

Abstract
Objectives
The aim of this study was to discern the role of the cardiac voltage-gated sodium ion channel SCN5A in the etiology of dilated cardiomyopathy (DCM).
Background
Dilated cardiomyopathy associates with mutations in the SCN5A gene, but the frequency, phenotype, and causative nature of these associations remain the focus of ongoing investigation.
Methods
Since 1991, DCM probands and family members have been enrolled in the Familial Cardiomyopathy Registry and extensively evaluated by clinical phenotype. Genomic deoxyribonucleic acid samples from 338 individuals among 289 DCM families were obtained and screened for SCN5A mutations by denaturing high-performance liquid chromatography and sequence analysis.
Results
We identified 5 missense SCN5A mutations among our DCM families, including novel mutations E446K, F1520L, and V1279I, as well as previously reported mutations D1275N and R222Q. Of 15 SCN5A mutation carriers in our study, 14 (93%) manifested arrhythmia: supraventricular arrhythmia (13 of 15), including sick sinus syndrome (5 of 15) and atrial fibrillation (9 of 15), ventricular tachycardia (5 of 15), and conduction disease (9 of 15).
Conclusions
Mutations in SCN5A were detected in 1.7% of DCM families. Two-thirds (6 of 9) of all reported DCM mutations in SCN5A localize to the highly conserved homologous S3 and S4 transmembrane segments, suggesting a shared mechanism of disruption of the voltage-sensing mechanism of this channel leading to DCM. Not surprisingly, SCN5A mutation carriers show a strong arrhythmic pattern that has clinical and diagnostic implications.
Keywords: arrhythmia, dilated cardiomyopathy, genetics, ion channels
Most genes associated with dilated cardiomyopathy (DCM) encode for structural proteins involved with contractile function and the cytoskeletal matrix (1). Mutations in these genes are believed to diminish overall structural integrity of myocytes leading to contractile filament disarray, cell death, and the development of fibrosis that is characteristic of dilation (2). Defects identified in calcium and potassium regulation argue for an alternative disease mechanism of dilation remodeling primarily driven by dysfunction in electrical excitability rather than a structural defect (3–6). Our discovery of a D1275N alteration in the ion channel gene SCN5A associated with DCM suggested that dysfunction in electrical excitability, caused by disruption of sodium channel function, also leads to dilation remodeling (7).
Olson et al. (8,9) further substantiated sodium channel involvement in dilation etiology with independent confirmation of the D1275N mutation in another branch of the same DCM family and through the identification of 3 additional SCN5A missense mutations (T220I, R814W, D1595H) and 1 frameshift mutation (2550-2551 ins TG) that associated with DCM. Hershberger et al. (10) recently reported 2 additional mutations associated with the dilation phenotype: I1835T and R222Q
With established evidence for mutations in SCN5A as a cause of DCM, we sought to determine the prevalence of SCN5A mutations in subjects from a large multicenter DCM cohort, the Familial Cardiomyopathy Registry. Results described herein identify additional disease-causing mutations that we hypothesize act primarily through related mechanisms of altered electrical excitability leading to pathological ion dysregulation. Significantly, mutations currently identified associating with DCM in SCN5A tend to target domains important for voltage-dependent activation. We conclude that mutations in voltage-sensitive regions of SCN5A (also known as Nav 1.5) lead to the eventual manifestation of DCM.
Methods
Patient cohort.
In all, 338 DCM subjects (including 289 probands) within the Familial Cardiomyopathy Registry were screened for SCN5A mutations. Informed consent was obtained from registrants under the institutional review board policies of participating institutions. Diagnosis for the affected status of DCM was made according to published criteria (11). Affected subjects and related family members were evaluated by investigators. Evaluations included a complete medical and family history, physical examination, 12-lead electrocardiogram, and echocardiography. Additional family members were interviewed, and their medical histories and medical records were reviewed to allow an accurate analysis of the phenotype. For deceased relatives, hospital records were examined when available, family physicians were interviewed, and multiple informants among close relatives were consulted for accuracy of diagnosis.
SCN5A mutation screening.
Genomic deoxyribonucleic acid (DNA) was extracted through either peripheral blood leukocytes or buccal swab sampling (epitope) according to standard protocols. The SCN5A coding exons 2 through 28 and related intronic boundaries were studied. Primers were designed utilizing publicly available genetic maps (12) and optimized with Primer 3 Input software (13). The DNA was amplified by polymerase chain reaction.
Genetic screening was accomplished through the use of denaturing high-performance liquid chromatography on a WAVE Fragment Analysis System (Transgenomic, Omaha, Nebraska). An ABI 377 DNA sequencer (Applied Biosystems, Foster City, California) was utilized for selective bidirectional DNA sequencing of variants manifesting abnormal elution profile on the WAVE system. Sequencing results were compared to published sequences in the National Center for Biotechnology Information (NCBI) using basic local alignment search tool (BLAST) algorithms (14). Criteria for the classification of a mutation as putatively disease causing included the following: predicted alteration of amino acid sequence, evolutionary conservation of the particular residue altered, probable effects on protein biophysics based on functional studies of conserved homologous regions within the voltage-gated ion channel family, segregation among affected family members, and absence in normal controls. For control data, we used the NCBI variation database (dbSNP) (15), data from reported literature and/or 300 ethnically matched control chromosomes. Specifically, for mutations V1279I and D1275N, we studied 300 ethnically-matched control chromosomes available in our laboratory. For mutation E446K, we used published data of >300 ethnically matched control chromosomes reported for the same exon (exon 10) (16). For mutation F1529L (exon 27), we used 500 ethnically matched controls previously published for the same exon (8). Finally, for R222Q, we used control data in an ethnically matched population for the same mutation as published by Hershberger et al. (10).
Results
Missense mutations.
The SCN5A mutation screening of our DCM population identified 5 missense mutations: D1275N, F1520L, V1279I, E446K, and R222Q. We reported identifying mutation D1275N previously (7). Here, we report finding our 4 additional mutations in subjects among a cohort of 289 DCM families (Figs. 1 and 2). Of these 4 additional mutations, 3 proved to be novel (E446K, F1520L, V1279I), and 1 (R222Q) has recently been identified within a separate DCM pedigree by other investigators (10). In our population, mutations D1275N, F1520L, V1279I, and R222Q occurred in conjunction with familial DCM whereas E446K occurred in an apparently sporadic form. Phenotype characteristics of SCN5A mutation carriers are detailed in Table 1. The SCN5A mutation carriers included subjects with overt DCM as well as their relatives with predominant arrhythmic phenotypes, in particular in the younger generations. Remarkably, 14 of 15 SCN5A mutations carriers (93%) manifested arrhythmia such as supraventricular arrhythmia (13 of 15), including sinus node dysfunction (5 of 15), atrial fibrillation (9 of 15), ventricular tachycardia (5 of 15), and conduction disease (9 of 15) (Fig. 3). Sinus node dysfunction (or sick sinus syndrome) in these patients included a range of electrophysiologic abnormalities, from sinus bradycardia and atrial standstill to atrial tachyarrhythmias (supraventricular tachycardia, atrial flutter, and atrial fibrillation) (17).
Figure 1. Pedigrees in the Familial Cardiomyopathy Registry Containing SCN5A Mutations.
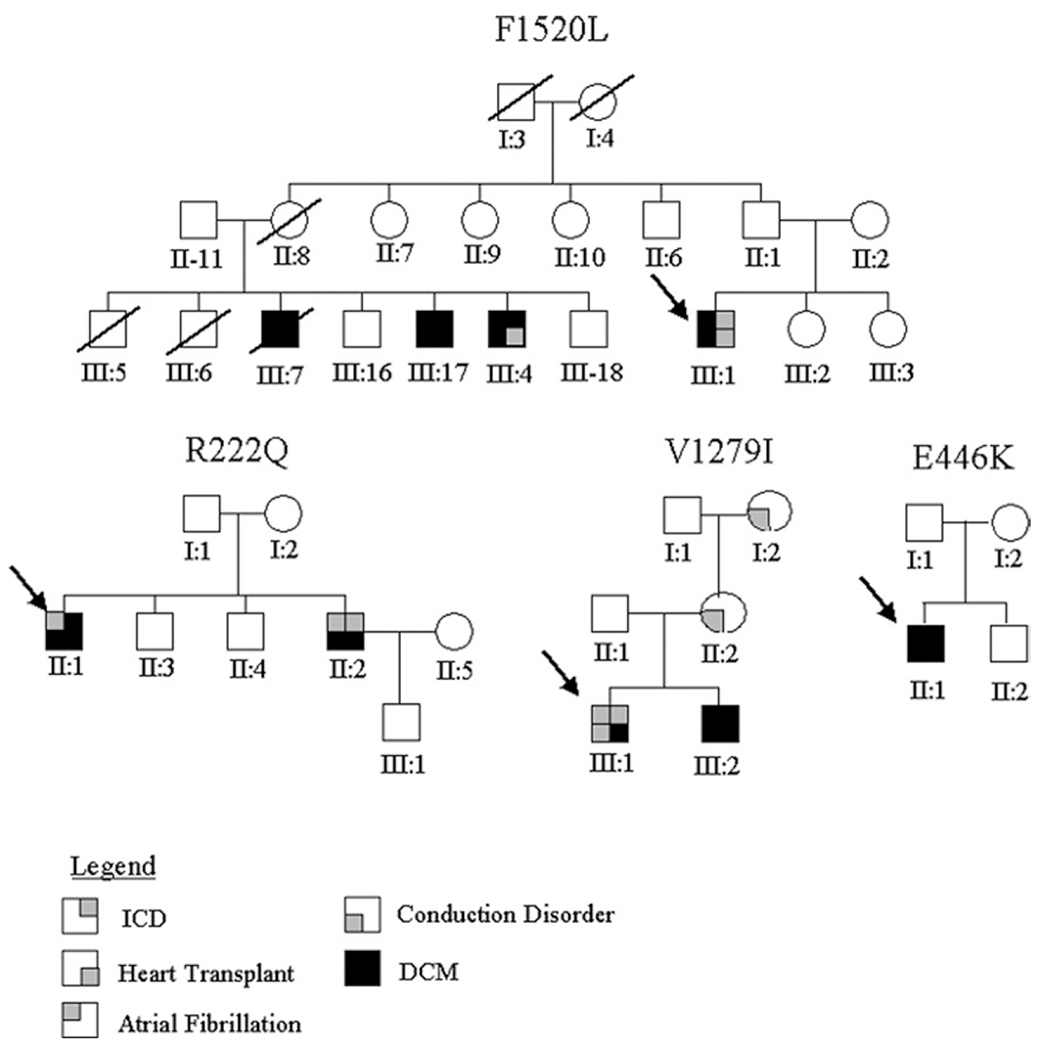
Missense mutations are listed as pedigree identifiers above related family trees. Generations are denoted by Roman numerals. Probands for each pedigree are designated by a black arrow. The legend for pertinent clinical information is described below the pedigrees. DCM = dilated cardiomyopathy; ICD = implantable cardioverter-defibrillator.
Figure 2. SCN5A Sequencing Results of the Familial Cardiomyopathy Registry.
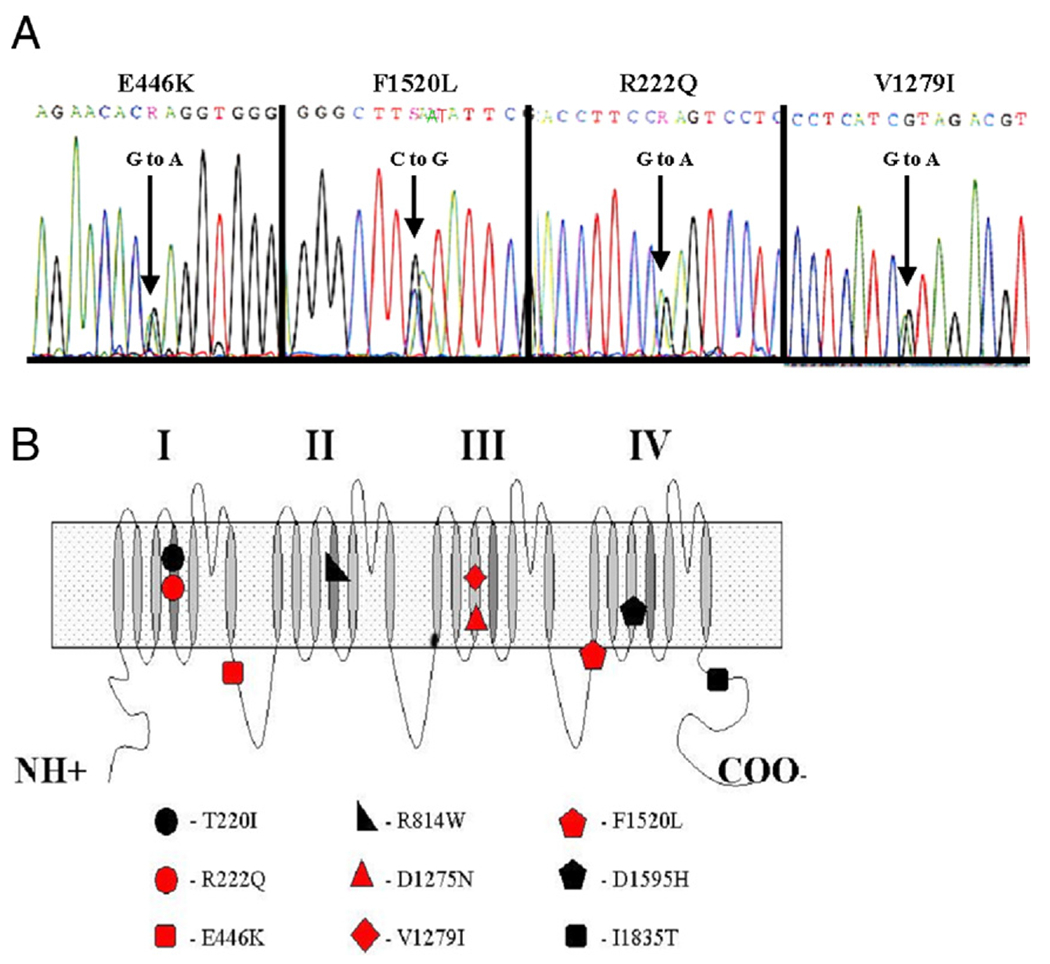
(A) Chromatograms demonstrating heterologous peaks in nucleotide sequence. For this report, samples manifesting abnormal elution profile on the WAVE system were sequenced, and 4 new missense mutations were identified among our dilated cardiomyopathy (DCM) population. (B) Relative locations of all currently reported DCM missense mutations of SCN5A superimposed on a schematic of the SCN5A protein (7,8,10). Red denotes 5 total missense mutations identified within the Familial Cardiomyopathy Registry, including the 4 new mutations reported here and D1275N identified previously (7).
Table 1.
Clinical Data for 5 Pedigrees Carrying SCN5A Mutations
| Subject | Pedigree ID* | Sex | Age at Onset, yrs† | Age at Diagnosis, yrs† | NYHA Functional Class | Symptoms | Arrhythmia | Conduction System Disease | LVEF, % | LVEDD, cm | Coronary Angiogram | Outcome | Follow-Up‡ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | R222Q | M | 25 | 26 | 1 | Palpitations | NSR, PVC >1,000/h, NSVT, PAC | No | 34 | 66 | NL | Stable, LVEF 46% | 172 |
| II-2 | R222Q | M | 17 | 19 | 1 | Tachycardia | AA, AF, PVC >1,400/h, NSVT >100 runs/h | No | 37 | 65 | NL | NYHA functional class II, LVEF 35%, ICD | 177 |
| III-1 | F1520L | M | 25 | 26 | 1 | Asymptomatic | NSR, PVC >100 runs/h, NSVT, PAC | No | 35 | 81 | N/A | ICD, transplant (38 yrs) | 138 |
| II-1 | E446K | M | 24 | 24 | 2 | DOE, fatigue, palpitations, CP | No | No | 22 | 68 | N/A | LVEF 40% | 39 |
| III-1 | V1279I | M | 20 | 50 | 1 | Asymptomatic | PAF, NSVT | LBBB | 51 | 61 | N/A | Stable, PSVT, LVEF 50%, ICD | 52 |
| II-6 | D1275N | M | 15 | 68 | 2 | DOE, syncope | AF, AFL | LAFB | (FS 23%) | N/A | N/A | Stable | 636 |
| III:4 | D1275N | M | 23 | 51 | 1 | Fatigue, syncope | AF | 3rd AVB | 50 | 55 | N/A | PM, stable | 336 |
| III:10 | D1275N | M | 22 | 41 | 1 | Palpitations | SSS, AF | 3rd AVB | 74 | 47 | N/A | PM, AV nodal ablation | 180 |
| III:14 | D1275N | M | 29 | 41 | 2 | Fatigue, palpitations | SSS, AF | 2nd AVB | (FS 20%) | 48 | N/A | PM, stable | 228 |
| III:15 | D1275N | M | 34 | 47 | 1 | Palpitations | SSS, SVT, AF | 1st AVB | 51 | 52 | N/A | PM, stable | 156 |
| III:17 | D1275N | F | 24 | 45 | 2 | Palpitations | Bradycardia | NL | (FS 30%) | N/A | N/A | Stable | |
| IV:2 | D1275N | M | 17 | 24 | 1 | Syncope | PVC, VT | 1st AVB, RBBB | 80 | 47 | N/A | PM, stable | 84 |
| IV:8 | D1275N | F | 14 | 20 | 1 | Syncope | SSS, AS, AF, SVT | RBBB | 56 | 35 | N/A | PM, stable | 72 |
| IV:9 | D1275N | F | 16 | 16 | 2 | DOE, palpitations, syncope | AF, AS | N/A | 50 | N/A | N/A | PM, DOE | 48 |
| IV:10 | D1275N | F | 13 | 18 | 1 | Palpitations | SSS | N/A | 58 | 44 | N/A | PM, stable | 60 |
Pedigree identifier (ID) identifies dilated cardiomyopathy (DCM) family by related SCN5A mutation and correlates to Figure 1, or in the case of D1275N, to the pedigree as previously reported (7).
Age in years.
Follow-up is months after diagnosis.
AA = atrial arrhythmias; AF = atrial fibrillation; AFL = atrial flutter; AS = atrial standstill; AVB = atrioventricular block; CP = chest pain; DOE = dyspnea on exertion; FS = fractional shortening; LAFB = left anterior fascicular block; LBBB = left bundle branch block; LVEDD = left ventricular end-diastolic dimension; LVEF = left ventricular ejection fraction; N/A = not available; NL = normal (absence of symptoms); No = no arrhythmia present; NSR = normal sinus rhythm; NSVT = nonsustained ventricular tachycardia; PAC = premature atrial contraction; PAF = paroxysmal atrial fibrillation; PM = pacemaker; PSVT = paroxysmal supraventricular tachycardia; PVC = premature ventricular contraction; RBBB = right bundle branch block; SSS = sick sinus syndrome; SVT = supraventricular tachycardia; VT = ventricular tachycardia.
Figure 3. Early Arrhythmia in SCN5A Mutation Carriers.
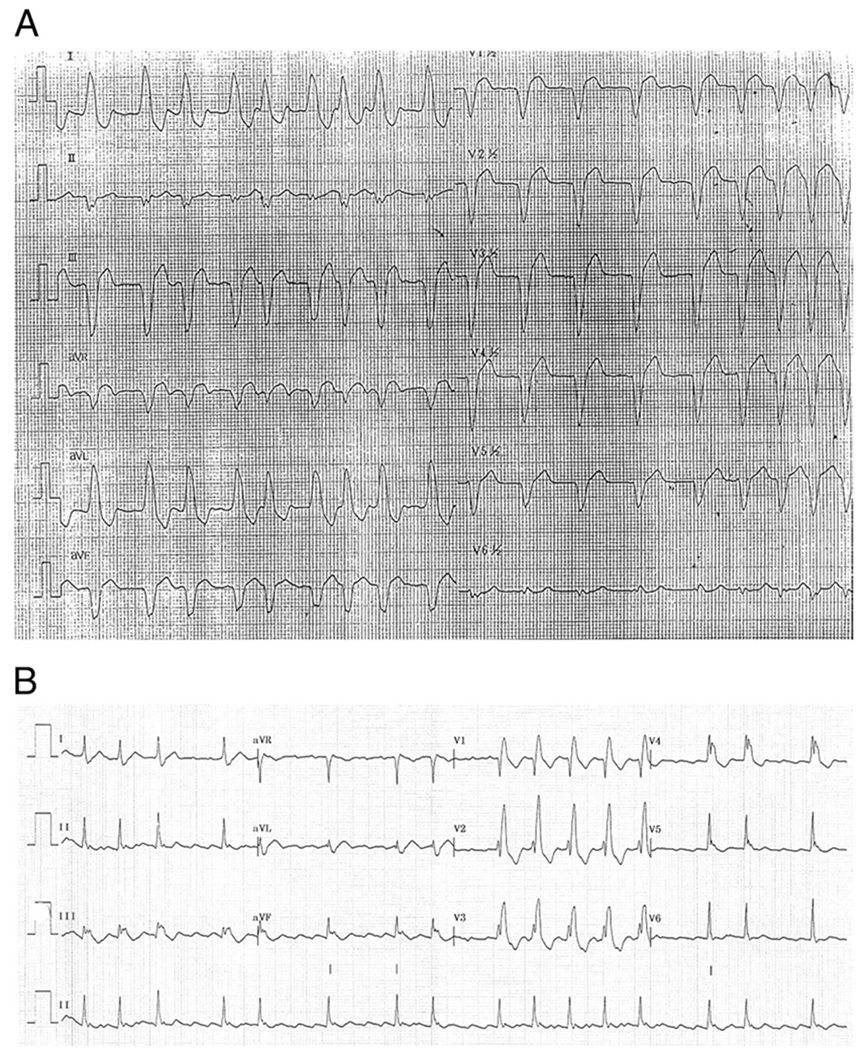
(A) Electrocardiogram of proband V1279I at the age of 47 years, showing atrial fibrillation and left bundle branch block. The left bundle branch block was accidentally found at a young age and followed for many years before the development of dilated cardiomyopathy. (B) Electrocardiogram of individual III-10 of family D1275N, showing atrial fibrillation when the patient had a normal left ventricular ejection fraction (74%) (see Table 1).
In 3 families (R222Q, F1520L, D1275N), a series of DCM genes had previously been excluded by screening analysis in our laboratory, including MYH7, MYH6, MYBPC3, ZASP, TNNT2, BAF, and EMD. Two families (E446K and V1279I) were recently enrolled and not previously screened for other DCM genes.
Mutation R222Q.
Two siblings carried the R222Q variation (c.665.G→A), and both shared the same phenotype, characterized by an early age of significant arrhythmic onset (19 and 25 years) with symptoms and signs including atrial fibrillation (II-1 and II-2 of R222Q pedigree) (Fig. 1 and Table 1). Neither demonstrated symptoms of heart failure (New York Heart Association [NYHA] functional class I). In a follow-up of >15 years, they remained stable with no or minimal additional symptoms. One of the siblings (II-2) received an implantable cardioverter-defibrillator on the basis of the severity of his ventricular arrhythmia. The mother was clinically investigated and found to be unaffected.
This R222Q variant alters a highly conserved, functionally critical, positively charged residue to an uncharged residue in domain I of the voltage-sensing transmembrane segment S4 (Fig. 2, Online Table 1) (18–20). Currently, there are 5 missense changes reported in this S4 transmembrane segment, and all associate with disease, including the T220I correlation to DCM identified by Olson et al. (8). The S4, R222 position is conserved across all domains of the family of voltage-gated ion channels regardless of species, including single-domain potassium channels and distantly related single-domain transient receptor protein channels (Online Table 1).
Mutation F1520L.
The proband of the F1520L (c.4560.C→G) pedigree demonstrated an early age of onset (26 years) with absence of heart failure symptoms at presentation (NYHA functional class I) (Fig. 1, Table 1). His abnormal electrocardiogram was discovered in a routine screening for competitive basketball players. The family history was characterized by a progressive worsening of phenotype leading ultimately to heart failure and heart transplantation for the proband and an affected family member (Fig. 1) despite optimal medical therapy performed in heart failure referral centers.
The novel variant F1520L changes the residue at this position from an aromatic ring to an aliphatic side chain. This change occurs in the domain III to domain IV (DIII-DIV) cytosolic linker very near the S1 transmembrane segment of DIV (Fig. 2) (18). This linker region encodes the fastinactivation gate, and missense mutations R1512W and K1527R are associated with Brugada syndrome and in close proximity to position F1520 (21,22). The F1520 residue is conserved across species within SCN5A but not among homologous voltage-gated sodium and calcium channel family members (Online Table 2).
Mutation E446K.
The index patient of the E446K (c.1336.G→A) pedigree had no clear family history and was considered a “sporadic” case (Fig. 1, Table 1). He demonstrated an early age of onset (24 years) and minimal symptoms of heart failure (NYHA functional class II). He presented with a history of palpitations, but no evidence of arrhythmia was found in routine electrocardiograms, and subsequently, no Holter monitoring was performed. His left ventricular ejection fraction dramatically improved from 24% to 40% over the 3 years of follow-up. His mother was clinically unaffected and did not carry the variant allele.
Novel variant E446K is found in the domain I to domain II (DI-DII) cytosolic linker at the exon-intron splicing boundary between exons 10 and 11 and alters the charge at this position from a negatively charged side chain to a positively charged one (Fig. 2) (18). The variant E446 resides in a region of the protein reported to harbor mutations associated with atrial fibrillation (E428K, H445D, L461V, N470K, and R481W) (16). The E446 residue is conserved across species among the entire family of homologous voltage-gated sodium channels but not in the population of paralogous voltage-gated calcium channels (Online Table 2).
Mutation V1279I.
The proband within the V1279I (c.3835.G→A) pedigree presented later in life at the age of 50 years and had no symptoms of heart failure (NYHA functional class I) (Fig. 1, Table 1). He manifested significant arrhythmia including atrial fibrillation and nonsustained ventricular tachycardia (Fig. 3A). He also showed a benign outcome and remained stable for >4 years of follow-up. Notably, in the family history exists a unique clinical pattern for the presence of conduction disease as indicated by left bundle branch block in the proband and a history of pacemakers in his mother and maternal grandmother.
This novel variant V1279I does not affect charge. The change occurs in the DIII-S3 transmembrane segment (Fig. 2) (18). The D1275N mutation identified in the CMD1E pedigree associated with conduction disorder, dilation, and arrhythmia represents the only other reported missense variant in this S3 transmembrane segment of this protein (7). Analogous S3 residue positions are almost universally conserved across domains II, III, and IV of the sodium and calcium voltage-gated ion channels regardless of species (Online Table 3).
Single nucleotide polymorphisms.
In addition to the novel missense variants reported above, a number of unreported single nucleotide polymorphisms (SNPs) were identified through sequencing analysis (Table 2). The intronic SNPs found in exons 10, 16, and 17 are adjacent to conserved splicing consensus sequence. Analysis of the relevant 3′ untranslated region sequence utilizing NNSPLICE 0.9 (23) showed no apparent or potentially disruptive splicing consensus sequence changes at these positions. All of the exonic SNPs listed represent synonymous changes.
Table 2.
Novel SCN5A Single Nucleotide Polymorphisms Identified in Familial Cardiomyopathy Registry Screening*
| Intronic | Exonic | |
|---|---|---|
| Exon 3 | −24C>T | |
| Exon 4 | +16G>C, +60A>T | |
| Exon 6 | +45C>A | V210V GTG>GTA |
| Exon 9 | +34G>A | P336P CCG>CCA |
| Exon 10 | −3C>A | |
| Exon 12 | −15T>C | P627P CCG>CCA |
| Exon 15 | +12 G>A | L771L CTC>CTT |
| Exon 16 | −5C>A, −13C>T | |
| Exon 17 | −6C>A | |
| Exon 24 | +38T>A, +43Ins |
The SCN5A screening identified a number of unreported nucleotide changes among our dilated cardiomyopathy population. Within coding regions, these changes are all consensus alterations or silent substitutions and are not believed to be associated with disease manifestation.
Discussion
This screening of a large cohort of DCM families demonstrates that SCN5A mutations may account for a modest proportion of DCM (1.7%) and provides further evidence that altered electrical excitability of the SCN5A protein can manifest as a dilation phenotype. Still, the specific mechanism producing this phenotype remains unknown. The cardiac sodium channel is responsible for the fast depolarization of the myocardium and is critical for the maintenance of impulse conduction in the heart (24). We note that the heterozygous (+/−) SCN5A knockout mice show a pattern of disease similar to that found among these DCM families including conduction dysfunction and age-dependent myocardial tissue changes with fibrosis (25).
The SCN5A DCM phenotype.
In the majority of cases we describe, conduction disturbances appear to be unrelated to the degree of left ventricular dysfunction (Table 1). In patient V1279I, left bundle branch block was the initial finding, presenting at young age and preceding, by many years, the diagnosis of DCM at the age of 46 years (Table 1 and Fig. 3A, showing left bundle branch block and atrial fibrillation at the age of 47 years). All but 1 index patient among these pedigrees had a DCM phenotype; in family D1275N, the index patient (IV-2) presented with an arrhythmic phenotype, and was evaluated because of his family history of DCM (7,8,26).
In family D1275N, as previously reported (7), conduction disease as well as arrhythmias were the presenting symptoms in 12 of 15 carriers (75%) and preceded the gradual development of left ventricular dysfunction (Fig. 3B) (7). In this family, the DCM phenotype predominantly manifested with age-related penetrance. As shown in Table 1, a dilated and dysfunctioning myocardium was present in the older generations, whereas at younger age, the arrhythmic trait predominated. Four family members (III-15, III-17, IV-8, and IV-10) (Table 1) had isolated arrhythmia and were identified though family screening. For these persons, mean age at diagnosis was 27 years. For comparison, the 5 D1275N mutation carriers with either dilation and/or LV dysfunction had a mean age of 41 years (7).
Strikingly, each of the 5 missense mutations we identified exhibit absolute conservation when compared to analogous positions within the cardiac sodium ion channels of varied species. For mutations D1275N, F1520L, V1279I, and R222Q, conserved identity extends across the entire family of voltage-gated sodium ion channels (Online Tables 1, 2, and 3). In addition, position V1279 is identically conserved across domains II, III, and IV of both voltage-gated sodium and voltage-gated calcium channels (Online Table 3). Conservation at position R222 is absolute across every alignment attempted, including all domains of related voltage-gated sodium, calcium, and potassium channels, and even the more distantly related transient receptor protein channels (Online Table 1).
With well over 160 disease-associated mutations reported for this protein, the screening of SCN5A has undoubtedly been thorough (24). A comprehensive review of the published literature showed no previous reports identifying novel mutations E446K, V1279I, or 1520L. In the case of position V1279, we evaluated an additional 300 ethnically matched chromosomes as position V1279 falls in the same exon for which controls were investigated for the adjacent D1275 position (7). The screening of 506 chromosomal controls for R222Q has also already been reported (10). Furthermore, we screened >550 chromosomes from the Familial Cardiomyopathy Registry for this report, and each of our mutations appeared only once. These data support the observation that these mutations are not merely rare polymorphisms.
SCN5A is a member of a family of voltage-gated sodium ion channels that express primarily in neurons and muscle fibers, and SCN5A expresses almost exclusively in the heart muscle (18). This channel initiates the upstroke of the action potential in myocyte depolarization, depolarizing membrane potentials by allowing a rapid influx of sodium ions. The resulting depolarizing current serves to signal downstream sarcomeric contraction within the myocyte. These depolarizations play a predominate role in the timing and initiation of global myocardium contraction by signaling through gap junctions to adjacent myocytes and along the specialized conduction pathways of the heart (27).
Heterozygous mutations in SCN5A have been shown to cause cardiomyopathies that are characterized by a strong conduction-related component. Functional studies of these mutations demonstrated both gain of function and loss of function variants that disrupt the natural rhythms of polarization and depolarization in the cardiac cycle. Gain of function mutations in the long-QT syndrome caused prolonged sodium current during depolarization due to deficits in the fastinactivation or inactivation-gated states leading to the observed arrhythmias (28–30). Mutant variants in Brugada syndrome shifted the voltage dependence of steady-state inactivation in the direction of more positive potentials, leading to an acceleration in recovery time from inactivation (31). In this form of Brugada syndrome, the resulting heterogeneity of the refractory period between the wild-type and mutant forms of the sodium channel has been suggested to result in an ideal electrical substrate for re-entrant arrhythmia (32). The SCN5A mutations associated with atrioventricular conduction block were shown to impair fast inactivation without producing non-inactivating currents (33). A reduction of sodium current density and an enhancement of slower inactivation components resulted in a significant reduction in myocardial conduction velocity, leading to the observed blocks (33).
Analysis of the clinical features of the mutation carriers in all 5 pedigrees we identified indicates a tendency for earlier age of onset (ranging from 15 to 34 years in all but 1 patient). Furthermore, carriers exhibit a predominant arrhythmic phenotype (93% of cases in this study) with supraventricular arrhythmias (86%) including sinus node dysfunction (33%), atrial fibrillation (60%), ventricular tachycardia (33%), and conduction defects (60%) (Table 1). These data complement the observations of Olson et al. (8), who reported finding 3 additional SCN5A mutations and a truncation in their patient population; in each instance cited, the SCN5A variation was associated with atrial fibrillation, impaired automaticity, and conduction delay. In the majority of our cases, carriers were asymptomatic or minimally symptomatic for heart failure and had a benign outcome over an extended period of follow-up (39 to 172 months) (Table 1). Only family F1520L appeared to have a more severe and progressive myocardial disease, with 2 patients undergoing transplantation (Fig. 1), indicating a variable expressivity of the phenotype. A limited number of patients were at risk of life-threatening events requiring primary prevention with implantable cardioverter-defibrillators (Table 1), and this was essentially related to the more severe degree of left ventricular dysfunction. No patients exhibited either long-QT syndrome or Brugada syndrome; specifically, no QT prolongation or shortening, T-wave abnormalities similar to those seen in long-QT syndrome, or “Brugada sign” were observed in our patients. Together, these findings support the hypothesis that the clinical features of DCM-related SCN5A channelopathies substantially and uniquely overlap (24).
Insights into the functional role of SCN5A mutations in DCM.
This report brings the total number of DCM-related SCN5A missense mutations to 9: T220I, R222Q, E446K, R814W, D1275N, V1279I, D1595H, F1520L, and I1835T (Fig. 2) (7,8,10). Of those 9, 6 localize to the S3 or S4 transmembrane segments (T220I, R222Q, and R814W, S4 regions in protein reference #Q14524 [Online Table 1]; and D1275N, V1279I, and D1595H, S3 regions in protein reference #Q14524 [Online Table 3]). Interestingly, recent crystallographic data on the related voltage-gated potassium channel Kv1.1 provides an estimate of the spatial relationship of the S3 and S4 regions. The crystallographic data were not able to resolve the S3 subunit and related linker loops. Instead, an alanine helix of appropriate length was inserted, and the S3 orientation was estimated based on its fit within the monomer and tetramer structures and within the physical length restrictions of interlinking amino acid loops (34,35). Although this model supports the functional hypothesis of an S3 to S4 charge-stabilizing interface, it does not provide enough information for actual physical characterization or molecular modeling of this relationship.
Variant T220I in domain I, transmembrane segment S4, of SCN5A has not been investigated electrophysiologically. In mutations R222Q and R814W, alterations occur in adjacent conserved arginine residues within S4 (domain I, S4, and domain II, S4, of SCN5A, respectively) (Fig. 2, Online Table 1) that contribute positive charge essential for voltage sensing across the cell membrane. These specific arginine residues within S4 have been investigated in the homologous voltage-gated potassium channel Shaker, and alter the gating function without modifying permeation (20,36–38). Analysis of R814W in SCN5A expressed in heterologous mammalian cell lines shows that this variant activates at a more negative membrane potential and opens with slower kinetics than the wild-type channel (39).
Variant V1279I in domain III, transmembrane segment S3, of SCN5A has not been investigated electrophysiologically. Two mutations also in S3 have been electrophysiologically analyzed; mutations D1275N and D1595H occur in analogous conserved aspartate residues within domain 3, S3, and domain 4, S3, respectively, of SCN5A (Fig. 2, Online Table 3). Alterations at this position in voltage-gated potassium channels affect gating without modifying permeation properties, mimicking affects similar to those mentioned in alterations of the conserved arginine residues of S4 (38,40). The D1595H SCN5A variant expressed in heterologous mammalian cell lines activates normally, but the kinetics of current decay are slower than wild-type channels (39). The D1275N SCN5A variant expressed in Xenopus oocytes results in a small decrease in recovery times from fast inactivation, with a depolarizing shift of 3.8 mV in voltage dependence of activation when compared to the wild-type channels (41).
Given the high rate at which DCM-associated mutations occur in S3 and S4 transmembrane regions of highly conserved homologous domains, and among charge clusters believed to interact directly in voltage-dependent gating, these data suggest that some instances of DCM are intrinsically tied to the disruption of voltage-sensing kinetics in SCN5A function.
Study limitations.
A study limitation is represented by the lack of segregation data in 3 of our families, because relatives were not available for genetic investigations. This limitation, common to human genetic studies and clinical genetic testing, is compensated by other consensus criteria, as described in the Methods section (42). Furthermore, more expression data to explore the mechanistic link of the new SCN5A mutations with DCM in mice models warrant future research.
Conclusions
Mutations in SCN5A in conjunction with DCM occur at a frequency of 1.7% within our study population and, as previously reported, may affect both familial and sporadic cases. The majority of DCM-related SCN5A mutations localize to highly conserved homologous S3 and S4 transmembrane segments, supporting the hypothesis for disruption of the sodium-channel voltage sensor in the etiology of dilation remodeling. Not surprisingly, SCN5A mutation carriers show a pattern of severe arrhythmia including atrial fibrillation and ventricular tachycardia, and a better understanding of the molecular basis of these symptoms will have important implications for the diagnosis and clinical management of DCM in these persons.
Supplementary Material
Acknowledgments
The authors thank the family members for their participation in these studies. Familial Dilated Cardiomyopathy Registry Research Group: University of Colorado, Denver, Colorado: Shelley Miyamoto, Jean Cavanaugh, Michael R. Bristow, Carlin Long, and Brian Lowes; Hospital and University of Trieste, Trieste, Italy: Francesca Brun, Marco Merlo, Michele Moretti, and Stylianos Pyxaras.
Funding sources are NIH/NHLBI 1 RO1 HL69071, NIH MO1 RR00051-1575, the American Heart Association 0150453N and 0250271N, and the Muscular Dystrophy Association PN0007-056. The authors have reported that they have no relationships to disclose.
Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- DNA
deoxyribonucleic acid
- NYHA
New York Heart Association
- SCN5A
cardiac voltage-gated sodium ion channel, alpha subunit
- SNP
single nucleotide polymorphism
Footnotes
APPENDIX
For supplementary Tables 1, 2, and 3, please see the online version of this article.
REFERENCES
- 1.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA. Genetic evaluation of cardiomyopathy—a Heart Failure Society of America practice guideline. J Card Fail 2009;15:83–97. [DOI] [PubMed] [Google Scholar]
- 2.Chen J, Chien KR. Complexity in simplicity: monogenic disorders and complex cardiomyopathies. J Clin Invest 1999;103:1483–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiso N, Stephan DA, Nava A, et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum Mol Genet 2001;10:189–94. [DOI] [PubMed] [Google Scholar]
- 4.Asahi M, Nakayama H, Tada M, Otsu K. Regulation of sarco(endo) plasmic reticulum Ca2+ adenosine triphosphatase by phospholamban and sarcolipin: implication for cardiac hypertrophy and failure. Trends Cardiovasc Med 2003;13:152–7. [DOI] [PubMed] [Google Scholar]
- 5.Schmitt JP, Kamisago M, Asahi M, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003; 299:1410–3. [DOI] [PubMed] [Google Scholar]
- 6.Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J Mol Cell Cardiol 2005;38:937–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNair WP, Ku L, Taylor MR, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004;110:2163–7. [DOI] [PubMed] [Google Scholar]
- 8.Olson TM, Michels W, Ballew JD, et al. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 2005;293:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson TM, Keating MT. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest 1996;97:528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hershberger RE, Parks SB, Kushner JD, et al. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci 2008;1:21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mestroni L, Maisch B, McKenna WJ, et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J 1999;20:93–102. [DOI] [PubMed] [Google Scholar]
- 12.National Center for Biotechnology Information [NCBI]. Available at: http://www.ncbi.nlm.nih.gov/. Accessed July 21, 2010.
- 13.Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press, 2000;365–86. [DOI] [PubMed] [Google Scholar]
- 14.BLAST Assembled RefSeq Genomes. Available at: www.ncbi.nlm.nih.gov/BLAST. Accessed July 21, 2010.
- 15.dbSNP. Available at: http://www.ncbi.nlm.nih.gov/SNP. Accessed July 21, 2010.
- 16.Darbar D, Kannankeril PJ, Donahue BS, et al. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008;117:1927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vijayaraman P, Ellenbogen KA. Bradyarrhythmias and pacemakers. In: Fuster V, O’Rourke RA, Walsh RA, et al. , editors. Hurst’s The Heart. 12th ed. New York: McGraw-Hill Medical, 2008:1020–54. [Google Scholar]
- 18.Gellens ME, George AL Jr., Chen LQ et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A 1992;89:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seoh SA, Sigg D, Papazian DM, Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 1996;16:1159–67. [DOI] [PubMed] [Google Scholar]
- 20.Aggarwal SK, MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 1996;16:1169–77. [DOI] [PubMed] [Google Scholar]
- 21.Rook MB, Bezzina Alshinawi C, Groenewegen WA, et al. Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the Brugada syndrome. Cardiovasc Res 1999;44:507–17. [DOI] [PubMed] [Google Scholar]
- 22.Yokoi H, Makita N, Sasaki K, et al. Double SCN5A mutation underlying asymptomatic Brugada syndrome. Heart Rhythm 2005;2:285–92. [DOI] [PubMed] [Google Scholar]
- 23.NNSPLICE 0.9. Available at: http://www.fruitfly.org/seq_tools/splice.html. Accessed July 21, 2010.
- 24.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Nat Rev Cardiol 2009;6:337–48. [DOI] [PubMed] [Google Scholar]
- 25.Royer A, van Veen TA, Le Bouter S, et al. Mouse model of SCN5A-linked hereditary Lenegre’s disease: age-related conduction slowing and myocardial fibrosis. Circulation 2005;111:1738–46. [DOI] [PubMed] [Google Scholar]
- 26.Greenlee PR, Anderson JL, Lutz JR, Lindsay AE, Hagan AD. Familial automaticity-conduction disorder with associated cardiomyopathy. West J Med 1986;144:33–41. [PMC free article] [PubMed] [Google Scholar]
- 27.Cohen SA, Barchi RL. Cardiac sodium channel structure and function. Trends Cardiovasc Med 1992;2:133–40. [DOI] [PubMed] [Google Scholar]
- 28.Bennett PB, Yazawa K, Makita N, George AL Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature 1995;376:683–5. [DOI] [PubMed] [Google Scholar]
- 29.Wei J, Wang DW, Alings M, et al. Congenital long-QT syndrome caused by a novel mutation in a conserved acidic domain of the cardiac Na+ channel. Circulation 1999;99:3165–71. [DOI] [PubMed] [Google Scholar]
- 30.Makita N, Shirai N, Nagashima M, et al. A de novo missense mutation of human cardiac Na+ channel exhibiting novel molecular mechanisms of long QT syndrome. FEBS Lett 1998;423:5–9. [DOI] [PubMed] [Google Scholar]
- 31.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998;392:293–6. [DOI] [PubMed] [Google Scholar]
- 32.Makita N, Shirai N, Wang DW, et al. Cardiac Na(+) channel dysfunction in Brugada syndrome is aggravated by beta(1)-subunit. Circulation 2000;101:54–60. [DOI] [PubMed] [Google Scholar]
- 33.Wang DW, Viswanathan PC, Balser JR, George AL Jr., Benson DW. Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation 2002;105:341–6. [DOI] [PubMed] [Google Scholar]
- 34.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005;309:897–903. [DOI] [PubMed] [Google Scholar]
- 35.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 2005;309:903–8. [DOI] [PubMed] [Google Scholar]
- 36.Bezanilla F The voltage sensor in voltage-dependent ion channels. Physiol Rev 2000;80:555–92. [DOI] [PubMed] [Google Scholar]
- 37.Papazian DM, Silverman WR, Lin MC, Tiwari-Woodruff SK, Tang CY. Structural organization of the voltage sensor in voltage-dependent potassium channels. Novartis Found Symp 2002;245:178–90, discussion 190–2, 261–4. [PubMed] [Google Scholar]
- 38.Silverman WR, Roux B, Papazian DM. Structural basis of two-stage voltage-dependent activation in K+ channels. Proc Natl Acad Sci U S A 2003;100:2935–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen TP, Wang DW, Rhodes TH, George AL Jr. Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res 2008;102:364–71. [DOI] [PubMed] [Google Scholar]
- 40.Planells-Cases R, Ferrer-Montiel AV, Patten CD, Montal M. Mutation of conserved negatively charged residues in the S2 and S3 transmembrane segments of a mammalian K+ channel selectively modulates channel gating. Proc Natl Acad Sci U S A 1995;92:9422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groenewegen WA, Firouzi M, Bezzina CR, et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res 2003;92:14–22. [DOI] [PubMed] [Google Scholar]
- 42.Taylor MRG, Slavov D, Ku L, et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2006;115:1244–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.