

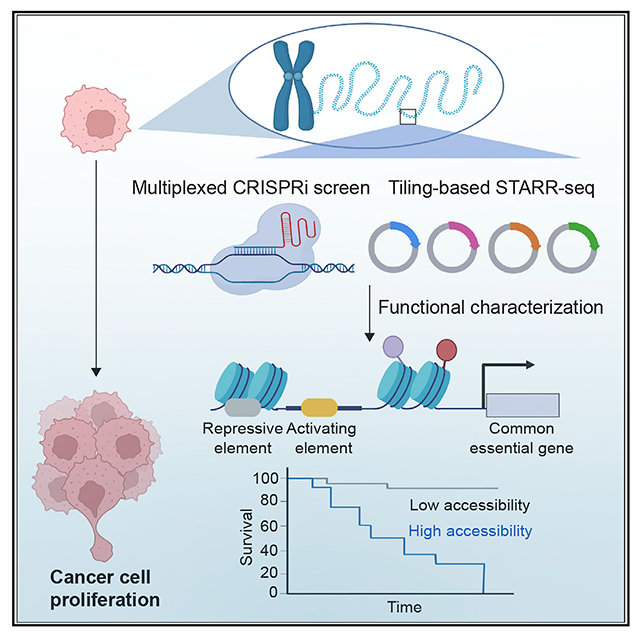
SUMMARY
A scarcity of functionally validated enhancers in the human genome presents a significant hurdle to understanding how these cis-regulatory elements contribute to human diseases. We carry out highly multiplexed CRISPR-based perturbation and sequencing to identify enhancers required for cell proliferation and fitness in 10 human cancer cell lines. Our results suggest that the cell fitness enhancers, unlike their target genes, display high cell-type specificity of chromatin features. They typically adopt a modular structure, comprised of activating elements enriched for motifs of oncogenic transcription factors, surrounded by repressive elements enriched for motifs recognized by transcription factors with tumor suppressor functions. We further identify cell fitness enhancers that are selectively accessible in clinical tumor samples, and the levels of chromatin accessibility are associated with patient survival. These results reveal functional enhancers across multiple cancer cell lines, characterize their context-dependent chromatin organization, and yield insights into altered transcription programs in cancer cells.
Graphical Abstract
In brief
Chen et al., carry out large-scale CRISPR screens to identify the enhancers required for cell fitness in multiple cancer cell lines. They further perform massively parallel reporter assays to characterize the structure of active enhancers, showing that they are modular and contain both activating and repressive DNA elements.
INTRODUCTION
Disruption of gene regulation is a major cause of human diseases ranging from congenital developmental disorders to cancers. Gene regulation patterns in each cell and tissue are dictated by the context-dependent interactions between cis-regulatory DNA elements in the genome and various transcriptional regulators. Millions of candidate cis-regulatory elements have been annotated in the human genome based on transcription factor (TF) binding, chromatin accessibility, histone modifications, and DNA hypomethylation (Hnisz et al., 2016; Kellis et al., 2014; Moore et al., 2020). Enhancers are often located at a distance from their target genes and may control gene expression through long-range chromatin contacts in both spatial and temporal manners (Schoenfelder and Fraser, 2019; Yu and Ren, 2017; Zheng and Xie, 2019; Bothma et al., 2014; Small et al., 1992). Due to the sparsity of functionally characterized enhancers in the human genome, how these elements contribute to human diseases is still incompletely understood.
Large-scale profiling studies from the Encyclopedia of DNA Elements, the Roadmap Epigenomics Projects, and The Cancer Genome Atlas program have identified millions of putative enhancers from >1,000 human cell/tissue types and begun to shed light on the altered chromatin landscapes in different cancer types (Chen et al., 2018a; Corces et al., 2018; Gorkin et al., 2020; Kundaje et al., 2015; Leung et al., 2015; Moore et al., 2020; Weinstein et al., 2013). In addition, a recent large-scale CRISPR-Cas9-based loss-of-function screens further uncovered ~2,000 genes essential for proliferation in diverse cancer cell lines (Ghandi et al., 2019). Most of these essential genes are necessary for cell proliferation/survival in multiple cancer types and involved in transcriptional and translational processes (Hart et al., 2015). While uncontrollable cell growth is a key feature during tumorigenesis, the functionality of putative enhancers that control essential genes remains to be determined, and this gap has hindered the elucidation of gene regulatory programs in cancer cells.
The advance in CRISPR-based perturbation assays coupled with sequencing has significantly improved our ability to identify functional enhancers from genomic regions bearing chromatin accessibility, histone modification, or other enhancer signatures (Fulco et al., 2019; Gasperini et al., 2019; Reilly et al., 2021). Due to diverse chromatin accessibility and histone modifications from different tissue/cell types, it is necessary to design and generate individual screening libraries for genes of interest in each corresponding cell line across various tissue types. For example, while abnormal activation of key oncogene c-MYC has been implicated in most types of human cancer (Dang, 2012; Meyer and Penn, 2008), only a few of functional MYC enhancers have been characterized in a handful of cancer cell lines, including K562 (Fulco et al., 2016), HCT116 (Hnisz et al., 2015), and A549 (Zhang et al., 2016). This limitation highlights the need for a high-throughput and unbiased approach to systematically test candidate cis-regulatory elements bearing chromatin accessibility or histone modifications from different cell lines/types. Here, we adopt a tiling-path library design, which utilizes paired-guide RNAs (pgRNAs) targeting broader genomic regions harboring the genes of interest. By coupling this tiling pgRNA library design with a CRISPR inference (CRISPRi)-based screen, we systematically identified and characterized enhancers necessary for cell proliferation and fitness (hereafter referred to as essential enhancers) for two key oncogenes, MYC and MYB, in ten human cancer cell lines representing six major cancer types. Based on the features of essential enhancers identified from the pilot study, we then carried out large-scale CRISPRi screens to interrogate the effect of >11,000 distal putative enhancers on cell fitness, finding hundreds of essential enhancers. Furthermore, we characterized the regulatory elements within the identified essential enhancers using a massively parallel reporter assay (STARR-seq) (Arnold et al., 2013), to reveal the organization principle of essential enhancers.
RESULTS
Characterization of pgRNA design for CRISPR-based functional perturbation screen in cancer cells
We previously utilized a CRISPR-Cas9-mediated deletion screen with a tiling-path pgRNA design to uncover functional enhancers within a given genomic region near the POU5F1 locus in human embryonic stem cells (Diao et al., 2017). To establish a functional framework to characterize essential enhancers required for cancer proliferation and survival, we first focused on the identification of essential enhancers near two key protooncogenes, MYC and MYB. Sustained MYC activation is required for tumorigenesis, and the partial suppression of MYC in cancer cells is sufficient to cause acute tumor regression due to the unusual transcriptional addiction in cancer (Bradner et al., 2017; Gabay et al., 2014). MYB, a cell-type-specific oncogene, is vital for tumorigenesis in leukemia, colorectal, and breast cancers partially through the regulation of key oncogenes in those cancer types (Mansour et al., 2014; Ramsay and Gonda, 2008). Although these oncogenic TFs have been well characterized and are recognized as critical cancer drivers, it remains challenging to develop small molecules to target them selectively in cancer cells while avoiding harming the proliferation of normal cells (Chen et al., 2018b; Pattabiraman and Gonda, 2013). Based on previous chromatin contact maps generated from different cell lines (Rao et al., 2014), we designed 13,373 pgRNAs targeting ~3.6 megabase (Mb) genomic interval within topologically associated domains around MYC and MYB loci and another set of 1,455 pgRNAs to use as negative controls (including non-targeting controls, which are the genomic sequence lacking protospacer-adjacent motifs for targeting by CRISPR-Cas9 or safe-targeting genomic loci, which are the validated genomic regions that do not cause growth defects in CRISPR-Cas9-based deletion screen (Morgens et al., 2017)) (Table S1). The mean genomic distance between the gRNAs in each pair was ~3 kilobase (kb), and the genomic spans of adjacent pgRNAs overlapped by 2.7 kb on average (Figure 1A). We reasoned that this tiling path library will interrogate candidate enhancers from cell lines/types with different chromatin accessibility or histone modifications. To compare the performance of our pgRNA library design to the common approaches, we also designed a single-guide RNA (sgRNA) library targeting all DNase-hypersensitive sites (DHSs) and H3K27ac peaks around those two key oncogenes identified from chronic myeloid leukemia K562 cells (Figure 1A). We next carried out a proliferation-based screen to characterize essential enhancers (Figure 1B) using a pgRNA library coupled with CRISPR-Cas9 nuclease (CRISPRn) or a sgRNA library coupled with CRISPR inference (CRISPRi). To identify essential enhancers from the CRISPR-based perturbation screens, we applied two computational methods, RELICS and CRISPY, which determine the perturbation effects based on the sgRNA or pgRNA read counts before and after 14 double times (see STAR Methods; Figure S2C). In addition, we considered a 500 bp genomic region up/downstream target sequence of each gRNA in the pair as the area of effect for a CRISPRi-based screen (Thakore et al., 2015). RELICS uses a generalized linear mixed model framework (Fiaux et al., 2020) to jointly describe the observed gRNA counts across time points under two models: a regulatory model (gRNAs target a regulatory sequence) and a background model (gRNAs do not target a regulatory sequence). CRISPY is a software pipeline based on a strategy we reported previously (Diao et al., 2017) that uses negative binomial statistics to identify the depleted gRNA species after 14 doubling times. Our results showed that several known essential enhancers for MYC (Fulco et al., 2016) are identified from the CRISPRi-sgRNA screen but not from the CRISPRn-pgRNA screen (Figure S1), indicating that applying tiling-based deletion to cancer cells could be confounded by aneuploidy of the cancer genome (Aguirre et al., 2016). Based on this observation, we adopted the CRISPRi-based screen together with the tiling design of pgRNAs. When we compared the effects of sgRNAs and pgRNAs using the CRISPRi-based cell proliferation screen in K562, we found that the tiling-path pgRNAs have comparable performance in detecting essential enhancers as the sgRNA screen (Figures 1C–1E and S2), indicating that both approaches provide similar resolution to uncover essential enhancers. Moreover, we observed that pgRNAs achieved greater effects on cell growth than sgRNAs after silencing of the same essential enhancers (Figure 1F).
Figure 1. Comparison of the performance of the CRISPRi screen using single or paired gRNAs.
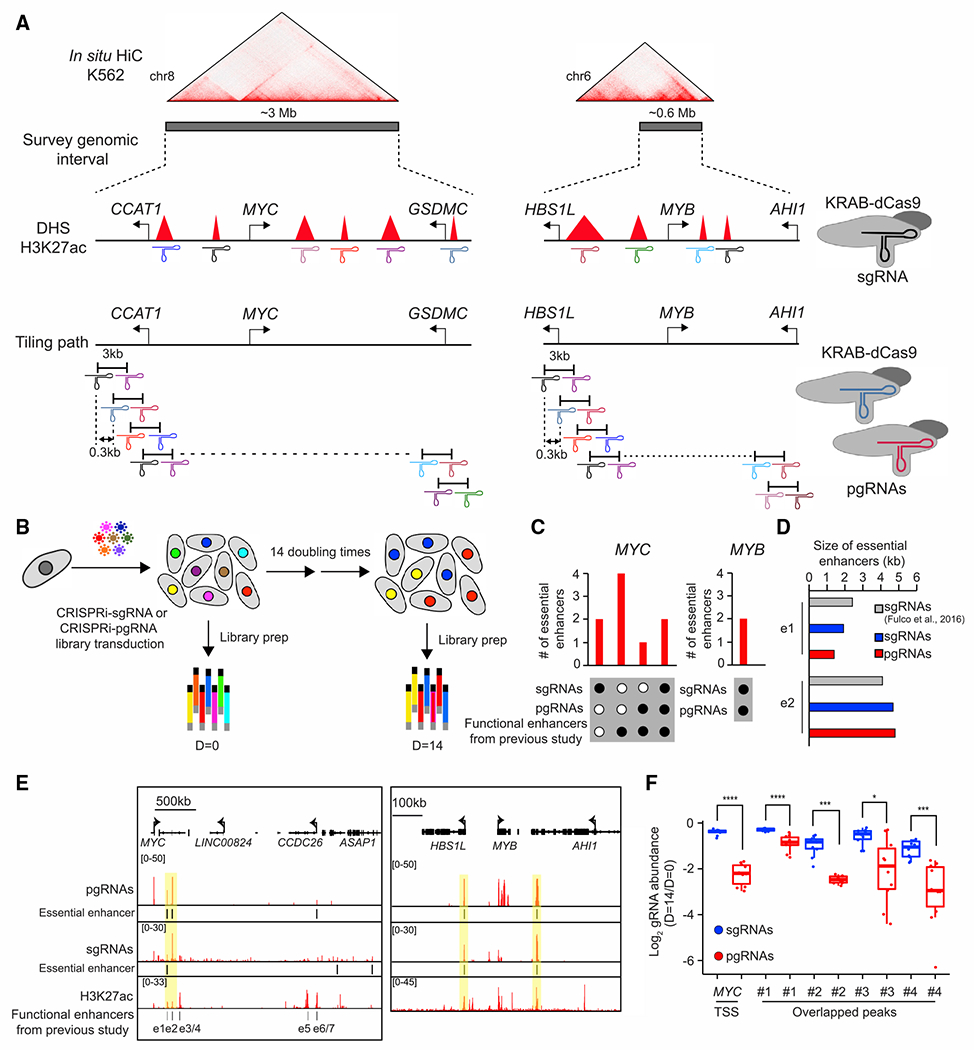
(A) Illustration of the design of the CRISPRi screen using single-guide (sgRNA) or paired guide RNA (pgRNA) libraries. We designed sgRNAs targeting every DNase hypersensitive site and a H3K27ac peak located within the 3- and 0.6-Mb MYC and MYB loci, or utilized tiling-based pgRNAs targeting the 3.6-Mb genomic region around MYC and MYB loci, respectively. Hi-C data in K562 cells were from a previous study (Rao et al., 2014).
(B) Schematic of the experimental strategy illustrates the identification of essential enhancers based on cell proliferation assay.
(C) Summary of essential enhancers for MYC or MYB identified from sgRNA- or pgRNA-based screen. Black dots and white dots indicate the presence and absence of essential enhancers identified from each category, respectively. Annotated functional enhancers were reported in a previous study (Fulco et al., 2016).
(D) Comparison between essential enhancers identified using sgRNA and pgRNA libraries at the MYC locus.
(E) The genome browser snapshot shows the CRISPRi scores (top) and essential enhancers (middle) identified from sgRNA and pgRNA libraries at the MYC (left) and MYB (right) loci. H3K27ac ChIP-seq signal, H3K27ac (bottom), and e1-e7 are the functional enhancers identified from the previous study (Fulco et al., 2016). The common essential enhancers identified from both screens are highlighted in yellow.
(F) The effect of perturbations using sgRNAs and pgRNAs in cell proliferation-based screen. The common peaks indicate essential enhancers identified from both sgRNA- and pgRNA-based screens. Common peaks 1 and 2 are previously identified enhancer e1 and e2, respectively. Fold changes represent the ratios between read counts after 14 doubling times (D = 14) to initial population (D = 0). p values were determined by two-sided Wilcoxon test (*p < 0.1; ***p < 0.001; ****p < 0.0001).
A previous CRISPRi-based proliferation screen identified seven functional enhancers (e1-e7) for MYC in K562 cells (Fulco et al., 2016), and our CRISPRi screen using the tiling pgRNA library only identified three of them (e1-2 and e6). We further tested the four (e3, e4, e5, and e7) previously reported enhancers by CRISPRi individually but did not observe significant growth defects in wild-type K562, except for a subtle reduction in MYC gene expression for three of them (e3-e4, e7) (Figures S3A–S3E). We also performed a second CRISPRi screen with the same pgRNA library using a genetically modified K562 cell line with a fluorescent reporter gene inserted downstream of the MYC locus (Figure S3F) and identified two additional functional enhancers (e3-e4) together with the three essential enhancers (e1-2, e6) based on MYC gene expression (Figure S3G). Depending on the type of readout chosen in the tiling pgRNA-based CRISPRi screen, the cell proliferation assay could identify essential enhancers that show detectable growth defects and strong effects on target gene expression in our experimental system.
The CRISPRi-pgRNA screen identifies essential enhancers in the MYC and MYB loci across multiple cancer cell lines
We next carried out a proliferation-based screen using CRISPRi with the above tiling pgRNA library to identify essential enhancers near the MYC and MYB loci in ten different human cancer cell lines representing six major cancer types (lung, breast, liver, colorectal, prostate, and leukemia) (Figure S5A). As expected, we found that the pgRNAs read counts were highly reproducible between biological replicates (Pearson’s R = 0.69–0.97), and the abundance of pgRNAs targeting the promoter of essential genes consistently decreased after 14 doubling times (Figure S4). Overall, we identified 24 robust and reproducible essential enhancers (Figures S5B and S5C; Table S4), which were found by both statistical approaches, including three and one previously characterized enhancers for MYC in K562 and HCT116, respectively (Fulco et al., 2016; Hnisz et al., 2015) (Figures 2A, S5B, and S5C). The majority of these essential enhancers were located hundreds of kilobases away from MYC (median genomic distance = 224,114 bp; Figure S5D) and exhibited chromatin contacts with the MYC promoter, as evidenced by proximity ligation-assisted ChIP-seq (PLAC-seq) experiments (Fang et al., 2016) with an H3K4me3 antibody (Figure 2B). We further verified that silencing the essential enhancers near the MYC and MYB loci individually by KRAB-dCas9 led to a significant reduction in target gene expression and cell proliferation (Figures 2C and S5E–S5H). In addition, our data showed that essential enhancers overlapped with ~4.3% and ~5% of DHSs or K27ac around target genes, respectively (Figure 2D). Further analysis revealed that these essential enhancers were strongly associated with active enhancer marks, such as DHS and H3K27ac, in the cell lines where they were identified (Figure 2E). Together, we established a functional framework that enables functional characterization of a large number of putative enhancers bearing chromatin accessibility or histone modifications across multiple cancer cell lines in a high-throughput, accurate, and reproducible manner.
Figure 2. Unbiased CRISPRi screen with a tiling pgRNA library identified essential enhancers around MYC and MYB oncogenes in 10 human cancer cell lines.
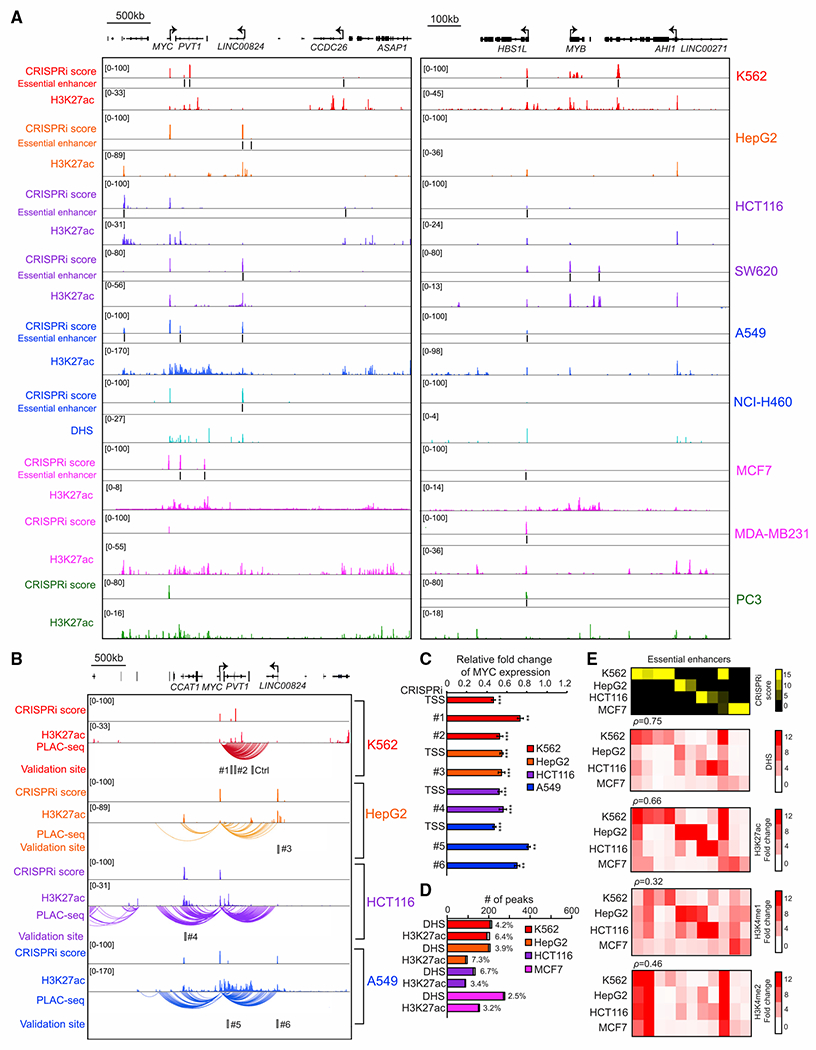
(A) The genome browser snapshot shows CRISPRi score (top), essential enhancers, and H3K27ac ChIP-seq signal, H3K27ac (middle), or DNase signal, DHS (bottom), from the indicated cell lines.
(B) Chromatin interactions identified by H3K4me3 PLAC-seq at the MYC locus across four different cancer cell lines. The gray box represents the selected genomic region for further validation.
(C) Gene expression measurement of the MYC gene by qRT-PCR after silencing the selected essential enhancers by CRISPRi in various cell lines (red, K562; orange, HepG2; purple, HCT116; blue, A549). Relative fold changes represent the ratios of gene expression from the CRISPRi silenced essential enhancer to the control samples. Data shown are mean ± SD from three biological replicates performed. p values were determined by a two-tailed Student’s t test after silencing individual enhancer compared with control cells (**p < 0.01, ***p < 0.001).
(D) The percentage of DHS or H3K27ac peaks that overlapped with essential enhancers from indicated cell lines. Gray bars indicate the number of DHS or H3K27ac peaks overlapped with essential enhancers (red, K562; orange, HepG2; purple, HCT116; blue, A549; pink, MCF7).
(E) Correlation between enhancer features (chromatin accessibility and histone modifications) and the function of essential enhancers across different cell types. The heatmap represents the chromatin accessibility and the signals of active enhancer marks across various cell lines. Each row represents the signal from the indicated dataset in each cell line and each column represents individual essential enhancers identified from the CRISPRi-pgRNA screen.
Large-scale identification of essential enhancers in colorectal cancer cells
Aberrant activation of enhancers is a crucial signature in colorectal cancers (Akhtar-Zaidi et al., 2012; Cohen et al., 2017), but only a small number of enhancers with validated function in colorectal cancer cells (CRCs) are known. Based on our findings that essential enhancers are strongly associated with active enhancer marks and spatially interact with target promoters from the pilot study (Figure 2), we used a pooled CRISPRi screen to interrogate the function of 6,642 putative distal enhancers, as determined by H3K27ac ChIP-seq signals and at least 3 kb apart from the known transcriptional start sites, located within 500 kb from 532 previously reported essential genes in the HCT116 CRC line (Hart et al., 2014, 2015). In addition, we also included additional 4,554 cell-line-specific active enhancers identified from HCT116 (Figure 3A). We designed five sets of pgRNAs for each distal putative enhancer and the promoter of 1,017 essential genes as positive controls, respectively. We reasoned that the utilization of multiple pgRNAs could minimize false negatives due to the potential low targeting efficiency from certain gRNA-targeting sequences. As negative controls, we designed 1,913 non-targeting pgRNAs and 5,215 pgRNAs that target 460 genomic control regions that do not show growth defects in previous CRISPR-Cas9 deletion-based proliferation screens (Morgens et al., 2017) and the promoters of 583 non-essential genes (Table S2). To identify candidate essential enhancers, we utilized a modified robust ranking aggregation algorithm (Kolde et al., 2012) from the MAGeCK method (Li et al., 2014), which is widely used to examine gene essentiality in genome-wide CRISPR-Cas9 knockout screen, and consider the effects on cell fitness from multiple pgRNAs targeting the same putative enhancer.
Figure 3. Large-scale cell fitness-based CRISPRi screen identified essential enhancers in a colorectal cancer cell line.
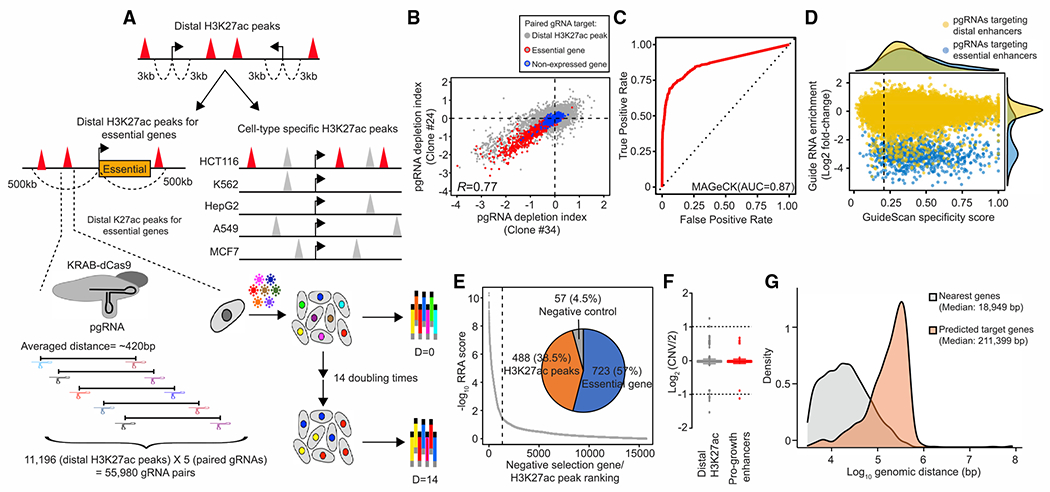
(A) Selection criteria for distal putative enhancers to be included in the screen and the design of experimental strategy. The red triangles represent distal H3K27ac peaks selected in this screen, and the gray triangles represent non-cell-type-specific H3K27ac peaks. The arrow indicates the transcription start site (TSS). Each distal H3K27ac peak is targeted with five sets of paired guide RNAs (pgRNAs), and the associated growth effect is measured from a pooled CRISPRi screen.
(B) Correlation of fitness effects in two independent KRAB-dCas9 stably expressed clones (Pearson’s R = 0.77). The paired gRNA depletion index shown is the average from two biological replicates in each clone. Red circled dots indicate pgRNAs targeting the promoter of essential genes, and blue circled dots indicate pgRNAs targeting the promoter of the non-essential genes. Gray dots represent pgRNAs targeting distal H3K27ac peaks.
(C) The selection of optimal thresholds to identify candidate targets from the screen. An empirical receiver operating characteristic (ROC) curve plots the true-positive rate against the false-positive rate for different possible cutoff points in negative selection. Totals of 1,017 essential genes and 583 non-essential genes with low expression level were used as positive and negative in the analysis. The area under the curve (AUC) shows the performance of the MAGeCK algorithm.
(D) Comparison of pgRNA fitness effect with specificity score. pgRNAs targeting all selected distal H3K27ac peaks are labeled as yellow, while pgRNAs targeting candidate essential enhancers identified from the screen (empirical FDR < 0.02) are labeled as blue. The specificity score of pgRNA was determined using the lowest specificity score from one of the two sgRNAs in each pair. Fold change represents the ratios between read counts in the D = 14 population and the control population (D = 0).
(E) Summary of candidate targets identified from the screen. Robust ranking aggregation (RRA) score is generated from the MAGeCK algorithm after including the effect from multiple pgRNAs targeting the same distal H3K27ac peak. A target with a smaller RRA score indicates a more substantial reduction in negative selection. Overall, 723 targets are essential genes, 488 targets are distal H3K27ac peaks, and 57 targets are non-essential genes or safe targets from the negative control group (empirical FDR < 0.02; 75% sensitivity).
(F) Comparison of DNA copy number at all selected distal H3K27ac peaks (n = 11,111) and essential enhancers (n = 488). CNV, copy number variation. No significant difference is observed (p = 0.11). p values were determined by the two-sided Wilcoxon test.
(G) Comparison of genomic distance between essential enhancers and nearest genes or predicted target genes. Significant longer genomic distance between predicted target genes and essential enhancers is observed (p = 6.6 × 10−23). p values were determined by the two-sided Wilcoxon test.
We first generated multiple stable KRAB-dCas9-expressed clones and observed a good correlation in H3K27ac levels with parental cells (Pearson’s R = 0.85), indicating that expression of KRAB-dCas9 had minimal non-specific effects on the activity of distal enhancers (Figure S6). Next, we performed a large-scale CRISPRi screen in two biological replicates from two independent KRAB-dCas9-expressed clones, respectively, to avoid the clonal-level bias. We obtained reproducible results evidenced by a strong correlation in the depletion index, which is the measured growth defects upon the perturbation in the negative selection screen, between the two clonal cell lines (Pearson’s R = 0.77). As expected, pgRNAs targeting the promoters of essential genes showed a greater reduction in fitness scores than pgRNAs targeting non-essential genes (Figure 3B). Furthermore, we used the read counts of non-targeting pgRNAs to estimate the null distribution when calculating the p values. To determine the optimal threshold to select candidate essential enhancers, we generated a receiver operating characteristic curve using 1,017 essential genes as positive controls along with 583 non-essential genes with low or undetectable levels of expression as negative controls. We identified candidate essential enhancers using a threshold that recovers 75% of the positive controls with empirical FDR < 0.02 (Figure 3C). The empirical FDR was calculated based on the percentage of candidate targets identified from negative controls above this selected threshold, including non-essential genes and safe-target control genomic regions to account for the possible toxicities from the silencing mediated by KRAB-dCas9. In general, we found that four out of five pgRNAs (on average) targeting candidate essential enhancers in the library can achieve detectable growth defects (Figure 3D; Table S4). Furthermore, we did not observe the enrichment of pgRNAs with low specificity scores (<0.2) targeting the candidate essential enhancers and no significant differences in the DNA copy number at essential enhancer loci (Figures 3D and 3F), affirming that the growth defects associated with the identified pgRNAs were unlikely due to potential off-target toxicity. Nevertheless, to identify essential enhancers at high confidence, we further excluded candidate essential enhancers targeted by multiple pgRNAs with low target specificity scores (<0.2) or were not selected by GuideScan software (Perez et al., 2017).
Overall, we identified 488 candidate essential enhancers (Figure 3E; Table S4). As expected, most essential enhancers are located within the 500 kb genomic range near the essential genes (observed = 334/expected = 290; hypergeometric test; p = 4.08 × 10−6). We further predicted their targets by integrating genome-wide maps of chromatin interactions centered at active or poised gene promoters determined using H3K4me3 PLAC-seq data and scores of gene dependence from previous genome-wide CRISPR-Cas9 knockout screens (see STAR Methods). We reasoned that the targets of essential enhancers are likely also crucial for cell survival/proliferation. Together, we predicted a total of 190 enhancer/gene pairs (E-G pairs) between 150 essential enhancers and 190 target genes (Table S4). Consistent with the notion that enhancers often control gene expression at tens to hundreds kilobases distant from their target genes, we observed significant longer genomic distances (median genomic distance = 211,399 bp) between essential enhancers and predicted target genes than the distances between the essential enhancers to the nearest genes (p = 6.6 × 10−23; Figure 3G). Interestingly, the predicted target genes tended to be ubiquitously expressed across various cell types (Figure S7A), with 94.4% of them found to be essential in 6 or more cell types (Figure S7B) (Hart et al., 2015), and participate in essential biological processes (Figure S7C). Furthermore, we also validated the role of essential enhancers to regulate several target genes by CRISPRi individually (Figure S7D).
The essential enhancers generally harbor both activating and repressive DNA elements
To further characterize the enhancer activity of the essential enhancers identified above and to delineate the cis-regulatory DNA elements within each of them, we performed massively parallel reporter assays, using the strategy known as STARR-seq (Arnold et al., 2013). We constructed a STARR-seq plasmid library consisting of 12,833 of 190-bp DNA fragments inserted downstream of a reporter gene driven by a minimum promoter made from the bacterial plasmid origin-of-replication (Muerdter et al., 2018). For each essential enhancer, we designed on average 23 of 190 bp-long DNA oligos to tile 1 kb upstream to 1 kb downstream with a 120 bp step size (Figure 4A). In addition, we included 60 oligonucleotides tiling 4 known MYC enhancers previously identified from HCT116 (Hnisz et al., 2015) and SV40 enhancer (Herr and Clarke, 1986; Schirm et al., 1987) as positive controls, and 2,095 oligonucleotides randomly selected from the yeast open reading frame (ORF) sequences as negative controls (see STAR Methods; Table S3). The plasmid library was transfected into HCT116 cells by electroporation in triplicate. Two days after transfection, we extracted DNA and RNA from the transfected cells and sequenced the plasmid DNA and reporter RNA transcripts containing the oligonucleotides in the library. We defined the regulatory activity of individual DNA element by comparing the RNA counts to the plasmid DNA counts (see STAR Methods) and showed that the RNA/DNA ratios were highly reproducible among three biological replicates (Pearson’s R > 0.9). Using a method described from the previous study (Yan et al., 2021), we determined DNA elements with enriched and depleted RNA counts using negative binomial regression statistics. With an empirically defined FDR threshold of 0.05 based on the set of yeast ORF oligonucleotides, we found 1,101 oligonucleotides with significant activating activity and 1,318 oligonucleotides with repressor activities (Figure 4B; see STAR Methods). Of the 488 HCT116 essential enhancers, 369 contained at least one activating element (median = 2), and 437 harbored at least one repressive element (median = 2) (Figures 4E and 4F).
Figure 4. Dissection of regulatory elements within the essential enhancers.
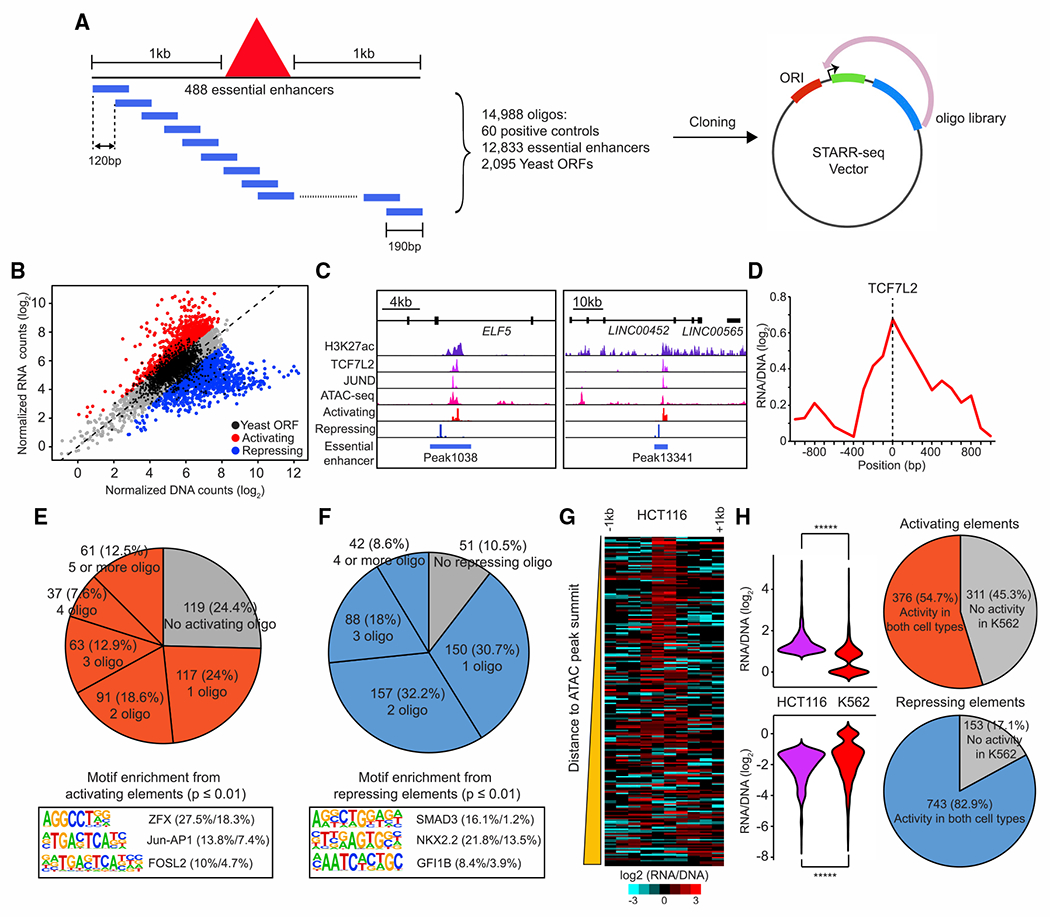
(A) A schematic diagram represents the design of a tiling-based STARR-seq library to test the enhancer activity of DNA elements within the 488 essential enhancers identified in HCT116 cells.
(B) Identification of activating or repressing DNA fragments within essential enhancers. Oligonucleotides with significant enhancer activities are labeled as red and oligonucleotides with significant repressive activities are shown as blue (empirical FDR < 0.05). An empirical FDR of 5%, calculated based on the rate of enriched negative control yeast oligonucleotides, was used to determine the activating and repressive elements (Yan et al., 2021).
(C) Examples of activating and repressing elements within the same essential enhancer. Genome browser snapshot shows the signals of active elements from STARR-seq overlapped with ChIP-seq signals of H3K27ac, TCF7L2, and JUND at accessible genomic regions while the signals of repressive elements are enriched in inaccessible chromatin regions at two representative essential enhancers.
(D) Aggregated plot shows enrichment of activating elements centered at the peak of TCF7L2 binding sites. A total of 125 TCF7L2 peaks overlapped with essential enhancers was used in this analysis.
(E and F) Top: the percentage of activating (E) and repressing (F) elements found in essential enhancers. Bottom: the top 3 enriched motifs associated with activating (E) and repressing (F) elements identified by HOMER are shown (Heinz et al., 2010). The percentage of enriched motifs from the foreground and background groups is shown nextto each motif. The foreground group contains genomic sequences with activating or repressing elements and the background group contains genomic sequences 1 kb up or downstream of the corresponding 488 essential enhancers. p values were determined using a hypergeometric test.
(G) Distribution of activating and repressing elements within the essential enhancers. The heatmap shows the enrichment of activating (left) or repressing (right) signals comprising the 1 kb surrounding the summit of ATAC-seq peaks overlapped with 488 essential enhancers. The essential enhancers are sorted from nearest to farthest according to the genomic distance of the activating elements to the summits of 229 ATAC-seq peaks overlapped with them.
(H) Comparison of the activity of activating (n = 687) and repressing elements (n = 896) between HCT116 and K562. p values were determined by the two-sided Wilcoxon test (*****p < 0.00001).
Unexpectedly, we found that 331 essential enhancers contained both activating and repressive elements. Using ATAC-seq data generated from HCT116, we found that the activating elements were strongly enriched at or near the accessible chromatin regions, while the repressive elements were broadly distributed outside of ATAC-seq peaks (Figures 4G and S8C). We also found that the activating DNA elements within the essential enhancers were centered at the binding sites of TCF7L2 (Figures 4C and 4D), which is a key transcriptional effector of Wnt/β-catenin signaling pathway (Frietze et al., 2012; He et al., 1998). Oncogenic mutations in APC and KRAS are frequently found in the majority of CRC (Cancer Genome Atlas Network, 2012) and are known to drive cancer proliferation by activating Wnt/β-catenin and Ras-Raf-MEK-ERK (MAPK/ERK) signaling pathways, respectively (Fearon and Vogelstein, 1990; Papke and Der, 2017; Pylayeva-Gupta et al., 2011). Through the motif analysis on activating elements, we identified motifs of Jun-AP1 and FOSL2 and chromatin binding of JunD, which acts downstream of the MAPK/ERK signaling pathway, were enriched at the activating DNA elements (Figures 4C, 4E, and S8A), indicating that activating elements within essential enhancers are crucial transcriptional hubs downstream of the signaling pathways driven by oncogenic mutations in CRC. Consistent with the hypothesis that Jun-AP1 plays key roles in cancer proliferation via the interaction with activating elements, inactivation of Jun-AP1 has been shown to attenuate intestinal cancer development in Apc Min mice (Nateri et al., 2005). Furthermore, our motif analysis also revealed that zinc-finger protein X-linked (ZFX), which was required for cell proliferation in HCT116 and the elevated expression was associated with poor outcomes in CRC (Jiang and Liu, 2015), was enriched at active elements within the essential enhancers (Figure 4E).
Interestingly, the TF binding motifs enriched at the repressive elements corresponded to TFs known as tumor suppressors or involved in the signaling pathway to suppress CRC proliferation (Figures 4F and S8B). The NK homeobox 2.2 (Nkx2.2) and growth factor-independent 1 (Gfi1) were both identified as tumor suppressor genes in CRC. Overexpression of those two TFs reduced cell proliferation in HCT116 in vitro and in vivo (Chen et al., 2019; He et al., 2020). In addition, Smad3 is one of the critical TFs downstream of the transforming growth factor β signaling pathway, which has been shown to inhibit cell proliferation, induce apoptosis, and suppress tumorigenicity in CRC (Massagué and Wotton, 2000; Wang et al., 2008). To determine whether activating elements within HCT116 essential enhancers were selectively activated in this cancer cell line, we further carried out the same STARR-seq assay in K562 cells. Our data indicated that 311 and 153 of activating and repressive elements were only active in HCT116, respectively (Figure 4H). Together, these results support a model that essential enhancers have modular structures containing both activating and repressive elements. They would be dynamically regulated by both transcriptional activating and repressor factors in the cell to control gene expression. Because the essential enhancers that we tested using STARR-seq assays were activating target genes to support cell proliferation and survival, they are more likely bound by transcriptional activators in these cells than repressors. Therefore, the chromatin accessibility within these enhancers would more likely occur at the binding sites of activators than the binding sites of the repressor proteins.
Essential enhancers have lineage-specific roles in cell fitness
The STARR-seq data indicated that activating elements within 202 HCT116 essential enhancers display regulatory activity in both K562 and HCT116, raising the question of whether those essential enhancers are also required for cell proliferation in K562. To characterize the function of essential enhancers, we interrogated the requirement of the same set of distal putative enhancers for cell proliferation in K562 cells by carrying out CRISPRi screens in duplicate with the same pgRNA library. As expected, we observed moderate correlation in the depletion index from biological replicates (Pearson’s R = 0.51) and a greater reduction in cell fitness when pgRNA targeting essential genes than non-essential genes. Using the same criteria to select candidate essential genes/enhancers as our previous screen in HCT116, we identified 617 essential genes and 81 essential enhancers from K562 cells (Figures 5A and 5B). Interestingly, only 8 of the 488 essential enhancers identified in HCT116 cells were also found to be essential in K562 cells, while a different set of 73 enhancers was determined to be necessary for cell fitness in K562 (Figure 5D). The K562 essential enhancers were enriched near genes previously shown to be indispensable in K562 (observed = 60/expected = 48; hypergeometric test; p = 3.04 × 10−3) and were generally found in open chromatin regions, displaying active enhancer marks, such as H3K4me1/me2 and H3K27ac, in K562 cells (Figure 5C). These results suggest that the essential enhancers regulate cell proliferation in a cell-lineage-dependent manner.
Figure 5. Cell-type-dependent requirements of essential enhancers.
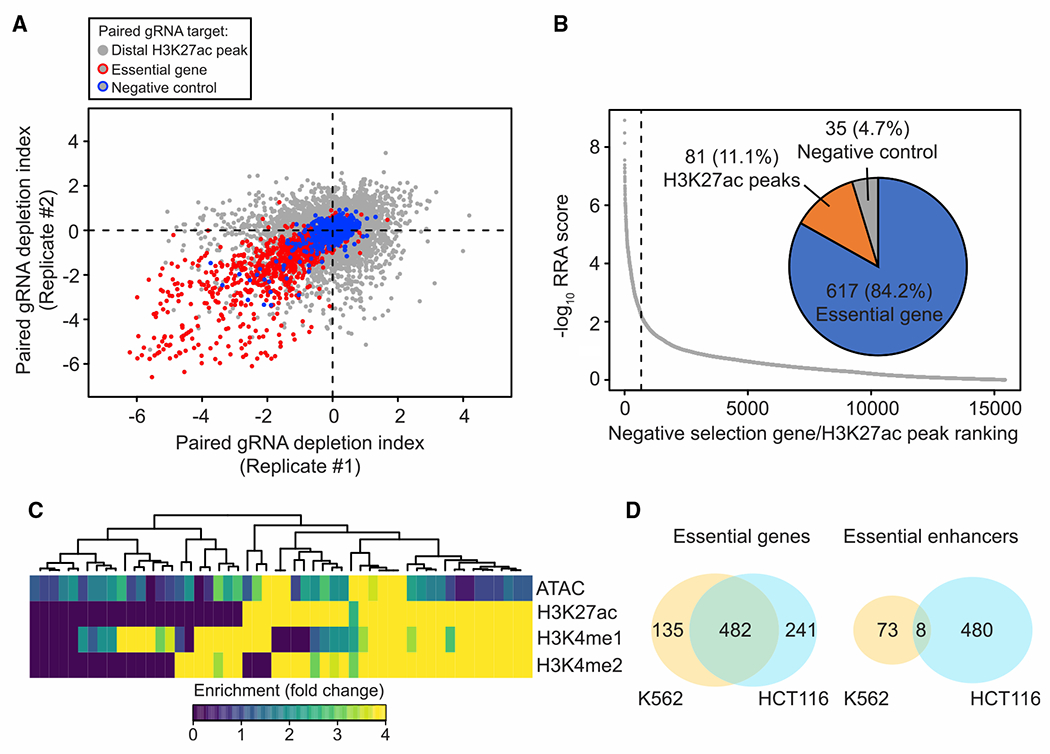
(A) Correlation of fitness effects in two biological replicates from CRISPRi screen in K562 (Pearson’s R = 0.51). Red circled dots indicate pgRNAs targeting the promoter of essential genes and blue circled dots indicate pgRNAs targeting the promoter of the non-expressed genes. Gray dots represent pgRNAs targeting distal H3K27ac peaks selected from HCT116.
(B) Summary of candidate targets identified from the K562 screen. Target with a smaller RRA score (identified by the MAGeCK algorithm) indicates a more substantial reduction in proliferation screen. Overall, 617 of the significant targets are essential genes, while 81 are distal H3K27ac peaks, and 35 are non-expressed genes or safe targets from negative control groups.
(C) Heatmap showing chromatin state of essential enhancers identified from K562. Each row represents the signal from the indicated dataset and each column represents individual essential enhancers identified in K562 cells.
(D) Venn diagram showing the number of overlapped essential genes (left) and essential enhancers (right) identified from K562 and HCT116 cells (yellow, K562; cyan, HCT116).
To understand the molecular mechanism that governs essential enhancers, we examined closely the essential enhancers near the MYC locus in ten human cancer cell lines. Enhancers are regulated by sequence-specific TFs to promote target gene expression during development (Buecker and Wysocka, 2012; Heinz et al., 2010; Spitz and Furlong, 2012). We found that the essential enhancers are occupied by lineage-specific TFs (Ghandi et al., 2019; Li et al., 2012; Nakshatri and Badve, 2007; van de Wetering et al., 2002) (Figure S9A). Our data further supported that the lineage-specific TFs were indeed required for the function of essential enhancers for MYC, as their knockdown using CRISPRi resulted in a cell-type-specific reduction of MYC gene expression (Figure S9B). Moreover, CRISPRi of either lineage-specific TFs or essential enhancers alone achieved comparable levels of effect on MYC expression as silencing both simultaneously (Figures S9C and S9D). These results, taken together, support the model that lineage-specific TFs worked through essential enhancers to regulate MYC gene expression in different cancer cell lines.
Chromatin accessibility of HCT116 essential enhancers in colorectal cancer samples
Uncontrolled cell growth through sustained proliferative signaling or evading growth suppressors is the hallmark of cancer (Hanahan and Weinberg, 2011). Our results showed that essential enhancers play critical roles in lineage-specific proliferation and are activated by oncogenic TFs. To investigate the chromatin state of essential enhancers during tumorigenesis, we interrogated chromatin accessibility of these elements using DNase-seq data generated from 13 primary embryonic and 9 adult colon tissues (Meuleman et al., 2020) and ATAC-seq data generated from a previous survey of chromatin accessibility in 38 colorectal cancer samples (Corces et al., 2018). Accessibility of a significant portion (observed = 227/expected = 155, hypergeometric test; p = 9.97 × 10−13) of essential enhancers was undetectable in human primary colon tissues using bulk DNase-seq (Meuleman et al., 2020) (Figure 6A). On the other hand, 72 of these 227 essential enhancers were in open chromatin from colorectal cancers samples while the remaining 155 essential enhancers were likely selectively activated under in vitro culture conditions in HCT116 cells. To further examine chromatin accessibility of essential enhancers in various cell types from primary colon tissues, we analyzed a comprehensive single-cell ATAC-seq atlas across 111 major cell types from 30 adult human tissues, including colon tissues from four healthy donors (Zhang et al., 2021). Our data validated that chromatin accessibility of those 72 colorectal cancer-specific essential enhancers is undetectable across 14 cell types identified from primary colon tissues (Figure 6B). Motif analysis of these colorectal cancer-specific essential enhancers showed enrichment of the same TFs, namely TCF7L2, AP1, and ZFX, that were enriched in the aforementioned activating elements within the essential enhancers (Figure 4E), indicating that oncogenic drivers in colorectal cancer samples are likely responsible for the activation of these de novo cancer-specific essential enhancers.
Figure 6. Chromatin accessibility of the essential enhancers in primary colorectal cancer samples.
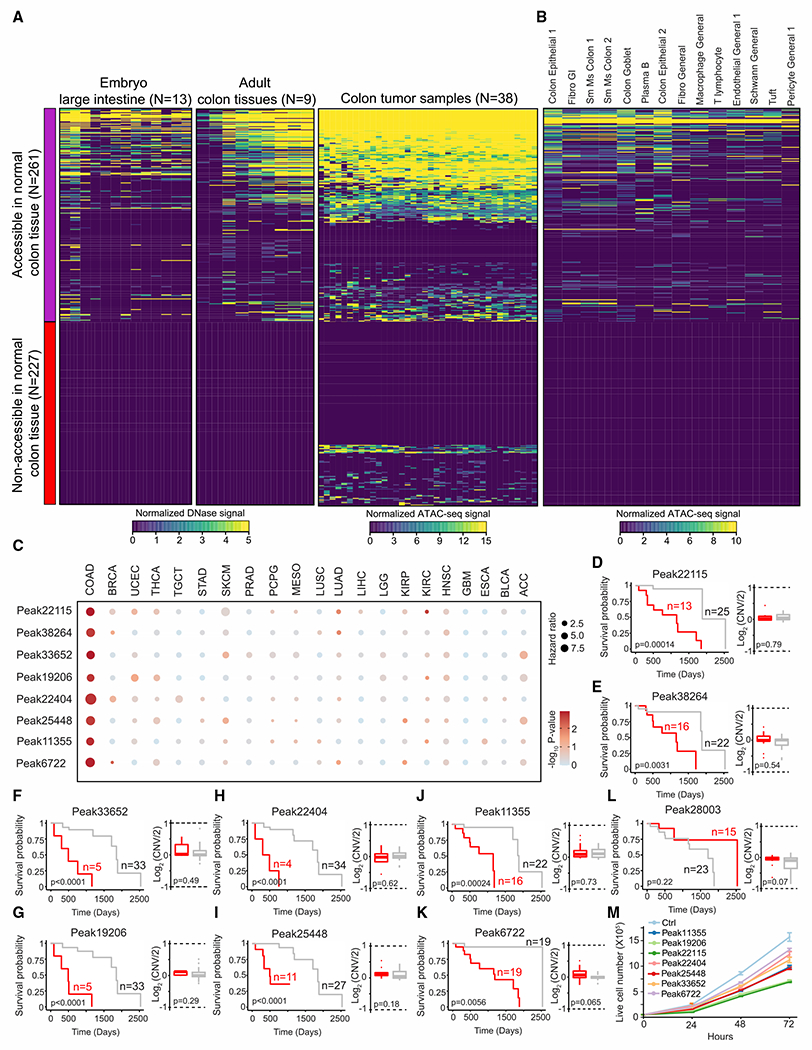
(A) Heatmap representation of chromatin accessibility of essential enhancers in primary colon tissues or colorectal tumor samples, as determined by DNase-seq or ATAC-seq, respectively. Left and middle: heatmap shows chromatin accessibility of 488 essential enhancers in primary colon tissues from 22 embryo or adult samples. Right: heatmap showing open chromatin identified from 38 colon cancer samples. Open chromatin regions, ATAC-seq peaks, were determined using MACS2 (Zhang et al., 2008) from individual colon cancer sample. Each row represents individual essential enhancers and each column represents individual samples. Seventy-two essential enhancers are overlapped with ATAC-seq peaks from colon cancer samples and are not accessible in primary colon tissues.
(B) Chromatin accessibility of essential enhancers in 14 distinct cell types identified from primary colon samples using single-cell ATAC-seq. Each row represents an individual essential enhancer and each column represents a cell type identified in primary colon tissues from four individuals. Single-cell ATAC-seq data were generated from the previous study (Zhang et al., 2021). Cell types are sorted by decreasing cell abundance from high to low.
(C) Cox proportional hazards regression analysis of essential enhancers across 21 different cancer types. Hazard ratio was determined using 528 ATAC-seq peaks, which are overlapped with 488 essential enhancers, identified in colon cancer samples. Eight essential enhancers were identified with significantly increased hazard ratio (hazard ratio > 3; multiple hypothesis testing; FDR < 0.1). FDR was estimated by a collection of p values in 528 ATAC-seq peaks generated from Cox proportional hazards regression analysis using qvalue (Storey and Tibshirani, 2003). ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LGG, low-grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma.
(D–K) Left: Kaplan-Meier analysis of overall survival of colon cancer samples based on chromatin accessibility at ATAC-seq peaks overlapped with selected essential enhancers. The red line indicates survival probability from the samples with higher chromatin accessibility at the indicated essential enhancers. p values were calculated using the log rank test. Right: DNA copy number at the indicated essential enhancer loci between high (red) and low (gray) accessibility groups. CNV, copy number variation. No significant difference in CNV is observed between high and low accessibility groups. p values were determined by the two-sided Wilcoxon test.
(L) Left: representation of Kaplan-Meier analysis of one randomly selected ATAC-seq peak overlapped with essential enhancers. p values were calculated using the log rank test. Right: DNA copy number at the indicated essential enhancer locus between high (red) and low (gray) accessibility groups. p values were determined by the two-sided Wilcoxon test.
(M) The effect of essential enhancers associated with poor clinical outcomes in cell proliferation. The measurement of cell proliferation after silencing essential enhancers with KRAB-dCas9 in HCT116 cells.
We further utilized the Cox proportional regression model to estimate the hazard ratio based on chromatin accessibility measured by the ATAC-seq assay from 528 pan-cancer ATAC-seq peaks (Corces et al., 2018), which overlapped with 488 HCT116 essential enhancers. Chromatin accessibility at 8 of HCT116 essential enhancers was strongly associated with a high hazard ratio (see STAR Methods; hazard ratio > 3, multiple hypothesis testing; FDR < 0.1) (Figure 6C). Consistently, high chromatin accessibility of those eight essential enhancers was strongly associated with lower survival probability in colorectal cancer patients (Figures 6D–6K). As control, we did not observe this association from randomly selected HCT116 essential enhancer (Figure 6L). In addition, chromatin accessibility at none of the K562 essential enhancers was associated with poor clinical outcomes in colorectal cancer samples. When we performed the same analysis using 528 randomly selected pan-cancer ATAC-seq peaks that overlapped with 11,111 distal enhancers included in our initial CRISPRi screen, we did not identify any distal enhancer associated with poor clinical outcomes. Furthermore, the association between the essential enhancer with poor clinical outcomes was specific to colorectal cancer (Figure 6C), consistent with the lineage-specific role of these essential enhancers in the gene-regulatory program. Interestingly, no significant difference in overall survival probability was observed when we used gene expression of the predicted target genes as predictors (Table S4). Moreover, we did not observe any significant change in DNA copy number at essential enhancers between the high and low accessibility groups (Figures 6D–6K). Finally, we validated the role of those essential enhancers in cell proliferation after the silencing using KRAB-dCas9 in HCT116 cells (Figure 6M).
DISCUSSION
Despite the rapid progress in identifying putative enhancers in the human genome over recent years, the function of most annotated putative enhancers is still untested. Here, we utilized a robust, unbiased, and high-throughput functional screen to systematically examine and characterize essential enhancers in human cancer cell lines. Recent studies have highlighted the importance of focal and extrachromosomal oncogene amplification to contribute to MYC expression and facilitate genetic heterogeneity in cancer (Shoshani et al., 2021; Turner et al., 2017; Wu et al., 2019). Furthermore, the enhancers located on extrachromosomal DNA are shown to spatially interact with MYC promoter and drive MYC overexpression (Hung et al., 2021). From the pilot screen, we also demonstrated that this screening approach is capable of identifying functional MYC enhancers from cancer cell lines with known DNA copy amplification (SW620; colorectal cancer, NCI-H460; lung cancer, MCF7; breast cancer) (Turner et al., 2017), further supporting the generalized usage of this approach to profile functional enhancers in various cancer types. In addition, this systematic functional perturbation assay is highly generalizable. This strategy can be readily applied to discover enhancers crucial for different physiological phenotypes, such as cellular differentiation, cellular responses to extracellular signaling, etc. Overall, our data revealed the landscape of functional enhancers and established a role for lineage-specific TFs as key TFs that regulate the essential enhancers across multiple cancer cell lines.
Through the high-throughput perturbation screen, we generated a catalog with >500 functional enhancers essential for proliferation and fitness in selected cancer cell lines. Furthermore, we dissected and characterized the regulatory elements within each of them with a massively parallel reporter assay (Ernst et al., 2016; Pang and Snyder, 2020). Interestingly, our data showed that essential enhancers generally harbor both activating and repressive elements, similar to the classical eve-skipped enhancers characterized from Drosophila (Small et al., 1992). Moreover, the activating and repressive elements are arranged characteristically relative to the open chromatin within each enhancer. While the activating elements are concentrated at the open chromatin regions and bound by oncogenic TFs in the cells analyzed in this study, the repressive elements are distributed outside the open chromatin and targeted by TFs with tumor suppressor function. These organization principles of essential enhancers might present an opportunity for designing targeted therapeutics in cancer research.
Another interesting finding of this study is that a significant fraction of the essential enhancers found in HCT116 cells displays chromatin accessibility only in CRCs. In addition, high chromatin accessibility at a subset of these essential enhancers is significantly associated with poor patient survival in CRC, raising the possibility that essential enhancers could serve as prognostic markers. Together, our study provides a systematic framework to decipher the functionality of cis-regulatory elements in cancer cells, delineates organization principles of transcriptional enhancers, and fills a current gap to uncover the gene regulatory program to control cell survival and proliferation in cancer cells.
Limitations of the study
To systematically determine and characterize the sequence features of essential enhancers, we first examined an ~3.6 Mb genomic interval around MYC and MYB loci and identified 24 essential enhancers from 10 cancer cell lines. While all identified essential enhancers were associated with the active enhancer marks, such as DHS and H3K27ac, in the cell lines where they were identified, the average size of identified essential enhancers was ~3 kb, which might contain multiple DHS or H3K27ac peaks in a given cell line. Due to large effect size (>1 kb) generated by KRAB-dCas9-mediated silencing, further experiments will be necessary to resolve the functional cis-regulatory elements within. Furthermore, this study reports a large number of essential enhancers required for cell proliferation in several cancer cell lines, including the CRC line HCT116 and the chronic myeloid leukemia cell line K562. One limitation of this result is that the cell lines used have been cultured in vitro for an extended period and might not fully recapitulate chromatin accessibility or histone modifications from the primary tumor samples. Future studies to examine the function of the essential enhancers in patient-derived xenograft models or genetically engineered animal models would further reveal the roles of essential enhancers in vivo. In addition, we found that essential enhancers contain regulatory elements with different transcriptional activities using a massively parallel reporter assay. However, an episomal reporter assay might not truly reflect the function of those regulatory elements in the native chromatin context. To eliminate this confounding factor, mutagenesis, such as prime editing (Anzalone et al., 2019; Gaudelli et al., 2017), would verify their impacts on essential enhancers. Moreover, we identified some essential enhancers only accessible in tumor samples or associated with significantly shorter patient survival using the existing ATAC-seq data from 38 colorectal cancer samples. However, tumor tissue samples are heterogeneous and contain both tumor cells and surrounding non-tumor cells. Future studies that profile chromatin accessibility from tumor samples using single-cell ATAC-seq would provide more in-depth information to determine and verify the potential application of essential enhancers as prognostic marks or therapeutic targets.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bing Ren (bren@ucsd.edu).
Materials availability
All unique reagents generated in this study will be available upon request. Distribution of materials may require signing Material Transfer Agreement (MTA) in accordance with policies of University California San Diego.
Data and code availability
Sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All dataset used in each figure are listed in Table S4.
This paper does not report original code. A combination of existing pipelines was used for data analysis. All software used for data analysis is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K4me3 | Millipore | Cat# 04-745; RRID:AB_1163444 |
| H3K27ac | Diagenode | Cat# C15200184; RRID:AB_2713908 |
| Bacterial and virus strains | ||
| Endura ElectroCompetent cells | Lucigen | Cat# 60242-1 |
| MAX Efficiency DH5 α Competent cells | Thermo Fisher | Cat# 18258012 |
| Chemicals, peptides, and recombinant proteins | ||
| Agencourt AMPure XP | Beckman Coulter | Cat# A63880 |
| SPRISelect Reagent | Beckman Coulter | Cat# B23319 |
| Herculase II Fusion DNA polymerase | Agilent | Cat# 600677 |
| KAPA HiFi HotStart ReadyMix | Roche | Cat# KK2601 |
| FuGene HD | Promega | Cat# E2311 |
| Lipofectamine 3000 | Thermo Fisher | Cat# L3000008 |
| FastDigest Esp3I | Thermo Fisher | Cat# FD0454 |
| FastAP | Thermo Fisher | Cat# EF0651 |
| MboI | NEB | Cat# R0147S |
| SalI-HF | NEB | Cat# R3138S |
| AgeI-HF | NEB | Cat# R3552S |
| Gibson Assembly Master Mix | NEB | Cat# E2611S |
| Quick ligation kit | NEB | Cat# M2200S |
| T4 PNK | NEB | Cat# M0201S |
| Protein A Magnetic Beads | NEB | Cat# S1425S |
| Protein G Magnetic Beads | NEB | Cat# S1430S |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat# M0530S |
| Puromycin | InvivoGen | Cat# ant-pr-1 |
| Blasticidin S HCl | Thermo Fisher | Cat# A1113903 |
| BX-795 (TBK1/IKK inhibitor) | Sigma-Aldrich | Cat# SML0694 |
| PKR inhibitor | Sigma-Aldrich | Cat# I9785 |
| Trizol | Thermo Fisher | Cat# 15596026 |
| KAPA SYBR Fast qPCR Kit | Roche | Cat# KK4600 |
| SuperScript III Reverse Transcriptase | Thermo Fisher | Cat# 18080044 |
| Critical commercial assays | ||
| Ultralow input library kit | Qiagen | Cat# 180492 |
| Qubit dsDNA HS Assay kit | Thermo Fisher | Cat# Q32851 |
| Oligotex mRNA mini kit | Qiagen | Cat# 70022 |
| M220 Focused-ultrasonicator | Covaris | Cat# 500295 |
| Deposited data | ||
| H3K27ac ChIP-seq | This study | GEO: GSE161874 |
| H3K4me3 PLAC-seq | This study | GEO: GSE161874 |
| CRISPRi screen | This study | Tables S1 and S2 |
| STARR-seq | This study | Table S3 |
| K562 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF945XHA.bigWig |
| HepG2 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF764VYK.bigWig |
| HCT116 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF126HQH.bigWig |
| SW620 H3K27ac ChIP-seq (Figure 2A) | GEO | GEO: GSE106923 |
| A549 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF256RBI.bigWig |
| NCI-H460 DNase-seq (Figure 2A) | ENCODE | ENCFF143RMC.bigWig |
| MCF7 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF226GBS.bigWig |
| MDA-MB231 H3K27ac ChIP-seq (Figure 2A) | GEO | GEO: GSE103887 |
| PC3 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF287SLL.bigWig |
| K562 DNase-seq (Figure 2E) | ENCODE | ENCFF413AHU.bigWig |
| HepG2 DNase-seq (Figure 2E) | ENCODE | ENCFF842XRQ.bigWig |
| HCT116 DNase-seq (Figure 2E) | ENCODE | ENCFF225OLI.bigWig |
| MCF7 DNase-seq (Figure 2E) | ENCODE | ENCFF949ANK.bigWig |
| K562 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF779QTH.bigWig |
| HepG2 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF764VYK.bigWig |
| HCT116 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF984WLE.bigWig |
| MCF7 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF515VXR.bigWig |
| K562 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF761XBZ.bigWig |
| HepG2 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF058GCZ.bigWig |
| HCT116 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF774BWO.bigWig |
| MCF7 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF275KBS.bigWig |
| K562 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF491AUC.bigWig |
| HepG2 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF109QAV.bigWig |
| HCT116 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF783QRO.bigWig |
| MCF7 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF299JCQ.bigWig |
| HCT116 Tcf7l2 ChIP-seq (Figure 4C) | ENCODE | ENCFF241JHM.bigWig |
| HCT116 JunD ChIP-seq (Figure 4C) | ENCODE | ENCFF199BDI.bigWig |
| HCT116 ATAC-seq (Figure 4C) | ENCODE | ENCFF259PSA.bigWig |
| HCT116 Tcf7l2 ChIP-seq (Figure 4D) | ENCODE | ENCFF296ZZB.bed |
| K562 ATAC-seq (Figure 5C) | ENCODE | ENCFF754EAC.bigWig |
| K562 H3K27ac ChIP-seq | ENCODE | ENCFF779QTH.bigWig |
| K562 H3K4me1 ChIP-seq | ENCODE | ENCFF761XBZ.bigWig |
| K562 H3K4me2 ChIP-seq | ENCODE | ENCFF491AUC.bigWig |
| Experimental models: Cell lines | ||
| K562 | ATCC | CCL-243 |
| HepG2 | ATCC | HB-8065 |
| HCT116 | ATCC | CCL-247 |
| SW620 | ATCC | CCL-227 |
| A549 | ATCC | CCL-185 |
| NCI-H460 | ATCC | HTB-177 |
| MCF7 | ATCC | HTB-22 |
| MDA-MB231 | ATCC | HTB-26 |
| DU-145 | ATCC | HTB-81 |
| PC3 | ATCC | CRL-1435 |
| 293FT | Thermo Fisher | R70007 |
| Oligonucleotides | ||
| Custom library Oligos for CRISPR screen | This study | Table S1 and S2 |
| Custom library Oligos for STARR-seq | This study | Table S3 |
| Custom PCR primers | This study | Table S4 |
| Custom Sequencing primers | This study | Table S4 |
| Recombinant DNA | ||
| pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro | Addgene | Plasmid# 71236 |
| pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-GFP | Addgene | Plasmid# 71236 |
| LentiGuide-Puro | Addgene | Plasmid# 52963 |
| PsPAX2 | Addgene | Plasmid# 12260 |
| pMD2.G | Addgene | Plasmid# 12259 |
| hSTARR-seq_ORI | Addgene | Plasmid# 99296 |
| Software and algorithms | ||
| BWA version 0.7.17 | (Li and Durbin, 2009) | https://github.com/lh3/bwa |
| Bowtie2 version 2.3.4.3 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools version 1.9 | (Li et al., 2009) | https://github.com/samtools/samtools |
| deepTools2 version 3.5.0 | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| GuideScan version 1.0 | (Perez et al., 2017) | http://www.guidescan.com |
| CRISPR-SE | (Li et al., 2021) | https://github.com/bil022/CRISPR-SE |
| edgeR version 3.12 | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R Bioconductor version 3.6.1 | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Survival version 3.2-7 | (Therneau and Grambsch, 2000) | https://cran.r-project.org/web/packages/survival/index.html |
| Survminer version 0.4.8 | https://rpkgs.datanovia.com/survminer/index.html | https://cran.r-project.org/web/packages/survminer/index.html |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/download.html |
| BEDTools | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/content/installation.html |
| MAGeCK version 0.5.9.2 | (Li et al., 2014) | https://sourceforge.net/projects/mageck/files/ |
| RELICS v1 | This study | https://github.com/patfiaux/RELICS/releases/tag/v1.0 |
| CRISPRY | This study | https://github.com/MichaelMW/crispy |
| MAPS version 1.0 | (Juric et al., 2019) | https://github.com/ijuric/MAPS |
| bigWigAverageOverBed | ENCODE | https://github.com/ENCODEDCC/kentUtils/blob/master/bin/linux.x86_64/bigWigAverageOverBed |
| SnapATAC | (Fang et al., 2021) | https://github.com/r3fang/SnapATAC |
| Scrublet | (Wolock et al., 2019) | https://github.com/swolock/scrublet |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell line
Human chronic myeloid leukemia cell line K562 was cultured in RPMI with 10% fetal bovine serum (FBS) (Gemini). Human liver cancer cell line HepG2, colon cancer cell line SW620, lung cancer cell line A549 and NCI-H460, breast cancer cell line MDA-MB231, prostate cancer cell line DU-145 and PC3 were cultured in DMEM/F12 media (Thermo Fisher) with 10% FBS. Human colorectal cancer cell line HCT116 was cultured in McCoy’s 5A Modified media (Thermo Fisher) with 10% FBS, and human breast cancer cell line MCF7 was cultured in DMEM (4500mg/L glucose) (Thermo Fisher) with 10% FBS. All human cancer cell lines were obtained from ATCC and tested negative for mycoplasma.
MYC reporter knock-in line generation
K562 cells were electroporated with CRISPR expression plasmids, and donor constructs from the previous study (Xiong et al., 2017) using cell line nucleofector Kit V (Lonza) following the manufacturer’s instructions. Cells were selected by puromycin (2 μg/mL) (InvivoGen) 3 days post-electroporation. After seven day selection, GFP positive cells were identified and isolated using a SH800S cell sorter (Sony) to one cell per well in a 96-well plate. The purity of the individual clone was examined by FACS and genotyped by PCR.
Generation of stable KRAB-dCas9 expressed clones in HCT116 cell line
HCT116 cells were transduced with lentivirus carrying KRAB-dCas9-BFP (Addgene #85969). Three days after transduction, BFP positive cells were isolated using a SH800S cell sorter (Sony) to one cell per well in a 96-well plate. The purity of the individual clones was examined by FACS, and several clones with the strongest BFP signal were selected for the following experiment.
METHOD DETAILS
Selection of genomic targets for CRISPRi screen
In the pilot study, we selected two key oncogenes, MYC and MYB, that are required for cellular proliferation in various cancer types. We followed a similar principle as the previous study (Fulco et al., 2016) to identify genomic regions for the tiling CRISPRi screen. Briefly, chromatin organization is known to play key roles in gene regulation, and the majority of enhancer and promoter interactions occur within topologically associated domains (TAD) (Dixon et al., 2012; Schoenfelder and Fraser, 2019). Because cell-type-specific chromatin interactions occur around the MYC locus, we combined Hi-C data from K562, HepG2, A549, HCT116, and selected genomic regions to include the entire TAD around MYC (3-Mb) and MYB (~600 kb) loci.
Selection of genomic targets for large-scale CRISPRi screen
We first identified essential enhancers sharing similar features as active enhancers from the pilot screen. That finding provided us the rationale to focus on H3K27ac peaks. Second, we narrowed it down to those H3K27ac peaks located within 500 kb around target genes because most three-dimensional (3D) promoter-based interactions occur within a 500 kb distance (Javierre et al., 2016; Jung et al., 2019). Third, we validated several essential enhancers identified from the pilot screen, indicating cell fitness is a reliable readout to reflect the reduction in gene expression of essential genes. However, we are mindful of the possible limitation in this screen’s sensitivity to identify weak essential enhancers, which only slightly or moderately regulate target gene expression. Last, our screen identified several context-dependent essential enhancers, suggesting that the activity of essential enhancers might be cell-type-specific. In summary, we selected 6,642 H3K27ac peaks located near the essential genes and 4,554 H3K27ac peaks based on cell-type-specific activity.
gRNA design for the pilot study in MYC and MYB loci
We used the guide RNA design tool, CRISPR-SE (Li et al., 2021), to identify gRNAs and select single guide RNA (sgRNA) or paired guide RNA (pgRNA) in two genomic loci (MYB locus — chr6: 134,923,863-135,478,863; MYC locus — chr8: 127,182,756-130,337,754). We designed 13,373 tiling pgRNAs targeting those two loci and another set of 1,455 pgRNAs as negative controls (non-targeting controls or safe-target genomic loci, which are the validated genomic regions that do not cause growth defects in Cas9-based screen (Morgens et al., 2017)). The mean genomic distance between the gRNAs in each pair was ~3 kb, and the genomic spans of adjacent pgRNAs overlapped by 2.7 kb on average. In addition, we also designed sgRNAs targeting all DHS and H3K27ac peaks located within the selected genomic regions. In the pgRNA library, we generated 11,800 and 1,573 pgRNAs for MYC and MYB loci, respectively. We also included 955 pgRNAs that lacked the PAM sequence targeting MYC locus and 500 pgRNAs targeting safe-targeting genomic loci as negative controls, and 170 pgRNAs targeting the promoter of essential genes (Gata1, Phb, Myb, and Myc). The guide RNA sequence is listed in Table S1.
Paired gRNA design for large-scale CRISPRi screen
We included 11,196 distal H3K27ac peaks and designed five pgRNAs targeting each peak. To maximize the genomic coverage of pgRNAs, we assigned sgRNAs to the same pair based on the relative location within each H3K27ac peak. For instance, we paired the first sgRNA closest to the start of the H3K27ac peak with the sixth sgRNA closest to the start of the same H3K27ac peak. In general, we selected the top 10 sgRNAs for each H3K27ac peak based on the improved criteria that we learned from the pilot tiling screen. Briefly, we found that ~40–60% sgRNAs located within our validated functional enhancers are effective with a higher on-target score (Doench et al., 2014). Furthermore, we observed the percentage of effective sgRNAs increasing to ~60–90% when we only considered sgRNAs identified from CRISPR-SE (Li et al., 2021) and FlashFry tools (McKenna and Shendure, 2018). To minimize potential false-negatives due to low targeting efficiency, we selected the top 10 sgRNAs, based on the improved selection criteria, targeting for each distal H3K27ac peak included in the screen. In addition, we used the same approach to design 2,300 sgRNA pairs targeting 460 safe-targeting genomic regions as negative controls. The safe-target genomic regions are the validated genomic regions that do not cause growth defects in the previous Cas9-based screen (Morgens et al., 2017). To assess the performance of large-scale screen in HCT116, we also designed five pgRNAs targeting 1,017 essential genes as positive controls and 583 non or low-expressed nonessential genes as negative controls using sgRNA rank score from previous CRISPRi genome-wide screen study (Horlbeck et al., 2016). Overall, we generated a large-scale sgRNA library containing 55,980 pgRNAs targeting 11,196 distal H3K27ac peaks, 5,391 pgRNAs targeting essential genes as positive controls, and 8,673 pgRNAs targeting safe-target, promoter of non- or low-expressed gene, and non-targeting control as negative controls. sgRNA information is listed in Table S2.
CRISPRi plasmid library construction
We designed, synthesized the pool of paired-guide RNA oligo (Agilent), and generated the pgRNA plasmid library as previously described (Diao et al., 2017) with the following modifications. Briefly, we amplified the oligo library by PCR with less than 20 cycles. After PCR, the pooled oligo library was purified with AMPure XP beads (Beckman Coulter). The lentiviral vector carrying dCas9-KRAB was obtained from Addgene (Plasmid #71236) and linearized with BsmBI followed by gel purification. We performed 5 or 20 Gibson assembly reactions for tiling or large-scale pgRNA library followed by manufacturer’s instructions (NEB) and purified DNA using ethanol precipitation. The purified DNA was electroporated into Endura competent cells (Lucigen) using 50–100 ng DNA per electroporation (we set up 6 or 24 replicate of electroporation for tiling or large-scale pgRNA library to maintain library complexity), and the colonies were harvested within 14 h at 30°C to minimize recombination activity in bacteria. We extracted the plasmids using the Plasmid Maxi prep kit (Macherey-Nagel).
Lentivirus generation
Briefly, 5ug of plasmid library was co-transfected with four ug PsPAX2 (Addgene #12260) and one ug pMD2.G (Addgene #12259) into a 10-cm dish of 293FT cells (Thermo Fisher) in DMEM containing 10% FBS using FuGene HD (Promega). Scale up the number of 293FT cells and transfection depending on the yield and library size. The growth medium was replaced 12 h after transfection, and the lentivirus was harvested 48 h post-transfection. The viral titer was determined for each individual cancer cell line using the survival cell number under puromycin selection from a serial dilution of lentivirus transduction.
Pooled CRISPRi screens for essentiality
We infected cells with lentiviral libraries at a low multiplicity of infection (MOI = 0.5) to ensure each infected cell got one viral particle. In general, we maintained cell numbers with at least 1000-fold coverage of the lentiviral library during the entire proliferation screen. For instance, we would have at least 15 million cells survived after puromycin selection for a pooled library with 15,000 paired-guide RNAs. We started puromycin selection (2 μg/mL) (InvivoGen) 48 h post-transduction for at least two days or until no survival cell was observed from the control group. After puromycin selection, we recovered and cultured cells in a lower puromycin concentration (0.2 μg/mL) for additional two days. To start the screen, we collected at least 15 million cells as “doubling time 0” and sub-cultured at least 15 million cells for 14 doubling times in a lower concentration of puromycin (0.2 μg/mL). The cell concentration, viability, and doubling time were examined every two days. We sub-cultured and split cells when they reached more than 90% confluency. In the end, at least 15 million cells that reached 14 doubling times were harvested. For the proliferation screen, we performed two replicate experiments for every human cancer cell line.
FACS-based CRISPR and CRISPRi screen
K562 MYC reporter cells were transduced with lentivirus carrying KRAB-dCas9 with tiling pgRNA pooled library using MOI = 0.3. Forty-eight hours post-transduction, cells were selected using blasticidin (8 μg/mL) (Thermo Fisher) for nine days. The cells were split in a 1 to 4 ratio every three days. We used at least 60 million survived cells after blasticidin selection for a pooled library with 15,000 paired-guide RNAs to start the screen. Cells were sorted into six different bins (Bin1-6) based on GFP signals using a SH800S cell sorter (Sony).
Generation of illumina sequencing library
Genomic DNA was isolated from proliferation-based (D = 0 and D = 14) or sorting-based (Bin #1- #6) screens and used to generate illumina sequencing libraries. Briefly, we used 400 ng genomic DNA as a template per PCR reaction. In 1st PCR, the library was PCR amplified using Herculase II (Agilent) in the 96-well plate to increase the coverage for 22 cycles with the following program (98°C for 5 min; 98°C for 35 s, 52°C for 30 s, 72°C for 1min and repeat for 21 cycles; 72°C for 5 min). After 1st PCR, we combined all reactions from the 96-well plate and used 2 ul of the mixture as the 2nd PCR amplification template. In 2nd PCR amplification, we amplified the library with Truseq index primers and prepared two PCR reactions per library. The 2nd PCR was amplified using KAPA Hi-Fi (Roche) for five cycles with the following program (95°C for 3 min; 98°C for 20 s, 65°C for 15 s, 72°C for 30 s and repeat for four cycles; 72°C for 1 min). The sequencing library was combined and gel-purified (~690bp). We generated 100 bp paired-end reads on Illumina Hiseq 4000. Primer information for illumina sequencing library is listed in Table S4.
Cloning individual sgRNAs
The lentiviral vector carrying dCas9-KRAB was obtained from Addgene (Plasmid #71236) and linearized with BsmBI followed by gel purification. sgRNA oligo (Table S4) was annealed and phosphorylated before the ligation. Individual sgRNA construct was verified using Sanger sequencing (Genewiz).
Cloning paired gRNAs
Paired gRNA cassette was PCR amplified from the gBlock template (Diao et al., 2017) containing tracRNA and mouse U6 promoter sequence using KAPA Hi-Fi (KAPA bioscience) with primers to add homology arms (Table S4) for Gibson assembly. We assembled a 20ng amplified cassette into a 50ng digested vector in a 20 μL Gibson reaction (NEB). The individual construct was verified using Sanger sequencing (Genewiz).
STARR-seq screen
Considering the heterochromatin spreading from KRAB-dCas9 in the CRISPRi screen, we extended 1 kb genomic region up and downstream of 488 essential enhancers and designed a tiling-oligonucleotide library. Each oligonucleotide is 190 bp and the genomic spans of adjacent oligonucleotide overlapped by 140 bp (except the first and last oligonucleotides only have 70 bp overlapped with adjacent oligonucleotide). In addition, we included 60 tiling oligonucleotides for the known enhancers and 2,095 negative controls that correspond to yeast open reading frames (ORFs) sequence (Table S3). Each oligonucleotide contains 190 bp of genomic sequence and 20 bp constant flanking sequences (upstream: 5′- ACACGACGCTCTTCCGATCT; downstream: AGATCGGAAGAGCACACGTC-3′) on both ends, which were used for amplification and cloning. The oligonucleotides were synthesized by Agilent. STARR-seq screen and library preparation were carried out as previously described (Arnold et al., 2013; Muerdter et al., 2018; Neumayr et al., 2019). In brief, we amplified a tiling-oligonucleotide library and cloned into the hSTARR-seq_ORI vector (Addgene #99296). The plasmid DNA was electroporated into HCT116 cells with TBK1/IKK and PKR inhibitors (Sigma). Cells were harvested 48 h after electroporation, and poly A+ RNA was extracted using oligotex mRNA kit (Qiagen). Due to the potential bias from low library complexity, we incorporated unique molecular identifiers (UMIs) to enable the quantification of reporter transcripts during sequencing library preparation. The libraries were sequenced with 2 × 100 paired-end cycles with Illumina NextSeq 2000 sequencer.
RNA extraction and quantitative RT-PCR
We used quantitative RT-PCR (RT-qPCR) to validate the effect of selected enhancers in gene regulation. In CRISPRi experiments, cells transduced with lentivirus carrying KRAB-dCas9 (Addgene #71236) and sgRNAs were harvested one week after the transduction. Total RNA was extracted using Trizol (Thermo Fisher) following the manufacturer’s instructions. Reverse transcription was performed for one hour using random priming (Promega). qPCR reactions (0.5 μL cDNA, 0.2 μM each primer, SYBR green Master Mix (Roche) were performed on a Roche LightCycler 480 Real-Time PCR detection system, using primers specific for each gene (Table S4). Data were normalized to loading controls (Gapdh).
Chromatin immunoprecipitation-sequencing (ChIP-seq)
ChIP-seq experiments for H3K27ac mark were performed as described in ENCODE experiment protocols (“Ren Lab ENCODE Chromatin Immunoprecipitation Protocol” in https://www.encodeproject.org/documents/) with minor modifications. The cells from ~80% confluent 10 cm dishes were crosslinked by adding fixation solution (1% formaldehyde, 0.1M NaCl, 1 mM EDTA, 50 mM HEPES•KOH pH 7.6) for 10 min at room temperature. Crosslinking was quenched with 125 mM Glycine for 5 min. We used 1.0 million cells for each ChIP sample. Shearing of chromatin was performed using truChIP Chromatin Shearing Reagent Kit (Covaris) according to the manufacturer’s instructions. Covaris M220 was used for sonication with the following parameters: 410 s duration at 20.0% duty factor, 75.0 peak power, 200 cycles per burst at 5–9°C temperature range. For immunoprecipitation, we used 50 μL Protein A or Protein G Magnetic beads (NEB) and washed twice with PBS with 5 mg/mL BSA and 4 μg of antibody coupled in 500 μL PBS with 5 mg/mL BSA overnight at 4°C. The magnetic beads were washed twice with ChIP buffer (20 mM Tris-HCl pH8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100), once with ChIP buffer including 500 mM NaCl, four times with RIPA buffer (10 mM Tris-HCl pH8.0, 0.25M LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% Na•Deoxycholate), and once with TE buffer (pH 8.0). Chromatin was eluted twice from washed beads by adding elution buffer (20 mM Tris-HCl pH8.0, 100 mM NaCl, 20 mM EDTA, 1% SDS) and incubating for 15 min at 65°C. The crosslinking was reversed at 65°C for 6 h, and RNase A (Sigma) was added for 1 h at 37°C followed by proteinase K (Ambion) treatment overnight at 50°C. ChIP-enriched DNA was purified using Phenol/Chloroform/Isoamyl alcohol extractions in phase-lock tubes. ChIP samples were end-repaired, A-tailed, and adaptor-ligated using QIAseq ultralow input library kit (Qiagen) according to the manufacturer’s instructions. Size selection using AMpure beads (Beckman Coulter) was performed to get 300–500 bp DNA, and PCR amplification (8–10 cycles) was performed. Library quality and quantity were measured using TapeStation (Agilent) and Qubit (Thermo Fisher). We generated 50 bp paired-end reads on Illumina Hiseq 4000.
Cell proliferation assay
We transduced cells with lentivirus carrying KRAB-dCas9 and sgRNA targeting candidate essential enhancers and performed puromycin (2 μg/mL) (InvivoGen) for three days post-electroporation to select against non-transduced cells. Seven days after the selection, the same number (4 × 105 cells for HCT116; 2 × 105 cells for A549) of viable cells was determined by trypan blue was split into 6-well plates triplicates. The number of viable cells was measured using trypan blue staining every 24 h in an automated cell counter (Bio-rad) for three days.
Proximity ligation ChIP-sequencing (PLAC-seq)
PLAC-seq libraries were prepared for K562, HepG2, HCT116, and A549 cells as previously described with minor modifications (Fang et al., 2016). In brief, cells were cross-linked for 15 min at room temperature with 1% formaldehyde and quenched for 5 min at room temperature with 0.2 M glycine (Thermo Fisher). The cross-linked cells were aliquot (~3 × 106 cells) and resuspended in 300 μL lysis buffer (10mM Tric-HCl pH 8.0, 10mM NaCl, 0.2% IGEPAL CA-630) and incubated on ice for 15 min. The suspension was then centrifuged at 2,500 xg for 5 min and the pellet was washed by resuspending in 300 μL lysis buffer and centrifuging at 2,500 xg for 5 min. The pellet was resuspended in 50 μL 0.5% SDS and incubated for 10 min at 62°C. 160 μL 1.56% Triton X-100 was added to the suspension and incubated for 15 min at 37°C. 25 μL of 10X NEBuffer 2 and 100 U MboI were added to digest chromatin for 2 h at 37°C with rotation (900 rpm). Enzymes were inactivated by heating for 20 min at 62°C. Fragmented ends were biotin labeled by adding 50 μL of a mix containing 0.3 mM biotin-14-dATP, 0.3 mM dCTP, 0.3 mM dTTP, 0.3 mM dGTP, and 0.8 U μl−1 Klenow and incubated for 60 min at 37°C with rotation (900 rpm). Ends were subsequently ligated by adding a 900 μL master mix containing 120 μL 10X T4 DNA ligase buffer (NEB), 100 μL 10% TritionX-100, 6 μL 20 mg mL−1 BSA, 10 μL 400 U μl−1 T4 DNA Ligase (NEB, high concentration formula) and 664 μL H2O and incubated for 120 min at 23°C with 300 rpm slow rotation. Nuclei were pelleted for 5 min at 4°C with centrifugation at 2,500 xg. For the ChIP, nuclei were resuspended in RIPA Buffer (10 mM Tris (pH 8.0), 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate) with proteinase inhibitors and incubated on ice for 10 min. Sonication was performed using a Covaris M220 instrument (Power 75W, duty factor 10%, cycle per bust 200, time 10 min, temperature 7°C) and nuclei were spun for 15 min at 14,000 rpm at 4°C. 5% of supernatant was taken as input DNA. To the remaining cell lysate was added anti-H3K4me3 antibody-coated Dynabeads M-280 Sheep anti-Rabbit IgG (5 μg antibody per sample, Millipore, 04–745), followed by rotation at 4°C overnight for immunoprecipitation. The sample was placed on a magnetic stand for 1 min and the beads were washed three times with RIPA buffer, two times with high-salt RIPA buffer (10 mM Tris pH 8.0, 300 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.1% deoxycholate), one time with LiCl buffer (10 mM Tris (pH 8.0), 250 mM LiCl, 1 mM EDTA, 0.5% IGEPAL CA-630, 0.1% sodium deoxycholate) and two times with TE buffer (10 mM Tris (pH 8.0), 0.1 mM EDTA). Washed beads were treated with 10 μg RNase A in extraction buffer (10 mM Tris (pH 8.0), 350 mM NaCl, 0.1 mM EDTA, 1% SDS) for 1 h at 37°C, and subsequently 20 μg proteinase K was added at 65°C for 2 h. ChIP DNA was purified with Zymo DNA clean & concentrator-5. For Biotin pull down, 25 μL of 10 mg mL−1 Dynabeads My One T1 Streptavidin beads was washed with 400 μL of 1X Tween Wash Buffer (5 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, 1 M NaCl, 0.05% Tween) and the supernatant removed after separation on a magnet. Beads were resuspended with 2X Binding Buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl), added to the sample and incubated for 15 min at room temperature. Beads were subsequently washed twice with 1X Tween Wash Buffer and in between heated on a thermomixer for 2 min at 55°C with mixing and washed once with 1X NEB T4 DNA ligase buffer. Library prep was prepared using QIAseq Ultralow Input Library Kit (Qiagen). KAPA qPCR assay was performed to estimate concentration and cycle number for final PCR. Final PCR was directly amplified off the T1 Streptavidin beads according to the qPCR results, and DNA was size selected with 0.5X and 1X SPRI Cleanup and eluted in 1X Tris Buffer and paired-end sequenced.
Analysis of tiling CRISPRi screen from the pilot study
The abundance of paired gRNAs from D = 0 and D = 14 was mapped to the initially designed sequences using BWA (Li and Durbin, 2009). First, we analyzed the data using RELICS v1, a method specifically designed to analyze tiling CRISPR screens. RELICS v1 uses a Generalized Linear Mixed Model (GLMM) (Gelman and Hill, 2007) to model gRNA counts across different pools. The output is a log Bayes Factor, which is calculated by comparing the ‘background’ model (a guide does not target a functional sequence) against a ‘functional sequence’ model (the guide targets a functional sequence). The functional model parameters are estimated by maximum likelihood from the observed guide counts for gRNAs targeting the MYC and PHB promoters (both are essential genes in cancer cells and used as positive controls in proliferation screen). In this study, the background model parameters were estimated by maximum likelihood from all the remaining guides. After computing the scores from both models, RELICS v1 calculates a RELICS score, also referred to as “CRISPRi score” in the CRISPRi-based screen, for each base pair by summing the log Bayes Factors of all paired gRNA overlapping genomic positions. A base pair is considered to be overlapped by gRNAs if it is within the ‘area of effect’ (AoE) for the CRISPR system used. We use 1kb as the AoE for a CRISPRi-based screen (Thakore et al., 2015). We selected genomic regions with an averaged CRISPRi score above 5 as candidate essential enhancers. This threshold was chosen to correspond to FDR <0.1 based on simulated data from CRSsim (Fiaux et al., 2020). We further selected the reproducible candidate enhancers identified from two independent pipelines, RELICS v1 and CRISPY, to generate high confidence candidate essential enhancers across different cell types. CRISPY is an improved pipeline developed from our previous study (Diao et al., 2017) and allows the flexibility of processing different types of screening data, data quality control, and peak calling for positive elements. RELICS v1 can be obtained from GitHub (https://github.com/patfiaux/RELICS/releases/tag/v1.0), and we used default settings for the analysis in this study. CRISPY can be obtained from GitHub (https://github.com/MichaelMW/crispy), and we performed the analysis with parameter -n 0.05, which only outputs the candidate peaks with FDR <0.05.
Analysis of large-scale CRISPRi screen
The abundance of pgRNAs from D = 0 and D = 14 was mapped to the initially designed sequences using BWA (Li and Durbin, 2009). We used MAGeCK (Li et al., 2014) to perform data quality assessment and identify candidate essential enhancers. For data quality assessment, we used the MAGeCK-mle module to calculate β-score (guide RNA depletion index) between biological replicates from two individual clones. A negative β-score indicates a target is negatively selected. Pearson correlation was performed to determine the correlation between β-score from two individual clones. Non-targeting pgRNAs were provided to generate the null distribution when calculating the p values. pgRNAs were ranked based on the p values, and we used the modified RRA algorithm from MAGeCK to obtain the RRA score for the individual target gene or H3K27ac peak. The threshold value for candidate essential enhancers is FDR <0.02 based on the empirical ROC curve with 75% sensitivity. The empirical FDR was calculated based on the percentage of candidate targets identified from negative controls above this threshold, including non-essential genes and safe-target control genomic regions to account for the possible toxicities from the silencing mediated by KRAB-dCas9. The information of selected candidate essential enhancers is listed in Table S4.
Removal of candidate essential enhancers targeting by low-specificity pgRNAs
We computed a specificity score for all pgRNAs included in the large-scale screen using GuideScan (Perez et al., 2017) from the webtool to evaluate the potential off-target mediated toxicity to affect cellular proliferation. For every pgRNA, we used the lowest specificity score from one of the two sgRNAs for the representation. A previous study (Tycko et al., 2019) has shown that sgRNAs with specificity score <0.2 tend to have substantial off-target toxicity in CRISPR-based screens. To minimize false-positive hints by off-target toxicity in our screen, we removed candidate essential enhancers targeted by more than two pgRNAs (40%) with a specificity score <0.2. Total, we further removed 70 candidate essential enhancers targeted by low-specificity pgRNAs (Table S4).
ChIP-seq data processing
Sequencing files were aligned to the human genome (hg38) with bowtie2 (Version 2.3.4.3) (Langmead and Salzberg, 2012). SAMtools (Version 1.9) (Li et al., 2009) and MarkDuplicates (Picard) were used to filter (MAPQ <30) and clean data post alignment. The reads were then converted to reads per kilobase per million in 200 bp bins using deepTools2 (Version 3.5.0) (Ramírez et al., 2016). We obtained reproducible H3K27ac peaks in HCT116 from ENCODE and filtered out H3K27ac peaks located within 3-kb around annotated TSS using bedtools. For each distal H3K27ac peaks (n = 22,744), we obtained ChIP-seq fold enrichment over input in each sample using bigWigAverageOverBed (https://github.com/ENCODE-DCC/kentUtils/blob/master/bin/linux.x86_64/bigWigAverageOverBed). Pearson correlation was performed to determine the correlation of H3K27ac signal between different samples.
Analysis of key epigenetic features with essential enhancer
For key epigenetic features analysis, each functional enhancer peak was then scored using read-depth normalized signals from DNase-seq, fold-change over input from ChIP-seq data, and the fitness score from tiling screen using bigWigAverageOverBed to extract averaged signal for essential enhancer peaks across different cell types. We used spearman’s rank correlation to determine the correlation between key features and essential enhancers across multiple cell types.
PLAC-seq data processing
PLAC-seq data were processed with MAPS (Juric et al., 2019) to normalize reads and identify long-range chromatin interactions. Specifically, MAPS aligned raw paired-end reads with BWA (Li and Durbin, 2009) to the reference genome hg38. Uniquely mapped reads were kept and split into intra-chromosomal reads and inter-chromosomal reads. We used the following steps to identify intra-chromosomal chromatin interactions. Each chromosome was first divided into 10 kb bins. Histone H3K4me3 ChIP-seq peaks (downloaded from ENCODE) were used as the anchor, and 10 kb bins overlapping with these ChIP-seq peaks were defined as the anchor bin. Depending on whether none, one, and two bins are the anchor bin, 10 kb bin pairs were further defined as ‘NOT’, ‘XOR’, and ‘AND’ sets. Only bin pairs in the ‘XOR’ and ‘AND’ sets were kept for downstream analysis. The raw contact frequency between two 10 kb bins in the ‘XOR’ and ‘AND’ sets was then fitted into a zero-truncated Poisson model to obtain the normalized contact frequency. Significant interactions were identified with FDR corrected p value cutoff of 0.01. Significant interactions were further grouped into clusters if two interactions were within 10 kb.
Differential chromatin interaction analysis
For differential interaction analysis in H3K4me3 PLAC-seq, the raw contact counts in 10 kb resolution bins were used as inputs, and we stratified the inputs into every 10-kb genomic distance to minimize the bias from a genomic distance. Since each input showed negative binomial distribution, we used edgeR (Robinson et al., 2010) to get the initial set of differential interactions. Next, we removed bins with less than 20 contact counts in each sample of two replicates from the downstream analysis. To avoid the antibody bias and TSS with differential H3K4me3 level, we further removed chromatin contacts overlapping with differential H3K4me3 ChIP-seq peaks at TSS (with fold-change decreased more than 50% than in HCT116). We only compared the chromatin contacts between different cell types at TSS of the predicted target genes with the same level of H3K4me3 ChIP-seq peaks.
Target gene prediction of essential enhancer
We used functional similarity and chromatin interaction to predict the targets of essential enhancers. From the tiling screen, we demonstrated the usage of cellular proliferation assay to identify strong functional enhancers for MYC oncogene across multiple cell types. We found that the distance between a majority of functional enhancers and target genes is within 500 kb. Thus, we first obtained a gene dependence score from the CRISPR-Cas9 knockout screen in HCT116 (Ghandi et al., 2019; Hart et al., 2015). We utilized chromatin interactions generated from H3K4me3 PLAC-seq to identify 155 enhancer and gene pairs (E-G pairs) between 115 essential enhancers and 115 essential genes. 145 of 155 enhancer and gene pairs (E-G pairs) were obtained from distal enhancers located near these essential genes, and 10 of E-G pairs were generated from distal enhancers that are associated with H3K27ac mark only in HCT116. In addition, we also connect 35 E-G pairs between 35 essential enhancers and 29 essential genes using the nearest essential gene. 4 of E-G pairs were generated from distal enhancers that are associated with H3K27ac mark only in HCT116. Overall, we identified 190 E-G pairs from 150 essential enhancers and predicted 144 essential genes, which included 100 essential genes initially included in the screen. The predicted E-G pairs are listed in Table S4.
STARR-seq data analysis
STARR-seq reads were aligned to the oligonucleotide library using BWA (Li and Durbin, 2009). Similar to the previous study (Yan et al., 2021), an oligonucleotide with DNA counts less than 25 reads in the plasmid DNA input library, and RNA counts less than 5 reads in at least three biological replicate libraries were removed for downstream analysis. We determined enriched or depleted oligonucleotides using negative binomial repression from the R package edgeR (Zhou et al., 2014). Biological dispersion was estimated using only yeast oligonucleotides where no real variation is expected. The resulting p values were adjusted by the Benjamini-Hochberg procedure, and the significance cut-off for candidate oligonucleotides was set to limit the rate of enriched yeast oligonucleotide to 5%.
Survival analysis
Clinical outcome data and normalized ATAC-seq counts were obtained from UCSC Xena (Goldman et al., 2020). Cox proportional hazards regression model and the hazard ratio were computed in R using package survival. Package survminer was used for drawing the Kaplan-Meier plots and defining the optimal threshold (surv_cutpoint) using the maximally selected rank statistics (Hothorn and Lausen, 2003; Lausen and Schumacher, 1992).
Motif enrichment analysis
We performed both de novo and known motif enrichment analysis using HOMER (Heinz et al., 2010).
sci-ATAC-seq dataset analysis
Seven sciATAC-seq data including four from the transverse colon and two from the sigmoid colon were used to identify cell types in the colon tissue (Zhang et al., 2021). (1) Quality control metrics: TSS enrichment and unique fragments. TSS positions were obtained from the GENCODE database v31 (Frankish et al., 2019). Tn5 corrected insertions were aggregated ±2000 bp relative (TSS strand-corrected) to each unique TSS genome-wide. Then this profile was normalized to the mean accessibility ± (1900–2000) bp from the TSS and smoothed every 11 bp. The max of the smoothed profile was taken as the TSS enrichment. We then filtered out all single cells that had fewer than 1,000 unique fragments and/or a TSS enrichment of less than 7 for all data sets. (2) Overall clustering strategy: we utilized two rounds of clustering analysis to identify cell clusters. The first round of clustering analysis was performed on individual samples. We divided the genome into 5kb consecutive windows and then scored each cell for any insertions in these windows, generating a window by cell binary matrix for each sample. We filtered out those windows that are generally accessible in all cells for each sample using a z-score threshold 1.65. Based on the filtered matrix, we then carried out dimension reduction using SnapATAC (Fang et al., 2021) and used the leiden algorithm (Traag et al., 2019) to identify cell clusters. We called peaks for each cluster using the aggregated profile of accessibility and then merged the peaks from all clusters to generate a union peak list. Based on the peak list, we generated a cell-by-peak count matrix and used Scrublet (Wolock et al., 2019) to remove potential doublets. Next, to carry out the second round of clustering analysis, we merged peaks called from all samples to form a reference peak list. We then generated a single binary cell-by-peak matrix using cells from all samples and again performed the dimension reduction followed by graph-based clustering to obtain the final cell clusters across the entire dataset.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed with R Bioconductor by using two-sided Wilcoxon test, two-tailed Student’s t-test, multiple hypothesis testing, or log rank test depending on the experimental settings. All details regarding the statistical test employed, the size of the samples and p values are all included in the figure legends.
Supplementary Material
Highlights.
Large-scale screen of enhancers required for cell fitness in multiple cancer cell lines
Essential enhancers are generally cell-type-specific
Essential enhancers generally contain both activating and repressive DNA elements
Chromatin accessibility of some essential enhancers is associated with poor prognosis
ACKNOWLEDGMENTS
We thank H. Zhao for MYC reporter knockin constructs; J.S. Weissman for lenti-KRAB-dCas9-BFP plasmid; C.A. Gersbach for lenti-KRAB-dCas9-sgRNA vector; A. Stark for hSTARR_ORI plasmid; and G. Li, Q. Zhu, M. Tastemel, and all members of the Ren lab for many productive and exciting discussions of this work. S.W. is supported by the Cancer Prevention and Research Institute of Texas (no. RR210034). The work is supported by NIH (UM1 HG009402, to B.R.) and the Ludwig Institute for Cancer Research.
Footnotes
DECLARATION OF INTERESTS
B.R. is a co-founder and consultant of Arima Genomics, Inc. and co-founder of Epigenome Technologies, Inc.; and P.S.M. is a co-founder and consultant of Boundless Bio, Inc. He has equity in the company and chairs the scientific advisory board, for which he is compensated. S.W. is a member of the scientific advisory board of Dimension Genomics Inc.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111630.
REFERENCES
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. (2016). Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 6, 914–929. 10.1158/2159-8290.cd-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. (2012). Epigenomic enhancer profiling defines a signature of colon cancer. Science 336, 736–739. 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, and Liu DR (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, and Stark A (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]
- Bothma JP, Garcia HG, Esposito E, Schlissel G, Gregor T, and Levine M (2014). Dynamic regulation of eve stripe 2 expression reveals transcriptional bursts in living Drosophila embryos. Proc. Natl. Acad. Sci. USA 111, 10598–10603. 10.1073/pnas.1410022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional addiction in cancer. Cell 168, 629–643. 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buecker C, and Wysocka J (2012). Enhancers as information integration hubs in development: lessons from genomics. Trends Genet. 28, 276–284. 10.1016/jztig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li C, Peng X, Zhou Z, Weinstein JN, and Liang H; Cancer Genome Atlas Research Network (2018a). A pan-cancer analysis of enhancer expression in nearly 9000 patient samples. Cell 173, 386–399.e12. 10.1016/j.cell.2018.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liu H, and Qing G (2018b). Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Targeted Ther. 3, 5. 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MS, Lo YH, Chen X, Williams CS, Donnelly JM, Criss ZK 2nd, Patel S, Butkus JM, Dubrulle J, Finegold MJ, and Shroyer NF (2019). Growth factor-independent 1 is a tumor suppressor gene in colorectal cancer. Mol. Cancer Res 17, 697–708. 10.1158/1541-7786.mcr-18-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AJ, Saiakhova A, Corradin O, Luppino JM, Lovrenert K, Bartels CF, Morrow JJ, Mack SC, Dhillon G, Beard L, et al. (2017). Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun 8, 14400. 10.1038/ncomms14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, et al. (2018). The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898. 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, et al. (2017). A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 14, 629–635. 10.1038/nmeth.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, and Root DE (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol 32, 1262–1267. 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Melnikov A, Zhang X, Wang L, Rogov P, Mikkelsen TS, and Kellis M (2016). Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat. Biotechnol 34, 1180–1190. 10.1038/nbt.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. (2021). Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun 12, 1337. 10.1038/s41467-021-21583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, and Ren B (2016). Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 26, 1345–1348. 10.1038/cr.2016.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, and Vogelstein B (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Fiaux PC, Chen HV, Chen PB, Chen AR, and McVicker G (2020). Discovering functional sequences with RELICS, an analysis method for CRISPR screens. PLoS Comput. Biol 16, e1008194. 10.1371/journal.pcbi.1008194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–d773. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frietze S, Wang R, Yao L, Tak YG, Ye Z, Gaddis M, Witt H, Farnham PJ, and Jin VX (2012). Cell type-specific binding patterns reveal that TCF7L2 can be tethered to the genome by association with GATA3. Genome Biol. 13, R52. 10.1186/gb-2012-13-9-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773. 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR,Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay M, Li Y, and Felsher DW (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med 4, a014241. 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, Jackson D, Leith A, Schreiber J, Noble WS, et al. (2019). A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377–390.e19. 10.1016/j.cell.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471. 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, and Hill J (2007). Data Analysis Using Regression and Multilevel/hierarchical Models (Cambridge University Press; ). [Google Scholar]
- Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, Barretina J, Gelfand ET, Bielski CM, Li H, et al. (2019). Next-generation characterization of the cancer cell line encyclopedia. Nature 569, 503–508. 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, Banerjee A, Luo Y, Rogers D, Brooks AN, et al. (2020). Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol 38, 675–678. 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, Li B, Chiou J, Wildberg A, Ding B, et al. (2020). An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751. 10.1038/s41586-020-2093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hart T, Brown KR, Sircoulomb F, Rottapel R, and Moffat J (2014). Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol. Syst. Biol 10, 733. 10.15252/msb.20145216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526. 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, and Kinzler KW (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512. 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- He Y, Liu XY, Gong R, Peng KW, Liu RB, and Wang F (2020). NK homeobox 2.2 functions as tumor suppressor in colorectal cancer due to DNA methylation. J. Cancer 11, 4791–4800. 10.7150/jca.43665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr W, and Clarke J (1986). The SV40 enhancer is composed of multiple functional elements that can compensate for one another. Cell 45, 461–470. 10.1016/0092-8674(86)90332-6. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Day DS, and Young RA (2016). Insulated neighborhoods: structural and functional units of mammalian gene control. Cell 167, 1188–1200. 10.1016/j.cell.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, Bradner JE, and Young RA (2015). Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 58, 362–370. 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, e19760. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, and Lausen B (2003). On the exact distribution of maximally selected rank statistics. Comput. Stat. Data Anal 43, 121–137. [Google Scholar]
- Hung KL, Yost KE, Xie L, Shi Q, Helmsauer K, Luebeck J, Schöpflin R, Lange JT, Chamorro González R, Weiser NE, et al. (2021). ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 600, 731–736. 10.1038/s41586-021-04116-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, et al. (2016). Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369–1384.e19. 10.1016/j.cell.2016.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, and Liu LY (2015). Zinc finger protein X-linked is overexpressed in colorectal cancer and is associated with poor prognosis. Oncol. Lett 10, 810–814. 10.3892/ol.2015.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung I, Schmitt A, Diao Y, Lee AJ, Liu T, Yang D, Tan C, Eom J, Chan M, Chee S, et al. (2019). A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet 51, 1442–1449. 10.1038/s41588-019-0494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric I,Yu M, Abnousi A, Raviram R, Fang R, Zhao Y, Zhang Y, Qiu Y, Yang Y, Li Y, et al. (2019). MAPS: model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments. PLoS Comput. Biol 15, e1006982. 10.1371/journal.pcbi.1006982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. (2014). Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 111, 6131–6138. 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R, Laur S, Adler P, and Vilo J (2012). Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 28, 573–580. 10.1093/bioinformatics/btr709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lausen B, and Schumacher M (1992). Maximally selected rank statistics. Biometrics 48, 73–85. 10.2307/2532740. [DOI] [Google Scholar]
- Leung D, Jung I, Rajagopal N, Schmitt A, Selvaraj S, Lee AY, Yen CA, Lin S, Lin Y, Qiu Y, et al. (2015). Integrative analysis of haplotype-resolved epigenomes across human tissues. Nature 518, 350–354. 10.1038/nature14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chen PB, and Diao Y (2021). CRISPR-SE: a brute force search engine for CRISPR design. NAR Genom. Bioinform. 3, lqab013. 10.1093/nargab/lqab013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. 10.1186/s13059-014-0554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Tuteja G, Schug J, and Kaestner KH (2012). Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell 148, 72–83. 10.1016/j.cell.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD, Etchin J, Lawton L, Sallan SE, Silverman LB, et al. (2014). Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377. 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, and Wotton D (2000).Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 19, 1745–1754. 10.1093/em-boj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, and Shendure J (2018). FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16, 74. 10.1186/s12915-018-0545-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuleman W, Muratov A, Rynes E, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Teodosiadis A, et al. (2020). Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244–251. 10.1038/s41586-020-2559-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, and Penn LZ (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990. 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710. 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Tsui CK, Haney MS, Hess GT, Han K, Jeng EE, et al. (2017). Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun 8, 15178. 10.1038/ncomms15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Boryń ŁM, Woodfin AR, Neumayr C, Rath M, Zabidi MA, Pagani M, Haberle V, Kazmar T, Catarino RR, et al. (2018). Resolving systematic errors in widely used enhancer activity assays in human cells. Nat. Methods 15, 141–149. 10.1038/nmeth.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakshatri H, and Badve S (2007). FOXA1 as a therapeutic target for breast cancer. Expert Opin. Ther. Targets 11, 507–514. 10.1517/14728222.11.4.507. [DOI] [PubMed] [Google Scholar]
- Nateri AS, Spencer-Dene B, and Behrens A (2005). Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 437, 281–285. 10.1038/nature03914. [DOI] [PubMed] [Google Scholar]
- Neumayr C, Pagani M, Stark A, and Arnold CD (2019). STARR-Seq and UMI-STARR-seq: assessing enhancer activities for genome-wide-high-and low-complexity candidate libraries. Curr. Protoc. Mol. Biol 128, e105. 10.1002/cpmb.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B, and Snyder MP (2020). Systematic identification of silencers in human cells. Nat. Genet 52, 254–263. 10.1038/s41588-020-0578-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke B, and Der CJ (2017). Drugging RAS: know the enemy. Science 355, 1158–1163. 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- Pattabiraman DR, and Gonda TJ (2013). Role and potential for therapeutic targeting of MYB in leukemia. Leukemia 27, 269–277. 10.1038/leu.2012.225. [DOI] [PubMed] [Google Scholar]
- Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, and Ventura A (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol 35, 347–349. 10.1038/nbt.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay RG, and Gonda TJ (2008). MYB function in normal and cancer cells. Nat. Rev. Cancer 8, 523–534. 10.1038/nrc2439. [DOI] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden L (2014).A3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SK, Gosai SJ, Gutierrez A, Mackay-Smith A, Ulirsch JC, Kanai M, Mouri K, Berenzy D, Kales S, Butler GM, et al. (2021). Direct characterization of cis-regulatory elements and functional dissection of complex genetic associations using HCR-FlowFISH. Nat. Genet 53, 1166–1176. 10.1038/s41588-021-00900-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirm S, Jiricny J, and Schaffner W (1987). The SV40 enhancer can be dissected into multiple segments, each with a different cell type specificity. Genes Dev. 1, 65–74. 10.1101/gad.1.1.65. [DOI] [PubMed] [Google Scholar]
- Schoenfelder S, and Fraser P (2019). Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet 20, 437–455. 10.1038/s41576-019-0128-0. [DOI] [PubMed] [Google Scholar]
- Shoshani O, Brunner SF, Yaeger R, Ly P, Nechemia-Arbely Y, Kim DH, Fang R, Castillon GA, Yu M, Li JSZ, et al. (2021). Chromothripsis drives the evolution of gene amplification in cancer. Nature 591, 137–141. 10.1038/s41586-020-03064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small S, Blair A, and Levine M (1992). Regulation of even-skipped stripe 2 in the Drosophila embryo. EMBO J. 11, 4047–057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, and Furlong EEM (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626. 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100, 9440–9445. 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, and Gersbach CA (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149. 10.1038/nmeth.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therneau TM, and Grambsch PM (2000). Modeling Survival Data: Extending the Cox Model (Springer; ). 978-1-4757-3294-8. [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233. 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. 10.1038/nature21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycko J, Wainberg M, Marinov GK, Ursu O, Hess GT, Ego BK, Li A, Truong A, Trevino AE, et al. (2019). Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat. Commun 10, 4063. 10.1038/s41467-019-11955-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. (2002). The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancercells. Cell 111, 241–250. 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- Wang J, Yang L, Yang J, Kuropatwinski K, Wang W, Liu XQ, Hauser J, and Brattain MG (2008). Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 68, 3152–3160. 10.1158/0008-5472.can-07-5348. [DOI] [PubMed] [Google Scholar]
- Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM, and Network CGAR (2013). The cancer genome atlas pan-cancer analysis project. Nat. Genet 45, 1113–1120. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9. 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703. 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Zhang Y, Yan J, Jain S, Chee S, Ren B, and Zhao H (2017). A scalable epitope tagging approach for high throughput ChIP-seq analysis. ACS Synth. Biol 6, 1034–1042. 10.1021/acssynbio.6b00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Qiu Y, Ribeiro Dos Santos AM, Yin Y, Li YE, Vinckier N, Nariai N, Benaglio P, Raman A, Li X, et al. (2021). Systematic analysis of binding of transcription factors to noncoding variants. Nature 591, 147–151. 10.1038/s41586-021-03211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, and Ren B (2017). The three-dimensional organization of mammalian genomes. Annu. Rev. Cell Dev. Biol 33, 265–289. 10.1146/an-nurev-cellbio-100616-060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Hocker JD, Miller M, Hou X, Chiou J, Poirion OB, Qiu Y, Li YE, Gaulton KJ, Wang A, et al. (2021). A single-cell atlas of chromatin accessibility in the human genome. Cell 184, 5985–6001.e19. 10.1016/j.cell.2021.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Choi PS, Francis JM, Imielinski M, Watanabe H, Cherniack AD, and Meyerson M (2016). Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet 48, 176–182. 10.1038/ng.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, and Xie W (2019). The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol 20,535–550. 10.1038/s41580-019-0132-4. [DOI] [PubMed] [Google Scholar]
- Zhou X, Lindsay H, and Robinson MD (2014). Robustly detecting differential expression in RNA sequencing data using observation weights. Nucleic Acids Res. 42, e91. 10.1093/nar/gku310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. All dataset used in each figure are listed in Table S4.
This paper does not report original code. A combination of existing pipelines was used for data analysis. All software used for data analysis is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K4me3 | Millipore | Cat# 04-745; RRID:AB_1163444 |
| H3K27ac | Diagenode | Cat# C15200184; RRID:AB_2713908 |
| Bacterial and virus strains | ||
| Endura ElectroCompetent cells | Lucigen | Cat# 60242-1 |
| MAX Efficiency DH5 α Competent cells | Thermo Fisher | Cat# 18258012 |
| Chemicals, peptides, and recombinant proteins | ||
| Agencourt AMPure XP | Beckman Coulter | Cat# A63880 |
| SPRISelect Reagent | Beckman Coulter | Cat# B23319 |
| Herculase II Fusion DNA polymerase | Agilent | Cat# 600677 |
| KAPA HiFi HotStart ReadyMix | Roche | Cat# KK2601 |
| FuGene HD | Promega | Cat# E2311 |
| Lipofectamine 3000 | Thermo Fisher | Cat# L3000008 |
| FastDigest Esp3I | Thermo Fisher | Cat# FD0454 |
| FastAP | Thermo Fisher | Cat# EF0651 |
| MboI | NEB | Cat# R0147S |
| SalI-HF | NEB | Cat# R3138S |
| AgeI-HF | NEB | Cat# R3552S |
| Gibson Assembly Master Mix | NEB | Cat# E2611S |
| Quick ligation kit | NEB | Cat# M2200S |
| T4 PNK | NEB | Cat# M0201S |
| Protein A Magnetic Beads | NEB | Cat# S1425S |
| Protein G Magnetic Beads | NEB | Cat# S1430S |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat# M0530S |
| Puromycin | InvivoGen | Cat# ant-pr-1 |
| Blasticidin S HCl | Thermo Fisher | Cat# A1113903 |
| BX-795 (TBK1/IKK inhibitor) | Sigma-Aldrich | Cat# SML0694 |
| PKR inhibitor | Sigma-Aldrich | Cat# I9785 |
| Trizol | Thermo Fisher | Cat# 15596026 |
| KAPA SYBR Fast qPCR Kit | Roche | Cat# KK4600 |
| SuperScript III Reverse Transcriptase | Thermo Fisher | Cat# 18080044 |
| Critical commercial assays | ||
| Ultralow input library kit | Qiagen | Cat# 180492 |
| Qubit dsDNA HS Assay kit | Thermo Fisher | Cat# Q32851 |
| Oligotex mRNA mini kit | Qiagen | Cat# 70022 |
| M220 Focused-ultrasonicator | Covaris | Cat# 500295 |
| Deposited data | ||
| H3K27ac ChIP-seq | This study | GEO: GSE161874 |
| H3K4me3 PLAC-seq | This study | GEO: GSE161874 |
| CRISPRi screen | This study | Tables S1 and S2 |
| STARR-seq | This study | Table S3 |
| K562 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF945XHA.bigWig |
| HepG2 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF764VYK.bigWig |
| HCT116 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF126HQH.bigWig |
| SW620 H3K27ac ChIP-seq (Figure 2A) | GEO | GEO: GSE106923 |
| A549 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF256RBI.bigWig |
| NCI-H460 DNase-seq (Figure 2A) | ENCODE | ENCFF143RMC.bigWig |
| MCF7 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF226GBS.bigWig |
| MDA-MB231 H3K27ac ChIP-seq (Figure 2A) | GEO | GEO: GSE103887 |
| PC3 H3K27ac ChIP-seq (Figure 2A) | ENCODE | ENCFF287SLL.bigWig |
| K562 DNase-seq (Figure 2E) | ENCODE | ENCFF413AHU.bigWig |
| HepG2 DNase-seq (Figure 2E) | ENCODE | ENCFF842XRQ.bigWig |
| HCT116 DNase-seq (Figure 2E) | ENCODE | ENCFF225OLI.bigWig |
| MCF7 DNase-seq (Figure 2E) | ENCODE | ENCFF949ANK.bigWig |
| K562 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF779QTH.bigWig |
| HepG2 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF764VYK.bigWig |
| HCT116 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF984WLE.bigWig |
| MCF7 H3K27ac ChIP-seq (Figure 2E) | ENCODE | ENCFF515VXR.bigWig |
| K562 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF761XBZ.bigWig |
| HepG2 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF058GCZ.bigWig |
| HCT116 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF774BWO.bigWig |
| MCF7 H3K4me1 ChIP-seq (Figure 2E) | ENCODE | ENCFF275KBS.bigWig |
| K562 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF491AUC.bigWig |
| HepG2 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF109QAV.bigWig |
| HCT116 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF783QRO.bigWig |
| MCF7 H3K4me2 ChIP-seq (Figure 2E) | ENCODE | ENCFF299JCQ.bigWig |
| HCT116 Tcf7l2 ChIP-seq (Figure 4C) | ENCODE | ENCFF241JHM.bigWig |
| HCT116 JunD ChIP-seq (Figure 4C) | ENCODE | ENCFF199BDI.bigWig |
| HCT116 ATAC-seq (Figure 4C) | ENCODE | ENCFF259PSA.bigWig |
| HCT116 Tcf7l2 ChIP-seq (Figure 4D) | ENCODE | ENCFF296ZZB.bed |
| K562 ATAC-seq (Figure 5C) | ENCODE | ENCFF754EAC.bigWig |
| K562 H3K27ac ChIP-seq | ENCODE | ENCFF779QTH.bigWig |
| K562 H3K4me1 ChIP-seq | ENCODE | ENCFF761XBZ.bigWig |
| K562 H3K4me2 ChIP-seq | ENCODE | ENCFF491AUC.bigWig |
| Experimental models: Cell lines | ||
| K562 | ATCC | CCL-243 |
| HepG2 | ATCC | HB-8065 |
| HCT116 | ATCC | CCL-247 |
| SW620 | ATCC | CCL-227 |
| A549 | ATCC | CCL-185 |
| NCI-H460 | ATCC | HTB-177 |
| MCF7 | ATCC | HTB-22 |
| MDA-MB231 | ATCC | HTB-26 |
| DU-145 | ATCC | HTB-81 |
| PC3 | ATCC | CRL-1435 |
| 293FT | Thermo Fisher | R70007 |
| Oligonucleotides | ||
| Custom library Oligos for CRISPR screen | This study | Table S1 and S2 |
| Custom library Oligos for STARR-seq | This study | Table S3 |
| Custom PCR primers | This study | Table S4 |
| Custom Sequencing primers | This study | Table S4 |
| Recombinant DNA | ||
| pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro | Addgene | Plasmid# 71236 |
| pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-GFP | Addgene | Plasmid# 71236 |
| LentiGuide-Puro | Addgene | Plasmid# 52963 |
| PsPAX2 | Addgene | Plasmid# 12260 |
| pMD2.G | Addgene | Plasmid# 12259 |
| hSTARR-seq_ORI | Addgene | Plasmid# 99296 |
| Software and algorithms | ||
| BWA version 0.7.17 | (Li and Durbin, 2009) | https://github.com/lh3/bwa |
| Bowtie2 version 2.3.4.3 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools version 1.9 | (Li et al., 2009) | https://github.com/samtools/samtools |
| deepTools2 version 3.5.0 | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| GuideScan version 1.0 | (Perez et al., 2017) | http://www.guidescan.com |
| CRISPR-SE | (Li et al., 2021) | https://github.com/bil022/CRISPR-SE |
| edgeR version 3.12 | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R Bioconductor version 3.6.1 | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Survival version 3.2-7 | (Therneau and Grambsch, 2000) | https://cran.r-project.org/web/packages/survival/index.html |
| Survminer version 0.4.8 | https://rpkgs.datanovia.com/survminer/index.html | https://cran.r-project.org/web/packages/survminer/index.html |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/download.html |
| BEDTools | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/content/installation.html |
| MAGeCK version 0.5.9.2 | (Li et al., 2014) | https://sourceforge.net/projects/mageck/files/ |
| RELICS v1 | This study | https://github.com/patfiaux/RELICS/releases/tag/v1.0 |
| CRISPRY | This study | https://github.com/MichaelMW/crispy |
| MAPS version 1.0 | (Juric et al., 2019) | https://github.com/ijuric/MAPS |
| bigWigAverageOverBed | ENCODE | https://github.com/ENCODEDCC/kentUtils/blob/master/bin/linux.x86_64/bigWigAverageOverBed |
| SnapATAC | (Fang et al., 2021) | https://github.com/r3fang/SnapATAC |
| Scrublet | (Wolock et al., 2019) | https://github.com/swolock/scrublet |