

SUMMARY
Methods for acquiring spatially resolved omics data from complex tissues use barcoded DNA arrays of low- to sub-micrometer features to achieve single-cell resolution. However, fabricating such arrays (randomly assembled beads, DNA nanoballs or clusters) requires sequencing barcodes in each array, limiting cost-effectiveness and throughput. Here, we describe a vastly scalable stamping method to fabricate polony gels, arrays of ~1-micrometer clonal DNA clusters bearing unique barcodes. By enabling repeatable enzymatic replication of barcode patterned gels, this method, compared with the sequencing-dependent array fabrication, reduced cost by at least 35-fold and time to approximately 7 hours. The gel stamping was implemented with a simple robotic arm and off-the-shelf reagents. We leveraged the resolution and RNA capture efficiency of polony gels to develop Pixel-seq, a single-cell spatial transcriptomic assay, and applied it to map the mouse parabrachial nucleus and analyze changes in neuropathic pain-regulated transcriptomes and cell-cell communication after nerve ligation.
Keywords: DNA array, DNA stamping, polony gel, Pixel-seq, spatial transcriptomics, parabrachial nucleus, chronic pain
In Brief:
Polony gels, arrays of micron-scale DNA clusters bearing unique barcodes, enable repeatable, gel-to-gel array replication and in situ capture of tissue RNAs with high resolution and efficiency. Pixel-seq, a polony gel-based, single-cell spatial transcriptomic assay, reveals neuronal and glial heterogeneity and chronic pain-induced changes in the mouse parabrachial nucleus.
Graphical Abstract
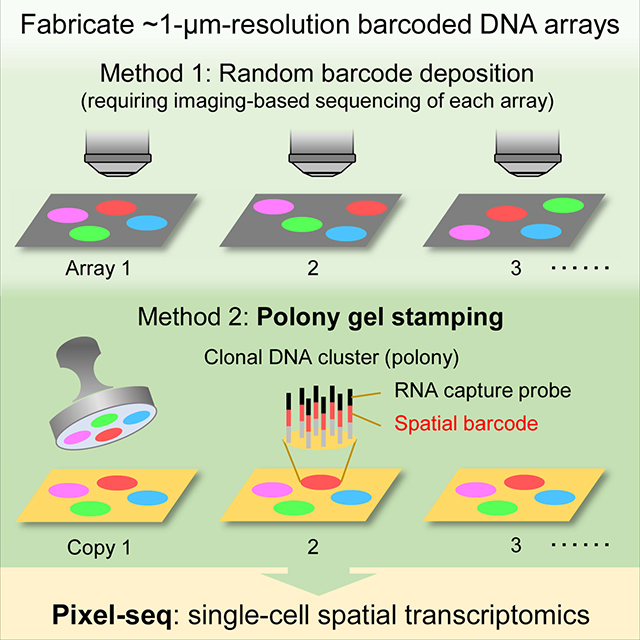
INTRODUCTION
Spatially barcoded DNA arrays are increasingly used for in situ capture and sequencing of RNAs and proteins to map the structure and function of heterogeneous tissues (Chen et al., 2022; Cho et al., 2021; Rodriques et al., 2019; Stahl et al., 2016; Stickels et al., 2021; Vickovic et al., 2019; Vickovic et al., 2022). To achieve single-cell resolution, DNA arrays require features significantly smaller than cells to delineate different shapes. Traditional spotting (DeRisi et al., 1996; Schena et al., 1995) or light-directed synthesis (Fodor et al., 1991) methods for the deposition or in situ synthesis of sequence-defined oligonucleotides at specific array positions on a substrate often generate features larger than mammalian cells (> 10 μm) with significant gaps to prevent feature merging. Recent advances of spatial transcriptomics utilized random arrays of smaller features such as DNA-coated beads (Rodriques et al., 2019; Vickovic et al., 2019), DNA nanoballs (Chen et al., 2022), and polymerase colonies (known as polonies (Gu et al., 2014) or DNA clusters (Cho et al., 2021)), all requiring decoding feature barcodes by sequencing each array in specialized flowcells. The barcode sequencing is a major cost- and rate-limiting factor of scaling up the array production; for example, sequencing barcodes in 38 tiles of 0.8 mm2 in an Illumina MiSeq flowcell (Cho et al., 2021) added a cost of ~$30 per mm2 and a time of 3–4 hours per run scaling linearly with barcode length and array size. It is desirable to develop sequencing-independent fabrication, which requires a paradigm shift in our underlying approach.
A possible method for simple and fast array fabrication is microcontact printing (Xia and Whitesides, 1998) using an elastomeric stamp to simultaneously copy arrayed molecules to a substrate. However, it has been an unsolved problem to construct a barcoded array on a stamp allowing consecutive printing without progressive decline of feature resolution and printed DNA amounts. Here, we report that polonies formed on the surface of an elastomeric, crosslinked polyacrylamide “stamp gel” as templates can be efficiently copied to many “copy gels” by DNA polymerase-catalyzed chain extension (Figure 1A). The gel-to-gel replication reliably achieved sub-micrometer resolution because all primers and templates are covalently attached to the gels to prevent DNA diffusion. Unlike traditional stamping requiring “re-inking” a stamp for consecutive printing (Lange et al., 2004), the enzymatic replication does not consume templates on the stamp. Notably, the stamping is also facilitated by DNA bridge amplification (Bentley et al., 2008) on gel surfaces to achieve a high copying efficiency and intensify faint prints. To obtain a spatial barcode map for a series of prints, only one or a few copy gels need to be sequenced. Additionally, copy gels can serve as stamps for next fabrication rounds. By utilizing polony gels, we demonstrated polony-indexed library-sequencing (Pixel-seq) for tissue mapping with high resolution and RNA capture efficiency (e.g., a mean of ~1,000 unique molecular identifiers (UMIs)/10 × 10 μm2 in mouse tissue).
Figure 1. Fabrication and characterization of polony gels.
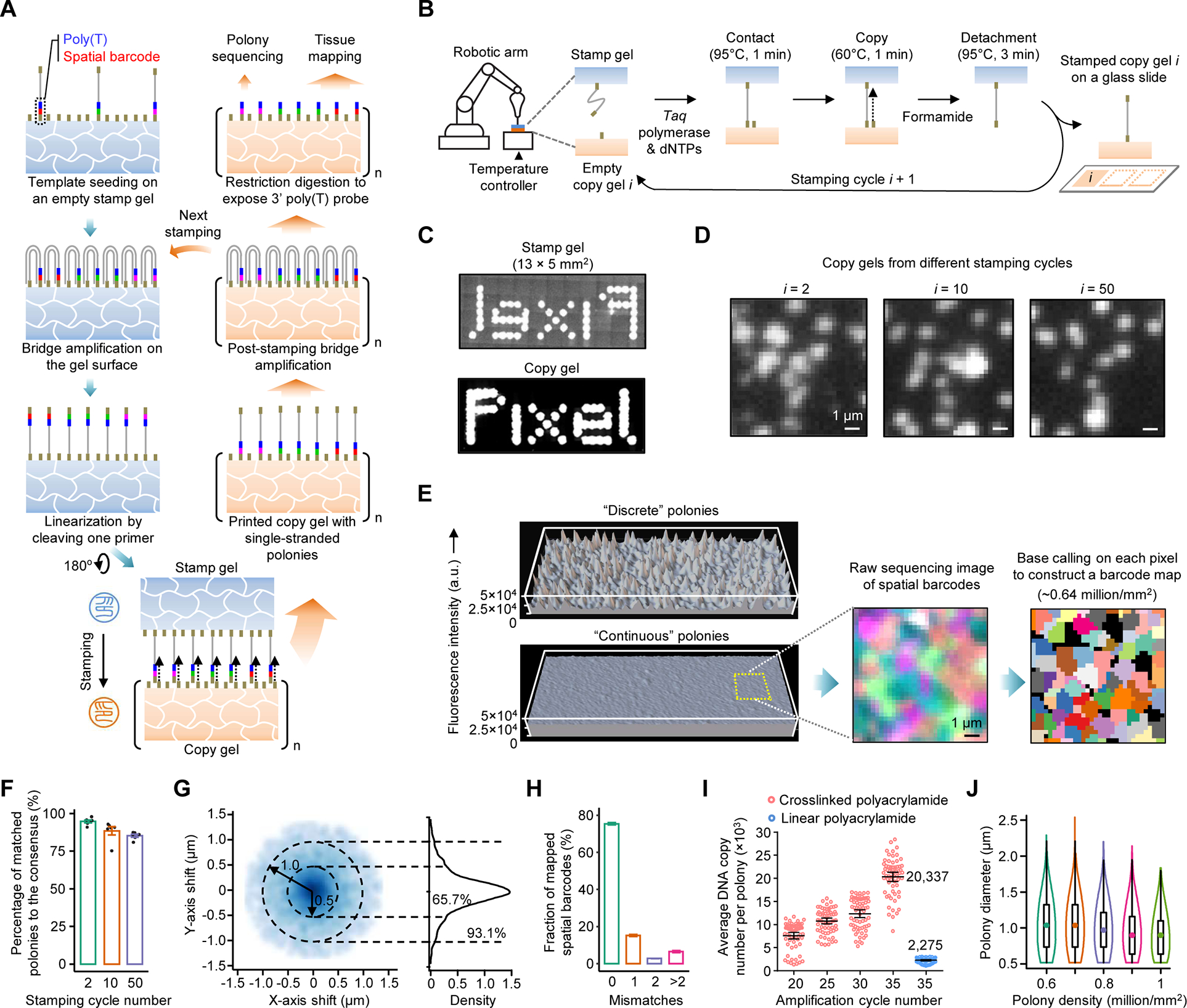
(A) Schematic of the amplifiable DNA stamping. USER-linearized single-stranded polonies are copied from a stamp to many copy gels. Copied DNAs are further bridge amplified to complete the replication. A few copy gels are used as the stamp for next fabrication or for polony sequencing to create a spatial barcode map; the majority are used for tissue mapping assays.
(B) A gel-to-gel DNA copying process automated with a stamping device.
(C) Millimeter-scale images of SYBR Green-stained polonies in a stamp and a copy gels. For comparison, templates were seeded on the masked, ~40-μm-thick stamp gel and amplified to polonies showing a pattern of the word “Pixel”.
(D) Images of SYBR Green-stained polonies from the 2nd, 10th, and 50th stamping cycles. The low-density polony gels (~105/mm2) were selected to facilitate visual comparison.
(E) 3D intensity profiles of SYBR Green-stained, discrete and continuous polonies amplified from templates seeded at the same density by 35 cycles.
(F) Four-channel sequencing images of two high-density polony gels (~8 × 105/mm2) from the 2nd and 10th stamping cycles. A spatial barcode map was generated by the pixel-level base calling.
(G) Box plot of the percentages of polonies in copy gels matched the consensus. Data represent means of six sampled gel positions each found with 195 to 332 polonies; error bars, standard deviation.
(H) 2D and 1D density plots of relative positions of polony centers in copy gels from the consensus. Dash circles denote center drifts of 0.5 and 1 μm. n = 4,521 and 11,441 for polonies at the low and high densities, respectively.
(I) Box plot of barcode error rates in eluted DNAs from two copy gels detected by Illumina sequencing.
(J) Comparison of polony bridge amplification efficiencies for two gel substrates. The linear polyacrylamide coating was prepared by a reported Illumina method.
(K) Violin plot of measured diameters of polonies at different densities. n = ~0.6 to 1 million.
We applied Pixel-seq to analyze the mouse parabrachial nucleus (PBN), a brain region in the pons for relaying sensory information (e.g., visceral malaise, taste, temperature, itch, and pain) to forebrain structures (Palmiter, 2018). Its heterogeneous structure and cell components remain poorly understood. By creating the first cell atlas of the PBN, we identified region-specific distributions of previously known and newly found neuron types. By precise anatomical and transcriptomic comparison of PBN neurons and glial cells, we analyzed changes in neuropathic pain-regulated gene expression and cell-cell communication in the homeostatic adult brain as important adaptations to chronic pain.
RESULTS
Polony gels enable amplifiable DNA stamping and show a continuous feature distribution
We selected the crosslinked polyacrylamide as stamp and copy gels allowing the low-pressure, conformal contact and the bridge amplification of template and copied DNAs. To automate the stamping process, a benchtop device was built with a robotic arm to position the stamp, a thermocycler to control the gel temperature, a digital balance to monitor the stamping pressure, and a fluidic system to amplify DNAs (Figures 1B and S1A). Different from previous methods that generated gel-embedded polonies (Gu et al., 2014; Mitra and Church, 1999), we amplified polonies on gel surfaces (Figure S1B) to facilitate DNA replication between gels. We first compared gels of varied thicknesses attached to different sized glass surfaces; the efficient DNA copying between large gel areas was observed at increased gel thicknesses (e.g., ≥ 40 μm; Figure 1C). To test reproducibility and robustness, the stamping was consecutively performed for 50 cycles. Feature patterns found on the copy gels were largely consistent (Figure 1D) and stable at varied stamping pressures (Figures S1C and S1D).
High-density polonies (≥ 0.6 million/mm2) often form a continuous DNA distribution with minimal feature-to-feature gaps, distinct from those amplified in Illumina nonpatterned flowcells showing a discrete, peak-shape distribution (Figure 1E). To understand the difference, we found that polonies on gel surfaces have a faster size expansion likely due to decreased gel constraints on the bridge amplification. These polonies appear to be easily accessible to restriction digestion; 93.6% of double-stranded DNAs were digested by TaqI to expose a 3’ poly(T) probe (Figure S1E). For spatial transcriptomic assays, the even distribution of poly(T) probes can minimize variations in RNA capture efficiency across DNA arrays. Although polonies are connected, they intend not to interpenetrate due to a polony exclusion effect (Aach and Church, 2004). Even at a high density, their borders were clearly delineated by polony sequencing (Figure 1F). Because polonies have varied sizes and shapes, to maximize the feature resolution, we developed a base-calling pipeline to determine the major barcode species in each pixel (0.325 × 0.325 μm2) of gel images to construct a spatial barcode map (Figure S1F).
The efficient replication of polony gels requires the post-stamping bridge amplification of copied DNAs which increases DNA densities and compensates for the inefficient copying in some gel areas. However, more amplification can cause polony size expansion and thus center drifts and introduce errors to spatial barcodes, compromising the resolution and accuracy, respectively. To assess this issue, we quantitively compared copy gels fabricated in a consecutive stamping experiment by analyzing feature patterns in multiple gel regions. Individual gels were compared with a consensus feature map constructed from aligned images of three copy gels. The repeated stamping is robust and only lost < 15% of features after 50 cycles, likely due to gradual template loss on the stamp (Figure 1G). Polony center drifts were found to decrease at a higher polony density; for example, the fraction of those below 0.5 μm increased from 65.7% at ~1 × 105/mm2 to 86.0% at ~8 × 105/mm2 (Figure 1H), possibly due to decreased polony sizes at the increased density. By sequencing 24-base pair spatial barcodes, 93.43 ± 0.04% of matched polonies in two gels were found with matched spatial barcodes with up to two mismatched bases (Figure 1I). Amplified polonies comprise ultradense capture probes; for example, the amplification yielded an average of 20,337 template copies per polony after 35 cycles, a ~9-fold increase from an Illumina method (Bentley et al., 2008) (Figures 1J, S1G, and S1H). With our sequencing imaging setup, we reliably fabricated gels with ~0.6 to 0.8 million features per mm2 passing filter and a mean feature diameter of 1.07 to 0.906 μm (Figure 1K). Fabricating higher resolution gels with smaller and denser features is possible because even more crowded polonies still show clear boundaries (Figure S1I) but sequencing them requires improved imaging resolution.
Demonstration of single-cell spatial transcriptomics on the mouse olfactory bulb (OB)
We developed Pixel-seq (Figure 2A) with a focus on translating the 1-μm feature resolution to the single-cell resolution of the assay for complex tissues such as the brain. To test assay conditions and compare the performance, we analyzed the mouse OB with morphologically diverse cells organized in a layered structure commonly used to validate spatial transcriptomic assays (Chen et al., 2022; Rodriques et al., 2019; Stahl et al., 2016; Stickels et al., 2021; Vickovic et al., 2019). We looked at two common issues of the array-based assays limiting the single-cell resolution, the lateral RNA diffusion between cells and the mixing of RNAs from multilayered cells found even in thin tissue slices. The polony gel-based RNA capturing, even without tissue fixation, yielded strong complementary DNA (cDNA) signals clearly delineating boundaries of neuronal cell bodies (Figure S2A). By comparing cell sizes detected by Pixel-seq and RNAscope, the median template drift was estimated to be ~0.86 μm (Figure S2B), smaller than the average polony size, suggesting that the gel-restrained diffusion does not severely compromise the feature resolution. Of note, the gels appeared to capture tissue RNAs from a single cell layer when frozen sections were placed on the dried gels; yielded cDNA signals were colocalized with stained nuclei in the gel-contacting cells not those in a deeper tissue (Figure 2B). The selective RNA capturing can be explained by fast occupancy of a gel surface by adjacent RNAs during the gel wetting by a tissue section (Figure S2C). The gel-based capture not only increased the resolution but also facilitated a fast preparation of cDNA sequencing libraries (~6 hours; see STAR Methods).
Figure 2. Pixel-seq-based single-cell spatial transcriptomics.
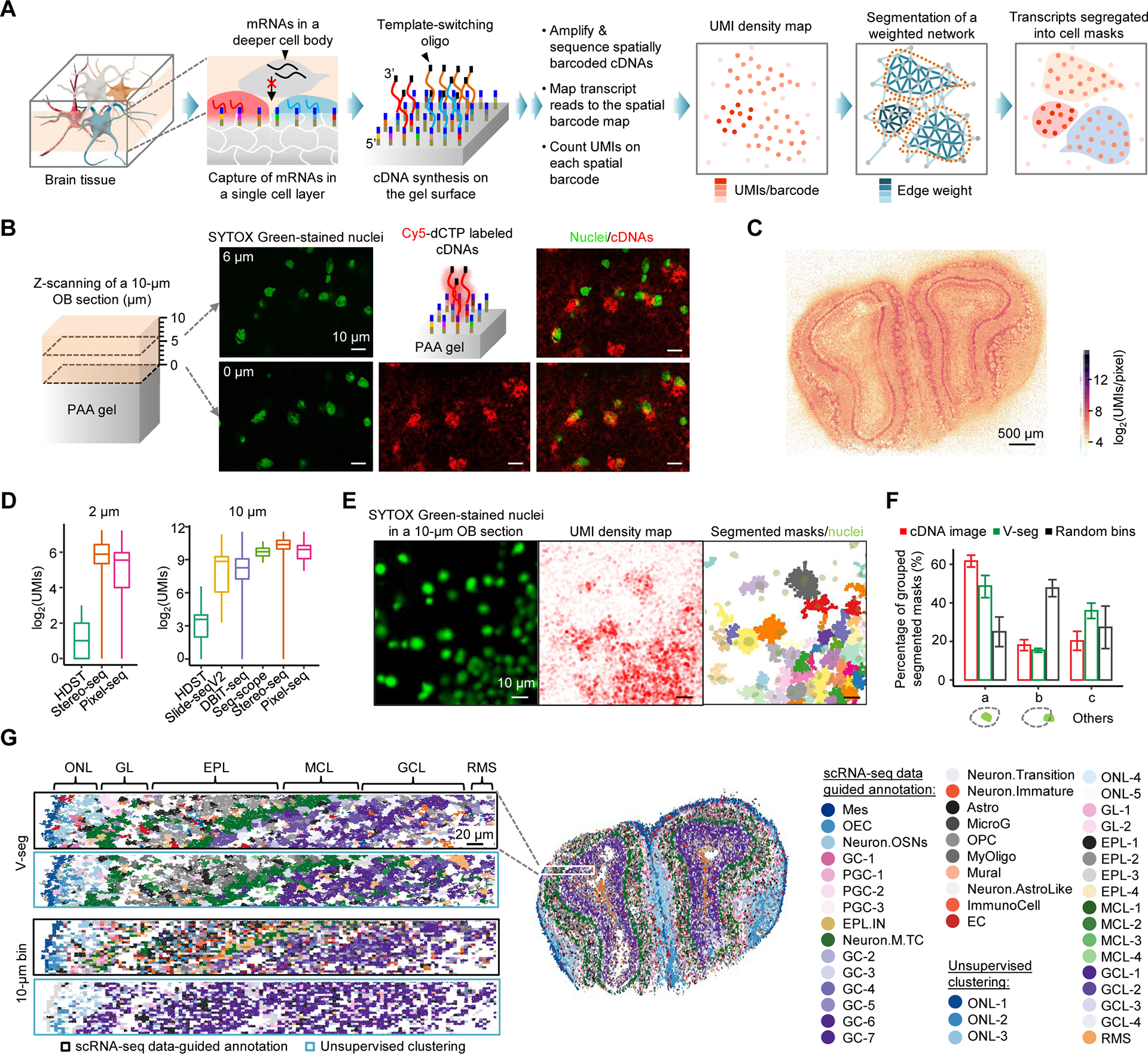
(A) Principle of Pixel-seq. RNAs are captured from the gel-touching cell layer in a cryosectioned tissue to synthesize barcoded cDNAs with a 3’ universal sequence introduced from a template-switching oligo for cDNA amplification. cDNAs are sequenced to associate RNAs to their gel locations to create a transcript map. A k-nearest neighbor network is built on the map where each barcode represents a node. Edge weights are calculated as a function of UMI counts, the distance and transcript similarity between two connected barcodes. The weighted network is segmented by a graph-based algorithm to create cell masks to aggregate transcripts for single-cell data analyses.
(B) Confocal analysis of stained nuclei in a mouse OB section attached to a gel and labelled cDNAs synthesized on the gel. Two detected layers of nuclei proximal (0 μm) and distal (6 μm) to the gel surface are overlaid with cDNA signals.
(C) Comparison of OB UMI density maps by RNA captures from a single (Pixel-seq) and multiple layered cells (Stereo-seq). i) Maps of total UMI densities measured on 2 × 2 μm2 bins. ii) Zoom-in comparison of the selected regions in i) (white dotted boxes). iii) Density plot of detected UMIs in the whole tissues. Means (dash lines): 45.6 and 60.5; maximums: 1,979 and 1,091.
(D) Comparison of selected gene expressions detected by Pixel-seq and the AMBA ISH data.
(E) Comparison of the capture efficiency of Pixel-seq and recent data from similar assays (see Table S1). The Pixel-seq OB UMIs were counted on bins of 7 × 7 (2 μm) and 33 × 33 (10 μm; a cutoff of 265) pixels.
(F) Validation of the V-seg result by overlaying segmented masks with stained nuclei in the same tissue. Immediately after placing the tissue on the gel pre-soaked with SYTOX Green, stained nuclei were imaged with the epifluorescence microscope used for polony gel sequencing.
(G) Comparison of the accuracy of V-seg, labelled cDNA image segmentation, and random bins of a size close to the average mask size. Results were validated by the colocalization with nuclei described in (F). a, masks containing single nuclei; b, masks partially overlapped with single nuclei; c, others including those overlapped with multiple or zero nuclei.
(H) Comparison of cell annotation outcomes of the segmentation by V-seg and random bins. Aggregated transcripts were analyzed by scRNA-seq data-guided annotation or unsupervised clustering. See full names of cell types in Figure S2G.
To assess the performance, we assayed 10-μm, coronal OB sections to obtain spatially resolved transcriptomes. Specifically, in a ~13-mm2 OB section, ~83% of raw reads were mapped to the barcode map to obtain ~82.5 million UMIs with a density range from 1 to 678 UMIs/barcode. The UMI density map displays a continuous, pixel-resolution, multi-layered structure (Figure 2C, panel i). The enlarged view shows marked density patterns rising from specific cell distributions in the ultrathin tissue layer, distinct from more even UMI distributions found by similar assays such as Stereo-seq (Figure 2C, panel ii), where RNAs released from multiple cell layers were likely captured under the assay condition. Although less RNAs were expected to be found in single than multiple cell layers, our mapped UMIs had a wider density range with a higher maximum (Figure 2C, panel iii), demonstrating the high capture efficiency. About 23,000 unique genes were detected with over 10 UMIs in at least one of three replicates; the data showed high correlation (R ≥ 0.968; Figure S2D). Detected OB layer-specific gene expressions agree with the in situ hybridization (ISH) data from the Allen Mouse Brain Atlas (AMBA) (Figure 2D). Together, compared with other assays, Pixel-seq achieved the high resolution and sensitivity (Figure 2E; Table S1).
With the high-resolution transcript maps in hand, we sought to segment mapped transcripts into single cells. Our simulation with seqFISH-mapped mouse cortex data (Eng et al., 2019) suggests that the 1-μm feature resolution is sufficient to separate regular cell bodies (Figure S2E). However, it is challenging to track all cell boundaries with standard staining methods and use the confocal images as “references” to guide cell segmentation. So far, array-captured brain transcripts were often randomly segregated in spatially aggregated pixels or random bins (Chen et al., 2022; Rodriques et al., 2019; Stahl et al., 2016; Stickels et al., 2021; Vickovic et al., 2019). The reference-independent segmentation is highly desirable, but available algorithms (e.g., Baysor (Petukhov et al., 2022)) were developed for imaging-based data on selected genes (Codeluppi et al., 2018; Moffitt et al., 2018) and cannot be directly applied to the global transcriptome data. Thus, we developed a volume-distance-based segmentation algorithm (V-seg) which constructs a nearest neighbor network from mapped transcripts, calculates edge weights, termed volume distances, based on UMI densities, the spatial distance and transcript similarity between two neighboring barcodes, and then segments the weighted network into masks representing single cells by a computationally efficient, graph-based community detection algorithm (Figures 2A (right three panels) and S2F).
We applied V-seg to segment the OB data, validated the results with the nuclear staining images, and compared the performance with image-based segmentation and random bins. In the OB section, V-seg segmented ~86% (~70.8 million) of mapped transcripts into 23,351 masks; 22,830 with UMIs ≥ 256 were selected for cell classification. Unsupervised clustering (Hao et al., 2021) of segregated transcripts recapitulated layer-specific distributions of major neuronal and non-neuronal cell types identified by single-cell RNA sequencing (scRNA-seq) (Tepe et al., 2018) (Figure S2G). To validate segmented cells, masks were aligned to stained nuclei in the same tissue (Figure 2F); to facilitate data registration, tissue images were acquired with the same microscope and magnification for the gel sequencing. Compared with random bins, V-seg and cDNA signal-guided segmentation, like the poly(A) staining-guided segmentation in other assays (Codeluppi et al., 2018; Moffitt et al., 2018), generated respectively 1.95- and 2.46-fold more masks containing a whole nucleus and 3.10- and 2.65-fold less masks partially overlapped with single nuclei (Figure 2G). About 36% of V-seg masks contained multiple or no nuclei partly because some nuclei were not in the gel-contacting cell layer (Figure 2B). The improved segmentation by V-seg is confirmed by the high similarity between the unsupervised clustering and scRNA-seq data-guided annotation results (Figure 2H), measured cell body sizes close to previous report (Pinching and Powell, 1971) (Figure S2H), and the consistency between mask shapes and marker gene distributions (Figure S2I). Segmented cells show cell-type specific UMI densities (Figure S2J) and their UMI counts typically increase with cell sizes, for example, the means of periglomerular type 1 (PGC-1; a mean diameter of 10.9 ± 4.6 μm) and mitral/tufted cells (M/TCs; 14.5 ± 4.8 μm) were 3,346 and 6,458 UMIs/cell, respectively. Our result reasonably agrees with the scRNA-seq data on gene expression (R = 0.722; Figure S2K) and cell type abundances (Figure S2L); the discrepancies are mostly due to the partial capture from cell pieces by Pixel-seq and cell losses in the dissociative assay.
Cell atlas of the PBN
We looked at the PBN packed with neuron clusters (or nuclei), a common structure in the brain distinct from the OB layered structure, which is hard to analyze without single-cell resolution. The PBN relays sensory information from the periphery to the forebrain, responding to internal and external stimuli, as well as maintaining homeostasis (Palmiter, 2018). Previous studies using unique genetic markers located neurons within the PBN that transmit distinct signals related to thermal sensation (Norris et al., 2021), pain (Huang et al., 2019), and appetite, visceral malaise, and threat detection (Campos et al., 2018). However, the identity of most cells in the PBN and their spatial organization were unknown.
We first analyzed PBN coronal sections in the middle (Bregma, −5.35 mm) with the largest cross section. With a sequencing depth of ~88%, each section yielded 21 ± 4.5 million UMIs located to a ~3 × 3 mm2 region centered on the PBN surrounded by the cerebellar cortex (CBX), trigeminal motor (V) and principal sensory nuclei (PSV), locus coeruleus (LC), and cuneiform nucleus (CUN) (Figure 3A). The UMI density map allows charting PBN subregions such as the lateral (PBNl) and medial (PBNm) divided by the superior cerebellar peduncle (scp), a large fiber tract showing distinctly fewer UMIs. Mapped transcripts were aggregated into 15,618 ± 1,093 cell masks per section. Unsupervised clustering by Seurat defined distinctive marker genes (Figure 3B), which were compared to the consensus in mousebrain.org (Zeisel et al., 2018) to identify 21 neuronal and non-neuronal cell types (Figure 3C). Further subclustering of neurons identified 18 subtypes (Figure 3D).
Figure 3. Single-cell spatial transcriptomic mapping of the PBN.
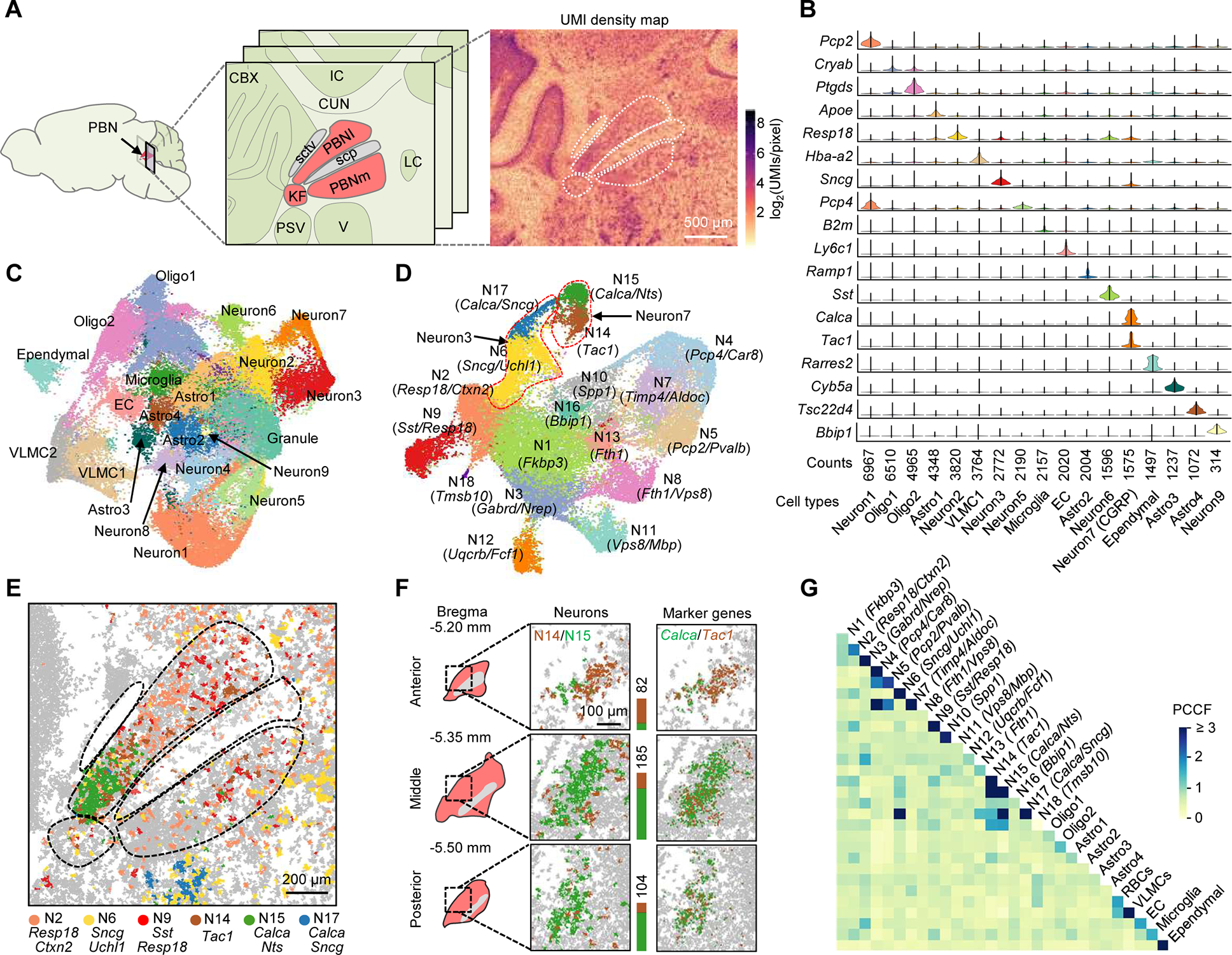
(A) Anatomical structure and UMI density map of a middle coronal section comprising the PBN (red) and neighboring regions. PBNI, lateral PBN; PBNm, medial PBN; KF, Kölliker-Fuse subnucleus; sctv, ventral spinocerebellar tract; scp, superior cerebellar peduncle; CBX, cerebellar cortex; IC, inferior colliculus; CUN, cuneiform nucleus; PSV, principal trigeminal sensory nucleus; V, trigeminal motor nucleus; LC, locus coeruleus.
(B) Violin plots of selected top genes showing differential expression in each cluster.
(C) Uniform Manifold Approximation and Projection (UMAP) clustering of the transcripts segregated into ~60,000 masks from four middle PBN sections passing quality control metrics. Astro, astrocyte; Oligo, oligodendrocyte; EC, endothelial cell; VLMC, vascular and leptomeningeal cell.
(D) UMAP clustering of the data representing 31,505 neuronal cells isolated from (C) with defined marker gene(s) for each cluster. Dotted lines highlight examples of separated subclusters from non-separated clusters in (C).
(E) Spatial distribution of major neuronal subtypes in the PBN and V.
(F) 3D mapping of PBN Tac1 (brown) and Calca (green) neurons. Stacked bars denote cell counts.
(G) Cell-cell contact heatmap of annotated clusters in (C) and (D). Cell contacts were quantified by a PCCF colocalization statistic.
To assess the robustness of the clustering, the spatial patterns of clustered cells were compared to the AMBA anatomic reference (Figures S3A). Most of clustered cells exhibit region-specific distributions correlated to the anatomical structure of the PBN and surrounding regions (Figures S3B and S3C). For example, ~81.0% and ~53.2% of clustered Calca/Nts+ and Tac1+ neuron subtypes were found in different subregions of the PBNl (Figure 3E), consistent with previous reports (Barik et al., 2018; Campos et al., 2018). The Calca+ neurons in the PBN and the trigeminal region were separated by differentially expressed markers (e.g., Sncg); the latter were correctly segmented from mixed trigeminal motor neurons (Sncg/Uchl1+) in the same region. Additionally, two unknown PBN neurons were identified: the Resp18/Ctxn2+ subtype in the PBNl’s dorsal and ventral subnuclei, the Sst/Resp18+ in the central subnucleus, and both also in the PBNm (Figure 3E). Their locations overlap with areas involved in a taste-guided behavior (Jarvie et al., 2021). Some non-neuronal cells also show region specificity; for example, the most abundant astrocyte subtype, Astro1, was enriched in the PBN and the neighboring pontine central gray region (Figure S3B).
To study the three-dimensional (3D) heterogeneity, we analyzed the anterior, middle, and posterior sections of the same PBN sample (Bregma, −5.20, −5.35, and −5.50 mm, respectively). Distinct changes along the rostral-caudal extent of the PBN were observed for distributions of major neuropeptide-expressing genes (Figure S3D) and validated by the AMBA ISH (Figure S3D) and RNAscope data (Figure S3E), implying transcriptomic and anatomical heterogeneity. We focused on the two known neuron subtypes in the same subnucleus, Calca/Nts+ and Tac1+. The clustering, as well as the spatial marker gene distributions, revealed their 3D organization: the Tac1+ cells are densely populated in the anterior position and surround the Calca/Nts+ in the middle position, and both are overlapped in the posterior position (Figure 3F).
Because the distance between cells affects their communication, we measured direct cell contacts in the PBN atlas. Adjacent cells were quantified using a pair cross-correlation function (PCCF) statistic (Philimonenko et al., 2000) to compare detected cell contacts (or colocalization) between the same or different subtypes to the probability of the random colocalization. The high colocalization between the same cell types agrees with observed cell aggregations; for example, the Purkinje (Pcp2/Pvalb+) and Bergmann (Timp4/Aldoc+) cells in the cerebellum and CGRP-expressing neurons (Calca/Nts+) in the PBN (Figures 3G and S3C). High neuron-neuron contacts were found for the Calca/Nts+ and Tac1+ in the PBNl and the Calca/Sncg+ and Sncg/Uchl1+ in the trigeminal. Typically, cells showing region-specific distributions were found with preferential contacts with specific neurons or non-neuronal cells.
Cell type- and subnucleus-specific transcript changes in response to chronic pain
After having the transcriptome reference map, we sought to discover if our method could detect changes in gene expression in response to stimuli. The precise analysis of activity-triggered adaptations in specific cells requires comparison of functionally identical cells (e.g., the same cell type in identical brain regions with similar connectivity) from different animals. To demonstrate the transcriptomic and anatomical accuracy of Pixel-seq for this application, we analyzed chronic pain-regulated changes in the PBN. The PBN is known to be a major hub to receive, process, and relay nociceptive signals (Palmiter, 2018; Sun et al., 2020). As part of adaptations to neuropathic pain, PBN cells are likely to mount complex transcriptional responses (Yap and Greenberg, 2018). However, such changes, as well as many others in different brain regions, are yet to be unveiled.
We assayed coronal PBN sections from animals that received either a sham operation or partial sciatic nerve ligation (SNL)-induced neuropathic pain (30th day post-surgery). To facilitate comparing cells in identical anatomical sites, we divided the sections into four subregions (two PBNl and two PBNm) and the trigeminal (V) (Figure 4A). To minimize variations caused by individual heterogeneity and the sectioning of brain samples, we focused on comparing two middle sections showing the highest cluster similarity (Figures 4B, S4A, and S4B). Unsupervised clustering of 32,377 cells pooled from the two sections identified 16 neuronal and 12 non-neuronal subtypes. A differential abundance analysis (Zhao et al., 2021) of the sham and SNL mouse data detected a remarkable imbalance of cell distributions (Figure 4C), which is corroborated by changed levels and spatial patterns of individual genes (Figure S4C). Differential gene-expression analysis found 487 genes in neurons and 181 in non-neurons with altered expression including 16 encoding secreted proteins (P < 0.05; Figure 4D); for example, 1.23 to 1.85-fold decreases (P < 0.05) of Apoe in glial subtypes, in contrast to the upregulation in other injury and disease models (Pfrieger and Ungerer, 2011), implying its multifaceted role in inflammation and pain modulation in the PBN. These genes in neuronal clusters were predicted to be differentially involved in neuron development, stress responses, inflammation, etc. (Figure 4E).
Figure 4. Chronic pain-regulated gene expression in PBN subnuclei.
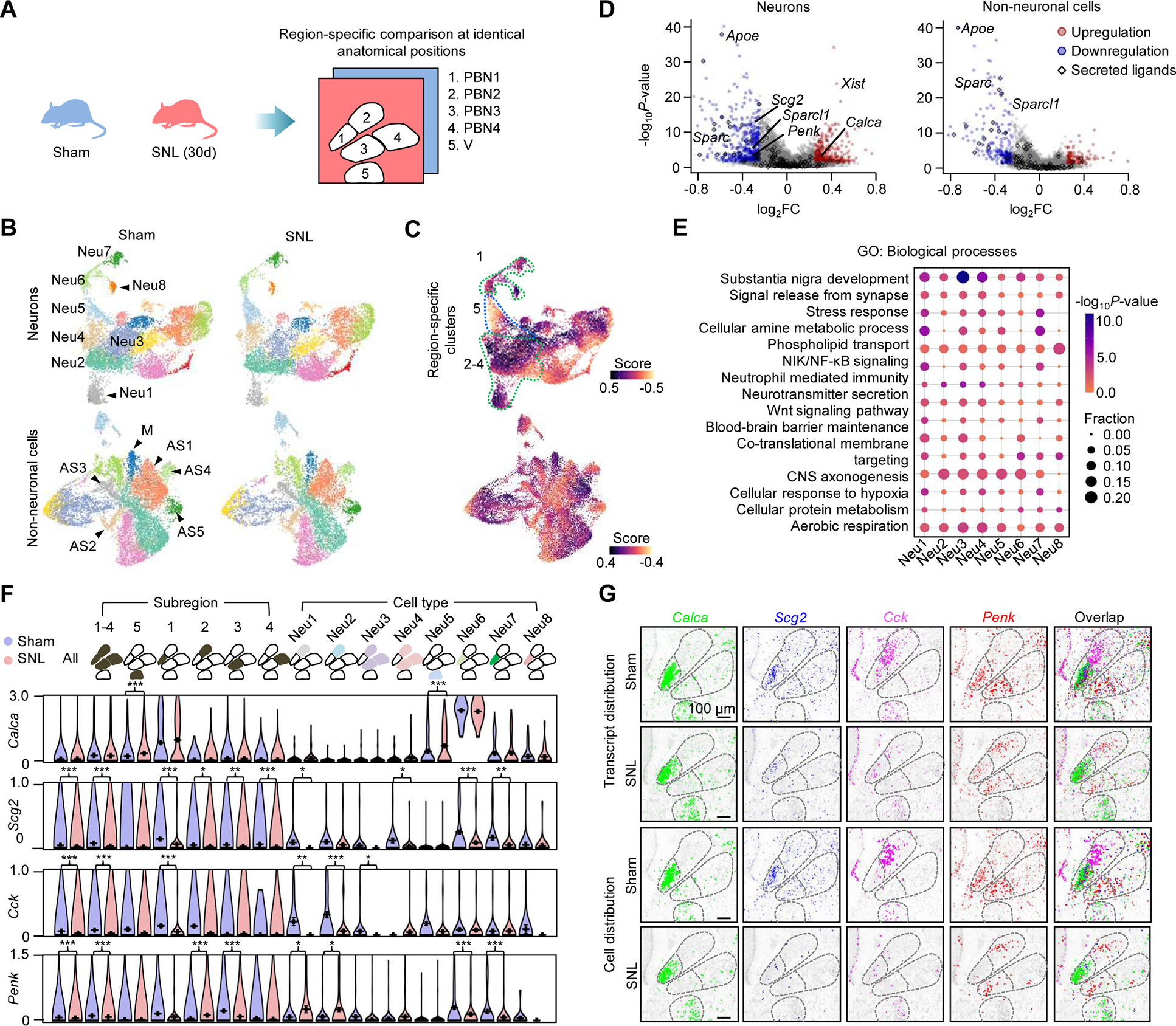
(A) Comparison of the Pixel-seq data of sham and pain mice at anatomically identical PBN and V subregions. SNL (30d), 30 days post partial sciatic nerve ligation.
(B) UMAP analysis to compare clusters in two middle coronal PBN sections from sham and SNL mice. Segmentation data representing 15,833 neurons and 16,473 non-neuronal cells are plotted. Major neurons, astrocytes, and microglia in the PBN and V are labelled.
(C) Differential abundance analysis of the data in (B). Dotted lines highlight the PBN and V region-specific neuronal clusters.
(D) Differential expression analysis of the data in (B). FC, fold change. Colored genes, |log2FC| ≥ 0.25 and P < 0.05, Wilcoxon rank-sum test. The upregulation of Xist was only found in female mice.
(E) Gene Ontology (GO) enrichment analysis of differentially expressed genes in the major neuronal clusters in the PBN and V. P-values, Fisher’s exact test.
(F) Comparison of region- and cell-type-specific expression of major neuropeptide genes. Data represent mean values of ≥ 217 cells in each group; error bars, standard error of mean. *P < 0.05, **P < 0.01, ***P < 0.001, Wilcoxon rank-sum test.
(G) Comparison of spatial patterns of the neuropeptide genes in (F) and associated cells.
We next asked how genes are regulated in specific cell types within subnuclei. Of particular interest is how neuropeptide gene expression responds to the pain condition. Thus, we analyzed transcriptional changes of peptide precursor genes within the chosen PBN sections. Calca, the gene encoding CGRP was slightly upregulated (1.55-fold, P = 6.07 × 10−6) in motor neurons in the trigeminal (Neu5), but not significantly in the PBN (Neu6) (Figures 4F and 4G). Scg2 and Cck were downregulated by 2.54- and 2.82-fold (P < 0.001), respectively, with regional specificity: Scg2 decreased across the PBN, but Cck changed mainly in the subregion 2 populated by Resp18/Ctxn2+ neurons. Notably, Penk, encoding an opioid precursor, showed decreased expression in subregions 1 and 3: 2.13- and 3.03-fold, respectively (P < 0.001), but a 3.07-fold increase in the subregion 2 (Neu1 and Neu2; P < 0.05). These examples along with all the other changes provide valuable clues for future functional experiments.
Cell-cell communication coordinated by transcriptional dynamics of microglia and astrocytes
Given the reference and pain-induced transcriptome maps, we asked how gene regulation affects local cell-cell communication. To quantitatively compare cell-signaling networks, we computed signaling likelihoods for each cell as “sender” or “receiver” using ligand and receptor transcript levels and spatial distances from other senders and receivers (Figure S5A) (Cang and Nie, 2020). Because transcripts are mainly detected in cell bodies and it is difficult to analyze the long-distance communication mediated by cell projections, neurons and non-neuronal cells were treated equally in this analysis. The comparison of signaling likelihoods between the sham and SNL mouse datasets indicates that the major changes in the PBN region were associated with microglia and astrocytes (Figure S5B) known to coordinate neuronal development and homeostasis (Vainchtein and Molofsky, 2020). A subcluster-level analysis of signaling between microglia (M), astrocytes (AS1-AS5), and major PBN neuron subtypes (N1-N4 and N6-N8) revealed subcluster-specific increases or decreases in microglial and astrocyte signaling likelihoods (Figure 5A). A detailed comparison of the contributions by individual ligand-receptor(s) pairs found that top contributors are neuropeptides, cytokines, glycoproteins, lipoproteins, and their receptors (Figure 5B). Furthermore, to understand the signaling heterogeneity, cells of a sender subcluster were profiled for the contribution by each ligand. Interestingly, the senders showed a bimodal (e.g., Mif in N2 and Apoe in AS1) or unimodal distribution (e.g., Spp1 in N3 and C1qb in M), and the pain-responsive and non-responsive subpopulations appeared to be separated in the bimodal distribution where the responsive cells had higher signaling likelihoods than the non-responsive (Figure 5C).
Figure 5. Chronic pain-associated cell-cell communication and glial transcriptome dynamics.
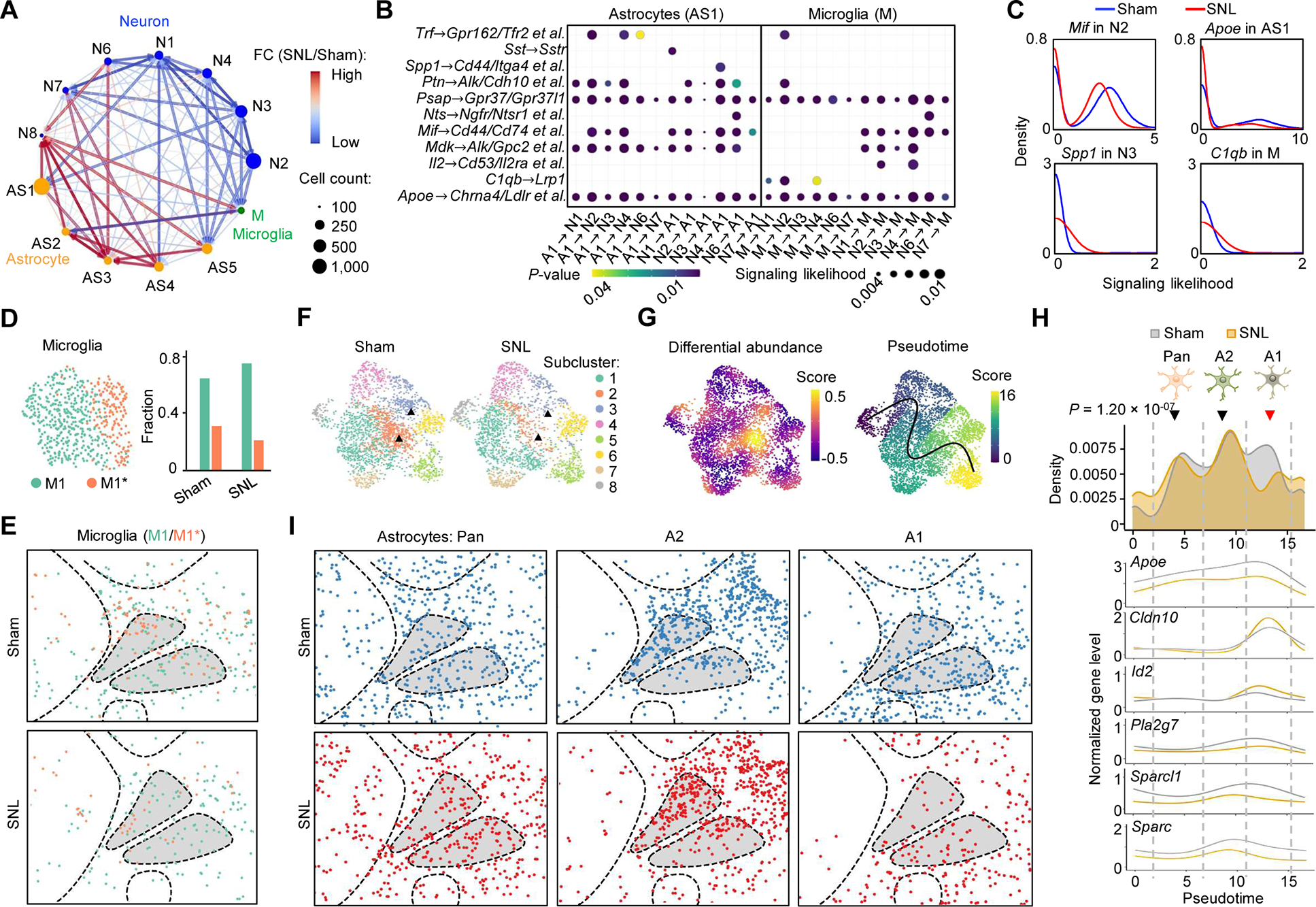
(A) Network representation of cell signaling likelihood changes for major neuronal, astrocyte and microglial clusters with significant distributions in the PBN. Signaling likelihoods were calculated by SpaOTsc.
(B) Dot plots of contributions of paired ligand-receptor genes to the changed signaling likelihoods in (A) in the most abundant astrocyte subtype (AS1) and microglia (M). P-value, Kolmogorov-Smirnov (KS) test.
(C) Density plots of signaling likelihoods of selected ligand genes in sender clusters.
(D) UMAP analysis of the microglia and the subtype proportions under the sham and pain conditions.
(E) Spatial distribution of the microglial subclusters. Segmentation data representing 584 cells are plotted.
(F) UMAP clustering of the astrocytes under the sham and pain conditions. Segmentation data representing 4,471 cells are plotted. Triangles denote the subclusters 2 and 3 showing the most significant changes.
(G) Differential abundance (left) and pseudotime (right) analyses of the data in (F). A pseudotime trajectory was inferred by Slingshot.
(H) Density plots of astrocyte subpopulations and the normalized expression of selected genes along the pseudotime trajectory in (G). P-value, KS test.
(I) Spatial patterns of the astrocyte subpopulations.
Given the signaling importance of microglia and astrocytes, we further investigated their transcriptomic heterogeneity. To date, microglial heterogeneity associated with physiologic roles such as supporting synaptic development and remodeling in the homeostatic adult brain is yet to be confirmed by scRNA-seq (Li et al., 2019), because microglial gene regulation is environmentally sensitive and can be easily disrupted by tissue dissociation (Gosselin et al., 2017). Here, 584 cells with microglial markers (e.g., C1qa and C1qb) were re-clustered into two subtypes, M1 and M1* (Figures 5D (left) and S5C). Despite their similarity, M1* was annotated with specific immune response-regulating marker genes (e.g., Mif and Sod1) involved in an interleukin-12 (Il-12)-mediated signaling pathway (P = 6.68 × 10−5) and neutrophil mediated immunity (P = 5.22 × 10−4) (Figure S5D). M1* is different from an activated neuroinflammatory state induced by lipopolysaccharide (Liddelow et al., 2017) due to the lack of three marker genes, Il1a, Tnf, and C1q. Considering a strong association between their marker genes in immune regulation, M1* could represent a transition state to the activated microglia. Under the pain condition, M1* decreased from 33.6% to 22.0% of the microglial population (Figure 5D (right)). Given the different ligand and receptor profiles of M1 and M1*, the decrease of M1* is associated with the changed communication in the signaling networks. Microglia showed a relatively even spatial distribution (Figure 5E) likely due to their high motility in the tissue.
Likewise, we sought to correlate transcriptomic and signaling heterogeneity of astrocytes identified from the initial clustering. 4,471 cells were re-clustered into eight subtypes annotated with marker genes (Figures 5F and S5E); most of the subclusters are connected, suggesting a continuum of transcriptomic states. As expected, some subtypes had region-specific distributions (Figure S5F). In the clustering outcome, the pain-induced major changes were found in the subtypes 2 and 3, which is supported by the differential abundance analysis (Figure 5G (left)). To understand the transcriptomic changes, we analyzed the pseudo-temporal ordering of all subtypes (Figure 5G (right)). Projection of the whole-cell population along a pseudotime trajectory revealed three separated groups, A1, A2, and Pan (Figure 5H), which can be correlated to three astrocyte states with specific physiological roles (Liddelow et al., 2017). For example, A1 and A2 astrocytes with differentially expressed Sparc and Sparcl1 are known to have destructive and protective roles, respectively, in maintaining homeostasis; thus, the increase of A1 is often associated with neuroinflammation. Here, a significant decrease of A1, mainly contributed by the subtypes 2 and 3, was found for the SNL condition (P = 1.20 × 10−7), suggesting that pain adaptation might involve an unknown neuronal protection mechanism. Remarkably, the comparison of astrocyte spatial distributions found that the A1 decrease was mostly in the PBN region, but the other two states had no obvious changes (Figure 5I). These results, together with the neuron-glia communication, provide important evidence of the region-specific glial transcriptomic dynamics supporting local neuronal activities.
DISCUSSION
Amplifying polonies atop crosslinked polyacrylamide gels brings advantages to the fabrication and application. The gel compatibility with microcontact printing and bridge amplification enabled the submicron-resolution polony replication, reducing the fabrication cost and time. For example, the consumable cost of fabricating a 7 × 7 mm2 array of > 30 million unique features decreased to ~$3 ($0.06/mm2; Table S2), a drastic reduction from those reported for DNA cluster and nanoball arrays (Table S3), and the time to ~7 hours (see STAR Methods). Unlike similar assays (Chen et al., 2022; Cho et al., 2021) whose major cost components were array costs, our assay cost is mainly determined by the commercial sequencing of barcoded cDNA libraries (e.g., mapping a 1-mm2 mouse brain area required ~20 million reads, a cost of ~$60 using an Illumina NovaSeq S4 flow cell). By lifting the burden of sequencing each array anew, the gel replication opens opportunities to break existing limitations. The sequencing requires placing DNAs in flowcells comprising glass or silicon surfaces suitable for optical scanning. Without the sequencing need, polony gels can be casted on other substrates for expanded assay flexibility. Gels with overly dense polonies (known as “overclustering”), which could improve the feature resolution but so far cannot be correctly sequenced, might become useful by copying different stamping gels with lower-density barcodes to the same copy gel. Finally, given the demonstrated sensitivity and resolution, crosslinked gels offer an ideal substrate for capturing tissue molecules. Their penetrable hydrophilic matrix appears to increase accessibility of densely patterned probes to tissue targets in a diffusion-constrained environment.
Pain, a multidimensional experience, involves sensory, affective, and cognitive components in the periphery and brain. So far, single-cell transcriptomics of pain-induced changes has been limited to sensory tissues, such as dorsal root ganglion (Kupari et al., 2021), but cells and responses in other components in pain processing are largely unknown. Despite the importance in elucidating pain mechanisms and developing new analgesics, research in this field has been hampered by the lack of suitable tools. In this regard, Pixel-seq directly addresses the unmet need. The first single-cell PBN atlas and the unveiled pain-regulated changes in the spatially resolved transcriptomes provide a basis for future mechanistic studies on the PBN’s roles in affective motivational and sensory discriminative pathways of pain and other processes. Although limited samples were analyzed, the unusual heterogeneity revealed by Pixel-seq highlights the necessity to analyze more anatomical positions at different time points to develop a complete view of the structural and functional landscape.
Limitations of the study
Due the timing of developing the stamping method, the OB and PBN data were collected with sequenced gels. Despite the improved feature resolution, DNA array-based spatial transcriptomic assays still face challenges to reliably achieve single-cell resolution. Compared our clustering results with those on dissociative scRNA-seq of brain tissues, Pixel-seq showed less optimal cell type separation. A major reason is that the feature resolution is insufficient to delineate small cell projections densely intertwined with brain cells. The reported highest feature resolution (0.22 μm; Table S1) probably reaching the limit of current array fabrication is still not enough for tracing axons and dendrites. Thus, alternative strategies such as tissue expansion (Chen et al., 2015) might be explored to push the resolution limit. In our data processing, decreased accuracy of V-seg was often found for closely aggregated cells especially of small sizes. Better accuracy could be achieved by machine learning-based algorithms (He et al., 2021; Littman et al., 2021; Park et al., 2021; Petukhov et al., 2022) and coupling the RNA data to cell boundary signals detected with fluorescently labeled or DNA-tagged affinity reagents. In this work, polony gels were only used for capturing RNA, but they should be applicable to protein detection with DNA-tagged antibodies (Liu et al., 2020; Vickovic et al., 2022) and possibly small-molecule analytes via affinity reagent innovation (Kang et al., 2019). They can also be designed with various probes for classical DNA array-based applications (Bumgarner, 2013). Pixel-seq detected subcellular transcript distributions in brain cells, which were not further investigated in our study. Fully exploiting the 1-μm spatial resolution requires the improved data analysis and validation and should be critical to revealing subcellular heterogeneity, such as protein localization, interaction, and modification.
STAR★METHODS
RESOURCE AVAILIABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Liangcai Gu (gulc@uw.edu).
Material availability
This study did not generate new unique reagents. All materials used for Polony gel making and Pixel-seq assay are listed in the key resources table and commercially available.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Adult mouse olfactory bulb | This study | N/A |
| Adult mouse parabrachial nucleus | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Contrad 70 | Decon Labs | Cat#1003 |
| 10N sodium hydroxide solution | Sigma-Aldrich | Cat#72068 |
| Hydrochloric acid | Fisher Scientific | Cat#A144-500 |
| 200 proof ethanol | Decon Labs | Cat#2701 |
| 3-(trimethoxysilyl)propyl methacrylate (Blind-Silane) | Sigma-Aldrich | Cat#M6514 |
| Acetic acid | Fisher Scientific | Cat#A35-500 |
| Acrylamide | Sigma-Aldrich | Cat#A9099 |
| N,N’-methylenebisacrylamide | Sigma-Aldrich | Cat#M7279 |
| Ammonium persulfate | Sigma-Aldrich | Cat#A3678 |
| UltraPure TEMED | Invitrogen | Cat#15524-010 |
| N-(5-bromoacetamidylpentyl) acrylamide (BRAPA) | Combi-blocks | Cat#HD-8626 |
| N, N-dimethylformamide | Sigma-Aldrich | Cat#D4551 |
| Potassium phosphate dibasic trihydrate | Sigma-Aldrich | Cat#P9666 |
| Potassium phosphate monobasic | Sigma-Aldrich | Cat#P9791 |
| 20× SSC | Invitrogen | Cat#AM9763 |
| Tween-20 | Sigma-Aldrich | Cat#P9416 |
| Taq2× master mix | New England Biolabs | Cat#M0270 |
| Trizma base | Sigma-Aldrich | Cat#93362 |
| Betaine | Sigma-Aldrich | Cat#B2629 |
| Ammonium sulfate | Sigma-Aldrich | Cat#A4418 |
| Magnesium sulfate | Sigma-Aldrich | Cat#M2773 |
| Triton X-100 | Sigma-Aldrich | Cat#T8787 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat#D8418 |
| Taq DNA polymerase | New England Biolabs | Cat#M0267X |
| dNTP mix (10 mM each) | GenScript | Cat#C01582-250 |
| Formamide (deionized) | Emdmillipore | Cat#4670-4L |
| Bst enzyme | New England Biolabs (or This study) | Cat#M0275 |
| USER enzyme | New England Biolabs | Cat#M5505 |
| TaqI-V2 | New England Biolabs | Cat#R0149 |
| Maxima H minus reverse transcriptase | Thermo Fisher Scientific | Cat#EP0753 |
| O.C.T. compound | Fisher Healthcare | Cat#4585 |
| RNase inhibitor | Invitrogen | Cat#10777019 |
| Ficoll PM 400 | Sigma-Aldrich | Cat# F4375 |
| Proteinase K | Qiagen | Cat#19131 |
| PKD buffer | Qiagen | Cat#1034963 |
| BSA | New England Biolabs | Cat#M9000S |
| Bst 2.0 WarmStart DNA polymerase | New England Biolabs | Cat#M0538L |
| Ultra II Q5 master mix | New England Biolabs | Cat#M0544L |
| Thermolabile exonuclease I | New England Biolabs | Cat#M0568L |
| Q5 HotStart polymerase | New England Biolabs | Cat#M0493L |
| Critical commercial assays | ||
| Nextera XT kit | Illumina | Cat#FC-131-1024 |
| HiSeq SBS kit v4 | Illumina | Cat#FC-401-4002 |
| RNAScope HiPlex assay | ACDBio | Cat#324409 |
| PhiX control library | Illumina | Cat#FC-110-3001 |
| Deposited data | ||
| Pixel-seq mouse OB and PBN data | GEO | GEO: GSE186097 |
| HDST mouse OB data | GEO | GEO: GSE130682 |
| Seq-scope mouse liver data | GEO | GEO: GES169706 |
| DBiT-seq mouse embryo data | GEO | GEO: GSE137986 |
| Slide-seqV2 mouse OB data | Broad | Stickels et al., 2021 |
| Visium mouse OB data | GEO | GEO: GSE153859 |
| scRNA-seq mouse OB data | GEO | GEO: GSE121891 |
| Stereo-seq mouse OB data | CNGB | CNGB:CNP0001543 |
| Oligonucleotides | ||
| BA(+): AATGATACGGCGACCACCGAGAUCTACAC | Integrated DNA Technologies | N/A |
| BA(−): CAAGCAGAAGACGGCATACGAGAT | Integrated DNA Technologies | N/A |
| PS-BA(+): /5PS/TTTTTTTTTTAATGATACGGCGACCACCGAGAUCTACAC | Integrated DNA Technologies | N/A |
| PS-BA(−): /5PS/TTTTTTTTTTCAAGCAGAAGACGGCATACGAGAT | Integrated DNA Technologies | N/A |
| Cy5-probe: /5Cy5/CTTGCGCTAGTCCGGCAACCTCGGTGG | Integrated DNA Technologies | N/A |
| UMI primer: CTACACGACGCTCTTCCGATCTNNNNNNNCCGCTAATACGACTCACTATAGGG | Integrated DNA Technologies | N/A |
| TruSeq sequencing primer: CCTACACGACGCTCTTCCGATC | Integrated DNA Technologies | N/A |
| TSO primer: /5’BIOSG/AAGCAGTGGTATCAACGCAGAGT | Integrated DNA Technologies | N/A |
| TruSeq libP5 primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT | Integrated DNA Technologies | N/A |
| Nextra index primer: CAAGCAGAAGACGGCATACGAGAT[INDEX]GTCTCGTGGGCTCGG | Integrated DNA Technologies | N/A |
| Polony sequencing primer: TTCCATCATTGTTCCCGGGTTCCTCATTCTC | Integrated DNA Technologies | N/A |
| Template-switching oligo: AAGCTGGTATCAACGCAGAGTGAATrG+GrG | Qiagen | 339414YCO0076714 |
| Software and algorithms | ||
| STAR | https://github.com/alexdobin/STAR | V2.7.0 |
| Cellpose | https://www.cellpose.org/ | V1.0 |
| Seurat | https://satijalab.org/seurat/ | V4.0.2 |
| iGraph | https://igraph.org/ | V1.3.0 |
| Flexbar | https://github.com/seqan/flexbar | V3.0 |
| Bowtie | http://bowtie-bio.sourceforge.net/ | V1.3.1 |
| DA-seq | https://github.com/KlugerLab/DAseq | V1.0.0 |
| Giotto | https://rubd.github.io/Giotto_site/ | V1.0.4 |
| Slingshot | https://github.com/kstreet13/slingshot | V2.2.0 |
| SpaOTsc | https://github.com/zcang/SpaOTsc | V1.0 |
| Dlight | https://github.com/GuLABatUW/Pixel-seq/tree/main/BaseCalling_DlightV2.3 | V2.3 |
| Micro-Manager | https://www.micro-manager.org | V1.4.22 |
| jSerialComm | http://fazecast.github.io/jSerialComm/ | N/A |
| Other | ||
| 4-axis robotic arm | DOBOT | Model: MG400 |
| CPAC HT 2-TEC Heating/cooling block | Inheco | PN:7000166 |
Date and code availability
Raw sequencing data and processed files have been deposited in Gene Expression Omnibus (accession number: GSE186097). All data were analyzed with standard programs and packages, as detailed in the Key Resource Table.
Data analysis code, along with a README, is available as a Github repository (https://github.com/GuLABatUW/Pixel-seq).
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal
Animal experiments were conducted according to US National Institutes of Health guidelines for animal research and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Washington. C57Bl/6J mice (Jackson Laboratory 000664) ranging from 3–4 months of age were used for all studies. The olfactory bulbs and PBN sections (Figure 3) were collected from male mice. For the chronic pain experiment, PBN sections were collected from two pairs of male and female mice under Sham and SNL conditions. Animals were housed in temperature- and humidity-controlled facilities with 12-h light/dark cycles with ad libitum access to standard chow diet (LabDiet 5053). Mice were anesthetized with Beuthansia (0.2 mL, i.p.; Merck) and decapitated. The brains were rapidly dissected, frozen on crushed dry ice, and stored at −80°C until cryosectioning in a Cryostat NX70 (Thermo Scientific).
METHOD DETAILS
Polony template construction
370-base pair (bp) DNA templates bearing a 24-bp random sequence were synthesized by Integrated DNA Technologies (IDT). They were PCR amplified (Taq 2 × master mix; New England Biolabs (NEB), M0270) with bridge PCR primers (BA(+) and BA(−)) for 15 cycles and size selected by 2% agar gel. Purified DNAs were quantified by Qubit 4 fluorometer (Thermo Fisher Scientific), diluted to 1 nM, and aliquoted for stamp gel preparation or storage at −20°C. The template sequence included:
5’-AATGATACGGCGACCACCGAGATCTACAC(P5 sequence)
TGCGGCCGCTAATACGACTCACTATAGGGATCT(T7 promoter)
NNNNNNNNNNNNNNNNNNNNNNNN(spatial barcode)
AGAGAATGAGGAACCCGGGAACAATGATGGAA(polony sequencing primer)
TTTTTTTTTTTTTTTTTTT (poly(T) probe)TCGA(TaqI site)
ACCACCGAGGTTGCCGGACTAGCGCAAG(Cy5-probe site)
TACTTGTCCATTCCTGAAGAAATATTATATTTATACAACTTACCCATAGAATCCTATTTACTAGGAAAGGAAAAGCCTCCTATTTATAAAAATTGGATAGAGCTTTCTCAACAACAGTGGAATATCAATGATAGAACAATTGCCGATTTATTAGATGGGGTCTTAATAATACCATCGA(TaqI site)
TCTCGTATGCCGTCTTCTGCTTG(P7 sequence)-3’
Gel casting
Polony gels were casted on 75 × 25 mm2 glass slides (Fisher Scientific, 12-550D) or 40-mm-diameter round coverslips (#1.5; Warner Instruments, 64–1696), which were cleaned by sonication in 5% Contrad 70, 0.5 M NaOH, 0.1 N HCl, and Milli-Q H2O, air dried in an AirClean PCR hood, and coated with Bind-silane (Sigma M6514) as previously described (Gu et al., 2014). To form a gel casting well on the slides, adhesive tapes (Grainer Carton Sealing Tape, 1.6 mil) with a punched center hole of 60 × 12 mm2 were attached to each slide. The casting of the crosslinked polyacrylamide was performed in an anaerobic chamber (Coy Lab) (Gu et al., 2014). Briefly, a gel casting solution comprising 8% acrylamide/bis-acrylamide (w/v, 19:2; Sigma-Aldrich, A9099 and M7279), 16 mg/mL N-(5-bromoacetamidylpentyl) acrylamide (Combi-blocks, HD-8626), 0.015% (w/v) ammonium persulfate (freshly dissolved; Sigma-Aldrich, A3678), and 0.025% (v/v) N,N,N ´,N ´-tetramethylethylenediamine (Thermo Fisher Scientific, 15524–010) was mixed and immediately pipetted into the well covered with a 60 × 24 mm cover glass (Corning, 2940–246). ~40 μL casting solution was typically used to cast a ~40-μm-thick gel. Gels were polymerized at room temperature (R.T.) in the anaerobic chamber for 2 hours before transferred to the PCR hood. After removing the coverslip, a gel-coated slide was assembled into a modified flowcell (BioSurface Technologies, FC 81-PC) forming a channel of 55 × 9 × 0.3 mm3 with a volume of ~150 μL. Gels were casted on the round coverslips for spatial barcodes sequencing. Typically, < 5-μm-thick gels were formed by placing a cut glass slide of 8 × 40 mm2 atop a gel casting solution applied onto the coverslip surface to form a uniform liquid layer between the coverslip and the top glass slide; the gels were polymerized in the anaerobic chamber at R.T. for 90 min. After removing the top glass, the gel-coated coverslip was assembled into a FCS2 flowcell (Bioptechs) with a 0.2-mm-thick gasket to form a channel of 9 × 35 mm2 with a volume of ~60 μL.
Primer grafting
A flowcell was washed with 1 mL milli-Q water and then 500 μL grafting buffer (10 mM potassium phosphate buffer, pH 7). Primer grafting was performed by incubating 25 μM each 5’ phosphorothioate-modified primers (PS-BA(+) and PS-BA(−)) in the grafting buffer at 50 °C for 1 hour. The flowcell was washed with 500 μL hybridization buffer (5 × SSC, 0.05% Tween-20; Invitrogen, AM9763 and Sigma-Aldrich, P9416) to remove non-hybridized primers and stored at 4 °C or used for the next step.
Preparation of stamp gels
To seed DNA templates on primer-grafted gels, 1 nM templates were denatured in freshly diluted 0.2 N sodium hydroxide (Sigma-Aldrich, 72068), neutralized with 200 mM Tris-HCl, pH 7, and diluted with ice-cold hybridization buffer to a working concentration of 8–12 pM, which typically resulted in a feature density of ~0.6 to 0.8 million per mm2. Template hybridization to the gel was performed by adding 500 μL diluted templates into a grafted flowcell, which was incubated on a heating/cooling block (CPAC HT 2-TEC, Inheco) at 75 °C for 2 min, air cooled to 40 °C, and washed with 500 μL amplification buffer (2 M betaine, 20 mM Tris-HCl (pH 8.8), 10 mM ammonium sulfate (Sigma-Aldrich A4418), 2 mM magnesium sulfate (Sigma-Aldrich, M2773), 0.1% (v/v) Triton-X-100 (Sigma-Aldrich T8787), and 1.3% (v/v) DMSO (Sigma-Aldrich, D8418)) to remove non-hybridized templates. To synthesize the first-strand DNAs anchored to the gel, the flowcell was added with 150 μL Taq DNA polymerase mixture (50 U/mL Taq DNA polymerase (NEB M0267) and 200 μM each dNTPs (GenScript C01582)) in the amplification buffer and incubated at 74 °C for 5 min before decreasing the temperature to 60 °C for bridge amplification. Polony amplification was performed with an automated fluidic device with a P625 pump set (Instech Laboratories). The amplification was performed at 60 °C with a HT 2-TEC heating/cooling block for 22 cycles, each including i) Denaturation: pump 500 μL deionized formamide (EMD Millipore, 4670) and stop for 1 min; ii) Annealing: pump 500 μL amplification buffer and stop for 2 seconds; iii) Extension: pump 500 μL Bst DNA polymerase mixture (80 U/mL Bst DNA polymerase (NEB, M0275 or lab purified with the same performance) and 200 μM each dNTPs) in the amplification buffer and stop for 1 min. The flow rate was 3 mL/min. After the amplification, double-stranded polony DNAs were linearized by washing the flowcell with 500 μL 1 × CutSmart buffer (NEB), adding 150 μL 100 U/mL USER (NEB M5505) in 1 × CutSmart buffer, and incubating the reaction at 37 °C for 1 hour. Gels with linearized DNAs were stored in 100% formamide or washed with 500 μL elution buffer (1 × SSC and 70% formamide (v/v)) for the stamping.
Gel stamping
Slides with primer-grafted gels were used as copy gels. Slides with stamp gels were cut by a glass cutter to the stamps of specific sizes (e.g., 7 × 7 mm2). A stamp was fixed by a double sided tape (3M, 468MP) to a flat surface (ɸ 8 mm) of a stamp holder connected to a desktop 4-axis robotic arm (Dobot, MG400) with the positioning repeatability of 0.05 mm. A stamping cycle included seven steps involving the placement of the stamp at four positions (1–4) (Figure. S1A): 1) In Position A, soak the stamp in formamide at 60 °C. 2) Move the stamp from Position A to Position B to wash the stamp with a stamping buffer (2 M betaine, 20 mM Tris-HCl (pH 8.8), 10 mM ammonium sulfate, 2 mM magnesium sulfate, 0.1% (v/v) Triton-X-100, and 1.3% (v/v) DMSO) at 60 °C for 10 seconds. 3) Move the stamp from Position B to Position C to soak the stamp in a stamp mix (100 U/mL Taq DNA polymerase and 200 μM each dNTPs in the stamping buffer) at 4 °C for 30 seconds; 4) Move the stamp from Position C to Position D to place the stamp on a specified copy gel position with a stamping pressure between 10 and 30 kPa at 95 °C for 1 min. Before the stamping, the copy gel was preincubated in the stamp mix at 95 °C. The pressure was monitored by an electronic balance (e.g., a weight of ~50 to 150 gram applied to the stamp of ~7 × 7 mm2; U.S. Solid, USS-DBS28). 5) In Position D, decrease the copy gel temperature to 60 °C and hold for 1 min. 6) In Position D, increase the copy gel temperature to 95 °C and meanwhile add 2 mL formamide to soak the stamp and copy gels for 3 min to dissociate the double-stranded DNAs. 7) Move the stamp from Position D to Position A to prepare for the next cycle. Meanwhile, wash the copy gel with 3 × 1 mL Milli-Q H2O to prepare it for stamping on the next gel position. Each stamping cycle took ~12 min. Six 7 × 7 mm2 arrays were stamped on a copy gel of 55 × 9 mm2 and then another copy gel was placed in Position D.
Post-stamping gel processing
After the stamping, copy gels were assembled to the flowcells and copied DNAs were bridge amplified for 20 cycles similarly as described in the stamp gel preparation. To expose 3’ poly(T) probe for RNA capturing, a flowcell was washed with 500 μL 1 × CutSmart buffer (NEB), added with 160 μL 2,000 U/mL TaqI (NEB, R0149) in 1 × CutSmart buffer, and incubated at 60 °C for 1.5 hours. TaqI-treated polony gels were stored in 100% formamide or washed with 500 μL elution buffer for Pixel-seq assays or polony sequencing. From gel casting to the completion of the TaqI digestion, the fabrication of a copy gel slide with six arrays ready for Pixel-seq assay took ~7 hours.
Polony Sequencing
To determine barcode sequences and distribution, gels were casted on a round coverslip as described above. The coverslip was assembled into a FCS2 flowcell for polony sequencing. TaqI-cleaved polonies were hybridized with a polony sequencing primer following the above described template hybridization protocol and then sequenced with a HiSeq SBS kit v4 (Illumina, FC-401-4002). Images were acquired using a Nikon Ti-E automated inverted epifluorescence microscope equipped with a perfect focus system, a Nikon CFI60 Plan Fluor 40 ×/1.3-NA oil immersion objective, a linear encoded motorized stage (Nikon Ti-S-ER), and an Andor iXon Ultra 888 EMCCD camera (16-bit dynamic range, 1,024 × 1,024 array with 13-μm pixels). A four-channel imaging setup comprised two laser lines (Laser Quantum GEM 532 nm (500 mW) and Melles Griot 85-RCA-400 660 nm (400 mWS)) and two filter cubes with an emission filter (610/60–730/60 or 555/40–685/20; Chroma Technology) and a 532/660 dichroic mirror (Chroma Technology). Sequencing regents were added to the flowcell by a fluidic system comprising a multi-position microelectric valve (Valco Instruments EMH2CA) and a multi-channel syringe drive pump (Kloehn V6 12K). The sequencing was automated by building an application in Java 1.6 using jSerialComm (http://fazecast.github.io/jSerialComm/) to control the fluidic system and Micro-Manager v1.4.22 (https://www.micro-manager.org/) to acquire images from selected stage positions. The sequencing was performed with reagents provided in the HiSeq SBS kit following a standard HiSeq sequencing protocol. Each sequencing cycle includes i) pre-cleavage wash with a cleavage buffer, ii) dye and protection group removal by a cleavage mix, iii) post-cleavage wash with a high salt buffer, iv) a pre-incorporation wash with an incorporation buffer, v) incorporation with an incorporation mix, and vi) imaging acquisition in a scan mix. Sequencing 24-bp barcodes on a gel of 22 × 9 mm2 on our platform took ~35 hours.
Pixel-seq assay
Tissue preparation
Mice were anesthetized with Beuthansia (0.2 mL, i.p.; Merck) and decapitated. The brain was rapidly dissected, frozen on crushed dry ice, and stored at −80 °C until cryosectioning.
Transcript capturing and cDNA synthesis
A polony gel on a coverslip or slide was disassembled from the flowcell, washed with milli-Q water and 3 × 200 μL wash buffer 1 (0.1 × SSC and 0.4 × Maxima H Minus RT buffer (Thermo Fisher, EP0753)), and dried in the PCR hood before use. For cryosectioning, a frozen tissue block was equilibrated at −20 °C in a Cryostat NX70 (Thermo Scientific) for 15 min, mounted onto a holder with O.C.T. (Fisher Healthcare, 4585), and sliced to 10-μm sections. Immediately after placing tissue sections on printed array positions on the dry gel surface, 50 μL tissue hybridization buffer (6 × SSC and 2 U/μL RNAseOUT (Thermo Fisher, 10777019)) was gently applied to immerse the sections and then the gel was incubated at R.T. for 15 min. After the hybridization, the buffer was removed by pipetting and the polony gel slide was assembled into multi-well reaction chambers (ProPlate; Grace Bio-Labs, 246868). cDNAs were synthesized by adding 100 μL reverse transcription (RT) mixture (5 μL Maxima H- reverse transcriptase (Thermo Fisher, EP0753), 20 μL 5 × Maxima RT buffer, 20 μL 20% Ficoll PM-400 (Sigma-Aldrich, F4375), 10 μL 10 mM each dNTPs, 5 μL 50 μM template-switching oligo (Qiagen, 339414YCO0076714), 2.5 μL RNAseOUT (40 U/μL), and 37.5 μL H2O) into each well and incubating the chambers at 42 °C for 1 hour. For some experiments, gel attached tissues were stained by SYTOX Green added by presoaking and drying the gels and imaged before adding the hybridization buffer. Tissues were imaged with a polony sequencing microscope described below or a Yokogawa spinning disc confocal system on a Nikon Ti-E stage with an Andor iXon Ultra 888 EMCCD camera (16-bit dynamic range, 1,024 × 1,024 array with 13-μm pixels) and a Nikon CFI60 Plan Fluor 40 ×/1.3-NA oil immersion objective under a FITC channel (excitation, 488 nm; emission filter, 525/50). A Cy5-dCTP-labeled cDNA assay was performed at the same cDNA synthesis condition except replacing the dNTPs with 500 μM each dATP/dGTP/dTTP, 12.5 μM dCTP, and 25 μM Cy5-dCTP (PerkinElmer, NEL577001EA).
Tissue cleanup
After the cDNA synthesis, the reaction buffer was removed, and the tissues were washed by 3 × 200 μL 0.1 × SSC. 100 μL proteinase K digestion solution (10 μL proteinase K (Qiagen, 19131) in 90 μL PKD buffer (Qiagen, 1034963)) was added to each chamber and incubated at 55 °C for 30 min. To remove digested proteins, genomic DNAs, and others, each chamber was washed with 3 × 200 μL elution buffer 2 (2 × SSC and 0.1% SDS) and 3 × 200 μL wash buffer 2 (0.1 × SSC).
Library construction and sequencing
Recovering spatially barcoded cDNAs from the gel and introducing unique molecular identifiers (UMIs) into cDNAs were achieved by the second-strand synthesis and primer extension, respectively. For example, 70 μL second-strand mix (7 μL 10 × isothermal amplification buffer (NEB, B0357), 7 μL 10 mM each dNTP mix, 3.5 μL 10 μM TSO primer, 0.5 μL 20 mg/mL BSA (NEB, B9000), 3 μL BST2.0 WarmStart DNA Polymerase (NEB, M0538), and 49 μL H2O) was added to each chamber and the chambers were sealed with a sealing film. After incubating the chambers at 65 °C for 15 min, the reagent was removed, and the gel was washed by 3 × 200 μL wash buffer 2. To elute the DNAs, a denature and elution mix (35 μL 0.08 M KOH) was added to each chamber, incubated at R.T. for 10 min, and neutralized by 5 μL Tris (1 M, pH 7.0). ~35 μL sample was transferred from each chamber into a tube and added with 65 μL UMI incorporation mix (50 μL 2 × Q5 Ultra II master mix (NEB, M0544), 2.5 μL 10 μM UMI primer, and 12.5 μL H2O). The UMI incorporation was performed by denaturing at 95 °C for 30 seconds, annealing at 65 °C for 30 seconds, and extension at 72 °C for 5 min in a PCR machine. Non-incorporated primer was removed by incubating the sample with 1 μL thermolabile exonuclease I (20 U; NEB, M0568) at 37 °C for 4 min and inactivating the exonuclease at 80 °C for 1 min. To amplify the cDNA library, the tube was added with 2 μL each 10 μM TSO and TruSeq sequencing primers and 1 μL Q5 HotStart polymerase (NEB M0493) for PCR amplification to obtain 5–10 ng DNA per sample. PCR reactions were performed as follows: annealing at 95 °C for 3 min, 4 amplification cycles each including 98 °C for 20 seconds, 65 °C for 45 seconds, and 72 °C for 3 min, 8 amplification cycles each including 98 °C for 20 seconds, 67 °C for 20 seconds, and 72 °C for 3 min, and a final incubation at 72 °C for 5 min. For example, we used 12 PCR cycles to amplify cDNAs from an OB section. Typically, after the amplification, ~1 ng DNA was used for sequencing library construction with a Nextera XT kit (Illumina FC-131-1024) and the TruSeq LibP5 primer, and Nextera index primers following the manufacturer’s protocol. From tissue cryosectioning to the completion of the sequencing library construction, the Pixel-seq assay took ~6 hours.
Nerve ligation surgery
Sciatic nerve surgeries were performed as previously described (Seltzer et al., 1990). Mice were anesthetized with 2% isoflurane at a flow rate of 1 L/min and a 1-cm long incision was made in the proximal one third of the lateral thigh. The sciatic nerve was exteriorized with forceps inserted under the nerve. A silk suture was then passed through approximately 1/2 of the nerve bundle, before being tightly ligated and crushed. For sham mice, the sciatic nerve was exteriorized using forceps and then returned to its normal position. The skin was then closed with sutures. All mice were euthanized for tissue extraction exactly 30 days post nerve ligation surgery.
RNAscope
10-μm-thick cryosectioned tissues were mounted on glass slides (Fisher Scientific, 12–550D). A RNAscope HiPlex assay (ACDBio, 324409) was performed following the manufacturer’s protocol. Briefly, slides were fixed in 4% paraformaldehyde, dehydrated in 50%, 70%, and 100% ethanol, then treated with Protease IV. Probes (e.g., Penk, Pdyn, Calca, and Tac1) were hybridized at 40 °C for 2 hours then amplified for detection. Cell nuclei were stained with DAPI then fluorescent images were acquired using a Keyence BZ-X700 microscope. Images were registered using HiPlex image registration software (ACDBio) and minimally processed in Fiji (ImageJ) to enhance brightness and contrast for optimal representation of the data.
Lateral diffusion analysis
To assess template lateral diffusion during RNA capturing, we compared cell sizes detected by RNAscope and Pixel-seq. Specifically, we analyzed an abundantly expressed neuropeptide gene (Gal) in the same cell type, Gal+ neurons, at identical anatomical positions adjacent to the PBN. Gal showed relatively strong, isolated signals which facilitated delineating individual cell shapes. The template diffusion was estimated by comparing the median distance of Pixel-seq-mapped Gal transcripts (n = 623) from calculated cell centroids and that calculated using the RNAscope data on identical PBN regions downloaded from Zenodo (DOI: 10.5281/zenodo.6707404).
Data Analysis
Polony image analysis
To compare stamped polonies on different gels, polony images were registered to the one with the highest signal-to-noise ratio using the imregcorr function in Matlab. Polonies were detected by a local-threshold method and their intensities, sizes, and centroids were measured using the regionprops function. We first built a consensus map of polonies with a normalized intensity > 0.1 and detected in at least two out of three copy gel images. Polony center shifts were calculated as the Euclidean distance between detected polonies and the consensus.
Base calling of polony sequencing
Raw sequencing tiff images were processed to extract intensities by Dlight, a custom-built suite in MATLAB. All images were registered to the merged images of the Cy3 and Cy5 channels from the first sequencing cycle using the imregcorr function. Next, a polony reference map was generated from images of the first eight sequencing cycles, termed template cycles. Polonies were identified by searching local signal thresholds (> median + 2 × standard deviation) in all template cycle images and then finding polony centroids with an optimal chastity value. A PhiX control library (Illumina FC-110–3001) was used to optimize Dlight parameters. Intensity values of polony centroids and image pixels were analyzed by a 3Dec base-caller (Wang et al., 2017) allowing the correction of the signal crosstalk between adjacent polonies. To find polony boundaries, unassigned image pixels were compared with spatial barcode-assigned polony centroids within a distance less than 5 pixels (Figure S1F). These pixels were assigned with adjacent assigned spatial barcodes with the highest signal correlation coefficient above 0.7. A final spatial barcode map was constructed by combining all sequenced gel positions into a single image.
Transcript mapping
After cDNA library sequencing, FASTQ files were processed to map transcripts to the spatial barcode map. Spatial barcode and UMI sequences were first extracted by Flexbar (Roehr et al., 2017). The index sequences were mapped back to the spatial barcode map by Bowtie (Langmead et al., 2009) allowing up to 2 mismatches. Paired-end reads of mapped indices were aligned to mouse transcriptome (GRCm38) using STARv2.7.0 (Dobin et al., 2013) with a default setting. Sequencing reads with the same transcriptome mapping locus, UMI, and spatial barcode were collapsed to unique records for subsequent analysis.
Transcript segmentation
A method, volume-distance-based segmentation of mapped transcripts (V-seg) implemented in R, was developed to aggregate feature-level UMIs to single cells. V-seg first used mapped transcript data to construct a rNN-network (nearest neighbors within a defined radius, r) by connecting spatial barcodes as nodes (Figure S2F). The network was constructed using a cutoff edge distance of 2 μm to ensure every index was connected to at least one different barcode and also decrease edge or connection redundancy. The edge weight was calculated as:
where Vi and Vj are two nodes or spatial barcodes, E(Vi, Vj) is an edge weight between Vi and Vj, D(Vi, Vj) is the spatial distance between Vi and Vj, UMI(Vi) is the UMI count in Vi, I(UMI(Vi), UMI(Vj)) is the UMI count of shared transcripts between Vj andVi, and ∑ UMI(V−i), is the sum of UMIs in all nodes connected to the node Vi. Next, the edge-weighted network was taken as an input for the graph-based segmentation with a Fast Greedy algorithm using an iGraph R package. Because the Fast Greedy algorithm is sensitive to the network size, to maximize a modularity score, it is necessary to optimize segmentation parameters for different tissues. In case of the OB data in Figure 2, the edge threshold was the weight < 3 and the spatial transcript map was divided to images of 800 × 800 pixels (1 pixel = 0.325 μm) for individual processing before stitching them together. In the PBN analysis in Figures 3 and 4, the segmentation was performed in two steps to separately process aggregated Calca cells and other cell types. Step 1: the edge threshold was the weight < 4 and the transcript map was divided to images of 600 × 600 pixels. Segmented cells were clustered by Seurat to separate Calca cells. Step 2: Other cells were re-segmented using the edge threshold of the weight < 4 and the image size of 800 × 800 pixels.
Segmentation validation
To evaluate the performance of V-seg, we collected the images of SYTOX Green-stained nuclei of the same mouse OB tissue as a reference at the beginning of a Pixel-seq assay. In parallel to the Pixel-seq assay, the Cy5-labelled cDNAs were also synthesized as described above and the images were acquired for cDNA image-based segmentation, similar to the poly(A) staining-guided segmentation used for imaging-based spatial transcriptomics (Codeluppi et al., 2018; Moffitt et al., 2018). The nuclear images were segmented by Cellpose (Stringer et al., 2021) and manually aligned to the cDNA images or the Pixel-seq-generated UMI density map. To quantitatively assess the accuracy of V-seg, V-seg-segmented masks were overlaid with the images of the stained nuclei in the same tissue and then divided into three scenarios: a) masks containing single nuclei, b) masks partially overlapped with single nuclei, and c) masks overlapped with multiple or zero nuclei. A higher ratio of a) indicates a higher segmentation accuracy. To demonstrate the improvement of V-seg compared with random bins commonly used for the array-based spatial transcriptome data analysis, this analysis was also performed for random bins with a regular size similar to the mean size of segmented masks.
Clustering analysis
For the OB clustering, 22,830 cells were analyzed. SCTransform normalization was applied in Seurat using 3,000 highly variable genes. After Principal Component Analysis (PCA), 18 PCs were used in the UMAP visualization, and the clustering graph was generated with a resolution of 0.8. For the clustering outcome shown in Figure 3C, 63,808 cells from four PBN sections were analyzed with 22 PCs and graphed with a resolution of 0.3. For the clustering outcome shown in Figure 3D, 31,505 neurons from Figure 3C were analyzed with 12 PCs and graphed with a resolution of 0.65. For the clustering in Figure 4B, 15,833 neurons from two PBN sections were analyzed with 18 PCs and graphed with a resolution of 0.7, and 16,544 non-neuronal cells were analyzed with 22 PCs and graphed with a resolution of 0.4. For the clustering outcome in Figure 5D, 584 microglia from the two PBN sections were analyzed with 14 PCs and graphed with a resolution of 0.4. For the clustering outcome in Figure 5F, 4,471 astrocytes were analyzed with 10 PCs and graphed with a resolution of 0.4. Marker genes for each cluster were identified by Seurat using a Wilcoxon test and then compared with the consensus in a published mouse brain cell type atlas (Zeisel et al., 2018).
scRNA-seq-guided annotation
Segmented OB cells were annotated with a published OB scRNA-seq dataset (GSE121891) (Tepe et al., 2018) as a reference to predict cell type compositions using scvi-tools (Gayoso et al., 2021). The top 3,000 variable genes were selected for the model training. Raw gene counts for training and testing datasets were scaled to 104. The training and prediction were run at max_epoches = 50. Cell types of segmented cells were determined by the predicted cell types with the highest ratios.
Differential abundance analysis
The clustering results in Figures 4C and 5G (left) were analyzed by DA-seq (Zhao et al., 2021) to identify differences between cell distributions in the UMAP space for the Sham and SNL conditions. Coordinates of the UMAP clustering outcomes and Sham and SNL-condition labels were maintained for the DA-seq analysis using a score vector of k = seq (50, 500, 50).
Spatial pattern gene analysis
To identify transcript distributions showing different spatial patterns in the Sham and SNL conditions, the cell-gene matrix and cell spatial coordinates were processed by Giotto (Dries et al., 2021). The analysis used a default setting (e.g., a minimum detected genes of 64 per cell and a scale factor of 6,400) to find spatially patterned genes by a “ranking” method. The top 200 spatially patterned genes were chosen for a GO enrichment analysis (Kuleshov et al., 2016). Selected genes belonging to representative functional groups are shown in Figure S4C.
PCCF statistic
We quantitatively compared cell contacts (or colocalization) between the same or different cell types using a pairwise cross-correlation function (PCCF) statistic which was previously applied to analyze polony colocalization (Gu et al., 2014). Here, to analyze cell colocalization, PCCF was defined as the ratio of a detected number of colocalization events to a random colocalization possibility. The random colocalization was simulated by a simplified model, where irregular cells were represented as round shapes with original sizes. After randomizing original cell positions, touched and overlapped cell masks were counted; 100 simulations were performed to calculate the average colocalization count as the random colocalization possibility. In Figure 3G, the PCCF was computed for all annotated cell clusters to generate the heatmap.
Pseudotime analysis
The pseudotime analysis of the astrocyte clusters in Figure 5G (right) was performed by Slingshot (Street et al., 2018) with parameters of stretch = 2 and thresh = 0.001. The P-value of the pseudotime values for the Sham and SNL conditions was calculated with a Kolmogorov-Smirnov test.
Cell-cell communication
The spatial profiling of signaling likelihoods of individual cells as senders and receivers was performed by SpaOTsc with default parameters (Cang and Nie, 2020). The calculation used an expression matrix of paired ligand and receptor genes and spatial locations of corresponding cells as inputs. For cluster-to-cluster communication, signaling likelihoods higher than 0.003 are shown in Figure 5A. In Figure 5B, a Kolmogorov-Smirnov test was used to compare cluster-level signaling likelihoods for paired ligand and receptor(s) genes between the Sham and SNL conditions.
Supplementary Material
(A) Automated gel stamping device comprises a desktop robotic arm to sequentially place the stamp in four positions in each stamping cycle: 1) formamide at 60 °C, 2) a stamping buffer at 60 °C, 3) a stamping mix including Taq polymerase and dNTPs at 4 °C, and 4) a specified position on a copy gel at 95 °C to 60 °C (see STAR Methods). The stamping pressure is monitored by an electronic balance.
(B) 3D reconstruction of z-stack images of a polony gel. Fluorescence polony signals were mainly located in a ~400-nm-thick layer at the gel surface.
(C) Comparison of polony patterns at equivalent locations of three copy gels stamped under the pressures of ~1, ~2, and ~3 × 104 Pa. Specifically. the force equivalent to the weights of ~50, ~100, and ~150 gram was applied to a stamp of ~7 × 7 mm2. The low-density polonies (~105/mm2) were replicated to facilitate visual comparison of feature patterns.
(D) Box plot of the percentages of polonies in copy gels in (C) matched to the consensus. Data represent mean values of six sampled gel positions found with 127 to 236 polonies; error bars, standard deviation.
(E) Measure of cleavage efficiency of double-stranded polonies by TaqI. (Left) Two polony gels prepared under the same condition were treated without or with TaqI (see STAR Methods), linearized by the USER digestion, and then probed with a Cy5-labelled oligo (magenta). (Right) The TaqI cleavage efficiency was measured by comparing Cy5 signals in eight randomly picked positions (1,024 × 1,024 pixels, 1 pixel = 0.325 × 0.325 μm2) in the TaqI-treated versus non-treated gels. Some residual polonies on the TaqI-treated gel are likely due to mutations in the TaqI cleavage site introduced by the polony amplification.
(F) Construction of a spatial barcode map from polony sequencing data. i) Raw sequencing images were processed to extract fluorescence intensities from all pixels. A barcode map was generated by base calling after channel, phase, and spatial crosstalk corrections. Correlation-based barcode correction on unmapped pixels were performed to analyze polony boundaries. ii) Spatial heatmap of the correlation coefficient of extracted intensities from all sequencing channels and cycles between the center pixel and neighboring pixels in the image. iii) Density plot of the correlation coefficient for all pixels in (ii). A cutoff of 0.7 was used to group pixels with an identical barcode. iv) Constructed pixel-level barcode map. Representative images of polonies amplified on (G) crosslinked polyacrylamide gels and (H) a linear polyacrylamide-coated glass surface following a reported Illumina method. DNA templates were seeded at similar densities and amplified with the indicated cycle numbers before SYBR Green staining. Images were acquired at the same imaging condition and subjected to background subtraction for fluorescence quantification in Figure 1J.
(I) Four-channel sequencing images of polonies at increased densities. Templates were seeded at specific densities and amplified by 35 cycles. Due to the polony exclusion effect, even at higher feature densities than typically sequenced (e.g., ~1.2 million/mm2 with an average feature size of ~0.5 μm), most polonies in different colors show clear boundaries. Some dark regions in the images were occupied by TaqI cleaved polonies without sequencing signals because some randomly generated barcodes contained TaqI cleavage site(s).
(A) Images of Cy5-labeled cDNA signals rising from gel-captured RNAs which delineate shapes of cell bodies in a 10-μm mouse OB-isocortex section. CTX, the cerebral cortex; AOB, the accessory olfactory bulb; AON, the anterior olfactory nucleus; MOB, the main olfactory bulb.
(B) Analysis of the template lateral diffusion by comparing cell sizes detected by RNAscope and Pixel-seq. It used the same cell type at identical anatomical positions. i) Detected signals of a neuronal peptide transcript (Gal) in Gal+ neurons adjacent to the PBN. Gal often gives relatively strong, isolated single cell signals and thus is suitable for delineating cell shapes. ii) Measure of template diffusion by comparing the median distances between Gal signals and calculated cell centroids. The diffusion distance is estimated to be ~0.86 μm.
(C) Illustration of RNA capture from a single layered cells on a dry polony gel.
(D) Comparison of three mouse OB datasets (OB1-3). R, Pearson correlation coefficient between detected gene expressions in two datasets.
(E) Computational simulation of separating cell bodies in a brain tissue by DNA array-based spatial transcriptomics with different feature sizes. i) The simulation uses a published mouse cortex seqFISH+ data with a pixel size of 0.103 μm (left) and overlays the single-cell data with simulated round features (right) with a diameter size from 10 to 1 μm. To consider feature variations in real experiments, features were simulated with a 5% size variation following a Gaussian distribution. In a 2D plane, they were progressively packed and then overlaid with the segmented transcriptomes to calculate the number of cells contacting a feature (right) and then aggregate touched transcripts by feature. 10 simulations were run for each diameter size. ii) Fraction of features that can be confidently assigned to single cells using a cutoff of 0.7 (i.e., 70% of transcripts in a feature from the same cell). Original and resized cortex cells with average diameters of 15.9 (left) and 7.95 μm (right), respectively, were used for the simulation.
(F) Principle of V-seg. i) Data pipeline. ii) A cutoff edge distance for the network construction was chosen based on the distribution of the distance between a barcode and the kth-nearest neighboring barcode. The cutoff of 2 μm (dotted line) was chosen to ensure every barcode was connected to at least one different barcode and avoid generating many redundant edges.
(G) Spatial patterns of the V-seg-processed mouse OB data annotated by i) guidance of scRNA-seq OB data and ii) unsupervised clustering. i) Mes, mesenchymal cell; OEC, olfactory ensheathing cell; Neuron.OSN, olfactory sensory neuron; GC, granule cell; PGC, periglomerular cell; EPL.IN, external plexiform layer interneuron; M/TC, mitral and tufted cell; Neuron.Transition, transitional neuron; Neuron.Immature, immature neuron; Astro, astrocyte; MicroG, microglia; OPC, oligodendrocyte precursor; MyOligo, myelinating oligodendrocyte; Mural, mural cell; Neuron.AstroLike, astrocyte like neuron; EC, endothelial cell. ii) ONL, olfactory neuron layer; GL, glomerular layer; EPL, external plexiform layer; MCL, mitral and tufted cell layers; GCL, granule cell layer; RMS, rostral migratory stream.
(H) Comparison of segmented mask sizes and aggregated UMIs for two cell types with distinct mask sizes, mitral/tufted (M/TC) and periglomerular type 1 cells (PGC). Data for 248 PGC and 1,723 M/TC cells were plotted with the total 22,830 cells detected in an OB section. M/TC and PGC mask sizes were measured to be 14.5 ± 4.8 and 10.9 ± 4.6 μm, respectively.
(I) Comparison of the UMI density map (top), the V-seg output (middle), and marker gene distributions (bottom).
(J) Violin plots of diameters (top) and UMI densities (bottom) of the masks annotated to specific cell types. Irregular masks were converted to round shapes to calculate diameters. Data for 5,565 to 135 masks for each cell type were plotted.
(K) Scatter plot of pseudo-bulk profiles between Pixel-seq and scRNA-seq mouse OB data. R, Pearson correlation coefficient.
(L) Comparison of relative abundances of major cell types detected by scRNA-seq and Pixel-seq (refer to (G), panel i).
(A) Spatial patterns of cell types in the PBN and neighboring regions. i) Anatomical reference. CBX, cerebellar cortex; MB, midbrain; CUN, cuneiform nucleus; sctv, ventral spinocerebellar tract; PBNl, parabrachial nucleus, lateral division; PBNm, parabrachial nucleus, medial division; KF, Kölliker-Fuse subnucleus; scp, superior cerebellar peduncle; V, trigeminal motor nucleus; SLC, subceruleus nucleus; LTD, laterodorsal tegmental nucleus; DTN, dorsal tegmental nucleus; PCG, pontine central gray; CENT, central lobule. ii) Spatial distributions of 21 cell types identified by Seurat clustering in a middle, coronal PBN section. Segmented masks are colored for two oligodendrocyte clusters (Oligo1 and Oligo2), ten neuronal clusters (Neuron1-Neuron9 and Granule), four astrocyte clusters (Astro1-Astro4), two vascular leptomeningeal clusters (VLMC1 and VLMC2), one endothelial cluster (EC), one ependymal cluster (Ependymal), and one microglia cluster (Microglia). iii) Spatial distributions of 18 re-clustered neuronal subclusters (N1-N18) with defined markers. Masks for non-neuronal cells are shown in grey.
(B) Spatial distributions of 21 cell types identified in the PBN section including 10 neuronal clusters (Neuron1-9 and Granule), two oligodendrocyte clusters (Oligo1 and Oligo2), four astrocyte clusters (Astro1-4), one microglial cluster (Microglia), one endothelial cluster (ECs), one ependymal cluster (Ependymal), and two vascular leptomeningeal clusters (VLMC1 and VLMC2). Cell masks are denoted by dots at their geometric centers.
(C) Spatial distributions of 18 neuronal clusters with defined marker genes.
(D) Pixel-seq-detected distribution of selected neuropeptide genes in anterior (Bregma, −5.20 mm), middle (−5.35 mm), and posterior (−5.50 mm) PBN sections. Neuropeptide transcripts (in red) are plotted atop all transcript reads (in gray). The result is compared with the AMBA ISH data.
(E) RNAscope-detected distribution of the neuropeptide genes in (D). Anatomical references (left) show subnuclei in the lateral (lateral (ls), dorsal (ld), center (lc), ventral (lv), central (lc), and external (le)) and medial PBN (medial (mm)) and superior cerebellar peduncles (scp).
(A) Spatial distributions of 28 cell types identified in a middle, coronal PBN section from a sham mouse, including 16 neuronal clusters (Neu1-Neu16) and 12 non-neuronal clusters (two oligodendrocyte (Oligo1 and Oligo2), one microglia (Microglia), five astrocyte (Astro1–5), two vascular leptomeningeal clusters (VLMC1 and VLMC2), one ependymal cluster (Ependymal), and one endothelial cluster (EC). Cell masks are plotted at their geometric centers.
(B) Spatial distributions of cell types in a PBN section anatomically matched to (A) from a SNL mouse.
(C) Comparison of spatial patterns of selected genes for i) secreted and ii) other ligands, iii) in GABAergic synapse, for iv) calcium ion binding and v) solute carrier superfamily proteins. Transcripts in purple are plotted to a UMI density map in grey. Each dot represents a spatial barcode with detected genes. Relative transcript levels were scaled to gray and purple color gradients.
(A) Spatial distributions of signaling likelihoods of sender and receiver cells in the same PBN section. Signaling likelihoods of cells as senders (left) and receivers (right) were calculated by SpaOTsc. Cell masks are plotted as filled circles at their geometric centers.
(B) Comparison of the signaling likelihoods between different cell types under the sham and SNL conditions. Astrocyte and microglial clusters are highlighted in orange and green, respectively, and others in gray.
(C) Feature plots of selected microglial marker genes.
(D) GO term enrichment analysis of marker genes of the two microglial subclusters, M1 and M1*. P-values, Fisher’s exact test.
(E) Feature plots of astrocyte marker genes for each subcluster.
(F) Spatial distributions of astrocyte subclusters in the PBN region. Astrocyte masks are plotted as filled circles at their geometric centers.
Highlights:
Polony gel stamping enables vastly scalable replication of DNA cluster arrays
Polony gels capture tissue RNAs with high spatial resolution and efficiency
Pixel-seq spatially maps single-cell transcriptomes of mouse brain tissues
Pixel-seq reveals chronic pain-induced changes in the parabrachial nucleus
ACKNOWLEDGMENTS
We thank B. Wang and W. Liang for contributions to a base calling algorithm, D. Liu and J. Cheng for the help to build the polony sequencer, Q. Tang for sharing mouse tissues, and S. Kang for discussing assay development. This work was supported by a University of Washington startup fund to L.G. and National Institutes of Health grants (R35GM128918, UG3CA268096, R21DA051555, and R21DA051194 to L.G., R41MH130299 to L.S., and R01DA024908 to R.D.P.).
Footnotes
DECLAEATION OF INTERESTS
Two patents on polony gel stamping and Pixel-seq have been filed by the University of Washington and TopoGene Inc. L.G. and L.S. are co-founders of TopoGene which aims to commercialize polony gels.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aach J, and Church GM (2004). Mathematical models of diffusion-constrained polymerase chain reactions: basis of high-throughput nucleic acid assays and simple self-organizing systems. J. Theor. Biol. 228, 31–46. [DOI] [PubMed] [Google Scholar]
- Barik A, Thompson JH, Seltzer M, Ghitani N, and Chesler AT (2018). A brainstem-spinal circuit controlling nocifensive behavior. Neuron 100, 1491–1503. [DOI] [PubMed] [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumgarner R (2013). Overview of DNA microarrays: types, applications, and their future. In Curr. Protoc. Mol. Biol. (101), 22.1.1–22.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos CA, Bowen AJ, Roman CW, and Palmiter RD (2018). Encoding of danger by parabrachial CGRP neurons. Nature 555, 617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang ZX, and Nie Q (2020). Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 11, 2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, Qiu X, Yang J, Xu J, Hao S, et al. (2022). Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792. [DOI] [PubMed] [Google Scholar]
- Chen F, Tillberg PW, and Boyden ES (2015). Optical imaging. Expansion microscopy. Science 347, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CS, Xi JY, Si YC, Park SR, Hsu JE, Kim M, Jun G, Kang HM, and Lee JH (2021). Microscopic examination of spatial transcriptome using Seq-Scope. Cell 184, 3559–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codeluppi S, Borm LE, Zeisel A, La Manno G, van Lunteren JA, Svensson CI, and Linnarsson S (2018). Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932–935. [DOI] [PubMed] [Google Scholar]
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, and Trent JM (1996). Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 14, 457–460. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dries R, Zhu Q, Dong R, Eng CHL, Li HP, Liu K, Fu YT, Zhao TX, Sarkar A, Bao F, et al. (2021). Giotto: a toolbox for integrative analysis and visualization of spatial expression data. Genome Biol. 22, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CHL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, Yun JN, Cronin C, Karp C, Yuan GC, and Cai L (2019). Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, and Solas D (1991). Light-directed, spatially addressable parallel chemical synthesis. Science 251, 767–773. [DOI] [PubMed] [Google Scholar]
- Gayoso A, Lopez R, Xing G, Boyeau P, Valiollah Pour Amiri V, Hong J, Wu K, Jayasuriya M, Melhman E, Langevin M, et al. (2022). A Python library for probabilistic analysis of single-cell omics data. Nat. Biotechnol. 40, 163–166. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Li C, Aach J, Hill DE, Vidal M, and Church GM (2014). Multiplex single-molecule interaction profiling of DNA-barcoded proteins. Nature 515, 554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zagar M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Tang X, Huang J, Zhou H, Chen K, Liu A, Ren J, Shi H, Lin Z, Li Q, et al. (2021). ClusterMap: Multi-scale Clustering Analysis of spatial gene expression. Nat. Commun. 12, 5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TW, Lin SH, Malewicz NM, Zhang Y, Zhang Y, Goulding M, LaMotte RH, and Ma QF (2019). Identifying the pathways required for coping behaviours associated with sustained pain. Nature 565, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvie BC, Chen JY, King HO, and Palmiter RD (2021). Satb2 neurons in the parabrachial nucleus mediate taste perception. Nat. Commun. 12, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Davidsen K, Gomez-Castillo L, Jiang H, Fu X, Li Z, Liang Y, Jahn M, Moussa M, DiMaio F, and Gu L (2019). COMBINES-CID: An efficient method for de novo engineering of highly specific chemically induced protein dimerization systems. J. Am. Chem. Soc. 141, 10948–10952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan QN, Wang ZC, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupari J, Usoskin D, Parisien M, Lou DH, Hu YZ, Fatt M, Lonnerberg P, Spangberg M, Eriksson B, Barkas N, et al. (2021). Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat. Commun. 12, 1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange SA, Benes V, Kern DP, Horber JK, and Bernard A (2004). Microcontact printing of DNA molecules. Anal. Chem. 76, 1641–1647. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Cheng ZL, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, et al. (2019). Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 101, 207–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Larke LEC, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman R, Hemminger Z, Foreman R, Arneson D, Zhang G, Gómez-Pinilla F, Yang X, and Wollman R (2021). Joint cell segmentation and cell type annotation for spatial transcriptomics. Mol. Syst. Biol. 17, e10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yang MY, Deng YX, Su G, Enninful A, Guo CC, Tebaldi T, Zhang D, Kim D, Bai ZL, et al. (2020). High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell 183, 1665–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra RD, and Church GM (1999). In situ localized amplification and contact replication of many individual DNA molecules. Nucleic Acids Res. 27, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Bambah-Mukku D, Eichhorn SW, Vaughn E, Shekhar K, Perez JD, Rubinstein ND, Hao J, Regev A, Dulac C, and Zhuang X (2018). Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris AJ, Shaker JR, Cone AL, Ndiokho IB, and Bruchas MR (2021). Parabrachial opioidergic projections to preoptic hypothalamus mediate behavioral and physiological thermal defenses. Elife 10, e60779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD (2018). The Parabrachial Nucleus: CGRP Neurons Function as a General Alarm. Trends Neurosci. 41, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Choi W, Tiesmeyer S, Long B, Borm LE, Garren E, Nguyen TN, Tasic B, Codeluppi S, Graf T, Schlesner., et al. (2021). Cell segmentation-free inference of cell types from in situ transcriptomics data. Nat. Commun. 12, 3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhov V, Xu RJ, Soldatov RA, Cadinu P, Khodosevich K, Moffitt JR, and Kharchenko PV (2022). Cell segmentation in imaging-based spatial transcriptomics. Nat Biotechnol. 40, 345–354. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, and Ungerer N (2011). Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 50, 357–371. [DOI] [PubMed] [Google Scholar]
- Philimonenko AA, Janacek J, and Hozak P (2000). Statistical evaluation of colocalization patterns in immunogold labeling experiments. J. Struct. Biol. 132, 201–210. [DOI] [PubMed] [Google Scholar]
- Pinching AJ, and Powell TPS (1971). Neuron types of glomerular layer of olfactory bulb. J. Cell Biol. 9, 305–345. [DOI] [PubMed] [Google Scholar]
- Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LLM, Chen F, and Macosko EZ (2019). Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehr JT, Dieterich C, and Reinert K (2017). Flexbar 3.0-SIMD and multicore parallelization. Bioinformatics 33, 2941–2942. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, and Brown PO (1995). Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470. [DOI] [PubMed] [Google Scholar]
- Seltzer Z, Dubner R, and Shir Y (1990). A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 43, 205–218. [DOI] [PubMed] [Google Scholar]
- Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82. [DOI] [PubMed] [Google Scholar]
- Stickels RR, Murray E, Kumar P, Li JL, Marshall JL, Di Bella DJ, Arlotta P, Macosko EZ, and Chen F (2021). Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat. Biotechnol. 39, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, and Dudoit S (2018). Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer C, Wang T, Michaelos M, and Pachitariu M (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106. [DOI] [PubMed] [Google Scholar]
- Sun L, Liu R, Guo F, Wen MQ, Ma XL, Li KY, Sun H, Xu CL, Li YY, Wu MY, et al. (2020). Parabrachial nucleus circuit governs neuropathic pain-like behavior. Nat. Commun. 11, 5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepe B, Hill MC, Pekarek BT, Hunt PJ, Martin TJ, Martin JF, and Arenkiel BR (2018). Single-cell RNA-seq of mouse olfactory bulb reveals cellular heterogeneity and activity-dependent molecular census of adult-born neurons. Cell Rep. 25, 2689–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainchtein ID, and Molofsky AV (2020). Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 43, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickovic S, Eraslan G, Salmen F, Klughammer J, Stenbeck L, Schapiro D, Aijo T, Bonneau R, Bergenstrahle L, Navarro JF, et al. (2019). High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickovic S, Lotstedt B, Klughammer J, Mages S, Segerstolpe A, Rozenblatt-Rosen O, and Regev A (2022). SM-Omics is an automated platform for high-throughput spatial multi-omics. Nat. Commun. 13, 795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Wan L, Wang A, and Li LM (2017). An adaptive decorrelation method removes Illumina DNA base-calling errors caused by crosstalk between adjacent clusters. Sci. Rep. 7, 41348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, and Whitesides GM (1998). Soft lithography. Angew. Chem. Int. Ed. Engl. 37, 550–575. [DOI] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-Regulated Transcription: Bridging the Gap between Neural Activity and Behavior. Neuron 100, 330–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular architecture of the mouse nervous system. Cell 174, 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jaffe A, Li H, Lindenbaum O, Sefik E, Jackson R, Cheng XY, Flavell RA, and Kluger Y (2021). Detection of differentially abundant cell subpopulations in scRNA-seq data. Proc. Natl. Acad. Sci. U. S. A. 118, e2100293118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Automated gel stamping device comprises a desktop robotic arm to sequentially place the stamp in four positions in each stamping cycle: 1) formamide at 60 °C, 2) a stamping buffer at 60 °C, 3) a stamping mix including Taq polymerase and dNTPs at 4 °C, and 4) a specified position on a copy gel at 95 °C to 60 °C (see STAR Methods). The stamping pressure is monitored by an electronic balance.
(B) 3D reconstruction of z-stack images of a polony gel. Fluorescence polony signals were mainly located in a ~400-nm-thick layer at the gel surface.
(C) Comparison of polony patterns at equivalent locations of three copy gels stamped under the pressures of ~1, ~2, and ~3 × 104 Pa. Specifically. the force equivalent to the weights of ~50, ~100, and ~150 gram was applied to a stamp of ~7 × 7 mm2. The low-density polonies (~105/mm2) were replicated to facilitate visual comparison of feature patterns.
(D) Box plot of the percentages of polonies in copy gels in (C) matched to the consensus. Data represent mean values of six sampled gel positions found with 127 to 236 polonies; error bars, standard deviation.
(E) Measure of cleavage efficiency of double-stranded polonies by TaqI. (Left) Two polony gels prepared under the same condition were treated without or with TaqI (see STAR Methods), linearized by the USER digestion, and then probed with a Cy5-labelled oligo (magenta). (Right) The TaqI cleavage efficiency was measured by comparing Cy5 signals in eight randomly picked positions (1,024 × 1,024 pixels, 1 pixel = 0.325 × 0.325 μm2) in the TaqI-treated versus non-treated gels. Some residual polonies on the TaqI-treated gel are likely due to mutations in the TaqI cleavage site introduced by the polony amplification.
(F) Construction of a spatial barcode map from polony sequencing data. i) Raw sequencing images were processed to extract fluorescence intensities from all pixels. A barcode map was generated by base calling after channel, phase, and spatial crosstalk corrections. Correlation-based barcode correction on unmapped pixels were performed to analyze polony boundaries. ii) Spatial heatmap of the correlation coefficient of extracted intensities from all sequencing channels and cycles between the center pixel and neighboring pixels in the image. iii) Density plot of the correlation coefficient for all pixels in (ii). A cutoff of 0.7 was used to group pixels with an identical barcode. iv) Constructed pixel-level barcode map. Representative images of polonies amplified on (G) crosslinked polyacrylamide gels and (H) a linear polyacrylamide-coated glass surface following a reported Illumina method. DNA templates were seeded at similar densities and amplified with the indicated cycle numbers before SYBR Green staining. Images were acquired at the same imaging condition and subjected to background subtraction for fluorescence quantification in Figure 1J.
(I) Four-channel sequencing images of polonies at increased densities. Templates were seeded at specific densities and amplified by 35 cycles. Due to the polony exclusion effect, even at higher feature densities than typically sequenced (e.g., ~1.2 million/mm2 with an average feature size of ~0.5 μm), most polonies in different colors show clear boundaries. Some dark regions in the images were occupied by TaqI cleaved polonies without sequencing signals because some randomly generated barcodes contained TaqI cleavage site(s).
(A) Images of Cy5-labeled cDNA signals rising from gel-captured RNAs which delineate shapes of cell bodies in a 10-μm mouse OB-isocortex section. CTX, the cerebral cortex; AOB, the accessory olfactory bulb; AON, the anterior olfactory nucleus; MOB, the main olfactory bulb.
(B) Analysis of the template lateral diffusion by comparing cell sizes detected by RNAscope and Pixel-seq. It used the same cell type at identical anatomical positions. i) Detected signals of a neuronal peptide transcript (Gal) in Gal+ neurons adjacent to the PBN. Gal often gives relatively strong, isolated single cell signals and thus is suitable for delineating cell shapes. ii) Measure of template diffusion by comparing the median distances between Gal signals and calculated cell centroids. The diffusion distance is estimated to be ~0.86 μm.
(C) Illustration of RNA capture from a single layered cells on a dry polony gel.
(D) Comparison of three mouse OB datasets (OB1-3). R, Pearson correlation coefficient between detected gene expressions in two datasets.
(E) Computational simulation of separating cell bodies in a brain tissue by DNA array-based spatial transcriptomics with different feature sizes. i) The simulation uses a published mouse cortex seqFISH+ data with a pixel size of 0.103 μm (left) and overlays the single-cell data with simulated round features (right) with a diameter size from 10 to 1 μm. To consider feature variations in real experiments, features were simulated with a 5% size variation following a Gaussian distribution. In a 2D plane, they were progressively packed and then overlaid with the segmented transcriptomes to calculate the number of cells contacting a feature (right) and then aggregate touched transcripts by feature. 10 simulations were run for each diameter size. ii) Fraction of features that can be confidently assigned to single cells using a cutoff of 0.7 (i.e., 70% of transcripts in a feature from the same cell). Original and resized cortex cells with average diameters of 15.9 (left) and 7.95 μm (right), respectively, were used for the simulation.
(F) Principle of V-seg. i) Data pipeline. ii) A cutoff edge distance for the network construction was chosen based on the distribution of the distance between a barcode and the kth-nearest neighboring barcode. The cutoff of 2 μm (dotted line) was chosen to ensure every barcode was connected to at least one different barcode and avoid generating many redundant edges.
(G) Spatial patterns of the V-seg-processed mouse OB data annotated by i) guidance of scRNA-seq OB data and ii) unsupervised clustering. i) Mes, mesenchymal cell; OEC, olfactory ensheathing cell; Neuron.OSN, olfactory sensory neuron; GC, granule cell; PGC, periglomerular cell; EPL.IN, external plexiform layer interneuron; M/TC, mitral and tufted cell; Neuron.Transition, transitional neuron; Neuron.Immature, immature neuron; Astro, astrocyte; MicroG, microglia; OPC, oligodendrocyte precursor; MyOligo, myelinating oligodendrocyte; Mural, mural cell; Neuron.AstroLike, astrocyte like neuron; EC, endothelial cell. ii) ONL, olfactory neuron layer; GL, glomerular layer; EPL, external plexiform layer; MCL, mitral and tufted cell layers; GCL, granule cell layer; RMS, rostral migratory stream.
(H) Comparison of segmented mask sizes and aggregated UMIs for two cell types with distinct mask sizes, mitral/tufted (M/TC) and periglomerular type 1 cells (PGC). Data for 248 PGC and 1,723 M/TC cells were plotted with the total 22,830 cells detected in an OB section. M/TC and PGC mask sizes were measured to be 14.5 ± 4.8 and 10.9 ± 4.6 μm, respectively.
(I) Comparison of the UMI density map (top), the V-seg output (middle), and marker gene distributions (bottom).
(J) Violin plots of diameters (top) and UMI densities (bottom) of the masks annotated to specific cell types. Irregular masks were converted to round shapes to calculate diameters. Data for 5,565 to 135 masks for each cell type were plotted.
(K) Scatter plot of pseudo-bulk profiles between Pixel-seq and scRNA-seq mouse OB data. R, Pearson correlation coefficient.
(L) Comparison of relative abundances of major cell types detected by scRNA-seq and Pixel-seq (refer to (G), panel i).
(A) Spatial patterns of cell types in the PBN and neighboring regions. i) Anatomical reference. CBX, cerebellar cortex; MB, midbrain; CUN, cuneiform nucleus; sctv, ventral spinocerebellar tract; PBNl, parabrachial nucleus, lateral division; PBNm, parabrachial nucleus, medial division; KF, Kölliker-Fuse subnucleus; scp, superior cerebellar peduncle; V, trigeminal motor nucleus; SLC, subceruleus nucleus; LTD, laterodorsal tegmental nucleus; DTN, dorsal tegmental nucleus; PCG, pontine central gray; CENT, central lobule. ii) Spatial distributions of 21 cell types identified by Seurat clustering in a middle, coronal PBN section. Segmented masks are colored for two oligodendrocyte clusters (Oligo1 and Oligo2), ten neuronal clusters (Neuron1-Neuron9 and Granule), four astrocyte clusters (Astro1-Astro4), two vascular leptomeningeal clusters (VLMC1 and VLMC2), one endothelial cluster (EC), one ependymal cluster (Ependymal), and one microglia cluster (Microglia). iii) Spatial distributions of 18 re-clustered neuronal subclusters (N1-N18) with defined markers. Masks for non-neuronal cells are shown in grey.
(B) Spatial distributions of 21 cell types identified in the PBN section including 10 neuronal clusters (Neuron1-9 and Granule), two oligodendrocyte clusters (Oligo1 and Oligo2), four astrocyte clusters (Astro1-4), one microglial cluster (Microglia), one endothelial cluster (ECs), one ependymal cluster (Ependymal), and two vascular leptomeningeal clusters (VLMC1 and VLMC2). Cell masks are denoted by dots at their geometric centers.
(C) Spatial distributions of 18 neuronal clusters with defined marker genes.
(D) Pixel-seq-detected distribution of selected neuropeptide genes in anterior (Bregma, −5.20 mm), middle (−5.35 mm), and posterior (−5.50 mm) PBN sections. Neuropeptide transcripts (in red) are plotted atop all transcript reads (in gray). The result is compared with the AMBA ISH data.
(E) RNAscope-detected distribution of the neuropeptide genes in (D). Anatomical references (left) show subnuclei in the lateral (lateral (ls), dorsal (ld), center (lc), ventral (lv), central (lc), and external (le)) and medial PBN (medial (mm)) and superior cerebellar peduncles (scp).
(A) Spatial distributions of 28 cell types identified in a middle, coronal PBN section from a sham mouse, including 16 neuronal clusters (Neu1-Neu16) and 12 non-neuronal clusters (two oligodendrocyte (Oligo1 and Oligo2), one microglia (Microglia), five astrocyte (Astro1–5), two vascular leptomeningeal clusters (VLMC1 and VLMC2), one ependymal cluster (Ependymal), and one endothelial cluster (EC). Cell masks are plotted at their geometric centers.
(B) Spatial distributions of cell types in a PBN section anatomically matched to (A) from a SNL mouse.
(C) Comparison of spatial patterns of selected genes for i) secreted and ii) other ligands, iii) in GABAergic synapse, for iv) calcium ion binding and v) solute carrier superfamily proteins. Transcripts in purple are plotted to a UMI density map in grey. Each dot represents a spatial barcode with detected genes. Relative transcript levels were scaled to gray and purple color gradients.
(A) Spatial distributions of signaling likelihoods of sender and receiver cells in the same PBN section. Signaling likelihoods of cells as senders (left) and receivers (right) were calculated by SpaOTsc. Cell masks are plotted as filled circles at their geometric centers.
(B) Comparison of the signaling likelihoods between different cell types under the sham and SNL conditions. Astrocyte and microglial clusters are highlighted in orange and green, respectively, and others in gray.
(C) Feature plots of selected microglial marker genes.
(D) GO term enrichment analysis of marker genes of the two microglial subclusters, M1 and M1*. P-values, Fisher’s exact test.
(E) Feature plots of astrocyte marker genes for each subcluster.
(F) Spatial distributions of astrocyte subclusters in the PBN region. Astrocyte masks are plotted as filled circles at their geometric centers.