

SUMMARY
Efforts to model the human gut microbiome in mice have led to important insights into the mechanisms of host-microbe interactions. However, the model communities studied to date have been defined or complex but not both, limiting their utility. Here, we construct and characterize in vitro a defined community of 104 bacterial species composed of the most common taxa from the human gut microbiota (hCom1). We then used an iterative experimental process to fill open niches: germ-free mice were colonized with hCom1 and then challenged with a human fecal sample. We identified new species that engrafted following fecal challenge and added them to hCom1, yielding hCom2. In gnotobiotic mice, hCom2 exhibited increased stability to fecal challenge and robust colonization resistance against pathogenic Escherichia coli. Mice colonized by hCom2 versus a human fecal community are phenotypically similar, suggesting that this consortium will enable mechanistic interrogation of species and genes on microbiome-associated phenotypes.
In brief
The development of a complex community of bacteria that represent the most common taxa from the human microbiome enables further mechanistic study of genes, pathways and species influence host physiology and health.
Graphical Abstract
INTRODUCTION
Experiments in which a microbial community is transplanted into germ-free mice have opened the door to studies of mechanism and causality in the microbiome. These efforts fall into two categories based on the nature of the transplanted community: complete, undefined communities (i.e., fecal samples) versus incomplete but defined communities (i.e., synthetic communities). Fecal transplantation studies have shown that the microbiome plays a role in a variety of host phenotypes including the response to cancer immunotherapy (Gopalakrishnan et al., 2018; Matson et al., 2018; Routy et al., 2018), caloric harvest (Ridaura et al., 2013), colonization resistance to enteric pathogens (Buffie et al., 2015), and neural development (Buffington et al., 2021; Sharon et al., 2019). While illuminating, a limitation of this format is that it is difficult to ‘fractionate’ an undefined community, making it challenging to discover which species are involved in a phenotype of interest.
Synthetic communities are less well developed as model systems for the gut microbiome (Blasche et al., 2017; Pacheco and Segrè, 2019; Walter et al., 2018; Widder et al., 2016; Xavier, 2011). Pioneering efforts have shown that a synthetic community can model the impact of diet on the microbiome (Faith et al., 2011), identified genes required for Bacteroides thetaiotaomicron growth in the mouse intestine in the presence of a 15-member community (Goodman et al., 2009), and demonstrated that complex communities composed of species isolated from a single donor can stably colonize mice (Goodman et al., 2011). More recent studies with defined communities have revealed mechanistic insights into immune modulation, glycan consumption, and other complex phenotypes driven by the microbiome (Faith et al., 2014; van der Lelie et al., 2021; Patnode et al., 2019; Wymore Brand et al., 2015). Although synthetic communities enable precise control over composition and manipulations such as strain dropouts and gene knockouts, the communities used are typically of low complexity (<20 strains), limiting their ability to model the biology of a native-scale microbiome.
An ideal model system for the gut microbiome would capture the advantages of both approaches: near-native complexity would allow a model microbiome to capture properties of an ecosystem that are missing from simpler model systems, including emergent phenomena such as resilience to perturbation (Dethlefsen and Relman, 2011; Ng et al., 2019) and cooperative metabolism (Morris et al., 2013). Moreover, complex consortia are a promising starting point for in vivo studies of the gut microbiome, for which they are better suited to model community-level phenomena such as immune modulation and the formation of structured multispecies biofilms.
Complete definition (i.e., communities composed entirely of known organisms) would enable reductionist experiments to probe mechanism. The ability to construct communities with defined composition is especially relevant in the context of experiments testing whether phenotypes can be transferred to germ-free mice via fecal transplant (Gopalakrishnan et al., 2018; Ridaura et al., 2013; Routy et al., 2018). At present, since transplanted communities are typically undefined, it is difficult to uncover the mechanisms underlying these phenomena. A defined model system of sufficient complexity would enable reductionist follow-up experiments, bringing the gut microbiome in line with other model systems in which mechanistic studies are possible.
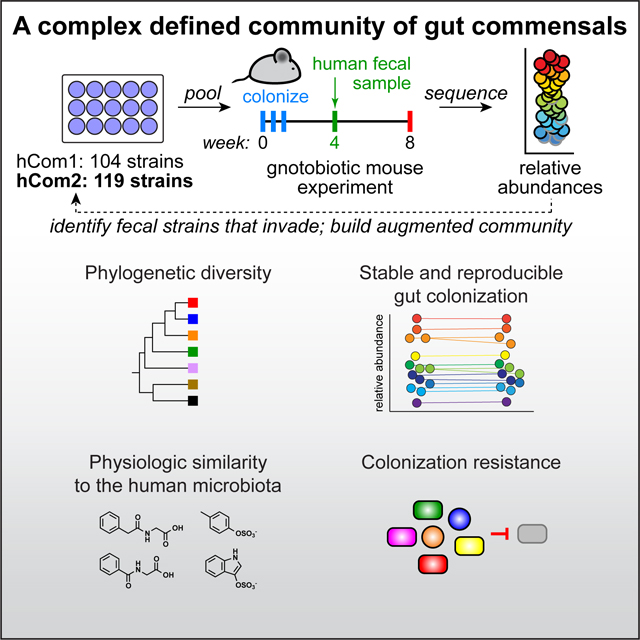
To this end, we sought to create a community that is defined, enabling precise manipulations, and complex enough to exhibit emergent features of a complete community such as stability upon engraftment and colonization resistance. We started by constructing a complex defined community that contains the most prevalent bacterial species in the human gut microbiome (hCom1). We demonstrate that the assembly of this 104-member community is reproducible even for very low abundance species. By systematically perturbing this community and its growth medium, we uncover strain-nutrient and strain-strain (e.g. syntrophic) interactions that underlie its composition. We then colonize germ-free mice with hCom1, showing that it adopts a stable, highly reproducible configuration in which its constituent species span six orders of magnitude of relative abundance. We augment the community by filling open niches using an iterative, ecology-based process, and show that the enlarged community (hCom2) is more resilient to perturbation and resistant to pathogen colonization. Finally, we demonstrate that mice colonized by hCom2 are phenotypically similar to mice harboring an undefined human fecal sample, suggesting that our consortium and augmentation process lay the foundation for developing complete, defined models of the human gut microbiome.
RESULTS
Designing and building a complex synthetic community
We set out to design a community composed of the most common bacterial species in the human gut microbiome. We analyzed metagenomic sequence data from the NIH Human Microbiome Project (HMP) to determine the most prevalent organisms—those that were present in the largest proportion of subjects, regardless of abundance. Although the HMP is not broadly representative of microbiomes from diverse geographies and ethnicities (Deschasaux et al., 2018; He et al., 2018; Sonnenburg and Sonnenburg, 2019), this data set was well suited to our purposes since it was sequenced at very high depth, enabling us to identify low-abundance organisms that are nevertheless highly prevalent (Kraal et al., 2014). After rank-ordering bacterial strains by prevalence, we found that ~20% (166/844) were present in >45% of the HMP subjects. Of these 166 strains, we were able to obtain 99 from culture collections or individual laboratories (Figure 1A; omitted strains are listed in Table S1). The profiled strains of three additional species were unavailable, so we used alternative strains of the same species (Lactococcus lactis subsp. lactis Il1403, Bacteroides xylanisolvens DSM 18836, and Megasphaera sp. DSM 102144). We added two additional strains to enable downstream experiments: Ruminococcus bromii ATCC 27255, a keystone species in polysaccharide utilization (Ze et al., 2012); and Clostridium sporogenes ATCC 15579, a model gut Clostridium species for which genetic tools are available (Dodd et al., 2017; Funabashi et al., 2020; Guo et al., 2019). These 104 strains—a community termed ‘hCom1’—are prevalent and abundant in Western human gut communities (Data S1). Notably, unlike other defined communities used to model the gut microbiome, our consortium is within ~2-fold of the estimated number of species in a typical human gut (STAR Methods) (Faith et al., 2013; Qin et al., 2010).
Figure 1: A complex gut bacterial community.

(A) A phylogenetic tree of the 104 strains in the community based on a multiple sequence alignment of conserved single-copy genes. The community was designed by identifying the most prevalent strains in sequencing data from the NIH Human Microbiome Project (HMP). Colored squares indicate the phylum of each strain: Firmicutes = red, Actinobacteria = blue, Verrucomicrobia = orange, Bacteroidetes = green, and Proteobacteria = purple. Also shown are the prevalence and relative abundances of each strain in the data set from the NIH HMP (n=81 subjects). The prevalence is the fraction of subjects in which the strain was detected. The distribution of log10(relative abundance) across subjects is shown with the mean denoted by a white line for each strain. Ruminococcus bromii ATCC 27255 and Clostridium sporogenes ATCC 15579 were added to the community despite low prevalence in the HMP samples. (B) The community reaches a stable configuration quickly. The community was propagated in vitro in SAAC medium to test the stability of its composition. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point. Strains are colored according to their rank-order abundance in the community at 48 h. By 12 h, the relative abundances of strains in the community spanned six orders of magnitude and remained largely stable through 48 h. (C) Communities generated from two inocula prepared on different days (i.e., biological replicates) have a similar architecture at 48 h. (D) Communities generated from the same inoculum (i.e., technical replicates) have a nearly identical composition at 48 h. In (C) and (D), the color of each circle represents the phylum of the corresponding species, and circles with gray outlines and faint colors represent strains whose presence could be explained by read mis-mapping.
A streamlined strain growth protocol simplified the assembly of hCom1 and single-strain dropouts (STAR Methods). We found that each of our 104 strains can be propagated in Mega Medium (MM), Chopped Meat Medium (CMM), or both (Key Resources Table). Growth rates, carrying capacities, and time of entry into stationary phase varied widely across strains and media. To simplify the process of community assembly while ensuring that slow-growing strains were actively dividing, each strain was inoculated from a frozen stock into liquid medium and passaged every 24 h for a total of 2–3 days. Before mixing individually cultured strains, we adjusted the volumes of each culture to achieve similar optical densities. A subset of the strains did not reach the diluted culture density of the remaining strains (STAR Methods); we added these cultures undiluted. We confirmed that our starting cultures were pure using metagenomic sequencing and high accuracy read mapping, as described in the next section.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Myeloid cells: anti-mouse Ly6c (HK1.4), FITC | BioLegend | Cat. #128006; RRID:AB_1186134 |
| Myeloid cells: anti-mouse CD11b (M1/70), PerCP/Cy5.5 | BioLegend | Cat. #101228; RRID: AB_893232 |
| Myeloid cells: anti-mouse CD103 (2E7), PE | BioLegend | Cat. #121406; RRID: AB_1133989 |
| Myeloid cells: anti-mouse CD11c (N418), PE-Cy7 | BioLegend | Cat. #117318; RRID: AB_493568 |
| Myeloid cells: anti-mouse CD317 (129C1), Alexa647 | BioLegend | Cat. #127106; RRID: AB_2067120 |
| Fixable Viability dye, APC-eFluor 780 | eBioscience | 65-0865-14 |
| Anti-mouse IgA (RMA-1), Biotin | BioLegend | Cat. #407004; RRID: AB_315079 |
| Streptavidin, BV421 | BioLegend | 405225 |
| Myeloid cells: anti-mouse I-A/I-E (M5/114.15.2), BV510 | BioLegend | Cat. #107636; RRID: AB_2734168 |
| T cells and epithelial cells: anti-mouse CD45 (30-F11), BV605 | BioLegend | Cat. #103155; RRID: AB_2650656 |
| Myeloid cells: anti-mouse F4/80 (BM8), BV650 | BioLegend | Cat. #123149; RRID: AB_2564589 |
| anti-mouse CD16/32 (2.4G2), FC block | BD Bioscience | Cat. #553141; RRID: AB_394655 |
| T cells: anti-mouse Helios (22F6), FITC | BioLegend | Cat. #137214; RRID: AB_10662745 |
| B and T cells: anti-mouse CD62L (MEL-14), PerCP/Cy5.5 | Biolegend | Cat. #104432; RRID: AB_2285839 |
| T cells: anti-mouse IL22 (Poly5164), PE | BioLegend | Cat. #516404; RRID: AB_2124255 |
| T cells: anti-mouse Foxp3 (FJK-16s), PE-Cy7 | eBioscience | Cat. #25-5773-82; RRID: AB_891552 |
| T cells: anti-mouse RORgt (B2D), APC | eBioscience | Cat. #17-6981-82; RRID: AB_2573254 |
| T cells: anti-mouse CD44 (IM7), BV421 | BioLegend | Cat. #103040; RRID: AB_2616903 |
| T cells: anti-mouse CD4 (RM4-5), BV510 | BioLegend | Cat. #100559; RRID: AB_2562608 |
| T cells: anti-mouse CD3e (145-2C11), BV605 | BioLegend | Cat. #100351; RRID: AB_2565842 |
| B cells: anti-mouse CD8a (53.6.7), BV650 | BioLegend | Cat. #100742; RRID: AB_2563056 |
| Myeloid cells: anti-mouse Ly6c (HK1.4), FITC | BioLegend | Cat. #128006; RRID:AB_1186134 |
| Myeloid cells: anti-mouse CD11b (M1/70), PerCP/Cy5.5 | BioLegend | Cat. #101228; RRID: AB_893232 |
| Myeloid cells: anti-mouse CD103 (2E7), PE | BioLegend | Cat. #121406; RRID: AB_1133989 |
| Bacterial and Virus Strains | ||
| Strain Name | Source | Media |
| Alistipes putredinis DSM 17216 | DSMZ | Chopped Meat Medium |
| Anaerotruncus colihominis DSM 17241 | DSMZ | Mega Medium |
| Bacteroides caccae ATCC 43185 | ATCC | Mega Medium |
| Bacteroides coprophilus DSM 18228 | DSMZ | Mega Medium |
| Bacteroides dorei 5_1_36/D4 | BEI | Mega Medium |
| Bacteroides eggerthii DSM 20697 | DSMZ | Mega Medium |
| Bacteroides finegoldii DSM 17565 | DSMZ | Mega Medium |
| Bacteroides fragilis 3_1_12 | BEI | Mega Medium |
| Bacteroides intestinalis DSM 17393 | DSMZ | Mega Medium |
| Bacteroides sp. 1_1_6 | BEI | Mega Medium |
| Bacteroides sp. 2_1_22 | BEI | Mega Medium |
| Bacteroides sp. 3_1_19 | BEI | Mega Medium |
| Bacteroides sp. 9_1_42FAA | BEI | Mega Medium |
| Bacteroides sp. 2_1_16 | BEI | Mega Medium |
| Bacteroides sp. D2 | BEI | Mega Medium |
| Bacteroides thetaiotaomicron VPI-5482 | ATCC | Mega Medium |
| Bacteroides xylanisolvens DSMZ 18836 | DSMZ | Mega Medium |
| Bacteroides uniformis ATCC 8492 | ATCC | Mega Medium |
| Bacteroides pectinophilus ATCC 43243 | ATCC | Chopped Meat Medium |
| Bacteroides plebeius DSM 17135 | DSMZ | Chopped Meat Medium |
| Bacteroides coprocola DSM 17136 | DSMZ | Chopped Meat Medium |
| Bacteroides stercoris ATCC 43183 | DSMZ | Mega Medium |
| Coprococcus eutactus ATCC 27759 | ATCC | Chopped Meat Medium |
| Eubacterium dolichum DSM 3991 | DSMZ | Mega Medium |
| Ruminococcus gnavus ATCC 29149 | BEI | Mega Medium |
| Eubacterium rectale ATCC 33656 | ATCC | Mega Medium |
| Clostridium methylpentosum DSM 5476 | DSMZ | Mega Medium |
| Clostridium nexile DSM 1787 | DSMZ | Mega Medium |
| Clostridium scindens ATCC 35704 | ATCC | Mega Medium |
| Clostridium sp. L2-50 | BEI | Chopped Meat Medium |
| Clostridium sp. M62/1 | BEI | Chopped Meat Medium |
| Clostridium asparagiforme DSM 15981 | DSMZ | Mega Medium |
| Clostridium bolteae ATCC BAA-613 | ATCC | Mega Medium |
| Clostridium hathewayi DSM 13479 | DSMZ | Mega Medium |
| Clostridium leptum DSM 753 | DSMZ | Chopped Meat Medium |
| Dorea formicigenerans ATCC 27755 | DSMZ | Mega Medium |
| Dorea longicatena DSM 13814 | DSMZ | Mega Medium |
| Coprococcus comes ATCC 27758 | ATCC | Mega Medium |
| Blautia hansenii DSM 20583 | DSMZ | Mega Medium |
| Bryantella formatexigens DSM 14469 | DSMZ | Mega Medium |
| Butyrivibrio crossotus DSM 2876 | DSMZ | Chopped Meat Medium |
| Ruminococcus torques ATCC 27756 | ATCC | Mega Medium |
| Parabacteroides merdae ATCC 43184 | DSMZ | Mega Medium |
| Subdoligranulum variabile DSM 15176 | DSMZ | Mega Medium |
| Parabacteroides johnsonii DSM 18315 | DSMZ | Chopped Meat Medium |
| Roseburia intestinalis L1-82 | ATCC | Mega Medium |
| Ruminococcus obeum ATCC 29174 | DSMZ | Mega Medium |
| Eubacterium ventriosum ATCC 27560 | DSMZ | Mega Medium |
| Faecalibacterium prausnitzii A2-165 | DSMZ | Chopped Meat Medium |
| Parabacteroides sp. D13 | BEI | Mega Medium |
| Eubacterium hallii DSM 3353 | DSMZ | Chopped Meat Medium |
| Roseburia inulinivorans DSM 16841 | DSMZ | Chopped Meat Medium |
| Prevotella buccalis ATCC 35310 | DSMZ | Chopped Meat Medium |
| Ruminococcus lactaris ATCC 29176 | ATCC | Chopped Meat Medium |
| Eubacterium eligens ATCC 27750 | DSMZ | Mega Medium |
| Holdemania filiformis DSM 12042 | DSMZ | Mega Medium |
| Bacteroides ovatus ATCC 8483 | ATCC | Mega Medium |
| Bacteroides vulgatus ATCC 8482 | ATCC | Mega Medium |
| Clostridium spiroforme DSM 1552 | DSMZ | Chopped Meat Medium |
| Eubacterium biforme DSM 3989 | DSMZ | Mega Medium |
| Blautia hydrogenotrophica DSM 10507 | DSMZ | Chopped Meat Medium |
| Clostridium saccharolyticum WM1 | DSMZ | Mega Medium |
| Parabacteroides distasonis ATCC 8503 | ATCC | Mega Medium |
| Eubacterium siraeum DSM 15702 | DSMZ | Chopped Meat Medium |
| Eggerthella lenta DSM 2243 | DSMZ | Chopped Meat Medium |
| Anaerostipes caccae DSM 14662 | DSMZ | Mega Medium |
| Bacteroides cellulosilyticus DSM 14838 | DSMZ | Mega Medium |
| Clostridium hylemonae DSM 15053 | DSMZ | Mega Medium |
| Acidaminococcus sp. D21 | BEI | Mega Medium |
| Catenibacterium mitsuokai DSM 15897 | DSMZ | Mega Medium |
| Collinsella aerofaciens ATCC 25986 | ATCC | Mega Medium |
| Acidaminococcus fermentans DSM 20731 | DSMZ | Mega Medium |
| Clostridium bartlettii DSM 16795 | DSMZ | Mega Medium |
| Ethanoligenens harbinense YUAN-3 | DSMZ | Chopped Meat Medium |
| Veillonella dispar ATCC 17748 | DSMZ | Chopped Meat Medium |
| Collinsella stercoris DSM 13279 | DSMZ | Chopped Meat Medium |
| Prevotella buccae D17 | BEI | Chopped Meat Medium |
| Mitsuokella multacida DSM 20544 | DSMZ | Mega Medium |
| Olsenella uli DSM 7084 | DSMZ | Chopped Meat Medium |
| Slackia heliotrinireducens DSM 20476 | DSMZ | Chopped Meat Medium |
| Bifidobacterium longum infantis ATCC 55813 | BEI | Mega Medium |
| Dialister invisus DSM 15470 | DSMZ | Mega Medium |
| Prevotella copri DSM 18205 | DSMZ | Chopped Meat Medium |
| Veillonella sp. 6_1_27 | BEI | Chopped Meat Medium |
| Slackia exigua ATCC 700122 | DSMZ | Chopped Meat Medium |
| Streptococcus thermophilus LMD-9 | ATCC | Chopped Meat Medium |
| Desulfovibrio piger ATCC 29098 | DSMZ | Chopped Meat Medium |
| Lactobacillus ruminis ATCC 25644 | ATCC | Mega Medium |
| Akkermansia muciniphila ATCC BAA-835 | DSMZ | Mega Medium |
| Bifidobacterium adolescentis L2-32 | BEI | Mega Medium |
| Bifidobacterium pseudocatenulatum DSM 20438 | DSMZ | Mega Medium |
| Solobacterium moorei DSM 22971 | DSMZ | Chopped Meat Medium |
| Anaerofustis stercorihominis DSM 17244 | DSMZ | Mega Medium |
| Lactococcus lactis DSMZ 20729 | DSMZ | Mega Medium |
| Granulicatella adiacens ATCC 49175 | DSMZ | Mega Medium |
| Clostridium sporogenes ATCC 15579 | ATCC | Mega Medium |
| Bacteroides dorei DSM 17855 | DSMZ | Mega Medium |
| Bifidobacterium catenulatum DSM 16992 | DSMZ | Mega Medium |
| Ruminococcus albus strain 8 | Laboratory of Robert Mackie | Chopped Meat Medium |
| Ruminococcus flavefaciens FD 1 | Laboratory of Robert Mackie | Chopped Meat Medium |
| Ruminococcus bromii ATCC (L2-63) | ATCC | Chopped Meat Medium |
| Veillonella sp. 3_1_44 | BEI | Chopped Meat Medium |
| Bifidobacterium breve DSM 20213 | DSMZ | Mega Medium |
| Megasphaera sp. DSMZ 102144 | DSMZ | Mega Medium |
| Adlercreutzia equolifaciens DSM 19450 | DSMZ | Chopped Meat Medium |
| Alistipes finegoldii DSM 17242 | DSMZ | Mega Medium |
| Alistipes ihumii AP11 | Laboratory of Emma Allen Vercoe | Chopped Meat Medium |
| Alistipes indistinctus YIT 12060 | DSMZ | Mega Medium |
| Alistipes onderdonkii DSM 19147 | DSMZ | Chopped Meat Medium |
| Alistipes senegalensis JC50 | DSMZ | Chopped Meat Medium |
| Alistipes shahii WAL 8301 | DSMZ | Chopped Meat Medium |
| Bacteroides rodentium DSM 26882 | DSMZ | Chopped Meat Medium |
| Bilophila wadsworthia ATCC 49260 | ATCC | Chopped Meat Medium |
| Blautia sp. KLE 1732 | BEI | Chopped Meat Medium |
| Blautia wexlerae DSM 19850 | DSMZ | Mega Medium |
| Burkholderiales bacterium 1_1_47 | Laboratory of Emma Allen Vercoe | Chopped Meat Medium |
| Butyricimonas virosa DSM 23226 | DSMZ | Mega Medium |
| Clostridiales bacterium VE202-03 | Laboratory of Kenya Honda | Mega Medium |
| Clostridiales bacterium VE202-14 | Laboratory of Kenya Honda | Mega Medium |
| Clostridiales bacterium VE202-27 | Laboratory of Kenya Honda | Chopped Meat Medium |
| Clostridium sp. VPI C48-50 | ATCC | Chopped Meat Medium |
| Intestinimonas butyriciproducens DSM 26588 | DSMZ | Mega Medium |
| Odoribacter splanchnicus DSM 20712 | DSMZ | Chopped Meat Medium |
| Oscillibacter sp. KLE 1728 | BEI | Chopped Meat Medium |
| Ruminococcus gauvreauii DSM 19829 | DSMZ | Mega Medium |
| Subdoligranulum sp. 4_3_54A2FAA | Laboratory of Emma Allen Vercoe | Chopped Meat Medium |
| Escherichia coli ATCC 43894 | ATCC | BHI |
| Escherichia coli MITI 27 | Laboratory of Michael Fischbach | BHI |
| Escherichia coli MITI 117 | Laboratory of Michael Fischbach | BHI |
| Escherichia coli MITI 135 | Laboratory of Michael Fischbach | BHI |
| Escherichia coli MITI 139 | Laboratory of Michael Fischbach | BHI |
| Escherichia coli MITI 255 | Laboratory of Michael Fischbach | BHI |
| Escherichia coli MITI 284 | Laboratory of Michael Fischbach | BHI |
| Enterobacter cloacae MITI 173 | Laboratory of Michael Fischbach | BHI |
| Eschericia coli S17-1 ƛ-pir | Laboratory of Michael Fischbach | BHI |
| Clostridium sporogenes ATCC 15579 Δotc | Laboratory of Michael Fischbach | Mega Medium |
| Clostridium sporogenes ATCC 15579 Δadi | Laboratory of Michael Fischbach | Mega Medium |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PBS | Gibco | 10010023 |
| Tryptone peptone | Difco | 211921 |
| Bacto yeast extract | Difco | 212750 |
| Magnesium sulfate heptahydrate | Sigma | M2773 |
| Sodium bicarbonate | Sigma | S5761 |
| Calcium chloride | Sigma | C7902 |
| Resazurin | Sigma | R7017 |
| Agar | Difco | DF0140-01-0 |
| Sodium acetate | Sigma | S2889 |
| Meat extract | Sigma | 70164 |
| D-glucose | Sigma | 47829 |
| L-cystine HCl | Sigma | C7477 |
| Potassium phosphate monobasic | Sigma | P5655 |
| Potassium phosphate dibasic | Sigma | P3786 |
| Vitamin K3 | Sigma | M5625 |
| Hematin | Sigma | H3281 |
| Tween 80 | Sigma | P4780 |
| Vitamin mix | ATCC | MD-VS |
| Trace mineral supplement | ATCC | MD-TMS |
| D-(+)-cellobiose | Sigma | C7252 |
| D-(+)-maltose monohydrate | Sigma | M5885 |
| D-(−)-fructose | Sigma | F0127 |
| Acetic acid, glacial | Sigma | A6283 |
| Propionic acid | Sigma | P5561 |
| Butyric acid | Sigma | B103500 |
| Isovaleric acid | Sigma | 129542 |
| Sterilized rumen fluid | Bar Diamond Ranch | #SRF |
| Chopped meat media | Hardy Diagnostics | K219 |
| Vitamin K2 | Sigma | V9378 |
| Ammonium sulfate | Sigma | A4418 |
| Nitrilotriacetic acid | Sigma | N9877 |
| Manganese(II) chloride tetrahydrate | Sigma | M5005 |
| Cobalt (II) hexahydrate | Sigma | C8661 |
| Calcium chloride dihydrate | Sigma | 223506 |
| Zinc chloride | Sigma | Z0152 |
| Copper chloride | Sigma | 451665 |
| Sodium molybdate dihydrate | Sigma | M1651 |
| Boric acid | Sigma | B6768 |
| Sodium selenite | Sigma | 214485 |
| Nickel chloride hexahydrate | Sigma | N6136 |
| Sodium tungstate dihydrate | Sigma | 72069 |
| L-alanine | Sigma | A7469 |
| L-arginine | Sigma | A5006 |
| L-asparagine | Sigma | A4159 |
| L-aspartic Acid | Sigma | A8949 |
| L-glutamic Acid | Sigma | 49449 |
| L-glutamine | Sigma | 49419 |
| L-glycine | Sigma | G7126 |
| L-histidine | Fisher | BP382 |
| L-isoleucine | TCI | I0181 |
| L-leucine | TCI | L0029 |
| L-lysine | Sigma | L5751 |
| L-methionine | Sigma | 64319 |
| L-phenylalanine | Sigma | P5482 |
| L-proline | Sigma | 81709 |
| L-serine | Sigma | S4500 |
| L-threonine | Sigma | 89179 |
| L-tryptophan | Sigma | T0254 |
| L-tyrosine | Sigma | 93829 |
| L-valine | Sigma | 94619 |
| T4 ligase | NEB | M0202T |
| AscI | NEB | R0558 |
| NotI | NEB | R0189 |
| Bacto tryptone | Thermo Fisher | 211701 |
| Sodium thioglycolate | Sigma | 1066910500 |
| D-cycloserine | Sigma | C6880 |
| Erythromycin | Sigma | 114-07-8 |
| Thiamphenicol | Sigma | T0261 |
| Luria Broth agar | Fisher | BP1425-500 |
| MacConkey agar | Sigma | M7408 |
| MacConkey sorbitol agar | Sigma | 88902 |
| Columbia agar with 5% sheep blood | BD | 221165 |
| Brain Heart Infusion broth | Fisher | CM1136B |
| Horse blood, defibrinated | Fisher | 50863761 |
| Glycerol | Fisher | PRH5433 |
| Potassium chloride | Sigma | P9541 |
| Magnesium chloride | Sigma | M1028 |
| Sodium phosphate dibasic | Sigma | S3264 |
| Sodium chloride | Sigma | S3014 |
| Uric acid | Sigma | U2625 |
| Glutathione | Sigma | G4251 |
| D-tryptophan | Sigma | T9753 |
| DMEM | Thermo Fisher | 10566024 |
| Percoll | Sigma | GE17-5445-01 |
| Methanol | Fisher | A456 |
| Formic acid | Sigma | 426229 |
| Ammonium bicarbonate | Sigma | 9830 |
| Ammonium formate | Sigma | 70221 |
| Acetonitrile | Fisher | A955 |
| 4-chloro-L-phenylalanine | Carbosynth | FC13398 |
| d4-cholic acid | Sigma | 614149 |
| Durapore PVDF 0.22-μm membrane | Millipore | UFC30GV00) |
| MultiScreen Solvinert 96 Well Filter Plate | Millipore | MSRLN0410 |
| Lithocholic acid | Sigma | L6250 |
| Murocholic acid | Steraloids | C0910-000 |
| Ursodeoxycholic acid | Sigma | U5127 |
| Hyodeoxycholic acid | Sigma | H3878 |
| Chenodeoxycholic acid | Sigma | c9377 |
| Deoxycholic acid | Sigma | D2510 |
| 7-oxocholic acid | Sigma | SMB00806 |
| Omegamuricholic acid | Steraloids | C1888-000 |
| Alphamuricholic acid | Steraloids | C1890-000 |
| Betamuricholic acid | Steraloids | C1895-000 |
| Gammamuricholic acid | Steraloids | C1850-000 |
| Cholic acid | Sigma | C1129 |
| 7-betacholic acid | TRC | U849900 |
| Cholic acid-2,2,4,4-d4 | Sigma | 614149 |
| Taurolithocholic acid | Sigma | T7515 |
| Tauroursodeoxycholic acid | Sigma | 580549 |
| Taurohyodeoxycholic acid | Steraloids | C0890-000 |
| Taurochenodeoxycholate | Sigma | T6260 |
| Taurodeoxycholic acid | Sigma | T0557 |
| Taurobetamuricholic acid | Steraloids | C1899-000 |
| Tauroomegamuricholic acid | Steraloids | C1889-000 |
| Taurocholic acid | Sigma | 86339 |
| Critical Commercial Assays | ||
| DNeasy Power Soil Kit | Qiagen | 12955-4 |
| Illumina NextSeq Kit | Illumina | NextSeq 500/550 v2.5 |
| Illumina NovaSeq kit | Illumina | NovaSeq 6000 S4 Reagent Kit v1.5 |
| Pico488 dsDNA quantification reagent | Lumiprobe | 92010 |
| ATP Determination Kit | Invitrogen | A22066 |
| Quick-DNA Fungal/Bacterial Miniprep Kit | Zymogen | D6005 |
| GentleMACS Lamina Propria Kit | Miltenyi Biotec | 130-097-410 |
| Macs SmartStrainers (100 um) | Miltenyi Biotec | 130-110-917 |
| GentleMACS C tubes | Miltenyi Biotec | 130-096-334 |
| MACS Buffer | Miltenyi Biotec | 130-091-222 |
| CK28 Hard Tissue Homogenizing Kit, Beads | VWR | 10144-556 |
| Foxp3/Transcription Factor Staining | eBioscience | 00-5523-00 |
| Creatinine Assay Kit | Abcam | ab204537 |
| Deposited Data | ||
| To be updated with public accession numbers | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 GF | Taconic Biosciences | N/A |
| Mouse: SW GF | Taconic Biosciences | N/A |
| Software and Algorithms | ||
| NinjaMap | This study | |
| Quast | (Gurevich et al. 2013) | v. 5.0.2 |
| SeqKit | (Shen et al. 2016) | v. 0.12.0 |
| GTDB-tk | (Chaumeil et al. 2019) | v. 1.2.0 |
| GTDB | (Parks et al. 2020) | release 89 (database) |
| CheckM | (Parks et al. 2015) | v. 1.1.2 |
| BBtools | https://jgi.doe.gov/data-andtools/bbtools/bbtools-user-guide/ | v. 38.37 |
| Unicycler | (Wick et al. 2017) | v. 0.4.8 |
| LRScaf | (Qin et al. 2018) | v. 1.1.9 |
| TGS-GapCloser | (Xu et al. 2019) | v. 1.0.1 |
| SPAdes | (Bankevich et al. 2012) | v. 3.13.1 |
| MetaBAT2 | (Kang et al. 2019) | v. 2.2.14 |
| Grinder | (Angly et al. 2012) | v. 0.5.4 |
| Bowtie2 | (Langmead and Salzberg 2012) | v. 2.3.5.1 |
| Samtools | (Li et al. 2009) | Samtools |
| MetaPhlan2 | (Truong et al. 2015) | MetaPhlan2 |
| Midas | (Nayfach et al. 2016) | Midas |
| Kraken2 | (Wood et al. 2019) | Kraken2 |
| Bracken | (Lu et al. 2017) | Bracken |
| Matlab | https://www.mathworks.com/products/matlab.html | |
| Other | ||
| 2.2-mL 96-well deep-well plates | Thomas Scientific | 1159Q92 |
| Silicone fitted plate mat | Thomas Scientific | SMX-DW96S20 |
| Corning 96-Well Clear Flat Bottom, Polystyrene, sterile | Corning | 3370 |
| Vinyl Tape | Coy | 1600330w |
| ACQUITY UPLC BEH C18 Column, 130Å, 1.7 μm, 2.1 mm×100 mm | Waters | 186002352 |
| ACQUITY UPLC BEH C18 VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 | Waters | 186003975 |
| ACQUITY UPLC BEH Amide VanGuard Pre-column, 130 Å, 1.7 μm, 2.1 | Waters | 186004799 |
| Waters ACQUITY UPLC BEH Amide Column, 130Å, 1.7 μm, 2.1 mm×150 mm | Waters | 186004802 |
| Kinetex C18 column (1.7 μm, 2.1×100 mm) | Phenomenex | N/A |
| Agilent 1290 Infinity II UPLC | Agilent 1290 Infinity II UPLC | N/A |
Development of a highly accurate metagenomic read-mapping pipeline
Having assembled a community of 104 species, we next addressed how to quantify the abundance of each strain accurately, a major challenge given our expectation that some strains would be present at low abundance. Various strains in the community have identical 16S hypervariable sequences in the V3-V4 region, ruling out 16S amplicon-based methods. We considered designing a custom amplicon-based pipeline, but such an approach would require the design and validation of new primer sets for future communities. As an alternative, we sought to use metagenomic sequencing to quantity community composition.
To test the performance of existing metagenomic analysis tools, we generated three ‘ground truth’ data sets. The first two consisted of simulated reads generated from the assembled genome sequences of each strain: one in which all 104 strains were equally abundant (to test sensitivity and specificity), and another in which strain abundance varied over six orders of magnitude (to test dynamic range). The third set consisted of actual reads derived from sequencing each strain individually using the same protocol as in subsequent community analyses. This data set allowed us to account for biases introduced by library construction and sequencing.
We found that metagenomic read mappers based on a combination of Bowtie2 (Langmead and Salzberg, 2012) and SAMtools (Li et al., 2009) were sensitive but inaccurate: there was substantial mis-mapping of reads from one strain to others, such that whole-genome sequencing data from an individual strain was often interpreted as having arisen from multiple strains. Read mis-mapping from any abundant strain could therefore create noise that exceeds signal from low-abundance strains, degrading accuracy. In contrast, algorithms that focus on a few universal genes or unique k-mers such as MetaPhlAn2 (Truong et al., 2015), MIDAS (Nayfach et al., 2016), Kraken2/Bracken (Lu et al., 2017; Wood et al., 2019), IGGsearch (Nayfach et al., 2019), or Sourmash (Titus Brown and Irber, 2016) were generally accurate to the species level, but since they only use a small fraction of the reads (<1%), their ability to detect low-abundance or closely related strains is limited.
To address these challenges, we developed a new algorithm, NinjaMap (Data S2). Taking advantage of the fact that every strain in our community has been sequenced (Table S2), NinjaMap can quantify strain abundances with high accuracy across six orders of magnitude (STAR Methods). In brief, NinjaMap considers every read from a sample. If a read does not match perfectly to any of the genomes in the community (typically 3–4% of the reads), it is tabulated but not assigned. If a read has a perfect match to only one strain, it is assigned unambiguously to that strain. If a read matches more than one strain perfectly, it is temporarily placed in escrow. After all unambiguous assignments are made, an initial estimate of the relative abundance of each strain is computed. Reads in escrow are then fractionally assigned in proportion to the relative abundance of each strain, normalized by the total size of the genomic regions available for unique mapping to avoid bias in favor of strains with large or phylogenetically distinct genome sequences. Finally, relative abundances are computed.
To assess the performance of NinjaMap, we conducted two tests. First, we assessed the degree of read mis-mapping from and into each strain’s ledger. We quantified how many reads from strain 1 were mis-assigned to strains 2–104 (which would underestimate the abundance of strain 1 in a community), and how many reads from strains 2–104 were mis-assigned to strain 1 (which would overestimate the abundance of strain 1). For simulated reads, most instances of these two types of read mis-mapping collectively resulted in relative abundance errors < ~10−5 (Data S2, Star Methods). For actual reads, mismapping was more frequent but still typically below a threshold of 10−4 (i.e., 0.01% relative abundance); mis-mapping likely arose either from deviations between the database genome sequence and the actual sequence of the strain in our collection, or from the process of sample preparation and sequencing (Data S2) (STAR Methods). The expected contribution to relative abundance from mismapping in a community context can be even lower for some strains (Data S2).
Second, we used NinjaMap to analyze simulated reads from a 104-strain community. We found that this tool can accurately quantify strains with abundances as low as 10−6 in the context of a mixed community of known composition (Data S2), in agreement with the analysis of single-isolate samples. Thus, NinjaMap is capable of quantifying strains accurately over a wide dynamic range of relative abundances.
Community construction is highly reproducible
We began by measuring the degree of reproducibility in community composition data by constructing and propagating the 104-member community multiple times in vitro. We included technical replicates to assess variation in bacterial growth, DNA extraction, and sequencing, and biological replicates to determine the impact of differences in the preparation of the inocula. We propagated the communities for 48 h and extracted DNA for sequencing at 0, 12, 24, and 48 h.
The range of cell densities at t=0 spanned multiple orders of magnitude (Figure 1B), with a mean log10(relative abundance) of −2.5±0.8 for all detectable strains. 95/104 strains were detectable at t=0; the remaining strains, which grew poorly when cultured individually, were below the limit of detection or had abundances that could potentially be explained by read mis-mapping. The communities reached a relatively stable configuration by 12 h (Figure 1B), with a remarkable degree of reproducibility among biological replicates (Figure 1C). Notably, very low-abundance strains (<10−4) were only slightly more variable than high-abundance strains. Technical replicates were even more similar (Figure 1D), indicating that community growth, DNA extraction, and sequencing contributed only modestly to variability. Taken together, these results indicate that community composition is robust to experimental variation.
A nutrient drop-out screen to map strain-nutrient interactions in the community
We next sought to explore the network of strain-nutrient interactions in the community. Although much is known about polysaccharide foraging by gut commensals (Martens et al., 2014), far less is known about amino acid utilization, so we performed the experiment in a defined growth medium (SAAC, STAR Methods) from which we could remove one amino acid at a time. Since amino acids are often utilized in pairs (Nisman, 1954; Smith and Macfarlane, 1997), eliminating one at a time from a complete background rather than adding one at a time to a null background has greater potential to reveal phenotypes relevant to community function. Moreover, performing this screen in the context of a diverse community (as opposed to the traditional practice of analyzing the growth of isolated strains) enables the potential study of community-dependent effects such as nutrient competition or mutualism-dependent nutrient utilization.
To map strain-amino acid interactions, we constructed the 104-member community (STAR Methods) and used it to inoculate 20 defined growth media, each deficient in a single amino acid, as well as complete SAAC (Figure 2A). Samples were taken at 48 h and metagenomic sequencing data were analyzed to determine the impact of amino acid deficiency on the relative abundance of each strain.
Figure 2: Systematic analysis of strain-amino acid interactions.

(A) Schematic of the amino acid dropout experiment. Frozen stocks of the 104 strains were used to inoculate cultures that were grown for 24 h, diluted to similar optical densities (to the extent possible), and pooled. The mixed culture was used to inoculate one of twenty defined media lacking one amino acid at a time. After 48 h, communities were sequenced and analyzed by NinjaMap to determine changes relative to growth in the complete defined medium. (B) Community composition is impacted by amino acid dropout. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point. Strains are colored according to their rank-order abundance in the community grown in complete defined medium (SAAC). Strains whose relative abundance could be explained by read mis-mapping from a more abundant strain in the same sample are plotted with a gray outline. Undetected strains were set to 10−7 for visualization. (C) Heat map showing the hierarchically clustered z-scores for each strain (x-axis) across amino acid dropouts (y-axis). The z-score was calculated based on the standard deviation of strain abundance across all samples except the cysteine dropout (STAR Methods). The Firmicutes L. lactis, C. sporogenes, and L. ruminis grew less robustly in the absence of Leu and Ile. Strains whose abundances could be explained by mis-mapping from a higher-abundance strain were not shown. (D) The effect of amino acid removal varies widely across amino acids. The fraction of strains with |z|>2 is shown for each amino acid dropout (n=66). (E) The absence of leucine or arginine leads to a large decrease in C. sporogenes relative abundance. Strains are colored according to their rank-order abundance in the community grown in complete defined medium. Only strains that were detected in at least one of the three samples were included (n=92). C. sporogenes is highlighted in black. L. lactis is highlighted in white. Undetected strains were set to 10−7 for visualization. (F) C. sporogenes growth in complete defined medium is dependent on the presence of arginine (Arg), and ornithine transcarbamoylase (otc) is partially responsible for Arg metabolism. Wild type C. sporogenes and a Δotc mutant were grown in complete defined medium +/− Arg. Growth curves depict the mean of 3 replicates. Error bars represent 1 standard deviation. (G) C. sporogenes requires otc to produce ATP from arginine. Intracellular ATP levels in C. sporogenes incubated in PBS containing 2 mM Arg are shown. (H) A proposed pathway for Arg metabolism in C. sporogenes. Based on these data, we propose that Arg is converted to citrulline by the putative Arg deiminase CLOSPO_00894; citrulline is then hydrolyzed to ornithine and carbamoyl phosphate by the putative ornithine transcarbamoylase CLOSPO_02415, leading to the production of ATP.
Global analysis of strain-amino acid interactions
To identify strain-amino acid interactions, we tabulated strains whose relative abundance deviated significantly from the mean across conditions, taking advantage of the fact that most amino acid dropouts had little effect on most strains (Figure 2B, STAR Methods). When the community was propagated in the complete defined medium, relative abundances spanned >6 orders of magnitude. 36% of the strains were present at 10−4–10−2 relative abundance, 8 strains were >10−2 and 50 were <10−4 (Figure 2B). In agreement with simulated results, NinjaMap was sensitive to strains with relative abundances as low as 10−6, enabling us to quantify the 56% of strains that were below the 10−3 limit of detection commonly used for metagenomic analyses (Franzosa et al., 2015). Our system is therefore capable of studying low-abundance microbes, some of which are known to have large biological impacts (Buffie et al., 2015; Funabashi et al., 2020).
To identify significant responses, we calculated the standard deviation of the relative abundance of each strain across experiments and computed z-scores (Figure 2C, STAR Methods). Strain-amino acid interactions that were previously identified in monoculture studies were also observed in our community format. Anaerostipes caccae, whose growth is stimulated by methionine (Soto-Martin et al., 2020), decreased in relative abundance in a community grown in methionine-deficient medium (z=−3.48). Likewise, C. sporogenes expansion was impeded by the absence of leucine (z=−2.56), a substrate it oxidatively decarboxylates to isovalerate to generate electrons (Guo et al., 2019). These observations demonstrate that even though >100 strains are competing for the same nutrients, the effects of eliminating one amino acid on the growth of one strain are readily observable in the context of a complex and diverse community.
Most strains responded to amino acid removal in ≤4 cases (Figure 2B). Moreover, relative abundances displayed low variability, with a mean standard deviation of log10(relative abundance) across strains <0.43. Only three strains, all of which are Firmicutes, were responsive to removal in >4 cases: Lactococcus lactis DSM 20729, Clostridium sporogenes ATCC 15579, and Lactobacillus ruminis ATCC 25644 (Data S3, Table S3). Thus, under these growth conditions, most strains are largely insensitive to amino acid removal while a small minority are highly responsive. We note that the response of a strain to amino acid removal may be direct (e.g. due to utilization for energy) or indirect (e.g. amino acid removal impacts an interacting strain).
Amino acids varied widely in terms of their impact on community composition (Figure 2D). More than half of the strains responded to cysteine removal, likely due to its effect as a reducing agent. More than 5% of the strains responded to methionine, histidine, isoleucine, arginine, valine, and tyrosine removal, while for eight amino acids there were no significant changes to the community at all (Figure 2D). Interestingly, there were large differences among similar amino acids: no strains responded to lysine removal, while 10.6% and 7.6% of the strains responded to histidine and arginine removal, respectively. The removal of isoleucine, leucine, and arginine had a particularly large impact on community structure: C. sporogenes and L. lactis, the two most abundant strains when grown in complete defined medium, decreased >500-fold in relative abundance when any of these amino acids were removed (Figure 2E); this sensitivity was also observed in a biological replicate experiment (Data S3). Taken together, our data suggest that certain amino acids are ‘keystone’ nutrients that play an important role in determining community composition.
C. sporogenes uses arginine to generate ATP
Among the 86 candidate strain-amino acid interactions revealed by our screen, we were particularly intrigued by those involving C. sporogenes. Although C. sporogenes can oxidize and reduce aromatic amino acids (Dodd et al., 2017), its relative abundance was unaffected by the removal of phenylalanine, tyrosine, or tryptophan (Data S3). In contrast, the removal of leucine, isoleucine, and arginine each had large impact on the fitness of C. sporogenes in the community. The second strongest phenotype was a decrease in relative abundance in the absence of arginine (Figures 2E, S2C); while C. sporogenes is known to metabolize arginine (Venugopal and Nadkarni, 1977; Wildenauer and Winter, 1986), no impact of arginine on growth or energy metabolism had been observed in prior work. To validate and characterize this interaction, we compared C. sporogenes growth in complete defined versus arginine-deficient medium. Although C. sporogenes grew well in complete defined medium, it exhibited a large growth defect in the absence of arginine (Figure 2F), indicating that this amino acid is an important substrate for growth.
C. sporogenes can use other amino acids as substrates to support ATP synthesis (Dodd et al., 2017). Hypothesizing that the same is true for arginine, we incubated wild-type C. sporogenes in a culture medium deficient in substrates for ATP synthesis. Upon addition of arginine, intracellular ATP levels rose sharply (Figure 2G), indicating that C. sporogenes generates ATP (directly or indirectly) from arginine.
To identify the enzymes involved in this process, we parsed the C. sporogenes genome for pathways known to capture energy from arginine. This search yielded candidate genes for each of the three steps in the arginine deiminase pathway (Figure 2H), which catalyzes the net conversion of arginine to ornithine plus CO2 and two equivalents of ammonium, generating one equivalent of ATP (Cunin et al., 1986). Using a method we recently developed to construct scarless deletions in C. sporogenes (Guo et al., 2019), we generated strains deficient in the putative arginine deiminase (CLOSPO_00894, Δadi) or ornithine carbamoyltransferase (CLOSPO_02415, Δotc). The Δotc mutant was unable to generate ATP in response to arginine provision, consistent with a role for the arginine deiminase pathway in C. sporogenes energy production (Figure 2G). In contrast, the Δadi mutant showed no defect in arginine-induced ATP production (Data S3), suggesting the possibility of an alternative pathway to generate citrulline from arginine. Consistent with these observations, the Δotc mutant (but not the Δadi mutant) was growth-deficient complete defined medium (Figure 2F, Data S3). The deficiency was partial, suggesting that an alternative pathway can generate energy from arginine under these conditions. Together, these results show that arginine metabolism by the arginine deiminase pathway contributes directly to the cellular ATP pool, augmenting our understanding of how amino acid metabolic pathways contribute to the fitness of a gut commensal within a complex community.
Attributes of a complex defined community in gnotobiotic mice
Our central goal in designing hCom1 was to enable mechanistic studies of the microbiome in the context of host colonization. As a starting point for in vivo work, we colonized germ-free Swiss-Webster (SW) mice with hCom1 (Figure 3A), which we prepared by propagating each strain individually and mixing OD-normalized cultures (STAR Methods). We sampled fecal pellets from the mice weekly for eight weeks, enumerated community composition in the inoculum and each fecal sample by metagenomic sequencing, and performed read analysis using NinjaMap.
Figure 3: Colonizing germ-free mice with a complex gut bacterial community.

(A) Schematic of the experiment. Frozen stocks of the 104 strains were used to inoculate cultures that were grown for 24 h, diluted to similar optical densities (to the extent possible, STAR Methods), and pooled. The mixed culture was used to colonize germ-free Swiss-Webster (SW) mice by oral gavage. Fecal samples were collected weekly at weeks 1–5 and week 8, subjected to metagenomic sequencing, and analyzed by NinjaMap to measure the composition of the community at each time point. (B) Relative abundances for most strains are tightly distributed. Each column depicts the relative abundance of an individual strain across all mice at week 4. (C) Average relative abundances of the inoculum versus the communities at week 4. Strains in the community spanned >6 orders of magnitude of relative abundance when colonizing the mouse gut. Dots are colored by phylum according to the legend in panel B. Data represent the average of all mice in the experiment. (D) hCom1 reaches a stable configuration by week 2. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point averaged over 5 mice co-housed in a cage. Strains are colored according to their rank-order relative abundance at week 4.
Our analysis yielded two main conclusions. First, almost all strains in the inoculum colonized the mouse gut (Figure 3B-C). We confirmed the presence of 103/104 strains in the inoculum; of these, 101 strains were detected in the mice at least once. The three strains we failed to detect in mice—Ethanoligenens harbinense YUAN-3, Clostridium methylpentosum DSM 5476, and Ruminococcus albus 8—were slow-growing and difficult to cultivate. While strain relative abundances spanned >6 orders of magnitude, nearly all strains exhibited low variation across 20 mice in four cages, with coefficient of variation (CV, standard deviation/mean) <0.4.
Second, the community quickly reached a stable configuration (Figure 3D). Averaged across mice, relative abundances remained largely constant two weeks after colonization, with Pearson’s correlation coefficient >0.95 at each time point with respect to the composition in week 8. After the first week, relative abundances stayed within a narrow range for the duration of the experiment (mean CV<0.2 across the 96 strains that remained above the limit of detection). Large shifts in relative abundance were rare: only 27/312 (8.7%) week-to-week strain-level changes were >10-fold.
An ecology-based process to fill open niches in the community
Although hCom1 is composed of prevalent species from the human gut microbiome, it is not as complex or phylogenetically rich as a human fecal community; the process that dictated its membership was not designed to ensure completeness by any functional or ecological criteria. To create a defined community that better models the gut microbiome, we sought to augment hCom1 by increasing the number of niches it fills in the gastrointestinal tract (Figure 4A). We designed an experimental strategy based on the principle of colonization resistance (Buffie and Pamer, 2013; Lawley and Walker, 2013), an ecological phenomenon in which resident organisms exclude invading species from occupied niches. We colonized germ-free mice for four weeks with hCom1, presumably filling the metabolic and anatomical niches in which its species reside. We then challenged these mice with one of three undefined fecal samples (Hum13), reasoning that invading species that would otherwise occupy a niche already filled by hCom1 would be excluded, whereas invading species whose niche was unfilled would be able to cohabit with hCom1. After four additional weeks, we used metagenomic sequencing to analyze community composition from fecal pellets.
Figure 4: Challenging hCom1 with human fecal communities to identify strains that fill open niches.

(A) Schematic of the experiment. Mice were colonized by freshly prepared hCom1 and housed for four weeks, presumably filling the metabolic and anatomical niches accessible to the strains in the community. At the beginning of week 5, the mice were challenged with one of three fecal communities from a healthy human donor or with PBS as a control; we reasoned that fecal strains that would otherwise occupy a niche already filled by hCom1 would be excluded, whereas fecal strains whose niche was unfilled would be able to cohabit with hCom1. After four additional weeks, we used metagenomic sequencing coupled with MIDAS to analyze community composition from fecal pellets collected at weeks 1–5 and 8. We then identified strains that colonized in the presence of hCom1 to augment the community to create hCom2, which were then used for another round of challenge experiments (Figure 5). (B) hCom1 is broadly but not completely resistant to fecal challenge. All plots represent MIDAS bins, a rough proxy for species-level taxa. Top row: blue squares in the waffle plots indicate species that derive from hCom1, and gray squares represent species from the fecal communities. Bottom row: pie charts representing the total relative abundance of MIDAS bins that derive from hCom1 versus the fecal communities. An average of 89% of the genome copies from week 8, comprising 58% of the MIDAS bins, derived from hCom1. The remaining 11% of the genome copies, and 42% of the MIDAS bins, represent new species that joined hCom1 from one of the fecal samples. (C) Despite the addition of new strains, the architecture of the community remains intact. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point averaged over the 5 co-housed mice that were challenged with fecal community Hum1. Strains are colored according to their rank-order relative abundance at week 4. Gray circles represent invading species derived from fecal community Hum1, defined as any species not present in weeks 1–4 in the group of mice shown. (D) The relative abundances of the hCom1-derived species present post-challenge are highly correlated with their pre-challenge levels. Pearson’s correlation coefficient with respect to the average relative abundance in weeks 2 and 3 are shown for the PBS control and 3 fecal community challenges, averaged across mice that received the same challenge. Correlation coefficients are shown for the 104 hCom1 species (solid lines) and for all species including invaders (dashed lines).
To determine which species from each fecal sample colonized in the presence of hCom1, we analyzed the composition of fecal pellets collected in weeks 5–8 to assign species as ‘input’ (hCom1-derived) or ‘invader’ (fecal sample-derived). For this analysis we used MIDAS (Nayfach et al., 2016), an enumeration tool that—unlike NinjaMap—does not require prior knowledge of the constituent strains. MIDAS and NinjaMap reported highly concordant relative abundance profiles using sequencing reads from hCom1-colonized mice, although—as expected—MIDAS was less sensitive since it utilizes only 1% of sequencing reads (Star Methods, Data S4). We used MIDAS for subsequent analyses of samples that were partially or completely undefined.
Using MIDAS, we cannot determine whether a strain present both pre- and post-challenge was derived from hCom1 (i.e., the original strain colonized persistently) or the fecal sample (i.e., a new strain displaced the original strain). To gain further insight into strain displacement versus persistence, we recruited reads from samples taken four weeks post-challenge (week 8) to a database composed of the hCom1 genome sequences, using only reads that were 100% identical to one or more of the genomes. We focused our analysis on genomes with high depth of coverage (≥10X). More than 60% of these strains were covered broadly (≥95%) by perfectly matching reads, indicating that most strains present pre- and post-challenge were either hCom1-derived or a closely related strain (Data S4).
As expected, mice challenged by saline instead of a fecal sample showed no evidence of new species post-challenge (Figure 4B). In hCom1-colonized mice challenged by a fecal sample, an average of 89% of the genome copies from week 8 (and 58% of the MIDAS bins, a rough proxy for species) derived from hCom1 (Figure 4B). The remaining 11% of the genome copies (and 42% of the MIDAS bins) represent new species that joined hCom1 from one of the fecal samples. Despite the addition of new species, the architecture of the community remained intact (Figure 4C): the relative abundances of the hCom1-derived species present post-challenge were highly correlated with their pre-challenge levels (Pearson’s r >0.85) (Figure 4D). Thus, hCom1 is broadly but not completely resilient to a human fecal challenge.
Designing and constructing an augmented community
The observation that only a small fraction of the post-challenge communities was composed of new species led us to hypothesize that we could improve the colonization resistance of hCom1 by adding the invading species, thereby improving its ability to fill niches in the gut. Twenty-four bacterial species entered hCom1 from ≥2 of the 3 fecal samples used as a challenge (Table S4); we focused on these species, reasoning that they were more likely to fill conserved niches in the community. We were able to obtain 22/24 from culture collections and we included all of them in the new community (hCom2). At the same time, we omitted seven species that either failed to colonize initially or were displaced in all three groups of mice (Figure S4), reasoning that they were incompatible with the rest of hCom1 or incapable of colonizing the mouse gut under the dietary conditions in which the experiment was performed. Thus, the new community contains 97 strains from hCom1 plus 22 new strains, for a total of 119 (Figure 4A, Figure S1, Table S2). These 22 strains are primarily Firmicutes or species of Alistipes. Many represent taxa that are phylogenetically under-represented in hCom1, suggesting that they might be able to occupy niches left open by the members of hCom1 (Figure S1).
We colonized four groups of germ-free SW mice with hCom2, collecting fecal pellets weekly (Figure 4A). As before, we measured community composition by analyzing metagenomic sequencing data with NinjaMap (Figure 5A, Table S4). The gut communities of hCom2-colonized mice rapidly reached a stable configuration (Pearson’s r with respect to week 8 >0.97) (Figure S2). 100 of the 119 strains were above the limit of detection; hCom1-derived strains colonized at similar relative abundances in the context of the augmented community (with similarly low CVs across mice) (Figure 5B). The species that were new to hCom2 exhibited a wide range of relative abundances; Bacteroides rodentium became the most abundant species, whereas the least abundant of the new species, Blautia sp. KLE 1732, had a mean abundance ~10−4 (Figure 5B).
Figure 5: An augmented community with improved resilience to fecal challenge.

(A) Comparing the architecture and strain-level relative abundances of hCom1 and hCom2. Each column depicts the relative abundance of an individual strain from hCom2 across all samples at week 4. 100 of the 119 strains were detected; those that are new to hCom2 are colored red. (B) Averaged relative abundances of the strains in hCom1 versus hCom2 at week 4. Strains that are new to hCom2 are indicated by a gray outline. Dots are colored by phylum according to the legend in panel B. (C) The architecture of hCom2 is largely unaffected by fecal challenge with Hum1–3. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point averaged over the 5 co-housed mice that were challenged with fecal community Hum1. Strains are colored according to their rank-order relative abundance at week 4. Gray circles represent invading species, defined as any species not present in weeks 1–4 in the group of mice shown. (D) Left: hCom2 is more resilient to fecal challenge than hCom1. Top row: blue squares in the waffle plots indicate MIDAS bins that derive from hCom2; gray squares represent MIDAS bins from the fecal communities. Bottom row: pie charts representing the percentage of MIDAS bins that derive from hCom2 versus the fecal communities. An average of 96% of the genome copies (and 81% of the MIDAS bins) come from hCom2 in the Hum1–3 challenges, demonstrating that the resilience of the community was improved markedly by augmentation with strains identified from the initial challenge (Figure 4). Right: hCom2 is broadly resilient to challenge by unrelated fecal samples (Hum4–6). In these challenges, an average of 81% of the genome copies (and 58% of the MIDAS bins) come from hCom2. (E) Nearly all invading strains at week 8 were repeat invaders from the first fecal challenge (Table S4). The dots representing invading strains are shown in full color; dots representing hCom2-derived strains are partially transparent. Dots that represent repeat invaders from the first fecal challenge experiment have a thick black border. (F) The relative abundances of the hCom2-derived species present post-challenge are highly correlated with their pre-challenge levels. Pearson’s correlation coefficient with respect to the average relative abundance in weeks 3 and 4 are shown for the PBS control and 3 fecal community challenges, averaged across mice that received the same challenge. Correlation coefficients are shown for the 119 species in hCom2 (solid lines) and for all species including invaders (dashed lines). (G) hCom2 resembles a fecal consortium more closely than hCom1. Averaged relative abundances of MIDAS bins are shown for hCom1- and hCom2-colonized mice versus mice colonized by a fecal community from one of three healthy human donors (Hum1–3). The phylum-level architecture of hCom2 is more closely correlated to that of humanized mice than hCom1 (Figure S3). (H) Pairwise correlation coefficients of phylum-level relative abundance vectors were higher between hCom2-colonized and Hum1–3 humanized mice than between hCom1-colonized and Hum1–3 humanized mice.
The augmented community is more resilient to human fecal challenge
Our goal in constructing hCom2 was to improve its completeness as assessed by its ability to occupy niches in the gut. To test whether hCom2 is more complete than hCom1, we challenged hCom2-colonized mice at the beginning of week 5 with the same fecal samples used to challenge hCom1, enabling us to compare results between the challenge experiments. Importantly, the 22 strains used to augment hCom1 were obtained from culture collections rather than the fecal samples themselves, reducing the likelihood that hCom2 and the fecal samples have overlapping membership at the strain level (Garud et al. 2019). Indeed, by recruiting sequencing reads to the genomes of the new organisms in hCom2, we found that 17/22 were covered broadly (≥95%) by perfectly matching reads, consistent with the view that they were derived from hCom2 and not the fecal challenge (Data S4).
An average of 96% of the genome copies (and 81% of the MIDAS bins) from week 8 derived from the strains in hCom2 (Figure 5C), demonstrating that the colonization resistance of hCom2 is markedly improved over hCom1 (Figure 5D). The remaining 4% of reads (and 19% of MIDAS bins) represent species that engrafted in the presence of hCom2 (Figures 5D, S2). Strikingly, nearly all of the species that invaded hCom2 also invaded hCom1 (Figure 5E, Table S4); we were either unable to obtain an isolate for inclusion in hCom2 or the species invaded hCom1 from only 1 of the 3 fecal samples used as a challenge, falling below our threshold for inclusion. These species represented virtually all of the remaining genome copies. We conclude that more extensive augmentation, based on the results of the first challenge experiment, would likely have enhanced colonization resistance further.
Moreover, compared to hCom1, the composition of hCom2 post-challenge was more similar to its pre-challenge state (Pearson’s r >0.95, Figure 5F). Taken together, these data show that hCom2 is more stable and complete than hCom1, and that the augmentation process is robust and fault-tolerant in identifying species that can occupy unfilled niches.
In the previous experiment, we challenged hCom2-colonized mice with Hum1–3, the same fecal communities used in the initial augmentation experiment (Figure 4). We next sought to determine whether hCom2 is resilient to challenge by unrelated fecal communities. hCom2-colonized mice were challenged with Hum4–6, which are compositionally distinct from Hum1–3 (Figure 4A). hCom2 was somewhat less stable to challenge by unrelated fecal samples: an average of 81% of the genome copies from week 8 (and 58% of the MIDAS bins) derived from hCom2 (Figure 5D). Thus, hCom2 is broadly but not completely resilient to challenge by unrelated fecal samples.
The architecture of hCom2 resembles that of a complete, undefined human fecal consortium
Our original goal in building a complex defined community was to develop a model system for the gut microbiome. Having demonstrated that hCom2 is stable and resilient to invasion, we sought to assess whether it has the functional attributes of a model system.
We started by asking how its architecture—the relative abundances of its constituent taxa— compares to that of a human fecal community. We colonized germ-free mice with three human fecal samples (Hum1–3; hereafter, ‘humanized’) and compared their community compositions to those of mice colonized with hCom2. The gut communities of hCom2-colonized and humanized mice were similar in three ways (Figures 5G-H, S3). First, relative abundances spanned at least five orders of magnitude, with some strains consistently colonizing at >10% and others at <0.001%. Second, the distribution of log relative abundances was centered at ~0.01%, indicating that the majority of strains in the community would be missed by enumeration tools that have a limit of detection of 0.1%. Third, relative abundances by taxon are similar down to the genus level (Figure S3). Thus, the architecture of hCom2 resembles that of a human fecal community in the mouse gut.
Reproducibility of colonization
We next addressed the question of biological reproducibility, which is a threshold requirement for an experimental model system. We started by analyzing data from the second fecal challenge experiment (with Hum1–3) to assess the technical reproducibility of community composition in mice colonized by hCom2. At week 4, strain abundances in 20 mice across 4 cages colonized by the same hCom2 inoculum were highly similar (pairwise Pearson’s correlation coefficients 0.96±0.01, Data S5).
Biological reproducibility was a greater concern. Given the complexity of hCom1 and hCom2, variability in the growth of individual strains could lead to substantial differences in the composition of inocula constructed on different days. To determine the extent to which this variability affects community architecture in vivo, we compared community composition in four groups of mice colonized by replicates of hCom2 constructed independently on different days (Figure 6A-B). The communities displayed a striking degree of similarity in relative abundance profiles after 4 weeks (Pearson’s correlation coefficient >0.95 between all pairs of biological replicates). We conclude that a relatively constant nutrient environment enables input communities with widely varying relative abundances to reach the same steady state configuration, consistent with ecological observations in other microbial communities (Aranda-Díaz et al., 2020; Goldford et al., 2018; Hibberd et al., 2017; Venturelli et al., 2018). This high degree of biological reproducibility will be enabling for the use of complex defined communities as experimental models.
Figure 6: hCom2-colonized mice are phenotypically similar to humanized mice.

(A) Schematic of the experiment. Germ-free SW mice were colonized with freshly prepared hCom2 or a fecal sample from a healthy human donor. One cohort of mice was sacrificed at two weeks for immune cell profiling; another was sacrificed at four weeks for targeted metabolite analysis. (B) The architecture of hCom2 in mice is highly reproducible. Left: community composition is highly similar across four biological replicates. Each dot is an individual strain; the collection of dots in a column represents the community at 4 weeks averaged over 5 mice co-housed in a cage. Strains are colored according to their average rank-order relative abundance across all samples. Right: Pearson’s pairwise correlation coefficients for technical and biological replicates. (C) hCom2-colonized, hCom1-colonized, and humanized mice have similar bacterial cell densities in vivo. Fecal samples from hCom2-colonized, hCom1-colonized, humanized, specific pathogen-free (SPF), or germ-free (GF) mice were homogenized and plated anaerobically on Columbia Blood Agar to enumerate colony forming units. (D) Immune cell types and numbers were broadly similar between hCom2-colonized and humanized mice. Colonic immune cells were extracted from hCom2-colonized, humanized, or germ-free mice (all C57BL/6), stained for cell surface markers, and assessed by flow cytometry. Statistical significance was assessed using a Student’s two tailed t-test (**: p<0.05). (E) hCom2-colonized mice and humanized mice have a similar profile of microbiome-derived metabolites. Urine samples from hCom2-colonized and humanized mice were analyzed by targeted metabolomics to measure a panel of aromatic amino acid metabolites by LC-MS. Statistical significance was assessed using a Student’s two tailed t-test (*: p<0.05; **: p<0.001). (F) Bile acids were extracted from fecal pellets collected from hCom2-colonized and humanized mice and were quantified by LC-MS. Statistical significance was assessed using a Student’s two tailed t-test (*: p<0.05; **: p<0.001).
To further investigate the potential for hCom2 to function as a model microbiome, we assessed its composition in a second strain of mice. Since the experiments to develop hCom2 used outbred SW mice, we chose 129/SvEv, an inbred mouse strain. We colonized germ-free 129/SvEv mice with hCom2 and collected fecal pellets after 4 weeks of colonization. Community composition was highly correlated with that of SW mice (Pearson correlation coefficient >0.95) (Data S5). These data indicate that hCom2, like the human gut microbiome (Rothschild et al., 2018), is robust to changes in host genotype.
hCom2-colonized mice are phenotypically similar to humanized mice
We performed three additional experiments to determine the degree to which hCom2-colonized mice resemble germ-free mice colonized by a human fecal community. Since our defined communities are composed of human fecal isolates, we colonized germ-free mice with hCom2 or an undefined human fecal community and assayed phenotypes after 4 weeks (Figure 6A). First, fecal pellets from each mouse were serially diluted and plated on Columbia blood agar to estimate the bacterial cell density in each community. Each group contained 1011-1012 colony forming units per gram of feces (Figure 6C), similar to previously reported estimates from humans and from conventional and humanized mice (Ley et al., 2006; Vandeputte et al., 2017). Thus, hCom2 colonizes the mouse gut to a similar extent as a normal murine or human fecal community.
Next, we sought to determine whether mice colonized by hCom2 harbor a similar immune cell profile to that of humanized mice. We extracted and stained colonic immune cells and assayed them by flow cytometry. Most immune cell subtypes, including CD4+ T cells, IgA+ B cells, macrophages, CD11b+ dendritic cells, and monocytes, were similarly abundant in humanized and hCom2-colonized mice (Figure 6D, Data S5), indicating that—at least in broad terms—hCom2-colonized mice are immunologically comparable to humanized mice.
Finally, to determine whether hCom2-colonized and humanized mice harbor a similar profile of microbiome-derived metabolites, we analyzed fecal pellets and urine samples using targeted metabolomics. Aromatic amino acid metabolite levels in urine (Figure 6E) and primary and secondary bile acid levels in feces (Figure 6F) were comparable between hCom2-colonized and humanized mice. Taken together, these data suggest that hCom2 is a reasonable model of gut microbial metabolism.
hCom2 exhibits robust colonization resistance against pathogenic Escherichia coli
To demonstrate its utility as a model system, we used hCom2 to study an emergent property of gut communities: their ability to resist colonization by pathogens and pathobionts (Buffie et al., 2015). To test whether hCom2 exhibits colonization resistance, we studied invasion by Escherichia coli ATCC 43894, an enterohemorrhagic E. coli (EHEC). We chose this strain for three reasons. First, EHEC is responsible for life-threatening diarrheal infections and hemolytic uremic syndrome, and enteric colonization by other E. coli strains has been linked to malnutrition and inflammatory bowel disease (Palmela et al., 2018; Pham et al., 2019). Second, colonization resistance to E. coli and other Enterobacteriaceae has been studied in detail (Litvak et al., 2019; Stromberg et al., 2018; Velazquez et al., 2019), but the commensal strains responsible and mechanisms by which they act are incompletely understood. Finally, hCom2 harbors no Enterobacteriaceae and only three species of Proteobacteria (Desulfovibrio piger, Bilophila wadsworthia, and Burkholderiales bacterium 1–1-47), so resistance to E. coli colonization would require a mechanism other than exclusion by a close relative occupying the same niche.
To test whether hCom2 is capable of resisting EHEC engraftment, we colonized germ-free SW mice with hCom2 or one of two other communities: a 12-member community (12Com) similar to one used in previous studies (McNulty et al., 2013) or an undefined fecal community from a healthy human donor (Figure 7A). hCom2 and 12Com do not contain any Enterobacteriaceae. To test whether non-pathogenic Enterobacteriaceae enhance colonization resistance to EHEC, we colonized two additional groups of mice with variants of hCom2 and 12Com to which a mixture of seven non-pathogenic Enterobacteriaceae strains were added (Escherichia coli MITI 27, Escherichia coli MITI 117, Escherichia coli MITI 135, Escherichia coli MITI 139, Escherichia coli MITI 255, Escherichia coli MITI 284, and Enterobacter cloacae MITI 173; termed ‘Enteromix’). After four weeks, we challenged with EHEC and assessed invasion by selective plating under aerobic growth conditions (Figure 7A).
Figure 7: hCom2 exhibits colonization resistance against enterohemorrhagic E. coli.

(A) Schematic of the experiment. We colonized germ-free SW mice with freshly prepared hCom2 or one of two other communities: a 12-member synthetic community (12Com) or a fecal community from a healthy human donor. hCom2 and 12Com do not contain any Enterobacteriaceae; to test whether non-pathogenic Enterobacteriaceae enhance colonization resistance to EHEC, we colonized two additional groups of mice with variants of hCom2 and 12Com to which a mixture of seven non-pathogenic Enterobacteriaceae strains were added (six E. coli and Enterobacter cloacae, Enteromix (EM)). After four weeks, we challenged with 109 colony forming units of EHEC and assessed the degree to which it colonized in two ways: by EHEC-selective plating under aerobic growth conditions, and by metagenomic sequencing with NinjaMap analysis. (B) hCom2 exhibits a similar degree of EHEC resistance to that of a fecal community in mice. Colony forming units of EHEC in mice colonized by the four different communities are shown. As expected, the fecal community conferred robust colonization resistance while 12Com did not. The addition of EM moderately improved the EHEC resistance of 12Com. Despite lacking Enterobacteriaceae, hCom2 exhibited a similar level of EHEC resistance to that of an undefined fecal community. (C) The architecture of hCom2 is stable following EHEC challenge. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point averaged over four co-housed mice. Strains are colored according to their phylum; EHEC is shown in black and members of the Enteromix community are shown in gray. (D) Schematic of the phylum dropout experiment. We colonized germ-free SW mice with four variants of hCom2, each one missing all species from the phyla Actinobacteria, Firmicutes, Proteobacteria, or Verrucomicrobia. After four weeks, we challenged with 109 colony forming units of EHEC and assessed the degree to which it colonized by EHEC-selective plating under aerobic growth conditions, and by metagenomic sequencing with NinjaMap analysis. (E) The ΔActinobacteria and ΔVerrucomicrobia communities retain the ability to resist EHEC invasion, while the ΔFirmicutes and ΔProteobacteria communities are sensitive to EHEC invasion. Right: a large survival difference in ΔFirmicutes-colonized mice compared with hCom2-colonized. (F) The architecture of the phylum dropout communities remains stable following EHEC challenge. Each dot is an individual strain; the collection of dots in a column represents the community at a single time point averaged over four co-housed mice. Strains are colored according to their phylum; EHEC is shown in black.
Consistent with previous reports (Mohawk and O’Brien, 2011; Stromberg et al., 2018), the undefined human fecal community conferred robust resistance against EHEC colonization (Figure 7B-C). In contrast, 12Com allowed much higher levels of EHEC growth; the addition of Enteromix to 12Com improved the phenotype but did not restore full EHEC resistance (Figure 7B). Despite lacking Enterobacteriaceae, hCom2 exhibited a similar level of EHEC resistance to that of an undefined fecal community (Figure 7B). Thus, hCom2 is sufficiently complete to exhibit comparable levels of colonization resistance to a native fecal community.
As a starting point for identifying which species in hCom2 are responsible for EHEC colonization resistance, we constructed four communities in which we dropped out, in turn, all of the species in the phyla Firmicutes, Verrucomicrobia, Actinobacteria, and Proteobacteria. We colonized mice with these phylum dropout communities and then challenged them with EHEC (Figure 7D). The ΔActinobacteria (missing 10 strains) and ΔVerrucomicrobia communities (missing 1 strain, Akkermansia muciniphila) resisted EHEC comparably to hCom2 (Figure 7E-F). However, the ΔProteobacteria and ΔFirmicutes communities were more susceptible. Thus, despite the lack of Enterobacteriaceae in hCom2, the absence of the three more distantly related species of Proteobacteria was sufficient to confer sensitivity to EHEC invasion.
The ΔFirmicutes community was highly sensitive to EHEC invasion (Figure 7E); the defect resulted in a large survival difference between hCom2-colonized and ΔFirmicutes-colonized mice (Figure 7E, right). These results indicate either that either Firmicutes play a role in EHEC resistance or that a change in community architecture induced by their removal renders the community sensitive to invasion. Further studies with more precise strain dropout experiments could uncover strains that confer resistance and may enable more targeted microbial therapy against EHEC colonization and infection.
DISCUSSION
By developing a community that is both defined and reasonably complex, we have generated a model system that captures much of the biology of a native microbiome. Future refinements are needed, including additional bacterial strains to occupy unfilled niches as well as archaea, fungi, and viruses, all of which are important components of the native ecosystem.
The computational pipeline we developed for read mapping makes it possible to analyze complex defined communities with high precision and sensitivity. Community structure can be quantified across six orders of magnitude in relative abundance, enabling the interrogation of low-abundance community members that play important roles in community function and dynamics (Buffie et al., 2015; Funabashi et al., 2020). The degree of technical and biological reproducibility (Figure 6B) is remarkable in a system this complex, which bodes well for future experimental efforts.
The process by which we augmented a defined community revealed two unexpected findings. First, a community composed of strains from >100 distinct donors can be stable in vivo. It remains to be seen whether there are appreciable differences in stability—or in fine-scale genomic and phenotypic adaptation—between communities composed of isolates from a single donor (in which strains have coexisted for years) versus multiple donors (in which strains have no prior history together). If a collection of strains with no common history can form a stable consortium, it will be interesting to determine the role of priority effects (i.e., order of arrival) and spatial and metabolic niche occupancy.
Second, the process we introduce here for filling open niches is surprisingly robust and fault tolerant. Most notably, nearly all of the fecal community-derived strains that invaded hCom1—Alistipes, Blautia, Bilophila, Oscilibacter, and Proteobacteria—were under-represented phylogenetically within hCom1 (Figure S1). Moreover, most of the strains that invaded hCom2 had previously invaded hCom1, indicating that niche filling is deterministic. Importantly, the augmentation process caused relatively little perturbation to the structure of the existing community (notable exceptions are shown in Table S4), suggesting that it will result in a progressive improvement of the community. While the augmentation process can only fill niches that are conserved from mice to humans, the observation that most of our human strains engrafted suggests that many niches are conserved.
If we had broadened our strain inclusion criteria, there is a reasonable likelihood we could have improved colonization resistance further after just one round of augmentation. To further enhance niche filling and stability, it would help to subject hCom2 to further rounds of augmentation using fecal samples from additional donors, ideally in the presence of a varying diet. It might also be possible to improve niche occupancy, for example, in the setting of intestinal inflammation by performing the augmentation process in a murine model of inflammatory bowel disease.
There is a pressing need for a common model system for the gut microbiome that is completely defined and complex enough to capture much of the biology of a full-scale community. We showed that hCom2 is a reasonable starting point for such a system: in spite of its complexity, it colonizes mice in a highly reproducible manner. Moreover, hCom2 faithfully models the carrying capacity, immune cell profile, and metabolic phenotypes of humanized mice. There remain some modest differences in metabolic and immune profiles, and the community is still missing certain taxa that will likely be important to add. Nonetheless, taken together, our findings suggest that hCom2 is a reasonable starting point for a model of the gut microbiome.
One of the most interesting possibilities for such a system would be to enable reductionist experiments downstream of a community transplantation experiment (e.g., to identify strains responsible for a microbiome-linked phenotype). Although we did not identify the strains responsible for colonization resistance to EHEC, we did find that removing species of Proteobacteria or Firmicutes rendered the community EHEC-sensitive. Follow-up experiments in which one or several strains at a time are eliminated from the community could narrow further from the phylum level to individual strains. Efforts to identify the strains responsible for other microbiome-linked phenotypes including response to cancer immunotherapy, caloric harvest, and neural development, would be of great interest.
Limitations of the study
Our study has three important limitations. First, while Com2 is stable to challenge with the fecal communities used to augment it, it is less stable to challenge with unrelated fecal communities. These data suggest that subsequent rounds of backfill—using a variety of unrelated fecal samples in series or in parallel—is a promising path toward an even stabler variant of hCom2.
Second, it is unclear how many more bacterial strains (or other components) may be necessary to model the full functional capacity of a native human microbiome. Prior estimates of the number of species in a typical human microbiome range from ~150–300 (Faith et al., 2013; Kraal et al., 2014; Qin et al., 2010). Nonetheless, the observation that a defined community of just 119 strains exhibits remarkable stability bodes well for future efforts. We estimate that hCom2 is within 2-fold of native-scale complexity (STAR Methods), so a full-scale system is experimentally feasible. As a starting point for efforts to build such a system, hCom2 will provide a standard for assessing the genomic and functional completeness of model communities, with the ultimate goal of modeling native-scale human microbiomes.
Third, strain-level variation among communities underlies some of the phenotypic differences conferred on the host by the microbiome (Campbell et al., 2020; Jin et al., 2022; Marcobal et al., 2011; McNulty et al., 2011). hCom2 represents just one consortium of strains, so neither hCom2 nor any other single community can model the impact of strain-level variation on host phenotype. However, we think that a defined community is a promising starting point for probing strain-level differences: a collection of communities that are identical but harbor different strains of a species of interest would be an ideal way to probe the impact of strain variation—or even individual genes—on phenotype.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michael Fischbach (fischbach@fischbachgroup.org).
Materials availability
C. sporogenes strains are available on request. The strains used in this study are available from the sources listed in the Key Resources Table.
Data and code availability
Metagenomic and whole-genome sequencing datasets generated for this study are available at the Sequence Read Archive. The ninjamap code used in this study can be found at the following github location: https://github.com/FischbachLab/ninjaMap/releases/tag/cheng_et_al and the associated docker containers are available at https://hub.docker.com/repository/docker/fischbachlab/ninjamap.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions
Bacterial strains were selected based on HMP sequencing data (Kraal et al., 2014). We obtained all species from publicly available repositories; the mean relative abundance and prevalence of each strain were quantified using the 81 samples from healthy human patients from North America. The 166 strains that appeared in ≥37 of the 81 samples were considered for inclusion in the community. We were able to obtain 104 of these strains from public repositories and academic laboratories; the origin of each strain is listed in the Key Resources Table.
Preparation of synthetic community for storage and for experiments
For all community experiments, strains were cultured in anaerobic conditions (10% CO2, 5% H2, 85% N2) in 2-mL 96-well plates for 24–48 h in their respective growth media (Key Resources Table): Mega Medium (Wu et al., 2015) supplemented with 400 μM vitamin K2, or Chopped Meat Medium supplemented with Mega Medium carbohydrate mix and 400 μM vitamin K2. For strain storage, 200 μL of liquid culture were aliquoted 1:1 into sterile 50% glycerol in a 1-mL 96-well plate. The plate was covered with an airtight silicone fitted plate mat, edges were sealed with O2-impervious yellow vinyl tape, and the plate was frozen at −80 °C. Each storage plate includes 3–4 “sentinel” wells containing only growth medium that were used to monitor potential contamination during revival.
Preparation of synthetic community for in vitro experiments
From frozen stocks in 96-well plates, 100 μL of each strain were used to inoculate 900 μL of fresh autoclave-sterilized media of the appropriate type for each strain in 2.2-mL 96-well deep well plates (Thomas Scientific, Cat. #1159Q92). All culturing was done in an anaerobic chamber (Coy Laboratories) at 10% CO2, 5% H2, and 85% N2 atmosphere. Strains were diluted 1:10 every 24 h for 2 days into fresh growth medium in 2.2-mL deep well plates, and then diluted 1:10 into 4 mL of the appropriate medium in 5-mL 48-well deep well plates (Thomas Scientific, Cat. #1223T83). After 24 h, the optical density at 600 nm (OD600) of each well was measured. As the spectrophotometer does not accurately measure OD values >1, individual strain cultures were diluted 1:10 to quantify OD600. Stocks were diluted to a final OD600 of 0.1 using fresh growth medium. Equal volumes of each stock were pooled to create a 104-member synthetic community. The community was centrifuged at 5000 × g for 5 min, washed, and resuspended in an equivalent volume of PBS to generate the pooled community working stock. SAAC medium (Dodd et al., 2017) was made containing all amino acids at 1 mM concentration except for cysteine, which was added at 4.126 mM (Table S6). Twenty similar media were made in which one amino acid at a time was removed. 1.6 mL of each medium were aliquoted in triplicate and inoculated with the pooled community at 1:100 dilution. Four 100-μL aliquots of each culture were collected at 48 h and processed for metagenomic sequencing.
Preparation of synthetic community for in vivo experiments
For all germ-free mouse experiments, strains were cultured and pooled in the following manner: From frozen stocks in 96-well plates, 100 μL of each strain were used to inoculate 900 μL of fresh autoclave-sterilized media of the appropriate type for each strain in 2.2-mL 96-well deep well plates (Thomas Scientific, Cat. #1159Q92). All culturing was done in an anaerobic chamber (Coy Laboratories) at 10% CO2, 5% H2, and 85% N2 atmosphere. Strains were diluted 1:10 every 24 h for 2 days into fresh growth medium in 2.2-mL deep well plates, and then diluted 1:10 into 4 mL of the appropriate medium in 5-mL 48-well deep well plates (Thomas Scientific, Cat. #1223T83). After 24 h, the OD600 of each well was measured after diluting individual strain cultures 1:10. Based on these measurements of OD600 and enumeration of colony forming units (CFUs), we found that an OD600 of 1.3 corresponds to ~109 cells/mL for E. coli. Using this estimate, we pooled appropriate volumes of each culture corresponding to 2 mL at OD600=1.3, centrifuged for 5 min at 5000 × g, and resuspended the pellet in 2 mL of 20% glycerol that had been pre-reduced for at least 48 h. For each inoculum preparation cycle, up to 18 of the 119 strains did not reach OD600~1.3. For these strains, the entire 4-mL culture volume was used for pooling (the following paragraph contains details on these 18 strains). Volumes were scaled up accordingly if more inoculum was required for an experiment. Following pooling and preparation, 1.2 mL of the synthetic community were aliquoted into 2-mL Corning cryovials (Corning, Cat. #430659), removed from the anaerobic chamber, and transported to the vivarium where each vial was uncapped and its contents orally gavaged into mice within 1 min of uncapping. Each mouse received 200 μL of the mixed community inoculum. For the initial augmentation experiments, we used freshly prepared inoculum; for all subsequent experiments, the inoculum was frozen in cryovials at −80 °C. On the day of the experiment, the inoculum was defrosted and administered by oral gavage. The target for the inoculation procedure was that each mouse should receive ~108 cells of each bacterial strain in a 200 μL volume, for a total of ~1010 bacterial cells since hCom1 and hCom2 harbor 104 and 119 strains, respectively.
Eighteen of the 119 strains did not always grow to a high enough OD to match the post-dilution OD of the other strains. We added these mono-cultures undiluted to the mixed culture. Of these 18 strains, four never reached the target culture density (Ethanoligenens harbinense DSMZ 18485, Slackia heliotrinireducens DSM 20476, Ruminococcus albus strain 8, and Ruminococcus flavefaciens FD-1). The remaining 14 strains (Clostridium sp. L2–50, Clostridium sp. M62/1, Clostridium leptum DSM 753, Butyrivibrio crossotus DSM 2876, Blautia hydrogenotrophica DSM 10507, Veillonella dispar ATCC 17748, Collinsella stercoris DSM 13279, Megasphaera sp. DSMZ 102144, Prevotella buccae D17, Slackia exigua ATCC 700122, Adlercreutzia equolifaciens DSM 19450, Alistipes ihumii AP11, Burkholderiales bacterium 1_1_47, and Blautia sp. KLE 1732) exhibited variable growth. When they did not meet the target OD, we added the complete undiluted monoculture to the pooled community mixture.
Of note, normalization by OD can be fraught given differences in cell size and shape. A titration curve relating CFUs to optical density would be more accurate. However, even with the OD-based method we used, our community data were reproducible in vitro (Figure 1C-D) and in vivo (Figure 6B).
Collection and preservation of human fecal samples
For all experiments, human fecal samples were preserved in the same manner for inoculation into germ-free or hCom1/2-colonized mice. Specifically, freshly voided human feces was collected in a sterile container and transported into the anaerobic chamber within 5–10 min. The fecal sample was weighed, mixed 1:1 with an equivalent volume of pre-reduced PBS, and stored at −80 °C.
Preparation of human fecal samples
For human fecal challenge experiments, a fecal mixture was defrosted in the anaerobic chamber and diluted 1:100 into pre-reduced PBS. One milliliter was aliquoted into pre-reduced 2-mL Corning cryovials, removed from the anaerobic chamber, and transported to the vivarium, where each vial was uncapped and orally gavaged into mice within 1 min of uncapping. Each mouse received 200 μL of the bacterial mixture. Feces contains ~1011 colony forming units per gram of feces (Vandeputte et al. 2017); based on the dilutions performed, we estimate that each mouse received 108-1010 bacterial cells in the fecal challenge.
For all non-challenge fecal colonization experiments, the preserved fecal mixture was defrosted in the anaerobic chamber and diluted 1:2 into pre-reduced PBS. One millilter of the resulting mixture was aliquoted into pre-reduced 2-mL Corning cryovials, removed from the anaerobic chamber, and transported to the vivarium, where each vial was uncapped and orally gavaged into mice within 1 min of uncapping. Each mouse received 200 μL of the bacterial mixture, equivalent to 1010–1011 bacterial cells per mouse.
Preparation of 12Com
Cultures of the 12 strains in 12Com (Bacteroides thetaiotaomicron VPI-5482, Bacteroides caccae ATCC 43185, Bacteroides ovatus ATCC 8483, Bacteroides uniformis ATCC 8492, Bacteroides vulgatus ATCC 8482, Clostridium scindens ATCC 35704, Collinsella aerofaciens ATCC 25986, Dorea longicatena DSM 13814, Eggerthella lenta DSM 2243, Eubacterium rectale ATCC 33656, Parabacteroides distasonis ATCC 8503, and Ruminococcus torques ATCC 27756) were prepared in their respective growth media and propagated anaerobically for 24 h to OD600~1.3. Two milliliters of each strain were pooled, centrifuged for 5 min at 5000 × g, and the pellet was resuspended in 2 mL of 20% pre-reduced glycerol and frozen in 1-mL aliquots in 2-mL Corning cryovials.
Preparation of Enteromix
Six strains of non-pathogenic Escherichia coli (strains MITI 27, MITI 117, MITI 135, MITI 139, MITI 255, MITI 284) and one strain of Enterobacter cloacae (MITI 173) were isolated from the fecal sample of a healthy human donor by mass spectrometry-guided enrichment culture. Strains were stored at −80 °C in 25% glycerol. To prepare cultures for mouse colonization, strains were grown overnight in BHI broth (Fisher Scientific, Cat. # B99070), diluted 1:10 into 5 mL BHI broth, and cultured to OD600=1.3. Two milliliters of each strain were pooled, centrifuged for 5 min at 5000 × g, and the pellet was resuspended in 200 μL of 20% pre-reduced glycerol. One hundred microliters of this mixture were added to a tube containing 1 mL of previously prepared hCom2 or 12Com inoculum to create hCom2+Enteromix or 12Com+Enteromix, respectively. Each mouse was orally gavaged with 220 μL of the appropriate community. The estimated amount of each Enteromix strain administered to mice was 109 cells per 20 μL dose.
METHOD DETAILS
Metagenomic sequencing
The same experimental pipeline was used for sequencing bacterial isolates and synthetic communities. Bacterial cells were pelleted by centrifugation under anaerobic conditions. Genomic DNA was extracted using the DNeasy PowerSoil HTP kit (Qiagen) and quantified in 384-well format using the Quant-iT PicoGreen dsDNA Assay Kit (Thermofisher). Sequencing libraries were generated in 384-well format using a custom low-volume protocol based on the Nextera XT process (Illumina). Briefly, the concentration of DNA from each sample was normalized to 0.18 ng/μL using a Mantis liquid handler (Formulatrix). If the concentration was <0.18 ng/μL, the sample was not diluted further. Tagmentation, neutralization, and PCR steps of the Nextera XT process were performed on a Mosquito HTS liquid handler (TTP Labtech), leading to a final volume of 4 μL per library. During the PCR amplification step, custom 12-bp dual unique indices were introduced to eliminate barcode switching, a phenomenon that occurs on Illumina sequencing platforms with patterned flow cells (Sinha et al. 2017). Libraries were pooled at the desired relative molar ratios and cleaned up using Ampure XP beads (Beckman) to achieve buffer removal and library size selection. The cleanup process was used to remove fragments <300 bp or >1.5 kbp. Final library pools were quality-checked for size distribution and concentration using a Fragment Analyzer (Agilent) and qPCR (BioRad). Sequencing reads were generated using a NovaSeq S4 flow cell or a NextSeq High Output kit, in 2×150 bp configuration. 5–10 million paired-end reads were targeted for isolates and 20–30 million paired-end reads for communities.
Constructing high quality genome assemblies
We obtained the latest RefSeq (O’Leary et al., 2016) assembly for each strain in our community and assessed its quality based on contig statistics from Quast v. 5.0.2 (Gurevich et al., 2013) and SeqKit v. 0.12.0 (Shen et al., 2016), using GTDB-tk v. 1.2.0 (Chaumeil et al., 2019) for taxonomic classification. A ‘combination score’ was calculated as a linear combination of the completeness and contamination scores (completeness–5×contamination) derived from the CheckM v. 1.1.2 lineage workflow (Parks et al., 2015); such a score has been used previously, along with the metrics described here (https://gtdb.ecogenomic.org/faq#gtdb_selection_criteria), to include or exclude genomes in the GTDB release 89 database (Parks et al., 2018, 2020). Genomes that contained any number of Ns, >100 contigs, GTDB lineage warnings, multiple matches, or had CheckM completeness <90, contamination >10, and combination score <90 were resequenced and reassembled.
Our hybrid assembly pipeline contains a workflow for de novo and reference-guided genome assembly using both Illumina short reads and PacBio or Nanopore long reads. The workflow has three main steps: read pre-processing, hybrid assembly, and contig post-processing. Read pre-processing included 1) quality trimming/filtering (bbduk.sh adapterFile=“adapters,phix” k=23, hdist=1, qtrim=rl, ktrim=r, entropy=0.5, entropywindow=50, entropyk=5, trimq=25, minlen=50), with adaptors and phix removed with kmer right trimming, kmer size of 23, Hamming distance 1 (allowing one mismatch), quality trimming of both sides of the read, filtering of reads with an average entropy <0.5 with entropy kmer length of 5 and a sliding window of 50, trimming to a Q25 quality score, and removal of reads with length <50 bp; 2) deduplication (bbdupe.sh); 3) coverage normalization (bbnorm.sh min=3) such that depth <3x was discarded; 4) error correction (tadpole.sh mode=correct); and 5) sampling (reformat.sh). All pre-processing was carried out using BBtools v. 38.37 for short reads. For long reads, we used filtlong v. 0.2.0 (fitlong --min_length 1000 --keep_percent 90 --length_weight 10) to discard any read <1 kb and the worst 10% of read bases, as well as to weigh read length as more important when choosing the best reads. Hybrid assembly was performed by Unicycler v. 0.4.8 (Wick et al., 2017) with default parameters using pre-processed reads. After assembly, the contigs from the assembler were scaffolded by LRScaf v. 1.1.9 (Qin et al., 2018) with default parameters. If the initial assembly did not produce the complete genome, gaps were filled by long reads TGS-GapCloser v. 1.0.1 (Xu et al., 2019) with default parameters.
If no long reads were available, short paired-end reads were assembled de novo using SPAdes v. 3.13.1 (Bankevich et al., 2012) with the --careful option to reduce the number of mismatches and short indels during assembly of small genomes. Assembly quality was assessed based on the CheckM v. 1.1.2 lineage. If contamination was detected, contigs corresponding to the genome of interest were extracted from the contaminated assembly using MetaBAT2 v. 2.2.14 (Kang et al., 2019) with default parameters.
Finally, the assembled genomes were evaluated using the same criteria as the RefSeq assemblies, and the assembly for each species with the best overall quality metrics was chosen as the reference assembly. This procedure resulted in the replacement of eight genomes: two from a PacBio/Illumina hybrid assembly, one from a Nanopore/Illumina hybrid assembly, one from a reference-guided Illumina assembly, and four from short-read assemblies of the respective isolate samples followed by binning (Table S2).
Generating and normalizing the NinjaMap database
The first step in the pipeline was to assess the uniqueness of each genome in the community. We generated error-free in silico reads such that each genome was uniformly covered at 10x depth. Each such genome read set was aligned to all genomes in the community. The uniqueness of a genome was defined as the fraction of the genome that did not have reads cross-mapped from another strain; uniqueness values were between 0 and 1, such that more unique genomes have a value closer to 1. The uniqueness value of a strain was used to normalize its final relative abundance in any community sample. All genome sequences were combined into one fasta file and a Bowtie2 v. 2.3.5.1 (Langmead and Salzberg, 2012) index was computed for future alignments. The database and strain weights were recomputed each time the community or a genome was updated.
NinjaMap alignment scoring
A primary goal of the NinjaMap algorithm is to analyze and tabulate every input read. A successful match was defined as a read aligned to a genome at 100% identity across 100% of the read length. If a read was uniquely matched to a single strain, its mate pair was also recruited as long as it had at least one match to the same strain. If exactly 1 strain was a perfect match for both reads, the pair was considered a “primary pair” and a score of 1 was given for each read. If >1 or 0 strains were a match for both reads, both reads were placed in escrow and analyzed separately as described below.
By prioritizing paired-read scoring, noise was significantly reduced while ensuring that as many reads as possible were considered for abundance estimates. Once preliminary strain abundances were calculated based on primary pairs, reads in escrow were then assigned fractionally to the strains to which they aligned perfectly. The fractional assignment was calculated based on the primary read abundances of each strain, normalized by the size of the unique region of each genome within the database, such that the total contribution for a read was 1. In some cases, an individual escrowed read matched to a strain without any matches to primary pairs; such reads were discarded and not used in the final estimates.
Finally, the total score for each strain in the database was normalized by the number of reads that aligned to the database, so that the relative abundances of all strains summed to 1.
Generating simulated sequencing reads
In silico data were generated to evaluate the Ninjamap algorithm in the absence of genome assembly errors and sequencing quality issues. Grinder v. 0.5.4 (Angly et al., 2012) was applied to each genome to generate error-free reads with the following parameters: -read_distribution 140, -insert_size 800, -mate_orientation FR, -delete_chars ‘-~*NX’, -mutation_dist uniform 0, -random_seed 1712, abundance_model uniform, -qual_levels 33 31, -fastq_output 1. The -coverage_fold parameter was adjusted based on the cases described below.
Uniform abundance isolate dataset
This dataset was created to test the sensitivity and specificity of the algorithm against our database of genomes. In silico data were generated for each genome with uniform coverage of 10x or 100x.
Variable abundance community dataset
In silico reads were generated for each genome at 10x, 0.1x, and 0.001x uniform coverage. Three datasets of mixed community reads were generated including every genome at a coverage randomly selected from the three levels. The observed relative abundance of each genome in our database was calculated using the NinjaMap algorithm and compared to the expected relative abundance based on coverage level, which ranged from ~3×10−6 to 0.03.
Augmenting the NinjaMap database
The additional genomes added to hCom1 to create hCom2 were evaluated using the same criteria as the RefSeq assemblies, and the assembly for each species with the best overall quality metrics was chosen as the reference assembly. This procedure resulted in the replacement of 85 genomes: two obtained from a PacBio/Illumina hybrid assembly, 69 from a Nanopore/Illumina hybrid assembly, one from a reference-guided Illumina assembly, and seven from short-read assemblies of the respective isolate samples followed by binning (Table S2).
Metagenomic read mapping
Paired-end reads from each sample were aligned to the hCom1 or hCom2 database using Bowtie2 with maximum insert length (-maxins) set to 3000, maximum alignments (-k) set to 300, suppressed unpaired alignments (--no-mixed), suppressed discordant alignments (--no-discordant), suppressed output for unaligned reads (--no-unal), required global alignment (--end-to-end), and using the “--very-sensitive” alignment preset (command: --very-sensitive -maxinsX 3000 -k 300 --no-mixed --no-discordant --end-to-end --no-unal). The output was piped into Samtools v. 1.9 (Li et al., 2009), which was used to convert the alignment output from SAM output stream to BAM format and then sort and index the BAM file by coordinates. Alignments were filtered to only keep those with >99% identity for the entire length of the read.
The median percentage of unaligned reads was 4.95% (range 4.10%−8.35%). To assess the origin of these reads, we performed a BLAST v. 2.11.0+ search through the ncbi/blast:latest docker image with parameters “-outfmt ‘6 std qlen slen qcovs sscinames staxids’ -dbsize 1000000, -num_alignments 100” from a representative sample against the ‘NCBI - nt’ database from 2021–02-16. We then filtered the BLAST results to obtain the top hits for a given query. Briefly, the script defined top hits as ones that had an e-value ≤1e-30, percent identity ≥99% and were within 10% of the best bit score for that query. To visualize and summarize the output, we used the ktImportTaxonomy script from the Krona package with default parameters. Reads were aggregated by NCBI taxon ID and separately by genus. We found that most of the hits were from taxa that are closely related to the organisms in our community, while others were from the mouse genome. We conclude that our experiments did not suffer from any appreciable level of contamination.
Sensitivity of NinjaMap
Our data provide several quantitative estimates of the sensitivity of NinjaMap: First, when considering the mismapping of sequencing data for a single isolate to other strains, error rates were typically 10−5-10−4 for both simulated and actual (Data S2) data. The expected contribution to relative abundance from mismapping in a community as calculated from the mismapping rates of isolates was also typically ~10−5-10−4 (Data S2). Thus, for a strain in a 100-member community with average relative abundance of 10−2, the contribution to relative abundance from mismapping is likely to be even lower (10−7-10−6).
Second, in strain dropout experiments that are not included in this version of manuscript, strains with average relative abundance ~10−5 (e.g., A. stercorihominis, S. heliotrinireducens, C. stercoris, A. putredinis), displayed similar coefficients of variation (standard deviation/mean) as more abundant strains, indicating that noise to due to mismapping was small. In addition, these strains were not detected by Ninjamap in their own dropouts, indicating that the sensitivity to them was well below 10−5. The maximum level of a strain in its own dropout that we think is real signal is 10−6.
Third, as our in silico data show (Data S2), mismapping does occur (for instance, due to inaccuracies in some genome assemblies such that a missing/contaminated sequence will result in the strain 1 assembly mapping to other strains that contain those sequences). In most cases we expect, based on our isolate sequencing data, that mismapping will contribute a very low fraction of a species’ reported relative abundance.
With those estimates in mind, we have set a permissive lower threshold for the NinjaMap data (10−7) and have adjusted all of our plots to make that the lower limit. We acknowledge that it is possible, in rare cases, for an abundant strain that displays an unusually high degree of mismapping to introduce noise that would interfere with real low-abundance strain signals. We expect that this problem will abate as some of our lower-quality genome assemblies are improved.
Amino acid dropout experiment and data analysis
Strains were passaged by diluting 1:10 into fresh growth medium every 24 h for 2–3 days. The day before amino acid dropout experiments, cultures were diluted 1:10 into 1 mL of fresh medium and grown for 24 h as inoculation working stocks. Strains were diluted 1:10 into 150 μL of the appropriate culture medium and a plate reader was used to measure absorbance at 600 nm. Stocks were diluted to a final OD600 of 0.1 using fresh growth medium. If a culture did not reach an OD600 of 0.1, the entire culture was used as the working stock for community assembly. Equal volumes of each stock were pooled to create a 104-member synthetic community. The community was centrifuged at 5000 × g for 5 min, washed, and resuspended in an equivalent volume of PBS to generate the pooled community working stock. SAAC medium (Dodd et al., 2017) was made containing all amino acids at 1 mM concentration except for cysteine, which was added at 4.126 mM (Table S5). Twenty similar media were made in which one amino acid at a time was removed. 1.6 mL of each medium were aliquoted in triplicate and inoculated with the pooled community at a 1:10 or 1:100 dilution. Four 100-μL aliquots of each culture were collected at 48 h and processed for metagenomic sequencing.
Read fractions were rescaled to sum to 1, thereby reflecting the relative abundances of reads mapped to one of the 104 genomes in our database. The effect of removal of an amino acid on a strain was estimated by calculating the z score , where Rk,j is the log10(relative abundance) of strain k in sample j and μk and σk are the mean and standard deviation, respectively, of log10(relative abundance) for strain k across all samples except the cysteine dropout. The cysteine dropout sample was excluded from the calculation of μk and σk because this sample was an obvious outlier. We expect that the outlier effect of cysteine dropout is likely due to its role in maintaining redox balance. We used z-scores rather than a direct comparison to the complete medium because most strains exhibited only small variations in relative abundance in most conditions. Data points that could be explained by mismapping were removed. Putative interactions were identified based on |zj,k|>2, i.e. amino acid dropouts that changed the log10(relative abundance) of strain k by ≥2 standard deviations relative to its mean. A few strains varied in relative abundance by several orders of magnitude; as a result, σk was large, so putative interactions would be missed using z-scores.
To identify clusters of strains that responded similarly or amino acids that elicited a similar response, we normalized Rk,j for each strain across samples by subtracting μk and performed hierarchical clustering of both strains and amino acid dropouts on a dataset including strains that were detected in all 20 amino acid dropout samples and in complete SAAC medium.
Constructing C. sporogenes mutants
C. sporogenes deletion mutants were constructed using a previously reported protocol (Guo et al., 2019); the strains and primers used for each mutant are listed in Table S6. In brief, from plasmids CS_OTC and CS_ADI, which harbor targeting and repair templates unique to each gene, we amplified DNA sequences encoding the gRNA locus (the gRNA plus adjacent elements and the repair template) and ligated the amplicon into the pMTL82254 backbone. These repair templates consist of 700- to 1200-bp sequences flanking the 40- to 100-bp sequence targeted for excision.
To construct the Δadi strain, a gRNA fragment was purchased from Quintara and amplified with primers fwd_pMTL82254_NotI and rev_gRNA_flank1. The two flanking regions were amplified from C. sporogenes genomic DNA using the primers 5rev_flank1 and 5fwd_flank1_flank2 for flank 1 and 5rev_flank1_flank2 and 5fwd_flank1_flank2 for flank 2. Next, the flanking regions were joined by amplifying with primers fwd_gRNA_flank1 and rev_flank2. The amplified gRNA fragment was attached to the joined flank construct by amplifying with primers fwd_pMTL82254_NotI and rev_pMTL82254_AscI. Finally, the pMTL82254 plasmid and the construct containing the gRNA, flank1, and flank2 regions were digested with NotI and AscI and ligated with T4 ligase (NEB). The final construct was named CS_ADI.
To make the Δotc strain, the gRNA fragment was purchased from Quintara and amplified with fwd_pMTL82254_NotI and rev_OTC_gRNA_flank1. The two flanking regions were amplified from C. sporogenes genomic DNA using the primers fwd_OTC_gRNA_flank1 and rev_OTC_flank1_flank2 for flank 1 and fwd_OTC_flank1_flank2 and rev_OTC_flank2 for flank 2. Next, the flanking regions were joined by amplifying with the primers fwd_OTC_gRNA_flank1 and rev_OTC_flank2. The amplified gRNA fragment was attached to the joined flank construct by amplifying with fwd_pMTL82254_NotI and rev_pMTL82254_AscI. Finally, the pMTL82254 plasmid and the construct containing the gRNA, flank1, and flank2 regions were digested with NotI and AscI and ligated with T4 ligase (NEB). The final construct was named CS_OTC.
CS_OTC or CS_ADI was electroporated into Escherichia coli S17 cells and conjugated into C. sporogenes strain ATCC 15579 using a previously described method (Guo et al. 2019). In brief, a single colony of wild-type C. sporogenes was used to inoculate 2 mL of TYG broth (3% (w/v) tryptone, 2% (w/v) yeast extract, 0.1% (w/v) sodium thioglycolate) and incubated anaerobically in an atmosphere consisting of 10% CO2, 5% H2, and 85% N2. E. coli S17 cells with CS_OTC or CS_ADI were grown in LB broth supplemented with 250 μg/mL erythromycin at 30 °C with shaking at 225 rpm. After 17–24 h, 1 mL of this culture was centrifuged at 1000 × g for 1 min and washed twice with 500 μL of PBS (40 mM potassium phosphate, 10 mM magnesium sulfate, pH 7.2). The pellet was transferred into the anaerobic chamber and 250 μL of C. sporogenes overnight culture were added and mixed with the cell pellet. Thirty-microliter aliquots of the mixture were plated on a pre-reduced TYG agar plate in eight spots. The plate was tilted to coalesce the spots and incubated for 24 h. Biomass from the plate was scraped using a sterile inoculation loop and suspended in 250 μL of pre-reduced PBS. One hundred microliters of the cell suspension were plated on TYG agar containing 10 μg/mL erythromycin and 250 μg/mL D-cycloserine to isolate single colonies. One colony was picked, sequence verified, and used as the starting point for the next conjugation.
In the second conjugation, E. coli S17 cells containing pMTL83153_fdx_Cas9 were grown in LB broth supplemented with 25 μg/mL chloramphenicol at 30 °C with shaking at 225 rpm. After washing, the pellet was moved into the anaerobic chamber and 250 μL of an overnight culture of C. sporogenes harboring the CS_OTC vector were thoroughly mixed with the E. coli cell pellet. Thirty-microliter aliquots of the mixture were plated on a pre-reduced TYG agar plate in eight spots. The plate was tilted to coalesce the spots and incubated for 72 h. Biomass from the plate was scraped using a sterile inoculation loop and resuspended in 250 μL of pre-reduced PBS. One hundred microliters of the cell suspension were plated on each of two pre-reduced TYG agar plates containing 10 μg/mL erythromycin, 15 μg/mL thiamphenicol, and 250 μg/mL D-cycloserine. C. sporogenes colonies typically appeared after 36–48 h, and 8–10 colonies were re-streaked on pre-reduced TYG agar plates containing 10 μg/mL erythromycin, 15 μg/mL thiamphenicol, and 250 μg/mL D-cycloserine to isolate single colonies. The isolated colonies were used to inoculate pre-reduced TYG broth supplemented with 10 μg/mL erythromycin and 15 μg/mL thiamphenicol, and genomic DNA was isolated using a Quick DNA fungal/bacterial kit (Zymo Research). Primers ADI_532_fwd and ADI_22_rev or OTC_5_up_fwd and OTC_930_down_rev (Table S6) were used to verify deletions.
ATP assay
An aliquot from a frozen stock of C. sporogenes was used to inoculate 5 mL of TYG broth and grown to stationary phase (~24 h). Cells were diluted 1:1000 into 20 mL of TYG broth and grown to late-log phase (~16 h). Cells were harvested by centrifugation (5,000 × g for 10 min at 4 °C) and washed twice with 20 mL of pre-reduced PBS. One hundred microliters of cells were seeded into rows of a 96-well microtiter plate (12 wells per condition). Two hundred microliters of pre-reduced 2 mM substrate (arginine) in phosphate washing buffer, or 200 μL of buffer alone, were dispensed into rows of a separate 96-well microplate. At t=0, 100 μL of substrate or buffer were added to the cells and mixed gently by pipetting. At t=−5 min, −1 min, 30 s, 1 min, 2 min, 5 min, 10 min, 20 min, 30 min, 45 min, 60 min, and 90 min, 10 μL of cells were extracted and mixed with 90 μL of DMSO to quench the reaction and liberate cellular ATP. For the time points t=−5 min and −1 min (prior to the addition of buffer or substrate), 5 μL of cell suspension were harvested and 5 μL of either buffer or substrate were added to the cell-DMSO mixture to bring the total volume to 100 μL. The ATP content from 10 μL aliquots of lysed cells was measured using a luminescence-based ATP determination kit (Invitrogen, Cat. #A22066). Absolute ATP levels were calculated using a calibration curve with known concentrations of ATP.
Reproducibility and colonization experiments
Groups of five 6- to 8-week-old female germ-free SW mice were colonized for 4 weeks with hCom1 or hCom2 and fecal pellets were sampled after 4 weeks. These fecal pellets were subjected to DNA extraction, metagenomic sequencing, and NinjaMap read mapping to estimate strain relative abundances.
Augmentation experiment
Individual strains were cultured in their respective media (Key Resources Table), normalized, and pooled to form the synthetic community as described in ‘Preparation of bacterial synthetic community.’ Mice were orally gavaged with a freshly prepared culture of the synthetic community three days in a row and were sampled weekly for 4 weeks. After 4 weeks, mice were orally gavaged with fecal sample from one of three healthy human donors (one donor per 5 mice) or PBS as a control.
For the fecal challenge experiment with samples Hum4–6, mice were orally gavaged only once with a frozen, then thawed culture of hCom2.
MIDAS analyses
MIDAS (Nayfach et al., 2016) was run using the database v. 1.2 with default parameters on each library. To determine which invading species to use in augmenting hCom1, a relative abundance threshold of 10−4 and minimum read count of 2 were applied. A species was selected to augment hCom1 if it was present above the threshold in ≥2 of the 3 challenge groups. For all other analyses, the MIDAS output was used without any filtering (STAR Methods).
MIDAS sensitivity analysis
To determine the sensitivity of MIDAS for analyses of strains in our communities, we generated error-free 150-bp paired-end reads in silico for each genome. Each simulated read set was individually processed by MIDAS. While most genomes were identified correctly and assigned to a single MIDAS bucket, 22 strains from hCom1 and hCom2 cross-mapped to multiple buckets. As expected, MIDAS was unable to separate closely related strains, with 14 MIDAS buckets from hCom1 and 17 from hCom2 recruiting reads from more than one strain (Table S7).
Analyzing strain displacement versus persistence
To determine the coverage of genomes from hCom1 and hCom2 in week 8 samples after a fecal challenge, reads were aligned to two Bowtie2 databases, hCom1 (version SCv1.2) and hCom2 (version SCv2.3). Each alignment file was filtered to only include alignments with 99% or 100% identity at 100% alignment length. Alignments at 99% identity were performed to recruit reads from any strain that was very similar but not identical. The breadth of coverage (i.e., the percentage of the genome covered by at least 1 read) and the depth of coverage (the average number of reads covering positions in the genome) was calculated for each organism in each sample at both identity thresholds.
Results from the MIDAS analysis of each sample were combined with MIDAS bucket strain contributions from the sensitivity analysis and strain coverage metrics. Most of the high abundance strains had high coverage depth and breadth of coverage at 99% and 100% identity, suggesting that the original strains (or highly similar variants) were present in the samples at week 8.
Bacterial load estimates
Six to 8-week-old female germ-free SW mice were colonized for 4 weeks with hCom1, hCom2, or one of two human fecal samples, and fecal pellets were sampled after 4 weeks. Female germ-free and conventional SW mice of the same age were sampled at the same time. Each colonization cohort contained 5 mice. For each mouse, two fecal pellets were collected in a pre-weighed 1.5-mL Eppendorf tube containing 200 μL of transport medium. After collection and weighing, the mass of the tube prior to sampling was subtracted to calculate fecal weight. Samples were transferred into the anaerobic chamber and each pellet was crushed with a 1-mL pipette tip and vortexed at maximum speed for 30 s to create a homogenous mixture. This mixture was serially diluted 1:10 twelve times; each dilution was plated on pre-reduced Columbia blood agar plates and incubated at 37 °C. After 24 h, colonies were counted for each dilution. Fecal pellets were also subjected to DNA extraction, metagenomic sequencing, and NinjaMap analysis to estimate strain relative abundances.
Immune profiling
Six to 8-week-old female germ-free C57BL/6 mice were colonized for 2 weeks with hCom2, a human fecal sample, or PBS as a negative control and fecal pellets were collected after 2 weeks. Mice were then sacrificed, colonic tissue was dissected, and immune cells were isolated using the Miltenyi Lamina Propria kit and Gentle MACS dissociator. Immune cells were stained using the antibodies listed in the Key Resources Table at 1:200 dilution and assessed using a LSRII flow cytometer. Fecal pellets were subjected to DNA extraction, metagenomic sequencing, and NinjaMap analysis to estimate strain relative abundances.
Metabolomics
Cohorts of 6–8-week-old female germ-free SW mice were colonized for 4 weeks with hCom1, hCom2, or one of two human fecal samples. Urine and fecal pellets were sampled after 4 weeks. Female germ-free and conventional SW mice of the same age were sampled at the same time. Fecal pellets were subjected to DNA extraction, metagenomic sequencing, and NinjaMap analysis to estimate strain relative abundances.
Sample preparation for LC/MS analysis
For urine samples, 5 μL of urine were diluted 1:10 with ddH2O and mixed with 50 μL of internal standard water solution (20 μM 4-chloro-L-phenylalanine and 2 μM d4-cholic acid). After centrifugation for 15 min at 4 °C and 18,000 × g, 50 μL of the resulting mixture were used for quantification of creatinine using a Creatinine Assay Kit (Abcam, Cat. #ab204537) as described in the manufacturer’s protocol. The remaining 50 μL were filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), and 5 μL were injected into the LC/MS.
For fecal pellets, ~40 mg wet feces were pre-weighed into a 2-mL screw top tube containing six 6mm ceramic beads (Precellys® CK28 Lysing Kit). Six hundred microliters of a mixture of ice-cold acetonitrile, methanol, and water (4/4/2, v/v/v) were added to each tube and samples were homogenized by vigorous shaking using a QIAGEN Tissue Lyser II at 25 Hz for 10 min. The resulting homogenates were subjected to centrifugation for 15 min at 4 °C and 18,000 × g. One hundred microliters of the supernatant were combined with 100 μL of internal standard water solution (20 μM 4-chloro-L-phenylalanine and 2 μM d4-cholic acid). The resulting mixtures were filtered through a Durapore PVDF 0.22-μm membrane using Ultrafree centrifugal filters (Millipore, UFC30GV00), or a MultiScreen Solvinert 96 Well Filter Plate (Millipore, MSRLN0410), and 5 μL were injected into the LC/MS.
Liquid chromatography/mass spectrometry (LC/MS)
For aromatic amino acid metabolites, analytes were separated using an Agilent 1290 Infinity II UPLC equipped with an ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 mm × 150 mm, Waters Cat. #186002352 and #186003975) and detected using an Agilent 6530 Q-TOF equipped with a standard atmospheric-pressure chemical ionization (APCI) source or dual Agilent jet stream electrospray ionization (AJS-ESI) source operating under extended dynamic range (EDR 1700 m/z) in negative ionization mode. For the APCI source, the parameters were as follows: gas temperature, 350 °C; vaporizer, 350 °C; drying gas, 6.0 L/min; nebulizer, 60 psig; VCap, 3500 V; corona, 20 μA; and fragmentor, 135 V. For the AJS-ESI source, the parameters were as follows: gas temperature, 350 °C; drying gas, 10.0 L/min; nebulizer, 40 psig; sheath gas temperature, 300 °C; sheath gas flow, 11.0 L/min; VCap, 3500 V; nozzle voltage, 1400 V; and fragmentor, 130 V. Mobile phase A was H2O with 6.5 mM ammonium bicarbonate, and B was 95% MeOH with 6.5 mM ammonium bicarbonate. Five microliters of each sample were injected via autosampler into the mobile phase, and chromatographic separation was achieved at a flow rate of 0.35 mL/min with a 10 min gradient condition (t=0 min, 0.5% B; t=4 min, 70% B; t=4.5 min, 98% B; t=5.4 min, 98% B; t=5.6 min, 0.5% B).
For bile acids, compounds were separated using an Agilent 1290 Infinity II UPLC equipped with a Kinetex C18 column (1.7 μm, 2.1 mm × 100 mm, Phenomenex, Cat. #00D-4475-AN) and detected using an Agilent 6530 Q-TOF equipped with a dual Agilent jet stream electrospray ionization (AJS-ESI) source operating under extended dynamic range (EDR 1700 m/z) in negative ionization mode. The parameters of the AJS-ESI source were as follows: gas temperature, 300 °C; drying gas, 7.0 L/min; nebulizer, 40 psig; sheath gas temp, 350 °C; sheath gas flow, 10.0 L/min; VCap, 3500 V; nozzle voltage, 1400 V; and fragmentor, 200 V. Mobile phase A was H2O with 0.05% formic acid, and B was acetone with 0.05% formic acid. Five microliters of each sample were injected via autosampler into the mobile phase and chromatographic separation was achieved at a flow rate of 0.35 mL/min with a 32 min gradient condition (t=0 min, 25% B; t=1 min, 25% B; t=25 min, 75% B, t=26 min, 100% B, t=30 min, 100% B, t=32 min, 25% B).
Online mass calibration was performed using a second ionization source and a constant flow (5 μL/min) of reference solution (119.0363 and 966.0007 m/z). The MassHunter Quantitative Analysis Software (Agilent, v. B.09.00) was used for peak integration based on retention time (tolerance of 0.2 min) and accurate m/z (tolerance of 30 ppm) of chemical standards. Quantification was based on a 2-fold dilution series of chemical standards spanning 0.05 to 100 μM (aromatic amino acid metabolites) or 0.001 to 100 μM (bile acids) and measured amounts were normalized by weights of extracted tissue samples (pmol/mg wet tissue) or creatinine level in the urine sample (μM/mM creatinine). The MassHunter Qualitative Analysis Software (Agilent, version 7.0) was used for targeted feature extraction, allowing mass tolerances of 30 ppm.
E. coli colonization resistance
6–8-week-old female germ-free SW mice were orally gavaged with 200 μL of hCom1, hCom2, a fecal sample from a healthy human donor, or 12Com, or with 220 μL of hCom2+Enteromix or 12Com+Enteromix, and fecal pellets were sampled weekly for 4 weeks. After 4 weeks, mice were orally gavaged with a 200-μL mixture containing 109 CFUs of EHEC and fecal pellets were sampled on days 0 (pre-EHEC infection), 2, 4, 6, and 14. After collection, all fecal samples were prepared aerobically. Specifically, fecal pellets were weighed and 10X (w/v) PBS was added to the tube. Each pellet was crushed with a 1-mL pipette tip and vortexed at maximum speed for 30 s to create a homogenous mixture. This mixture was serially diluted 1:10 six successive times and 5 μL of each dilution were plated on McConkey-Sorbitol agar. Plates were incubated at 37 °C for 16–18 h. The resulting colonies were enumerated and verified to be EHEC by metagenomic sequencing. Fecal pellets were also subjected to DNA extraction, metagenomic sequencing, and NinjaMap analysis to estimate strain relative abundances.
Estimation that hCom2 is within two-fold of native-scale complexity
We came to this estimate in two ways, both of which have important caveats but generally support our claim.
A compilation of estimates from the literature.
Historic (1970–1980s) estimates were based on traditional culture-based techniques (Guarner and Malagelada, 2003). For example, Moore et al attempted to Gram-stain and culture (aerobically and anaerobically) all of the organisms from 20 healthy human stool samples (Holdeman, 1975). This attempt yielded 1147 unique strains and 113 morphologically and metabolically distinct organisms, which (per their statistical estimate) accounted for 94% of the viable cells in volunteer stool biomass.
More recent metagenomic sequencing analyses have expanded upon these diversity estimates. One study performed metagenomic sequencing on 124 European volunteers with species-level resolution, and uncovered 1000–1150 unique bacterial species, 18 of which were detected in all individuals, 57 in ≥90% and 75 in ≥50% of individuals (the authors termed these the ‘common bacterial core species’) (Qin et al., 2010). An analysis of the human microbiome metagenomic sequencing database involving 81 healthy US volunteers with strain-level resolution showed that there were 79 shared strains in 100% of individuals and 525 unique strains (Kraal et al., 2014). Interestingly an analysis of the supplemental data showed that the 79 shared strains from the analysis in (Kraal et al., 2014) encompass all 75 strains of the set of “common bacterial core species” in (Qin et al., 2010). Further analysis of the supplemental information and tables from (Kraal et al., 2014) showed that metagenomic sequencing uncovered 108–348 unique strains per individual.
These metagenomic observations have been recapitulated with 16S sequencing. Faith et al performed low-error amplicon 16S sequencing (LEA-Seq) of the V4 region in combination with metagenomic sequencing of 37 stool microbiomes from healthy US individuals (Faith et al., 2013). This study had strain-level resolution, and review of the supplemental information and tables showed that study individuals harbored 195–243 unique strains; the authors posited that “…on average 60% of the approximately 200 microbial strains harbored in each adult’s intestine is retained in their host over the course of a five-year sampling period.”
The caveats of these estimates are that three elements varied in each case: 1) the samples assessed, 2) the methods used to make the estimate, and 3) the level of resolution at which a taxon was called. Thus, the literature examples lack internal consistency.
Our own estimate.
Using MIDAS, we performed an analysis of the average number of species-level bins in each of the samples included in this study, as shown below:
| Sample | Number of MIDAS bins |
|---|---|
| hCom1 | 59 |
| hCom2 | 79 |
| H1-FMT (humanized mice) | 85 |
| H2-FMT (humanized mice) | 87 |
| H3-FMT (humanized mice) | 94 |
| H1-fecal (fecal sample) | 145 |
| H2-fecal (fecal sample) | 199 |
| H3-fecal (fecal sample) | 180 |
The number of MIDAS bins identified in fecal samples from mice colonized with hCom1 or hCom2 was between 63% (59/94) and 93% (79/85) of the number of MIDAS bins in mice colonized with Hum1–3, and between 30% (59/199) and 54% (79/145) of the number of MIDAS bins in Hum1–3 fecal samples.
The most important caveat of this analysis is that it is based on the taxonomic resolution of a MIDAS ‘bin’, which corresponds roughly to the species level. As a consequence, strain-level variation (including multiple strains of a species) is not taken into account, and any species that are not present in the MIDAS database are not counted.
Having noted those caveats, both estimates are consistent with the view that hCom2 is within ~2-fold of the species-level complexity of a native community.
QUANTIFICATION AND STATISTICAL ANALYSIS
For the analysis of communities in vitro, the statistical details of experiments can be found in the figure legends. Reported n values are the total samples (cultures) per group. Unless otherwise stated, p-values were not corrected for multiple hypothesis testing. Benjamini-Hochberg corrections, hypergeometric tests, Student’s t-tests (unpaired or two-tailed), and Kruskal-Wallis tests were performed in MATLAB.
For the analysis of communities in vivo, relative abundances were calculated from the output of NinjaMap or MIDAS without rarefying the total number of reads across samples. Relative abundances at each time point were averaged across the 4–5 mice that were co-housed in the same isolator and subjected to the same fecal challenge. Correlation coefficients were calculated after setting undetected bins to a minimum value (10−6 and 10−7 for MIDAS and NinjaMap, respectively) and performing a log10 transformation. Mice were not considered in fecal challenge analyses if sequence reads in a sample from any week were of poor quality or abnormally variable. This filtering affected one of five mice in all groups except for fecal challenge experiment 1, Hum3 (2 mice affected) and fecal challenge experiment 2, Hum1 (0 mice affected). Further details of statistical analyses can be found in the corresponding figure legends. All statistical analyses and tests were performed in MATLAB, and scripts for analyses are available at https://github.com/FischbachLab.
Supplementary Material
Data S5. hCom2 as a model system, related to Figure 6.
Data S3: Discovery and elucidation of a strain-amino acid interaction and its molecular mechanism, related to Figure 2.
Table S7: MIDAS sensitivity analysis, related to Figure 4 and 5.
MIDAS species bins (sample_id) and associated hCom1 and hCom2 strain abundances. Sensitivity was determined by processing simulated metagenomic sequencing reads for each hCom1 or hCom2 strain using MIDAS and assessing the distribution of reads among MIDAS species bins.
Data S2: Standardization and benchmarking of NinjaMap, related to STAR Methods.
Data S1: Composition and function of hCom1 and hCom2, related to Figure 1.
Table S6: Strains and primers used to generate Clostridium sporogenes adi and otc mutants, related to Figure 2.
Table S5: Modified Standard Amino Acid Complete (SAAC) medium recipe, related to Figure 2.
SAAC media is a minimal medium commonly used for assessment of bacterial metabolism of amino acids (Dodd et al., 2017). SAAC complete contains all amino acids at 1 mM concentration, with the exception of cysteine at 4.126 mM.
Table S2: Genome assemblies from strains in the synthetic communities hCom1 and hCom2, related to Figure 1.
Unique genome assemblies utilized or completed for this manuscript, with the type of assembly and the libraries involved. This procedure resulted in the replacement of eight genomes: two obtained from a PacBio and Illumina hybrid assembly and six from short-read assembly of the respective isolate samples followed by binning.
Table S3: Strain relative abundances in amino acid dropout experiments, related to Figure 2 and S2.
Relative abundances were measured after 48 h of community outgrowth in each dropout media. The data shown are from one of two biological replicates.
Table S4: Invading strains from the two fecal challenge augmentation experiments and hCom1 species whose relative abundance was highly impacted by fecal challenge, related to Figure 4, 5, and Data S1.
For each augmentation iteration, strains that invaded from ≥1 of 3 donor stools are shown. Also shown are the seven hCom1 strains that were left out of hCom2 as they did not persist after fecal challenge. For impacted strains from hCom1, red MIDAS bins denote species that changed by >10-fold in response to >1 fecal challenge.
Table S1: Omitted strains, related to Figure 1 and S1.
62 strains fall above the >45% prevalence cutoff used for inclusion in hCom1 but were not included for one of the following reasons: phylogenetic redundancy at the species level, inability to obtain them from a commercial strain bank or from a laboratory source, or literature suggesting the organism is predominantly part of the oral microbiome.
Figure S1: Phylogenetic tree of strains from hCom1 and hCom2, related to Figures 4 and 5.
Strains in black are present in hCom1 and hCom2. Strains in red are only in hCom2, and strains in blue are only in hCom1.
Figure S2: Stability and invading strains from the first and second fecal challenge experiments, related to Figures 4 and 5.
(A) Average relative abundances of MIDAS bins for hCom2-colonized mice over all 8 weeks of the experiment duration. (B) Microbiome compositions in hCom1- and hCom2-colonized mice. Week 4 and 8 species distributions are shown for each group. Week 8 distributions are split into two groups: on the left are input species and on the right are invading species, as depicted in the right inset. Invading species from weeks 5–8 that are common to all three groups are outlined in bold (Table S4).
Figure S3: The architecture of hCom2 more closely resembles that of a human fecal consortium than hCom1, related to Figure 4 and 5.
(A) Phylum-level relative abundances from mice colonized with hCom1, hCom2, or healthy human fecal samples Hum1–3 (n=5 mice per group). Bray-Curtis (BC) dissimilarities (B) and correlation coefficients (C) at each taxonomic level between fecal pellets from germ-free mice colonized with hCom2, human fecal samples Hum1, Hum2, and Hum3 (H1-fecal, H2-fecal, and H3-fecal), and fecal pellets from germ-free mice colonized by Hum1, Hum2, and Hum3 at week 4 (H1-FMT, H2-FMT, and H3-FMT). The average similarity among the fecal or humanized fecal samples is shown as bold or dashed black lines, respectively. (D) Composition as determined by MIDAS of hCom1-colonized mice, hCom2-colonized mice, the three original samples from Hum1-FMT, Hum2-FMT, and Hum3-FMT (labeled 1–3), and four additional humanized mouse samples from human fecal samples unrelated to Hum1–3 (labeled A-D). (E) Principal coordinate analysis of BC dissimilarity at the species, family, and phylum levels. Each circle denotes a human fecal sample and is connected by a line to a square that represents the corresponding sample from humanized mice at week 4.
HIGHLIGHTS.
We introduce hCom1, a defined community of 104 gut bacterial species
We fill open niches in vivo to form hCom2, a defined community of 119 species
In gnotobiotic mice, hCom2 exhibited robust colonization resistance against E. coli
Mice colonized by hCom2 versus a human fecal community are phenotypically similar
ACKNOWLEDGMENTS
We are deeply indebted to members of the Fischbach and Huang labs for helpful discussions, and to Rod Mackie (UIUC) for bacterial strains used in this study. A.A.-D. is a Howard Hughes Medical Institute International Student Research fellow, a Stanford Bio-X Bowes fellow, and a Siebel Scholar. This work was supported by a Dean’s Postdoctoral Fellowship (to P.-Y.H.), NIH F32GM143859 (to P.-Y.H.), Human Frontier Science Program award LT000493/2018-L (to K.N.), a Fellowship from the Astellas Foundation for Research on Metabolic Disorders (to K.N.)., the Stanford Microbiome Therapies Initiative (to M.A.F. and K.C.H.), NIH grants DP1 DK113598 (to M.A.F.), P01 HL147823 (to M.A.F.), R01 DK101674 (to M.A.F.), RM1 GM135102 (to K.C.H.), and R01 AI147023 (to K.C.H.), NSF grant EF-2125383 (to K.C.H. and M.A.F.), the Helmsley Charitable Trust (to M.A.F.), the Bill and Melinda Gates Foundation (to M.A.F.), an HHMI-Simons Faculty Scholars Award (to M.A.F.), the Leducq Foundation (to M.A.F.), the Stanford-Coulter Translational Research Grants Program (to M.A.F.), MAC3 Impact Philanthropies (to M.A.F.), and the Allen Discovery Center at Stanford on Systems Modeling of Infection (to K.C.H.). K.C.H. and M.A.F. are Chan Zuckerberg Biohub Investigators.
Stanford University and the Chan Zuckerberg Biohub have patents pending for microbiome technologies on which the authors are co-inventors. M.A.F. is a co-founder and director of Federation Bio and Kelonia, a co-founder of Revolution Medicines, and a member of the scientific advisory boards of NGM Bio and Zymergen. A.G.C. and K.N. have been paid consultants to Federation Bio. A.R.B. has been an employee of Federation Bio.
Footnotes
DECLARATION OF INTERESTS
The other authors have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
REFERENCES
- Angly FE, Willner D, Rohwer F, Hugenholtz P, and Tyson GW (2012). Grinder: a versatile amplicon and shotgun sequence simulator. Nucleic Acids Res. 40, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Díaz A, Ng KM, Thomsen T, Real-Ramírez I, Dahan D, Dittmar S, Gonzalez CG, Chavez T, Vasquez KS, Nguyen TH, et al. (2020). High-throughput cultivation of stable, diverse, fecal-derived microbial communities to model the intestinal microbiota. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasche S, Kim Y, Oliveira AP, and Patil KR (2017). Model microbial communities for ecosystems biology. Current Opinion in Systems Biology 6, 51–57. [Google Scholar]
- Buffie CG, and Pamer EG (2013). Microbiota-mediated colonization resistance against intestinal pathogens. Nat. Rev. Immunol. 13, 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffington SA, Dooling SW, Sgritta M, Noecker C, Murillo OD, Felice DF, Turnbaugh PJ, and Costa-Mattioli M (2021). Dissecting the contribution of host genetics and the microbiome in complex behaviors. Cell 184, 1740–1756.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin W-B, Guo C-J, Violante S, et al. (2020). Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil P-A, Mussig AJ, Hugenholtz P, and Parks DH (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunin R, Glansdorff N, Piérard A, and Stalon V (1986). Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50, 314–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschasaux M, Bouter KE, Prodan A, Levin E, Groen AK, Herrema H, Tremaroli V, Bakker GJ, Attaye I, Pinto-Sietsma S-J, et al. (2018). Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531. [DOI] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA 108 Suppl 1, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, et al. (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, McNulty NP, Rey FE, and Gordon JI (2011). Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 333, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. (2013). The long-term stability of the human gut microbiota. Science 341, 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Ahern PP, Ridaura VK, Cheng J, and Gordon JI (2014). Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Sci. Transl. Med. 6, 220ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJM, and Huttenhower C (2015). Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci USA 112, E2930–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabashi M, Grove TL, Wang M, Varma Y, McFadden ME, Brown LC, Guo C, Higginbottom S, Almo SC, and Fischbach MA (2020). A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldford JE, Lu N, Bajić D, Estrela S, Tikhonov M, Sanchez-Gorostiaga A, Segrè D, Mehta P, and Sanchez A (2018). Emergent simplicity in microbial community assembly. Science 361, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, and Gordon JI (2011). Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 108, 6252–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C-J, Allen BM, Hiam KJ, Dodd D, Van Treuren W, Higginbottom S, Nagashima K, Fischer CR, Sonnenburg JL, Spitzer MH, et al. (2019). Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, and Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wu W, Zheng H-M, Li P, McDonald D, Sheng H-F, Chen M-X, Chen Z-H, Ji G-Y, Zheng Z-D-X, et al. (2018). Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat. Med. 24, 1532–1535. [DOI] [PubMed] [Google Scholar]
- Hibberd MC, Wu M, Rodionov DA, Li X, Cheng J, Griffin NW, Barratt MJ, Giannone RJ, Hettich RL, Osterman AL, et al. (2017). The effects of micronutrient deficiencies on bacterial species from the human gut microbiota. Sci. Transl. Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W-B, Li T-T, Huo D, Qu S, Li XV, Arifuzzaman M, Lima SF, Shi H-Q, Wang A, Putzel GG, et al. (2022). Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, and Wang Z (2019). MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraal L, Abubucker S, Kota K, Fischbach MA, and Mitreva M (2014). The prevalence of species and strains in the human microbiome: a resource for experimental efforts. PLoS ONE 9, e97279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, and Walker AW (2013). Intestinal colonization resistance. Immunology 138, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lelie D, Oka A, Taghavi S, Umeno J, Fan T-J, Merrell KE, Watson SD, Ouellette L, Liu B, Awoniyi M, et al. (2021). Rationally designed bacterial consortia to treat chronic immune-mediated colitis and restore intestinal homeostasis. Nat. Commun. 12, 3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Peterson DA, and Gordon JI (2006). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. [DOI] [PubMed] [Google Scholar]
- Litvak Y, Mon KKZ, Nguyen H, Chanthavixay G, Liou M, Velazquez EM, Kutter L, Alcantara MA, Byndloss MX, Tiffany CR, et al. (2019). Commensal Enterobacteriaceae Protect against Salmonella Colonization through Oxygen Competition. Cell Host Microbe 25, 128–139.e5. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Breitwieser FP, Thielen P, and Salzberg SL (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science 3, e104. [Google Scholar]
- Marcobal A, Barboza M, Sonnenburg ED, Pudlo N, Martens EC, Desai P, Lebrilla CB, Weimer BC, Mills DA, German JB, et al. (2011). Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Kelly AG, Tauzin AS, and Brumer H (2014). The devil lies in the details: how variations in polysaccharide fine-structure impact the physiology and evolution of gut microbes. J. Mol. Biol. 426, 3851–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, Luke JJ, and Gajewski TF (2018). The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, Henrissat B, Oozeer R, Cools-Portier S, Gobert G, et al. (2011). The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Transl. Med. 3, 106ra106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty NP, Wu M, Erickson AR, Pan C, Erickson BK, Martens EC, Pudlo NA, Muegge BD, Henrissat B, Hettich RL, et al. (2013). Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol. 11, e1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohawk KL, and O’Brien AD (2011). Mouse models of Escherichia coli O157:H7 infection and shiga toxin injection. J. Biomed. Biotechnol. 2011, 258185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris BEL, Henneberger R, Huber H, and Moissl-Eichinger C (2013). Microbial syntrophy: interaction for the common good. FEMS Microbiol. Rev. 37, 384–406. [DOI] [PubMed] [Google Scholar]
- Nayfach S, Rodriguez-Mueller B, Garud N, and Pollard KS (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 26, 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Shi ZJ, Seshadri R, Pollard KS, and Kyrpides NC (2019). New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Aranda-Díaz A, Tropini C, Frankel MR, Van Treuren W, O’Loughlin CT, Merrill BD, Yu FB, Pruss KM, Oliveira RA, et al. (2019). Recovery of the Gut Microbiota after Antibiotics Depends on Host Diet, Community Context, and Environmental Reservoirs. Cell Host Microbe 26, 650–665.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisman B (1954). The Stickland reaction. Bacteriol. Rev. 18, 16–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco AR, and Segrè D (2019). A multidimensional perspective on microbial interactions. FEMS Microbiol. Lett. 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, Barnich N, Ng SC, and Colombel J-F (2018). Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 67, 574–587. [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, and Tyson GW (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, and Hugenholtz P (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. [DOI] [PubMed] [Google Scholar]
- Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, and Hugenholtz P (2020). A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086. [DOI] [PubMed] [Google Scholar]
- Patnode ML, Beller ZW, Han ND, Cheng J, Peters SL, Terrapon N, Henrissat B, Le Gall S, Saulnier L, Hayashi DK, et al. (2019). Interspecies Competition Impacts Targeted Manipulation of Human Gut Bacteria by Fiber-Derived Glycans. Cell 179, 59–73.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T-P-T, Tidjani Alou M, Bachar D, Levasseur A, Brah S, Alhousseini D, Sokhna C, Diallo A, Wieringa F, Million M, et al. (2019). Gut microbiota alteration is characterized by a proteobacteria and fusobacteria bloom in kwashiorkor and a bacteroidetes paucity in marasmus. Sci. Rep. 9, 9084. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin M, Wu S, Li A, Zhao F, Feng H, Ding L, Chang Y, and Ruan J (2018). Lrscaf: improving draft genomes using long noisy reads. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, et al. (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215. [DOI] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. [DOI] [PubMed] [Google Scholar]
- Sharon G, Cruz NJ, Kang D-W, Gandal MJ, Wang B, Kim Y-M, Zink EM, Casey CP, Taylor BC, Lane CJ, et al. (2019). Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 177, 1600–1618.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Le S, Li Y, and Hu F (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 11, e0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EA, and Macfarlane GT (1997). Dissimilatory amino Acid metabolism in human colonic bacteria. Anaerobe 3, 327–337. [DOI] [PubMed] [Google Scholar]
- Sonnenburg ED, and Sonnenburg JL (2019). The ancestral and industrialized gut microbiota and implications for human health. Nat. Rev. Microbiol. 17, 383–390. [DOI] [PubMed] [Google Scholar]
- Soto-Martin EC, Warnke I, Farquharson FM, Christodoulou M, Horgan G, Derrien M, Faurie J-M, Flint HJ, Duncan SH, and Louis P (2020). Vitamin Biosynthesis by Human Gut Butyrate-Producing Bacteria and Cross-Feeding in Synthetic Microbial Communities. MBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromberg ZR, Van Goor A, Redweik GAJ, Wymore Brand MJ, Wannemuehler MJ, and Mellata M (2018). Pathogenic and non-pathogenic Escherichia coli colonization and host inflammatory response in a defined microbiota mouse model. Dis. Model. Mech. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus Brown C, and Irber L (2016). sourmash: a library for MinHash sketching of DNA. JOSS 1. [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- Vandeputte D, Kathagen G, D’hoe K, Vieira-Silva S, Valles-Colomer M, Sabino J, Wang J, Tito RY, De Commer L, Darzi Y, et al. (2017). Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511. [DOI] [PubMed] [Google Scholar]
- Velazquez EM, Nguyen H, Heasley KT, Saechao CH, Gil LM, Rogers AWL, Miller BM, Rolston MR, Lopez CA, Litvak Y, et al. (2019). Endogenous Enterobacteriaceae underlie variation in susceptibility to Salmonella infection. Nat. Microbiol. 4, 1057–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli OS, Carr AC, Fisher G, Hsu RH, Lau R, Bowen BP, Hromada S, Northen T, and Arkin AP (2018). Deciphering microbial interactions in synthetic human gut microbiome communities. Mol. Syst. Biol. 14, e8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal V, and Nadkarni GB (1977). Regulation of the arginine dihydrolase pathway in Clostridium sporogenes. J. Bacteriol. 131, 693–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Maldonado-Gómez MX, and Martínez I (2018). To engraft or not to engraft: an ecological framework for gut microbiome modulation with live microbes. Curr. Opin. Biotechnol. 49, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick RR, Judd LM, Gorrie CL, and Holt KE (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widder S, Allen RJ, Pfeiffer T, Curtis TP, Wiuf C, Sloan WT, Cordero OX, Brown SP, Momeni B, Shou W, et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. 10, 2557–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildenauer FX, and Winter J (1986). Fermentation of isoleucine and arginine by pure and syntrophic cultures of Clostridium sporogenes. FEMS Microbiol. Lett. 38, 373–379. [Google Scholar]
- Wood DE, Lu J, and Langmead B (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, McNulty NP, Rodionov DA, Khoroshkin MS, Griffin NW, Cheng J, Latreille P, Kerstetter RA, Terrapon N, Henrissat B, et al. (2015). Genetic determinants of in vivo fitness and diet responsiveness in multiple human gut Bacteroides. Science 350, aac5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymore Brand M, Wannemuehler MJ, Phillips GJ, Proctor A, Overstreet A-M, Jergens AE, Orcutt RP, and Fox JG (2015). The altered schaedler flora: continued applications of a defined murine microbial community. ILAR J. 56, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier JB (2011). Social interaction in synthetic and natural microbial communities. Mol. Syst. Biol. 7, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Guo L, Gu S, Wang O, Zhang R, Fan G, Xu X, Deng L, and Liu X (2019). TGSGapCloser: fast and accurately passing through the Bermuda in large genome using error-prone third-generation long reads. BioRxiv. [Google Scholar]
- Ze X, Duncan SH, Louis P, and Flint HJ (2012). Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 6, 1535–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S5. hCom2 as a model system, related to Figure 6.
Data S3: Discovery and elucidation of a strain-amino acid interaction and its molecular mechanism, related to Figure 2.
Table S7: MIDAS sensitivity analysis, related to Figure 4 and 5.
MIDAS species bins (sample_id) and associated hCom1 and hCom2 strain abundances. Sensitivity was determined by processing simulated metagenomic sequencing reads for each hCom1 or hCom2 strain using MIDAS and assessing the distribution of reads among MIDAS species bins.
Data S2: Standardization and benchmarking of NinjaMap, related to STAR Methods.
Data S1: Composition and function of hCom1 and hCom2, related to Figure 1.
Table S6: Strains and primers used to generate Clostridium sporogenes adi and otc mutants, related to Figure 2.
Table S5: Modified Standard Amino Acid Complete (SAAC) medium recipe, related to Figure 2.
SAAC media is a minimal medium commonly used for assessment of bacterial metabolism of amino acids (Dodd et al., 2017). SAAC complete contains all amino acids at 1 mM concentration, with the exception of cysteine at 4.126 mM.
Table S2: Genome assemblies from strains in the synthetic communities hCom1 and hCom2, related to Figure 1.
Unique genome assemblies utilized or completed for this manuscript, with the type of assembly and the libraries involved. This procedure resulted in the replacement of eight genomes: two obtained from a PacBio and Illumina hybrid assembly and six from short-read assembly of the respective isolate samples followed by binning.
Table S3: Strain relative abundances in amino acid dropout experiments, related to Figure 2 and S2.
Relative abundances were measured after 48 h of community outgrowth in each dropout media. The data shown are from one of two biological replicates.
Table S4: Invading strains from the two fecal challenge augmentation experiments and hCom1 species whose relative abundance was highly impacted by fecal challenge, related to Figure 4, 5, and Data S1.
For each augmentation iteration, strains that invaded from ≥1 of 3 donor stools are shown. Also shown are the seven hCom1 strains that were left out of hCom2 as they did not persist after fecal challenge. For impacted strains from hCom1, red MIDAS bins denote species that changed by >10-fold in response to >1 fecal challenge.
Table S1: Omitted strains, related to Figure 1 and S1.
62 strains fall above the >45% prevalence cutoff used for inclusion in hCom1 but were not included for one of the following reasons: phylogenetic redundancy at the species level, inability to obtain them from a commercial strain bank or from a laboratory source, or literature suggesting the organism is predominantly part of the oral microbiome.
Figure S1: Phylogenetic tree of strains from hCom1 and hCom2, related to Figures 4 and 5.
Strains in black are present in hCom1 and hCom2. Strains in red are only in hCom2, and strains in blue are only in hCom1.
Figure S2: Stability and invading strains from the first and second fecal challenge experiments, related to Figures 4 and 5.
(A) Average relative abundances of MIDAS bins for hCom2-colonized mice over all 8 weeks of the experiment duration. (B) Microbiome compositions in hCom1- and hCom2-colonized mice. Week 4 and 8 species distributions are shown for each group. Week 8 distributions are split into two groups: on the left are input species and on the right are invading species, as depicted in the right inset. Invading species from weeks 5–8 that are common to all three groups are outlined in bold (Table S4).
Figure S3: The architecture of hCom2 more closely resembles that of a human fecal consortium than hCom1, related to Figure 4 and 5.
(A) Phylum-level relative abundances from mice colonized with hCom1, hCom2, or healthy human fecal samples Hum1–3 (n=5 mice per group). Bray-Curtis (BC) dissimilarities (B) and correlation coefficients (C) at each taxonomic level between fecal pellets from germ-free mice colonized with hCom2, human fecal samples Hum1, Hum2, and Hum3 (H1-fecal, H2-fecal, and H3-fecal), and fecal pellets from germ-free mice colonized by Hum1, Hum2, and Hum3 at week 4 (H1-FMT, H2-FMT, and H3-FMT). The average similarity among the fecal or humanized fecal samples is shown as bold or dashed black lines, respectively. (D) Composition as determined by MIDAS of hCom1-colonized mice, hCom2-colonized mice, the three original samples from Hum1-FMT, Hum2-FMT, and Hum3-FMT (labeled 1–3), and four additional humanized mouse samples from human fecal samples unrelated to Hum1–3 (labeled A-D). (E) Principal coordinate analysis of BC dissimilarity at the species, family, and phylum levels. Each circle denotes a human fecal sample and is connected by a line to a square that represents the corresponding sample from humanized mice at week 4.
Data Availability Statement
Metagenomic and whole-genome sequencing datasets generated for this study are available at the Sequence Read Archive. The ninjamap code used in this study can be found at the following github location: https://github.com/FischbachLab/ninjaMap/releases/tag/cheng_et_al and the associated docker containers are available at https://hub.docker.com/repository/docker/fischbachlab/ninjamap.