

Abstract

Protamines are arginine-rich proteins that condense DNA in sperm. Despite their importance in reproduction, information on protamine structure is scarce. We, therefore, used molecular dynamics to examine the structures of salmon, bull P1, and human P1 protamines. The sizes and shapes of each protamine varied widely, indicating that they were disordered with structures covering a broad conformational landscape, from hairpin loop structures to extended coils. Despite their general disorder, the protamines did form secondary structures, including helices and hairpin loops. In eutherians, hairpins may promote disulfide bonding that facilitates protamine-DNA condensation, but the specifics of this bonding is not well established. We examined inter-residue distances in the simulations to predict residue pairs likely to form intramolecular bonds, leading to the identification of bonding pairs consistent with previous results in bull and human. These results support a model for eutherian protamine structures where a highly charged center is surrounded by disulfide-bond-stabilized loops.
Introduction
Protamines are arginine-rich nuclear proteins that package DNA molecules in many plants and animal sperm cells.1 The sequences and structures of protamines are specialized for tight DNA packaging, allowing these proteins to pack DNA to a final volume ∼6 to 7× smaller than the nuclei of somatic cells that use histones to compact DNA.2−6 It is thought that this ultratight packaging of DNA in sperm cells both facilitates efficient genetic delivery and protects DNA from attack by mutagens or nucleases.3,7 It is well known that protamine dysfunction is associated with higher rates of damage in DNA, high rates of infertility, and poor reproductive outcomes, making protamines essential to the reproductive process.6,8,9
DNA condensation by protamines has been the subject of several experimental investigations.10−14 These studies often focus on the use of piscine protamine, such as salmon, or mammalian protamine such as bull P1 protamine. A significant difference between the protamines found in fish and mammals is the presence of cysteines in mammalian protamines. Cysteine residues in mammalian protamines undergo oxidation resulting in the formation of intramolecular and intermolecular disulfide bonds.15,16 In bull, intramolecular disulfide linkages seem to result in the stabilization of hairpin-like structures.15 These cysteines play an essential role in DNA condensation in eutherian mammals. For instance, Vilfan et al. showed that bull protamine-DNA condensates prepared in vitro yielded more compact, uniform, and spherical particles that resembled native sperm cell chromatin compared to condensates prepared using salmon protamine.10 As the arginine-rich DNA-binding regions in salmon and bull protamine are similar, it was hypothesized that the tighter DNA packaging by bull protamines in vitro is a result of the folded hairpin structure. A later X-ray diffraction study by Hutchison et al. showed that DNA packing density in isolated bull sperm nuclei is slightly less than that in salmon nuclei.11 However, the DNA packaging in bull sperm chromatin was highly dependent on the cysteine–cysteine linkages. Full reduction of disulfide bonds in bull protamine led to a significant decrease in packing density.11 While the residues engaged in intramolecular disulfide bonding in bull protamine have been previously identified,15 the disulfide bond pattern in other mammalian protamines such as human P1 protamine remains to be established. However, some models17,18 of intramolecular bonding in human P1 protamine have been proposed.
While investigations into protamine-DNA condensation are helpful, we believe it is also necessary to examine protamine structure in the absence of DNA. Protamines belong to the family of intrinsically disordered proteins (IDPs), likely due to the high number of positively charged arginine residues in protamine sequences (∼50 to 70% arginine). Experimental techniques such as NMR and SAXS can be employed to study IDPs, but the structural information they provide is limited.19 Computational approaches are thus an attractive complementary approach to studying IDPs.20−22 In this paper, we use molecular dynamics (MD) simulations to explore the conformational landscape of three highly studied10,11,15,18,23−25 protamine molecules: chum salmon, bull P1, and human P1 protamines. The objective of our study is twofold: (i) to generate representative conformations of free protamine molecules in solution that can be used to further explore DNA-protamine condensation and (ii) to identify candidate residues for intramolecular bonding in human P1 based on conformations sampled by MD simulations. As our simulations reveal, protamine structures sample a range of different conformations, as expected of proteins with disordered structures. These conformations could be used to understand how protamine conformations change after binding with DNA. We use our simulations to identify potential candidate residues for intramolecular bonding in bull and in human P1. The residue pairs identified as the most likely bonding candidates from the simulations are consistent with both intramolecular disulfide bond pairs that have been previously identified in bull15 and some bonding models17,18 that have been proposed for human P1.
Methods
Molecular Dynamics Simulation Details
We perform molecular dynamics simulations using three protamine sequences: salmon protamine (UNIPROT P69014),26 human P1 protamine from McKay et al.,27 and bull P1 protamine from Balhorn et al.15 (Table 1). For each sequence, 13 separate simulations were performed (randomized using different seed values). The initial protamine structures were built using the sequence command of the tleap module of AMBER 18.28 No disulfide bonds were incorporated. Table S1 provides some details about the simulations.
Table 1. Protamine Sequences Used for MD Simulation.

As building chain structures in this way results in extended conformations, we began each simulation with an implicit solvent simulation using the Generalized Born solvation model. First, the system was heated for 2 ns from 0 to 300 K and then continued for at least 30 ns. The radius of gyration (Rg) values in these implicit solvent simulations were monitored, and the simulations were continued for up to 320 ns, until the Rg of the chain became stable (see Figures S1–S6). Final structures from implicit solvent simulations were then used to start independent explicit solvent simulations.
In the explicit solvent simulations, the protamines were solvated in a TIP3P water box and Cl– counterions were added to neutralize the charges of the protamine molecules. The ff14SB29 force field was used for both explicit and implicit solvent simulations. The Cl– counterions were the only salt ions present. The protamine structures were held fixed while water and ions were minimized with 10 000 cycles of steepest descent followed by 10 000 cycles of conjugate gradient. Next, the entire system was minimized unrestrained for 5000 cycles of steepest descent and then by 5000 cycles of conjugate gradient. The system was heated at NVT conditions from 0 to 300 K with restraints on the protein for 200 ps and further equilibrated by a restrained NPT equilibration at a pressure of 1 atm for 5 ns. The production simulations were performed for an additional 200 ns (without any restraints) and the trajectories were saved every 2 ps. The frames from the final 80 ns of simulation were used for most analyses. During the production run, the temperature was held constant at 300 K using Langevin dynamics with a collision frequency of 1.0 ps–1. The particle mesh Ewald method was used to treat long-range electrostatic interactions with a 10 Å cutoff.30 The integration time-step of the simulations was 2 fs. The PMEMD engine of AMBER, mostly run with CUDA acceleration, was used to perform simulations.31−33 The CPPTRAJ and PYTRAJ modules of AMBER 18 were used for trajectory analysis and VMD was used for trajectory visualization.34−36 Secondary structure was assessed using the DSSP algorithm via CPPTRAJ.34,37
Characterization of Arginine Residue Arrangement and Shape in Protamine Molecules
All protamines are strongly basic due to the presence of multiple arginine (R) and occasional lysine (K) residues. The arrangements of arginine residues in protamine sequences were characterized using the sequence order parameter (λ) value, used previously by Ziebarth et al.38 to characterize copolymer sequences. We treated each arginine residue as monomer A, and each nonarginine residue as monomer B, and defined λ as λ = PAA + PBB – 1. PMN is the conditional probability of an M monomer immediately following an N monomer. λ ranges from −1 for an alternating polymer, where PBB = PAA = 0, to 0 for a random polymer, where PAB = PAA = PA, to +1 for a diblock copolymer where PBB = PAA = 1.
The average arginine stretch length for a given protamine sequence was calculated according to the formula
where Li is the length of the ith arginine stretch in a sequence and n is the total number of arginine stretches on a sequence.
Additionally, we monitored the progress of our simulations using the following quantities: radius of gyration (Rg), end-to-end distance Ree, and the ratio between the two Rs = Ree2/Rg2, which has been referred to as shape factor.39 This ratio or the shape factor, Rs, reflects the chain conformation of the macromolecule. If Rs = 6, the chain adopts a Gaussian conformation. If Rs is close to 10, it adopts a rod conformation, and if Rs is close to 2, it adops a collapsed coil. Rs is different from shape descriptors generated using Rg tensor values. We will refer to Rs as the shape ratio to distinguish it from other shape factors frequently used in different contexts. Figures S1–S6 show how Rg and Rs vary for each trajectory, from implicit solvent simulations to explicit solvent simulations.
Disulfide Bond Analysis
No disulfide bonds were set in the protamines during the simulations. Sulfur atoms with the potential for disulfide bonding remained bonded to hydrogen atoms during simulations. Residue pair distance values during trajectories were used to predict the potential for disulfide bonding. For the disulfide bond predictions, distances between every cysteine–cysteine pair were calculated. One cysteine–tyrosine pair in human P1, labeled “:6:14Y” (residue #6 and residue #14), was additionally included because it has been suggested to potentially form an intramolecular bond in a previous human P1 model.18 For both bull and human P1 protamines, three separate distance criteria were used to generate a final list of potential pairs between nonconsecutive residues. For each residue pair, the number of simulation snapshots that met all three criteria was counted, and the most likely bonding pairs were identified as the residue pairs that met the criteria in most snapshots. The first distance criterion was that the sulfur atom to sulfur atom distances (which will be referred to as S–S distance in later instances) for cysteine–cysteine pairs (sulfur–oxygen distance for the cysteine–tyrosine pair) was less than 10 Å. To get an idea of the distance between sulfur atoms of nearby cysteine residues, the S–S distances between consecutive cysteines were measured in the bull and human P1 trajectories. The sulfur atoms of consecutive cysteine residues were exclusively within 10 Å, and therefore, we used an S–S cutoff of 10 Å to identify nonconsecutive cysteine residues with sulfur atoms that were relatively close together. The two other criteria used to identify residues in conformations that may lead to disulfide bonding related to Cα–Cα and Cα–Cβ distances, as these distances were recently used by Gao et al. to develop a machine learning algorithm to predict disulfide bond sites.40 Gao et al. collected information about disulfide bond pairs to train their machine learning algorithm.40 Using their collected data (specifically the PISCES40,41 dataset), we generated a histogram to identify permitted Cα–Cβ distance ranges for disulfide bonding (see Figure S7). Based on that histogram (and an additional Cα–Cα histogram generated by Gao et al.), we used Cα–Cα distances less than 7.5 Å and Cα–Cβ distances less than 6.5 Å as additional cutoffs for our disulfide bond predictions.40 Thus, nonconsecutive residue pairs with sulfur–sulfur/sulfur–oxygen distance less than 10 Å, Cα–Cα distance less than 7.5 Å, and Cα–Cβ distance less than 6.5 Å in the same frame were identified as potential pairs for intramolecular bonding.
Results and Discussion
Conformation Landscape of Protamine Molecules
The sequences of the salmon, bull P1, and human P1 protamines studied here are provided in Table 1. Salmon is an example of a fish protamine and both bull and human P1 are eutherian mammal P1 protamines. The sequences are similar in that, as is typical for protamines, a large proportion of all three sequences are arginines; however, they have notable differences that could impact their structure and interactions with DNA. First, the charge density along the salmon sequence is higher, with arginines making up two-thirds of the residues in salmon protamine, compared to less than half of the residues in the human P1 protamine. Second, the arrangement of the arginines into clusters of 3–7 consecutive arginines vs single arginines (i.e., arginine residues in-between two nonarginine residues) has noticeable differences for the three sequences. In human P1 protamine, over half of the arginine residues are singles, while the arginines in salmon and bull protamines are much more likely to be clustered. To quantify this behavior, we calculated the sequence order parameter (λ), as described in Methods, and the mean length of arginine stretches (Table 2) for the three protamine sequences. The λ values of the salmon and bull sequences are similar and positive, reflecting that most of the arginine residues in both sequences are located within arginine clusters. In contrast, human protamine, with its many single arginines, has a negative λ value. Consistent with our calculated λ values, the lengths of arginine clusters, on average, are >3 for salmon and bull and only ∼1.5 for human P1. Thus, despite human and bull P1 protamines having similar lengths and numbers of arginine residues, the arrangements of the arginines along these sequences are very different. Third, eutherian P1 protamine sequences can be separated into a central domain (residues ∼13 to 42), that has been hypothesized to be the DNA-binding region, and flanking regions.1,18 Both bull and human P1 protamines fit this model, as the central domain region of each sequence contains the long clusters of consecutive arginines, while all of the arginines in the flanking regions are singles or instances of only two consecutive arginines. In contrast, the arginine clusters in salmon protamine occur throughout the sequence.
Table 2. Properties of the Three Protamine Sequences.
| length | number of arginines | % positive charge | sequence order parameter (λ) | mean arginine stretch length | number of cysteines | |
|---|---|---|---|---|---|---|
| salmon | 33 | 21 | 64 | +0.25 | 3.5 | 0 |
| bull P1 | 49 | 27 | 55 | +0.28 | 3.0 | 7 |
| human P1 | 50 | 24 | 48 | –0.31 | 1.5 | 6 |
To see if the above sequence features (i.e., charge density, λ value, and a hypothesized central DNA-binding region for eutherian P1 protamines) are typical of fish and eutherian P1 protamines, we analyzed 34 fish sequences and 145 eutherian P1 sequences gathered by Powell et al.18 As expected, piscine protamines generally have higher charge densities than eutherian P1 protamines (Figure S8), a pattern also reflected in the salmon, bull, and human P1 protamines studied here. Figure S9 shows our calculated λ values for piscine and eutherian P1 protamines. Fish protamines mostly have positive λ values, whereas eutherian P1 protamines have a spectrum of negative and positive λ values. Salmon protamine has a λ value (0.25) close to the average fish λ value (0.21), whereas bull (λ = 0.28) and human P1 (λ = −0.31) protamines represent opposing extremes of the eutherian P1 λ spectrum. Figure S10 shows general locations of single arginines and arginine clusters in fish and eutherian P1 protamines. As reflected in our salmon, bull, and human P1 sequences, arginine clusters tend to occur throughout fish sequences as opposed to only the central regions in eutherian P1 sequences.
Figure 1 shows a plot of the protamine radius of gyration (Rg) vs shape ratio (Rs) values for all 13 salmon simulation trajectories, with the different trajectories denoted by different colored dots (Figure S11 shows data for each simulation separately). Rs tends to increase with Rg—indicating as the chain gets bigger, it becomes more extended, resulting in Ree increasing more than Rg. The values of Rs have a strikingly large range, from Rs ∼2 to ∼10. This heterogeneity is consistent with what is known about IDPs. Unlike folded proteins, IDPs do not have well-defined equilibrium conformations, and we, therefore, expect molecular dynamics of IDPs to sample a broader range of different conformations. While it may be surprising to see low Rs values for such a highly charged molecule as the salmon protamine, it is usually the formation of hairpin loops that lead to such low Rs values (discussed later).
Figure 1.

Scatter plot of shape ratio (Rs) vs radius of gyration (Rg) for salmon protamine. Each data point represents Rg and Rs values in single frames that were collected every 200 ps. The 13 individual trajectories are coded by different colors. Conformation images for numbered snapshots are shown on the right.
While there is substantial overlap between simulations, the different trajectories in Figure 1 tend to sample different regions of the salmon protamine’s conformational space. Several of the trajectories are centered around Rg ∼20 (and Rs ∼6), but there are some trajectories where the protamine has high Rs (dark blue, for example) or low Rs (purple) conformations for a substantial amount of the simulation time. We believe that this ability to sample a wider range of conformations is an advantage of the simulation approach of performing multiple simulations instead of a single long simulation, in which the protein conformation could be artificially confined to a local minimum.42 However, for each salmon, human P1, and bull P1, we continued one trajectory for an extended period. As Figures S12–S17 show, Rg and Rs values generally followed the same pattern beyond 200 ns.
Representative conformations throughout the conformational space are shown on the right side of Figure 1. These reveal the formation of hairpin loops in more than one instance. We noticed that some simulation trajectories have persistent hairpin loops while others do not. To examine this more closely, we calculated the average inter-residue distance for every residue pair in every simulation trajectory. Figure 2 shows distances for simulation trajectories 6 and 9 (represented by purple dots and gray dots in Figure 1, respectively), where one has a persistent hairpin loop and the other does not. Based on low off-diagonal distances in Figure 2A, we can tell that the hairpin structure in Figure 1 (purple dots, image #3) is persistent throughout that trajectory. In contrast, Figure 2B (represented by gray dots, images #6 and 8) shows no low off-diagonal values and thus no persistent hairpin loop. Figure S18 shows distance heatmaps for every salmon trajectory. Persistent hairpin loop structures seem to form in 3 out of 13 trajectories, based on off-diagonal patterns. Hairpin loop formation in salmon protamine, which, unlike mammalian protamines, does not have cysteines and does not form intramolecular crosslinks, is unexpected. We believe these persistent hairpin loops are stabilized by hydrogen bonds between multiple atom pairs within the loop.
Figure 2.

Heatmap of average distances between centers of mass of each residue pair in salmon protamine in two separate trajectories. Heatmap (A) is from simulation trajectory #6 (purple dots and image #3 in Figure 1). Heatmap (B) is from simulation trajectory #9 (gray dots and images #6 and 8 in Figure 1). Distance values in Angstrom are color-coded according to the bar on the right.
Plots of Rg vs Rs for bull and human P1 are shown in Figures 3 and 4, respectively (Figures S19 and S20 show how they vary trajectory by trajectory) and show many of the same features observed for the salmon protamine. The sizes and shapes of human and bull protamines vary significantly, indicating that the proteins are disordered and take on many distinct conformations. The individual simulation trajectories also occupy different subsets of the overall conformational space, with some trajectories having a large amount of overlap and others that appear as outliers. The trajectory with low Rs values represented by pink dots in Figure 3, for example, is relatively isolated on the plot. The conformation image that represents that trajectory (image #1, Figure 3) shows two simultaneous hairpin loops. Because of these hairpin loops, end-to-end distances are low and, thus, Rs values are low. The distance heatmap (shown in Figure S21, simulation #7) has off-diagonal values consistent with two persistent hairpin loops. Distance heatmaps for a few other bull and human P1 trajectories (Figures S21 and S22, respectively) also have low off-diagonal values that indicate hairpin loops.
Figure 3.

Scatter plot of shape ratio (Rs) vs radius of gyration (Rg) for bull P1 protamine. Each data point represents Rg and Rs values in single frames that were collected every 200 ps. The 13 individual trajectories are coded by different colors. Conformation images for numbered snapshots are shown on the right.
Figure 4.

Scatter plot of shape ratio (Rs) vs radius of gyration (Rg) for human P1 protamine. Each data point represents Rg and Rs values in single frames that were collected every 200 ps. The 13 individual trajectories are coded by different colors. Conformation images for numbered snapshots are shown on the right.
Figure 5 compares the distributions of Rs, Rg, and end-to-end distance (Ree) for the three protamines. Except for shoulders at low Rs values, bull P1 and salmon have nearly identical Rs distributions, while human P1 tends to have higher Rs values (Figure 5A). The average Rs for human P1 is 7.3, and the average Rs for bull and salmon are 5.5 and 5.6, respectively. The similarity between Rs distributions of salmon and bull protamines mirrors the similarity between the arrangement of arginine residues along their sequences, as both salmon and bull sequences have λ values near +0.25. Human P1, meanwhile, has a negative λ value. The larger Rs in human P1 is somewhat surprising, as the larger number of consecutive arginines in bull and salmon may have been expected to increase electrostatic repulsion, and lead to higher Rs values. This is not the case in our simulations, as human P1, with its more alternating arrangement of arginine residues, more frequently adopted conformations with high Rs values. However, the charged guanidinium groups on arginine residues are relatively far from the protein backbone, and therefore, the impact of having neighboring arginines on the chain conformation is not as large as in other polyelectrolyte chains (such as polyethyleneimine or PEI43) where charged groups are along the polymer chain backbone. We also note that the connection between λ and Rs values is based on only three sequences, and further studies need to be conducted to thoroughly examine this relationship.
Figure 5.

Histograms of (A) shape ratio (Rs), (B) radius of gyration (Rg), and (C) end-to-end distance (Ree) values, from all 13 simulations for salmon, bull P1, and human P1 protamines.
As for Rg values (Figure 5B), salmon has fewer residues (33) than either of the P1 protamines, hence not surprisingly has the smallest Rg distribution. Although bull P1 and human P1 sequences have similar lengths, bull P1 has higher Rg values. Oskolkov et al. previously reported hydrodynamic radii of 3 and 4 nm for salmon and human P1 protamines, respectively.17 These hydrodynamic radii values are higher than the average Rg values of salmon and human P1 seen in our simulations (∼1.9 and ∼2.4 nm, respectively), an expected result as hydrodynamic radii are normally higher than Rg. However, the ratio of human to salmon Rg values in the simulations is similar to that of the hydrodynamic radii reported by Oskolkov et al.
While protamines are IDPs, they can and do develop secondary structures in our simulations, such as the helices found in many of the snapshots of the bull and human simulations shown in Figures 3 and 4 (respectively). To quantify the secondary structure observed in the MD simulations, we calculated the overall fractions of helices, sheets, and coils for each protamine (Figure 6). All three protamines have high fractions of random coil, consistent with the protamines being IDPs that often form flexible, random coil structures,44 and very low amounts of sheet formation. The amount of helices observed varied for the three protamines, with human P1 having the most helical content (up to ∼40%). Given that human P1 has ∼50% arginines, we did not originally expect human P1 to have such high helical content; however other protein regions with similar amounts of arginine residues, such as the ARM region in the HIV-1 Rev protein, are also believed to form helices.45 Oskolkov et al. reported some helix formation in salmon and human P1 protamines.17 To compare with simulation results, we used two computational webservers, namely, PSIPRED46 and PredictProtein,47 that predict secondary structure based on protein sequence, on all three protamines. Both webservers predicted that the proteins would form helices, with helical percentages similar to what was observed in the simulations.46,47 We additionally looked at the salmon (UNIPROT26P69014), bull P1 (UNIPROT26P02318), and human P1 (UNIPROT26P04553) structures in the AlphaFold48,49 Protein Structure Database. The salmon structure is in an extended coiled conformation, with little helix formation; the bull and human P1 structures have more helices but were predicted with low confidence.
Figure 6.
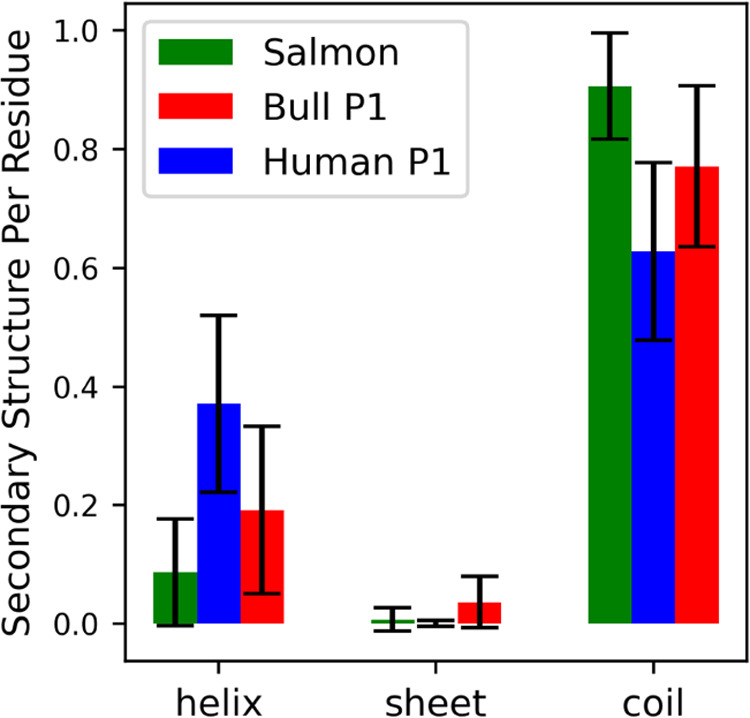
Average helix, sheet, and coil content per residue for all three protamines. Secondary structure categories were identified using DSSP “α,” “3–10”, and “Pi” were combined and called “helix”; “Anti” and “Para” were combined and called “sheet”; and “Bend,” “Turn,” and “None” were combined and called “coil”.
To investigate the relationship between protamine size and secondary structure, we sorted frames from all simulation trajectories in order of increasing Rg and then plotted the fraction of residues that formed secondary structures (i.e., “Para,” “Anti,” “3–10,” “α,” “Pi,” “Turn”, and “Bend”) in these sorted frames. The plot is shown in Figure 7A for human P1 protamine and in Figures S23 and S24 for salmon and bull, respectively. For all three protamines, there is a noticeable decrease in the amount of secondary structure as the chain gets larger (Rg increases). For example, in human P1, the ∼5% of conformations with the smallest Rg values have a mean secondary structure per residue per frame of ∼0.8, while the ∼5% conformations with the largest Rg values have around half that amount. This result is not surprising, as secondary structure is associated with interactions between residues (e.g., hydrogen bonds) that tend to decrease the size of the chain. We also observe shifts in the locations along the sequence that have secondary structure as the chain size varies. In human P1, small conformations commonly include significant helical formation between residues ranging from 15 to 35, while the helices are more often found between residues 5 and 20 for conformations with high Rg (Figure 7B). Additionally, in both human and bull P1 protamines, α helices are more likely to be found at the N-terminal end of the chain than the C-terminal end, even though the amount of charged residues at both chain ends is relatively low for both sequences.
Figure 7.

Secondary structure vs Rg for human P1 protamine (every 200 ps). (A) Rg values and the percent of residues that were classified as having secondary structure (i.e., they were classified by DSSP as any category other than “None”). (B) Secondary structure for each residue in each frame according to the color bar. Frames in both panels are sorted in order of increasing Rg values from all 13 simulations.
We notice changes in secondary structure (especially helices) happening within the 200 ns trajectory timeframe. Helices break, form, and/or remain constant throughout trajectories. Figure S25, for instance, shows secondary structure dynamics across a single bull trajectory, and for residues ∼15 to 25, there are α helices present initially that disappear later. Figure S26 shows how secondary structure varies in another bull trajectory. In Figure S26, there are some α helices that persist throughout the trajectory time, some that disappear and some that form—all happening within the 200 ns timeframe. Variable secondary structure dynamics are seen in all three protamine structures. However, there are trajectories where the secondary structures do not change from start to finish, such as the human P1 trajectory shown in Figure S27.
Candidate Residues for Intramolecular Bonding in Human P1 Protamine
Last, we examine the possibility of intramolecular bond formation through cysteine–cysteine or cysteine–tyrosine linkages in mammalian P1 protamines. Although our attention is primarily on cysteine pairs for intramolecular disulfide bonding, a potential cysteine–tyrosine crosslink has been proposed recently in some mammalian protamines.18 Hence, we include one cysteine–tyrosine pair (labeled:6:14Y, where “Y” refers to tyrosine) in human P1 in our analysis. We argue that simulations can be used to investigate the likelihood that a pair of cysteine residues is involved in a disulfide bond, as disulfide bond formation requires that pair of residues first get close together in the right orientation.50−52 We note that our investigation is solely focused on intramolecular bonds, as our simulations contained single protamine molecules and are not suitable for the investigation of intermolecular interactions. By monitoring distances between pairs of cysteine residues in our simulations, we identify residue pairs that are more likely to form disulfide bonds: disulfide bond candidate pairs will be close together with the proper orientation more often than pairs of cysteines that are not likely to bond. We analyze distances between cysteine residues in both bull P1 (in which intramolecular disulfide bonds have been previously15 identified), and human P1 simulations even though our primary goal is to identify candidate residue pairs in human P1.
To begin this analysis, we measured the distances between the sulfur atoms of all pairs of cysteine residues in bull and human P1 protamine during the simulations (Figure 8). There are seven cysteine residues in bull protamine, so 21 residue pair combinations were considered. Human P1 protamine has six cysteine residues, and one cysteine–tyrosine pair of special interest. Hence, 16 residue pair combinations were considered for human P1. In both human and bull, there are two pairs of neighboring cysteine residues, and we notice that these neighboring cysteines both get close to the same distant cysteines, potentially giving these residues multiple means to form disulfide bonds. For example, residues 38 and 39 both commonly come near residue 47 in both bull and human, making disulfide bond formation between residue 47 and either residue 38 or 39 possible. To investigate the distances between cysteine residue pairs more fully, we monitored and visualized distances between cysteine residue pairs that get close during the simulations and found that, in many cases, close residues are brought together by the formation of hairpin loop conformations. Figure 9 shows a representative example, showing the distance between residues 39 and 47 in a single trajectory for human P1. These residues stay relatively close together throughout the simulation and have periods when they are very close together (within 5 Å). The closeness of these residues is associated with the formation of a hairpin loop conformation that brings residues 39 and 47 together and remains stable throughout the simulation, as shown by the images of the protein structure shown in Figure 9. This hairpin loop is thus a potential precursor to an intramolecular disulfide bond.
Figure 8.

Violin plots of S–S distances between cysteine–cysteine residue pairs for (A) bull P1 and (B) human P1 protamines. Pairs are identified according to residue numbers in the sequences in Table 1 (A). The dotted vertical line separates consecutive pairs from nonconsecutive pairs. 6:14Y pair indicates a cysteine–tyrosine pair included in human P1 analysis. The dotted horizontal line indicates S–S cutoff. Pairs that meet all three distance criteria as described in the Methods section (S–S distance <10 Å, Cα–Cα distance <7.5 Å and Cα–Cβ distance <6.5 Å in the same frame) are identified.
Figure 9.

Example of hairpin loop formation and the associated variation in S–S distance of cysteine residues 39:47. Partial conformation images with residues 39 and 47 shown as balls and the other residues shown as ribbons are shown for the three indicated time points.
In addition to a close approach between the sulfur atoms of cysteine residues, disulfide bond formation has also been suggested to be promoted when other atom pairs get close.40 Specifically, having low Cα–Cβ and Cα–Cα distances has been shown to be a common feature of pairs of cysteine residues that form disulfide bonds.40 Therefore, we monitored Cα–Cβ and Cα–Cα distances between cysteine residues in both the bull and human P1 simulations (Figures S28–S31). In general, residue pairs with low sulfur–sulfur distances also have low Cα–Cα/Cα–Cβ distances. However, as highlighted in Figure 8, there are some residue pairs that have low S–S atom distances but not Cα–Cα/Cα–Cβ distances, and, thus, are not likely to be in the correct orientation for disulfide bonding (e.g., 22–39 in bull P1, 29–47 in human P1).
To make a final list of potential disulfide bond candidates from the simulations, we calculated percentages of snapshots where a pair of residues satisfies all three distance criteria simultaneously: S–S distance is less than 10 Å, Cα–Cα distance is less than 7.5 Å and the Cα–Cβ distance is less than 6.5 Å. Table 3 lists the pairs that satisfy all three criteria at some point for bull P1 and human P1 protamines. Seven pairs were found to satisfy all three criteria in bull P1, and five in human P1. Interestingly, the top three pairs (i.e., pairs with percent snapshots >1%) are the same residues in both bull and human P1. Because other identified candidate pairs have much lower snapshot percentages in both bull and human P1, we do not think they are as important as the top three pairs. Intramolecular disulfide bonds reportedly form between 6–14 and 39–47 in bull P1.15 These are two of our top three identified candidate pairs for intramolecular disulfide bonding in bull P1, with the third being 38–47 which is a possible alternative to 39–47. In other words, the list of candidate pairs identified in our simulations for bull includes pairs that have been previously identified in bull.15 Surprisingly, for human P1, the residue pair 6-14Y is one of the top three pairs identified. Residue 14 is a tyrosine in human P1 protamine and would not normally be considered for intramolecular bond formation, but it was recently observed in multiple sequence alignment studies that some mammalian protamines substitute cysteines with tyrosines in certain positions, and position 14 in human P1 is one of those positions.18 In human P1 protamine, Powell et al.18 proposed intramolecular bonds between 6-14Y and 39–47, and Oskolkov et al.17 proposed intramolecular bonds between 6–29 and 39–47. Our simulation results do not identify 6–29 as a potential candidate residue pair for intramolecular disulfide bonding (because they do not get close in our simulations), but they do predict the two other proposed pairs. We speculate that Oskolkov et al. did not consider 6-14Y because position 14 is a tyrosine. Table 3 provides a full list of potential candidate pairs for bull and human P1. Figure 10 provides a diagrammatic illustration of our top three residue pairs for bull and human P1, and includes previously identified pairs for bull and proposals for human P1. Figures S32 and S33 identify the frames from Table 3 (i.e., frames where all three cutoff conditions are met) within their respective trajectories, in both the bull and human P1 simulations. As Figures S32 and S33 show, the top three residue pairs in both bull and human P1 protamine simulations usually meet their cutoff conditions in multiple trajectories and/or meet them persistently within a single trajectory.
Table 3. Candidate Pairs for Disulfide/Crosslink Formation That Meet All Cutoff Conditions.

Colored residue pairs are the top candidates, with red indicating that the pair has been previously identified or proposed in the literature.
“Y” indicates tyrosine.
Figure 10.

Schematic of (A) bull P1 and (B) human P1 protamine sequences showing candidate residue pairs for disulfide/crosslink formation. Only the top three pairs from Table 3 for bull and human P1 are shown here. Red lines connect residue pairs that were both predicted by the MD simulation and also have been suggested by at least one previous model, while blue lines connect residue pairs predicted by only the MD simulation. The dotted green line connects a residue pair proposed by a previous model that is not predicted by our MD simulation.
Concluding Remarks
We employ MD simulations to explore the structures of free protamine molecules in solution because detailed structural information of protamines is lacking in the current literature. Specifically, we examine one fish (salmon) and two mammalian (bull P1 and human P1) protamines, three protamines that are frequently used in experimental studies. One of the key differences between fish and mammalian protamines is that mammalian protamines contain cysteine residues and, therefore, can form intra- and intermolecular disulfide bonds.1,3,11,15 The formation of intramolecular disulfide bonds has been associated with hairpin loop structures in mammalian protamines.11,18 As disulfide bond patterns in protamines have not been firmly established, we used our simulations to examine the relationship between protamine sequences, hairpin loop structures, and the possibility of intramolecular bond formation. First, we observed that all three protamines adopt compact structures (i.e., structures with low Rs values) that were commonly associated with the formation of long-lasting hairpin loops. This result was somewhat surprising in the case of salmon protamine, as fish protamines lack cysteines. But there were hydrogen bonding interactions between multiple atom pairs within the salmon hairpin loop. Thus, our simulations indicate that the formation of hairpins is not solely limited to eutherian protamines, but may instead be a general feature of protamine structures.
Second, we monitored the distances between potential intramolecular bond pairs in the bull P1 and human P1 simulations to further investigate the possibility for intramolecular bonding. We note that the simulations did not explicitly include any intramolecular bonds, and, therefore, any cysteine–cysteine interactions in the simulations were spontaneous. By making use of three distance cutoffs, we identified residue pairs that were most likely to be in the proper configuration for the formation of an intramolecular bond. The residue pairs 6–14 and 39–47, which have been previously identified to form intramolecular disulfide bonds in bull P1 protamine,15 are among the top three potential residue pairs predicted in our simulations. The top three pairs predicted in our MD simulations (6–14/6-14Y, 38–47, and 39–47) are the same for bull and human P1, even though bull and human P1 are different sequences. This is despite the fact that human P1 has a tyrosine substitution in the 14th position, making 6-14Y a cysteine–tyrosine crosslink rather than a cysteine–cysteine disulfide bond. Tyrosine substitutions have been noted recently in some mammalian protamines in multiple sequence alignment studies, and 6-14Y has been proposed to form a crosslink in human P1 in that study.18 Thus, our simulations provide results that are markedly consistent with previously proposed bonding pairs in both human and bull. As Figure 10 illustrates, our top predicted crosslinks would cause both ends of the protamines to form loops that surround a highly charged central region.11 We think this finding is interesting and future MD simulations could be used to explore whether this is true in other mammalian protamines. Specifically, future MD simulations could be used to explore the structures of other eutherian P1 protamines to see if residue pairs 6–14 and 39–47 get close, to potentially form bond-stabilized hairpin loops and isolate a highly charged central region. These positions are mainly cysteine residues in eutherian P1 protamines. Multiple sequence alignment studies by Powell et al.18 showed cysteines to be highly conserved among eutherian P1 protamine sequences.
We also attempted to provide a general characterization of the sizes and shapes of the protamines in the simulations. As protamines are highly charged molecules without well-defined equilibrium structures, we treated them as flexible polyelectrolytes and characterized their structures using polymer physics principles. Specifically, our approach used shape and size estimators to characterize protamine conformation. The simulation results support this approach, as the broad distributions in the scatter plots of Rs against Rg (Figures 1, 3, and 4) show that the protamines are flexible with a heterogeneous array of conformations, ranging from compact coils to extended rod-like structures. The polyelectrolyte character of the protamines is the result of the many arginine residues along their sequences. While all protamines have a high percentage of arginine residues, the arrangement of the arginines along the sequence can be very different. We used the sequence order parameter (λ) to quantify the arrangement of arginine residues along protamine sequences. We noticed that salmon and bull P1 protamines, which have similar λ values, have almost identical Rs distributions. We thus speculate that a relationship between λ and Rs values may exist. A future study could explore whether this relationship is true. Additionally, the tools used in this study, such as Rs, Rg, and Ree, could potentially be combined with other shape descriptors, such as relative shape anisotropy, to study other disordered proteins.
All three protamines form secondary structures in our simulations. Human P1 has the highest amount of helix per residue per frame, followed by bull, followed by salmon. This leads us to think of salmon as the most disordered of the three protamine structures, and human P1 the least. We compared our protamine structures to those in the AlphaFold48,49 Protein Structure Database. AlphaFold predictions were largely comparable to ours, with salmon being almost entirely coiled, and varying degrees of helices in the bull and human P1 structures. The salmon structure was deemed a confident prediction whereas the bull and human P1 predictions were of lower confidence. Interestingly, low-confidence regions are not uncommon in the AlphaFold Protein Structure Database, and disordered regions tend to overlap with low-confidence regions.53 Thus, AlphaFold and MD simulation seem to concur that these protamine structures are disordered, even though AlphaFold uses a very different approach to structure prediction (Artificial Intelligence/Deep Learning).53 However, AlphaFold did not provide us the landscape of conformations of the disordered proteins that MD simulations did.
Our simulations identified protamine conformations in solution alone. As protamines meet and condense DNA, it is likely that interaction with DNA affects protamine conformation in a nontrivial manner, which we do not cover in our study. We think the impact of interactions with DNA on protamine structures is an interesting avenue to explore in future studies.
Acknowledgments
J.E.D. acknowledges financial support from the National Science Foundation (MCB-1453168).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c04227.
Further technical details of MD simulations (Table S1); radius of gyration (Rg) and shape ratio (Rs) in each implicit solvent and explicit solvent trajectory for salmon, bull, and human P1 protamines (Figures S1–S6); histogram of inter-cysteine Cα–Cβ distances in disulfide bonds, based on previously published datasets (Figure S7); charge densities in fish and eutherian P1 protamine sequences (Figure S8); sequence order parameter values (λ) in fish and eutherian P1 sequences (Figure S9); locations of single arginine residues and clustered arginine residues in fish and eutherian P1 sequences (Figure S10); scatter diagrams of shape ratio (Rs) against radius of gyration (Rg) for each salmon trajectory (Figure S11); radius of gyration (Rg) and shape ratio (Rs) values for a single salmon, bull, and human P1 trajectories continued for an extended period beyond 200 ns (Figures S12–S17); heatmap of average inter-residue distance in each salmon trajectory (Figure S18); scatter diagrams of shape ratio (Rs) against radius of gyration (Rg) for each bull and human P1 trajectory (Figures S19 and S20); heatmaps of average inter-residue distance in each bull and human P1 trajectory (Figures S21 and S22); secondary structure against radius of gyration (Rg) for salmon and bull simulations, all trajectories combined (Figures S23 and S24); secondary structure progression in specific bull and human P1 trajectories (Figures S25–S27); violin plots of Cα–Cα distances and Cα–Cβ distances for all cysteine pairs in bull and human P1 protamine (and one cysteine–tyrosine pair in human P1 protamine), for all trajectories combined (Figures S28–S31); scatter plots identifying frames where disulfide bond cutoff conditions are met, within their respective trajectories, for both bull and human P1 protamines (Figures S32 and S33) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Balhorn R. The Protamine Family of Sperm Nuclear Proteins. Genome Biol. 2007, 8, 227. 10.1186/gb-2007-8-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathke C.; Baarends W. M.; Awe S.; Renkawitz-Pohl R. Chromatin Dynamics during Spermiogenesis. Biochim. Biophys. Acta 2014, 1839, 155–168. 10.1016/j.bbagrm.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Oliva R. Protamines and Male Infertility. Hum. Reprod. Update 2006, 12, 417–435. 10.1093/humupd/dml009. [DOI] [PubMed] [Google Scholar]
- McLay D.; Clarke H. Remodelling the Paternal Chromatin at Fertilization in Mammals. Reproduction 2003, 125, 625–633. 10.1530/rep.0.1250625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney A. P.; Zhang J. Rapid Evolution of a Primate Sperm Protein: Relaxation of Functional Constraint or Positive Darwinian Selection?. Mol. Biol. Evol. 1999, 16, 706–710. 10.1093/oxfordjournals.molbev.a026153. [DOI] [PubMed] [Google Scholar]
- Champroux A.; Torres-Carreira J.; Gharagozloo P.; Drevet J. R.; Kocer A. Mammalian Sperm Nuclear Organization: Resiliencies and Vulnerabilities. Basic Clin. Androl. 2016, 26, 17. 10.1186/s12610-016-0044-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R.; Dixon G. H. Vertebrate Protamine Genes and the Histone-to-Protamine Replacement Reaction. Prog. Nucleic Acid Res. Mol. Biol. 1991, 40, 25–94. 10.1016/S0079-6603(08)60839-9. [DOI] [PubMed] [Google Scholar]
- Akmal M.; Aulanni’am A.; Widodo M. A.; Sumitro S. B.; Purnomo B. B.; Widodo The Important Role of Protamine in Spermatogenesis and Quality of Sperm: A Mini Review. Asian Pac. J. Reprod. 2016, 5, 357–360. 10.1016/j.apjr.2016.07.013. [DOI] [Google Scholar]
- Wang T.; Gao H.; Li W.; Liu C. Essential Role of Histone Replacement and Modifications in Male Fertility. Front. Genet. 2019, 10, 962. 10.3389/fgene.2019.00962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilfan I. D.; Conwell C. C.; Hud Nv. Formation of Native-like Mammalian Sperm Cell Chromatin with Folded Bull Protamine. J. Biol. Chem. 2004, 279, 20088–20095. 10.1074/jbc.M312777200. [DOI] [PubMed] [Google Scholar]
- Hutchison J. M.; Rau D. C.; DeRouchey J. E. Role of Disulfide Bonds on DNA Packaging Forces in Bull Sperm Chromatin. Biophys. J. 2017, 113, 1925–1933. 10.1016/j.bpj.2017.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer L. R.; Corzett M.; Balhorn R. Protamine-Induced Condensation and Decondensation of the Same DNA Molecule. Science 1999, 286, 120–123. 10.1126/science.286.5437.120. [DOI] [PubMed] [Google Scholar]
- Miller D.; Brinkworth M.; Iles D. Paternal DNA Packaging in Spermatozoa: More than the Sum of Its Parts? DNA, Histones, Protamines and Epigenetics. Reproduction 2010, 139, 287–301. 10.1530/REP-09-0281. [DOI] [PubMed] [Google Scholar]
- Toma A. C.; de Frutos M.; Livolant F.; Raspaud E. DNA Condensed by Protamine: A “Short” or “Long” Polycation Behavior. Biomacromolecules 2009, 10, 2129–2134. 10.1021/bm900275s. [DOI] [PubMed] [Google Scholar]
- Balhorn R.; Corzett M.; Mazrimas J.; Watkins B. Identification of Bull Protamine Disulfides. Biochemistry 1991, 30, 175–181. 10.1021/bi00215a026. [DOI] [PubMed] [Google Scholar]
- Emelyanov A. V.; Fyodorov Dv. Thioredoxin-Dependent Disulfide Bond Reduction Is Required for Protamine Eviction from Sperm Chromatin. Genes Dev. 2016, 30, 2651–2656. 10.1101/gad.290916.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskolkov N.; Bar-Shir A.; Chan K. W. Y.; Song X.; van Zijl P. C. M.; Bulte J. W. M.; Gilad A. A.; McMahon M. T. Biophysical Characterization of Human Protamine-1 as a Responsive CEST MR Contrast Agent. ACS Macro Lett. 2015, 4, 34–38. 10.1021/mz500681y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C. D.; Kirchoff D. C.; DeRouchey J. E.; Moseley H. N. B. Entropy Based Analysis of Vertebrate Sperm Protamines Sequences: Evidence of Potential Dityrosine and Cysteine-Tyrosine Cross-Linking in Sperm Protamines. BMC Genomics 2020, 21, 277. 10.1186/s12864-020-6681-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha U. R.; Juneja P.; Zhang Q.; Gurumoorthy V.; Borreguero J. M.; Urban V.; Cheng X.; Pingali S. V.; Smith J. C.; O’Neill H. M.; Petridis L. Generation of the Configurational Ensemble of an Intrinsically Disordered Protein from Unbiased Molecular Dynamics Simulation. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 20446–20452. 10.1073/pnas.1907251116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley N.; Esteban-Martín S.; de Fabritiis G. Progress in Studying Intrinsically Disordered Proteins with Atomistic Simulations. Prog. Biophys. Mol. Biol. 2015, 119, 47–52. 10.1016/j.pbiomolbio.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Duong V. T.; Chen Z.; Thapa M. T.; Luo R. Computational Studies of Intrinsically Disordered Proteins. J. Phys. Chem. B 2018, 122, 10455–10469. 10.1021/acs.jpcb.8b09029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D.; Luo R.; Chen H.-F. The IDP-Specific Force Field Ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. 10.1021/acs.jcim.7b00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRouchey J. E.; Rau D. C. Role of Amino Acid Insertions on Intermolecular Forces between Arginine Peptide Condensed DNA Helices. J. Biol. Chem. 2011, 286, 41985–41992. 10.1074/jbc.M111.295808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino Y.; Takahashi Y.; Kawarasaki M.; Akita R.; Enari H.; Yamamoto S. The Suppressive Effect of Protamine from Chum Salmon Milt on Lipid Absorption in Humans. Nippon Shokuhin Kagaku Kogaku Kaishi 2008, 55, 360–366. 10.3136/nskkk.55.360. [DOI] [Google Scholar]
- Hud N. V.; Milanovich F. P.; Balhorn R. Evidence of Novel Secondary Structure in DNA-Bound Protamine Is Revealed by Raman Spectroscopy. Biochemistry 1994, 33, 7528–7535. 10.1021/bi00190a005. [DOI] [PubMed] [Google Scholar]
- Bateman A.; Martin M.-J.; Orchard S.; Magrane M.; Agivetova R.; Ahmad S.; Alpi E.; Bowler-Barnett E. H.; Britto R.; Bursteinas B.; Bye-A-Jee H.; Coetzee R.; Cukura A.; da Silva A.; Denny P.; Dogan T.; Ebenezer T.; Fan J.; Castro L. G.; Garmiri P.; Georghiou G.; Gonzales L.; Hatton-Ellis E.; Hussein A.; Ignatchenko A.; Insana G.; Ishtiaq R.; Jokinen P.; Joshi V.; Jyothi D.; Lock A.; Lopez R.; Luciani A.; Luo J.; Lussi Y.; MacDougall A.; Madeira F.; Mahmoudy M.; Menchi M.; Mishra A.; Moulang K.; Nightingale A.; Oliveira C. S.; Pundir S.; Qi G.; Raj S.; Rice D.; Lopez M. R.; Saidi R.; Sampson J.; Sawford T.; Speretta E.; Turner E.; Tyagi N.; Vasudev P.; Volynkin V.; Warner K.; Watkins X.; Zaru R.; Zellner H.; Bridge A.; Poux S.; Redaschi N.; Aimo L.; Argoud-Puy G.; Auchincloss A.; Axelsen K.; Bansal P.; Baratin D.; Blatter M.-C.; Bolleman J.; Boutet E.; Breuza L.; Casals-Casas C.; de Castro E.; Echioukh K. C.; Coudert E.; Cuche B.; Doche M.; Dornevil D.; Estreicher A.; Famiglietti M. L.; Feuermann M.; Gasteiger E.; Gehant S.; Gerritsen V.; Gos A.; Gruaz-Gumowski N.; Hinz U.; Hulo C.; Hyka-Nouspikel N.; Jungo F.; Keller G.; Kerhornou A.; Lara V.; le Mercier P.; Lieberherr D.; Lombardot T.; Martin X.; Masson P.; Morgat A.; Neto T. B.; Paesano S.; Pedruzzi I.; Pilbout S.; Pourcel L.; Pozzato M.; Pruess M.; Rivoire C.; Sigrist C.; Sonesson K.; Stutz A.; Sundaram S.; Tognolli M.; Verbregue L.; Wu C. H.; Arighi C. N.; Arminski L.; Chen C.; Chen Y.; Garavelli J. S.; Huang H.; Laiho K.; McGarvey P.; Natale D. A.; Ross K.; Vinayaka C. R.; Wang Q.; Wang Y.; Yeh L.-S.; Zhang J.; Ruch P.; Teodoro D. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay D. J.; Renaux B. S.; Dixon G. H. The Amino Acid Sequence of Human Sperm Protamine P1. Biosci. Rep. 1985, 5, 383–391. 10.1007/BF01116555. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. I.; Cruzeiro V. W. D.; Darden T.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Huang Y.; Izadi S.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Salomon-Ferrer R.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER 2018; University of California: San Francisco, 2018.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Götz A. W.; Poole D.; le Grand S.; Walker R. C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- le Grand S.; Götz A. W.; Walker R. C. SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. 10.1016/j.cpc.2012.09.022. [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Nguyen H.; Roe D. R.; Swails J.; Case D. A.. PYTRAJ: Interactive Data Analysis for Molecular Dynamics Simulations, 2016, 10.5281/zenodo.44612. [DOI]
- Kabsch W.; Sander C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Ziebarth J. D.; Williams J.; Wang Y. Selective Adsorption of Heteropolymer onto Heterogeneous Surfaces: Interplay between Sequences and Surface Patterns. Macromolecules 2008, 41, 4929–4936. 10.1021/ma800212n. [DOI] [Google Scholar]
- Rieloff E.; Skepö M. The Effect of Multisite Phosphorylation on the Conformational Properties of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2021, 22, 11058. 10.3390/ijms222011058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Dong X.; Li X.; Liu Z.; Liu H. Prediction of Disulfide Bond Engineering Sites Using a Machine Learning Method. Sci. Rep. 2020, 10, 10330 10.1038/s41598-020-67230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Dunbrack R. L. PISCES: A Protein Sequence Culling Server. Bioinformatics 2003, 19, 1589–1591. 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- Yan S.; Peck J. M.; Ilgu M.; Nilsen-Hamilton M.; Lamm M. H. Sampling Performance of Multiple Independent Molecular Dynamics Simulations of an RNA Aptamer. ACS Omega 2020, 5, 20187–20201. 10.1021/acsomega.0c01867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallops C. E.; Yu C.; Ziebarth J. D.; Wang Y. Effect of the Protonation Level and Ionic Strength on the Structure of Linear Polyethyleneimine. ACS Omega 2019, 4, 7255–7264. 10.1021/acsomega.9b00066. [DOI] [Google Scholar]
- Wang J.; Cao Z.; Li S. Molecular Dynamics Simulations of Intrinsically Disordered Proteins in Human Diseases. Curr. Comput.-Aided Drug Des. 2009, 5, 280–287. 10.2174/157340909789577865. [DOI] [Google Scholar]
- Watts N. R.; Eren E.; Zhuang X.; Wang Y.-X.; Steven A. C.; Wingfield P. T. A New HIV-1 Rev Structure Optimizes Interaction with Target RNA (RRE) for Nuclear Export. J. Struct. Biol. 2018, 203, 102–108. 10.1016/j.jsb.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292, 195–202. 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Bernhofer M.; Dallago C.; Karl T.; Satagopam V.; Heinzinger M.; Littmann M.; Olenyi T.; Qiu J.; Schütze K.; Yachdav G.; Ashkenazy H.; Ben-Tal N.; Bromberg Y.; Goldberg T.; Kajan L.; O’Donoghue S.; Sander C.; Schafferhans A.; Schlessinger A.; Vriend G.; Mirdita M.; Gawron P.; Gu W.; Jarosz Y.; Trefois C.; Steinegger M.; Schneider R.; Rost B. PredictProtein - Predicting Protein Structure and Function for 29 Years. Nucleic Acids Res. 2021, 49, W535–W540. 10.1093/nar/gkab354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; Evans R.; Pritzel A.; Green T.; Figurnov M.; Ronneberger O.; Tunyasuvunakool K.; Bates R.; Žídek A.; Potapenko A.; Bridgland A.; Meyer C.; Kohl S. A. A.; Ballard A. J.; Cowie A.; Romera-Paredes B.; Nikolov S.; Jain R.; Adler J.; Back T.; Petersen S.; Reiman D.; Clancy E.; Zielinski M.; Steinegger M.; Pacholska M.; Berghammer T.; Bodenstein S.; Silver D.; Vinyals O.; Senior A. W.; Kavukcuoglu K.; Kohli P.; Hassabis D. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M.; Anyango S.; Deshpande M.; Nair S.; Natassia C.; Yordanova G.; Yuan D.; Stroe O.; Wood G.; Laydon A.; Žídek A.; Green T.; Tunyasuvunakool K.; Petersen S.; Jumper J.; Clancy E.; Green R.; Vora A.; Lutfi M.; Figurnov M.; Cowie A.; Hobbs N.; Kohli P.; Kleywegt G.; Birney E.; Hassabis D.; Velankar S. AlphaFold Protein Structure Database: Massively Expanding the Structural Coverage of Protein-Sequence Space with High-Accuracy Models. Nucleic Acids Res. 2022, 50, D439–D444. 10.1093/nar/gkab1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis A. K.; Freedman R. B. Assisting Oxidative Protein Folding: How Do Protein Disulphide-Isomerases Couple Conformational and Chemical Processes in Protein Folding?. Top. Curr. Chem. 2013, 328, 1–34. 10.1007/128_2011_171. [DOI] [PubMed] [Google Scholar]
- Hudson D. A.; Gannon S. A.; Thorpe C. Oxidative Protein Folding: From Thiol–Disulfide Exchange Reactions to the Redox Poise of the Endoplasmic Reticulum. Free Radical Biol. Med. 2015, 80, 171–182. 10.1016/j.freeradbiomed.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P. J.; Bulleid N. J. Mechanisms of Disulfide Bond Formation in Nascent Polypeptides Entering the Secretory Pathway. Cells 2020, 9, 1994. 10.3390/cells9091994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff K. M.; Pappu Rv. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208 10.1016/j.jmb.2021.167208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.