

Abstract
A detailed understanding of where bacteria localize is necessary to advance microbial ecology and microbiome-based therapeutics. The site-specialist hypothesis predicts that most microbes in the human oral cavity have a primary habitat type within the mouth where they are most abundant. We asked whether this hypothesis accurately describes the distribution of the members of the genus Streptococcus, a clinically relevant taxon that dominates most oral sites. Prior analysis of 16S rRNA gene sequencing data indicated that some oral Streptococcus clades are site-specialists while others may be generalists. However, within complex microbial populations composed of numerous closely related species and strains, such as the oral streptococci, genome-scale analysis is necessary to provide the resolution to discriminate closely related taxa with distinct functional roles. Here we assess whether individual species within this genus are specialists using publicly available genomic sequence data that provides species-level resolution. We chose a set of high-quality representative genomes for human oral Streptococcus species. Onto these genomes, we mapped shotgun metagenomic sequencing reads from supragingival plaque, tongue dorsum, and other sites in the oral cavity. We found that every abundant Streptococcus species in the healthy human oral cavity showed strong site tropism and that even closely related species such as S. mitis, S. oralis, and S. infantis specialized in different sites. These findings indicate that closely related bacteria can have distinct habitat distributions in the absence of dispersal limitation and under similar environmental conditions and immune regimes. Substantial overlap between the core genes of these three species suggests that site-specialization is determined by subtle differences in genomic content.
Keywords: biogeography, spatial organization, spatial structure, metagenome, pangenome
Summary
Patterns of microbial distribution, and prospects for modulating the microbiome, are determined by poorly understood rules governing where microbes can grow and thrive. In the human mouth, closely related bacteria specialize for different sites despite the ready transport of bacteria throughout the mouth by saliva, giving rise to polymicrobial biofilms with compositions specific to different regions of the mouth. Site-tropism is an important component of the ecology of the oral microbiome as it influences the set of taxa that bacteria are likely to encounter in short-range interactions. However, abundant taxa from the clinically relevant genus Streptococcus appeared to be site generalists based on marker gene data. Using the high resolution provided by genome sequences and metagenomic data, we tested for site specialization of closely related Streptococcus species. We found that every abundant species displayed a preference for one of the major oral sites of buccal mucosa, tongue dorsum, or dental plaque, validating the site-specialist hypothesis. Examining the gene content and functions of three closely related Streptococcus species, each localized to a different region, revealed only modest differences. These results indicate that subtle differences in genome content can result in dramatically different spatial distributions within the microbiome.
Introduction
Accurate information about the spatial arrangement of bacteria is necessary in order to discover the rules governing which bacteria colonize which host sites in the human microbiome and to realize the potential of the microbiome as a therapeutic target. The Human Microbiome Project (HMP) was designed to establish a high-resolution baseline for similarities and differences in microbiome composition from individual to individual and site to site (Turnbaugh et al., 2007). Together with previous cultivation-based and cultivation-independent studies, the HMP demonstrated that bacteria occupy characteristic habitats: bacteria that are most abundant in the human gut tend to be rare on the skin or in the mouth, and vice versa (Costello et al., 2009; HMP Consortium, 2012). Thus, most bacteria are found predominantly in one broad habitat type. Not yet clear, however, is what features of the habitat determine which bacteria can thrive, how finely subdivided are the habitats, and what range of micro-habitats each bacterium can occupy.
For addressing these questions, the human oral cavity provides a natural experiment with many replicates and built-in controls. The distinct surfaces in the mouth (including enamel as well as keratinized, non-keratinized, and specialized mucosa) represent distinct potential microbial habitats that are spatially adjacent, with minimal barriers to microbial dispersal (Proctor and Relman, 2017; Mark Welch et al., 2020). Each human individual is an island whose mouth has undergone the process of colonization. The bacteria inhabiting the same mouth are exposed to the same host diet, behavior, and immune regime, controlling for many of the variables that might influence microbial community composition. The composition of the oral microbiome is relatively well-understood, with a curated database (Dewhirst et al., 2010) identifying ~700 bacterial species resident in the mouth. The majority of these species can be cultivated in the laboratory. The HMP (HMP Consortium, 2012), as well as independent research efforts, has generated sequenced genomes for most of the cultivable oral microbes as well as shotgun metagenomic sequence data sampled from a variety of sites within the mouth for several hundred individuals. Thus, the knowledge base exists to support a systems-level study of the habitat distribution of the oral microbiota.
The site-specialist hypothesis for the oral microbiota was developed based on 16S rRNA gene sequence data from the HMP as well as prior cultivation and cultivation-independent studies (Gibbons et al., 1963; Gibbons et al., 1964 A; Gibbons et al., 1964 B; Gordon and Gibbons, 1966; Gordon and Jong, 1968; Frandsen et al., 1991; Mager et al., 2003; Aas et al., 2005; Zaura et al., 2009; HMP Consortium, 2012; Huse et al., 2012; Peterson et al., 2013; Mark Welch et al., 2014; Eren et al., 2014; Hall et al., 2017; Bernardi et al., 2020). These studies showed that whether a taxon appears to be a generalist or a specialist depends on the resolution of the analysis: at the genus level, most oral bacteria have representatives throughout the mouth, but at the species level they are site-specialists; most species preferentially colonize certain regions of the mouth (Mark Welch et al., 2019). While site-specialists may be present with a low abundance across other oral sites, the site-specialist hypothesis predicts that their relative abundance will be significantly greater in their preferred habitat than at these other sites. This primary habitat for a species can sometimes be narrowly defined; for example, some bacteria are abundant only on the keratinized gingiva (Eren et al., 2014) and one, Simonsiella mulleri, appears to live exclusively on the hard palate (Aas et al., 2005; Caselli et al., 2020). Often, however, the primary habitat is broader and consists of a group of sites; for example, many bacteria are specialists for both supra and subgingival dental plaque, others for the tongue dorsum, palatine tonsils, and throat (Eren et al., 2014; Mark Welch et al., 2019). These distribution patterns suggest specialized adaptation for a subset of sites within the mouth.
A major exception to this pattern is found in the genus Streptococcus, which contains both specialist and apparent generalist taxa. Streptococcus is the most abundant genus in the oral cavity (Mager et al., 2003; Segata et al., 2012). As primary colonizers, oral streptococci play important roles in biofilm formation (Jenkinson, 1994; Li et al., 2004). Some members of the genus contribute to the progression of disease while others help maintain the health of their host (Abranches et al., 2018). Thus, the spatial distribution of this genus is a critical feature of oral ecology. Some species of oral streptococci are so closely related that short regions of the 16S rRNA gene fail to distinguish them. 16S sequences are notably insufficient for differentiating species within the Mitis group (Jensen et al., 2016; Croxen et al., 2018; Velsko et al., 2019). Consequently, many studies relying on 16S sequencing data only distinguish between a handful of oral Streptococcus operational taxonomic units (OTU) (Zaura et al., 2009; Huse et al., 2012; Eren et al., 2014; Hall et al., 2017). The analysis of Eren et al. (2014) distinguished 11 OTUs for around 30 known oral Streptococcus species and indicated that the Streptococcus genus includes both site-specialist taxa and an apparent generalist, the group containing the abundant oral commensal S. mitis and its close relatives S. pneumoniae, S. oralis, S. infantis, S. cristatus, and S. australis (Mark Welch et al., 2019).
Here, we test the site-specialist hypothesis for each human oral Streptococcus species using isolate genome sequences combined with shotgun metagenomic sequence data. From the many sequenced genomes available at NCBI, we selected a set of reference genomes and used short-read mapping from metagenomic samples to demonstrate localization patterns and site-tropism of species. We then carried out pangenome analysis to determine whether there are genes or functions that distinguish the specialists for each site. Our results show that each of the major oral species demonstrates site-tropism within the mouth. These findings indicate that closely related bacteria can have distinct habitat distributions in the absence of dispersal limitation and under similar dietary and immune regimes. Further, distinct distributions occur despite whole-genome analysis showing only small differences in gene content and functional annotation, indicating that subtle differences in genomic content beyond the presence and absence of genes can have ecologically significant effects.
Materials and Methods
We used a workflow adapted from Delmont and Eren (2018) to perform metapangenomic analyses in the anvi’o v7 platform (Eren et al., 2021) with Python v3.7.9.
Reference Genomes and Metagenomes.
Following previous authors (Delmont and Eren, 2018; Almeida et al., 2019), from among the available reference genomes we selected a set of genomes, each of which shared no more than a given percentage average nucleotide identity (ANI), in this case, 95% with any other genome in the set. We used NCBI Reference Sequence Database (RefSeq) genomes from the named Streptococcus species and unnamed Streptococcus “human microbial taxa” (HMT) in the eHOMD (http://www.homd.org ). We also included genomes sequenced from human isolates if there was evidence of their presence in the human oral cavity (Shen et al., 2002; Huch et al., 2013; Tetz et al., 2019; Bernardi et al., 2020). The Genome Taxonomy Database (GTDB) groups RefSeq genomes into clusters sharing ≥ 95% ANI (Parks et al., 2020). We chose one representative from each group (Table S1) that had a completeness of ≥ 90% estimated by CheckM (Parks et al., 2014). When choosing representatives, we also preferentially selected type strains and strains available from culture collections as well as genomes with high completeness and low contamination scores estimated by CheckM. Where possible within these constraints, we chose the representative genome identified by the GTDB. We added two additional genomes for eHOMD human microbial taxa (HMT) that were sequenced after the creation of the GTDB and substituted two GTDB cluster representatives for more recently sequenced genomes from the same strain that was more complete.
We downloaded the metagenomes used in this study from the Human Microbiome Project (HMP) Data Portal. These metagenomes consisted of 101-bp paired-end reads sequenced from samples collected from nine oral sites in phases I and II of the HMP. We downloaded all metagenomes uploaded through 2016 for oral sites that had at least 100 samples uploaded through this date and downloaded all metagenomes uploaded through 6/1/2021 for the other sites.
Data Cleaning.
We used the anvi’o program ‘anvi-compute-genome-similarity’ to calculate the ANI between all the genomes and clustered them based on these ANI values. This script used the program pyANI and the ANI BLAST algorithm (Pritchard et al., 2016). The S. mitis 4928STDY7071560 genome (GCF_902159415.1) was eliminated from the reference genome set as it shared no more than 85% ANI with any other Streptococcus spp. genome. S. periodonticum KCOM 2412 (GCF_003963555.1) was eliminated because it shared an ANI of > 95% with the S. anginosus type strain sequence, S. anginosus NCTC10713 (GCF_900636475.1). To avoid downstream problems, contigs smaller than 200 nucleotides were dropped from the reference genomes with the ‘anvi-script-reformat-fasta’ and all IUPAC ambiguity codes were replaced with “N”s.
Before the genomes were made publicly available, likely human reads had been removed from the samples. We performed additional quality-filtering of the metagenomic reads using ‘iu-filter-quality-minoche’ (https://github.com/merenlab/illumina-utils ) a program that implements the recommendations of Minoche et al. (2011) for improving the quality of Illumina sequencing data (Eren et al., 2013).
Reference Genome Annotation.
With ‘anvi-gen-contigs-database,’ we identified predicted protein-coding genes using a k-mer size of 4 and Prodigal v2.6.3 (Hyatt et al., 2010). First, we used ‘anvi-run-hmms’ to search for Hidden Markov Models (HMMs) against four default HMM sources using hmmscan from HMMER v3.2.1 (Eddy, 2009). Then, we used ‘anvi-run-pfams’ to match gene clusters with functions from the European Bioinformatics Institute’s Pfam database with hmmsearch from HMMER v3.2.1. Finally, we used ‘anvi-run-cogs’ to match gene clusters with functions from the updated 2020 version of NCBI's Clusters of Orthologous Groups database (Tatusov et al., 2000) with NCBI’s Protein-Protein BLAST v2.10.1+ (Altschul et al., 1990). We annotated amino acid sequences, which were exported from the contigs database with ‘anvi-get-sequences-for-gene-calls,’ using eggNOG-mapper v2 with precomputed eggNOG v5 clusters through the online interface (http://eggnog5.embl.de/#/app/emapper) and imported the annotations into the contigs database with ‘anvi-script-run-eggnog-mapper’ (Huerta-Cepas et al., 2017; Huerta-Cepas et al., 2019). For each source, the function most frequently annotated for the amino acid sequences in that gene cluster was considered the representative function for the gene cluster.
Phylogenomics.
To check the genomes’ NCBI species designations, we used ‘anvi-gen-phylogenomic-tree’ and FastTree v2.1.3 SSE3 (Price et al., 2010) to generate a phylogenomic tree with the Streptococcus spp. reference genomes and a Lactobacillus crispatus genome included as an outgroup. The tree was based on the amino acid sequences of 205 single-copy core genes present in all 154 Streptococcus spp. genomes acquired with ‘anvi-get-sequences-for-gene-clusters’ and aligned with MUSCLE. FastTree calculated local support values using the Shimodaira-Hasegawa test with 1,000 resamples. To differentiate between S. mitis, S. pneumoniae, and S. pseudopneumoniae genomes, we aligned S. pneumoniae and S. pseudopneumoniae species-specific marker sequences identified by Croxen et al. (2018) to all the genomes with BLASTn (Zhang et al., 2000). We plotted the dendrograms using the ‘ape’ and ‘dendextend’ R packages (Galili, 2015; Paradis and Schliep, 2019).
Mapping Specificity Test.
To evaluate the specificity of mapping to the reference genome set, we generated a set of simulated paired-end read samples using the program ‘reads-for-assembly’ (https://github.com/merenlab/reads-for-assembly ). Each sample used a single genome from one of the following three categories as a template for the simulated reads – (1) oral streptococci type strain genomes from the reference genome set; (2) oral streptococci genomes not in the reference genome set but that had ≥ 95% ANI to a genome in the reference genome set, and (3) type strain genomes from other major human oral genera. The samples contained 100 bp long reads which had a mean offset of 30 bp with a standard deviation of 1 bp. The reads covered their template genome to a mean depth of 100 reads and simulated sequencing error was introduced so that the reads had an average base substitution error rate of 0.5%. This error rate falls within the expected range for Illumina reads quality-filtered by low-quality end trimming; the insertion-deletion error rate would be expected to be negligible for these reads (Minoche et al., 2011; Schirmer et al., 2016). To reduce non-specific mapping, we competitively mapped the reads to the reference genomes set with bowtie2 v2.4.1 (Langmead and Salzberg, 2012), so that each read was mapped only to the one genome that provided the closest match. Using bowtie2, we first generated a reference index for mapping and then mapped the reads to the genome set using bowtie2 v2.4.1 with the “--very-sensitive,” “--end-to-end,” and “--no-unal” flags. We used Samtools v1.9 (Li et al., 2009) to sort and index the read alignment data generated by bowtie2. Using ‘anvi-single-profile,’ we used the BAM files output by Samtools to create an anvi’o single-profile database for each metagenome’s alignment data. With ‘anvi-merge-profile,’ we merged the single-profile databases for all metagenomes. We used ‘anvi-summarize’ to calculate the mean depth of coverage of the reads from each sample averaged across each genome (total mean depth of coverage) and the mean depth of coverage across nucleotide positions in the 2nd and 3rd quartiles when the nucleotides are ranked by their depth of coverage (Q2Q3 mean depth of coverage). To assess the total read recruitment to each species, we summed the total mean depth of coverage and the Q2Q3 mean depth of coverage for the genomes from the same species.
Metagenomics.
To assess the representation of the oral streptococci in the HMP metagenomes, we competitively mapped the HMP metagenomes to the reference genome set as we did in the specificity test. Because the specificity test indicated using Q2Q3 mean depth of coverage excludes much of the cross-mapping of reads from one species to genomes of another species from the mapping results (see supplemental materials), we measured the abundance of each genome, relative to the whole reference genome set, by dividing the Q2Q3 mean depth of coverage for that genome by the sum of the Q2Q3 mean depth of coverage for the whole reference genome set. We measured the abundance of a species relative to the abundance of all oral streptococci species by dividing the sum of the Q2Q3 mean depth of coverage for all genomes of that species by the sum of the Q2Q3 mean depth of coverage for the whole reference genome set.
Analysis of HMP metagenome-assembled genomes.
To check the species level designations of the putative Streptococcus spp. metagenome-assembled genomes (MAGs) that Pasolli et al. (2019) assembled and binned from the oral HMP metagenomes, we calculated the ANI between each MAG and each of our reference genomes with anvi-compute-genome-similarity.’
Statistics.
For each species included in Fig. 4B, we evaluated differences in mean relative abundance between the buccal mucosa, tongue dorsum, and supragingival plaque metagenomes with a Kruskal-Wallis test followed by Dunn's test (Dunn, 1964) using the FSA v0.9.1 R package (Ogle et al., 2021). We chose these nonparametric tests as the data did not meet the assumptions of normality and homogeneous variance of the equivalent parametric tests. We adjusted the Dunn's p-values using the Bonferroni correction to maintain a false discovery rate of 5% across the multiple comparisons for each species.
Figure 4: Mapping of whole-genome sequence data reveals site-tropism of species that cannot be distinguished based on 16S rRNA gene sequencing data.
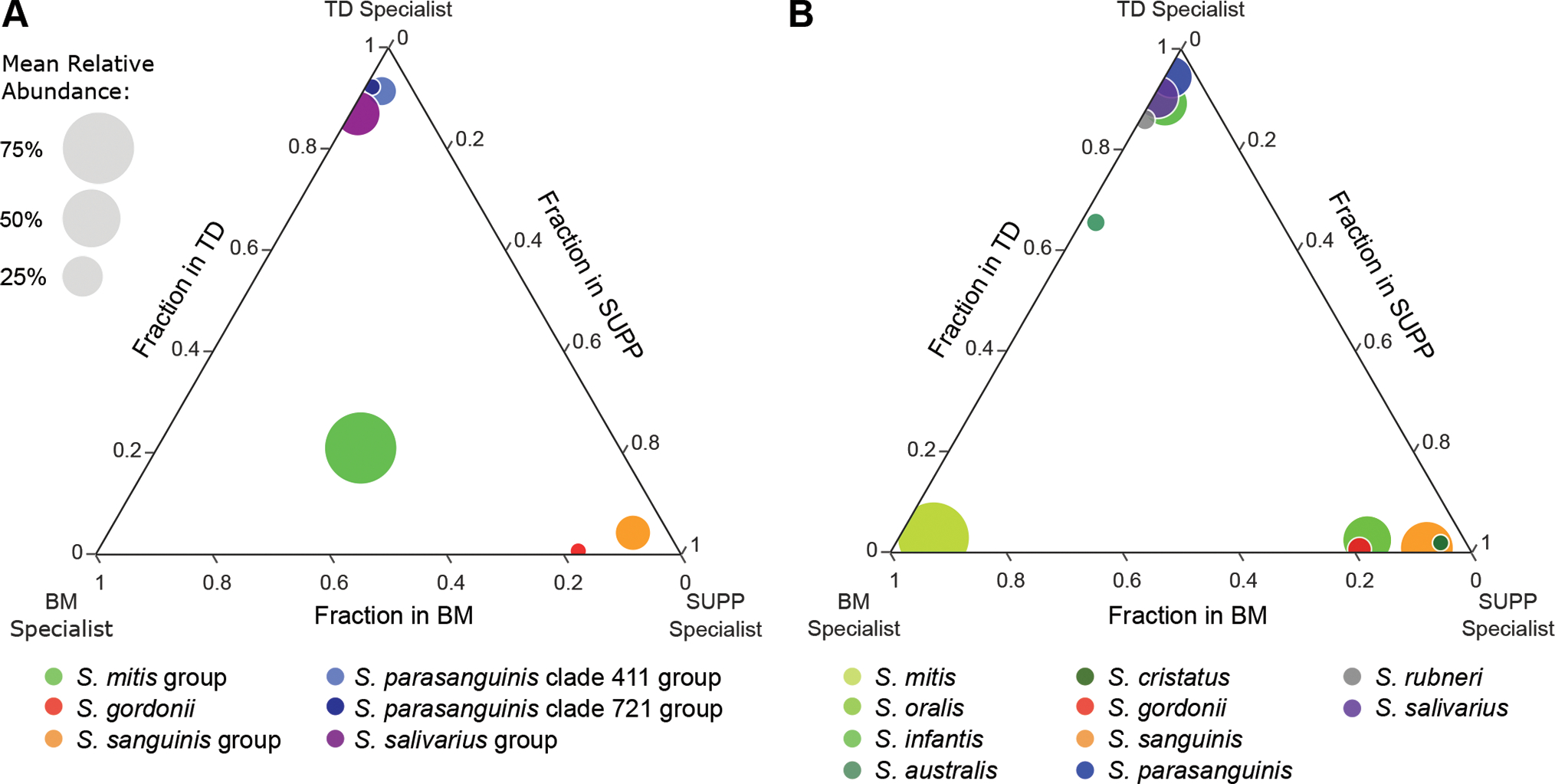
These ternary plots show the mean relative abundance of streptococci across three oral sites – buccal mucosa (BM), tongue dorsum (TD), and supragingival plaque (SUPP) – estimated via (A) oligotyping analysis of 16S rRNA gene sequencing data using the V1-V3 region of the 16S rRNA gene and (B) mapping whole-genome shotgun sequencing reads to the reference genome set. Bubbles are color-coded by taxon. Species in B and the groups they were lumped into in A are shown in different hues of the same color. Bubble size is proportionate to the mean relative abundance in the oral sites where the taxon is most abundant. The fraction of mapping to a site is calculated by dividing the mean relative abundance for a taxon in that site by the sum of the mean relative abundances of that taxon in all three sites. The bubbles of site-specialist taxa cluster near a corner of the plot. A is adapted from Mark Welch et al. (2019) and based on data from Eren et al. (2014). Species shown are those with a mean relative abundance ≥ 3% averaged across all samples from at least one of the three sites.
Pangenomics.
To evaluate the distribution of genes within and between the human oral Streptococcus species, we used ‘anvi-pan-genome’ to construct an anvi’o pangenome database from the annotated reference genomes. This program first used Protein-Protein BLAST v2.10.1+ to find similar gene calls throughout all the genomes and used MUSCLE v3.8.425 (Edgar, 2004) to align the genes. The gene calls were clustered based on the homology of their translated amino acid sequences with the Markov Cluster Algorithm (MCL) using an MCL-inflation parameter of 10 while weak matches were eliminated using a minimum bit score or “minbit” heuristic of 0.5 (Van Dongen and Abreu-Goodger, 2012). Finally, the genomes were hierarchically clustered based on the frequencies of the gene clusters they contained using Euclidean distances with Ward’s method, and the gene clusters themselves were hierarchically clustered based on their presence or absence within the genomes using Euclidean distances with Ward’s method. We used ‘anvi-compute-functional-enrichment’ to calculate the fraction of the genomes from each species annotated with that function and to select a representative function from each of the three annotation sources for each gene cluster based on which function was annotated most frequently. We created a more targeted pangenome with just the S. mitis, S. oralis, and S. infantis genomes as above, except we used a minbit heuristic of 0.8 due to the narrower taxonomic scope of this pangenome.
Results
Identification of representative genomes and species-level groups.
Estimation of species abundance by metagenomic read mapping requires careful selection of a reference genome set. Problems arise when sequence reads from one species find their best match in a genome from a different species. This can occur not only when mobile elements and other highly conserved sequences are present, but also if the species are closely related with diverse and complex populations and the reference genome set includes denser representation from one taxon than another. Therefore, we selected a set of genomes (Table S1) that were accurately identified to species and distributed as evenly as possible across sequence space (Delmont and Eren, 2018; Almeida et al., 2019). From genomes of oral streptococci in the RefSeq database, we chose a set (Table S1) in which each genome shared no more than 95% ANI with any other genome using selection criteria detailed in the Materials and Methods. For some species, all sequenced genomes available at NCBI shared an ANI > 95%; these species, therefore, were each represented by a single genome in our set (e.g., S. mutans, S. pyogenes, S. agalactiae, and S. salivarius in Fig. 1). Other species were more genomically diverse and therefore were represented by multiple genomes. As has been previously reported, many genomes deposited into RefSeq for the Mitis group streptococci have questionable species designations, due to factors including high intra-species diversity relative to inter-species diversity as well as frequent horizontal gene transfer and recombination events between species (Chi et al., 2007; Donati et al., 2010; Jensen et al., 2016; Croxen et al., 2018; Velsko et al., 2019). Therefore, we checked the species identifications of the provisional reference genomes by constructing a phylogenomic tree based on the concatenated amino acid sequences of 205 single-copy core genes present in all the genomes and by evaluating the ANI between all genomes. Numerous Mitis group genomes clustered within a species different than their NCBI designation (Fig. S1). We therefore re-assigned these genomes to corrected species designations reflecting their clade in the phylogenomic tree (Table S1), thus establishing accurate sets of species genomes for metapangenomic and pangenomic analysis.
Figure 1: Clustering of genomes based on single-copy genes is consistent with clustering based on average nucleotide identity.
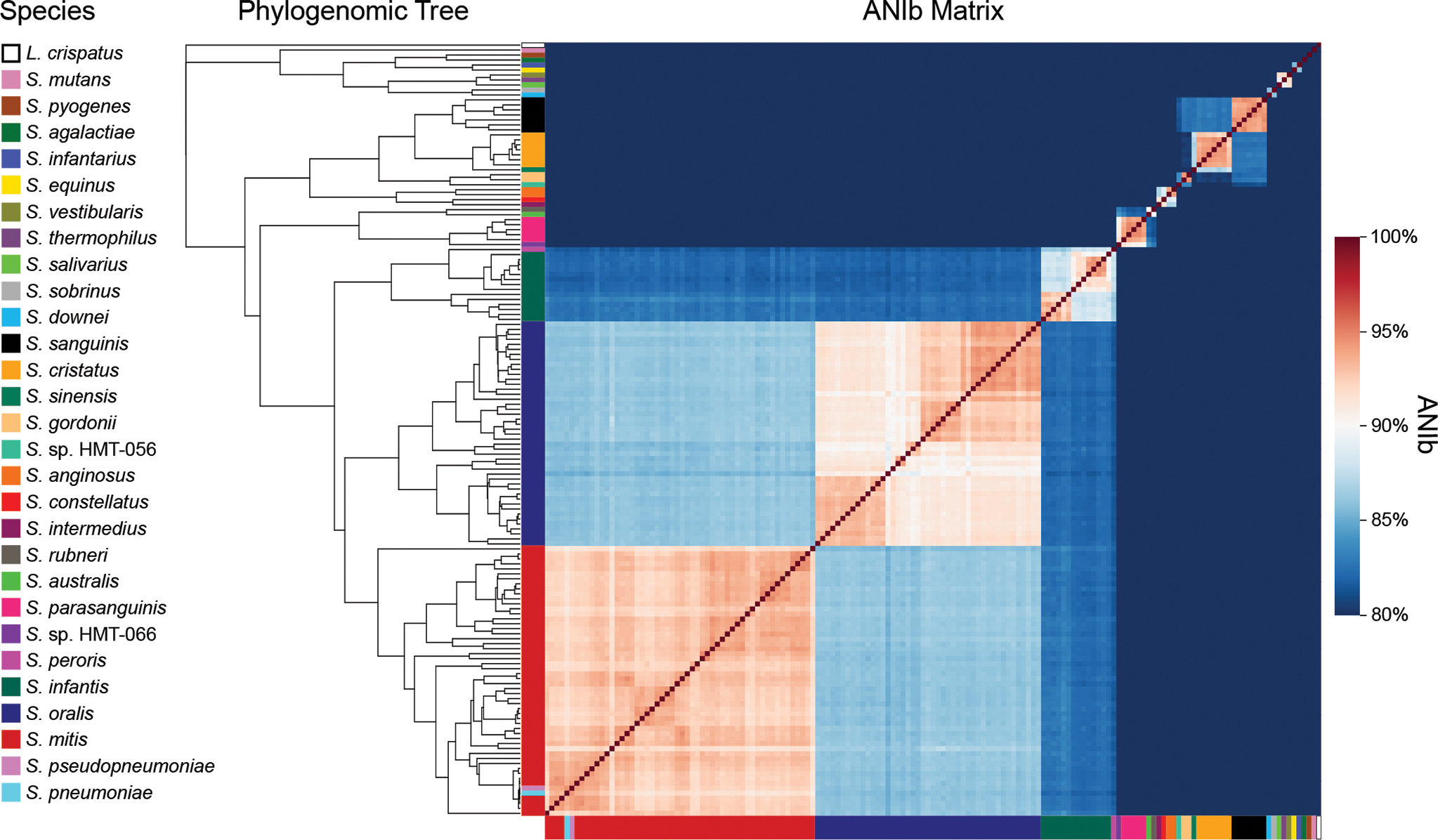
A phylogenomic tree of 155 genomes across 28 Streptococcus species and 1 outgroup species was constructed using 205 single-copy genes core to the oral streptococci. The matrix displays the ANI calculated with the BLAST method (ANIb) between each genome in the tree and every other genome in the tree. The genomes are color-coded by species and arranged according to their placement in the phylogenomic tree.
Genomes of different species segregated into discrete groups rather than falling along a continuum of relatedness, even among the closely related S. mitis, S. oralis, and S. infantis. The phylogeny was consistent both with relatedness as indicated by the ANI values and with prior phylogenies constructed with genomes identified as Mitis group species in NCBI (Figs. 1, S1; Table S2). The genomes of most species formed monophyletic clades that shared 90–95% ANI. Exceptions included the S. pneumoniae and S. pseudopneumoniae type strain sequences, which fell within the S. mitis clade, and the S. peroris type strain sequence which was placed within the S. infantis clade consistent with phylogenies constructed for members of the Mitis group (Chi et al., 2007; Jensen et al., 2016; Kilian and Tettelin, 2019). The combination of ANI and phylogenomics was insufficient to distinguish S. pneumoniae and S. pseudopneumoniae genomes from S. mitis because S. pneumoniae and S. pseudopneumoniae are effectively sub-clades within S. mitis (Jensen et al., 2016; Croxen et al., 2018; Velsko et al., 2019) and both species share > 93% ANI with some S. mitis strains (Fig. 1) (Croxen et al., 2018). To identify S. pneumoniae and S. pseudopneumoniae genomes, we aligned species-specific marker sequences for S. pneumoniae and S. pseudopneumoniae (Croxen et al., 2018) to all the reference genomes. This alignment resulted in the identification of a single genome representing S. pneumoniae and a single genome representing S. pseudopneumoniae, the type strain in each case.
Metagenomic read mapping reveals taxon site-tropism and ecological relevance of reference genomes.
To assess the distribution and abundance of streptococci across the oral cavity we used metagenomic short reads sequenced from oral samples and mapped them competitively to our selected oral Streptococcus spp. reference genomes. We mapped a total of 706 quality-filtered metagenomic samples containing 34.4 billion paired-end Illumina reads (Table S3). These samples had been collected from nine sites (buccal mucosa, keratinized gingiva, hard palate, tongue dorsum, throat, palatine tonsils, supragingival plaque, subgingival plaque, and saliva) in 144 volunteers and shotgun sequenced as part of the HMP (Lloyd-Price et al., 2017). Using the anvi’o microbial ‘omics data analysis platform (Eren et al., 2021) we assessed the abundance of genes and genomes within each sample, and we aggregated the data from genomes within the same species to generate species-level information.
Read mapping showed that each Streptococcus species preferentially colonized a subset of oral sites. Generally, the relative abundance of the streptococci in the buccal mucosa resembled that in the keratinized gingiva (Fig. 2A; Table S4). Their relative abundance on the tongue dorsum resembled that on the throat and palatine tonsils, and their relative abundance in the supragingival plaque resembled that in the subgingival plaque. The majority of the HMP samples come from three sites – buccal mucosa, tongue dorsum, and supragingival plaque – which represent the three major categories of host tissue found in the oral cavity: non-keratinized mucosa, keratinized mucosa, and enamel. Among these three sites, each species that was abundant enough for its distribution to be measured had several-fold greater relative abundance in one of these three sites than in the others.
Figure 2: Mapping shows site-tropism of species and differing abundance of strains within a species.
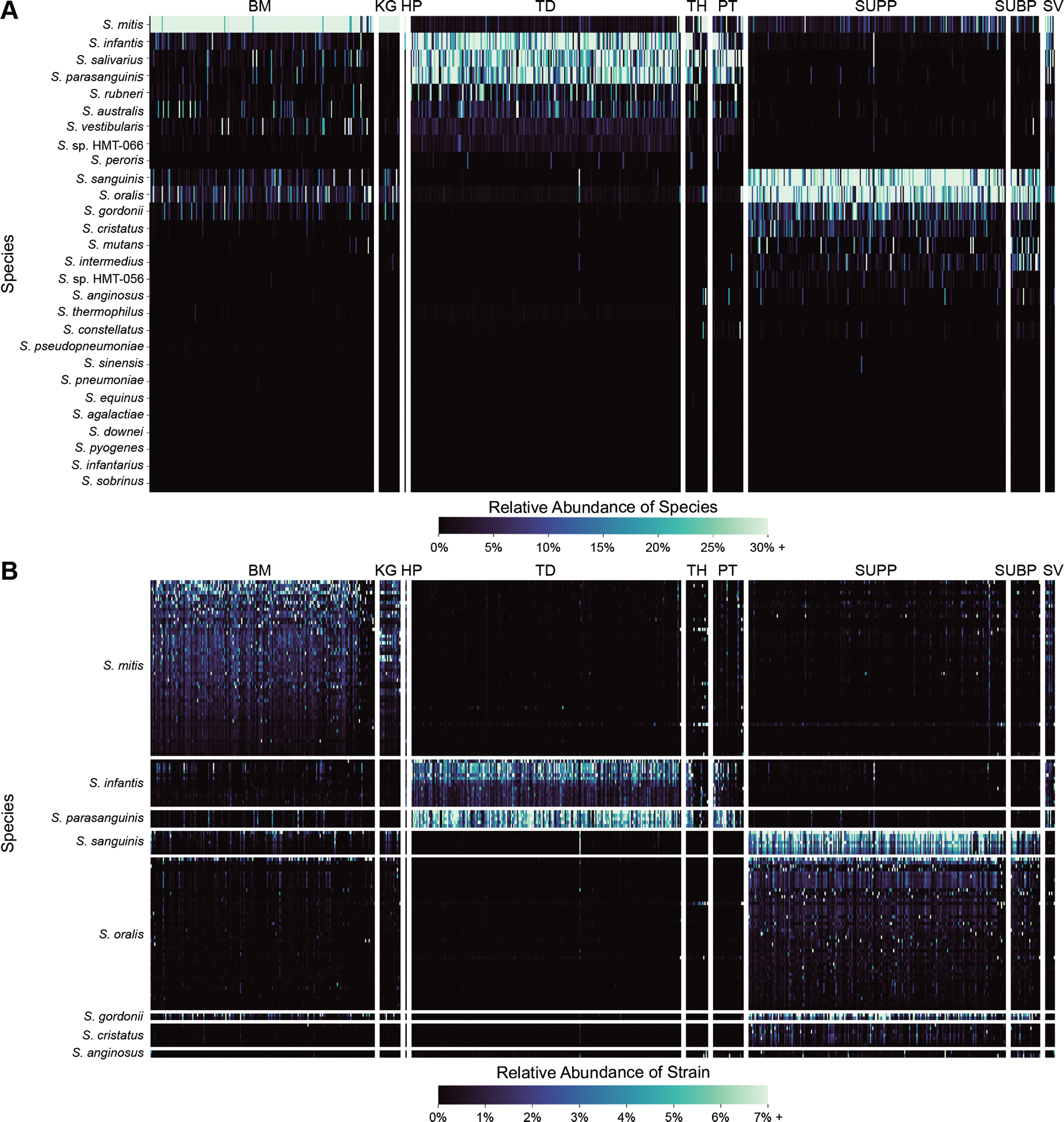
The heatmaps show the relative abundance of oral streptococci in each of the metagenomes sampled across nine oral sites. Fig. 2A shows relative abundance of species; Fig. 2B shows relative abundance of individual strains from species with more than one representative genome. The rows and columns correspond to individual species and samples, respectively. There are 183 buccal mucosa (BM), 23 keratinized gingiva (KG), 1 hard palate (HP), 220 tongue dorsum (TD), 21 throat (TH), 31 palatine tonsils (PT), 209 supragingival plaque (SUPP), 32 subgingival plaque (SUBP), and 8 saliva (SV) samples. The samples are grouped by site and then ranked by descending number of total reads. The strains are grouped first by species and then ranked by descending mean Q2Q3 relative abundance across the site (BM, TD, or SUPP) where they are most abundant.
The closely related species S. mitis, S. infantis, and S. oralis showed distinct localization patterns. S. mitis was most abundant on the buccal mucosa and keratinized gingiva, while S. infantis was most abundant on the tongue, throat, and palatine tonsils, and S. oralis was most abundant in dental plaque. In addition to assessing relative abundance based on depth of mapping, we examined the breadth of mapping, the percentage of nucleotides in the genome that recruited at least one read. This breadth metric validates the abundance calculation by confirming whether species that recruit a high depth of coverage also show high breadth, indicating that the coverage is genome-wide and not due to cross-mapping from related species to a small fraction of the genome. The breadth metric revealed the same distribution patterns (Fig. S2; Table S5): S. mitis recruited a high breadth of coverage in the buccal mucosa and keratinized gingiva, S. infantis in the tongue dorsum, and S. oralis in dental plaque. Thus, the higher resolution afforded by this whole-genome analysis made it possible to distinguish the mapping patterns of these taxa, which cannot be clearly resolved in analyses that rely on the 16S rRNA gene alone.
Whereas the results above provide species-level analysis by summing the reads that mapped to each of the representative genomes from a species, separating the mapping results for each reference genome shows that not all strains of a given species are equally represented in the oral cavities of a large set of subjects (Fig. 2B). While the individual S. mitis, S. oralis, and S. infantis reference genomes that recruited the most reads differed between individuals, some genomes had a high depth of coverage across most samples while others had low depths of coverage across most samples, indicating some strains from these species are consistently common in the sampled population while others are rare. The differences in genome-level mapping indicate which sequenced genomes are most representative of the populations in the healthy mouth in these subjects. Overall, genomes within a species showed similar distribution patterns across the oral sites, providing no evidence for subspecialization of strains within a named species for different sites.
Analysis of the breadth of coverage at the gene level confirms differential site-tropism among closely related taxa by showing whether the gene content of a given sequenced genome matches the gene content of the population in the mouth. We examined the breadth of coverage across all genes within individual genomes for each of the major oral streptococci in samples from each of the three major oral sites (Figs. 3, S3). We considered genes detected if they had a breadth of coverage of at least 90% to account for hypervariable regions where the gene sequences in the reference genome might differ from the sequence in the population in the metagenomes. Among the closely-related S. infantis, S. mitis, and S. oralis, most of the genes in the genome were detected primarily in samples from a single habitat as indicated by the depth of coverage results: tongue dorsum for S. infantis, buccal mucosa for S. mitis, and supragingival plaque for S. oralis (Fig. 3). For each of the other Streptococcus species that were sufficiently abundant to be detectable with this analysis, strong site-tropism was also observed (Fig. S3).
Figure 3: Breadth of coverage validates site tropisms and indicates how well the sequenced genome matches the gene content of the population in the mouth.
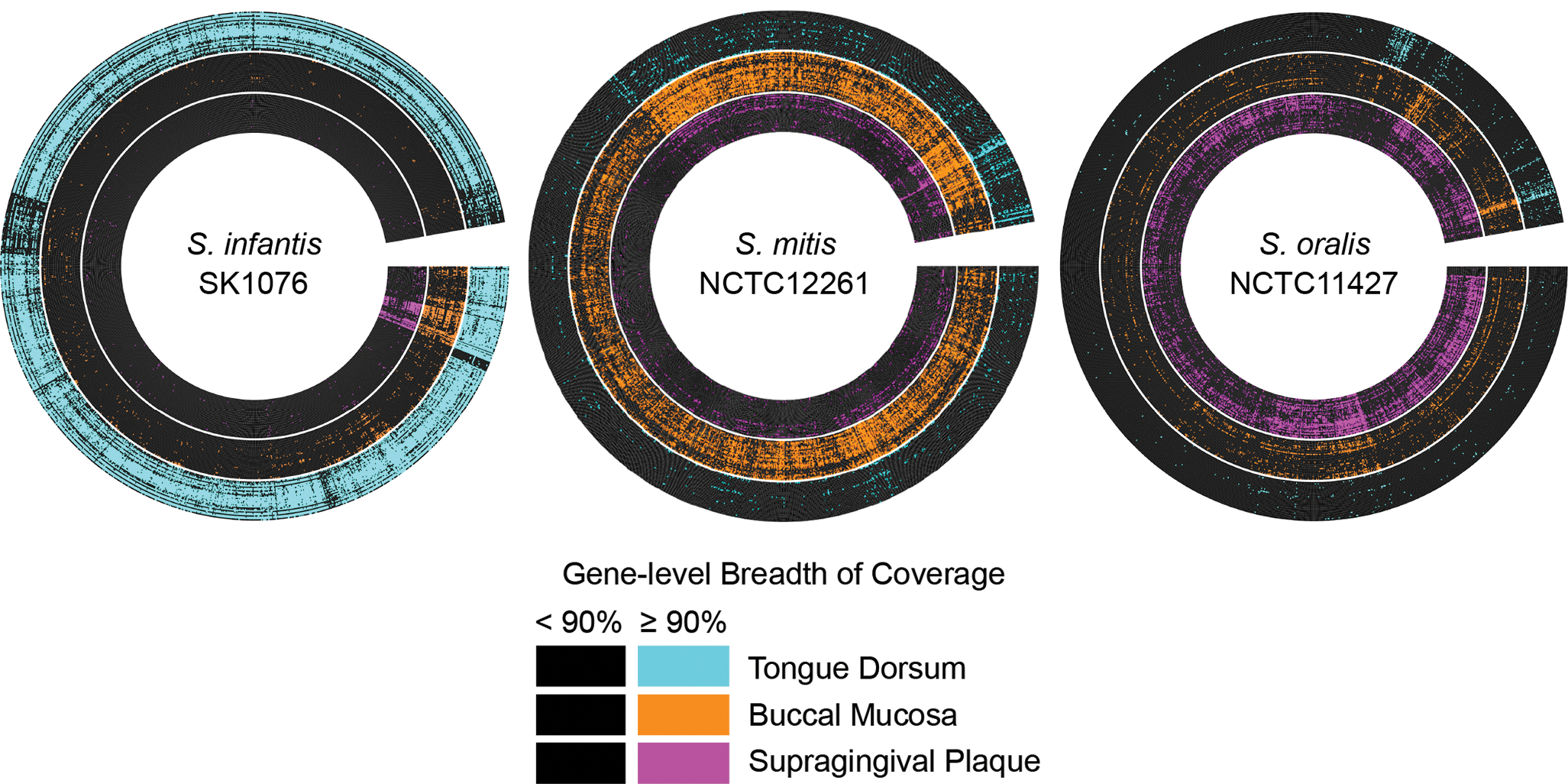
The radial heatmap displays the breadth of coverage of the predicted genes from a representative genome by the 30 buccal mucosa, tongue dorsum, and supragingival plaque samples with the most quality-filtered reads. Each radius represents a predicted gene. Each concentric ring represents a metagenomic sample. Genes are black if their breadth of coverage is < 90% and color-coded by site if their breadth of coverage is ≥ 90%. The genes are arranged by breadth of coverage. Site tropisms of S. infantis, S. mitis, and S. oralis were confirmed, as most genes of S. infantis were detected in most samples from tongue dorsum, most genes of S. mitis were detected in samples from buccal mucosa, and most genes of S. oralis were detected in samples from supragingival plaque. The genomes displayed here are the genomes from each species with the greatest Q2Q3 mean depth of coverage averaged across all metagenomes and whose species designation at NCBI matched our corrected species designations.
As an additional check on our finding of different site tropisms for closely related oral streptococci, we analyzed a set of published metagenome-assembled genomes (MAGs) that were assembled and binned from the HMP metagenomes by Pasolli et al. (2019). As a MAG can be assembled only from a metagenome in which the taxon is represented by many sequence reads, the site from which a MAG can be assembled is an indicator of where these species are highly abundant. For the 188 MAGs that Pasolli et al. assembled from an oral metagenome and identified as Streptococcus, we checked the species designation by calculating the ANI shared between our reference genomes and the MAGs (Table S6). Using these ANI values, we assigned some of the MAGs new species-level designations according to their similarity to the reference genomes. This process yielded 33 MAGs we identified as S. mitis, all of which were assembled from buccal mucosa or keratinized gingiva samples; 37 MAGs identified as S. oralis, all assembled from supragingival or subgingival plaque; and 7 MAGs identified as S. infantis, all assembled from the tongue dorsum (Table S7). Thus, the identity of MAGs assembled from the HMP metagenomes validates the site tropisms we detected by metagenomic mapping to reference genomes.
Whereas prior results using short regions of the 16S rRNA gene had suggested that a cluster of Streptococcus species contained oral generalists, genomic read mapping resolved this cluster into individual species that primarily localize to different sites. The diagram in Fig. 4A, modified from Mark Welch et al. (2019), shows habitat specialization based on oligotyping data in which the genus was divided into subsets, most of which were clusters of related species. The cluster containing S. mitis, S. oralis, and S. infantis appeared to be a generalist. Mapping shotgun sequencing reads identified a dozen Streptococcus species (Fig. 4B). These species include buccal mucosa specialist S. mitis, tongue dorsum specialists S. infantis, S. australis, S. parasanguinis, S. rubneri, and S. salivarius, as well as supragingival plaque specialists S. oralis, S. cristatus, S. gordonii, and S. sanguinis. Each of these species had a significantly greater mean relative abundance in the metagenomes from their preferred site (p ≤ 4.472 x 10-26, Table S8). These data further show the importance of the species-level resolution provided by shotgun sequencing read mapping. Taken alone the 16S rRNA data classified the Mitis group as an apparent generalist taxon; however, the shotgun sequencing mapping data show that the species previously lumped into the Mitis group (S. mitis, S. infantis, S. australis, S. oralis, and S. cristatus) are specialists with preferences for either the buccal mucosa, tongue dorsum, or supragingival plaque.
Functional annotation of S. mitis, S. oralis, and S. infantis reveals no species-specific core functions that could drive localization to different sites.
Pangenomics, which entails the identification of essential core and nonessential accessory genes for a set of related microbial genomes, can be used to identify genes involved in adaptation to distinct microhabitats that may give rise to the spatial distribution patterns revealed by metagenomics (Scholz et al., 2016; Nayfach et al., 2016; Delmont and Eren, 2018). Seeking to identify genes underlying these differential distribution patterns, we constructed a pangenome of the genus Streptococcus using the reference genome set generated above. The visualization of the pangenome shows gene clusters, groups of genes with high amino acid sequence similarity, clustered according to their prevalence across the genomes and the genomes clustered according to their gene content (Fig. 5). Arranging the genomes based on the 18,895 gene clusters of the pangenome gave results that were broadly consistent with the phylogenomic tree: both analysis methods grouped the same genomes into species-level clusters with multiple genomes and placed the S. pneumoniae and S. pseudopneumoniae genomes within the S. mitis clade and the S. peroris genome within the S. infantis clade (Figs. S1, S4). A set of 606 core gene clusters, constituting 27–38% of the clusters in each genome, was found across all the reference genomes (Fig. 5).
Figure 5: Pangenome of the genus Streptococcus in the human oral cavity.
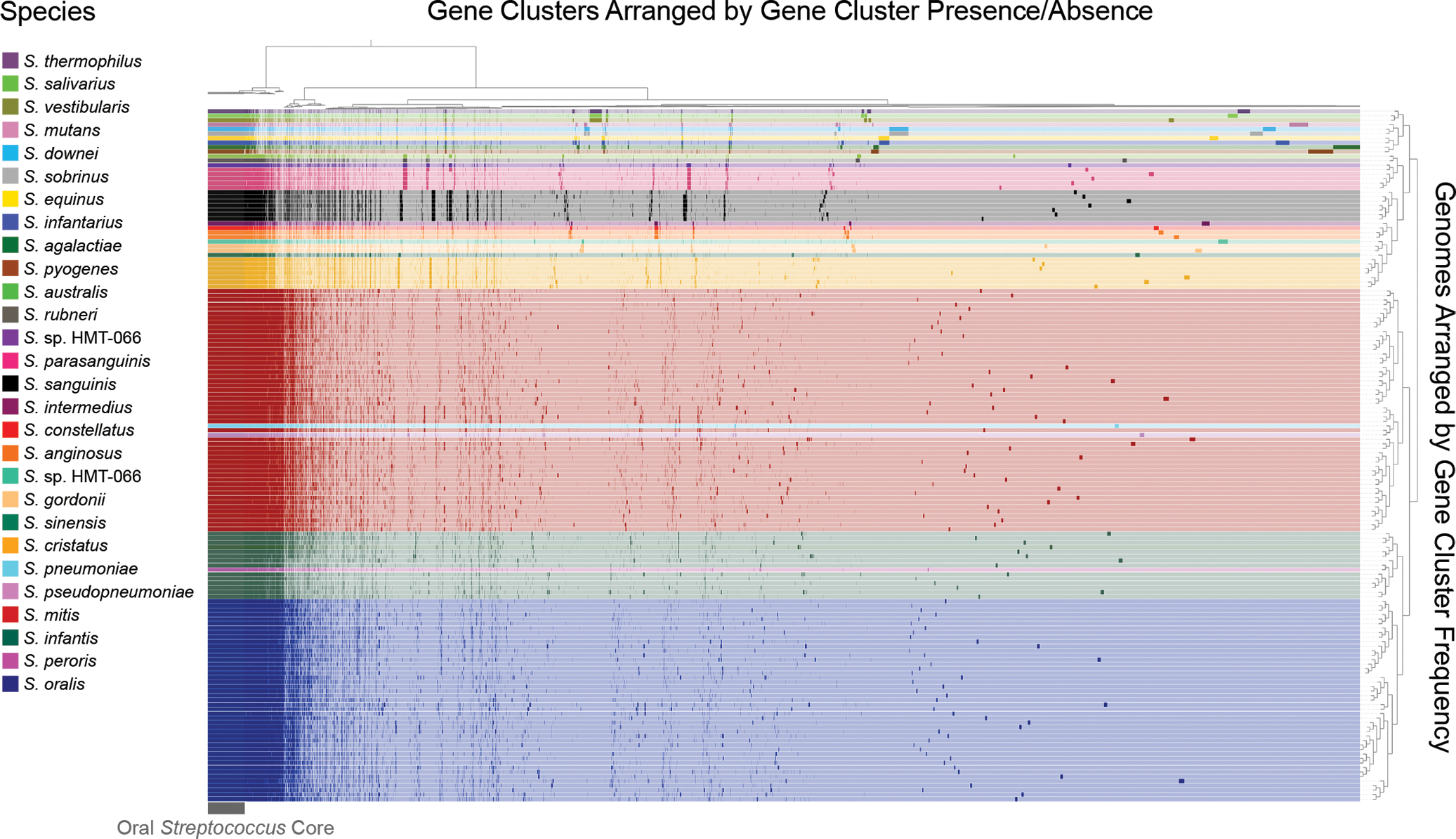
The genomes are clustered by the frequencies of their gene clusters and color-coded by species. The gene clusters are clustered according to their presence or absence in each genome; presence is denoted by a dark shade and absence by a light shade of the color representing each species. These 154 genomes of oral streptococci have shared ANI values of < 95%, so species with more genomic nucleotide-level diversity have a larger number of representative genomes. The number of representatives is also affected by the availability of genomes for a species. The 18,895 distinct gene clusters of the pangenome include 606 core genes that occur in every genome. Large sets of species-specific core genes distinguish several of the species.
Although some Streptococcus species in the pangenome possess large blocks of species-specific gene clusters, others – notably S. mitis, S. oralis, and S. infantis – do not. Inspection of the pangenome shows blocks of gene clusters characteristic of individual species such as S. cristatus, S. sanguinis, and S. parasanguinis, as well as blocks characteristic of groups of closely related species such as S. salivarius, S. vestibularis, and S. thermophilus (Fig. 5). S. sanguinis, for example, has a well-defined block of species-specific core genes that account for 2.3–2.5% of the gene clusters in its genome. By contrast, and consistent with the results of a similar pangenome constructed by Velsko et al. (2019), S. mitis, S. oralis, and S. infantis appear to share many genes and do not have major blocks of gene clusters unique to each species. The apparent similarity of the species-specific core for these three species contrasts with the observed differences in their distribution.
The genomic diversity within the Mitis group was explored at higher resolution by constructing a targeted pangenome with only S. mitis, S. infantis, and S. oralis genomes. In the targeted pangenome, constructed using a more stringent value of the “minbit” parameter for eliminating clusters with low amino acid sequence similarity, modest blocks of species-specific core genes were detected for each of S. mitis, S. oralis, and S. infantis (Fig. 6; Table S9). S. mitis and S. oralis also shared 44 core gene clusters, while S. infantis shared 1 core gene cluster with S. mitis and 2 with S. oralis. To determine whether these gene clusters had unique and potentially niche-defining functions, we carried out functional annotation of each called gene using the Pfam, NCBI COG, and eggNOG databases. The results indicated that nearly all the species-specific core gene clusters were annotated with functions found in all three species. Depending on the annotation source, each species had from zero to two annotated functions that were both unique and core to the species. No annotated function was unique and core to two species. Thus, using a more stringent clustering parameter revealed a set of species-specific core genes for each taxon but these were distinguished by amino acid divergence and not by functional divergence, as discerned using current annotation databases.
Figure 6: Pangenome of S. mitis, S. oralis, and S. infantis.
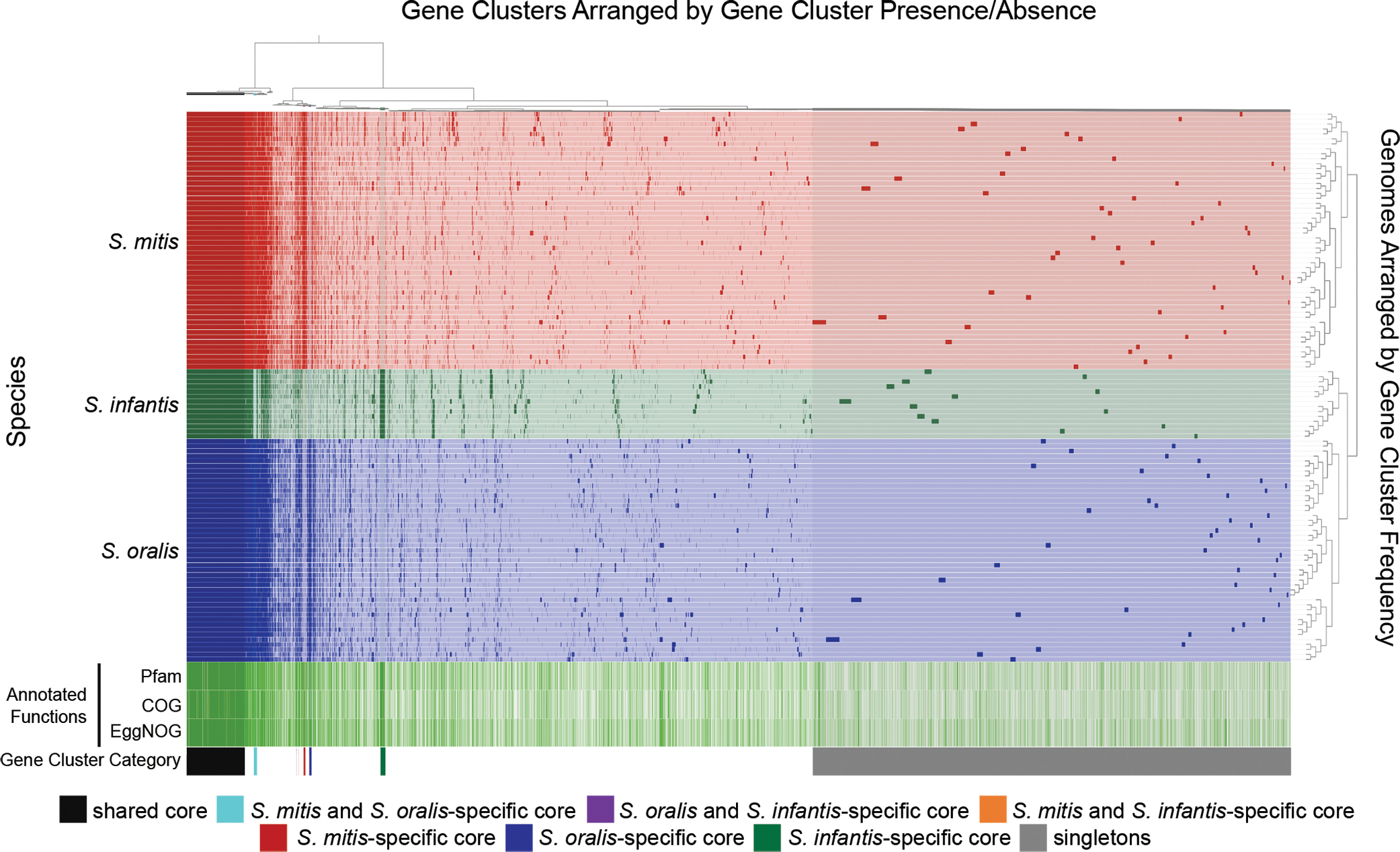
This pangenome is prepared with a more stringent clustering parameter than that of Fig. 5 (see Methods) and shows small blocks of species-specific core genes in addition to a large block of core genes shared among all three species. Genes and genomes are clustered as in Fig. 5. Gene clusters present in all genomes are marked shared core. Gene clusters present in all genomes of one or two species and none of the genomes of the other species are marked as species-specific core. Gene clusters present in a single genome are marked as singletons. The “Annotated Functions” layers indicate whether the gene cluster has (green) or has not (white) been assigned a Pfam, COG, or eggNOG function. In all cases where a species-specific core gene received a functional annotation, genes in the other two species were also assigned that functional annotation, with these exceptions: in S. mitis “PrsW family intramembrane metalloprotease” (Pfam) and “membrane proteinase PrsW, cleaves anti-sigma factor RsiW, M82 family (PrsW)” (COG); in S. oralis "TraX protein” (Pfam); and in S. infantis “UPF0126 domain” and “Competence protein” (Pfam) and “Uncharacterized membrane protein YeiH (YadS) (PDB:5WUC)” and “DNA uptake channel protein ComEC, N-terminal domain (ComEC) or DNA uptake channel protein ComEC C-terminal domain, metallo-beta-lactamase superfamily (ComEC)” (COG). Thus, although species-specific core genes could be detected at the level of protein sequence, functional annotation was similar for all three species.
Discussion
To analyze the distribution of the oral streptococci it was first necessary to define the boundaries circumscribing these species. Streptococcus mitis, S. oralis, and S. infantis each possess unusually high within-species genomic divergence as measured by ANI, which raises the question of whether the species as currently defined are biologically meaningful, or whether their genomic diversity should be recognized as additional species. While there is not a standard prokaryotic species definition, bacterial species are generally considered to consist of collections of strains that are genomically coherent; they share a greater gene content and sequence similarity with each other than with other species (Konstantinidis and Tiedje, 2005). Intraspecies genomic coherence is maintained through gene exchange, and barriers to recombination have been proposed as the limits to bacterial and archaeal species (Bobay and Ochman, 2017). A genomic distance of around 95% ANI is often recommended as a species boundary as this similarity score circumscribes most recognized species (Konstantinidis and Tiedje, 2005; Jain et al., 2018; Olm et al., 2020; Parks et al., 2020). However, the members of multiple recognized Streptococcus Mitis group species share mean ANIs between 90% and 95% (Jensen et al., 2016). Therefore, when Parks et al. (2020) proposed a new taxonomy for the RefSeq genomes in the GTDB using a 95% ANI species boundary, they subdivided these species into as many as 50 species clusters. Our phylogenomic analysis supports the idea that the current named oral Streptococcus species, including S. mitis, S. oralis, and S. infantis, are genomically coherent. The Streptococcus spp. genomes we analyzed formed distinct clusters with respect to ANI that corresponded to existing species classifications. In addition to genomic coherence, biologically meaningful species are expected to share consistent phenotypes (Konstantinidis and Tiedje, 2005). Mapping indicated that members of a named species shared a common localization phenotype, which differed between closely related species like S. mitis, S. oralis, and S. infantis. These results support the validity of the recognized oral streptococci species and highlight the difficulty of selecting a universal genomic similarity threshold to circumscribe all prokaryote species.
For complex microbial populations composed of numerous closely-related species and strains, genome-scale analysis provides the resolution necessary to demonstrate site-tropism. We determined that all the major oral Streptococcus species were site-specialists. Using the greater resolution provided by mapping whole-genome sequencing data, we could determine the localization for Streptococcus species and assess how the site-specialist hypothesis applied to closely related species, not distinguishable by their 16S rRNA gene sequence. Following a common trend for oral taxa (Mager et al., 2003; Segata et al., 2012), each Streptococcus species was most abundant in one of three groups of sites containing either the buccal mucosa, tongue dorsum, or supragingival plaque. S. sanguinis, S. cristatus, and S. gordonii were among the most abundant species in supragingival plaque, confirming prior findings based on 16S rRNA gene sequencing data (Huse et al., 2012; Peterson et al., 2013; Eren et al., 2014; Hall et al., 2017). S. australis, S. salivarius, and S. parasanguinis were among the most abundant species on the tongue dorsum, likewise confirming the findings of 16S rRNA gene studies (Aas et al., 2005; Mark Welch et al., 2014; Eren et al., 2014; Bernardi et al., 2020). Our analysis indicates S. rubneri, a more recently identified species not included in earlier studies, is also a tongue dorsum specialist. Whole-genome sequencing data differentiated between S. mitis, S. oralis, and S. infantis, which could not be distinguished in 16S rRNA gene studies (Zaura et al., 2009; Huse et al., 2012; Mark Welch et al., 2014; Eren et al., 2014; Hall et al., 2017). When the attempt was made to distinguish between these species, S. oralis and S. infantis were either scarce or undetected (Aas et al., 2005; Peterson et al., 2013). Our test with simulated data indicates that mapping whole-genome sequencing reads can distinguish between all oral Streptococcus species allowing for a more accurate comparison of the abundance of species between sites. The results of mapping HMP whole-genome sequencing data indicated that the three closely related species S. mitis, S. oralis, and S. infantis preferentially localized to different sites. The distribution of these species demonstrates that taxonomy is not always a clear indicator of the spatial niche where oral species specialize.
The HMP metagenomes we analyzed were only sampled from healthy subjects. Common oral diseases like caries and periodontitis substantially alter the biochemistry of the oral cavity, potentially changing the most favorable habitats for oral streptococci. For example, the increased acidification associated with carious lesions corresponds to increases in the abundance of S. mutans and a decrease in S. sanguinis in supragingival plague (Gross et al., 2012; Richards et al., 2017). The inflammation and gum recession at the periodontal pocket accompanying periodontitis have been found to correspond with an increase in the abundance of S. constellatus, S. intermedius, S. mutans and S. sp. HMT-071 and a decrease in S. sanguinis in the subgingival plaque (Abusleme et al., 2013; Rams et al., 2014; Dani et al., 2016; Ai et al., 2017). Thus, we would not necessarily expect to find the same patterns of site-tropism we found in healthy subjects under conditions of oral disease.
Although each of the major oral streptococcus species was most abundant at one primary region, many species were also detected in a subset of samples at the other sites. Detection of site-specialists outside their favored sites could indicate the presence of a strain or sub-population with a site specialization different from the rest of the species. While the species Haemophilus parainfluenzae contains strains that apparently specialize in different sites (Utter et al., 2020), the mapping results for individual strains of oral streptococci showed no indication of differential site-tropism of strains. Instead, detection of species like S. mitis outside their primary sites might be due to the colonization of favorable microhabitats within unfavorable oral sites. For example, the supragingival plaque biofilm is heterogeneous and contains various complex structures (Mark Welch et al., 2016) in which specialized microhabitats for otherwise rare oral microbes may exist. A variety of habitats may also be created by temporal succession, as the abrasion of the tooth surface and the shedding of old host cells would be expected to create a fresh substrate for new biofilm formation, creating a shifting mosaic steady state in which supragingival plaque in both the initial and the late stages of successional development coexist. S. mitis is a primary colonizer of tooth surfaces and is abundant in new plaque (Nyvad and Kilian, 1990; Frandsen et al., 1991; Li et al., 2004); however, as dental plaque matures it begins to be supplanted by other taxa (Ramberg et al., 2003). Our detection of low abundances of S. mitis in supragingival plaque samples may correspond to the detection of S. mitis in patches of initial plaque. The low abundance of cells that primarily localize to other sites may also correspond to the detection of bacterial “tourists,” bacteria deposited at the site where the conditions are unfavorable for colonization and growth. Finally, our conclusions about the distribution of Streptococcus species are based on our analysis of metagenomic samples, which may have been biased by sampling methodology (McInnes and Cutting, 2010). Cells are shed into the saliva from all oral sites and dispersed throughout the oral cavity by salivary flow. Because the HMP sampling protocols did not include precautions to exclude saliva, the samples may include cells shed from other sites.
The distribution of the detectable Streptococcus species suggests specialized adaptation for different spatial niches within the oral cavity. To better understand the adaptation of Streptococcus species to different spatial niches within the mouth, we constructed a pangenome with the S. mitis, S. oralis, and S. infantis genomes to look for differences between the genes core to each species that might explain their spatial distribution. Yet we found only small differences in gene content and functional annotation between the species. Thus, phenotypic differences between the species may be due to subtle differences in genomic content – like small sequence differences in conserved genes, differences in gene expression, or differences in gene copy number. Lefébure and Stanhope (2007) previously found that numerous core genes shared between several other Streptococcus species, especially those related to colonization and biofilm formation, were subject to positive selection, supporting the idea that differences between protein-coding genes sharing similar functions may contribute to niche partitioning. One phenotype that would be reasonably expected to distinguish the three species is the capacity to adhere to different substrates. To resist the shearing force of salivary flow and remain in a preferred environment, non-motile streptococci must adhere to that site. Oral streptococci possess many adhesins that mediate highly specific adhesion to components of the acquired salivary pellicle, extracellular matrix, host cells, and other microbes (Nobbs et al., 2009). Interestingly, the S. mitis species-specific core included a gene cluster that received the Pfam annotation of “putative adhesin”, and that same annotation was assigned to a species-specific gene cluster present in 90% of the S. oralis genomes and two species-specific gene clusters that together were present in 93% of the S. infantis genomes. Putative adhesins such as these might contribute to differences in the localization of the species. This hypothesis is supported by the prior finding that, relative to other streptococci, S. mitis adheres better to the buccal mucosa and teeth and more poorly to the tongue dorsum (Liljemark and Gibbons, 1972). One limitation of this analysis is the limits to the accuracy and specificity of the tools presently available for functional annotation. Some of the core gene clusters specific to one or two species received no annotation, while others received annotations that were either vague or based on functions characterized for proteins from taxa as distant as eukaryotes.
To summarize, we demonstrated that all the major oral streptococci were site-specialists; they were most abundant in one of three different regions including either the buccal mucosa, tongue dorsum, or dental plaque. Even the closely related species S. mitis, S. oralis, and S. infantis displayed preferences for different oral sites. The substantial overlap in the core genes and gene functions between these three species suggests that subtle differences in genomic content may be sufficient to determine their different localization. Partitioning of ecological niches has been established as one mechanism maintaining biodiversity by permitting the coexistence of different species (Chesson, 2000). Evidence of nutrient partitioning has been found within the gut microbiome and shown to permit species coexistence (Tuncil et al., 2017; Goyal et al., 2018; Brochet et al., 2021). Spatial niche partitioning, like that exhibited by the oral streptococci, might likewise be a mechanism facilitating species coexistence in the oral microbiome. With a range of complex, polymicrobial biofilms distributed across a range of environments, the human oral microbiome provides a natural experiment for further investigation of spatial niche partitioning in the healthy microbiome.
Supplementary Material
Acknowledgments
We thank A. Murat Eren, Jonathan Giacomini, and Matthew Ramsey for helpful discussions and Rich Fox for expert systems administration and assistance with using the Josephine Bay Paul Center servers at the Marine Biological Laboratory. This work was supported by NIH National Institute of Dental and Craniofacial Research grants R01 DE022586 and R01 DE030136 (to G.G.B.) and R01 DE 027958 (to J.M.W.).
Footnotes
Conflict of interest
No, there is no conflict of interest.
References
- Aas JA, Paster BJ, Stokes LN, Olsen I, & Dewhirst FE (2005). Defining the normal bacterial flora of the oral cavity. Journal of Clinical Microbiology. 43(11),5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abranches J, Zeng L, Kajfasz JK, Palmer SR, Chakraborty B, Wen ZT, Richards VP, Brady LJ & Lemos JA (2018). Biology of oral streptococci. Microbiology Spectrum. 6(5),10.1128/microbiolspec.GPP3–0042-2018. doi: 10.1128/microbiolspec.GPP3-0042-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, & Diaz PI (2013). The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. The ISME Journal. 7(5),1016–1025. doi: 10.1038/ismej.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai D, Huang R, Wen J, Li C, Zhu J, & Xia LC (2017). Integrated metagenomic data analysis demonstrates that a loss of diversity in oral microbiota is associated with periodontitis. BMC Genom. 18:1041. doi: 10.1186/s12864-016-3254-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, Lawley TD, & Finn RD (2019). A new genomic blueprint of the human gut microbiota. Nature. 568(7753),499–504. doi: 10.1038/s41586-019-0965-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, & Lipman DJ (1990). Basic local alignment search tool. Journal of Molecular Biology. 215(3),403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Bernardi S, Karygianni L, Filippi A, Anderson AC, Zürcher A, Hellwig E, Vach K, Macchiarelli G, & Al-Ahmad A (2020). Combining culture and culture-independent methods reveals new microbial composition of halitosis patients’ tongue biofilm. Microbiologyopen. 9(2),e958. doi: 10.1002/mbo3.958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobay LM & Ochman H (2017). Biological species are universal across life's domains. Genome Biology and Evolution. 9(3),491–501. doi: 10.1093/gbe/evx026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet S, Quinn A, Mars RAT, Neuschwander N, Sauer U, & Engel P (2021). Niche partitioning facilitates coexistence of closely related honey bee gut bacteria. eLife. 10,e68583. doi: 10.7554/eLife.68583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caselli E, Fabbri C, D’Accolti M, Soffritti I, Bassi C, Mazzacane S, & Franchi M (2020). Defining the oral microbiome by whole-genome sequencing and resistome analysis: the complexity of the healthy picture. BMC Microbiology. 20(1),120. doi: 10.1186/s12866-020-01801-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesson P (2000). Mechanisms of maintenance of species diversity. Annual Review of Ecology, Evolution, and Systematics. 31(1),343–366. doi: 10.1146/annurev.ecolsys.31.1.343 [DOI] [Google Scholar]
- Chi F, Nolte O, Bergmann C, Ip M, & Hakenbeck R (2007). Crossing the barrier: evolution and spread of a major class of mosaic pbp2x in Streptococcus pneumoniae, S. mitis and S. oralis. International Journal of Medical Microbiology. 297(7–8),503–512. doi: 10.1016/j.ijmm.2007.02.009 [DOI] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, & Knight R (2009). Bacterial community variation in human body habitats across space and time. Science. 326(5960),1694–1697. doi: 10.1126/science.1177486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxen MA, Lee TD, Azana R, & Hoang LM (2018). Use of genomics to design a diagnostic assay to discriminate between Streptococcus pneumoniae and Streptococcus pseudopneumoniae. Microbial Genomics. 4(7),e000175. doi: 10.1099/mgen.0.000175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani S, Prabhu A, Chaitra KR, Desai NC, Patil SR, & Rajeev R (2016). Assessment of Streptococcus mutans in healthy versus gingivitis and chronic periodontitis: A clinico-microbiological study. Contemp Clin Dent 7(4),529–534. doi: 10.4103/0976-237X.194114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmont TO & Eren AM (2018). Linking pangenomes and metagenomes: the Prochlorococcus metapangenome. PeerJ. 6,e4320. doi: 10.7717/peerj.4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu WH, Lakshmanan A, & Wade WG (2010). The human oral microbiome. Journal of Bacteriology. 192,5002–5017. doi: 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati C, Hiller NL, Tettelin H, Muzzi A, Croucher NJ, Angiuoli SV, Oggioni M, Dunning Hotopp JC, Hu FZ, Riley DR, Covacci A, Mitchell TJ, Bentley SD, Kilian M, Ehrlich GD, Rappuoli R, Moxon ER, & Masignani V (2010). Structure and dynamics of the pan-genome of Streptococcus pneumoniae and closely related species. Genome Biology. 11(10),R107. doi: 10.1186/gb-2010-11-10-r107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn OJ (1964). Multiple comparisons using rank sums. Technometrics. 6,241–252. [Google Scholar]
- Eddy SR (2009). A new generation of homology search tools based on probabilistic inference. Genome Informatics. 23(1),205–211. doi: 10.1142/9781848165632_0019 [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 32(5),1792–1797. doi: 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Vineis JH, Morrison HG, & Sogin ML (2013). Correction: A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS One. 8(6),10.1371/annotation/afa5c40d-c604–46ae-84c4–82cb92193a5e. doi: 10.1371/annotation/afa5c40d-c604-46ae-84c4-82cb92193a5e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Borisy GG, Huse SM, & Mark Welch JL (2014). Oligotyping analysis of the human oral microbiome. Proceedings of the National Academy of Sciences of the United States of America. 111(28),E2875–E2884. doi: 10.1073/pnas.1409644111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren AM, Kief E, Shaiber A, Veseli I, Miller SE, Schechter MS, Fink I, Pan JN, Yousef M, Fogarty EC, Trigodet F, Watson AR, Esen ÖC, Moore RM, Clayssen Q, Lee MD, Kivenson V, Graham ED, Merrill BD, … Willis AD (2021). Community-led, integrated, reproducible multi-omics with anvi'o. Nature Microbiology. 6(1),3–6. doi: 10.1038/s41564-020-00834-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen EV, Pedrazzoli V, & Kilian M (1991). Ecology of viridans streptococci in the oral cavity and pharynx. Oral Microbiology and Immunology. 6(3),129–133. doi: 10.1111/j.1399-302x.1991.tb00466.x [DOI] [PubMed] [Google Scholar]
- Galili T (2015). dendextend: an R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics. 31(22),3718–3720. doi: 10.1093/bioinformatics/btv428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons RJ, Socransky SS, Sawyer S, Kapsimalis B, & MacDonald JB (1963). The microbiota of the gingival crevice area of man—II: The predominant cultivable organisms. Archives of Oral Biology. 8(3),281–289. doi: 10.1016/0003-9969(63)90020-7 [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, Socransky SS, de Araujo WC, & van Houte J (1964). Studies of the predominant cultivable microbiota of dental plaque. Archives of Oral Biology. 9(3),365–370. doi: 10.1016/0003-9969(64)90069-X [DOI] [PubMed] [Google Scholar]
- Gibbons RJ, Kapsimalis, & Socransky B, S. S. (1964). The source of salivary bacteria. Archives of Oral Biology. 9,101–103. doi: 10.1016/0003-9969(64)90052-4 [DOI] [PubMed] [Google Scholar]
- Gordon DF Jr. & Jong BB (1968). Indigenous flora from human saliva. Journal of Applied Microbiology. 16(2),428–29. doi: 10.1128/am.16.2.428-429.1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DF Jr. & Gibbons RJ (1966). Studies of the predominant cultivable micro-organisms from the human tongue. Archives of Oral Biology. 11(6),627–632. doi: 10.1016/0003-9969(66)90229-9 [DOI] [PubMed] [Google Scholar]
- Goyal A, Dubinkina V, & Maslov S (2018). Multiple stable states in microbial communities explained by the stable marriage problem. The ISME Journal. 2(12),2823–2834. doi: 10.1038/s41396-018-0222-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross EL, Beall CJ, Kutsch SR, Firestone ND, Leys EJ, & Griffen AL (2012). Beyond Streptococcus mutans: dental caries onset linked to multiple species by 16S rRNA community analysis. PLoS One 7(10),e47722. doi: 10.1371/journal.pone.0047722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MW, Singh N, Ng KF, Lam DK, Goldberg MB, Tenenbaum HC, Neufeld JD, Beiko RG, & Senadheera DB (2017). Inter-personal diversity and temporal dynamics of dental, tongue, and salivary microbiota in the healthy oral cavity. npj Biofilms and Microbiomes. 3,2. doi: 10.1038/s41522-016-0011-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch M, De Bruyne K, Cleenwerck I, Bub A, Cho GS, Watzl B, Snauwaert I, Franz CMAP, & Vandamme P (2013). Streptococcus rubneri sp. nov., isolated from the human throat. International Journal of Systematic and Evolutionary Microbiology. 63(11),4026–4032. doi: 10.1099/ijs.0.048538-0 [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, & Bork P (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular Biology and Evolution. 34(8),2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, von Mering C, & Bork P (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research. 47(D1),D309–D314. doi: 10.1093/nar/gky1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse SM, Ye Y, Zhou Y, & Fodor AA (2012). A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One. 7(6),e34242. doi: 10.1371/journal.pone.0034242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium. (2012). Structure, function and diversity of the healthy human microbiome. Nature. 486(7402),207–214. doi: 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, & Hauser LJ (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 11,119. doi: 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, & Aluru S High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. (2018). Nature Communications. 9(1),5114. doi: 10.1038/s41467-018-07641-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson HF (1994). Adherence and accumulation of oral streptococci. Trends in Microbiology. 2(6),209–212. doi: 10.1016/0966-842x(94)90114-k [DOI] [PubMed] [Google Scholar]
- Jensen A, Scholz CFP, & Kilian M (2016). Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. International Journal of Systematic and Evolutionary Microbiology. 66(11),4803–4820. doi: 10.1099/ijsem.0.001433 [DOI] [PubMed] [Google Scholar]
- Kilian M & Tettelin H (2019). Identification of virulence-associated properties by comparative genome analysis of Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, three S. oralis subspecies, and S. infantis. mBio. 10(5),e01985–19. doi: 10.1128/mBio.01985-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT & Tiedje JM (2005). Genomic insights that advance the species definition for prokaryotes. Proceedings of the National Academy of Sciences of the United States of America. 102(7),2567–2572. doi: 10.1073/pnas.0409727102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolenbrander PE, & London J (1993). Adhere today, here tomorrow: oral bacterial adherence. Journal of Bacteriology. 175(11),3247–3252. doi: 10.1128/jb.175.11.3247-3252.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B & Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods. 9,357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefébure T & Stanhope MJ (2007). Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biology. 8(5),R71. doi: 10.1186/gb-2007-8-5-r71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, & Durbin R (2009). The sequence alignment/map format and SAMtools. Bioinformatics. 25(16),2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Helmerhorst EJ, Leone CW, Troxler RF, Yaskell T, Haffajee AD, Socransky SS, & Oppenheim FG (2004). Identification of early microbial colonizers in human dental biofilm. Journal of Applied Microbiology. 97(6),1311–1318. doi: 10.1111/j.1365-2672.2004.02420.x [DOI] [PubMed] [Google Scholar]
- Liljemark WF & Gibbons RJ (1972). Proportional distribution and relative adherence of Streptococcus miteor (mitis) on various surfaces in the human oral cavity. Infection and Immunity. 6(5),852–859. doi: 10.1128/IAI.6.5.852-859.1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, Brady A, Creasy HH, McCracken C, Giglio MG, McDonald D, Franzosa EA, Knight R, White O, & Huttenhower C (2017). Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 550(7674),61–66. doi: 10.1038/nature23889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DL, Ximenez-Fyvie LA, Haffajee AD, & Socransky SS (2003). Distribution of selected bacterial species on intraoral surfaces. Journal of Clinical Periodontology. 30(7),644–654. doi: 10.1034/j.1600-051x.2003.00376.x [DOI] [PubMed] [Google Scholar]
- Mark Welch JL, Utter DR, Rossetti BJ, Mark Welch DB, Eren AM, & Borisy GG (2014). Dynamics of tongue microbial communities with single-nucleotide resolution using oligotyping. Frontiers in Microbiology. 5,568. doi: 10.3389/fmicb.2014.00568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Welch JL, Rossetti BJ, Rieken CW, Dewhirst FE, & Borisy GG (2016). Biogeography of a human oral microbiome at the micron scale. Proceedings of the National Academy of Sciences of the United States of America. 9:113(6),E791–800. doi: 10.1073/pnas.1522149113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Welch JL, Dewhirst FE, & Borisy GG (2019). Biogeography of the oral microbiome: The site-specialist hypothesis. Annual Review of Microbiology. 73,335–358. doi: 10.1146/annurev-micro-090817-062503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Welch JL, Ramírez-Puebla ST, & Borisy GG (2020). Oral microbiome geography: Micron-scale habitat and niche. Cell Host & Microbe. 28(2),160–168. doi: 10.1016/j.chom.2020.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Pérez C, Greening C, Bay SK, Lappan RJ, Zhao Z, De Corte D, Hulbe C, Ohneiser C, Stevens C, Thomson B, & Stepanauskas R (2022). Phylogenetically and functionally diverse microorganisms reside under the Ross Ice Shelf. Nat. Commun. 13(1):1–5. doi: 10.1038/s41467-021-27769-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes P & Cutting M (2010). Manual of procedures for Human Microbiome Project, Version 12.0
- Minoche AE, Dohm JC, & Himmelbauer H (2011). Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biology. 12(11),R112. doi: 10.1186/gb-2011-12-11-r112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Rodriguez-Mueller B, Garud N, & Pollard KS (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Research. 26(11),1612–1625. doi: 10.1101/gr.201863.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobbs AH, Lamont RJ, & Jenkinson HF (2009). Streptococcus adherence and colonization. Microbiology and Molecular Biology Reviews. 73(3),407–450. doi: 10.1128/MMBR.00014-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyvad B & Kilian M (1990). Comparison of the initial Streptococcal microflora on dental enamel in caries-active and in caries-inactive individuals. Caries Research. 24(4),267–272. doi: 10.1159/000261281 [DOI] [PubMed] [Google Scholar]
- Ogle DH, Doll JC, Wheeler P, & Dinno A (2021). FSA: Fisheries stock analysis. R package version 0.9.1, https://github.com/droglenc/FSA. [Google Scholar]
- Olm MR, Crits-Cristoph A, Diamond S, Lavy A, Matheus Carnevali PB, & Banfield JF (2020). Consistent metagenome-derived metrics verify and define bacterial species boundaries. mSystems. 5(1),e00731–00719. doi: 10.1128/mSystems.00731-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E & Schliep K (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics. 35(3),526–528. doi: 10.1093/bioinformatics/bty633 [DOI] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, & Tyson GW (2014). Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research. 25(7),1043–1055. doi: 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Chuvochina M, Chaumeil PA, Rinke C, Mussig AJ, & Hugenholtz P (2020). Complete domain-to-species taxonomy for bacteria and archaea. Nature Biotechnology. 38(9),1079–1086. doi: 10.1038/s41587-020-0501-8 [DOI] [PubMed] [Google Scholar]
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P, Collado MC, Rice BL, DuLong C, Morgan XC, Golden CD, Quince C, Huttenhower C, & Segata N (2019). Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176(3),649–662.e20. doi: 10.1016/j.cell.2019.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SN, Snesrud E, Liu J, Ong AC, Kilian M, Schork NJ, & Bretz W (2013). The dental plaque microbiome in health and disease. PLoS One. 8(3),e58487. doi: 10.1371/journal.pone.0058487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, & Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 5(3),e9490. doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard L, Glover RH, Humphris S, Elphinstone JG, & Toth IK (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Analytical Methods. 8(1),12–24. doi: 10.1039/C5AY02550H [DOI] [Google Scholar]
- Proctor DM and Relman D (2017). The landscape ecology and microbiota of the human nose, mouth, and throat. Cell Host & Microbe. 21(4),421–432. doi: 10.1016/j.chom.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rams TE, Degener JE, & van Winkelhoff AJ (2014). Antibiotic resistance in human chronic periodontitis microbiota. J. Periodontol. 85(1),160–169. doi: 10.1902/jop.2013.130142 [DOI] [PubMed] [Google Scholar]
- Ramberg P, Sekino S, Uzel NG, Socransky S, & Lindhe J (2003). Bacterial colonization during de novo plaque formation. Journal of Clinical Periodontology. 30(11),990–995. doi: 10.1034/j.1600-051x.2003.00419.x [DOI] [PubMed] [Google Scholar]
- Richards VP, Alvarez AJ, Luce AR, Bedenbaugh M, Mitchell ML, Burne RA, & Nascimento MM (2017). Microbiomes of site-specific dental plaques from children with different caries status. Infect. Immun. 85(8):e00106–e00117. doi: 10.1128/IAI.00106-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M, D'Amore R, Ijaz UZ, Hall N, & Quince C (2016). Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics. 17,125. doi: 10.1186/s12859-016-0976-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, & Segata N (2016). Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nature Methods. 13(5),435–438. doi: 10.1038/nmeth.3802 [DOI] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, & Huttenhower C (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nature Methods. 9(8),811–814. doi: 10.1038/nmeth.2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Samaranayake LP, Yip HK, & Dyson JE (2002). Bacterial and yeast flora of root surface caries in elderly, ethnic Chinese. Oral Diseases. 8(4),207–217. doi: 10.1034/j.1601-0825.2002.01796.x [DOI] [PubMed] [Google Scholar]
- Sieradzki ET, Morando M, & Fuhrman JA (2021). Metagenomics and quantitative stable isotope probing offer insights into metabolism of polycyclic aromatic hydrocarbon degraders in chronically polluted seawater. mSystems. 6(3),e00245–21. doi: 10.1128/mSystems.00245-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa S, Mende DR, Zeller G, Izquierdo-Carrasco F, Berger SA, Kultima JR, Coelho LP, Arumugam M, Tap J, Nielsen HB, Rasmussen S, Brunak S, Pedersen O, Guarner F, de Vos WM, Wang J, Li J, Doré J, Ehrlich SD, Stamatakis A, & Bork P (2013). Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 10(12),1196–1199. doi: 10.1038/nmeth.2693 [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, & Koonin EV (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Research. 28(1),33–36 doi: 10.1093/nar/28.1.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetz G, Vikina D, Brown S, Zappile P, Dolgalev I, Tsirigos A, Heguy A, & Tetz V (2019). Draft genome sequence of Streptococcus halitosis sp. nov., isolated from the dorsal surface of the tongue of a patient with halitosis. Microbiology Resource Announcements. 8(4),e01704–e01718. doi: 10.1128/MRA.01704-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncil YE, Xiao Y, Porter NT, Reuhs BL, Martens EC, & Hamaker BR (2017). Reciprocal prioritization to dietary glycans by gut bacteria in a competitive environment promotes stable coexistence. mBio. 8(5),e01068–17. doi: 10.1128/mBio.01068-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, & Gordon JI (2007). The human microbiome project. Nature. 449(7164),804–810. doi: 10.1038/nature06244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utter DR, Borisy GG, Eren AM, Cavanaugh CM, & Mark Welch JL (2020). Metapangenomics of the oral microbiome provides insights into habitat adaptation and cultivar diversity. Genome Biology. 21(1),293. doi: 10.1186/s13059-020-02200-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dongen S & Abreu-Goodger C (2012). Using MCL to extract clusters from networks. Methods in Molecular Biology. 804,281–295. doi: 10.1007/978-1-61779-361-5_15 [DOI] [PubMed] [Google Scholar]
- Velsko IM, Perez MS, & Richards VP (2019). Resolving phylogenetic relationships for Streptococcus mitis and Streptococcus oralis through core- and pan-genome analyses. Genome Biology and Evolution. 11(4),1077–1087. doi: 10.1093/gbe/evz049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, & Hong PY (2020). Genome-resolved metagenomics and antibiotic resistance genes analysis in reclaimed water distribution systems. Water. 12(12),3477. doi: 10.3390/w12123477 [DOI] [Google Scholar]
- Zaura E, Keijser BJF, Huse SM, & Crielaard W (2009). Defining the healthy "core microbiome" of oral microbial communities. BMC Microbiology. 9,259. doi: 10.1186/1471-2180-9-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, & Miller W (2000). A greedy algorithm for aligning DNA sequences. Journal of Computational Biology. 7(1–2),203–214. doi: 10.1089/10665270050081478 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.