

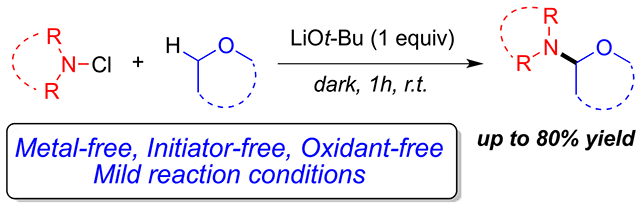
Abstract
Herein we describe a metal-free regioselective α-amination of ethers mediated by N-chloroimides in ethereal solvents in the presence of lithium tert-butoxide. This reactivity of N-chloroimides leads to the synthesis of hemiaminal ethers in good to excellent yields at room temperature. This C–H functionalization is achieved without the use of a light, heat source, or external radical initiators. Initial mechanistic work indicates that the reaction proceeds through a radical pathway.
Graphical Abstract
The hemiaminal ether moiety1 is embedded within numerous natural products2 and pharmaceutical agents (Figure 1).3 The bioactivity of such compounds is wide and ranges from anticancer (tegafur) to anti-HIV (zalcitabine and didanosine) activity, and some also exhibit lyase and hepatitis C inhibition. Traditional approaches to generating hemiaminal ethers proceed via metal-catalyzed hydroamination of enol ethers4 or through the substitution of a leaving group in the α-position of an ether substrate.5 These methods possess limitations as enol ethers are acid-sensitive functional groups and α-halogenated ethers are inherently unstable.
Figure 1.
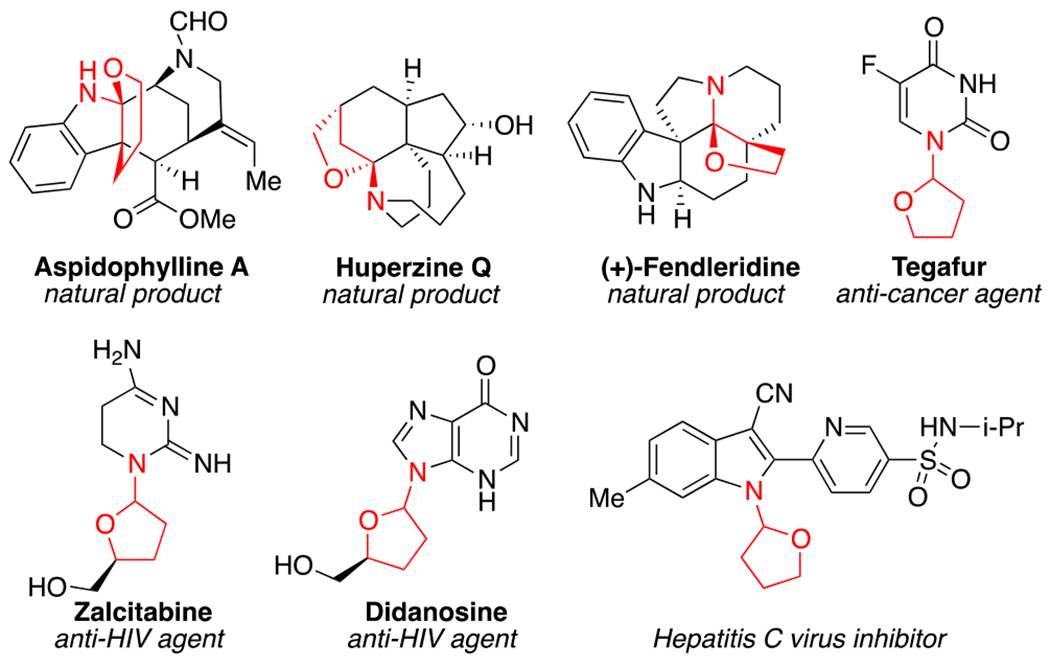
Selected examples of natural products and bioactive agents containing hemiaminal ether skeletons.
More recently, direct C(sp3)–H amination of ethers has represented a more efficient approach that enables late-stage functionalization of complex molecules.6,7 Methods that access α-aminated ethers through such C(sp3)–H activation processes can be divided into three main categories that depend on the nature of the aminating agent employed: (i) transition-metal-catalyzed nitrene insertion,8 (ii) metal-free organonitrenoid insertion,9 and (iii) radical-based10 or metal-catalyzed11 cross-coupling reaction of NH-amines (Figure 2).
Figure 2.
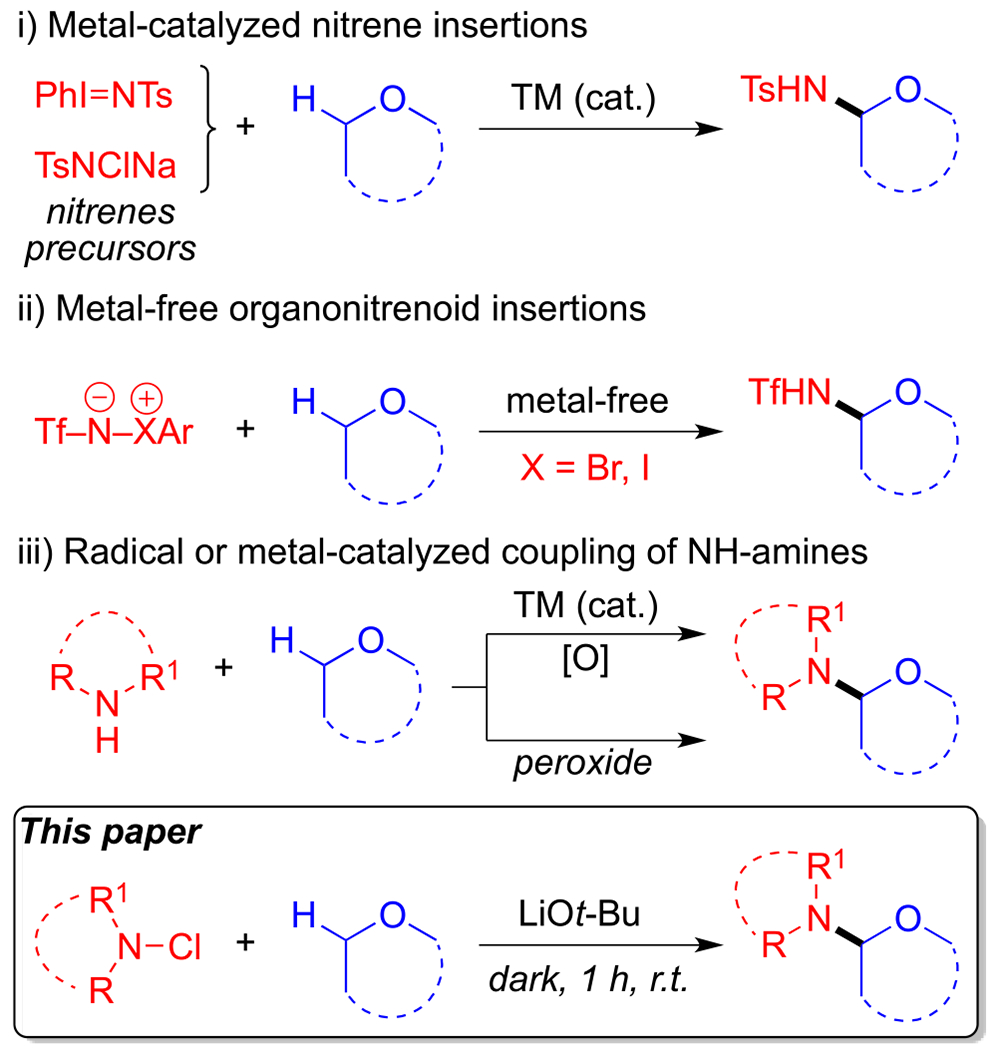
Different methods to generate hemiaminal ethers.
Despite the advances made toward the regioselective α-amination of C(sp3)–H bonds utilizing the methods mentioned above, there still remains the challenge of developing more environmentally friendly and atom-economical reactions that do not require the use of strong oxidants or external radical initiators. Herein, we describe a novel metal-free, regioselective α-amination of cyclic and acyclic ethers using stable N-chloroimides as the aminating agent. Our method generates the desired products in good yields at room temperature without the need for transition metal catalysts, external radical initiators, heat, or light. We found that stable N-chloroimides can selectively aminate in the presence of lithium tert-butoxide at room temperature (Figure 2).
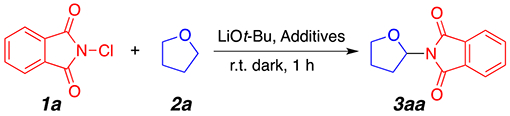
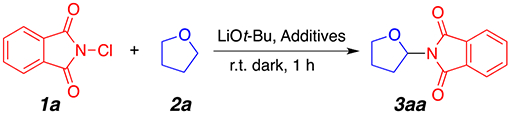
To begin our study, we chose commercially available N-chlorophthalimide 1a and tetrahydrofuran (THF) 2a as model substrates. Optimal reaction conditions (Table 1, entry 1) were obtained when using 1 equiv of LiOt-Bu at room temperature in the dark for 1 h. The lithium counterion appears essential for optimal reactivity since the use of sodium or potassium tert-butoxide (Table 1, entries 2 and 3) provided the desired product in significantly lower yields. Importantly, a control experiment in the absence of a base did not provide the desired product (Table 1, entry 4). Mild heating of the reaction at 40 °C did not have a deleterious effect on product formation (entry 5). Extensive screening of other inorganic (Table 1, entries 6–10) and organic (Table 1, entries 11 and 12) bases did not provide the desired product in satisfactory useful yields. An important observation from our optimization study was the deleterious effect of oxygen. Lower yields were obtained when the reaction was performed in an open-to-air microwave vial (Table 1, entry 13). Presumably, in the open-to-air reactions, we surmise that atmospheric oxygen is responsible for the quenching of the generated radical species, leading to reduced overall yields. Also, we hypothesize that covering the reaction flask in aluminum foil would prevent light-induced decomposition of N-chlorophthalimide, but such path seems to be minimal. Indeed, laboratory lighting did not have a considerable effect on product formation (Table 1, entry 14). As such, it was not surprising that the optimal reaction conditions were achieved in the absence of oxygen in an argon atmosphere. The use of reagent-grade THF (nonanhydrous and stabilized with 0.025% butylated hydroxytoluene) also afforded the aminated THF product in only 62% NMR yield (Table 1, entry 15), corroborating further the radical nature of this transformation. Finally, while reactions in neat THF (1 mL) were consistently optimal, the use of a 1:1 (by volume) mixture of dichloroethane (DCE) and THF at 50 °C also gave the desired product in 72% NMR yield (Table 1, entry 17).
Table 1.
Reaction Optimization and Influence of Bases on Reaction Outcomea
| ||||
|---|---|---|---|---|
|
| ||||
| entry | base | temperature (°C) | additives | yield (%)b |
| 1 | LiOt-Bu | 26 (rt) | 80c (86) | |
| 2 | NaOt-Bu | rt | 44 | |
| 3 | KOt-Bu | rt | 20 | |
| 4 | none | rt | 0 | |
| 5 | LiOt-Bu | 40 | 76 | |
| 6 | CS2CO3 | rt | 15 | |
| 7 | K2CO3 | rt | 0 | |
| 8 | LiOH·H2O | rt | 19 | |
| 9 | LiOAc | rt | 0 | |
| 10 | LiOMe | rt | 41 | |
| 11 | Et3N | rt | 19 | |
| 12 | pyridine | rt | 0 | |
| 13d | LiOt-Bu | rt | air | 22 |
| 14e | LiOt-Bu | rt | light | 78 |
| 15f | LiOt-Bu | rt | 62 | |
| 16g | LiOt-Bu | 50 | CH2Cl2 | 47 |
| 17g | LiOt-Bu | 50 | DCE | 72 |
All reactions were performed using 1 mL of anhydrous, degassed, nonstabilized THF, 0.14 mmol of N-chlorophthalimide, and 0.14 mmol of LiOt-Bu under an argon atmosphere, with constant stirring in the dark, and at room temperature, unless stated otherwise.
NMR yield obtained using dibromomethane as an internal standard.
Isolated yield.
The reaction was performed in an open atmosphere.
The reaction was performed without covering the reactor in aluminum foil to allow ambient light in the reaction flask.
Stabilized reagent-grade THF was used without further purification and without degassing.
The reaction was performed using a 1:1 ratio of THF and the additive solvent (total volume 1 mL).
With the established optimal conditions in hand, we proceeded to first examine the scope of ether substrates 2 compatible with our reaction conditions. Gratifyingly, the protocol was efficient for the α-amination of a diverse set of ethers (Scheme 2). Cyclic ethers such as tetrahydrofuran and tetrahydropyran were found to react smoothly to generate the coupling products 3aa and 3ab in good isolated yields. Linear ethers were also tolerated and afforded the corresponding products 3ac, 3ad, 3ae, and 3af in moderate to good yields. In a previously published article, diethyl ether 2d and methyl tert-butyl ether 2f substrates failed to undergo amination due to the poor solubility of the reagents used.10e This amination reaction may therefore be complementary to existing amination strategies. The reaction of 2-methyltetrahydrofuran provided an interesting regioselectivity assessment experiment. Interestingly, the aminated product 3ag was found as a major regioisomer favoring the less sterically hindered C–H bond at the 4 position (3ag, Scheme 2) in 72% yield (dr = 1:1.5). This result mirrors previous radical amination strategies independently developed by the Hu group10d and the Du group.10f Other unsymmetrical substrates, such as dimethoxyethane (DME), also provided the expected product 3ae as single regioisomers in the internal C(sp3)–H bond. Dioxane and acetals were also tolerated and provided the desired products 3ah and 3ai in good yields. These substrates required longer reaction times (4 h instead of 1 h) to achieve a sufficient amount of the desired aminated products (Figure 3). Tetrahydrothiophene 3aj also reacted under the current reaction conditions, but the transformation only provided the product in moderate yields. An important limitation of the current method remains the poor reactivity of benzyl ethers. As shown in Scheme 2, compounds 3ak and 3al were only provided in trace amounts. This, presumably, may be due to decomposition of the reactive intermediates into aldehydes under our reaction conditions.12
Scheme 2.
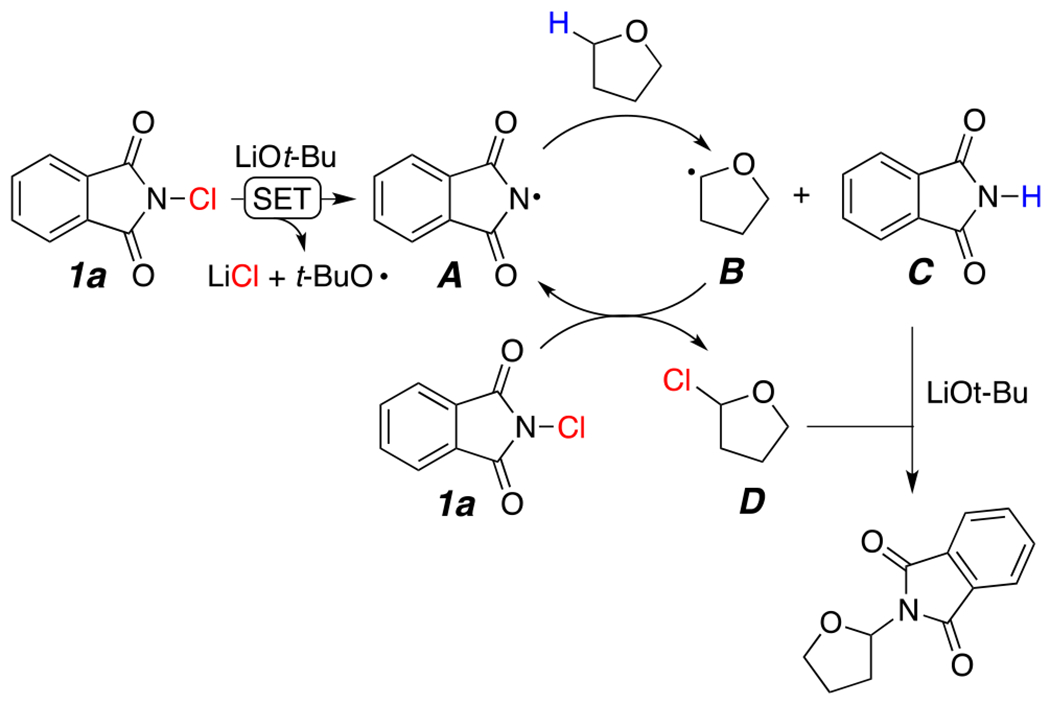
Proposed Mechanism
Figure 3.
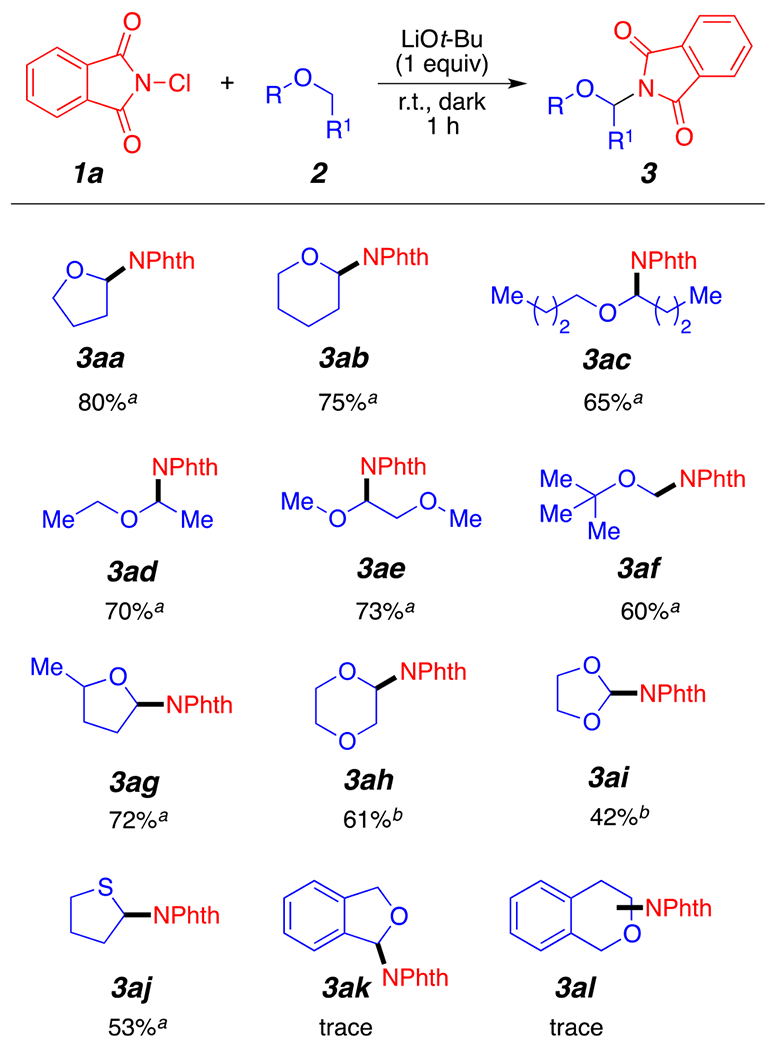
Reaction scope. All reactions were performed under an argon atmosphere in 1 mL of solvent, 1a (0.55 mmol), and LiOt-Bu (0.55 mmol), unless otherwise specified. (See Experimental Section.) (a) Refers to the reaction conditions shown in Table 1, entry 1 (General Method). (b) Reaction stirred for 4 h. (See Experimental Section.) Yields reported are isolated yields.
Next, we examine the compatibility of our optimal reaction conditions with different imide agents (1b–1h) using THF 2a as the preferred ether substrate. As shown in Figure 4A, the method enabled the α-amination of THF using some of the tested N-chloroimide reagents in moderate yields. N-Chlorosuccinimide (NCS) provided the desired product 3ba in a low yield; however, N-bromosuccinimide (NBS) and N-iodosuccinimide (NIS) did not provide the desired product in synthetically useful yields. Presumably, the greater reactivity of NBS and NIS lead to premature decomposition of the reagents. On the other hand, N-chloro-3,3-dimethyl-glutarimide (33DMNCG, product 3ca) provided the desired product in a good yield. We surmise that the difference in reactivity between succinimide reagents (NCS, NBS, and NIS) and glutarimide reagents (33DMNCG) arises from the decomposition pathways of these reagents (Figure 4B). Indeed, under radical reaction conditions, succinimide-based reagents generally undergo a radical ring opening after the formation of the N-centered radical, while 33DMNCG is a more stable N-centered radical and does not decompose via ring opening.13 Unfortunately, the corresponding aminated product for 2-chloro-5-nitroisoindoline-1,3-dione and 2,4,5,6,7-pentachloroisoindoline-1,3-dione was generated in only trace amounts under our reaction conditions. The lack of product formation with these electron-withdrawing N-chloroimides was quite surprising to us, and we can only surmise at this moment that the N-centered radical intermediates of these species were either not generated under our reaction conditions, are not reactive enough to perform a C–H abstraction, or are not stable.
Figure 4.
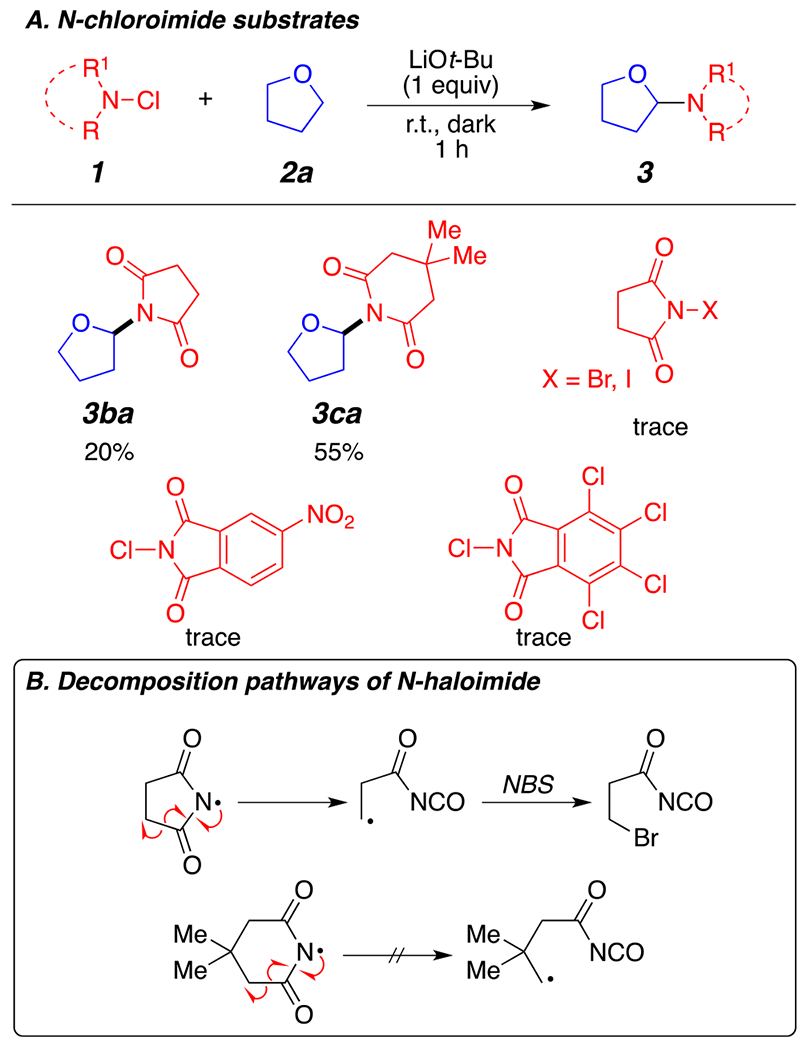
(A) Scope of imide reagents. (B) Radical decomposition pathways of N-halosuccinimide versus 3,3-dimethyl-N-haloglutarimide.
Of major interest, N-chlorosulfonimides also generated the desired product 3da in a good yield (Figure 5). Similarly, 1-chlorobenzotriazole provided a separable mixture of two regioisomeric products 3ea/3e’a in a good yield as well. Similar regioisomeric mixtures from reactive benzotriazole reagents have been reported in the literature.10f Interestingly, 1-chloroindoline-2,3-dione did not aminate THF under our reaction conditions.
Figure 5.
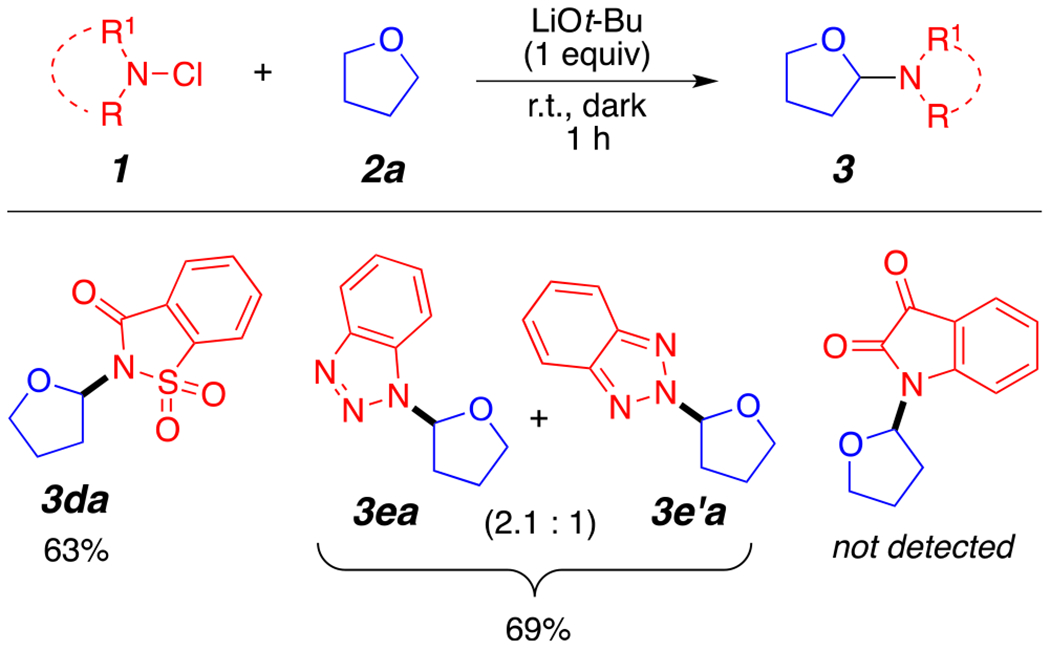
Scope of other aminating agents.
To gain insight into the possible mechanism of this reaction, we carried out a number of experiments, some of which are illustrated in Table 2. An important observation from these experiments was the relationship between the equivalents of base used and the overall yield of the reaction. When substoichiometric amounts of LiOt-Bu (e.g., 0.5 equiv) were used, the desired product was generated in only 48% yield (Table 2, entry 1). Similarly, superstoichiometric amounts of a base (1.5 equiv) was also detrimental to the reaction forming the desired product in 65% yield (Table 2, entries 2).
Table 2.
Mechanistic Studiesa
| |||
|---|---|---|---|
|
| |||
| entry | LiOt-Bu (equiv) | additives | yield (%)b |
| 1 | 0.5 | 48 | |
| 2 | 1.5 | 65 | |
| 3 | 1 | TEMPO (1 equiv) | 0 |
| 4 | 1 | TEMPO (0.5 equiv) | 0 |
All reactions were performed using 1 mL of anhydrous, degassed, nonstabilized THF, 0.14 mmol of N-chlorophthalimide, and 0.14 mmol of LiOt-Bu under an argon atmosphere, with constant stirring in the dark, and at room temperature, unless stated otherwise.
NMR yield obtained using dibromomethane as an internal standard.
In the absence of a base, no desired product formation was observed via NMR analysis, but GC–MS analysis of this crude reaction mixture revealed the formation of chlorinated THF and phthalimide. (See Supporting Information.) Similar halogenation of ethers using N-halosuccinimide reagents is well-documented in the literature.12 Addition of 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as a radical trapping reagent into the reaction mixture led to complete reaction shutdown (Table 2, entries 3 and 4). Importantly, neither the desired product nor the chlorinated THF byproducts were observed through GC–MS analysis of these crudes.
In an attempt to trap radical intermediates generated during the reaction, we used 1,1-diphenylethylene (4) as a reaction additive.14 (See Supporting Information.) To our delight, we observed the formation of products 5a, 5b, and 5c by GC–MS (Scheme 1). This result suggests the formation of chlorine, tert-butoxy, and phthalimide radicals. Formation of 5a and 5c could come from homolytic cleavage of the N–Cl bond15 of 1a in the ethereal solvent. Such an initiation step is supported by the presence of chlorinated THF in the absence of a base.12 Formation of 5b and 5c supports a single-electron transfer (SET) step between LiOt-Bu and 1a. Literature precedents for this initiation step are documented.16
Scheme 1.
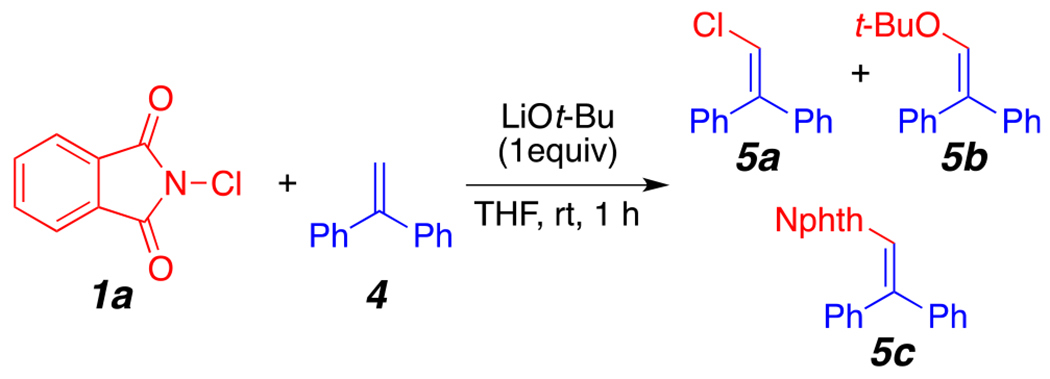
Radical Trapping Experiment
On the basis of the preliminary mechanistic investigations above and previous literature on the functionalization of ethers,6 we propose a plausible mechanism involving both a radical step and a two-electron substitution (Scheme 2).
A single-electron transfer (SET) process between LiOt-Bu and N-chlorophthalimide would generate tert-butoxyl radical and phthalimidyl radical A (Scheme 2). Literature precedents for this initiation step are documented16 and in our case supported by the generation of products 5b and 5c. Regioselective abstraction of α-hydrogen from THF by radical intermediate A forms a stable carbon-centered ethereal radical B and phthalimide C. Stable imidyl radicals have been shown to react as good hydrogen abstractors.13a In the propagation step, radical intermediate B reacts with reagent 1a to form chlorinated THF D and regenerates the imidyl radical A. Similar radical chlorination mechanisms have been reported with N-chlorosulfamate esters.17 Finally, the base deprotonates phthalimide C to generate an imide anion that subsequently reacts with D via SN2 to afford the desired product (Scheme 2).
In summary, we have discovered a mild and metal-free method for the formation of hemiaminal ethers. This new approach enables the regioselective α-amination of various cyclic and acyclic ethers with different N-chloroimides such as phthalimides, sulfonimides, and triazoles. Preliminary mechanistic investigations suggest the reaction is initiated via, first, a radical chlorination of the ether substrate followed by a substitution reaction with the nucleophilic nitrogen source to afford the desired aminated product. This method provides rapid access to many hemiaminal ethers that may serve as useful synthetic or biologically active small molecule intermediates for further functionalization.
EXPERIMENTAL SECTION
General Considerations.
All reagents and solvents were purchased and used without further purification unless otherwise noted. All reactions were performed under an inert atmosphere unless otherwise stated. Room temperature refers to 26 °C.
Moisture-sensitive reactions were performed using flame-dried glassware under an atmosphere of dry argon (Ar). Air- and water-sensitive reactions were setup in a Vacuum Atmosphere GENESIS glovebox held under an atmosphere of argon gas (working pressure 2–6 mbar).
Flame-dried equipment was stored in a 130 °C oven before use and either allowed to cool in a cabinet desiccator or assembled hot and allowed to cool under an inert atmosphere. Air- and moisture-sensitive liquids and solutions were transferred via a plastic or glass syringe.
Chromatographic purification of products was accomplished using flash column chromatography Silicycle Silica flash F60 (particle size 40–63 μm, 230–400 mesh).
Thin-layer chromatography was performed on EMD Millipore silica gel 60 F254 glass-backed plates (layer thickness 250 μm, particle size 10–12 μm, impregnated with a fluorescent indicator). Visualization of the developed chromatogram was accomplished by fluorescence quenching under shortwave UV light and/or by staining with phosphomolybdic acid, p-anisaldehyde, or KMnO4 stains.
Instrumentation.
For NMR spectrometry, NMR spectra were obtained on Bruker spectrometers operating at 400 or 500 MHz for 1H NMR and 101 or 126 MHz for 13C{1H} NMR. The data were reported in the following order: chemical shifts (δ ppm), multiplicity (s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet), coupling constant, (Hz), relative integral made in reference to NMR solvent signals.
For mass spectrometry, gas chromatograph–mass spectrometry was obtained using a Hewlett-Packard GC System HP 6890 Series coupled with a HP 5973 Mass Selective Detector. High-resolution mass spectra were obtained using an Agilent Technologies 6520 Accurate-Mass Q-TOF LC/MS with electrospray ionization (ESI).
Optimization Method.
General Method.
On a bench top, a 10 mL microwave vial was charged with the appropriate N-chloroimide (0.55 mmol), covered with a Kim wipe, and tightened with a rubber band. The vial was then brought into an argon-filled glovebox before adding the base (0.55 mmol) and capped with 20 mm microwave crimp caps with septa in the glovebox. The vial was removed from the glovebox, covered completely with aluminum foil, and 2 mL of the corresponding ether solvent was added. The reaction was then stirred at room temperature for 1 h unless otherwise stated. After completion, the reaction mixture was prepared directly for chromatography without workup.
Compounds Synthesis and Characterization.
1-Chloro-4,4-dimethylpiperidine-2,6-dione (1c).
A 25 mL round-bottom flask was flame-dried and allowed to cool to room temperature. The flask was then cooled to 0 °C and charged with 3,3,-dimethylglutarimide (240 mg, 1.70 mmol, 1 equiv). Ten mL of anhydrous DCM was subsequently added to dissolve the 3,3,-dimethylglutarimide. Isocyanuric chloride (500 mg, 2.15 mmol, 1.30 equiv) was added in one portion, and the resulting solution was stirred at 0 °C for 4 h. After this time, the reaction mixture was prepared for flash chromatography (solid deposition) without any workup. The desired product was isolated as a white solid in 92% yield. Spectra data matched reported data.18 1H NMR (400 MHz, CDCl3): δ 1.15 (s, 6H), 2.73 (s, 4H). 13C{1H} NMR (100 MHz, CDCl3): δ 27.4, 29.5, 47.4, 167.4.
2-(Tetrahydrofuran-2-yl)isoindoline-1,3-dione (3aa).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydrofuran), product 3aa was isolated in 80% yield (96 mg) after column chromatography (Rf = 0.39, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.90–2.07 (m, 1H), 2.24–2.33 (m, 1H), 2.35–2.44 (m, 1H), 2.51–2.59 (m, 1H), 3.92–3.97 (m, 1H), 4.17–4.23 (m, 1H), 6.04 (dd, J = 4.84, 4.96 Hz, 1H), 7.71–7.73 (m, 1H), 7.82–7.85 (m, 1 H). 13C{1H} NMR (100 MHz, CDCl3): δ 26.0, 29.1, 69.8, 80.9, 123.3, 132.0, 134.1. 167.8.
2-(Tetrahydro-2H-pyran-2-yl)isoindoline-1,3-dione (3ab).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydropyran), product 3ab was isolated in 75% yield (96 mg) after column chromatography (Rf = 0.41, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.55–1.80 (m, 4 H), 2.04–2.08 (m, 1H), 2.74–2.80 (m, 1H), 3.65–3.71 (m, 1H), 4.11–4.15 (m, 1H), 5.35 (dd, J = 11.4, 2.2 Hz, 1H), 7.34–7.56 (m, 2H), 7.87–7.90 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 23.6, 24.9, 27.9, 68.9, 79.4, 123.5, 131.8, 134.2, 167.4.
2-(1-Butoxybutyl)isoindoline-1,3-dione (3ac).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of dibutyl ether), product 3ac was isolated in 65% yield (99 mg) after column chromatography (Rf = 0.28, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 0.87 (t, J = 7.36 Hz, 3H), 0.96 (t, J = 7.40 Hz, 3H), 1.21–1.59 (m, 3H), 1.41–1.50 (m, 1H), 1.51–1.59 (m, 1H), 2.10–2.15 (m, 1H), 2.22–2.31 (m, 1H), 3.41–3.51 (m, 2H), 5.38 (t, J = 7.04 Hz, 1H), 7.74–7.78 (m, 2H), 7.86–7.89 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 13.6, 13.7, 18.8, 19.2, 31.4, 34.6, 68.8, 82.0, 123.5, 131.7, 134.2, 168.1.
2-(1-Ethoxyethyl)isoindoline-1,3-dione (3ad).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of diethyl ether), product 3ad was isolated in 70% yield (85 mg) after column chromatography (Rf = 0.50, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.15 (t, J = 7.04 Hz, 3H), 1.76 (d, J = 6.32 Hz, 3H), 3.46–3.52 (m, 2H), 5.56 (q, J = 6.32 Hz, 1H), 7.71–7.74 (m, 2H), 7.83–7.85 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 14.9, 19.3, 64.2, 78.0, 123.4, 131.7, 134.2, 168.9.
2-(1,2-Dimethoxyethyl)isoindoline-1,3-dione (3ae).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of 1,2-dimethoxyethane), product 3ae was isolated in 73% yield (95 mg) after column chromatography (Rf = 0.35, hexane: ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 3.31 (s, 3H), 3.33 (s, 3H), 3.88–3.98 (m, 2H), 5.39 (t, J = 6.52 Hz, 1H), 7.67–7.69 (m, 2H), 7.78–7.81 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 56.9, 59.2, 70.9, 81.4, 123.6, 131.7, 134.3, 168.0.
2-(tert-Butoxymethyl)isoindoline-1,3-dione (3af).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of methyl tert-butyl ether), product 3af was isolated in 60% yield (77 mg) after column chromatography (Rf = 0.43, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.22 (s, 9H), 5.04 (s, 2H), 7.65–772 (m, 2H), 7.80–7.84 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 27.8, 61.3, 123.9, 134.2, 134.7, 167.6.
2-(5-Methyltetrahydrofuran-2-yl)isoindoline-1,3-dione (3ag).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of 2-methyltetrahydrofuran), product 3ag was isolated in 72% yield (92 mg) as a mixture of diastereoisomers after column chromatography (Rf = 0.52, hexane/ethyl acetate, 3:1, dr = 1:1.5). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.20 (d, J = 6.08 Hz, 1.9H), 1.29 (d, J = 6.0 Hz, 1.2 Hz), 1.50–1.58 (m, 0.8 H), 1.94–2.02 (m, 0.4H), 2.05–2.15 (m, 0.4H), 2.20–2.28 (m, 1H), 2.30–2.37 (m, 0.8H), 2.43–2.50 (m, 0.4H), 2.54–2.63 (m, 0.7H), 4.01–4.07 (m, 0.3H), 4.46–4.51 (m, 0.6H), 5.90 (dd, J = 3.76, 3.68 Hz, 0.3H), 6.03 (dd, J = 5.80, 3.64 Hz, 0.6 H), 7.63–7065 (m, 2H), 7.76–7.78 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 20.3, 20.8, 29.6, 29.8, 32.5, 33.9, 80.3, 80.7, 123.3, 132.0, 134.1, 168.0.
2-(1,4-Dioxan-2-yl)isoindoline-1,3-dione (3ah).
Using the General Method and stirring for 4 h, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of 1,4-dioxane), product 3ah was isolated in 61% yield (78 mg) after column chromatography (Rf = 0.26, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10d 1H NMR (400 MHz, CDCl3): δ 3.78–3.82 (m, 3H), 3.97–4.00 (m, 2H), 4.62 (dd, J = 10.2, 10.2 Hz, 1H), 5.57 (dd, J = 2.84, 2.84 Hz, 1H), 7.77–7.80 (m, 2H), 7.89–7.92 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 65.8, 66.4, 67.5, 76.2, 123.7, 131.6, 134.5, 167.1.
2-(1,3-Dioxolan-2-yl)isoindoline-1,3-dione (3ai).
Using the General Method and stirring for 4 h, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of 1,3-dioxolane), product 3ai was isolated in 42% yield (51 mg) after column chromatography (Rf = 0.40, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 4.09–4.13 (m, 2H), 4.46–4.49 (m, 2H), 6.80 (s, 1H), 7.75–7.77 (m, 2H), 7.87–7.89 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 66.6, 100.7, 123.6, 131.9, 134.4, 166.8.
2-(Tetrahydrothiophen-2-yl)isoindoline-1,3-dione (3aj).
Using the General Method, (100 mg of N-chlorophthalimide, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydrothiophene), product 3aj was isolated in 53% yield (68 mg) after column chromatography (Rf = 0.57, hexane/ethyl acetate, 3:1). 1H NMR (400 MHz, CDCl3): δ 1.93–2.02 (m, 1H), 2.22–2.29 (m, 1H), 2.39–2.53 (m, 2H), 2.86–2.91 (m, 1H), 3.29–3.35 (m, 1H), 5.99 (dd, J = 5.52, 5.68 Hz, 1H), 7.63–7.65 (m, 2H), 7.45–7.77 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 31.9, 34.36, 34.47, 57.5, 123.8, 132.0, 134.1, 167.4. Low-resolution MS (EI): 233 (M+), 200, 186, 174, 158, 149, 130, 117, 104, 86, 76, 59, 50. HRMS (ESI): [M + Na]+ calcd for C12H12NNaO2S, 256.0403; found, 256.0414.
1-(Tetrahydrofuran-2-yl)pyrrolidine-2,5-dione (3ba).
Using the General Method, (73.6 mg of N-chlorosuccinimide, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydrofuran), product 3ba was isolated in 20% yield (19 mg) after column chromatography (Rf = 0.41, hexane/ethyl acetate, 1:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.86–1.97 (m, 1H), 2.06–2.15 (m, 1H), 2.20–2.27 (m, 1H), 2.31–2.38 (m, 1H), 2.60 (s, 4H), 3.81–3.87 (m, 1H), 4.08 (q, J = 6.80 Hz, 1H), 5.80 (dd, J = 5.0, 5.0 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 26.1, 28.2, 28.5, 70.2, 81.6, 176.7.
4,4-Dimethyl-1-(tetrahydrofuran-2-yl)piperidine-2,6-dione (3ca).
Using the General Method, (100 mg of 1-chloro-4,4-dimethylpiperidine-2,6-dione, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydrofuran), product 3ca was isolated in 55% yield (62 mg) after column chromatography (Rf = 0.29, hexane/ethyl acetate, 3:1). Spectra data matched reported data.10f 1H NMR (400 MHz, CDCl3): δ 1.00 (s, 6H),1.85–1.90 (m, 1H), 2.04–2.12 (m, 1H), 2.17–2.25 (m, 1H), 2.42 (s, 4H), 3.79 (td, J = 4.60 Hz, 4.14 Hz, 1H), 4.14 (q, J = 7.12 Hz, 1H), 6.35 (dd, J = 4.64, 3.09 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 26.4, 27.6, 28.9, 29.0. 29.6, 47.3, 70.0, 83.0, 172.0.
2-(Tetrahydrofuran-2-yl)benzo[d]isothiazol-3(2H)-one 1,1-dioxide (3da).
Using the General Method, (100 mg of N-chlorosaccharin, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydrofuran), product 3da was isolated in 63% yield (73 mg) after column chromatography (Rf = 0.31, hexane/ethyl acetate, 3:1). Spectra data matched reported data.6 1H NMR (400 MHz, CDCl3): δ 2.02–2.09 (m, 1H), 2.30–2.43 (m, 1H), 2.69–2.76 (m, 1H), 3.98–4.03 (m,1 H), 4.25–4.31 (m, 1H), 6.11 (dd, J = 3.68, 3.36 Hz, 1H), 7.81–7.90 (m, 3H), 8.03 (d, J = 7.16 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 24.6, 30.3, 70.1, 85.8, 120.7, 125.1, 126.8, 134.2, 135.0, 138.4, 159.2.
1-(Tetrahydrofuran-2-yl)-1H-benzo[d][1,2,3]triazole (3ea) and 2-(Tetrahydrofuran-2-yl)-2H-benzo[d][1,2,3]triazole (3e’a).
Using the General Method, (100 of mg 1-chlorobenzotriazole, 44 mg of lithium tert-butoxide, and 2 mL of tetrahydrofuran), products are isolated in 69% yield (85 mg, combined) after column chromatography. Spectra data matched reported data.19 3ea (47% yield, 58 mg) 1H NMR (400 MHz, CDCl3): δ 2.05–2.16 (m, 1H), 2.26–2.38 (m, 1H), 2.40–2.50 (m, 1H), 3.03–3.10 (m, 1H), 3.94–4.05 (m, 2H), 6.45 (dd, J = 2.61 Hz, 4.81 Hz, 1H), 7.29–7.33 (m, 1H), 7.40–7.44 (m, 1H), 7.65 (d, J = 8.82 Hz, 1H), 8.00 (d, J = 8.82 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ 24.4, 30.8, 69.2, 87.9, 110.4, 119.7, 124.1, 127.4, 132.8, 146.3. 3e’a (22% yield, 27 mg) 1H NMR (400 MHz, CDCl3): δ 2.04–2.11 (m, 1H), 2.39–2.50 (m, 2H), 2.64–2.72 (m, 1H), 4.07 (q, J = 6.32 Hz, 1H), 4.24–4.30 (m, 1H), 6.52 (dd, J = 2.21, 4.46 Hz, 1H), 7.29–7.33 (m, 2H), 7.79–7.82 (m, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 24.3, 32.3, 70.2, 94.2, 118.5, 126.6, 144.3.
Radical Trapping: Formation of Compounds 5a–c.
A microwave vial was charged with 250 mg of N-chlorophthalimide. The vial was then brought into a glovebox, and 110 mg of lithium tert-butoxide was added; the vial was capped and removed from the glovebox. The vial was completely covered with aluminum foil before adding 500 μL of 1,1-diphenylethylene in 500 μL of THF. The resulting solution was stirred at room temperature for 1 h, after which the crude mixture was filtered through a short pad of silica gel. GC–MS and NMR analysis of the crude mixture showed the formation of products 5a, 5b, and 5c. (See Supporting Information for the GC–MS spectrum.)
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge IUPUI for financial support. Z.W.T. is also grateful for financial support from the Center for Research and Learning (CRL) at IUPUI (Undergraduate Research Opportunities; MURI Projects). Prof. J.L. Roizen of Duke University and Prof. M.H. Nantz of the University of Louisville are thanked for their insights. The authors also acknowledge J.T. Floreancig for his help in completing the project.
Funding
This publication was made possible, in part, with support from the Indiana Clinical and Translational Sciences Institute funded in part by grant no. UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Start-up funding from Indiana University–Purdue University Indianapolis (IUPUI) was also used to support this research.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b00824.
1H and 13C{1H} NMR spectra for all aminated products and GC–MS spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Ricci A Amino Group Chemistry, From Synthesis to the Life Sciences; Wiley-VCH: Weinheim, 2008. [Google Scholar]; (b) Pirrung MC Book Review of Amino Group Chemistry: From Synthesis to the Life Sciences. J. Am. Chem. Soc 2008, 130, 8567–8567. [Google Scholar]
- (2).(a) Subramaniam G; Hiraku O; Hayashi M; Koyano T; Komiyama K; Kam T-S Biologically Active Aspidofractinine, Rhazinilam, Akuammiline, and Vincorine Alkaloids from Kopsia. J. Nat. Prod 2007, 70, 1783–1789. [DOI] [PubMed] [Google Scholar]; (b) Zu L; Boal BW; Garg NK Total Synthesis of (±)-Aspidophylline A. J. Am. Chem. Soc 2011, 133, 8877–8879. [DOI] [PubMed] [Google Scholar]; (c) Tan CH; Ma XQ; Chen GF; Zhu DY Two Novel Lycopodium Alkaloids from Huperzia serrata. Helv. Chim. Acta 2002, 85, 1058–1061. [Google Scholar]; (d) Nakayama A; Kogure N; Kitajima M; Takayama H Asymmetric Total Synthesis of a Pentacyclic Lycopodium Alkaloid: Huperzine Q. Angew. Chem., Int. Ed 2011, 50, 8025–8028. [DOI] [PubMed] [Google Scholar]; (e) Campbell EL; Zuhl AM; Liu CM; Boger DL Total Synthesis of (+)-Fendleridine (Aspidoalbidine) and (+)-1-Acetylaspidoalbidine. J. Am. Chem. Soc 2010, 132, 3009–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Tan SH; Banwell MG; Willis AC; Reekie TA Application of a Raney-Cobalt-Mediated Tandem Reductive Cyclization Protocol to Total Syntheses of the Aspidosperma Alkaloids (±)-Limaspermidine and (±)-1-Acetylaspidoalbidine. Org. Lett 2012, 14, 5621–5623. [DOI] [PubMed] [Google Scholar]
- (3).Bonnac LF; Mansky LM; Patterson SE Structure-Activity Relationships and Design of Viral Mutagens and Application to Lethal Mutagenesis. J. Med. Chem 2013, 56, 9403–9414. [DOI] [PubMed] [Google Scholar]
- (4).Cheng X; Hii KK Palladium-Catalyzed Addition of R2NH to Double Bonds. Synthesis of α-Amino Tetrahydrofuran and Pyran Rings. Tetrahedron 2001, 57, 5445–5450. [Google Scholar]
- (5).Eliel EL; Daignault RA Reductions with Metal Hydrides. XVI. Reduction of Some 2-Tetrahydropyranylamines. J. Org. Chem 1965, 30, 2450–2451. [Google Scholar]
- (6).Sun K; Wang X; Li G; Zhu Z; Jiang Y; Xiao B Efficient Imidation of C(sp3)-H Bonds Adjacent to Oxygen Atoms of Aryl Ethers under Metal-Free Conditions. Chem. Commun 2014, 50, 12880–12883. [DOI] [PubMed] [Google Scholar]
- (7).For a review on hemiaminal ether synthesis via C–H activation, see:; Dian L; Quingyu X; Daisy Z-N; Yunfei D Direct Functionalization of Alkyl Ethers to Construct Hemiaminal Ether Skeletons (HESs). Org. Biomol. Chem 2018, 16, 4384–4398. [DOI] [PubMed] [Google Scholar]
- (8).(a) Yu X-Q; Huang J-S; Zhou X-G; Che C-M Amidation of Saturated C-H Bonds Catalyzed by Electron-Deficient Ruthenium and Manganese Porphyrins. A Highly Catalytic Nitrogen Atom Transfer Process. Org. Lett 2000, 2, 2233–2236. [DOI] [PubMed] [Google Scholar]; (b) Albone DP; Challenger S; Derrick AM; Fillery SM; Irwin JL; Parsons CM; Takada H; Taylor PC; Wilson DJ Amination of Ethers using Chloramine-T Hydrate and a Copper(I) Catalyst. Org. Biomol. Chem 2055, 3, 107–111. [DOI] [PubMed] [Google Scholar]; (c) Fructos MR; Trofimenko S; Diaz-Requejo MM; Pérez PJ Facile Amine Formation by Intermolecular Catalytic Amidation of Carbon-Hydrogen Bonds. J. Am. Chem. Soc 2006, 128, 11784–11791. [DOI] [PubMed] [Google Scholar]; (d) He L; Yu J; Zhang J; Yu X-Q α-Amidation of Cyclic Ethers Catalyzed by Simple Copper Salt and a Mild and Efficient Preparation Method for α,ϖ-Amino Alcohols. Org. Lett 2007, 9, 2277–2280. [DOI] [PubMed] [Google Scholar]; (e) Bhuyan R; Nicholas KM Efficient Copper-Catalyzed Benzylic Amidation with Anhydrous Chloramine-T. Org. Lett 2007, 9, 3957–3959. [DOI] [PubMed] [Google Scholar]; (f) Cano I; Nicasio MC; Pérez PJ Nitrene Transfer Reactions Catalysed by Copper(I) Complexes in Ionic Liquid using Chloramine-T. Dalton Trans. 2009, 0, 730–734. [DOI] [PubMed] [Google Scholar]; (g) Tubaro C; Biffis A; Gava R; Scattolin E; Volpe A; Basato M; Díaz-Requejo MM; Perez PJ Polynuclear Copper(I) Complexes with Chelating Bis and Tris N Heterocyclic Carbene Ligands: Catalytic Activity in Nitrene and Carbene Transfer Reactions. Eur. J. Org. Chem 2012, 2012, 1367–1372. [Google Scholar]; (h) Gava R; Biffis A; Tubaro C; Zaccheria F; Ravasio N Heterogeneous Copper-Based Catalysts for the Amidation of Activated C-H Bonds. Catal. Commun 2013, 40, 63–65. [Google Scholar]; (i) Abedi Y; Biffis A; Gava R; Tubaro C; Chelucci G; Stoccoro S Cu-iminopyridine Complexes as Catalysts for Carbene and Nitrene Transfer Reactions. Appl. Organomet. Chem 2014, 28, 512–516. [Google Scholar]; (j) Wang H; Li Y; Wang Z; Lou J; Xiao Y; Qiu G; Hu X; Altenbach H-J; Liu P Iron-Catalyzed Efficient Intermolecular Amination of C(sp3)-H bonds with Bromamine-T as Nitrene Source. RSC Adv. 2014, 4, 25287–25290. [Google Scholar]; (k) Beltrán Á; álvarez E; Díaz Requejo MM; Pérez PJ Direct Synthesis of Hemiaminal Ethers via a Three Component Reaction of Aldehydes, Amines and Alcohols. Adv. Synth. Catal 2015, 357, 2821–2826. [Google Scholar]
- (9).Ochiai M; Yamane S; Hoque MM; Saito M; Miyamoto K Metal-Free α-C-H Amination of Ethers with Hypervalent Sulfonylimino-λ3-bromane that Acts as an Active Nitrenoid. Chem. Commun 2012, 48, 5280–5282. [DOI] [PubMed] [Google Scholar]
- (10).(a) Guo H-M; Xia C; Niu H-Y; Zhang X-T; Kong S-N; Wang D-C; Qu G-R Intermolecular Hydrogen Abstraction Reaction between Nitrogen Radicals in Purine Rings and Alkyl Ethers: A Highly Selective Method for the Synthesis of N 9 Alkylated Purine Nucleoside Derivatives. Adv. Synth. Catal 2011, 353, 53–56. [Google Scholar]; (b) Muramatsu W; Nakano K Efficient C(sp3)-H Bond Functionalization of Isochroman by AZADOL Catalysis. Org. Lett 2015, 17, 1549–1552. [DOI] [PubMed] [Google Scholar]; (c) Campos J; Goforth SK; Crabtree RH; Gunnoe TB Metal-Free Amidation of Ether sp3 C-H Bonds with Sulfonamides using PhI(OAc)2. RSC Adv. 2014, 4, 47951–47957. [Google Scholar]; (d) Buslov I; Hu X Transition Metal Free Intermolecular α C-H Amination of Ethers at Room Temperature. Adv. Synth. Catal 2014, 356, 3325–3330. [Google Scholar]; (e) Pan Z; Fan Z; Lu B; Cheng J Halogen Bond Promoted α C-H Amination of Ethers for the Synthesis of Hemiaminal Ethers. Adv. Synth. Catal 2018, 360, 1761–1767. [Google Scholar]; (f) Dian L; Wang S; Zhang-Negrerie D; Du Y; Zhao K Organocatalytic Amination of Alkyl Ethers via n-Bu4NI/t-BuOOH-Mediated Intermolecular Oxidative C(sp3)-N Bond Formation: Novel Synthesis of Hemiaminal Ethers. Chem. Commun 2014, 50, 11738–11741. [DOI] [PubMed] [Google Scholar]
- (11).Pan S; Liu J; Li H; Wang Z; Guo X; Li Z Iron-Catalyzed N-Alkylation of Azoles via Oxidation of C-H Bond Adjacent to an Oxygen Atom. Org. Lett 2010, 12, 1932–1935. [DOI] [PubMed] [Google Scholar]
- (12).Mayhoub AS; Talukdar A; Cushman M An Oxidation of Benzyl Methyl Ethers with NBS that Selectively Affords Either Aromatic Aldehydes or Aromatic Methyl Esters. J. Org. Chem 2010, 75, 3507–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Lüning U; Seshadri S; Skell PS Glutarimidyl Chemistry: Substitution Reactions. Mechanism of Ziegler Brominations. J. Org. Chem 1986, 51, 2071–2077. [Google Scholar]; (b) Chow YL; Zhao D-C Photodecomposition of N-Bromosuccinimide. Radical Chain Carriers and Their Interrelations. J. Org. Chem 1987, 52, 1931–1939. [Google Scholar]
- (14).For the precedent for the use of 1,1-diphenylethylene as a radical trapping agent, see:; Sha W; Zhang W; Ni S; Mei H; Han J; Pan Y Photoredox-Catalyzed Cascade Difluoroalkylation and Intramolecular Cyclization for Construction of Fluorinated γ-Butyrolactones. J. Org. Chem 2017, 82, 9824–9831. [DOI] [PubMed] [Google Scholar]
- (15).(a) Percec V; Grigoras C N Chloro Amides, Aactams, Carbamates, and Imides. New Classes of Initiators for the Metal Catalyzed Living Radical Polymerization of Methacrylates. J. Polym. Sci., Part A: Polym. Chem 2005, 43, 5283–5299. [Google Scholar]; (b) Wang X-Y; Chang L-Q; Zhou H; Zhang K-D N Chlorosuccinimide (NCS): A Novel Initiator for Atom Transfer Radical Polymerization of Methyl Methacrylate. Chin. J. Chem 2006, 24, 1214–1218. [Google Scholar]; (c) O’reilly RJ; Karton A; Radom L N-H and N-Cl Homolytic Bond Dissociation Energies and Radical Stabilization Energies: An Assessment of Theoretical Procedures through Comparison with Benchmark Quality W2w Data. Int. J. Quantum Chem 2012, 112, 1862–1878. [Google Scholar]; (d) Bernofsky C; Bandara BM; Hinojosa O; Strauss SL Hypochlorite-Modified Adenine Nucleotides: Structure, Spin-Trapping, and Formation by Activated Guinea Pig Polymorphonuclear Leukocytes. Free Radical Res. Commun 1990, 9, 303–315. [DOI] [PubMed] [Google Scholar]; (e) Hawkins CL; Davies MJ Hypochlorite-Induced Damage to Nucleosides: Formation of Chloramines and Nitrogen-Centered Radicals. Chem. Res. Toxicol 2001, 14, 1071–1081. [DOI] [PubMed] [Google Scholar]
- (16).Barham JP; Coulthard G; Emery KJ; Doni E; Cumine F; Nocera G; John MP; Berlouis LEA; McGuire T; Tuttle T; Murphy JA KOtBu: A Privileged Reagent for Electron Transfer Reactions? J. Am. Chem. Soc 2016, 138, 7402–7410. [DOI] [PubMed] [Google Scholar]
- (17).Short MA; Blackburn JM; Roizen JL Sulfamate Esters Guide Selective Radical-Mediated Chlorination of Aliphatic C-H Bonds. Angew. Chem., Int. Ed 2018, 57, 296–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).De Luca L; Giacomelli G; Nieddu G A Simple Protocol for Efficient N-Chlorination of Amides and Carbamates. Synlett 2005, 2, 223–226. [Google Scholar]
- (19).Singh MK; Akula HK; Satishkumar S; Stahl L; Lakshman MK Ruthenium-Catalyzed C-H Bond Activation Approach to Azolyl Aminals and Hemiaminal Ethers, Mechanistic Evaluations, and Isomer Interconversion. ACS Catal. 2016, 6, 1921–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.