursor solution increased carrier lifetimes, photoluminescence (PL) quantum efficiencies, and film stability in the air by reducing the formation of tin‐rich clusters, which are susceptible to oxidation.[ 103 ]
One of the earliest heterovalent doping studies by Takahashi et al. was based on artificially incorporating Sn4+ within the THP structure. The Sn4+ incorporation induced additional V Sn and increased conductivity.[ 104 ] Another heterovalent dopant candidate is Bi3+, which Zhang et al. incorporated into (PEA)2SnBr4. The Bi3+ incorporation resulted in a slight blue shift of the PL spectrum and reduced emission from the low energy tail.[ 105 ] Zhou et al. also incorporated Bi3+ into the crystal lattice of CsSnI3. The authors demonstrated that the air instability of black phase CsSnI3 can be greatly lessened by Bi3+ dopant by restricting the direct conversion of γ‐CsSnI3 into 0D Cs2SnI6.[ 90 ] The improved stability was also revealed through DFT calculations. The lattice contraction suppressed V Sn formation and increased the energy barrier between the transformation of γ‐CsSnI3 to Cs2SnI6 (Figure 7d,e). Therefore, the introduction of an appropriate extrinsic dopant can achieve further modulate optoelectronic properties. However, heterovalent doping may not always work for all types of THPs. A combined theoretical and experimental study on the incorporation of monovalent metal cation (M+) into (PEA)2SnI4 revealed that the calculated formational energy of M+ doped‐(PEA)2SnI4 was too high and resulted in destabilized octahedral cage layer.[ 106 ] Instead, experimental results showed that monovalent metal iodide existed separately within the film along the grain boundaries but was not incorporated into the lattice. Thus, monovalent metal iodides as additives enhanced the device's performance as a grain boundary passivating agent.[ 106 , 107 ] Interestingly, molecular doping, which controls the amount of charge by bringing dopant molecules closer to the perovskite lattice, is also being studied. A more in‐depth study is needed on whether this molecular doping in THP occurs through the same charge transfer as in organic semiconductors.
The effect of electrical doping of THPs is necessary to expand our knowledge of the optoelectronic and electronic properties and their tunability. Extensive theoretical and experimental studies are required to visualize the effect of intrinsic or extrinsic atomic movement on the lattice structure and the device's performance. Although doping research on LHPs is widely investigated, THPs have yet been fully explored.
3. Chemical Oxidation of THP
Generally, most THP films for solar cells and FETs are fabricated by spin‐coating process. To achieve good film quality, each precursor component, such as MAI, FAI, and SnX2 must be highly soluble in the chosen solvent. However, selecting the solvent to match the solubility of various components and to obtain high optoelectronic and electrical properties remains a challenge. For example, the solubility of Sn2+ compounds differs according to their halide component.[ 108 ] SnCl2 and SnF2 are highly soluble in polar protic solvents such as water but are not suitable for making THP films. For the fabrication of high‐quality THP films, polar aprotic solvents are preferred because of their chemical coordination ability with all components. Because of this early research on THPs, the polar aprotic solvent N,N‐dimethylformamide (DMF) was used to fabricate THP films, despite the fact that the resulting film had poor surface morphology and limited performance when used in solar cells.[ 68 ] In a follow‐up study, Hao et al. achieved better THP films by replacing DMF with dimethyl sulfoxide (DMSO) because DMSO intermediate phase controls the crystallization kinetics of the THP films.[ 109 ] However, Saidaminov et al. found the irreversible redox reaction forms between dimethylsulfide (DMS) and Sn4+ in DMSO solution using nuclear magnetic resonance (1H NMR) and X‐ray absorption near‐edge spectroscopy (Figure 8a–c).[ 110 ] In another study reported at about the same time, Pascual et al. confirmed that Sn4+ tends to be prominent when a solution of FASnI3 in DMSO was heated at 100°C for 30 min.[ 111 ] Eventually, the presence of Sn4+ in the precursor was identified as the main cause of defects and induces p‐doping in each THP film, which undesirably affects the photovoltaic performance.[ 112 ] Hence, the double‐sided effects of DMSO must be carefully engineered to benefit from forming the intermediate phase with perovskite precursors and improving crystallite quality by slowing down crystallization while compensating for the accelerated Sn oxidation. In a recent study, Girolamo et al. identified 16 non‐sulfoxide solvents, 12 of which can form stable perovskite precursors at 100°C for FASnI3. The 12 solvents identified containing either amide, diamide, carbamate, or protic bifunctional groups were able to form SnI2‐solvent complexes and retard Sn oxidation to some extent. However, only mixture of N,N‐diethylformamide:DMPU solvent system showed some photovoltaic performances.[ 113 ]
Figure 8.
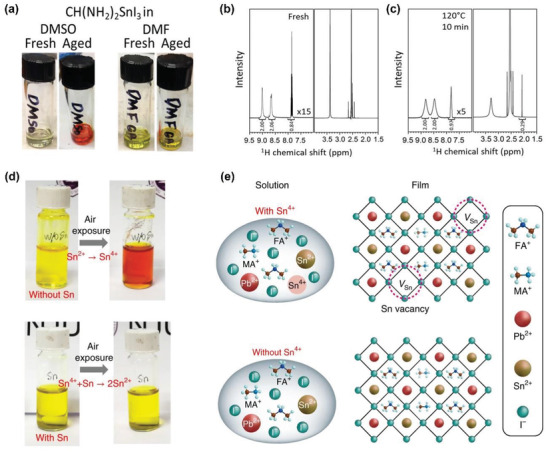
Sn oxidation in THP precursors. a) Images of fresh and aged FASnI3 solutions in DMSO solvent (left) and in DMF solvent (right). 1H NMR spectra of FASnI3 solutions in DMSO solvent b) before and c) after thermal treatment (120°C, 10 min). The peak at 2.07 ppm indicates the presence of DMS. a–c) Reproduced with permission.[ 110 ] Copyright 2020, American Chemical Society. d) Image of Sn oxidation of Sn2+ to Sn4+ in THP solution in ambient air (top), and reduction of Sn4+ to Sn2+ with the addition of metallic Sn powder. e) Schematic representation of Sn vacancy formation due to presence of Sn4+ in mixed Sn‐Pb perovskite precursor solution (top) and suppression of Sn vacancy formation in Sn‐reduced precursor solution with the absence of Sn4+. (d,e) Reproduced with permission.[ 128 ] Copyright 2019, Nature Publishing Group.
The issue of Sn2+ oxidation into Sn4+ not only occurs from the chemical interaction with the solvent, but also from air exposure.[ 114 , 115 ] Upon the exposure, the highly active 5s 2 electrons of Sn2+ allow easy oxidation into Sn4+ to form V Sn in the perovskite lattice, leaving shallow traps close to the valence band. The facile oxidation and surplus of Sn4+ lead to high density p‐type doping. This phenomenon is a critical issue for TPSCs, as oxidized perovskite layers tend to show reduced carrier lifetime and result in higher non‐radiative carrier recombination rate. Together with these dual aspects, TPSCs result in low photovoltaic performance. The easy oxidation of Sn2+ is also detrimental to FETs, as excess p‐type doping prohibits reaching an OFF‐state with extremely high hole density. Thus, to reduce the Sn4+ contents, several approaches have been explored, such as addition of tin halide‐derivatives (SnX2), [ 75 , 87 , 116 , 117 , 118 , 119 , 120 ] metallic Sn powder,[ 67 , 121 ] hydrazine,[ 122 , 123 , 124 ] and other Lewis base reducing reagents.[ 125 , 126 , 127 ]
The fluoride in SnF2 increases the Sn2+ content of the THP films by reducing the amount of Sn4+ by stabilizing the oxidation caused by DMSO.[ 129 ] The higher electronegativity of F or Cl can interact with the adjacent Sn2+ in the perovskite lattice to prevent the Sn2+ from losing its electron pair. Sn(CH3COO)2 has the same effect as SnF2 and can prevent Sn2+ oxidation and yield even higher photovoltaic response in TPSCs.[ 130 ] Nevertheless, note that excess SnF2, SnCl2, or Sn(CH3COO)2 might induce phase segregation in THP films. Hence, an optimal amount of such additives for the fabrication of high‐quality THP films is much necessary. Lin et al. added metallic Sn powder as a reducing agent into an already oxidized, Sn4+ filled solution.[ 128 ] The authors specifically chose Sn powder because it is insoluble in precursor solution on its own but still contributes to the perovskite lattice once oxidized by Sn4+ to generate Sn2+. Before the film fabrication, the leftover Sn residues were removed by filtering the solution. In the absence of Sn powder, the as‐prepared yellow perovskite precursor solution rapidly turned orange‐red when exposed to ambient air (Figure 8d). This color change indicates Sn2+ oxidation into Sn4+ in the solution. As previously discussed, conventional SnF2 additive has certain limitations in preventing Sn2+ oxidation, and it does not sufficiently reduce Sn4+ back to Sn2+ once oxidized. Conversely, metallic Sn readily reduces Sn4+ into Sn2+ via comproportionation reaction.
| (2) |
After adding metallic Sn powder into oxidized perovskite precursor solution, the orange‐red solution returned to yellow even in ambient air, indicating the successful reduction of Sn4+ into Sn2+. The addition of Sn powder can suppress the formation of V Sn by reducing the presence of Sn4+, forming a Sn4+free precursor (Figure 8e). This method enhances the air stability of the precursor solution itself and reduces V Sn inside the grains of the respective THP films, which has been successfully demonstrated in THP‐based FETs in a following study by Zhu et al.[ 131 ]
In an alternate approach, Nakamura et al. introduced a novel Sn4+ scavenger method by adding a tetramethyldihydropyrazine derivative (TM‐DHP) to form in situ metallic tin nanoparticles. When 1 mol% of TM‐DHP was added to the perovskite precursor solution of FA0.75MA0.25SnI3 in DMSO with 10 mol% of SnF2, the TM‐DHP reacts with the SnF2 rapidly and retards to Sn (0) nanoparticles. The subsequent THP film showed better film coverage with larger grains and impressive certified photovoltaic response.[ 132 ] Also, Song et al. introduced hydrazine as a strong reducing reagent through vapor atmosphere, ultimately suppressing Sn4+ formation.[ 124 ] The highly volatile hydrazine can be easily introduced and removed without thermal treatment at high temperatures. Following this report, many studies utilized hydrazine‐based materials to prohibit Sn2+ oxidation.[ 122 , 123 ] Alternative reducing agents with Lewis base groups, such as P—O, S—O, and C—O bonds can also form coordination interaction with Sn2+ in THP, inhibiting further oxidation.[ 125 , 126 ] For example, Tai et al. introduced hydroxybenzene sulfonic acid along with SnCl2 additive treatment as an anti‐oxidant in TPSC, showing effective control of the oxidation through aging time.[ 127 ]
THP devices typically used encapsulation methods for reliable operation in ambient air. The THP encapsulation methods include the coating of polymer layers, such as poly(methyl methacrylate) or Cytop, and physical methods, such as glass encapsulation.[ 133 , 134 , 135 , 136 ] However, these encapsulation processes can undoubtedly increase production difficulties and manufacturing costs. Based on the chemical understanding, we would like to address the following key facts that could be considered for the fabrication of THP films:
-
1)
Solvent choice: To prevent the Sn4+ oxidation in the precursor solvent, a solvent system with low Sn2+ oxidation should be considered. This can be achieved by a single solvent or by mixing several solvents.
-
2)
Sn4+ scavenger: To reduce the Sn4+ content in the THP films, techniques for removing Sn4+ from THP films should be evaluated.
-
3)
High surface coverage: Poor film coverage occurs due to the fast film formation rate and rapid oxidation of Sn2+ to Sn4+. High film coverage can be achieved through proper solvent selection, additive addition, and control of the composition ratio of precursors.
To solve these issues, many approaches have been introduced for the development of THP‐based devices. In the following sections, we review the key development that addressed these issues for the development of TPSCs and FETs.
4. Application of THP for Solar Cells
4.1. Operation Mechanism of TPSCs
Generally, using PCE, the performance of solar cells is evaluated. The PCE of a solar cell can be obtained from the current–voltage (J–V) curves. Under solar light irradiation with a power intensity of P in. The J–V curves provide vital information on short‐circuit current density (J SC), open‐circuit voltage (V OC), and the fill factor (FF), which reveals the PCE. Briefly, PCE can be obtained by the following equation:
| (3) |
At short‐circuit conditions, the photogenerated charge carrier flows inside the solar cell, and under illumination, the total current density can be measured by incident photon flux density Jphoton (λ):
| (4) |
where λ max and λ min represent the maximum and minimum wavelength of the absorbed photons by the absorber layer, respectively, and IPCE is defined as the incident monochromatic photon‐to‐electron conversion efficiency. The IPCE of a solar cell can be described by the following equation:
| (5) |
where α(λ) is the light‐harvesting efficiency and ηc(λ) is the charge collection efficiency and R(λ) is the total reflectivity of the solar cell. From the above equations, it is eminent that all these aspects must be considered to obtain a high J SC. To obtain high light‐harvesting efficiency α(λ), the bandgap of THP film is the most critical because the photons can be absorbed only when their energy is higher than the bandgap. Additionally, for efficient photon absorption, the THP film must be thick enough with smooth surface morphology. The charge collection efficiency, ηc(λ) is determined by the carrier diffusion length and the charge‐carrier extraction capability of the adjacent charge transport layers. As mentioned in the previous sections, the THP films go through the rapid oxidation process resulting in severe V Sn, which can contribute to the lower α(λ), ηc(λ) and result in lower J SC than the theoretical limit.
Another critical parameter for obtaining high PCE in TPSCs is the V OC, which can be described from the ideal photodiode equation:
| (6) |
where Jph is the reverse saturation current, which is determined by the IPCE spectrum. Therefore, to obtain high V OC, increased absorption of incident photons is necessary. To increase the photon absorption, the THP layer in the TPSC should have low bulk and surface defects. Unfortunately, the bulk and surface defects in THPs are severe due to the rapid oxidation of the Sn2+ states. Additionally, the V OC of the TPSCs can be correlated to the quasi‐fermi level splitting (QFLS). Mostly, QFLS can be obtained using PL quantum yield (PLQY):
| (7) |
| (8) |
where E Fe corresponds to the electron quasi‐fermi level and E Fh corresponds to the hole quasi‐fermi level. To obtain a minimal difference between the E Fe and E Fh, the energetic match between the THP layer and the adjacent charge transport layers is very critical. The CBM and the VBM must match well with the adjacent electron and hole charge‐carrier layers, respectively, to obtain a high V OC. The QFLSrad is the radiative limit of the THP layer that is associated with the VOC in zero non‐radiative recombination.[ 137 ] According to Equation (8), a high PLQY can assist in obtaining high V OC. Therefore, a high PLQY of the THP layer and a suitable charge‐carrier layers with matched energy levels are a probable solution to obtaining high V OC for TPSCs.[ 138 ]
The FF, as shown in equation 3 is an influential parameter to obtain a high PCE of TPSCs. The FF is mostly determined by the charge flow within the TPSC and charge transport layers. Usually, the FF correlates with the balance between the series resistance (R S) and shunt resistance (R sh) of TPSCs. To obtain a high FF, R s should be low, and R sh should be as large as possible to lower the power loss of the device.[ 139 ] From a theoretical understanding, it is understandable that for achieving high‐performing TPSCs, it is important to consider all aspects including the THP layer fabrication with low defects and respective charge‐carrier layers with matched energy levels. Some approaches have been summarized in the following sections to obtain a high photovoltaic response.
4.2. Structures of TPSCs
Among the existing structures for PSCs applications, TPSCs are fabricated with mesoporous and planar (n‐i‐p) and inverted planar (p‐i‐n) designs. In an n‐i‐p structure, the THP layer is sandwiched between a compact TiO2 and a mesoporous TiO2 layer, which acts as the electron transport layer (ETL) and a hole transport layer (HTL) with a metal electrode (usually Ag and Au) (Figure 9a). The compact TiO2 layer prevents direct contact between the two selective contacts besides being an electron transport material, whereas the mesoscopic layer serves as a basis for the nucleation and development of the THP layer, promoting charge transfer and electron collection.[ 140 , 141 ] To enable the injection of electrons, the conduction band energy level of the ETL layer must be lower than the THP layer. In this concern, apart from TiO2, other metal oxides such as SnO2 and Nb2O5 can be a suitable alternative, although they have seen limited success in TPSCs. However, the presence of oxygen vacancies at the ETL in this structure might accelerate the Sn oxidation. Conversely, the HTL layer should extract the photogenerated holes from the THP layer effectively while blocking the electrons. Generally, in Pb‐based PSCs, C81H68N4O8 (Spiro‐OMeTAD) has been widely adopted to fabricate high‐performing PSCs. However, Spiro‐OMeTAD as the HTL requires additional dopants such as lithium bis(trifluoromethanesulfonyl) imide (Li‐TFSI), 4‐tert‐Butylpyridine to extract hole efficiently from the THP layer. However, the presence of such dopants has a high influence on dissolving the THP layer. Although dopant‐free HTLs may be a probable solution for TPSC fabrication, Zhu et al. first discovered that the p‐i‐n structure is more suitable for TPSC fabrication.[ 142 ] The p‐i‐n structure offers the advantage of depositing a dopant‐free ETL layer on top of the perovskite (Figure 9b). TPSC fabricated with the p‐i‐n structure exhibits less hysteresis, high PCE, and light soaking stability due to the shorter diffusion length and higher charge mobility of the THP film.[ 48 , 118 , 143 ] In the p‐i‐n structure of TPSCs, the THP layer is fabricated on top of an HTL layer. Generally, poly(3,4‐ethylenedioxythiophene (PEDOT:PSS)[ 144 ] is applied as an HTL layer with a VB level of −5.20 eV, which matches well with the VB of that THP layer.
Figure 9.
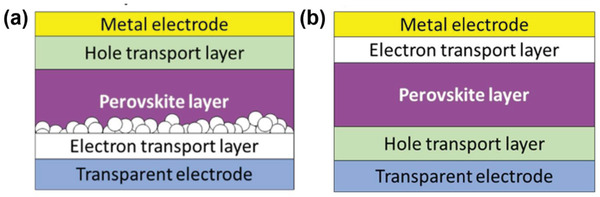
Structures of PSCs a) n‐i‐p structure and b) p‐i‐n structure.
To reduce the V OC loss and obtain high PCE, it is important to choose suitable HTL and ETLs according to the CB and VB. Conversely, the adjacent ETL layer should have a CBM level of −4.50 eV or lower. In this aspect, [6,6]‐Phenyl C61 butyric acid methyl ester (PCBM) (CB = −3.90 eV)‐, C60 (CB = −4.50 eV)‐, and ICBA (−3.70 eV)‐based TPSCs have shown reproducible performances with impressive PCEs (Figure 10 ). Nevertheless, note that in the respective TPSCs, the CB level of the corresponding THP was tuned by either structural modification, additive engineering, or cation displacement. Therefore, for obtaining A high PCE, a higher or lower shift of the CB or VB is expected when the THP layer is modified, and appropriate energy matching HTL or ETL is required.
Figure 10.
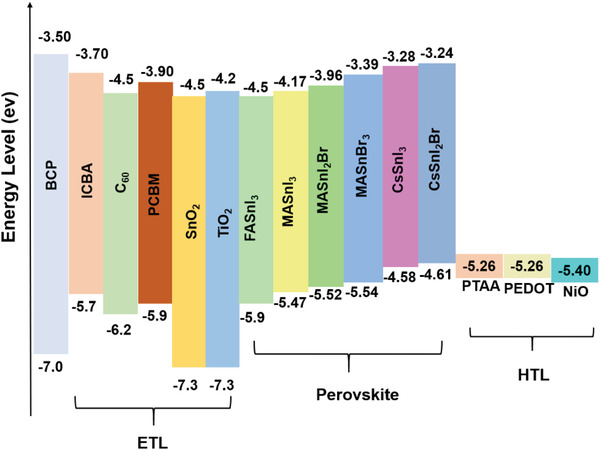
Energy level diagram of THPs with ETL and HTL.
4.3. Progress of TPSCs
4.3.1. MASnX3‐Based Solar Cells
The early days of TPSCs applications were conducted using MASnI3 as the perovskite absorber due to the structural similarity with the Pb‐based counterpart of MAPbI3. The energy bandgap of MASnI3 is 1.30 eV, which is perfect for single p–n junction solar cells. The first MASnI3‐based PSC was reported with a mesoporous structure and showed a PCE of 6.4%.[ 68 ] The MASnI3 layer is deposited by the one‐step spin‐coating from a DMF precursor. The MASnI3 films showed high mobility and conductivity, but a substantial dark carrier concentration appeared due to severe self‐doping due to the rapid oxidation of Sn2+ to Sn4+. To reduce the dark carrier concentration and induce slower crystallization, DMSO was studied as a solvent to generate the SnI2·3DMSO intermediate phase.[ 109 ] The DMSO‐based MASnI3 films exhibited a smoother surface morphology (Figure 11 ) and high surface coverage, although their respective PSC showed significantly low PCE. Song et al. highlighted the importance of the vapor atmosphere during MASnI3 film fabrication and introduced hydrazine vapor to further improve the surface morphology of the spin‐coated MASnI3 films and reduced the Sn4+/Sn2+ ratios by 20%.[ 124 ] However, the respective TPSC showed a low PCE of 3.89% in an n‐i‐p structured PSC. The low performance of the TPSCs was attributed to the lower V OC of the devices due to the bulk defects in the THP layer. Kim et al. suggested that the large V OC loss of MASnI3‐based TPSCs is due to the surface recombination at the interface rather than bulk recombination of the perovskite, which originates from the Sn2+ oxidation.[ 145 ] A further study confirmed that the introduction of toluene instead of the diethyl ether as the antisolvent could not retard the recombination of the MASnI3 perovskite due to unintentional hole doping with the nonpolar solvents.[ 146 ] Ke et al. proposed an alternate method by introducing ethylenediammonium (en) in the MASnI3 framework with a hypothesis on hollow MASnI3 perovskite. These {en}MASnI3 films showed a much lower electron‐hole recombination ratio because of the high surface coverage on top of the mesoporous TiO2 layer and the respective TPSCs attained a PCE of 6.63%.[ 78 ] A‐cation exchange approach through a two‐step deposition process was proposed to reduce the background carrier density of the MASnI3 films. In the first step, a hydrazinium tin iodide (N2H5SnI3) layer was deposited by a simple spin‐coating technique, which was followed by a transformation into MASnI3 in a MA gas atmosphere through an organic cation displacement approach.[ 147 ] The two‐step deposition method reduced the electron‐hole recombination of the MASnI3 film and resulted in dense and uniform MASnI3 film with micrometer‐sized grains and high crystallization and resulted in PSCs with PCE > 7%. Besides the solution‐processed mesoporous TPSCs, vapor‐assisted PSCs were evaluated by a low‐temperature vapor‐assisted solution process but showed poor photovoltaic performance.[ 148 ]
Figure 11.

Surface morphology observed by SEM of CH3NH3SnI3 perovskite layer on mesoporous TiO2 from different solvents, a) DMF, b) NMP, and c,d) DMSO with different magnifications. a–d) Reproduced with permission.[ 109 ] Copyright 2017, American Chemical Society.
In an alternate attempt to reduce the bulk defect and obtain high V OC, ion exchange/insertion reactions between solid‐state SnF2 and gaseous methylammonium iodide was adopted for the preparation of MASnI3 films. Contrary to the traditional approach, the p‐i‐n was adopted where PEDOT:PSS was applied as the HTL. The respective MASnI3 films showed a highly uniform, pinhole‐free coverage with excess SnF2, and the respective film yielded a low content of Sn4+. This high‐quality perovskite film enables the realization of a PCE of 7.78%.[ 149 ] Despite numerous efforts, the PCE of MASnI3‐based TPSCs is still low compared with other TPSCs. Table 1 summarizes the notable performances of MASnI3‐based PSCs (Table 2 ).
Table 2.
Development of photovoltaic performances of MASnI3‐based PSCs
| Structure | Method | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
|---|---|---|---|---|---|---|
| n‐i‐p | Solvent‐mediated crystallization by DMSO | 21.40 | 0.32 | 46 | 3.15 | [109] |
| n‐i‐p | Hydrazine vapor atmosphere | 19.92 | 0.377 | 51 | 3.89 | [124] |
| n‐i‐p | Hollow({en}MASnI3) perovskite as absorber | 24.28 | 0.42 | 63.72 | 6.63 | [78] |
| n‐i‐p | Cation exchange by HASnI3 with MASnI3 | 22.91 | 0.486 | 64 | 7.13 | [147] |
| p‐i‐n | Ion exchange/insertion reactions between solid‐state SnF2 and gaseous methylammonium iodide | 20.68 | 0.57 | 0.66 | 7.78 | [149] |
4.3.2. FASnX3‐Based Solar Cells
Significant progress was achieved for TPSCs when MA cation is replaced by FA. FA cation can stabilize the crystal structure more than the MA and lead to higher photovoltaic performances.[ 78 , 79 ] Furthermore, the p‐i‐n structure is more suitable for FASnX3‐based TPSC fabrication. Zhu et al. compared the performance of n‐i‐p and p‐i‐n structures for FASnI3‐based TPSCs. Their findings highlight that the photovoltaic performance of p‐i‐n structured FASnI3‐based TPSCs (PCE = 7.09%) outperforms the n‐i‐p structured TPSCs (PCE = 4.34%) due to favorable energetic matching with their respective charge transport layers (ETLs and HTLs).[ 142 ] Since then, almost all FASnI3‐based TPSCs studies have been performed utilizing the p‐i‐n structure. Hence, we only discuss here the development of FASnI3‐based TPSCs for p‐i‐n structure.
Coadditive Engineering
Coadditive engineering has shown promising aspects to retard the Sn2+ oxidation when introducing coadditives with SnF2 as they tend to form a complex within the perovskite system, which can encapsulate the perovskite grains and modify the crystal structure. In the coadditive engineering approach, SnF2 is often used as the main additive and “other” coadditives were added. For simplicity of discussion, only “other” coadditives are discussed where mentioned. To retard the oxidization of Sn2+, a few reducing agents such as Catechin,[ 150 ] ethylenediammonium dihypophosphite,[ 151 ] and hypo‐phosphoric acid[ 152 ] have been evaluated, but they failed to show a high PCE. Hydrazinium chloride showed a remarkable performance to reduce the Sn2+ oxidation by 20% (Figure 12a,b). Besides the reduction capability of the hydrazinum, Cl− contributes to achieving a pinhole‐free smooth surface morphology. The corresponding TPSCs showed 5.4% PCE and retained 65% of their initial performance after 1000 h in the N2 environment.[ 122 ] It must be noted that by utilizing hydrazinium chloride, the shelf‐life stability for a more extended period was first observed for TPSCs. By utilizing the antioxidant properties of hydrazine, other hydrazine alternatives, such as hydrazine dihydrochloride,[ 153 ] trihydrazine dihydriodide (THDH),[ 154 ] and phenylhydrazine hydrochloride (PHCl) were evaluated for TPSCs. Trihydrazine dihydriodide coadditive engineered FASnI3 TPSCs showed improved PCE of 8.48%.[ 154 ] PHCl as a coadditive showed a robust capacity to reduce the Sn2+ oxidation and shifted the VBM by 0.984 eV (Figure 12c), which influenced obtaining a high J SC of 23.54 mA cm−2 and a remarkable V OC of 0.76 V (Figure 12d). The presence of a hydrazino group and a hydrophobic phenyl group provided a bifunctional benefit to retard the surface defects and matched VBM with the PEDOT:PSS. The corresponding TPSCs showed PCE of 11.4% and showed stable photovoltaic performance in an N2 environment for 110 days.[ 155 ] Interestingly, the authors noted that the incorporation of PHCl induced the self‐repairing ability of the FASnI3 films. Trace amount (1 mol%) of gallium acid (GA) and an excess of SnCl2 can lead to the formation of an amorphous GA–SnCl2 complex and result in the self‐encapsulated phase of the FASnI3 perovskite, which increases the stability of TPSCs. The large bandgap of GA‐SnCl2 based FASnI3 prohibits the transfer of both charge carriers from FASnI3 to the adjacent charge transport layers in TPSCs. The FASnI3‐based TPSCs fabricated by this complex showed a PCE of 9.03% with stable photovoltaic behavior without any degradation for 1500 h in N2 environment.[ 156 ] Consecutively, Trimethyl thiourea (3T), with structural features of both a Lewis base and H‐bond donor can influence the fabrication of high‐quality FASnI3 films.[ 157 ] The unique bifunctional properties of 3T as a co‐additive can successfully enhance the morphology and texture of FASnI3 films by spreading and joining individual crystal grains. With such attributes, the charge carrier lifetime of FASnI3 perovskite was increased up to 123 ns leading to low V OC loss (only 0.2 V lower than the theoretical limit) in the respective TPSCs. Due to the high V OC of 0.92 V, J SC of 20.4 mA cm−2 and FF of 0.76, the 3T‐FASnI3‐based TPSCs showed a high PCE of 14.05%. It is noticeable that even with staggering V OC, the J SC remained low, which was attributed to the low perovskite film thickness rather than the charge‐carrier diffusion length. However, even increasing the film thickness did not result in higher J SC , rather resulted in lower V OC .
Figure 12.
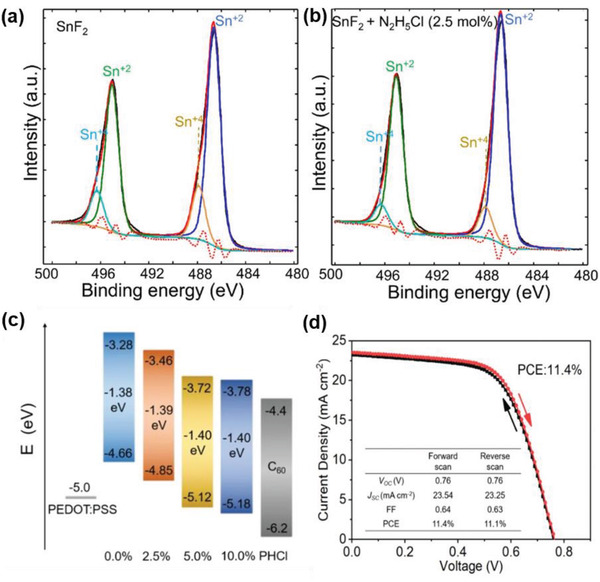
Coadditive engineering of FASnI3. a) High‐resolution XPS spectra (Sn 3d) of FASnI3 + SnF2 and b) FASnI3 + SnF2 + N2H5Cl (2.5 mol%) films a,b) Reproduced with permission.[ 122 ] Copyright 2017, American Chemical Society. c) Potential energy diagrams (energies in eV with respect to vacuum) of PEDOT:PSS, C60, FASnI3 with 0.0%, 2.5%, 5.0%, and 10.0% of PHCl. d) The J–V curves of the best device with 5.0% of PHCl measured using forward and reverse scan modes under 100 mW cm−2 AM 1.5G irradiation c,d) Reproduced with permission.[ 155 ] Copyright 2020, Wiley‐VCH.
Controlled Crystallization
Numerous studies on TPSCs development highlight the rapid oxidation of Sn2+ in FASnI3‐based perovskite due to the rapid crystallization of the perovskite layer. Even if the precursor solution is heat treated at 100 °C in a glovebox filled with PPM level oxygen before spin‐coating and antisolvent treatment, the organocation FA+ and iodide ion (I−) are easily volatilized and are not properly integrated into the perovskite structure. This phenomenon induces rapid oxidation and results in high crystal defects of the FASnI3 film. To decelerate the crystallization process, Jokar et al. highlighted ethylenediammonium diiodide (EDAI2) in trace amounts can lower the crystallization rate and promote the growth of highly crystalline FASnI3 films more slowly. During the slow crystallization process, EDAI2 can retard the V Sn inside the FASnI3 crystal by decelerating crystal growth through a kinetic balance between nucleation and crystal growth. The 1 mol% EDAI2‐FASnI3‐based TPSCs can achieve a PCE of 8.5% and exhibit stable performance for 1400 h in an N2 environment.[ 158 ] The π‐conjugated Lewis‐base molecule 2‐cyano‐3‐[5‐[4‐(diphenylamino)phenyl]‐2‐thienyl]‐propenoic acid (CDTA) shows electron‐accepting properties to reduce the oxidation and binding to the SnI2 at the precursor stage. This strong electron‐accepting behavior contributes to a more stable Lewis adduct during the nucleation process. The CDTA‐based FASnI3 films suppressed the in‐plane and out‐of‐plane rotation of [SnI6]4− and reduced the carrier recombination of the FASnI3 films. Consequently, the corresponding TPSCs showed a high PCE of 10.32%, which is attributed to a 0.13 V increase in V OC and a 10% increase in FF compared to the pristine TPSC. Controlling the crystallization via electron‐accepting small molecules of CDTA attained the first time observation of light soaking stability of the TPSCs at maximum power point tracking (MPPT) for 1000 h.[ 159 ] Another molecule poly(vinyl alcohol) (PVA) with a high density of hydroxyl groups can make a strong O—H—I− hydrogen bond with the FASnI3 system leading to nucleation sites which induce lowered crystallization rate. As a result, the FASnI3 perovskite crystals are more oriented and can inhibit the migration of the iodide ions to the adjacent charge transport layers. With these attributes, the V OC value is increased from 0.55 V (for pristine TPSC) to 0.63 V (PVA based TPSC) and resulted in long‐term stable photovoltaic performance with a PCE of 8.9%.[ 160 ] By taking the advantage of the slower nucleation of the H—I bond and inducing highly crystalline films, a bifunctional compound, hydroxylamine hydrochloride (HaHc) has recently been applied to reduce electronic defects of the FASnI3 perovskites. The hydroxyl group in the HaHc formed a hydrogen bond with iodide ion in the FASnI3 perovskite system to slow the crystallization, and the Cl− ion coordinated with the under coordinated Sn2+ ions of the FASnI3 structure. With these bifunctional aspects of the HaHc, the corresponding HaHc‐FASnI3 based THP films showed lower trap assisted recombination, bimolecular recombination, and trap density as compared to the pristine FASnI3. With such beneficial improved electronic properties, the V Sn was reduced and improved V OC up to 0.676 V for the HaHc‐FASnI3 based TPSCs was observed with 500 h light soaking stability.[ 161 ] In terms of V Sn reduction, the H—I bond inclusion seems to be an effective approach, although the resultant TPSCs performance is still lower when compared to coadditive engineering approaches. Meng et al. proposed that the crystallization of FASnI3 perovskite is more vulnerable to the solution air surface. By using a tailormade fluorinated organic cation, pentafluorophen‐oxy‐ethylammonium iodide (FOEI) in the FASnI3 precursor solution, the surface energy of the solution/air surface was reduced, which simultaneously served as a template and resulted in highly crystalline films. When the FOEI‐FASnI3 films were measured using depth‐dependent grazing incident X‐ray diffraction measurements at different incident angles (Figure 13a,b), the perpendicular growth at (100) plane was retarded significantly (Figure 13c). The prepared FASnI3‐FOEI films showed very low surface roughness and threefold increased carrier lifetime as compared to the pristine FASnI3 film. As a result, the FASnI3‐FOEI based TPSCs showed improved V OC of 0.67 V, a J SC of 21.59 mA cm−2, and a FF of 0.75, yielding a certificated PCE of 10.16%.[ 162 ]
Figure 13.
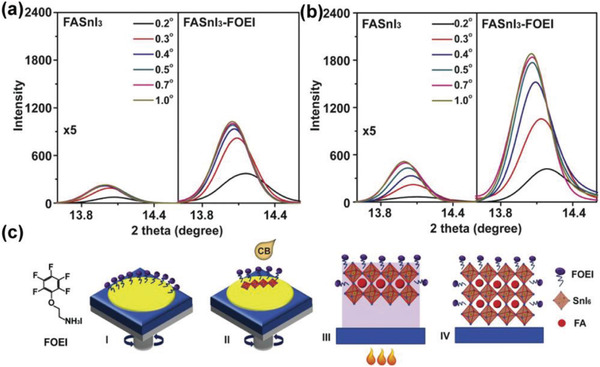
Depth‐dependent grazing incident X‐ray diffraction patterns of the FASnI3 and FASnI3‐FOEI perovskite films after antisolvent dripping a) without or b) with thermal annealing at different incident angles. The ×5 in (a) left and (b) left means that the original intensity is multiplied by 5 in this image to clearly show the tendency. c) Possible scenarios of the surface‐controlled growth of the FASnI3–FOEI perovskite films. a–c) Reproduced with permission.[ 162 ] Copyright 2020, Elsevier.
Amine complex CH3NH3I·3CH3NH2 (MAI·3MA) was evaluated as potential to further stabilize the structure of the FASnI3 perovskite by Dai et al. When compared to the conventionalFASnI3:xSnF2 precursor, the MAI·3MA: FASnI3:xSnF2 precursor can induce chemical stability owing to due to the extra donor electron groups in the amine complex. Due to the complex formation, the crystallization rate from precursor to THP film is significantly reduced resulting in highly crystalline THP films with low Sn2+ oxidation and non‐radiative recombination centers. As a result, the V Sn is reduced and the corresponding TPSC can result in V OC of 0.65 V, leading to a PCE of 9.53% with a light soaking stability of 1000 h.[ 163 ] However, in a recent breakthrough study, Jiang et al. highlighted the importance of SnI2 complex for regulating the crystal growth of FASnI3 perovskite.[ 164 ] Typically SnI2 complexes are formed using two step synthesis process (TSS). However, SnI2 complexes prepared by TSS method result in SnI2 segregation. The segregation causes unsolvated edge‐sharing SnI2 clusters which limit the crystal modulation of the THP growth. To promote smooth, compact FASnI3 layer, and to retard the SnI2 segregation, a one‐step synthesis (OSS) of SnI2. (DMSO) x colloidal complexes were developed, where in situ, tin metal and I2‐DMSO in solution complex are obtained. The SnI2.(DMSO) x synthesized by the OSS method can be well dispersed to obtain SnI2 precursor without the formation of any edge‐sharing clusters. With these beneficial aspects, the stable SnI2. (DMSO) x colloidal complexes can assist to bind with the cations of FAI and phenylethylamonium bromide (PEABr) and promote the growth of highly crystalline and smooth THP film. As a result, the electron diffusion length of the THP film was improved to 290 ± 20 nm (versus pristine = 210 ± 20 nm). As a result, the corresponding TPSCs show a high V OC of 0.91 V, J SC of 20.6 mA·cm−2, and a FF of 0.77; leading to a PCE of 14.63%. It must be noted that by this approach TPSCs showed first time photovoltaic performance toward 15% PCE.
Template‐Assisted Growth
Throughout the THPs progress the solvent to film conversion rate has been highlighted as a potential source for Sn2+ oxidation. In recent reports, template‐assisted seed growth has shown more success to fabricate high‐quality FASnI3 film for TPSCs application. In a seeded growth (SG) approach proposed by Cao et al., a layer of perovskite film is first deposited by an antisolvent method, and the same step is repeated. In this method, the first spin‐coated FASnI3 layer acts as a template to promote the growth of uniform THP layer with low grain boundaries and highly crystalline perovskite films with compact and large grains and a longer carrier lifetime. TPSCs fabricated by the SG method showed a V OC of 0.49 V, which is far below other conventional reported methods.[ 165 ] To facilitate template assisted growth, poly(ethylene‐co‐vinyl acetate) (EVA) self‐sealing polymer with the ability of complex formation with SnI2 was tested and verified. The C=O groups of EVA act as a powerful Lewis acid–base and forms a SnI2 complexation with uncoordinated tin atoms in perovskite grains, which influences the improvement of the grain size, optimized grain orientation, and decrease the surface defects of FASnI3 films. Furthermore, The FASnI3.EVA based THPs show a self‐encapsulation effect which can repeal the moisture and oxygen ingression and result in humidity (60%) stable TPSCs.[ 166 ]
Treatment of FASnI3
Since the FASnI3 surface is more prone to Sn2+ oxidation and leads to extreme surface defects, the post‐treatment method was considered a possible solution. Chowdhury et al. introduced a postdeposition vapor annealing method (PDVA), where after the antisolvent treatment, the FASnI3 films were treated in MACl enclosed vapor system. The PDVA method induced the growth of high‐quality, defect‐free FASnI3 film and showed a PCE of 8.48% when implemented in TPSCs.[ 167 ] A solution approach for post‐treatment by bidentate amine‐edamine was evaluated by Kamaruddin et al. Before the antisolvent treatment, edamine treatment was introduced on top of the FASnI3 films. The addition of edamine which has a free electron pair acts as an electron donating group and facilitates the co‐ordination with the under‐coordinated Sn2+ of the FASnI3 system (Figure 14a,b). As a result, the respective THP films showed ideal crystallographic orientation near the grain boundaries and improves the surface morphology with improved grain size. With such beneficial aspects of the edamine post‐treatment, the TPSCs J SC was improved up to 23.09 mA cm−2 with V OC of 0.60 and FF of 0.73, leading to a PCE of 10.18%[ 168 ] Recently, Liu et al. reported the highest certified PCE of 11.73% (by an authorized public test center) with 1000 h light soaking stability under MPPT conditions by the pretreatment method of FASnI3 film (Figure 14c,d) In this method, prior to the annealing of the THP film, right after the dripping of antisolvent n‐propylammonium iodide (PAI) from a mixed solvent of chloroform and DMSO was spin‐coated on top of the pre‐nucleated FASnI3 film.[ 169 ] The PAI treatment facilitated the recrystallization of the FASnI3 films by forming an intermediate PAI‐FASnI3 phase. Although DMSO itself is known to dissolve the perovskite layer, however it was claimed that 1% v of DMSO with 100% v chloroform can successfully assist to form highly oriented FASnI3 crystals. With these extraordinary features, the non‐radiative recombination centers of the FAsnI3 film were reduced leading to high electron diffusion length. As a result, the V Sn of the THP film was reduced and respective TPSCs showed a high V OC of 0.73 V, leading to a PCE of 11.22%.
Figure 14.
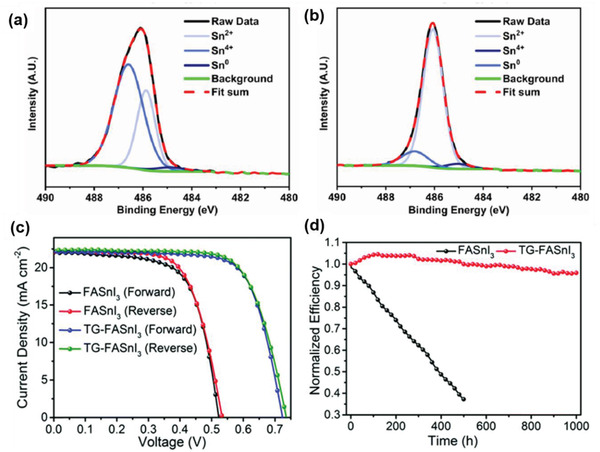
Post‐treatment–narrow XPS spectra after 10 s of argon etching of a) 0 mm edamine and b) 0.05 mm edamine‐passivated FA0.98EDA0.01SnI3 samples. a,b) Reproduced with permission.[ 168 ] Copyright 2021, American Chemical Society. c) Pretreatment—The J–V curves of FASnI3 and TG‐FASnI3‐based TPSCs. d) The stability test of the corresponding device under simulated AM 1.5 G (100 mW cm−2) operating at MPPT c,d) Reproduced with permission.[ 169 ] Copyright 2021, Royal Society of Chemistry.
Ionic Liquid
Ionic liquids (ILs) are well known for their very low vapor pressure, making them ideal candidates for slower crystallization. When a small amount is added to the perovskite precursor, it tends to remain in the resulting perovskite film after all the solvent has been evaporated during the annealing process. With controlled crystal growth, the surface defects at the perovskite grain boundaries can be reduced effectively. Additionally, ILs can form a dipole at the perovskite/charge transport layer interface, which can improve the energy level alignment due to the reduced interfacial energy barriers.[ 170 ] Lin et al. introduced 1‐butyl‐3‐methylimidazolium bromide (BMIBr) as IL to induce Ostwald ripening effect of FASnI3 films. During the thermal annealing of perovskite films, BMIBr IL domains generated by a lower melting point act as Ostwald ripening agents for dissolving the perovskite grains. The FASnI3 films fabricated by inclusion of BMIBr showed larger grain size with fewer grain boundaries which eventually reduces the defect states. The successful suppression of defect states reduces the V Sn of the FASnI3 films and the corresponding TPSCs showed V OC up to 0.68 V, with J SC of 19.63 mA cm−2 and FF of 0.72 (PCE = 10.09%).[ 171 ] In an alternate approach, IL formamidine acetate (FAAc) improved the FASnI3‐based TPSCs performance up to 9.96%. The FAAc contains CH3COO− a group, which can form an intermediate phase and slow the nucleation of the FASnI3 film. Even after the thermal annealing treatment, the FAAc remained within the perovskite system and retarded the cationic vacancies of the respective THP film, helping to retain 82% of the initial PCE during the 1500 h light soaking stability test.[ 172 ] n‐butylammonium acetate (BAAc) IL showed coordination with specific O…Sn chelating bonds and N‐H…X hydrogen bonds in the FASnI3 system retarded the Sn2+ oxidation in the precursor solution. Contrary to the other reports, the BAAc can suppress precursor Sn2+ oxidation even when heated at 100°C for 2 h and can form highly crystalline FASnI3 films with fewer surface defects. The BAAc‐FASnI3‐based TPSCs exhibited a PCE of 10.4% and maintained 80% of their initial photovoltaic performance for up to 100 h at 85°C.[ 173 ] Note that this is the first report on TPSCs showing stable photovoltaic performance at elevated temperatures.
Charge Extraction Engineering
Due to the less charge extraction ability at the perovskite/ETL interface, the V OC of the TPSCs is often low.[ 174 ] Abdoul‐Sahkour et al. highlighted the importance of the charge collection loss of the FASnI3/ETL interface and introduced diaminomaleonitrile (DAMN) in the FASnI3 system to collect the photogenerated charges more effectively. The DAMN with two cyano groups which perform as an electron‐withdrawing group can successfully extract electrons effectively from the FASnI3 layer and transfer them to the adjacent C60 ETL, which was confirmed by a 42% increment of electron mobility. Consequently, the V OC of the DAMN‐FASnI3‐based TPSC improved by 0.5 V compared with the pristine FASnI3. Due to the better charge management in the FASnI3 system, the DAMN‐FASnI3‐based TPSC showed stable photovoltaic behavior at MPPT conditions for 300 h.[ 175 ] In a recent report, Chen et al. highlighted the importance of the charge transport at the HTL/FASn0.9Ge0.1I3 interface.[ 176 ] To promote efficient charge transport at the HTL/FASnI3, the FASnI3 perovskite was doped with 1 mol% GeI2. Unlike the usual dopants or additives which contribute to the surface of the perovskite layer, the GeI2 induces in situ formation of a thin (≈3 nm) amorphous interfacial GeO2 layer at the NiO X /FASn0.9Ge0.1I3 interface. The GeO2 layer prevents the Sn2+ oxidation at the interface, provides more robust mechanical bonding, and promotes effective charge transportation in the TPSC. As a result, the corresponding TPSCs showed a high PCE of 10.43%.
Cationic Displacement
Cationic or anionic displacement of FASnI3 can tune the bandgap, push the VBM to a more shallower region, increase the light‐harvesting capability of the THP films and provide a basis for high‐performance TPSC fabrication.[ 177 ] Compositional engineering by cationic displacement for FASnI3‐based TPSCs was first evaluated by Zhao et al. where MA cation was partially introduced in the FASnI3 system. The increasing content of MA cation in the FASnI3 perovskite system shifts the VBM level and induced a favorable VBM level with the adjacent PEDOT:PSS HTL (Figure 15b). The (FA)0.75(MA)0.25SnI3‐based TPSCs showed PCE of 8.12%, which were higher than the MASnI3 (PCE = 4.29%)‐ and FASnI3 (PCE = 6.60%)‐based TPSCs. The improved photovoltaic response was attributed to the improved V OC by 0.15 V compared with the MASnI3 or FASnI3.[ 178 ] Liu et al. discussed the importance of the appropriate antisolvent for the fabrication of FA X MA X SnI3 films and revealed that chlorobenzene (CB) is the more suitable antisolvent. When compared with the other antisolvents of diethyl ether and toluene, CB as an antisolvent could assist the formation of a dense, compact FA0.75MA0.25SnI3 layer and the respective TPSCs showed 9.06% PCE.[ 179 ]
Figure 15.
a) Schematic illustration of the device structure. b) Band alignment diagram. c) Cross‐sectional scanning electron microscope (SEM) image of a completed device (scale bar: 500 nm). a–c) Reproduced under the terms of the Creative Commons CC‐BY license.[ 178 ] Copyright 2017, The Authors. Published by Wiley‐VCH.
In an alternate approach by vacuum‐assisted self‐healing of defects in FA0.75MA0.25SnI3 perovskite films to suppress the nonradiative recombination, losses were reported by Wan et al. Before the antisolvent treatment of the pre‐annealed FA0.75MA0.25SnI3 perovskite films were placed under high vacuum of 5 × 10−4 Pa. The introduction of vacuum treatment before the annealing in order to reduce the trap density reduced the nonradiative recombination and promoted interfacial charge transfer in the respective TPSCs leading to a V OC of 0.631 V and FF of 0.75 (PCE of 10.3%).[ 180 ] Inclusion of Diethylammonium iodide (DEAI) as a cation in the (DEA x FA1– x )0.98EDA0.01SnI3 perovskite system can shift the VBM to a much deeper level (from −4.97 to −5.15 eV) and reduce the deep trap states of the respective THP film with low defect densities. With such favorable VBM level, the hole transportation between the PEDOT:PSS and the (DEA x FA1– x )0.98EDA0.01SnI3 layer is more efficient and the V OC loss is reduced in the respective TPSC. With an optimal 10% DEA in this mixed cation‐based TPSC a high V OC of 0.67 V (versus pristine = 0.59 V) was attained.[ 181 ] By introducing a trace amount of ethylammonium (EA) as the cation to the FASnI3 systems, a noteworthy improvement in TPSCs was observed by Nishimura et al. The FA0.98EA0.01SnI3 based THP showed a shift of 0.30 eV in VBM and 0.15 eV shift in CBM. The favorable shift of both ends facilitated obtaining a high V OC of 0.62 V while maintaining a high J SC of 23.05 mA cm−2 leading to a PCE of 10.80%. The inclusion of EA as a cation could successfully repress the trap states of the THP layer which increased charge carrier mobility. Further surface treatment of the TPSCs with EDAI elevated the V OC up to 0.84 V and reached a PCE of 13.24%.[ 182 ] However, after EDAI surface treatment, the J SC decreased to 20.32 from 23.05 mA cm−2, which highlights the double‐sided effect of the surface treatment process for mixed cation THP perovskites. To overcome the drawbacks of the reduced J SC, the same research group recently introduced trimethylsilyl bromide (Me3SiBr) as a surface passivation agent instead of EDAI.[ 183 ] Upon surface treatment with 0.15 mm Me3SiBr on top of the FA0.98EA0.01SnI3 perovskite, the surface morphology was improved, which showed lower defect densities, larger grains, and higher carrier density. As a result, the J SC of 24.11 mA cm−2 can be maintained even after surface treatment. However, the V OC of the respective TPSC was reduced to 0.70 V, leading to a PCE of 12.22%. Additionally, the Me3Si+ remains as the counter cation on the surface and forms a hydrophobic protective layer which protects the FA0.98EA0.01SnI3 film from oxidation which showed stable photovoltaic performance in a N2 glovebox for 92 days of storage in N2 filled glovebox by maintaining 80% of its initial efficiency.
Halide Exchange
By mixing the halide content of the THP, the bandgap can be tuned further. Weber et al. partially replaced the I with Br and reported that MA0.75FA0.15PEA0.1Sn(Br0.33 I77)3 can increase the bandgap from 1.29 to 1.50 eV. Although a significant improvement in TPSCs performance was expected, their fabricated TPSCs showed a low PCE of 4.63%.[ 184 ] The bandgap of FASnI3 films was further enhanced to 1.61 eV by the addition of GuBr by Chen et al. with an optimized composition of GA0.06(FA0.8Cs0.2)0.94SnI2Br, which showed matched energy level alignment with the adjacent charge transport layer and reduced the trap density (both surface and bulk). The best performing TPSC showed VOC of 0.64 V, J SC of 15.16 mA cm−2, and FF of 0.72 after aging.[ 185 ] In a recent report, Br mixed perovskite system of EDA0.01(GA0.06(FA0.8Cs0.2)0.94)0.98SnI2Br, the PCE was further enhanced up to 8.66%.[ 186 ] However, the improved photovoltaic performance of these wide bandgap TPSCs was attributed to the influence of replacement of PEDOT:PSS by a monolayer of 2PACZ rather than the influence of the wide bandgap THP layer (Table 3 ).[ 186 ]
Table 3.
Summary of photovoltaic performances of FASnI3‐based PSCs
| Coadditive engineering | ||||||
|---|---|---|---|---|---|---|
| Additive | Coadditive | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
| SnF2 | N2H5Cl | 17.63 | 0.455 | 0.673 | 5.4 | [122] |
| SnF2 | 5‐AVAI | 18.89 | 0.592 | 0.623 | 7.0 | [187] |
| SnCl2 | KHSQA | 17.64 | 0.552 | 0.694 | 6.76 | [127] |
| SnF2 | THDH | 22.12 | 0.54 | 0.71 | 8.48 | [154] |
| SnF2 | PHCl | 23.5 | 0.76 | 0.64 | 11.43 | [155] |
| SnCl2 | GA | 19.75 | 0.64 | 0.714 | 9.03 | [156] |
| SnF2 | 3T | 20.4 | 0.92 | 0.76 | 14.3 | [157] |
| Controlled crystallization | ||||||
|---|---|---|---|---|---|---|
| Additive | Compound for crystallization control | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
| SnF2 | EDAI2 | 21.3 | 0.583 | 0.718 | 8.9 | [158] |
| SnF2 | CDTA | 21.83 | 0.64 | 0.739 | 10.32 | [159] |
| SnF2 | PVA | 20.37 | 0.632 | 0.693 | 8.92 | [160] |
| SnF2 | HaHc | 19.40 | 0.676 | 0.70 | 9.18 | [161] |
| SnF2 | FOEI | 21.59 | 0.670 | 0.75 | 10.84 | [162] |
| SnF2 and NH4SCN | SnI2·(DMSO) x adduct | 20.6 | 0.91 | 0.77 | 14.63 | [164] |
| Template‐assisted growth | ||||||
|---|---|---|---|---|---|---|
| Additive | Compound for template‐assisted growth | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
| SnF2 | EVA | 22.80 | 0.523 | 0.646 | 7.72 | [166] |
| SnF2 | SG‐FASnI3 | 22.52 | 0.490 | 0.662 | 7.30 | [165] |
| Ionic liquid | ||||||
|---|---|---|---|---|---|---|
| Additive | Ionic liquid | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
| SnF2 | BMIBr | 19.86 | 0.70 | 0.723 | 10.09 | [171] |
| SnF2 | FAAc | 23.20 | 0.59 | 0.727 | 9.96 | [172] |
| SnF2 | BAAc | 22.20 | 0.65 | 0.716 | 10.40 | [173] |
| Cation displacement | ||||||
|---|---|---|---|---|---|---|
| Additive/method | Perovskite compound | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
| HAT method | (FA)0.75(MA)0.25SnI3 | 19.40 | 0.55 | 0.670 | 7.20 | [188] |
| SnF2 | (FA)0.75(MA)0.25SnI3 | 21.20 | 0.61 | 0.627 | 8.12 | [178] |
| Solvent engineering | (FA)0.75(MA)0.25SnI3 | 24.30 | 0.55 | 0.673 | 9.06 | [179] |
| SnF2 + TM‐DHP | (FA)0.75(MA)0.25SnI3 | 22.00 | 0.76 | 0.690 | 11.5 | [132] |
| Vacuum‐Assisted growth | (FA)0.75(MA)0.25SnI3 | 21.62 | 0.63 | 0.755 | 10.3 | [180] |
| Surface treatment with EDAI2 | FA0.98EA0.01SnI3 | 20.32 | 0.84 | 0.78 | 13.24 | [182] |
| Surface treatment with Me3SiBr | FA0.98EA0.01SnI3 | 24.11 | 0.70 | 0.72 | 12.22 | [183] |
4.3.3. CsSnX3‐Based Solar Cells
Because of the absence of hydrophilic organic cations of MA and FA, CsSnX3 perovskites offer unique advantages in terms of structural and thermal stability. With an exciton binding energy of 12–18 meV, CsSnX3‐based perovskites are comparable with LHPs.[ 61 , 189 , 190 ] However, when implemented in TPSCs, CsSnX3 has shown lower performances when compared with FASnI3‐based TPSCs. The low performance of CsSnX3‐based TPSCs is attributed to the V Sn, which generates severe crystal defects during the formation state of films. Distinctively, when the Sn content is increased, the p‐type conductivity reduces because of the reduction of accepter defects, which can influence the increase of shunt resistance and result in high‐performance TPSCs.[ 73 ] To experimentally address these phenomena, Kumar et al. reported that adding 20% SnF2 can decrease the carrier density from 1019 to 1017 cm−3, which increases the formation energy of V Sn.[ 75 ] Consequently, the TPSCs fabricated in n‐i‐p configuration showed a PCE of 2.02% with a V OC of 0.24 V. An improved V OC of 0.55 V with a PCE of 2.76% was obtained in p‐i‐n structured TPSCs when excess SnI2 was added to prepare the CsSnI3 films.[ 191 ] In an alternate study, Song et al. evaluated the role of excess SnI2 in hydrazine vapor atmosphere and highlighted the impact on the surface morphology and crystallinity. The inclusion of excess SnI2 induced homogenous CsSnX3 film with smooth surface morphology without influencing the crystallinity. With an optimized CsI/SnI2 molar ratio of 0.4, a PCE of 4.8% was attained with n‐i‐p structured PSCs.[ 120 ] A systematic investigation of the addition of excess SnX2 (X = Cl, Br, I, and F) revealed that SnCl2 is the most suitable compound to obtain smooth surface morphology and maintain the B‐γ phase of CsSnI3 for TPSCs applications with PCE of 3.56%. However, the partial replacement of Cl and I was not observed; rather, an ultrathin layer of SnCl2 layer formed on top of the perovskite layer and provided assistance to achieve stability.[ 192 ]
Although the introduction of SnX2 additives in the CsSnI3 framework induced some photovoltaic response but the PCE was significantly lower than 5%. The SnX2 additives failed to address the critical Sn2+ oxidation even in an N2 atmosphere with a trace amount of oxygen (1 ppm). To increase the photovoltaic response, taking for example from FASnI3 perovskites, the coadditive strategy was adopted for CsSnI3 perovskite films. The addition of 2‐aminopyrazine with 20% SnF2 remarkably suppressed the Sn2+ oxidation, which was attributed to the electron‐donating capability of the amino group in the aminopyrazine and the formation of the aminopyrazine–SnF2 complex. Upon spin‐coating, the precursor followed by antisolvent and thermal annealing at 100°C resulted in smooth CsSnI3 perovskite films with compact grains. Although the V Sn of the respective TPSC was not reduced which can be observed from the low V OC of 0.40 V.[ 193 ] A higher PCE of 7.50% was attained by utilizing the lone electron pairs of −NH and −CO functional groups of methylenebis(acrylamide) (MBAA) (Figure 16a), which can coordinate with Sn2+ and prevent oxidation and eventually reduce the V Sn of the B‐γ CsSnI3 films.[ 194 ] Remarkably, the TPSCs showed stability for 120 h under continuous 1 sun illuminations. The lone electron pairs of –NH and two —CO units can form a trigeminal coordination bonding with Sn2+, which reduces the defect density of the CsSnI3 film. With such properties, when phthalimide (PTM) was used as a coadditive with SnF2, the electron density of CsSnI3 was successfully passivated, and smooth pinhole‐free surface morphology was observed (Figure 16b). The respective TPSCs showed the highest PCE of 10.1% for CsSnI3‐based TPSCs with V OC of 0.64 V, J SC of 21.81 mA cm−2, and FF of 0.721.[ 195 ]
Figure 16.
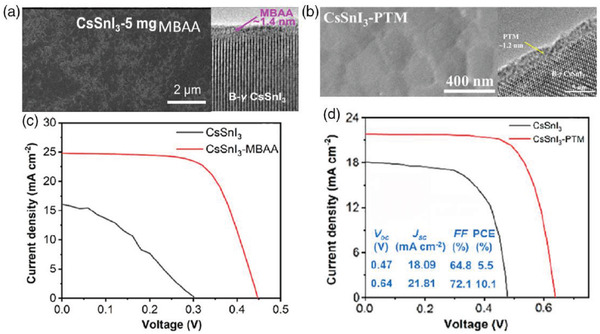
SEM and HRTEM images of a) CsSnI3‐MBAA and b) CsSnI3‐PTM. J–V curves of the device fabricated with c) CsSnI3‐MBAA and d) CsSnI3‐PTM. a,c) Adapted with permission.[ 194 ] Copyright 2021, American Chemical Society. b,d) Adapted with permission.[ 195 ] Copyright 2021, American Chemical Society.
In an alternate approach, B‐site engineering of the CsSnI3 was evaluated to tune VBM and CBM. To maintain the concept of low toxicity, Ge element with a similar electronic structure to Sn was considered and evaluated for CsSnI3. DFT calculations revealed that 50% of Ge inclusion in the CsSnI3 can induce a direct bandgap with VBM and CBM can induce a high absorption coefficient and low exciton binding energy.[ 196 ] However, Ge incorporation into the precursor and thin film fabrication may not be a feasible approach for the fabrication of CsSn0.5Ge0.5I3‐based perovskites. Chen et al. introduced a CsSn0.5Ge0.5I3 perovskite powder synthesis method by solid‐state reaction in an evacuated Pyrex glass tube at 450°C.[ 197 ] The as‐synthesized CsSn0.5Ge0.5I3 perovskite powder showed high PL intensity, a bandgap of 1.50 eV, and upshifted VBM. Interestingly, when the CsSn0.5Ge0.5I3 powders were evaporated to fabricate perovskite films, upon exposure to air, the formation of a Ge4+‐rich native‐oxide layer on the surface of the perovskite film was observed. The native‐oxide layer effectively protected the film surface, suppressed the charge recombination, and enhanced the holes extraction at the perovskite/HTL interface in an n‐i‐p‐structured TPSC. The respective TPSCs fabricated with CsSn0.5Ge0.5I3 achieved a PCE of 7.11% with V OC of 0.63 V, J SC of 18.61 mA cm−2, and FF of 0.606 with high ambient air stability of 500 h in an N2 environment.[ 197 ]
Apart from the typical solution processing methods, the evaporation‐assisted solution (EAS) method and the vacuum flash‐assisted solution processing (VASP) method have been evaluated for the fabrication of CsSnI3 films. The EAS method combines the solution process followed by vacuum evaporation of the perovskite films. In the EAS method, first, a precursor containing SnI2/SnF2 is deposited on top of the substrate by one step of spin coating and annealed at 100°C. Before annealing, CsI is thermally evaporated at 150°C for 10 min to obtain CsSnI3 films. The n‐i‐p‐structured TPSCs fabricated using EAS‐assisted CsSnI3 showed a PCE of 2.23%.[ 198 ] The fabrication of CsSnI3 films was evaluated using the VASP method, where the CsSnI3 precursor solution is first deposited on top of the indium tin oxide (ITO) substrate and immediately transferred to the vacuum chamber to remove the solvent. The introduction of such a fast solvent removal technique improved the crystallization and surface properties but attained only a PCE of 3.8%.[ 199 ] Yin et al. proposed a sequential vapor deposition strategy, which combined the advantages of both the vapor deposition and the surface passivation. Thiosemicarbazide (TSC) with S‐C‐N functional groups was introduced as a passivator in addition to the SnI2 and CsI evaporation source. The vapor deposition was performed sequentially by depositing SnI2, TSC, and CsI sequentially and annealing at 120°C to form a (B‐γ) CsSnI3 with large grain size and uniform coverage. The fabrication of extraordinary (B‐γ) CsSnI3 films prepared using PASVD was attributed to the strong electrostatic attachment and coordination interaction, which leads to electron cloud density and reduced the V Sn. When TPSCs were fabricated in a p‐i‐n configuration, the V OC enhancement from 0.47 to 0.63 V was observed which is ascribed to the reduced deep level trap‐state density of the successfully passivated THP films. Interestingly, by adoption of this approach, a PCE > 8.0% was noted for CsSnI3‐based TPSCs with stable photovoltaic performance up to 500 h (Table 4 ).[ 200 ]
Table 4.
Development of photovoltaic performances of CsSnI3‐based PSCs
4.3.4. 2D/3D Solar Cells
2D/3D THP films exhibited a superior crystal quality when compared with 3D THP films due to the decreased background ions and carriers. To further improve the MASnI3 film quality, the conventional 2D/3D approach was examined. Perovskites with a Goldschmidt tolerance factor (t) between 0.8 and 1.0 show a photoactive black phase, which can induce high‐quality MASnI3 film. Ji et al. adopted EA cation into MASnI3 to induce a preferential orientation perpendicular to the substrate resulting in a 2D/3D MASnI3‐based PSC with PCE of 9.24%.[ 201 ] The (EA X MA(0.98− X )2D/3D)SnI3 films could successfully passivate the hole traps, induce preferential crystallization rate, produce a uniform and smooth surface, and suppress the trap‐assisted nonradiative recombination. Shao et al. introduced a lower concentration of PEAI (0.16 M) to FASnI3 to obtain 2D/3D structures and verified that the crystallinity of 3D THPs is significantly better when compared with the 3D FASnI3‐based THPs, which increased the V OC of the respective TPSCs by 0.67 V when compared with the 3D FASnI3‐based TPSC and showed PCE of 9%.[ 202 ] In follow‐up work, the authors evaluated the importance of stoichiometry of 2D/3D PEA0.08FA x SnI3 (where x refers to the concentration of the FA cation in the case of fixed PEA concentration). It was observed the concentration of FA cation significantly impacts the crystallinity of the 2D/3D films.[ 203 ] A hierarchical 2D‐quasi‐2D–3D structure using NH4SCN as an additive was proposed to regulate the crystal growth of PEA x FA1‐ x SnI3 THP films by Wang et al.[ 204 ] With regulated crystal growth, the quasi‐2D/3D THP films showed higher carrier mobility and reduced the carrier density resulting in a PCE of 9.41% in the respective TPSCs. They hypothesize that a thin layer of 2D layer of PEA2SnI4 on top of the 3D FASnI3 film can provide resilience against ambient air and moisture. Because of this, the respective TPSCs can maintain 90% of their initial PCE even after 600 h storage in air. Chen et al. added a bulky divalent organic cation, 4‐(aminomethyl)‐piperidinium (4‐AMP) to FASnI3 system. Interestingly, the addition of 15 mol% of 4‐AMP with FASnI3 did not influence 2D/3D formation but rather remained at the surface and grain boundaries. Interestingly, this concentration was not enough to create detectable low‐dimensional phases.[ 205 ] To further elevate the PCE, Jiang et al. proposed that for the development of 2D/3D aside from the THP layer the adjacent charge carriers play a pivotal role. They replaced the ETL (PCBM) with ICBA and HTL (NiO X ) with PEDOT:PSS in the PEA x FA1‐ x SnI3+NH4SCN‐based TPSCs and increased the V OC up to 0.94 V, leading to a remarkable PCE of 12.4%.[ 206 ] Such high V OC was attributed to the matched energy level of the HTL/perovskite and perovskite/ETL interface.[ 206 ] By using the same HTL and ETLs, the best performing TPSCs with 14.81% were reported with 4‐fluoro‐phenethylammonium cations (FPEABr) in FASnI3 system. It is well known that the fluorinated cations can create stronger Van der Waals interaction or hydrogen bonding reducing the Sn2+ oxidation and creating a 2D microstructure to influence the 3D phase growth. Additionally, fluorinated compounds provide the unique characteristics of hydrophobicity due to the presence of fluorine atoms, which resulted in the high PCE‐based TPSCs with longer stability.[ 207 ] Despite the significant efforts by numerous research groups, the V OC loss of TPSCs was minimized up to 0.45 V. Wang et al. proposed a vacuum treatment method to establish a 2D/3D stratified heterojunction which can enable charge separation at the interface. Prior to the 2D/3D stratified heterojunction formation, guanidinium thiocyanate (GuaSCN) was introduced as a co‐additive to improve the crystallinity of the 2D perovskite phase. By the synergetic effects of the vacuum treatment and GuaSCN, the respective THP films showed high hole mobility, passivated the surface traps, and prolonged carrier lifetime up to 140 ns (the highest carrier lifetime for TPSCs). The energy level diagram of the pure quasi‐2D perovskite (n = 2) film shows a similar valence band level E V (−5.08 eV) to 3D FASnI3 film (−5.10 eV) while its conduction band level E C (−3.26 eV) is much higher than that of FASnI3 (−3.71 eV), which can prohibit electron transfer across the quasi‐2D/3D mixed phase. With such beneficial aspects, the TPSCs fabricated by this method with PEA2FA n − 1Sn n I3 n + 1(n = 10) showed a high V OC of 1.01 V, which minimizes the V OC loss of any type of TPSCs to 0.39 V (Table 5 ).[ 208 ]
Table 5.
Notable progress of photovoltaic performances of 2D/3D TPSCs
| Perovskite compound | 2D material | J SC [mA cm−2] | V OC [V] | FF | PCE [%] | Ref. |
|---|---|---|---|---|---|---|
| FASnI3 | Evaporated PEAI | 20.07 | 0.47 | 0.74 | 6.98 | [209] |
| FASnI3 | PEABr treatment | 22.64 | 0.54 | 0. 64 | 7.86 | [210] |
| FASnI3 | PEAI0.08 | 24.10 | 0.525 | 0.71 | 9.0 | [202] |
| FASnI3 | PEAI‐ NH4SCN | 22.00 | 0.61 | 0.70 | 9.41 | [204] |
| FA3Sn4I13 | BA0.5PEA0.5I | 21.82 | 0.60 | 0.67 | 8.82 | [211] |
| FASnI3 | PEACl0.12 | 22.06 | 0.59 | 0.69 | 9.1 | [212] |
| FASnI3 | PEASCN0.15 | 17.40 | 0.94 | 0.75 | 12.4 | [206] |
| FA n − 1Sn n I3 n + 1 (n = 10) | PEAI2 | 20.32 | 1.01 | 0.67 | 13.79 | [208] |
| FASnI3 | FPEABr0.10 | 24.91 | 0.84 | 0.76 | 14.81 | [207] |
4.4. Upscaling of TPSCs
Despite the progress in TPSCs in terms of photovoltaic performance and stability, note that most of the performances are evaluated with a small size aperture area of 0.02–0.1 cm2. To realize the scalability of all types of solar cells, authorized public test centers recommend evaluating the photovoltaic behavior with an aperture area of 1 cm2 or more.[ 213 ] However, because of the difficulty of fabricating THPs over a large aperture area, TPSCs with an aperture area greater than 1 cm2 have been very rarely reported. Chowdhury et al. highlighted that only a traditional approach may not be suitable for scaling of TPSCs.[ 167 ] After the antisolvent treatment on the FASnI3 film, the PDVA process assisted by methylammonium chloride was introduced. The subsequent FASnI3 films fabricated by this method depicted uniform, continuous, pinhole‐free, and highly crystalline morphology, which leads to a longer PL carrier lifetime over a large aperture area. The respective TPSCs fabricated by this PDVA method showed PCE of 6.33% with J SC = 19.59 mA cm−2, V OC = 0.53 V, and FF = 0.61 with a stable performance after 200 h of storage in an N2 atmosphere (Figure 17a).[ 167 ] A conjugated organic cation containing a large volume of amines‐3‐phenyl‐2‐propen‐1‐amine (PPA) was tested for the fabrication of cm2‐sized TPSC by Ran et al.[ 214 ] The addition of PPA to FASnI3 could promote the grain size, passivate the grains, reduce trap density, extract photogenerated charges, and induce preferential orientation of the perovskite film. Consequently, a PCE of 7.08% with photovoltaic parameters of V OC of 0.56 V, J SC of 17.57 mA cm−2, and FF of 0.72 (Figure 17b). Compared with the previously reported 1 cm2 TPSC based on the PDVA process, the conjugated PPA introduction within the FASnI3 framework showed a higher FF due to the better charge management of the TPSC.[ 214 ] In a recent report, instead of adopting the one‐step antisolvent method, a two‐step method was evaluated as a potential route to solve the scaling issue of TPSCs. In this method, the SnI2 layer with a 10% SnF2 in DMSO was first deposited by a spin‐coating method followed by the deposition of FAI in a 2‐methyl‐2‐butanol solvent. The implementation of two‐step deposition successfully induced coverage over a large surface area with lower defects and showed PCE of 10.09% with J SC of 19.96 mA cm−2, V OC of 0.77 V, and FF values and 0.667% (Figure 17c).[ 215 ] Compared with the previously reported 1 cm2‐sized PSCs, the TPSCs fabricated by the two‐step method showed a higher V OC of ≈0.21 V, which was attributed to the steric hindrances of 2‐methyl‐2‐butanol to hydroxyl introduced at the second step. By lowering the crystallization process, a uniform surface morphology over a large surface area was achieved which increased the PL lifetime and influenced the photovoltaic performance.
Figure 17.
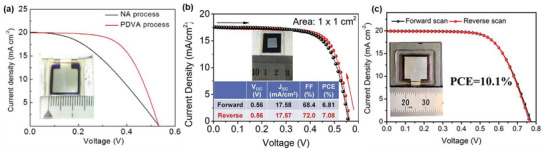
Development of 1 cm2‐sized TPSCs—a) PDVA processed first 1 cm2 TPSC. Reproduced with permission.[ 167 ] Copyright 2019, Wiley‐VCH. b) Conjugated organic cation based 1 cm2 TPSC. Reproduced with permission.[ 214 ] Copyright 2019, Elsevier. c) Two‐step deposition based on highest performance 1 cm2 TPSC. Reproduced with permission.[ 215 ] Copyright 2022, American Chemical Society.
4.5. Challenges and Future Perspective
Recent reports show various approaches to overcome some key bottlenecks of the TPSCs development. Nevertheless, obtaining all aspects of an efficient TPSC such as J SC, V OC, and FF remains a challenge. The literature review presented above shows that when a high V OC is achieved, the J SC remains low and when a favorable J SC is achieved the V OC remains low. With such characteristics, the PCE of TPSCs is now in the range of ≈14%, which is significantly lower than the 33% of the theoretical limit. Although numerous researches have shown promising aspects to creating a balance between the photovoltaic parameters by the approaches mentioned above, the real intrinsic mechanism to achieve high photovoltaic performances remains unknown. As can be observed, there is a significant lack of understanding of various THP material properties. Especially for high‐performing FASnI3 materials, many physical and electrical properties remain unknown. Therefore, apart from TPSC development, future research should be focused on understanding the THP material's optoelectronic properties, chemical behavior, and film properties. Since the complexity of TPSCs fabrication by solution‐processable methods was also highlighted, other alternative fabrication methods must be evaluated.
5. Application of THP for FETs
5.1. Operation Mechanism of FETs
FETs are the fundamental units in microelectronics for amplifying and switching electronic signals through modulating the electric field.[ 128 , 218 ] A typical FET is constructed with four main components: gate, dielectric, active layer, and electrodes (Figure 18a). The dielectric layer separates the gate electrode from the active semiconducting layer. The most commonly used dielectric in FETs is silicon dioxide (SiO2).[ 219 , 220 ] However, it shows a high driving voltage because of the low dielectric constant, when the thickness is reduced, the charge carriers penetrate thin layers of SiO2, resulting in a high gate leakage current, which is a critical fault of device downscaling.[ 221 ] Hence, materials with high dielectric constant may replace SiO2 in the future for the downscaling of FETs. The active layer is where charge‐carrier transport takes place and determines the field‐effect conductivity of the devices. For the n‐type active layer, amorphous metal oxides, especially indium gallium zinc oxide (IGZO), have succeeded in commercialization for backplane TFT of the large‐area organic light‐emitting display.[ 222 , 223 ] For the p‐type active layer, various materials such as silicon (Si) and other inorganic III‐V semiconductors have been developed until now but suffer from the high cost and complexity of fabrication.[ 224 , 225 ] Although the search for alternative semiconductors beyond inorganics has taken its place in organic materials with low cost, convenience, and scalability of fabrication, its low carrier transport mobility still limits the performance.[ 226 , 227 ] Recently, MHP stood out as a promising semiconductor candidate for FET active layer due to suitable features, such as high charge‐carrier transport mobility and solution processability (Figure 18b).[ 228 , 229 , 230 , 231 ] The charge‐carrier transport begins when a bias voltage is applied to the gate and metal contacts, which are also called electrodes. Metals that have suitable work functions when matched with the active layer can be used as source and drain electrodes. The distance between two electrodes is called the channel length L and the width is called channel width W.
Figure 18.
Structure and operational mechanism of FETs. a) Cross‐sectional schematic of a typical FET. Reproduced with permission.[ 216 ] Copyright 2016, Wiley‐VCH. b) FET structure of hybrid perovskite FET. Reproduced with permission.[ 217 ] Copyright 2016, Nature Publishing Group. c) Schematic illustrations of the four types of common FET configurations: (from left) bottom‐gate bottom‐contact (BGBC), bottom‐gate top‐contact (BGTC), top‐gate bottom‐contact (TGBC), and top‐gate top‐contact (TGTC). d) Schematic representations of FET operating regions: i) initial state, ii) linear region, iii) depletion region, and iv) saturation region.
As illustrated in Figure 18c, there are typically four types of transistor configurations depending on the positions of contact electrodes and gate: bottom‐contact bottom‐gate (BCBG), bottom‐contact top‐gate, top‐contact top‐gate (TCTG), and top‐contact bottom‐gate.[ 232 , 233 ] The FET performance may vary significantly based on its fabricated configuration due to differences in interface contacts, particularly one between the active layer and the contacts, and the other between the active layer and the dielectric layer. Bottom‐gate devices can achieve a good interface between the active layer and the dielectric layer and high‐quality surface. However, the active layer is directly exposed to the environment and susceptible to degradation. In comparison, top‐gate devices can reduce potential degradation from exposure; however, the active layer may be influenced by the deposition of dielectric or contact layers.[ 234 ]
In a typical FET, a bias voltage is applied to the gate and drain electrodes, whereas the source electrode is grounded. Gate voltage (V g) creates a potential difference between gate and source, which is a driving electric force for charge carriers to accumulate at the channel between source and gate electrode in enhancement mode devices. Drain voltage generates a potential difference between source and drain that induces the accumulated charge carriers at the channel to transport from source to drain. Gate and drain voltage are applied simultaneously and form a controllable current between source and drain. Thus, the magnitude of applied gate voltage can change the carrier density participating in transport through the channel, as well as the total current. The magnitude of applied source–drain voltage affects the charge‐carrier concentration gradient at the channel.[ 218 ]
The operation of FET with polycrystalline or amorphous semiconducting films can be divided into four main regions, as illustrated in Figure 18d: i) The device state before any bias voltage is applied. ii) The applied positive(negative) V g drives electrons(holes) at the insulator/active layer interface. Some of the induced charge carriers fill in the trap states during the transport, the others are mobile carriers. The carrier density at the channel is related to the magnitude of V g and capacitance (C) of the insulator. When there are sufficient mobile carriers at the active channel, V g should be higher than V Th (V g − V Th > 0). If only V g is applied without source–drain voltage, the charge carriers at the channel are uniformly distributed. iii) When a small source–drain voltage (V DS << V g) is applied, a linear gradient of carrier concentration is formed from source to drain electrode. V DS will be increased to the point of V DS = V g − V Th, where a pinch‐off point exists at the transport channel adjacent to the drain (source) electrode. This is also called the depletion region, at which the difference between V g and V DS matches V Th. At this point, the electric field is not sufficient to induce additional mobile carriers to accumulate at the channel near the drain electrode. iv) When V DS is further increased (V DS > V g − V Th), the same effect is enhanced, whereas the transport channel length is reduced. This is also called the saturation region, where the carriers can be driven across the narrow depletion zone and form a space‐charge‐limited current. Further boost of V DS expands the depletion region, which cannot substantially increase source–drain current I DS and saturates at a certain level called I D,sat.
Some of the important parameters for quantifying the FET performance include field‐effect mobility (µ FE), on/off current ratio, threshold voltage (V Th), and subthreshold swing (SS). µ FE can explain the carrier transport capacity and can be extracted from the linear region of the transfer curve. On/off current ratio characterizes the ability to control channel current with V g and can be calculated by the ratio of I DS at on‐ and off‐state. V Th describes the interface charge traps and SS shows the ability to switch on and off the device.[ 235 , 236 ]
As discussed above, FETs are building blocks for modern electronic technologies. Through the discovery of promising semiconductor materials, innovative device structures, and electric circuit applications, FETs have successfully obtained a highly mature level of fabrication. Among the incumbent and emerging semiconductor materials, THPs offer excellent optoelectronic properties, including low effective mass, high carrier mobility, long charge‐carrier lifetime, and defect tolerance character.[ 237 , 238 , 239 ] These attractive properties enabled the development of high‐performance, highly reliable THP‐based FETs over the last two decades, which will be discussed in the next section.
5.2. Recent Progress in THPs in FETs
5.2.1. 2D THP: (PEA)2SnI4
2D layered THPs are highly stable with long‐chain organic bulky ligand in between the inorganic slabs to suppress ion migration and moisture penetration. Additionally, this family of materials is especially attractive for allowing versatile structure and tunable optoelectronic properties through compositional engineering of organic cations.[ 241 , 242 ] Because of these advantages, the first‐ever made perovskite transistor was based on 2D THP than any other 3D perovskites or LHPs. Almost two decades ago, Kagan et al. used (PEA)2SnI4 as the channel layer for BGBC FET, as illustrated in Figure 19a.[ 240 ] The device showed pure p‐channel characteristics with mobility of µ h = 0.62 cm2 V−1 s−1 and a reasonable on/off current ratio of 104 at room temperature (Figure 19b,c). This work highlighted the potential for fabricating MHP‐based FETs with a cost‐effective solution process. Through spin‐coating, the perovskite layer showed a high orientation in the (001) direction, which favors hole transport in the 2D crystal plane parallel to the device channel (Figure 19d). Additionally, the microstructures of perovskite film, including crystallinity and grain boundaries, are critical for device performance. Mitzi et al. introduced (PEA)2SnI4‐based BGBC FETs fabricated through melt process with silicon and polyimide substrates.[ 243 ] The melt‐processed channel layer exhibited considerably enlarged grain size relative to spin‐coated films. Consequently, the fabricated device showed both improved saturation and linear regime hole mobilities of 2.6 and 1.7 cm2 V−1 s−1, respectively, at room temperature. Despite the initial success in 1999, research based on (PEA)2SnI4 accelerated only in the recent few years encouraged by remarkable advances in solar cells and LEDs. Recently, the performance of 2D THP‐based FETs remarkably developed through precise engineering of film morphology, device structure/interface engineering, crystal formation, and organic spacer synthesis.
Figure 19.
2D (PEA)2SnI4‐based FETs. a) Schematic illustration of a BGBC FET structure with a layered (PEA)2SnI4 channel. b) Corresponding transfer characteristics at V D = −100 V. c) Corresponding extracted hole mobility. d) XRD spectrum of a layered (PEA)2SnI4 thin film. a–d) Reproduced with permission.[ 240 ] Copyright 1999, American Association for the Advancement of Science. e) Schematic representation of a TGTC FET structure with (PEA)2SnI4 semiconducting channel, SAM‐treated substrate, and MoO x hole‐injection layer. f) Corresponding FET output curves. g) Corresponding transfer characteristics. e–g) Reproduced with permission.[ 238 ] Copyright 2016, Wiley‐VCH.
Matsushima et al. first fabricated (PEA)2SnI4 by vacuum deposition on top of octadecyltricholorosilane (OTS)‐treated Si/SiO2 substrates.[ 244 , 245 ] By optimizing the growth rate and temperature of OTS on substrates, the film morphology was much improved and the fabricated FETs exhibited hole mobility of µ h = 0.78 cm2 V−1 s−1 and an on/off current ratio of 105 at room temperature. The same group proposed a significant development in solution‐processed (PEA)2SnI4 FETs by applying a series of optimization processes, including a self‐assembled monolayer (SAM)‐treatment on substrates and the addition of MoO x as a hole‐injection layer between the channel and the electrode (Figure 19e).[ 238 ] Specifically, NH3I‐SAM treatment induced more well‐developed perovskite crystallites and reduced the amount of residual starting materials and ions. Also, TGTC configuration was formed with Cytop as a dielectric, which reduced resistance for charge carriers to travel through an alternating small‐bandgap inorganic layer and a large‐bandgap organic layer, and diminished hole trap density near the dielectric/perovskite interface. The optimized transistor exhibited high hole mobility of µ h = 15 cm2 V−1 s−1 and a high on/off current ratio of 106 at room temperature (Figure 19f,g). The authors also investigated the intrinsic µ h of (PEA)2SnI4‐based FET by increasing L to reduce the contact resistance (R c) between the perovskite semiconductor and source/drain electrodes while using the same TCTG device configuration.[ 246 ] With a negligible contribution of R c relative to the total resistance, the hole mobility increased and saturated to a value of 26 cm2 V−1 s−1.
The performance of polycrystalline film devices is often intrinsically limited by grain boundaries, where carrier transport is slowed down or prohibited. To solve this issue, Matsushima et al. fabricated a BGBC transistor with exfoliated crystals of (PEA)2SnI4.[ 247 ] Due to the reduced structural disorder and amount of grain boundaries, the fabricated device exhibited tremendously high hole mobility of 40 cm2 V−1 s−1 at room temperature. Despite such high performance, the restrictions on reproducing good quality crystals led to less than 1% yield of an operating transistor. Furthermore, their efforts in discovering the unique nature of (PEA)2SnI4 expanded to fabricating n‐type TGTC FETs.[ 248 ] By inserting C60 as a buffer layer in between the semiconductor and the low‐work‐function metal (Al) electrodes, the authors presented an n‐type transistor with electron mobility of µ e = 2.1 cm2 V−1 s−1 and an on/off current ratio of 104 at room temperature. This work presented the possibility of ambipolar charge transport with (PEA)2SnI4 with a newly designed device structure.
Additionally, interface engineering was often approached to enhance the performance of (PEA)2SnI4 transistors. Zhang et al. fabricated a solution‐processed (PEA)2SnI4 transistor with PVA and cross‐linking poly(4‐vinylphenol) (CL‐PVP) as the dielectric and ITO as the gate electrode. The optimized device exhibited hole mobility of 0.28 and 0.33 cm2 V−1 s−1 in the forward and reverse voltage scan, and an on/off current ratio of 103 at room temperature.[ 249 ] The negligible hysteresis was highlighted due to the beneficial properties of PVA/CL‐PVP dielectric, including its high quality, good compatibility with perovskite, and suppressed ion migration in perovskite film.
Although various approaches have enhanced the device performance of (PEA)2SnI4 FETs, the fragility of the material and other unexplored factors limited the overall operating device yield. Recently, Zhu et al. demonstrated a systematic study from starting materials to film and finally to FETs. The authors provided a set of universal methods for highly reproducible, high‐performance (PEA)2SnI4 FET with hole mobility of 3.51 cm2 V−1 s−1 and an on/off current ratio of 106.[ 131 ] The methods included self‐passivation of grain boundaries by using excess PEAI, control of grain crystallization by adding Lewis‐base adducts, and passivation of iodide vacancy through oxygen treatment (Figure 20a). The passivation of grain boundaries and enlargement of grain size enabled the reproducibility and reliability of FET performance (Figure 20b). Notably, the authors fabricated the first perovskite‐based complementary inverters with optimized p‐channel (PEA)2SnI4 FET and n‐channel IGZO FETs, which showed a high gain of over 30 with an excellent noise margin (Figure 20c). Following this work, a wide range of solvent and additive engineering techniques have been applied to (PEA)2SnI4 FETs.[ 106 , 107 , 250 , 251 ]
Figure 20.
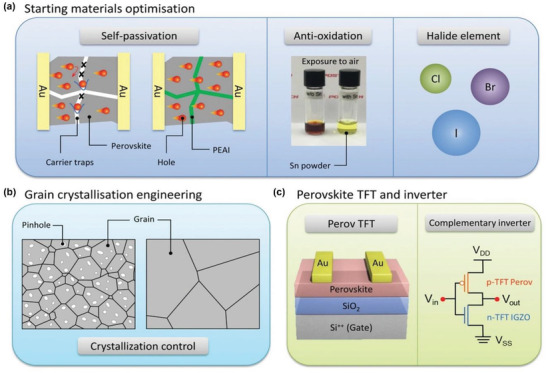
A systematic study on 2D (PEA)2SnI4 FET development. a) Optimization of starting materials: i) self‐passivation by adding slightly excess PEAI, ii) anti‐oxidation using metallic Sn powder, and iii) effect of halide element. b) Grain crystallization engineering through an adduct approach. c) Device fabrication, trace oxygen doping effect on FETs, and the first perovskite‐IGZO FET‐based complementary inverter. a–c) Reproduced with permission.[ 131 ] Copyright 2020, Wiley‐ICH.
(PEA)2SnI4 is also one of the first THPs to be studied as a channel layer for a phototransistor.[ 252 ] Phototransistors are a special type of transistors, as well as being categorized into photodetectors, with the capability to modulate channel conductance through electrical bias and irradiated photons.[ 253 ] THPs are an especially attractive material for phototransistors, owing to their large optical absorption coefficient, long exciton diffusion length, and high charge carrier mobility.[ 254 , 255 , 256 ] The detailed mechanism of the operation of halide perovskite phototransistors and photodetectors is thoroughly covered in a few previous review papers.[ 33 , 256 , 257 , 258 ] Throughout this work, some of the important achievements made in THP‐based phototransistors will be highlighted for several THPs. Zhu et al. significantly improved the photo detecting ability of (PEA)2SnI4‐based phototransistor by incorporating binary solvent of DMF and ethyl acetate (EA).[ 250 ] The antisolvent addition smoothened the film formation through nucleation and consecutive oriented grain ripening, allowing efficient charge transport through full‐coverage film. Through amplified transistor function and photogate properties, the DMF/EA incorporated (PEA)2SnI4 phototransistor showed photoresponsivity of 1.6 × 105 A W−1 and remarkably high detectivity of 3.2 × 1017 Jones.
5.2.2. 2D THPs: Synthesis of New Organic Spacers
Although extensive efforts were made to achieve high‐performance (PEA)2SnI4‐based FETs, most of these techniques focused on device engineering rather than the perovskite material itself. One of the earliest attempts in molecular engineering of 2D THPs was by Mitzi et al., who reported a study on tuning the electronic properties of (PEA)2SnI4 by fluorination of PEA ligand.[ 27 ] By substituting a hydrogen atom on the phenyl ring of PEA with a fluorine atom, the authors composed m‐fluorophenylethyl ammonium tin iodide (m‐FPEA)2SnI4 (m = 2, 3, and 4, representing the position of the fluorine atom) (Figure 21a). The different positions of fluorinated atoms induced subtle changes in Sn—I—Sn bond angle and fabricated BGBC FET hole mobilities: 156.41° and 0.56 cm2 V−1 s−1 (m = 4), 154.21° and 0.51 cm2 V−1 s−1 (m = 3), and 153.31°, and 0.24 cm2 V−1 s−1 (m = 2). (Figure 21b). Although the device performance did not show evident improvement, this study was one of the earliest approaches to associate cation molecular engineering with electronic devices.
Figure 21.
2D organic spacer synthesis and FET fabrication. a) (4‐FPEA)2SnI4 crystal structure, viewed along the (011) axis. b) Corresponding BGBC FET transfer characteristics. a,b) Reproduced with permission.[ 27 ] Copyright 2001, American Chemical Society. c) Chemical structure of 4Tm. d) Side‐view of (4Tm)2SnI4 crystal structure. e) Corresponding FET transfer characteristics. f) Chemical structure of TT. g) Side‐view of (TT)2SnI4 crystal structure. h) Corresponding FET transfer characteristics. i) PL images of (TT)2SnI4 thin films after thermal annealing treatment at 155°C for 4 min (top) and 15 min (bottom) (scale bar = 20 µm). j) Schematic representation of carrier transport in small grain (top) and large grain (bottom)‐based (TT)2SnI4 thin film. c–j) Reproduced with permission.[ 259 ] Copyright 2021, American Chemical Society.
Recently, Gao et al. proposed a breakthrough in novel organic spacer synthesis for 2D THPs.[ 260 ] The authors modulated the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital energy levels of the ligands, which altered the overall quantum well structure as well as the charge injection. Inspired by organic semiconductors, polythiophene, for example, the authors featured linear π‐conjugated oligothiophene ligand, 2‐(3‴,4′‐dimethyl‐[2,2′:5′,2″:5″,2‴‐quaterthiophen]‐5‐yl)ethan‐1‐ammonium, also known as 4Tm (Figure 21c). The novel 4Tm ligand exhibited a suitable HOMO level that matched the work function of Au better than PEA or BA and reduced contact resistance to enhance hole injection. Also, 4Tm was incorporated into 2D THP layered structure (4Tm)2SnI4 and applied as a p‐channel layer to BGTC FETs, exhibiting hole mobility of µ h = 2.32 cm2 V−1 s−1 (Figure 21d,e). This performance was achieved without additional treatments, which highlighted the excellence of the material itself. Moreover, the incorporation of 4Tm increased grain size up to a micrometer scale, which reduced charge scattering at grain boundaries. Furthermore, the device held remarkable air stability for up to 30 days, in contrast to the survival of pristine (PEA)2SnI4‐based FET for less than 1 day. The air stability greatly increased due to the hydrophobic and bulky properties of the π‐conjugated spacer, which can restrict oxygen and moisture penetration. The strong intermolecular interactions between the staggering conjugated 4Tm ligands also increased the intrinsic stability of the material.
The same group extended their studies to synthesizing thienothiophene derivative cation, 2‐(4′‐methyl‐5′‐(5‐(3‐methylthiophen‐2‐yl)thieno[3,2‐b]thiophen2‐yl)‐[2,2′‐bithiophen]‐5yl) ethan‐1‐ammonium, also known as TT (Figure 21f).[ 259 ] Their champion device of (TT)2SnI4 exhibited hole mobility of µ h = 9.35 cm2 V−1 s−1 and an on/off current ratio of 106 (Figure 21g,h). The performance enhancement was induced by extended π‐conjugation and increased planarity of the TT ligand, which slowed down the nucleation process and significantly enlarged grain size to almost a millimeter scale (Figure 21i,j). Their variety of works in molecular engineering of organic spacers extended the potential application of highly stable 2D THPs in electronic devices.
5.2.3. 2D/3D Hybrid THPs
From the first works of THPs to the recent extension of studies, 2D THPs demonstrated remarkable enhancement in device performance and air stability. Despite its progress, the fundamental intrinsic limits of 2D perovskites are yet to be solved. Due to the quantum and dielectric confinement effect in 2D perovskites, the charge transport is mostly confined in the corner‐sharing inorganic octahedral cage layer, whereas the charge carrier perpendicular to the inorganic layer is strongly restricted. Contrary to 2D perovskites, 3D perovskites have a higher potential for faster charge transport in absence of the quantum and dielectric confinement effect. However, 3D THPs are notorious for high trap density and high self‐p‐doping. Until the works of Shao et al., there were no reports that used 3D THPs as the semiconducting layer for FETs. The authors successfully reduced the hole density of 3D FASnI3 by incorporating a small amount of 2D (PEA)2SnI4.[ 97 ] The addition of (PEA)2SnI4 greatly enhanced the crystallinity and orientation of the 3D perovskite phase, which was critical for FET application (Figure 22a,b). The highly crystalline 2D/3D film exhibited a much smaller amount of trap states than its 3D counterpart. The reduced trap states were particularly Sn vacancies, which are the dominant source of high p‐doping in 3D Sn‐based perovskite (Figure 22c). Through the control of crystallinity and orientation, along with reduced trap density, (PEA)2SnI4‐incorporated FASnI3‐based FET exhibited hole mobility of 0.21 cm2 V−1 s−1 and an on/off current ratio of 104 (Figure 22d).
Figure 22.
2D/3D hybrid FASnI3/(PEA)2SnI4 FET. Schematic representation of phase distribution and orientation in a) only 3D film. b) 2D/3D film. Transfer characteristics of BGBC FET with c) only 3D FASnI3 as an active layer d) 2D/3D hybrid (PEA)2SnI4/FASnI3 as an active layer with optimized thickness. a–d) Reproduced with permission.[ 97 ] Copyright 2021, Wiley‐VCH. e) Schematic illustration of grain morphologies in cross‐sectional view of 2D (top‐left), 3D (top‐right), 2D/3D core–shell (bottom‐left) and 2D/3D mixture (bottom‐right) Sn‐based perovskite thin films. f) Transfer characteristics of 2D/3D core–shell FET with 2D/3D ratio of 1:9 and vacuum treatment. e,f) Reproduced under the terms of the Creative Commons CC‐BY license.[ 261 ] Copyright 2022, The Authors. Published by Wiley‐VCH.
Recently, Kim et al. also reported a study on the incorporation of 2D (PEA)2SnI4 into 3D FASnI3 from an alternative perspective.[ 261 ] The authors presented a new concept of 2D–3D core–shell structure, where the 3D core is fully isolated by the 2D counterpart. The suggested structure provided two major benefits: independent control of V Th by 2D component and significantly improved grain boundary resistance by the 2D/3D interface. The 2D (PEA)2SnI4 FETs suffer from abnormally saturated on‐current caused by large series resistance at the grain boundaries, whereas 3D THP‐based FET exhibits the excessive carrier density that only permits on‐state (Figure 22e). This 2D–3D core–shell simultaneously solves both issues by first controlling the on‐state by 2D component and forming a smoother match between 2D/3D lattices. By adding SnF2 to facilitate the formation of core–shell structure and vacuum treatment, (PEA)2SnI4‐FASnI3‐based FET showed hole mobility of 25 cm2 V−1 s−1 and an on/off current ratio of 106 (Figure 22f). Additionally, the authors combined the optimized p‐channel FET with n‐channel IGZO FET to fabricate CMOS with a gain of over 200. Moreover, Shen et al. proposed a synthesis of 2D incorporated 3D Cs‐based THP crystal. A millimeter‐sized (PEA)2CsSn2I7 crystal was applied as a channel layer to BCBG FET, which exhibited hole mobility of 34 cm2 V−1 s−1 at a low temperature of 77 K.[ 262 ]
In another perspective, FASnI3 itself has high hole concentration and hole mobility due to self p‐doping nature of 3D THP perovskite, which is outstanding to be used as a phototransistor. Recently, Liu et al. developed a high‐performance FASnI3‐based phototransistor with a high responsivity of ≈105 A W−1 at a low operating voltage.[ 263 ] The authors also incorporated FASnI3/PEDOT:PSS heterojunction due to the enhanced photogate effect from the heterojunction, reaching a maximum responsivity of 2.6 × 106 A W−1.[ 264 ]
5.2.4. 3D THP: CsSnI3
Groundbreaking development in p‐type tin perovskite‐based transistors was recently accomplished by our group through developing a 3D CsSnI3‐based channel layer.[ 20 ] The fabricated champion device achieved a record performance with hole mobility over 50 cm2 V−1 s−1 and on/off current ratios exceeding 108 (Figure 23a–e). These values meet the requirements for the backplane of high‐end displays and integrated logic circuits. Additionally, the optimized CsSnI3‐based TFTs highlighted high operational stability and reproducibility (Figure 23f). Using bias–stress test, the authors discovered a strikingly fast performance recovery within 1 min after the measurement. This unique feature of CsSnI3‐based TFTs is a drastic improvement from the commercialized p‐type polycrystalline Si or n‐type metal oxide‐based TFTs, which require hour‐long recovery time or extra thermal treatments.[ 265 ] The authors developed a series of methods to accentuate the fast carrier transport and compensate for the fundamental self‐p‐doping issues of 3D THPs. The methods include SnF2 incorporation in CsI‐rich precursor and a small portion of SnI2 substitution with PbI2. SnF2 incorporation has already demonstrated efficient suppression of Sn2+ oxidation and control of crystallization in THPs.[ 75 , 86 , 89 ] Additionally, CsSnI3 solution with slight excess CsI improved film morphology, as well as electrical performance. Moreover, due to the lower Lewis acidity of Pb2+ compared with Sn2+, a small amount of Pb substitution in the precursor slowed down the conversion to the perovskite phase, ultimately improving the film crystalline quality (Figure 23g).[ 266 , 267 , 268 , 269 , 270 ] Hall‐effect measurements were conducted to further evaluate the electrical properties of the film. CsI‐rich films demonstrated lower hole concentration but significantly increased Hall mobilities due to improved film uniformity/crystallinity and reduced defect density, such as V Sn (Figure 23h,i). This series of techniques can be a stepping stone toward the development of high‐performance p‐type THP‐based transistors and complementary electronics.
Figure 23.
3D CsSnI3‐based FET. a) Schematic illustration of BGTC TFT structure. b) µ FE and I on/I off of CsSnI3‐based TFTs with different molar ratios of CsI/SnI2. c) µ FE and I on/I off of CsI‐rich (x = 1.25) precursors with various ratios of Pb substitution. d) Transfer characteristics of optimized CsSnI3‐based TFT (x = 1.25, 10 mol% Pb substitution, and 7 mol% of SnF2). e) Corresponding output curves. f) Transfer curves of one optimized CsSnI3‐based TFT under negative bias–stress measurement for various durations and the recovery behavior at V GS = V DS = −40 V. g) XRD patterns of perovskite films deposited from precursors with different ratios of CsI/SnI2 and that from CsI‐rich (x = 1.25) precursor with 10 mol% Pb substitution and 7 mol% SnF2. Hall measurement derived parameters, including h) hole concentration of thin films deposited from precursors with different CsI/SnI2 ratios (without SnF2) and that from optimized CsI‐rich (x = 1.25), 10 mol% Pb substitution, and 7 mol% SnF2 precursor and i) corresponding Hall mobility. a–i) Reproduced with permission.[ 20 ] Copyright 2022, Nature Publishing Group.
5.2.5. 3D THP: MASnI3
The first demonstration of p‐channel perovskite transistors based on 3D MASnI3 was recently reported as shown in Figure 24a.[ 95 ] By rationalizing the effects of halide (I/Br/Cl) anion engineering on the improvement of film morphology and suppression of tin/iodide vacancy, the authors developed high‐performance hysteresis‐free TFTs with a high hole mobility of 20 cm2 V−1 s−1 and on/off ratio exceeding 107 (Figure 24b). The optimized device demonstrated a threshold voltage of 0 V, which is an ideal enhancement mode where no applied bias voltage is required to turn off the transistor. This behavior is a highly desirable trait for simplifying circuit design and minimizing power consumption in electronic applications.[ 236 , 271 ] The authors revealed that contrary to previous studies on LHPs, ion migration had a negligible contribution to hysteresis in THP‐based transistors, but minority carrier traps induced by iodine vacancy V I were the primary cause. The deep electron traps induced by V I defects were significantly reduced by Br/Cl co‐substitution with strong binding affinities to V I sites and stabilized the overall lattice structure (Figure 24c–e). Furthermore, operational stability is another crucial figure of merit for practical applications. Through on/off switching stability and bias–stress stability test, the I‐pristine device suffered from serious carrier trapping, whereas the I/Br/Cl optimized device showed much‐improved stability, almost comparable to those of stable organic and amorphous silicon‐based FETs (Figure 24f,g).[ 239 , 272 ] By further combining the optimized p‐channel perovskite FETs with n‐channel IGZO FETs, the authors developed monolithically integrated high‐gain complementary inverters and highlighted their high compatibility and processability for electronic applications.
Figure 24.
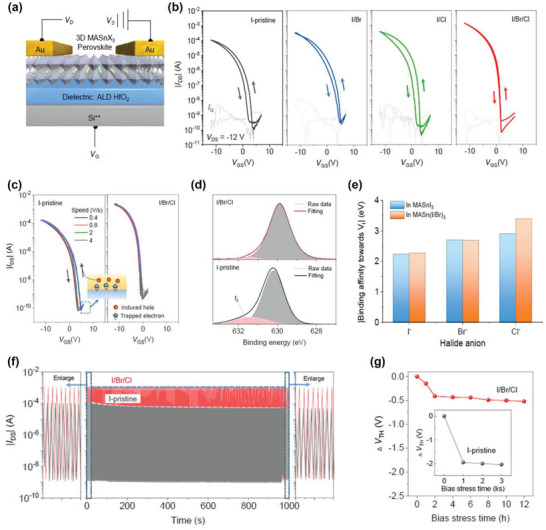
3D MASnI3‐based FET: Removal of hysteresis through anion engineering. a) Transistor structure. b) Transfer characteristics with different perovskite channel layers. c) Transfer characteristics of I‐pristine and I/Br/Cl transistors measured at various scan speeds. d) I 3d 3/2 core level XPS spectra. e) Calculated binding affinities of halide anions with V I‐site in MASnI3 and MASn(I/Br)3. f) On/off switching stability of I‐pristine and I/Br/Cl transistors. g) Variation of V Th under bias–stress. a–g) Reproduced with permission.[ 95 ] Copyright 2022, Nature Publishing Group.
5.2.6. Sn2+‐Free THP: Vacancy‐Ordered Double Perovskite
Despite the extensive efforts to improve the air stability of THP film, its application in an inert atmosphere remains limited.[ 69 , 273 ] An alternative approach to developing an air‐stable THP device is to modulate the perovskite material composition itself. Vacancy‐ordered halide double perovskites are attractive alternatives for their toxic lead and unstable Sn2+‐based counterparts.[ 91 , 93 , 274 ] Despite the early discovery of this composition and continuous theoretical studies, the application on electronic devices was rare until a recent report by Liu et al.[ 92 ] The authors demonstrated a synergetic solution process method to modulate the film quality of Cs2SnI6 and its feasibility in FETs (Figure 25a,b). The incorporation of slight excess SnI4 in a mixed DMF/DMSO solvent‐based precursor generated improved crystallinity, uniform morphology, and high electrical properties (Figure 25c). Also, the authors used Mn2+ doping to further tune the electrical properties for TFT application and achieved electron mobility of µ e = 1.2 cm2 V−1 s−1 and an on/off current ratio of 104 (Figure 25d,e). Most importantly, Cs2SnI6‐based TFT exhibited ambient air stability with reliable operation for more than 1 week, which is an impressive improvement from previous 2D THP‐based TFTs (Figure 25f). The dramatic enhancement in air stability benefited from the attractive chemical compositions of Cs2SnI6, including strong Sn—I covalent bonding and a stable Sn tetravalent state.[ 275 , 276 ] Thus, the authors highlighted Cs2SnI6 as a promising alternative material for high‐performance and high‐stability THP‐based transistors. Furthermore, the authors expanded the potential electronic applications of n‐channel Cs2SnI6 TFT by combining it with p‐channel (PEA)2SnI4 TFT to develop the first all‐perovskite complementary inverter with a high‐gain voltage over 38 (Figure 25g–i).
Figure 25.
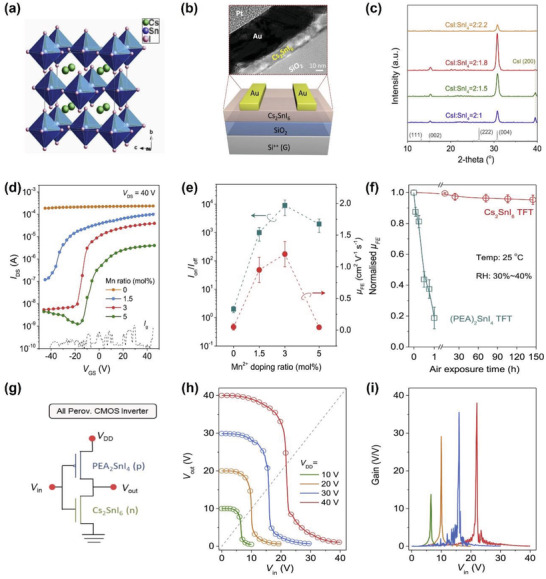
Sn2+‐free perovskite FET. a) The crystal lattice structure of vacancy‐ordered double perovskite Cs2SnI6. b) TFT structure and corresponding cross‐sectional TEM image. c) X‐ray diffraction peak patterns of Cs2SnI6 as a function of the CsI:SnI4 ratio in the precursor solution. d) Transfer characteristics of Cs2SnI6 TFT with various ratios of Mn2+ doping. e) Summary of electron mobility µ e and I on/I off of TFTs. f) Normalized µ FE of Sn2+‐based (PEA)2SnI4 TFT and Sn(IV)‐based Cs2SnI6 TFTs as a function of ambient air exposure time. g) Schematic diagram of CMOS inverter with combined p‐channel (PEA)2SnI4 TFT and n‐channel Mn2+‐doped Cs2SnI6 TFT. Corresponding h) voltage transfer and i) gain voltage curves at different V DDs. a–i) Reproduced with permission.[ 92 ] Copyright 2022, Cell Press.
5.3. Summary and Future Perspective
From the first‐ever reported (PEA)2SnI4‐based transistor to recent success in CsSnI3‐based transistor, two decades of research in THPs have shown a striking increase in device performance, along with efforts to enhance its stability. Table 6 and Figure 26 illustrate the extension of studies on THP‐based FETs with major milestones from 2D to 3D materials, achieved by multiple research groups. The majority of the reported devices are solution‐processed, and only a few used other methods, such as exfoliated crystals grown from solution or melting process from grown crystals. Only one report has used vacuum deposition to fabricate the THP layer. Each of these methods has its advantages and disadvantages. For example, solution process is a simple and cost‐effective fabrication method, advantageous for industrial applications. However, the solution process has relatively low reproducibility because the quality of the solution has a critical effect on the fabricated film and patterning issue. The recent achievement of high‐performance THP‐based FETs with hole mobility as high as over 50 cm2 V−1 s−1 by Liu et al. was also fabricated via a solution process. Through this method, the work highlighted a great potential in the industrial application of THP‐based p‐type transistors, with comparable performance with commercialized n‐type transistors. The next step in the development of THP‐based FETs will be to greatly increase the reproducibility of high performances through vacuum deposition. As shown through the success of the OLED industry, vacuum deposition can fabricate highly reproducible devices in mass production. THP devices may follow the pathway toward commercialization with such high performance and reproducibility.
Table 6.
Summary of device performance of THP‐based transistors
| Material | Dielectric/contact | Structure | Deposition/treatment | µ h [cm2 V−1 s−1] | µ e [cm2 V−1 s−1] | I on/I off | T | Ref. |
|---|---|---|---|---|---|---|---|---|
| (PEA)2SnI4 | SiO2/Au | BGBC | – | 0.62 | ≈104 | R.T. | [240] | |
| PEASnI4 | Cytop/Au | TCTG | NH3I‐SAM, MoO x HIL | 15 | ≈106 | R.T. | [238] | |
| PEASnI4 | SiO2/Au | BGTC | Vacuum deposition, OTS‐SAM | 0.78 | ≈105 | R.T. | [244] | |
| PEASnI4 | Cytop/Al | TCTG | NH3I‐SAM, C60 EIL | 2.1 | ≈104 | R.T. | [248] | |
| (PEA)2SnI4 | SiO2/Au | BGTC | MoO x HIL | 7.9 | ≈107 | R.T. | [135] | |
| (PEA)2SnI4 | SiO2/Au | BGBC | Single crystal, MoO x HIL | 40 | ≈106 | R.T. | [247] | |
| (PEA)2SnI4 | PVA/Cl‐PVP/Au | BGTC | Polymer dielectric | 0.28/0.33 | 102≈103 | R.T. | [249] | |
| (PEA)2SnI4/PVP | Cl‐PVP/Au | BGTC | Polymer dielectric | 0.31 | ≈103 | R.T. | [277] | |
| (PEA)2SnI4/PVP, PEO | Cl‐PVP/Au | BGTC | Polymer mixed with precursor | 0.013 | 0.0068 | ≈104 | R.T. | [278] |
| (PEA)2Sn x Pb1‐ x I4 | PVA/Cl‐PVP/Au | BGTC | Sn/Pb ratio | 0.02 (x = 0.7) | ≈102 | R.T. | [279] | |
| (PEA)2SnI4/CNT | SiO2/Au | BGTC | CNT mixed with precursor | 1.51 | ≈105 | R.T. | [280] | |
| (PEA)2SnI4 | SiO2/Au | BGTC | Oxygen treatment | 3.51 | ≈106 | R.T. | [131] | |
| (PEA)2SnI4/CB/EA | SiO2/Au | BGTC | Binary solvent | 3.8 | ≈105 | R.T. | [250] | |
| (PEA)2SnI4/Urea | SiO2/Au | BGTC | Urea mixed with precursor | 4 | ≈105 | R.T. | [251] | |
| (PEA)2SnI4/CuI | SiO2/Au | BGTC | CuI mixed with precursor | 2.61 | ≈106 | R.T. | [106] | |
| (4‐MeO‐PEA)2SnI4 | SiO2/Polyimide/Au | BGBC | Melt process | 2.6 | ≈106 | R.T. | [243] | |
| (m‐FPEA)2SnI4 | SiO2/Pd | BGBC | – | 0.2 ≈ 0.6 | ≈105 | R.T. | [27] | |
| (PEA)2CsSnI7 | SiO2/Cr/Au | BGBC | Single crystal | 34 | – | 77 K | [262] | |
| (4Tm)2SnI4 | SiO2/Au | BGTC | – | 2.32 | ≈106 | R.T. | [260] | |
| (TT)2SnI4 | SiO2/Au | BGTC | – | 10 | ≈106 | R.T. | [259] | |
| (STm)2SnI4 | SiO2/Au | BGTC | – | 1.52 | ≈106 | R.T. | [281] | |
| FASnI3/(PEA)2SnI4 | PMMA/Al2O3/Au | TGBC | – | 0.21 | ≈104 | R.T. | [97] | |
| FASnI3/(PEA)2SnI4 | SiO2/Pt | BGBC | Vacuum treatment | 25 | ≈106 | R.T. | [261] | |
| MASnI3 | HfO2/Au | BGTC | I/Br/Cl anion engineering | 20 | ≈107 | R.T | [95] | |
| Cs2SnI6 | SiO2/Au | BGTC | Mn2+ doping | 1.2 | ≈104 | R.T. | [92] | |
| CsSnI3 | SiO2/Au | BGTC | Small Pb substitution | 55 | ≈108 | R.T. | [20] |
Channel layers are solution‐processed unless mentioned otherwise, µ h: Hole mobility, µ e: Electron mobility, I on/I off: On/off current ratio, T: Temperature
Figure 26.
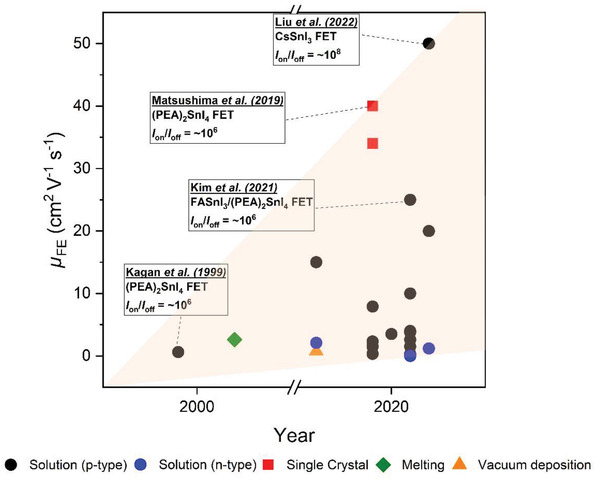
Development of THP‐based FETs over the past two decades.
6. Conclusion
To conclude, we reviewed the structural property relationship, device performance, and stability of several THPs (MASnI3, FASnX3, and CsSnX3) and presented their current issues and perspective opportunities. Thus far, despite the high device performance of the potential of THPs, there has been no report showing the ambient stability for the commercialization level. Therefore, to further improve our standing on device performance and its stability, we must indeed pay attention to overcoming several challenges including i) pure and non‐oxidized initial chemicals and non‐oxidizing solvents, ii) fully understanding of film formation dynamics, iii) precise control of cation and anion defects in the film, iv) suppression of Sn2+ oxidation, v) commercial‐grade device operation stability in the air ambient, and vi) better energy band alignment. The vital difference between THPs and its Pb‐based counterparts originated from the instability of Sn2+ in 3D structure and the tendency to realize V Sn. Thus, to aid our discussion, we hence also review the mechanism governing Sn oxidation, as well as the efficient route to reduce Sn4+ and the encapsulation approach to alleviating the poor stability, especially in ambient air. It is understood that the under‐performance of THP‐based devices is due to the formation of V Sn; thus, a greater understanding of the Sn oxidation would lead to better‐performing THP‐based devices. To date, numerous techniques have been introduced to reduce such effects, namely, the use of additives to act as reducing agents, control crystallization, partial ion substitution, and reduced dimensionality, and we indeed believe that an important breakthrough will be reported in the near future. Henceforward, finding new, stable, and effective solvents for THP‐based devices, having a deep understanding of the Sn chemistry via film formation, interfacial engineering to better match the energy bands of THPs, and stabilizing Sn2+ through device fabrication procedures are greatly important. Alternatively, the implementation of high‐performing devices using vacuum‐deposited THPs can be another breakthrough to fundamentally escape from the issues of various solubility of precursors and additives or stable and effective solvents. Last, although huge progress has recently been made in THP‐based optoelectronics devices, a deep understanding of the photo‐physics and photo‐chemistry processes is hardly obtained. Many approaches have been introduced to improve the performance of those devices including employing reducing agents, morphological control, compositional engineering, device engineering as well as interfacial engineering, but the impact of these approaches on the structural properties and device performance requires more systematic investigation. This will ultimately improve not only device performance but also device stability, which is critical to commercialization.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgements
T.H.C. and Y.R. contributed equally to this work. This study was supported by the Ministry of Science and ICT through the National Research Foundation, funded by the Korean government (NRF‐2021R1A2C3005401 and NRF‐2020M3D1A1110548), LG Display Co.
Biographies
Towhid H. Chowdhury is a researcher who works with the Department of Chemical Engineering, Pohang University of Science & Technology (POSTECH). Before joining POSTECH, he used to work with National Institute for Materials Science (NIMS), Japan (2018–2021), and Kyoto University, Japan (2021) as a postdoctoral researcher. He has a keen research interest in the development of low toxic perovskite semiconductors for optoelectronics and electrics applications.

Youjin Reo received her B.S. degree in Chemical Engineering from the Pohang University of Science and Technology (POSTECH) in 2020. She is currently a Ph.D. candidate in the Department of Chemical Engineering at POSTECH under the supervision of Prof. Yong‐Young Noh. Her research interests include the development of perovskite‐based semiconductors through solution‐process and thermal deposition, as well as their applications in transistors and circuits.

Abd. Rashid Mohd Yusoff received his Ph.D from Prof. Ivo Hümmelgen's group in the Departamento de Física at Universidade Federal do Paraná, Brazil. Then, he worked as a Postdoc Fellow, followed by Research Professor in Prof. Jin Jang's group at the Department of Information Display, Kyung Hee University. In 2018, he joined the Sêr SAM group at Swansea University as a Senior Research Fellow with a focus on perovskite photovoltaics until 2020. He is now at Pohang University of Science and Technology as Assistant Research Professor focusing on halide field effect transistor and light emitting diode.

Yong‐Young Noh is chair professor of the Department of Chemical Engineering, Pohang University of Science and Technology (POSTECH). He received Ph.D. in 2005 from GIST and then worked at the Cavendish Laboratory in Cambridge, UK, as a postdoctoral associate. Afterward, he worked at ETRI, as a senior researcher, at Hanbat National University as an assistant professor, and at Dongguk University as an associate professor. His research interest is the field of developing printable semiconductors including organic, carbon nanotubes, perovskite, metal halide and metal oxide for field‐effect transistors, photodiodes, and light‐emitting diodes.

Chowdhury T. H., Reo Y., Yusoff A. R. B. M., Noh Y.‐Y., Sn‐Based Perovskite Halides for Electronic Devices. Adv. Sci. 2022, 9, 2203749. 10.1002/advs.202203749
Contributor Information
Abd Rashid Bin Mohd Yusoff, Email: abdrashid@postech.ac.kr.
Yong‐Young Noh, Email: yynoh@postech.ac.kr.
References
- 1. Wang Z., Zhang Z., Xie L., Wang S., Yang C., Fang C., Hao F., Adv. Opt. Mater. 2022, 10, 2101822. [Google Scholar]
- 2. Kim H. P., Vasilopoulou M., Ullah H., Bibi S., Gavim A. E. X., Macedo A. G., da Silva W. J., Schneider F. K., Tahir A. A., Teridi M. A. M., Nanoscale 2020, 12, 7641. [DOI] [PubMed] [Google Scholar]
- 3. Vasilopoulou M., Yusoff M., Daboczi M., Conforto J., Gavim A. E. X., da Silva W. J., Macedo A. G., Soultati A., Pistolis G., Schneider F. K., Dong Y., Jacoutot P., Rotas G., Jang J., Vougioukalakis G. C., Chochos C. L., Kim J.‐S., Gasparini N., Nat. Commun. 2021, 12, 4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang H. P., Li S., Liu X., Shi Z., Fang X., He J. H., Adv. Mater. 2021, 33, 2003309. [DOI] [PubMed] [Google Scholar]
- 5. Zhang L., Miao J., Li J., Li Q., Adv. Funct. Mater. 2020, 30, 2003653. [Google Scholar]
- 6. Dong H., Zhang C., Liu X., Yao J., Zhao Y. S., Chem. Soc. Rev. 2020, 49, 951. [DOI] [PubMed] [Google Scholar]
- 7. Vasilopoulou M., Kim B. S., Kim H. P., da Silva W. J., Schneider F. K., Mat Teridi M. A., Gao P., Mohd Yusoff A. R. b., Nazeeruddin M. K., Nano Lett. 2020, 20, 5081. [DOI] [PubMed] [Google Scholar]
- 8. Park H., Ha C., Lee J.‐H., J. Mater. Chem. A 2020, 8, 24353. [Google Scholar]
- 9. Tian J., Xue Q., Yao Q., Li N., Brabec C. J., Yip H. L., Adv. Energy Mater. 2020, 10, 2000183. [Google Scholar]
- 10. Jeong J., Kim M., Seo J., Lu H., Ahlawat P., Mishra A., Yang Y., Hope M. A., Eickemeyer F. T., Kim M., Yoon Y. J., Choi I. W., Darwich B. P., Choi S. J., Jo Y., Lee J. H., Walker B., Zakeeruddin S. M., Emsley L., Rothlisberger U., Hagfeldt A., Kim D. S., Grätzel M., Kim J. Y., Nature 2021, 592, 381. [DOI] [PubMed] [Google Scholar]
- 11. Kang D. H., Park N. G., Adv. Mater. 2019, 31, 1805214. [Google Scholar]
- 12. Aydin E., De Bastiani M., De Wolf S., Adv. Mater. 2019, 31, 1900428. [DOI] [PubMed] [Google Scholar]
- 13. Wolff C. M., Caprioglio P., Stolterfoht M., Neher D., Adv. Mater. 2019, 31, 1902762. [DOI] [PubMed] [Google Scholar]
- 14. Liu W. W., Wu T. H., Liu M. C., Niu W. J., Chueh Y. L., Adv. Mater. Interfaces 2019, 6, 1801758. [Google Scholar]
- 15. Raman R. K., Gurusamy Thangavelu S. A., Venkataraj S., Krishnamoorthy A., Renewable Sustainable Energy Rev. 2021, 151, 111608. [Google Scholar]
- 16. Gao W., Chen C., Ran C., Zheng H., Dong H., Xia Y., Chen Y., Huang W., Adv. Funct. Mater. 2020, 30, 2000794. [Google Scholar]
- 17. Mariyappan P., Chowdhury T. H., Subashchandran S., Bedja I., Ghaithan H. M., Islam A., Sustainable Energy Fuels 2020, 4, 5042. [Google Scholar]
- 18. Chiara R., Morana M., Malavasi L., ChemPlusChem 2021, 86, 879. [DOI] [PubMed] [Google Scholar]
- 19. Abdel‐Shakour M., Chowdhury T. H., Matsuishi K., Bedja I., Moritomo Y., Islam A., Sol. RRL 2021, 5, 2000606. [Google Scholar]
- 20. Liu A., Zhu H., Bai S., Reo Y., Zou T., Kim M.‐G., Noh Y.‐Y., Nat. Electron. 2022, 5, 78. [Google Scholar]
- 21. Scaife D. E., Weller P. F., Fisher W. G., J. Solid State Chem. 1974, 9, 308. [Google Scholar]
- 22. Clark S. J., Flint C. D., Donaldson J. D., J. Phys. Chem. Solids 1981, 42, 133. [Google Scholar]
- 23. Parry D. E., Tricker M. J., Donaldson J. D., J. Solid State Chem. 1979, 28, 401. [Google Scholar]
- 24. Yamada K., Kuranaga Y., Ueda K., Shusaku G., Tsutomu O., Yoshihiro F., Bull. Chem. Soc. Jpn. 1998, 71, 127. [Google Scholar]
- 25. Yamada K., Matsui T., Tsuritani T., Okuda T., Ichiba S., Z. Naturforsch., A: Phys. Sci. 1990, 45, 307. [Google Scholar]
- 26. Yamada K., Nose S., Umehara T., Okuda T., Ichiba S., Bull. Chem. Soc. Jpn. 1988, 61, 4265. [Google Scholar]
- 27. Mitzi D. B., Dimitrakopoulos C. D., Kosbar L. L., Chem. Mater. 2001, 13, 3728. [Google Scholar]
- 28. Stoumpos C. C., Malliakas C. D., Kanatzidis M. G., Inorg. Chem. 2013, 52, 9019. [DOI] [PubMed] [Google Scholar]
- 29. Dong H., Ran C., Gao W., Sun N., Liu X., Xia Y., Chen Y., Huang W., Adv. Energy Mater. 2022, 12, 2102213. [Google Scholar]
- 30. Imran T., Rauf S., Raza H., Aziz L., Chen R., Liu S., Wang J., Ahmad M. A., Zhang S., Zhang Y., Liu Z., Chen W., Adv. Energy Mater. 2022, 12, 2200305. [Google Scholar]
- 31. Li M., Li F., Gong J., Zhang T., Gao F., Zhang W.‐H., Liu M., Small Struct. 2022, 3, 2100102. [Google Scholar]
- 32. Wang J., Gao Z., Yang J., Lv M., Chen H., Xue D.‐J., Meng X., Yang S., Adv. Energy Mater. 2021, 11, 2102131. [Google Scholar]
- 33. Cao F., Li L., Adv. Funct. Mater. 2021, 31, 2008275. [Google Scholar]
- 34. Gao Y., Pan Y., Zhou F., Niu G., Yan C., J. Mater. Chem. A 2021, 9, 11931. [Google Scholar]
- 35. Kieslich G., Sun S., Cheetham A. K., Chem. Sci. 2015, 6, 3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Weng D.‐F., Wang Z.‐M., Gao S., Chem. Soc. Rev. 2011, 40, 3157. [DOI] [PubMed] [Google Scholar]
- 37. Goldschmidt V. M., Naturwissenschaften 1926, 14, 477. [Google Scholar]
- 38. Mitzi D. B., J. Chem. Soc., Dalton Trans. 2001, 1. [Google Scholar]
- 39. Shannon R. D., Acta Crystallogr., Sect. A: Found. Adv. 1976, 32, 751. [Google Scholar]
- 40. Green M. A., Ho‐Baillie A., Snaith H. J., Nat. Photonics 2014, 8, 506. [Google Scholar]
- 41. Kieslich G., Sun S., Cheetham A. K., Chem. Sci. 2014, 5, 4712. [Google Scholar]
- 42. Gao P., Bin Mohd Yusoff A. R., Nazeeruddin M. K., Nat. Commun. 2018, 9, 5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Correa‐Baena J.‐P., Saliba M., Buonassisi T., Grätzel M., Abate A., Tress W., Hagfeldt A., Science 2017, 358, 739. [DOI] [PubMed] [Google Scholar]
- 44. Gao W., Ran C., Li J., Dong H., Jiao B., Zhang L., Lan X., Hou X., Wu Z., J. Phys. Chem. Lett. 2018, 9, 6999. [DOI] [PubMed] [Google Scholar]
- 45. Mitzi D., Wang S., Feild C., Chess C., Guloy A., Science 1995, 267, 1473. [DOI] [PubMed] [Google Scholar]
- 46. Even J., Pedesseau L., Jancu J. M., Katan C., Phys. Status Solidi RRL 2014, 8, 31. [Google Scholar]
- 47. Takahashi Y., Obara R., Lin Z.‐Z., Takahashi Y., Naito T., Inabe T., Ishibashi S., Terakura K., Dalton Trans. 2011, 40, 5563. [DOI] [PubMed] [Google Scholar]
- 48. Umari P., Mosconi E., De Angelis F., Sci. Rep. 2014, 4, 4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laurita G., Fabini D., Stoumpos C., Kanatzidis M., Seshadri R., Chem. Sci. 2017, 8, 5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parrott E. S., Milot R. L., Stergiopoulos T., Snaith H. J., Johnston M. B., Herz L. M., J. Phys. Chem. Lett. 2016, 7, 1321. [DOI] [PubMed] [Google Scholar]
- 51. Sharma V. K., Kanchana V., Gupta M. K., Mittal R., J. Solid State Chem. 2020, 290, 121541. [Google Scholar]
- 52. Dawson M., Ribeiro, C. , Morelli, M. R. , Mater. Res. 2022, 25, e20210441. [Google Scholar]
- 53. Mitzi D. B., Liang K., J. Solid State Chem. 1997, 134, 376. [Google Scholar]
- 54. Schueller E. C., Laurita G., Fabini D. H., Stoumpos C. C., Kanatzidis M. G., Seshadri R., Inorg. Chem. 2018, 57, 695. [DOI] [PubMed] [Google Scholar]
- 55. Kahmann S., Nazarenko O., Shao S., Hordiichuk O., Kepenekian M., Even J., Kovalenko M. V., Blake G. R., Loi M. A., ACS Energy Lett. 2020, 5, 2512. [Google Scholar]
- 56. Rameez M., Lin E. Y.‐R., Raghunath P., Narra S., Song D., Lin M.‐C., Hung C.‐H., Diau E. W.‐G., ACS Appl. Mater. Interfaces 2020, 12, 21739. [DOI] [PubMed] [Google Scholar]
- 57. Chung I., Song J.‐H., Im J., Androulakis J., Malliakas C. D., Li H., Freeman A. J., Kenney J. T., Kanatzidis M. G., J. Am. Chem. Soc. 2012, 134, 8579. [DOI] [PubMed] [Google Scholar]
- 58. Kubicki D. J., Prochowicz D., Salager E., Rakhmatullin A., Grey C. P., Emsley L., Stranks S. D., J. Am. Chem. Soc. 2020, 142, 7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. da Silva E. L., Skelton J. M., Parker S. C., Walsh A., Phys. Rev. B 2015, 91, 144107. [Google Scholar]
- 60. Borriello I., Cantele G., Ninno D., Phys. Rev. B 2008, 77, 235214. [Google Scholar]
- 61. Huang L.‐y., Lambrecht W. R. L., Phys. Rev. B 2013, 88, 165203. [Google Scholar]
- 62. Stroppa A., Di Sante D., Barone P., Bokdam M., Kresse G., Franchini C., Whangbo M.‐H., Picozzi S., Nat. Commun. 2014, 5, 5900. [DOI] [PubMed] [Google Scholar]
- 63. Giustino F., Snaith H. J., ACS Energy Lett. 2016, 1, 1233. [Google Scholar]
- 64. Shockley W., Queisser H. J., J. Appl. Phys. 1961, 32, 510. [Google Scholar]
- 65. Drago R. S., J. Phys. Chem. 1958, 62, 353. [Google Scholar]
- 66. Prasanna R., Gold‐Parker A., Leijtens T., Conings B., Babayigit A., Boyen H.‐G., Toney M. F., McGehee M. D., J. Am. Chem. Soc. 2017, 139, 11117. [DOI] [PubMed] [Google Scholar]
- 67. Hao F., Stoumpos C. C., Cao D. H., Chang R. P. H., Kanatzidis M. G., Nat. Photonics 2014, 8, 489. [Google Scholar]
- 68. Noel N. K., Stranks S. D., Abate A., Wehrenfennig C., Guarnera S., Haghighirad A.‐A., Sadhanala A., Eperon G. E., Pathak S. K., Johnston M. B., Petrozza A., Herz L. M., Snaith H. J., Energy Environ. Sci. 2014, 7, 3061. [Google Scholar]
- 69. Xiao Z., Song Z., Yan Y., Adv. Mater. 2019, 31, 1803792. [DOI] [PubMed] [Google Scholar]
- 70. Xie G., Xu L., Sun L., Xiong Y., Wu P., Hu B., J. Mater. Chem. A 2019, 7, 5779. [Google Scholar]
- 71. Xiao Z., Yan Y., Adv. Energy Mater. 2017, 7, 1701136. [Google Scholar]
- 72. Euvrard J., Yan Y., Mitzi D. B., Nat. Rev. Mater. 2021, 6, 531. [Google Scholar]
- 73. Xu P., Chen S., Xiang H.‐J., Gong X.‐G., Wei S.‐H., Chem. Mater. 2014, 26, 6068. [Google Scholar]
- 74. Shi T., Zhang H.‐S., Meng W., Teng Q., Liu M., Yang X., Yan Y., Yip H.‐L., Zhao Y.‐J., J. Mater. Chem. A 2017, 5, 15124. [Google Scholar]
- 75. Kumar M. H., Dharani S., Leong W. L., Boix P. P., Prabhakar R. R., Baikie T., Shi C., Ding H., Ramesh R., Asta M., Graetzel M., Mhaisalkar S. G., Mathews N., Adv. Mater. 2014, 26, 7122. [DOI] [PubMed] [Google Scholar]
- 76. Xiao Z., Meng W., Wang J., Mitzi D. B., Yan Y., Mater. Horiz. 2017, 4, 206. [Google Scholar]
- 77. Meggiolaro D., Ricciarelli D., Alasmari A. A., Alasmary F. A. S., De Angelis F., J. Phys. Chem. Lett. 2020, 11, 3546. [DOI] [PubMed] [Google Scholar]
- 78. Ke W., Stoumpos C. C., Spanopoulos I., Mao L., Chen M., Wasielewski M. R., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 14800. [DOI] [PubMed] [Google Scholar]
- 79. Ke W., Stoumpos C. C., Zhu M., Mao L., Spanopoulos I., Liu J., Kontsevoi O. Y., Chen M., Sarma D., Zhang Y., Wasielewski M. R., Kanatzidis M. G., Sci. Adv. 2017, 3, e1701293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Leijtens T., Prasanna R., Gold‐Parker A., Toney M. F., McGehee M. D., ACS Energy Lett. 2017, 2, 2159. [Google Scholar]
- 81. Freysoldt C., Grabowski B., Hickel T., Neugebauer J., Kresse G., Janotti A., Van de Walle C. G., Rev. Mod. Phys. 2014, 86, 253. [Google Scholar]
- 82. Haque M. A., Hernandez L. H., Davaasuren B., Villalva D. R., Troughton J., Baran D., Adv. Energy Sustainability Res. 2020, 1, 2000033. [Google Scholar]
- 83. Mitzi D. B., Feild C. A., Schlesinger Z., Laibowitz R. B., J. Solid State Chem. 1995, 114, 159. [Google Scholar]
- 84. Takahashi Y., Hasegawa H., Takahashi Y., Inabe T., J. Solid State Chem. 2013, 205, 39. [Google Scholar]
- 85. Gupta S., Bendikov T., Hodes G., Cahen D., ACS Energy Lett. 2016, 1, 1028. [Google Scholar]
- 86. Gupta S., Cahen D., Hodes G., J. Phys. Chem. C 2018, 122, 13926. [Google Scholar]
- 87. Liao W., Zhao D., Yu Y., Grice C. R., Wang C., Cimaroli A. J., Schulz P., Meng W., Zhu K., Xiong R.‐G., Yan Y., Adv. Mater. 2016, 28, 9333. [DOI] [PubMed] [Google Scholar]
- 88. Milot R. L., Klug M. T., Davies C. L., Wang Z., Kraus H., Snaith H. J., Johnston M. B., Herz L. M., Adv. Mater. 2018, 30, 1804506. [DOI] [PubMed] [Google Scholar]
- 89. Savill K. J., Ulatowski A. M., Farrar M. D., Johnston M. B., Snaith H. J., Herz L. M., Adv. Funct. Mater. 2020, 30, 2005594. [Google Scholar]
- 90. Zhou S., Zhu S., Guan J., Wang R., Zheng W., Gao P., Lu X., J. Phys. Chem. Lett. 2021, 12, 10996. [DOI] [PubMed] [Google Scholar]
- 91. Lee B., Stoumpos C. C., Zhou N., Hao F., Malliakas C., Yeh C.‐Y., Marks T. J., Kanatzidis M. G., Chang R. P. H., J. Am. Chem. Soc. 2014, 136, 15379. [DOI] [PubMed] [Google Scholar]
- 92. Liu A., Zhu H., Reo Y., Kim M.‐G., Chu H. Y., Lim J. H., Kim H.‐J., Ning W., Bai S., Noh Y.‐Y., Cell Rep. Phys. Sci. 2022, 3, 100812. [Google Scholar]
- 93. Saparov B., Sun J.‐P., Meng W., Xiao Z., Duan H.‐S., Gunawan O., Shin D., Hill I. G., Yan Y., Mitzi D. B., Chem. Mater. 2016, 28, 2315. [Google Scholar]
- 94. Wang A., Yan X., Zhang M., Sun S., Yang M., Shen W., Pan X., Wang P., Deng Z., Chem. Mater. 2016, 28, 8132. [Google Scholar]
- 95. Zhu H., Liu A., Shim K. I., Jung H., Zou T., Reo Y., Kim H., Han J. W., Chen Y., Chu H. Y., Lim J. H., Kim H.‐J., Bai S., Noh Y.‐Y., Nat. Commun. 2022, 13, 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hsu S.‐N., Zhao W., Gao Y., Akriti, Segovia M., Xu X., Boudouris B. W., Dou L., Nano Lett. 2021, 21, 7839. [DOI] [PubMed] [Google Scholar]
- 97. Shao S., Talsma W., Pitaro M., Dong J., Kahmann S., Rommens A. J., Portale G., Loi M. A., Adv. Funct. Mater. 2021, 31, 2008478. [Google Scholar]
- 98. Begum R., Parida M. R., Abdelhady A. L., Murali B., Alyami N. M., Ahmed G. H., Hedhili M. N., Bakr O. M., Mohammed O. F., J. Am. Chem. Soc. 2017, 139, 731. [DOI] [PubMed] [Google Scholar]
- 99. Liu W., Lin Q., Li H., Wu K., Robel I., Pietryga J. M., Klimov V. I., J. Am. Chem. Soc. 2016, 138, 14954. [DOI] [PubMed] [Google Scholar]
- 100. Saliba M., Matsui T., Domanski K., Seo J.‐Y., Ummadisingu A., Zakeeruddin S. M., Correa‐Baena J.‐P., Tress W. R., Abate A., Hagfeldt A., Grätzel M., Science 2016, 354, 206. [DOI] [PubMed] [Google Scholar]
- 101. Zhao W., Yao Z., Yu F., Yang D., Liu S., Adv. Sci. 2018, 5, 1700131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hou L., Zhu Y., Zhu J., Gong Y., Li C., J. Mater. Chem. C 2020, 8, 8502. [Google Scholar]
- 103. Bowman A. R., Klug M. T., Doherty T. A. S., Farrar M. D., Senanayak S. P., Wenger B., Divitini G., Booker E. P., Andaji‐Garmaroudi Z., Macpherson S., Ruggeri E., Sirringhaus H., Snaith H. J., Stranks S. D., ACS Energy Lett. 2019, 4, 2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Takahashi Y., Obara R., Nakagawa K., Nakano M., Tokita J.‐y., Inabe T., Chem. Mater. 2007, 19, 6312. [Google Scholar]
- 105. Zhang R., Mao X., Cheng P., Yang Y., Yang S., Wumaier T., Deng W., Han K., J. Energy Chem. 2019, 36, 1. [Google Scholar]
- 106. Reo Y., Zhu H., Go J.‐Y., Shim K. I., Liu A., Zou T., Jung H., Kim H., Hong J., Han J. W., Noh Y.‐Y., Chem. Mater. 2021, 33, 2498. [Google Scholar]
- 107. Go J.‐Y., Zhu H., Reo Y., Kim H., Liu A., Noh Y.‐Y., ACS Appl. Mater. Interfaces 2022, 14, 9363. [DOI] [PubMed] [Google Scholar]
- 108. Smith P. J., Chemistry of Tin, Springer, New York: 2012. [Google Scholar]
- 109. Hao F., Stoumpos C. C., Guo P., Zhou N., Marks T. J., Chang R. P. H., Kanatzidis M. G., J. Am. Chem. Soc. 2015, 137, 11445. [DOI] [PubMed] [Google Scholar]
- 110. Saidaminov M. I., Spanopoulos I., Abed J., Ke W., Wicks J., Kanatzidis M. G., Sargent E. H., ACS Energy Lett. 2020, 5, 1153. [Google Scholar]
- 111. Pascual J., Nasti G., Aldamasy M. H., Smith J. A., Flatken M., Phung N., Di Girolamo D., Turren‐Cruz S.‐H., Li M., Dallmann A., Avolio R., Abate A., Mater. Adv. 2020, 1, 1066. [Google Scholar]
- 112. Zhou Y., Poli I., Meggiolaro D., De Angelis F., Petrozza A., Nat. Rev. Mater. 2021, 6, 986. [Google Scholar]
- 113. Di Girolamo D., Pascual J., Aldamasy M. H., Iqbal Z., Li G., Radicchi E., Li M., Turren‐Cruz S.‐H., Nasti G., Dallmann A., De Angelis F., Abate A., ACS Energy Lett. 2021, 6, 959. [Google Scholar]
- 114. Cao J., Yan F., Energy Environ. Sci. 2021, 14, 1286. [Google Scholar]
- 115. Wang T., Yan F., Chem. ‐ Asian J. 2020, 15, 1524. [DOI] [PubMed] [Google Scholar]
- 116. Chung I., Lee B., He J., Chang R. P. H., Kanatzidis M. G., Nature 2012, 485, 486. [DOI] [PubMed] [Google Scholar]
- 117. Koh T. M., Krishnamoorthy T., Yantara N., Shi C., Leong W. L., Boix P. P., Grimsdale A. C., Mhaisalkar S. G., Mathews N., J. Mater. Chem. A 2015, 3, 14996. [Google Scholar]
- 118. Ma L., Hao F., Stoumpos C. C., Phelan B. T., Wasielewski M. R., Kanatzidis M. G., J. Am. Chem. Soc. 2016, 138, 14750. [DOI] [PubMed] [Google Scholar]
- 119. Marshall K. P., Walker M., Walton R. I., Hatton R. A., Nat. Energy 2016, 1, 16178. [Google Scholar]
- 120. Song T.‐B., Yokoyama T., Aramaki S., Kanatzidis M. G., ACS Energy Lett. 2017, 2, 897. [Google Scholar]
- 121. Gu F., Ye S., Zhao Z., Rao H., Liu Z., Bian Z., Huang C., Sol. RRL 2018, 2, 1800136. [Google Scholar]
- 122. Kayesh M. E., Chowdhury T. H., Matsuishi K., Kaneko R., Kazaoui S., Lee J.‐J., Noda T., Islam A., ACS Energy Lett. 2018, 3, 1584. [Google Scholar]
- 123. Li F., Zhang C., Huang J.‐H., Fan H., Wang H., Wang P., Zhan C., Liu C.‐M., Li X., Yang L.‐M., Song Y., Jiang K.‐J., Angew. Chem., Int. Ed. 2019, 58, 6688. [DOI] [PubMed] [Google Scholar]
- 124. Song T.‐B., Yokoyama T., Stoumpos C. C., Logsdon J., Cao D. H., Wasielewski M. R., Aramaki S., Kanatzidis M. G., J. Am. Chem. Soc. 2017, 139, 836. [DOI] [PubMed] [Google Scholar]
- 125. Cao J., Tai Q., You P., Tang G., Wang T., Wang N., Yan F., J. Mater. Chem. A 2019, 7, 26580. [Google Scholar]
- 126. Li W., Li J., Li J., Fan J., Mai Y., Wang L., J. Mater. Chem. A 2016, 4, 17104. [Google Scholar]
- 127. Tai Q., Guo X., Tang G., You P., Ng T.‐W., Shen D., Cao J., Liu C.‐K., Wang N., Zhu Y., Lee C.‐S., Yan F., Angew. Chem., Int. Ed. 2019, 58, 806. [DOI] [PubMed] [Google Scholar]
- 128. Lin R., Xiao K., Qin Z., Han Q., Zhang C., Wei M., Saidaminov M. I., Gao Y., Xu J., Xiao M., Li A., Zhu J., Sargent E. H., Tan H., Nat. Energy 2019, 4, 864. [Google Scholar]
- 129. Pascual J., Flatken M., Félix R., Li G., Turren‐Cruz S.‐H., Aldamasy M. H., Hartmann C., Li M., Di Girolamo D., Nasti G., Hüsam E., Wilks R. G., Dallmann A., Bär M., Hoell A., Abate A., Angew. Chem., Int. Ed. 2021, 60, 21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dai Z., Lv T., Barbaud J., Tang W., Wang T., Qiao L., Chen H., Zheng R., Yang X., Han L., Sci. China Mater. 2021, 64, 2645. [Google Scholar]
- 131. Zhu H., Liu A., Shim K. I., Hong J., Han J. W., Noh Y.‐Y., Adv. Mater. 2020, 32, 2002717. [DOI] [PubMed] [Google Scholar]
- 132. Nakamura T., Yakumaru S., Truong M. A., Kim K., Liu J., Hu S., Otsuka K., Hashimoto R., Murdey R., Sasamori T., Kim H. D., Ohkita H., Handa T., Kanemitsu Y., Wakamiya A., Nat. Commun. 2020, 11, 3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Li D., Cheng H.‐C., Wang Y., Zhao Z., Wang G., Wu H., He Q., Huang Y., Duan X., Adv. Mater. 2017, 29, 1601959. [DOI] [PubMed] [Google Scholar]
- 134. Li F., Ma C., Wang H., Hu W., Yu W., Sheikh A. D., Wu T., Nat. Commun. 2015, 6, 8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Matsushima T., Terakawa S., Leyden M. R., Fujihara T., Qin C., Adachi C., J. Appl. Phys. 2019, 125, 235501. [Google Scholar]
- 136. Sutherland L. J., Weerasinghe H. C., Simon G. P., Adv. Energy Mater. 2021, 11, 2101383. [Google Scholar]
- 137. He Y., Abdellaoui I., Abdel‐Shakour M., Chowdhury T. H., Kamarudin M. A., Nogueira A. F., Shen Q., Hayase S., Islam A., Sakurai T., Jpn. J. Appl. Phys. 2021, 60, SBBF13. [Google Scholar]
- 138. Zhang X., Wang S., Zhu W., Cao Z., Wang A., Hao F., Adv. Funct. Mater. 2022, 32, 2108832. [Google Scholar]
- 139. Wu T., Liu X., Luo X., Lin X., Cui D., Wang Y., Segawa H., Zhang Y., Han L., Joule 2021, 5, 863. [Google Scholar]
- 140. Shi Z., Jayatissa A. H., Materials 2018, 11, 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Song Z., Watthage S. C., Phillips A. B., Heben M. J., J. Photonics Energy 2016, 6, 022001. [Google Scholar]
- 142. Zhu Z., Chueh C. C., Li N., Mao C., Jen A. K. Y., Adv. Mater. 2018, 30, 1703800. [DOI] [PubMed] [Google Scholar]
- 143. Dixit H., Boro B., Ghosh S., Paul M., Kumar A., Singh T., Phys. Status Solidi RRL 2022, 219, 2100823. [Google Scholar]
- 144. Huang D., Goh T., Kong J., Zheng Y., Zhao S., Xu Z., Taylor A. D., Nanoscale 2017, 9, 4236. [DOI] [PubMed] [Google Scholar]
- 145. Kim H. D., Miyamoto Y., Kubota H., Yamanari T., Ohkita H., Chem. Lett. 2017, 46, 253. [Google Scholar]
- 146. Handa T., Yamada T., Kubota H., Ise S., Miyamoto Y., Kanemitsu Y., J. Phys. Chem. C 2017, 121, 16158. [Google Scholar]
- 147. Li F., Zhang C., Huang J. H., Fan H., Wang H., Wang P., Zhan C., Liu C. M., Li X., Yang L. M., Angew. Chem., Int. Ed. 2019, 58, 6688. [DOI] [PubMed] [Google Scholar]
- 148. Yokoyama T., Cao D. H., Stoumpos C. C., Song T.‐B., Sato Y., Aramaki S., Kanatzidis M. G., J. Phys. Chem. Lett. 2016, 7, 776. [DOI] [PubMed] [Google Scholar]
- 149. Wang P., Li F., Jiang K.‐J., Zhang Y., Fan H., Zhang Y., Miao Y., Huang J.‐H., Gao C., Zhou X., Wang F., Yang L.‐M., Zhan C., Song Y., Adv. Sci. 2020, 7, 1903047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yang W.‐F., Cao J.‐J., Dong C., Li M., Tian Q.‐S., Wang Z.‐K., Liao L.‐S., Appl. Phys. Lett. 2021, 118, 023501. [Google Scholar]
- 151. Shahbazi S., Li M.‐Y., Fathi A., Diau E. W.‐G., ACS Energy Lett. 2020, 5, 2508. [Google Scholar]
- 152. Dang Y., Zhou Y., Liu X., Ju D., Xia S., Xia H., Tao X., Angew. Chem., Int. Ed. 2016, 55, 3447. [DOI] [PubMed] [Google Scholar]
- 153. You J., Wang M., Xu C., Yao Y., Zhao X., Liu D., Dong J., Guo P., Xu G., Luo C., Zhong Y., Song Q., Sustainable Energy Fuels 2021, 5, 2660. [Google Scholar]
- 154. Li F., Fan H., Zhang J., Huang J.‐H., Wang P., Gao C., Yang L.‐M., Zhu Z., Jen A. K.‐Y., Song Y., Jiang K.‐J., Sol. RRL 2019, 3, 1900285. [Google Scholar]
- 155. Wang C., Gu F., Zhao Z., Rao H., Qiu Y., Cai Z., Zhan G., Li X., Sun B., Yu X., Zhao B., Liu Z., Bian Z., Huang C., Adv. Mater. 2020, 32, 1907623. [DOI] [PubMed] [Google Scholar]
- 156. Wang T., Tai Q., Guo X., Cao J., Liu C.‐K., Wang N., Shen D., Zhu Y., Lee C.‐S., Yan F., ACS Energy Lett. 2020, 5, 1741. [Google Scholar]
- 157. Zhu Z., Jiang X., Yu D., Yu N., Ning Z., Mi Q., ACS Energy Lett. 2022, 7, 2079. [Google Scholar]
- 158. Jokar E., Chien C.‐H., Fathi A., Rameez M., Chang Y.‐H., Diau E. W.‐G., Energy Environ. Sci. 2018, 11, 2353. [Google Scholar]
- 159. Wu T., Liu X., He X., Wang Y., Meng X., Noda T., Yang X., Han L., Sci. China: Chem. 2020, 63, 107. [Google Scholar]
- 160. Meng X., Lin J., Liu X., He X., Wang Y., Noda T., Wu T., Yang X., Han L., Adv. Mater. 2019, 31, 1903721. [DOI] [PubMed] [Google Scholar]
- 161. Chowdhury T. H., Kaneko R., Kaneko T., Sodeyama K., Lee J.‐J., Islam A., Chem. Eng. J. 2022, 431, 133745. [Google Scholar]
- 162. Meng X., Wang Y., Lin J., Liu X., He X., Barbaud J., Wu T., Noda T., Yang X., Han L., Joule 2020, 4, 902. [Google Scholar]
- 163. Dai Z., Tang W., Wang T., Lv T., Luo X., Cui D., Sun R., Qiao L., Chen H., Zheng R., Yang X., Han L., Sci. China Mater. 2021, 64, 1849. [Google Scholar]
- 164. Jiang X., Li H., Zhou Q., Wei Q., Wei M., Jiang L., Wang Z., Peng Z., Wang F., Zang Z., Xu K., Hou Y., Teale S., Zhou W., Si R., Gao X., Sargent E. H., Ning Z., J. Am. Chem. Soc. 2021, 143, 10970. [DOI] [PubMed] [Google Scholar]
- 165. Cao K., Cheng Y., Chen J., Huang Y., Ge M., Qian J., Liu L., Feng J., Chen S., Huang W., ACS Appl. Mater. Interfaces 2020, 12, 41454. [DOI] [PubMed] [Google Scholar]
- 166. Liu G., Liu C., Lin Z., Yang J., Huang Z., Tan L., Chen Y., ACS Appl. Mater. Interfaces 2020, 12, 14049. [DOI] [PubMed] [Google Scholar]
- 167. Chowdhury T. H., Kayesh M. E., Lee J.‐J., Matsushita Y., Kazaoui S., Islam A., Sol. RRL 2019, 3, 1900245. [Google Scholar]
- 168. Kamarudin M. A., Hirotani D., Wang Z., Hamada K., Nishimura K., Shen Q., Toyoda T., Iikubo S., Minemoto T., Yoshino K., Hayase S., J. Phys. Chem. Lett. 2019, 10, 5277. [DOI] [PubMed] [Google Scholar]
- 169. Liu X., Wu T., Chen J.‐Y., Meng X., He X., Noda T., Chen H., Yang X., Segawa H., Wang Y., Han L., Energy Environ. Sci. 2020, 13, 2896. [Google Scholar]
- 170. Seo J.‐Y., Matsui T., Luo J., Correa‐Baena J.‐P., Giordano F., Saliba M., Schenk K., Ummadisingu A., Domanski K., Hadadian M., Hagfeldt A., Zakeeruddin S. M., Steiner U., Grätzel M., Abate A., Adv. Energy Mater. 2016, 6, 1600767. [Google Scholar]
- 171. Lin Z., Su Y., Dai R., Liu G., Yang J., Sheng W., Zhong Y., Tan L., Chen Y., ACS Appl. Mater. Interfaces 2021, 13, 15420. [DOI] [PubMed] [Google Scholar]
- 172. Xu R., Dong H., Li P., Cao X., Li H., Li J., Wu Z., ACS Appl. Mater. Interfaces 2021, 13, 33218. [DOI] [PubMed] [Google Scholar]
- 173. Li G., Su Z., Li M., Yang F., Aldamasy M. H., Pascual J., Yang F., Liu H., Zuo W., Di Girolamo D., Iqbal Z., Nasti G., Dallmann A., Gao X., Wang Z., Saliba M., Abate A., Adv. Energy Mater. 2021, 11, 2101539. [Google Scholar]
- 174. Boehm A. M., Liu T., Park S. M., Abtahi A., Graham K. R., ACS Appl. Mater. Interfaces 2020, 12, 5209. [DOI] [PubMed] [Google Scholar]
- 175. Abdel‐Shakour M., Chowdhury T. H., Matsuishi K., Karim M. A., He Y., Moritomo Y., Islam A., ACS Appl. Energy Mater. 2021, 4, 12515. [Google Scholar]
- 176. Chen M., Dong Q., Xiao C., Zheng X., Dai Z., Shi Y., Luther J. M., Padture N. P., ACS Energy Lett. 2022, 7, 2256. [Google Scholar]
- 177. Nguyen B. P., Jung H. R., Ryu K. Y., Kim K., Jo W., J. Phys. Chem. C 2019, 123, 30833. [Google Scholar]
- 178. Zhao Z., Gu F., Li Y., Sun W., Ye S., Rao H., Liu Z., Bian Z., Huang C., Adv. Sci. 2017, 4, 1700204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Liu X., Yan K., Tan D., Liang X., Zhang H., Huang W., ACS Energy Lett. 2018, 3, 2701. [Google Scholar]
- 180. Wan Z., Ren S., Lai H., Jiang Y., Wu X., Luo J., Wang Y., He R., Chen Q., Hao X., Wang Y., Wu L., Constantinou I., Zhang W.‐H., Zhang J., Zhao D., Adv. Mater. Interfaces 2021, 8, 2100135. [Google Scholar]
- 181. Zhang Z., Kumar Baranwal A., Razey Sahamir S., Kapil G., Sanehira Y., Chen M., Nishimura K., Ding C., Liu D., Li H., Li Y., Akmal Kamarudin M., Shen Q., Ripolles T. S., Bisquert J., Hayase S., Sol. RRL 2021, 5, 2100633. [Google Scholar]
- 182. Nishimura K., Kamarudin M. A., Hirotani D., Hamada K., Shen Q., Iikubo S., Minemoto T., Yoshino K., Hayase S., Nano Energy 2020, 74, 104858. [Google Scholar]
- 183. Zhang Z., Wang L., Kumar Baranwal A., Razey Sahamir S., Kapil G., Sanehira Y., Akmal Kamarudin M., Nishimura K., Ding C., Liu D., Li Y., Li H., Chen M., Shen Q., Ripolles T. S., Bisquert J., Hayase S., J. Energy Chem. 2022, 71, 604. [Google Scholar]
- 184. Weber S., Rath T., Kunert B., Resel R., Dimopoulos T., Trimmel G., Monatsh. Chem. 2019, 150, 1921. [Google Scholar]
- 185. Chen M., Kamarudin M. A., Baranwal A. K., Kapil G., Ripolles T. S., Nishimura K., Hirotani D., Sahamir S. R., Zhang Z., Ding C., Sanehira Y., Bisquert J., Shen Q., Hayase S., ACS Appl. Energy Mater. 2021, 4, 5615. [Google Scholar]
- 186. Chen M., Kapil G., Wang L., Razey Sahamir S., Baranwal A. K., Nishimura K., Sanehira Y., Zhang Z., Akmal Kamarudin M., Shen Q., Hayase S., Chem. Eng. J. 2022, 436, 135196. [Google Scholar]
- 187. Kayesh M. E., Matsuishi K., Kaneko R., Kazaoui S., Lee J.‐J., Noda T., Islam A., ACS Energy Lett. 2018, 4, 278. [Google Scholar]
- 188. Liu J., Ozaki M., Yakumaru S., Handa T., Nishikubo R., Kanemitsu Y., Saeki A., Murata Y., Murdey R., Wakamiya A., Angew. Chem., Int. Ed. 2018, 57, 13221. [DOI] [PubMed] [Google Scholar]
- 189. Konstantakou M., Stergiopoulos T., J. Mater. Chem. A 2017, 5, 11518. [Google Scholar]
- 190. Chen Z., Yu C., Shum K., Wang J. J., Pfenninger W., Vockic N., Midgley J., Kenney J. T., J. Lumin. 2012, 132, 345. [Google Scholar]
- 191. Marshall K. P., Walton R. I., Hatton R. A., J. Mater. Chem. A 2015, 3, 11631. [Google Scholar]
- 192. Marshall K., Walker M., Walton R., Hatton R., Nat. Energy 2016, 1, 16178. [Google Scholar]
- 193. Ban H., Zhang T., Gong X., Sun Q., Zhang X.‐L., Pootrakulchote N., Shen Y., Wang M., Sol. RRL 2021, 5, 2100069. [Google Scholar]
- 194. Ye T., Wang K., Hou Y., Yang D., Smith N., Magill B., Yoon J., Mudiyanselage R. R. H. H., Khodaparast G. A., Wang K., Priya S., J. Am. Chem. Soc. 2021, 143, 4319. [DOI] [PubMed] [Google Scholar]
- 195. Ye T., Wang X., Wang K., Ma S., Yang D., Hou Y., Yoon J., Wang K., Priya S., ACS Energy Lett. 2021, 6, 1480. [Google Scholar]
- 196. Ju M.‐G., Dai J., Ma L., Zeng X. C., J. Am. Chem. Soc. 2017, 139, 8038. [DOI] [PubMed] [Google Scholar]
- 197. Chen M., Ju M.‐G., Garces H. F., Carl A. D., Ono L. K., Hawash Z., Zhang Y., Shen T., Qi Y., Grimm R. L., Pacifici D., Zeng X. C., Zhou Y., Padture N. P., Nat. Commun. 2019, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Zhu P., Chen C., Gu S., Lin R., Zhu J., Sol. RRL 2018, 2, 1700224. [Google Scholar]
- 199. Li J., Huang J., Zhao A., Li Y., Wei M., J. Mater. Chem. C 2020, 8, 8840. [Google Scholar]
- 200. Li B., Di H., Chang B., Yin R., Fu L., Zhang Y.‐N., Yin L., Adv. Funct. Mater. 2021, 31, 2007447. [Google Scholar]
- 201. Ji L., Liu D., Wang Y., Zhang T., Chen H., Li Y., Zheng H., Yang Y., Chen Z. D., Yang W., Chem. Eng. J. 2020, 402, 125133. [Google Scholar]
- 202. Shao S., Liu J., Portale G., Fang H.‐H., Blake G. R., ten Brink G. H., Koster L. J. A., Loi M. A., Adv. Energy Mater. 2018, 8, 1702019. [Google Scholar]
- 203. Shao S., Nijenhuis M., Dong J., Kahmann S., ten Brink G. H., Portale G., Loi M. A., J. Mater. Chem. A 2021, 9, 10095. [Google Scholar]
- 204. Wang F., Jiang X., Chen H., Shang Y., Liu H., Wei J., Zhou W., He H., Liu W., Ning Z., Joule 2018, 2, 2732. [Google Scholar]
- 205. Chen M., Dong Q., Eickemeyer F. T., Liu Y., Dai Z., Carl A. D., Bahrami B., Chowdhury A. H., Grimm R. L., Shi Y., Qiao Q., Zakeeruddin S. M., Grätzel M., Padture N. P., ACS Energy Lett. 2020, 5, 2223. [Google Scholar]
- 206. Jiang X., Wang F., Wei Q., Li H., Shang Y., Zhou W., Wang C., Cheng P., Chen Q., Chen L., Ning Z., Nat. Commun. 2020, 11, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yu B.‐B., Chen Z., Zhu Y., Wang Y., Han B., Chen G., Zhang X., Du Z., He Z., Adv. Mater. 2021, 33, 2102055. [DOI] [PubMed] [Google Scholar]
- 208. Wang T., Loi H.‐L., Cao J., Qin Z., Guan Z., Xu Y., Cheng H., Li M. G., Lee C.‐S., Lu X., Yan F., Adv. Sci. 2022, 9, 2200242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Ran C., Xi J., Gao W., Yuan F., Lei T., Jiao B., Hou X., Wu Z., ACS Energy Lett. 2018, 3, 713. [Google Scholar]
- 210. Liao M., Yu B.‐B., Jin Z., Chen W., Zhu Y., Zhang X., Yao W., Duan T., Djerdj I., He Z., ChemSusChem 2019, 12, 5007. [DOI] [PubMed] [Google Scholar]
- 211. Qiu J., Xia Y., Zheng Y., Hui W., Gu H., Yuan W., Yu H., Chao L., Niu T., Yang Y., Gao X., Chen Y., Huang W., ACS Energy Lett. 2019, 4, 1513. [Google Scholar]
- 212. Li M., Zuo W.‐W., Yang Y.‐G., Aldamasy M. H., Wang Q., Cruz S. H. T., Feng S.‐L., Saliba M., Wang Z.‐K., Abate A., ACS Energy Lett. 2020, 5, 1923. [Google Scholar]
- 213. Chen W., Wu Y., Yue Y., Liu J., Zhang W., Yang X., Chen H., Bi E., Ashraful I., Grätzel M., Science 2015, 350, 944. [DOI] [PubMed] [Google Scholar]
- 214. Ran C., Gao W., Li J., Xi J., Li L., Dai J., Yang Y., Gao X., Dong H., Jiao B., Spanopoulos I., Malliakas C. D., Hou X., Kanatzidis M. G., Wu Z., Joule 2019, 3, 3072. [Google Scholar]
- 215. Liu X., Wu T., Luo X., Wang H., Furue M., Bessho T., Zhang Y., Nakazaki J., Segawa H., Han L., ACS Energy Lett. 2022, 7, 425. [Google Scholar]
- 216. Kang B., Ge F., Qiu L., Cho K., Adv. Electron. Mater. 2017, 3, 1600240. [Google Scholar]
- 217. Li D., Wang G., Cheng H.‐C., Chen C.‐Y., Wu H., Liu Y., Huang Y., Duan X., Nat. Commun. 2016, 7, 11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Zaumseil J., Sirringhaus H., Chem. Rev. 2007, 107, 1296. [DOI] [PubMed] [Google Scholar]
- 219. McKee R. A., Walker F. J., Chisholm M. F., Phys. Rev. Lett. 1998, 81, 3014. [Google Scholar]
- 220. Meiners L. G., J. Vac. Sci. Technol. 1981, 19, 373. [Google Scholar]
- 221. Green M. L., Sorsch T. W., Timp G. L., Muller D. A., Weir B. E., Silverman P. J., Moccio S. V., Kim Y. O., Microelectron. Eng. 1999, 48, 25. [Google Scholar]
- 222. Liu A., Zhu H., Sun H., Xu Y., Noh Y.‐Y., Adv. Mater. 2018, 30, 1706364. [DOI] [PubMed] [Google Scholar]
- 223. Nomura K., Ohta H., Takagi A., Kamiya T., Hirano M., Hosono H., Nature 2004, 432, 488. [DOI] [PubMed] [Google Scholar]
- 224. Liu A., Zhu H., Noh Y.‐Y., Mater. Sci. Eng., R 2019, 135, 85. [Google Scholar]
- 225. Sirringhaus H., Adv. Mater. 2014, 26, 1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Khim D., Han H., Baeg K.‐J., Kim J., Kwak S.‐W., Kim D.‐Y., Noh Y.‐Y., Adv. Mater. 2013, 25, 4302. [DOI] [PubMed] [Google Scholar]
- 227. Sirringhaus H., Kawase T., Friend R. H., Shimoda T., Inbasekaran M., Wu W., Woo E. P., Science 2000, 290, 2123. [DOI] [PubMed] [Google Scholar]
- 228. Giorgi G., Fujisawa J.‐I., Segawa H., Yamashita K., J. Phys. Chem. Lett. 2013, 4, 4213. [DOI] [PubMed] [Google Scholar]
- 229. Ponseca C. S., Savenije T. J., Abdellah M., Zheng K., Yartsev A., Pascher T., Harlang T., Chabera P., Pullerits T., Stepanov A., Wolf J.‐P., Sundström V., J. Am. Chem. Soc. 2014, 136, 5189. [DOI] [PubMed] [Google Scholar]
- 230. Zhu H., Liu A., Noh Y.‐Y., Nat. Electron. 2020, 3, 662. [Google Scholar]
- 231. Zhu H., Liu A., Noh Y.‐Y., J. Inf. Disp. 2021, 22, 257. [Google Scholar]
- 232. Liu Y., Chen P.‐A., Hu Y., J. Mater. Chem. C 2020, 8, 16691. [Google Scholar]
- 233. Zhang C., Chen P., Hu W., Small 2016, 12, 1252. [DOI] [PubMed] [Google Scholar]
- 234. Kim M., Ryu S. U., Park S. A., Choi K., Kim T., Chung D., Park T., Adv. Funct. Mater. 2020, 30, 1904545. [Google Scholar]
- 235. Yang T., Wu Q., Dai F., Huang K., Xu H., Liu C., Chen C., Hu S., Liang X., Liu X., Noh Y.‐Y., Liu C., Adv. Funct. Mater. 2020, 30, 1903889. [Google Scholar]
- 236. Zhu H., Shin E.‐S., Liu A., Ji D., Xu Y., Noh Y.‐Y., Adv. Funct. Mater. 2020, 30, 1904588. [Google Scholar]
- 237. Lin Y.‐H., Pattanasattayavong P., Anthopoulos T. D., Adv. Mater. 2017, 29, 1702838. [DOI] [PubMed] [Google Scholar]
- 238. Matsushima T., Hwang S., Sandanayaka A. S. D., Qin C., Terakawa S., Fujihara T., Yahiro M., Adachi C., Adv. Mater. 2016, 28, 10275. [DOI] [PubMed] [Google Scholar]
- 239. Senanayak S. P., Abdi‐Jalebi M., Kamboj V. S., Carey R., Shivanna R., Tian T., Schweicher G., Wang J., Giesbrecht N., Nuzzo D. D., Beere H. E., Docampo P., Ritchie D. A., Fairen‐Jimenez D., Friend R. H., Sirringhaus H., Sci. Adv. 2020, 6, eaaz4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Kagan C. R., Mitzi D. B., Dimitrakopoulos C. D., Science 1999, 286, 945. [DOI] [PubMed] [Google Scholar]
- 241. Shi E., Gao Y., Finkenauer B. P., Akriti, Coffey A. H., Dou L., Chem. Soc. Rev. 2018, 47, 6046. [DOI] [PubMed] [Google Scholar]
- 242. Zhao W., Hsu S.‐N., Boudouris B. W., Dou L., ACS Appl. Electron. Mater. 2021, 3, 5155. [Google Scholar]
- 243. Mitzi D. B., Dimitrakopoulos C. D., Rosner J., Medeiros D. R., Xu Z., Noyan C., Adv. Mater. 2002, 14, 1772. [Google Scholar]
- 244. Matsushima T., Fujita K., Tsutsui T., Jpn. J. Appl. Phys. 2004, 43, L1199. [Google Scholar]
- 245. Matsushima T., Yasuda T., Fujita K., Adachi C., J. Appl. Phys. 2016, 120, 233301. [Google Scholar]
- 246. Matsushima T., Hwang S., Terakawa S., Fujihara T., Sandanayaka A. S. D., Qin C., Adachi C., Appl. Phys. Express 2017, 10, 024103. [Google Scholar]
- 247. Matsushima T., Leyden M. R., Fujihara T., Qin C., Sandanayaka A. S. D., Adachi C., Appl. Phys. Lett. 2019, 115, 120601. [Google Scholar]
- 248. Matsushima T., Mathevet F., Heinrich B., Terakawa S., Fujihara T., Qin C., Sandanayaka A. S. D., Ribierre J.‐C., Adachi C., Appl. Phys. Lett. 2016, 109, 253301. [Google Scholar]
- 249. Zhang F., Zhang H., Zhu L., Qin L., Wang Y., Hu Y., Lou Z., Hou Y., Teng F., J. Mater. Chem. C 2019, 7, 4004. [Google Scholar]
- 250. Zhu H., Liu A., Kim H., Hong J., Go J.‐Y., Noh Y.‐Y., Chem. Mater. 2021, 33, 1174. [Google Scholar]
- 251. Zhu H., Liu A., Zou T., Jung H., Heo S., Noh Y. Y., Mater. Today Energy 2021, 21, 100722. [Google Scholar]
- 252. Chen C., Zhang X., Wu G., Li H., Chen H., Adv. Opt. Mater. 2017, 5, 1600539. [Google Scholar]
- 253. Xie C., Liu C.‐K., Loi H.‐L., Yan F., Adv. Funct. Mater. 2020, 30, 1903907. [Google Scholar]
- 254. Gu H., Chen S.‐C., Zheng Q., Adv. Opt. Mater. 2021, 9, 2001637. [Google Scholar]
- 255. Huang X., Guo Y., Liu Y., Chem. Commun. 2021, 57, 11429. [DOI] [PubMed] [Google Scholar]
- 256. Li Y., Shi Z., Liang W., Ma J., Chen X., Wu D., Tian Y., Li X., Shan C., Fang X., Mater. Horiz. 2021, 8, 1367. [DOI] [PubMed] [Google Scholar]
- 257. Li X., Gao X., Zhang X., Shen X., Lu M., Wu J., Shi Z., Colvin V. L., Hu J., Bai X., Yu W. W., Zhang Y., Adv. Sci. 2021, 8, 2003334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Zhang Y., Ma Y., Wang Y., Zhang X., Zuo C., Shen L., Ding L., Adv. Mater. 2021, 33, 2006691. [DOI] [PubMed] [Google Scholar]
- 259. Liang A., Gao Y., Asadpour R., Wei Z., Finkenauer B. P., Jin L., Yang J., Wang K., Chen K., Liao P., Zhu C., Huang L., Boudouris B. W., Alam M. A., Dou L., J. Am. Chem. Soc. 2021, 143, 15215. [DOI] [PubMed] [Google Scholar]
- 260. Gao Y., Wei Z., Yoo P., Shi E., Zeller M., Zhu C., Liao P., Dou L., J. Am. Chem. Soc. 2019, 141, 15577. [DOI] [PubMed] [Google Scholar]
- 261. Kim J., Shiah Y.‐S., Sim K., Iimura S., Abe K., Tsuji M., Sasase M., Hosono H., Adv. Sci. 2022, 9, 2104993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Shen H., Li J., Wang H., Ma J., Wang J., Luo H., Li D., J. Phys. Chem. Lett. 2019, 10, 7. [DOI] [PubMed] [Google Scholar]
- 263. Liu C.‐K., Tai Q., Wang N., Tang G., Loi H.‐L., Yan F., Adv. Sci. 2019, 6, 1900751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Liu C.‐K., Tai Q., Wang N., Tang G., Hu Z., Yan F., ACS Appl. Mater. Interfaces 2020, 12, 18769. [DOI] [PubMed] [Google Scholar]
- 265. Fortunato E., Barquinha P., Martins R., Adv. Mater. 2012, 24, 2945. [DOI] [PubMed] [Google Scholar]
- 266. Hao F., Stoumpos C. C., Chang R. P. H., Kanatzidis M. G., J. Am. Chem. Soc. 2014, 136, 8094. [DOI] [PubMed] [Google Scholar]
- 267. Klug M. T., Milot R. L., Patel J. B., Green T., Sansom H. C., Farrar M. D., Ramadan A. J., Martani S., Wang Z., Wenger B., Ball J. M., Langshaw L., Petrozza A., Johnston M. B., Herz L. M., Snaith H. J., Energy Environ. Sci. 2020, 13, 1776. [Google Scholar]
- 268. Wang J., Datta K., Li J., Verheijen M. A., Zhang D., Wienk M. M., Janssen R. A. J., Adv. Energy Mater. 2020, 10, 2000566. [Google Scholar]
- 269. Xiao M., Huang F., Huang W., Dkhissi Y., Zhu Y., Etheridge J., Gray‐Weale A., Bach U., Cheng Y.‐B., Spiccia L., Angew. Chem., Int. Ed. 2014, 53, 9898. [DOI] [PubMed] [Google Scholar]
- 270. Zuo F., Williams S. T., Liang P.‐W., Chueh C.‐C., Liao C.‐Y., Jen A. K.‐Y., Adv. Mater. 2014, 26, 6454. [DOI] [PubMed] [Google Scholar]
- 271. Rim Y. S., Chen H., Zhu B., Bae S.‐H., Zhu S., Li P. J., Wang I. C., Yang Y., Adv. Mater. Interfaces 2017, 4, 1700020. [Google Scholar]
- 272. Park S., Kim S. H., Choi H. H., Kang B., Cho K., Adv. Funct. Mater. 2020, 30, 1904590. [Google Scholar]
- 273. Xi J., Loi M. A., ACS Energy Lett. 2021, 6, 1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Ullah S., Wang J., Yang P., Liu L., khan J., Yang S.‐E., Xia T., Guo H., Chen Y., Sol. RRL 2021, 5, 2000830. [Google Scholar]
- 275. Chu Y., Hu Y., Xiao Z., J. Phys. Chem. C 2021, 125, 9688. [Google Scholar]
- 276. Ke J. C.‐R., Lewis D. J., Walton A. S., Spencer B. F., O'Brien P., Thomas A. G., Flavell W. R., J. Mater. Chem. A 2018, 6, 11205. [Google Scholar]
- 277. Zhang F., Zhang Q., Liu X., Hu Y., Lou Z., Hou Y., Teng F., ACS Appl. Mater. Interfaces 2021, 13, 24272. [DOI] [PubMed] [Google Scholar]
- 278. Zhang F., Zhang Q., Liu X., Qin L., Hu Y., Lou Z., Hou Y., Teng F., J. Mater. Chem. A 2021, 9, 22842. [Google Scholar]
- 279. Qin C., Zhang F., Qin L., Liu X., Ji H., Li L., Hu Y., Lou Z., Hou Y., Teng F., Adv. Electron. Mater. 2021, 7, 2100384. [Google Scholar]
- 280. Zhu H., Liu A., Luque H. L., Sun H., Ji D., Noh Y.‐Y., ACS Nano 2019, 13, 3971. [DOI] [PubMed] [Google Scholar]
- 281. Wei Z., Wang K., Zhao W., Gao Y., Hu Q., Chen K., Dou L., Chem. Commun. 2021, 57, 11469. [DOI] [PubMed] [Google Scholar]