

Abstract
Enteric pathogens are exposed to a dynamic polymicrobial environment in the gastrointestinal tract1. This microbial community has been shown to be important during infection, but there are few examples illustrating how microbial interactions can influence virulence of invading pathogens2. Here, we show that expansion of a group of antibiotic-resistant, opportunistic pathogens in the gut, the enterococci, enhances fitness and pathogenesis of C. difficile. Through a parallel process of nutrient restriction and cross-feeding, enterococci shape the metabolic environment in the gut and reprogram C. difficile metabolism. Enterococci provide fermentable amino acids, including leucine and ornithine, which increase C. difficile fitness in the antibiotic perturbed gut. Parallel depletion of arginine by enterococci through arginine catabolism provides a metabolic cue for C. difficile that facilitates increased virulence. Evidence of microbial interaction between these two pathogenic organisms is observed in multiple mouse models of infection and C. difficile-infected patients. These findings provide mechanistic insights into the role that pathogenic microbiota have in susceptibility to and severity of C. difficile infection.
Enteric infections are polymicrobial by nature, as pathogens become exposed to a rich microbial ecosystem and complex metabolic environment during invasion of the gastrointestinal tract. The study of pathogen-microbiota interactions during infection is central to our ability to understand and treat enteric infections. One of the most significant enteric pathogens globally is Clostridioides difficile3, but little is known about how C. difficile cooperates with the rich collection of microorganisms in the gastrointestinal (GI) tract.
Enterococci are enriched in the C. difficile-infected gut and vancomycin-resistant Enterococcus (VRE) frequently co-infects patients with C. difficile 2,4–10. However, the impact of Enterococcus on susceptibility to C. difficile infection (CDI) and clinical outcomes has not been explored. To further define the association between Enterococcus and C. difficile during infection, we quantified Enterococcus burdens in pediatric patients. Consistent with studies in adults, we found an enrichment of enterococci in the stools of pediatric patients with CDI (Extended Data Fig. 1a) and a positive correlation between enterococcal and C. difficile burdens (Spearman ρ = 0.551; n = 19) (Extended Data Fig. 1b). These data confirm that enterococci are highly abundant in the CDI gut and positively correlate with C. difficile burden.
Enterococcal – C. difficile interactions
To determine a causal role for enterococci in susceptibility to CDI, mice were infected with C. difficile following antibiotic-mediated depletion of endogenous enterococci. Mice given cefoperazone, which does not target enterococci, showed robust C. difficile colonization 1-day post-infection (Fig 1a; Extended Data Fig. 1c). However, enterococcal depletion with cefoperazone and vancomycin resulted in a C. difficile colonization delay (Fig 1a; Extended Data Fig. 1c). To test if this was a direct effect of enterococci, we introduced Enterococcus faecalis (strain OG1RF) immediately preceding CDI. Introduction of E. faecalis into the enterococcal-depleted gut recovered early C. difficile colonization on day 1 post-infection (Fig. 1a). This indicated that enterococci may alter the gastrointestinal environment following antibiotic treatment and support C. difficile during early colonization events. To test the reciprocal impact of C. difficile on enterococcal fitness in the gut, we infected mice with C. difficile deficient in toxin production (tcdA−tcdB−). Enterococci outgrow in the antibiotic treated gut independent of toxin; however, burdens were significantly increased in the presence of C. difficile toxin (Extended Data Fig. 1d). These data are consistent with previous studies that demonstrate the ability of enterococci to bloom following antibiotic perturbation11,12 and further demonstrate that C. difficile toxin-mediated damage provides an added fitness advantage.
Fig. 1. Enterococci promote C. difficile fitness and pathogenesis.
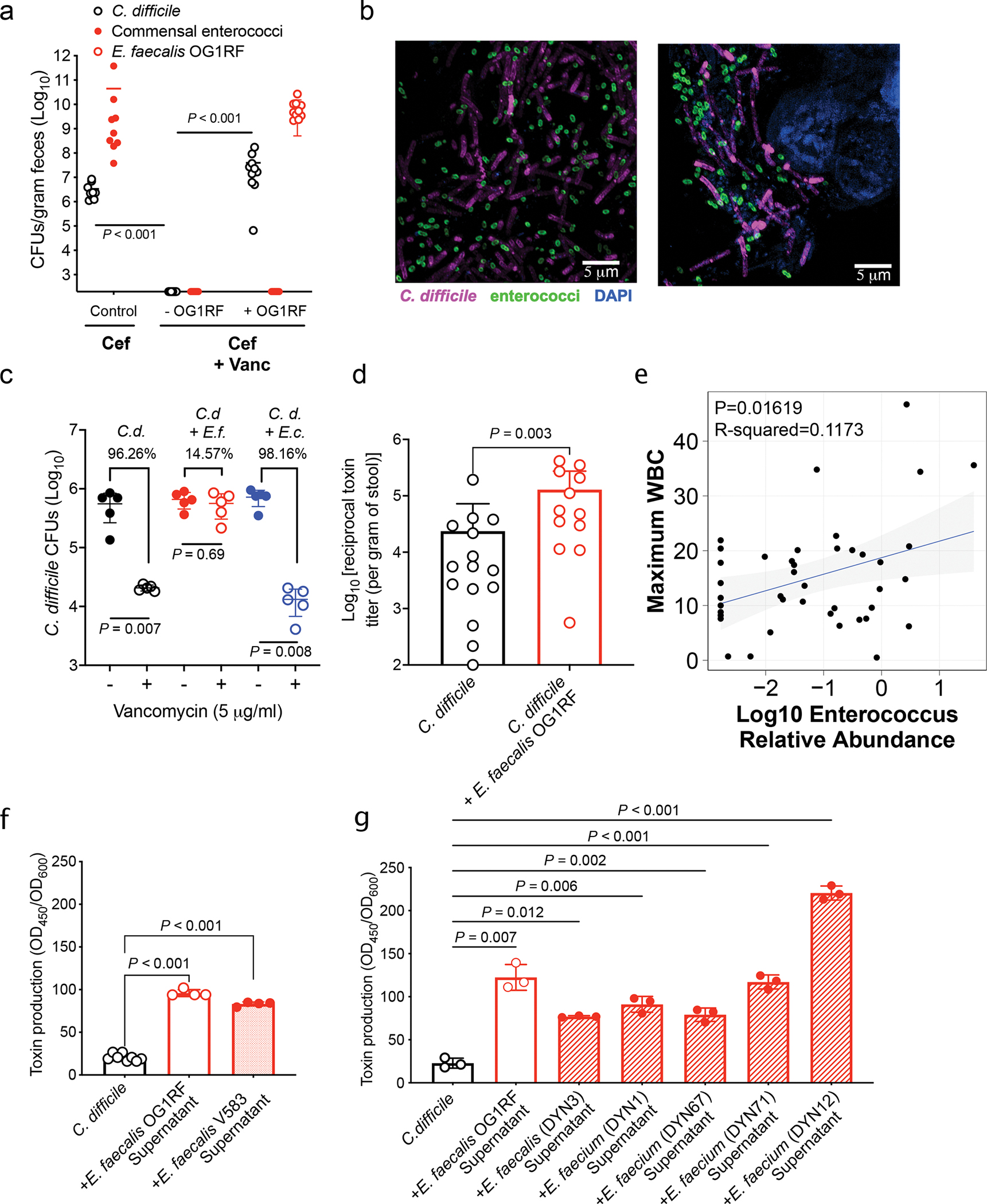
(a) Bacterial burdens (CFUs) for endogenous enterococci measured day of C. difficile inoculation. C. difficile burdens measured on first day post-inoculation to determine success of initial colonization. Burdens calculated following cefoperazone (cef) or cef + vancomycin (vanc) treatment (mean ± s.d., n = 10/group, two-sided Mann-Whitney for each comparison, P<0.001). (b) FISH image in lumen (left) or associated with the mucosa (right) during mouse infection. (c) CFUs from C. difficile CD196 (C.d.) monoculture or co-culture (E. faecalis = E.f., E. coli = E.c.) biofilms. Biofilms treated with vehicle (−) or vancomycin (+). Percent killing shown vs. untreated (mean ± s.d., n=5, two-sided Mann-Whitney test, C.d.: P = 0.007; C.d. + E.f:. P = 0.69; C.d. + E.c.: P = 0.008). (d) C. difficile toxin titers from mice infected with C. difficile or C. difficile + E. faecalis OG1RF following cef + vanc treatment (mean ± s.d., n = 15 for C. difficile only and n = 12 for C. difficile + E. faecalis, two-sided Mann-Whitney test, P=0.003). (e) Linear regression relating peripheral WBC to Log10 Enterococcus 16S rRNA relative abundance (95% confidence interval shown, n = 41, P = 0.017). Two-sided Spearman correlation was also performed and showed a positive correlation between WBC and Enterococcus relative abundance (Spearman ρ = 0.252; n = 41). (f) C. difficile toxin levels measured by ELISA following growth with E. faecalis supernatants (mean ± s.d., n = 8 for C. difficile only and n = 4 for E. faecalis OG1RF and E. faecalis V583, one-way Welch ANOVA, P<0.001). (g) C. difficile toxin ELISA following introduction of cell-free supernatants from enterococci isolated from pediatric patients with IBD + CDI (mean ± s.d., n = 3/group, one-way Welch ANOVA, P<0.02, exact P values in Supplementary Table 5).
To test if enterococci interact with C. difficile during infection, we first performed fluorescent in situ hybridization (FISH) during CDI in mice. Enterococci co-localize with C. difficile in the lumen (Fig. 1b, left) and in biofilm-like aggregates against the host epithelium (Fig. 1b, right). To examine the potential role of dual species biofilms, we performed biofilm assays in vitro. C. difficile readily forms biofilms with E. faecalis and this markedly enhances C. difficile survival during vancomycin treatment (Fig. 1c). This phenomenon was not observed in dual species biofilms with Escherichia coli, another member of the microbiota (Fig. 1c). Notably, transposon mutants in genes associated with E. faecalis biofilm structure (fsrB::Tn; OG1RF_11528, prsA::Tn; OG1RF_10423)13,14 no longer provided significant protection for C. difficile in the presence of vancomycin (Extended Data Fig. 2a–b). This demonstrates that E. faecalis biofilm structure is important to enhance C. difficile survival following antibiotic exposure and suggests that dual species biofilms may promote persistence during infection.
Biofilms provide ideal conditions for horizontal gene transfer between bacterial species15. We postulated that these two organisms may share mobile genetic elements (MGEs) in the gut. Analysis of genomes from C. difficile and vancomycin-resistant E. faecium and E. faecalis clinical isolates from co-colonized patients resulted in the detection of six sequence clusters predicted to encode MGEs. The largest gene cluster encoded a Tn916-like tetracycline resistance-carrying transposon16 (Extended Data Fig. 2c). Surprisingly, we also observed a cluster encoding a transposon carrying a predicted collagen-binding surface adhesin that resembled the CD0386 adhesin, a known C. difficile surface protein and substrate of Sortase B17 (Extended Data Fig 2c). When expressed in E. faecalis OG1RF, CD0386 enhanced binding to collagen-coated plates, suggesting that horizontal transfer of genes from C. difficile to enterococci can support the fitness of this opportunistic pathogen in the gut (Extended Data Fig. 2d). These data provide evidence of robust MGE transfer between C. difficile and VRE during infection.
Enterococci enhance pathogenesis
To examine the impact of enterococci on C. difficile pathogenesis, we quantified C. difficile toxin titers from the stool of mice pre-inoculated with E. faecalis OG1RF. Mice co-infected with E. faecalis showed significantly higher titers of toxin in stool compared to those infected with just C. difficile (Fig. 1e). To examine the clinical relevance of these findings, we evaluated the relationship between Enterococcus relative abundance based on 16S rRNA gene sequencing and maximum peripheral white blood cell (WBC) count, a measure of systemic inflammation, in an adult cohort of CDI. Enterococcus was positively associated with and inflammation based on a linear regression model, with serum WBCs increasing by 3.03 thousand cells per microliter (standard error 1.20; P = 0.017) (Fig. 1d). Spearman correlation analysis further showed a positive correlation between WBCs and Enterococcus (Spearman ρ = 0.252; n = 33). Based on these trends, we hypothesized that enterococci directly impacts C. difficile virulence. To explore this mechanistically, we quantified C. difficile toxin production in the presence of enterococci in vitro. Toxin gene expression and toxin production was significantly enhanced following growth with E. faecalis OG1RF in liquid culture (Extended Data Fig. 3a–b). Notably, C. difficile toxin production was also enhanced following growth in media supplemented with cell-free supernatants from E. faecalis, demonstrating a role for soluble factors in enhanced virulence (Fig. 1f, Extended Data Fig. 3c). Increased toxin observed following exposure to a pathogenic VRE strain of E. faecalis (V583) (Fig. 1f, Extended Data Fig. 3c), as well as a range of E. faecalis and E. faecium strains from pediatric patients with CDI (Fig. 1g). This effect was not observed when C. difficile was exposed to cell-free supernatants from many non-enterococcal strains isolated from the same patients (Extended Data Fig. 3d). Together, these results illustrate that enterococci increase C. difficile pathogenesis by enhancing toxin production.
Enterococci reshape metabolome
To shed light on the molecular mechanisms of enterococcal – C. difficile interactions, we performed RNA sequencing of co-cultures. C. difficile transcripts associated with carbohydrate uptake, amino acid utilization, and Stickland fermentation were significantly altered in the presence of E. faecalis (Fig. 2a, Extended Data Fig. 4a, and Supplementary Table 1), suggesting significant metabolic reprogramming. Transcripts associated with metal uptake, arginine uptake and metabolism, and carbohydrate uptake and metabolism were altered in E. faecalis (Extended Data Fig. 4b–c and Supplementary Table 2). To contextualize these transcriptional data, we leveraged a metabolic network reconstruction of C. difficile (iCdR703)18,19. Following integration of transcriptome data20, we generated distinct context-specific models for C. difficile and simulated bacterial growth (Extended Data Fig. 4d). Growth was iteratively simulated under near optimal context-specific conditions to generate distributions of possible activity range for all metabolic reactions. Flux sampling for the biomass objective function revealed a significant increase in the predicted growth rate of C. difficile during co-culture conditions, suggesting enterococci aid C. difficile growth (Fig. 2b). Analyses of sampled optimal metabolic flux states predicted that C. difficile core metabolism is significantly altered in the presence of enterococci and that differences in amino acid and carbohydrate-related catabolism were largely responsible for this change, with L-ornithine amino-lyase being the most important feature (Fig. 2c–d). Targeted analysis of exchange fluxes associated with substrates and byproducts supported a shift to amino acid import and fermentation in co-culture (Fig. 2d, Extended Data Figs. 4a, 5a–c), with a predicted increase in uptake and consumption of ornithine, D-alanine, L-leucine, and L-valine and increased efflux of the fermentation end products of isoleucine (2-methylbutyrate) and proline (5-aminovalerate) (Fig. 2e–j). Modeling also predicted increased efflux of acetate and N-acetyl-D-glucosamine when co-cultured (Fig. 2k–l).
Fig. 2. Context-specific GENRE analysis of C. difficile CD196 reveals significant metabolic shifts when in coculture with Enterococcus.
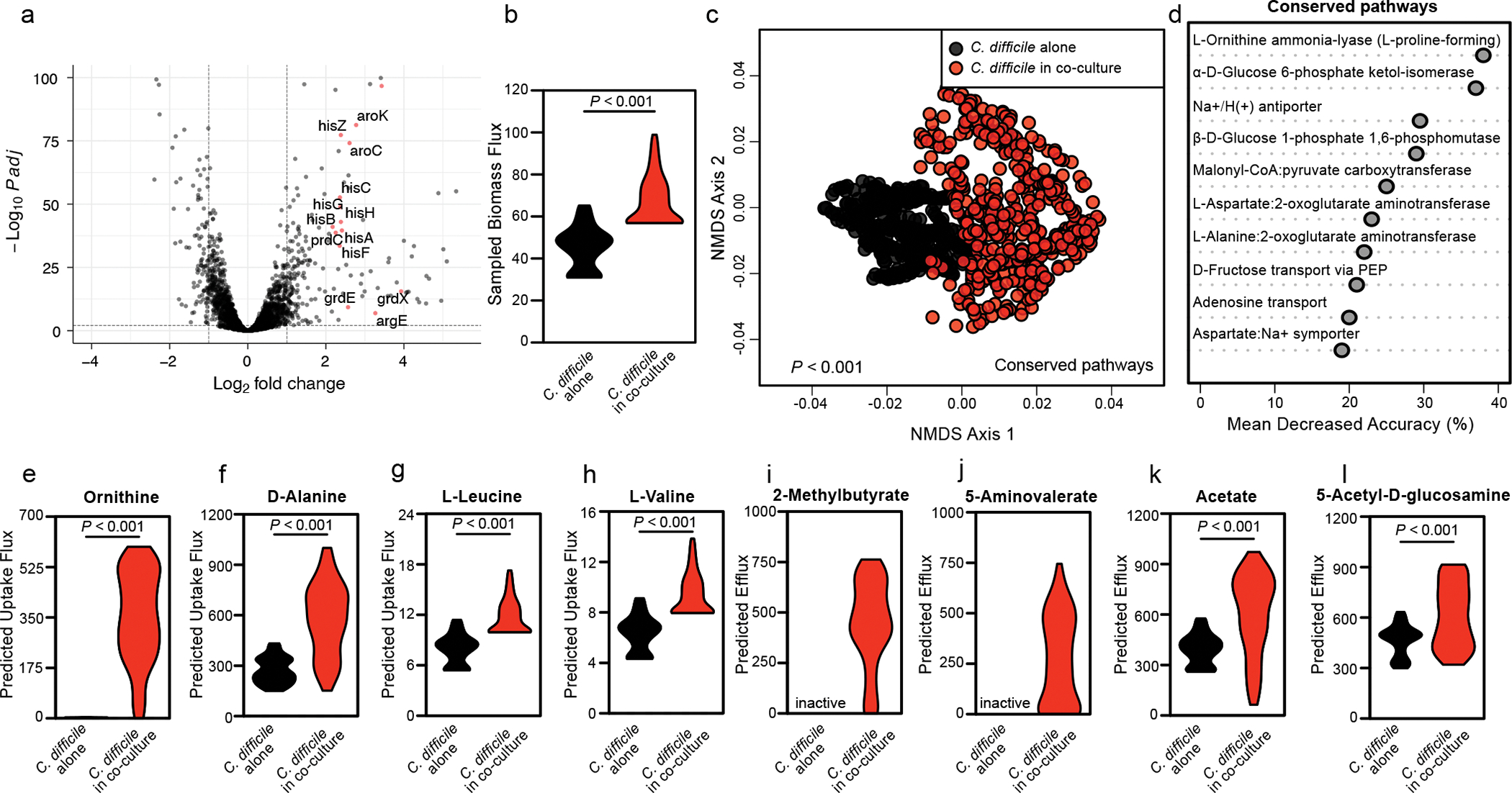
(a) Volcano plot showing C. difficile transcripts altered following co-culture measured by RNA sequencing. Significance determined using two-sided Wald test and corrected for multiple comparisons using the Benjamini-Hochberg method. Red points denote genes associated with amino acid metabolism. (b) Samples of metabolic flux states with biomass synthesis as the objective function in each context-specific model. Significant difference calculated by two-sided Wilcoxon rank-sum test (P<0.001). (c) Non-metric Multidimensional Scaling of Bray-Curtis dissimilarities for flux samples of all shared reactions across both experimental contexts. Significant difference determined by one-way PERMANOVA (P=0.001). (d) AUC-Random Forest supervised machine learning results highlighting reactions that differentiate flux distributions during C. difficile growth and C. difficile growth in the context of E. faecalis (k = 10; OOB = 0%). (e-h) Difference in simulated uptake of (e) L-ornithine (P<0.001), (f) D-alanine (P<0.001), (g) L-leucine (P<0.001), or (h) L-valine (P<0.001) across context-specific models. (i-l) Difference in (i) 2-methylbutyrate, (j) 5-aminovalerate, (k) acetate (P<0.001), and (l) N-acetyl-D-glucosamine (P<0.001) efflux across context-specific models. Significance determined by two-sided Wilcoxon rank-sum test.
Based on systems analyses, we predicted that E. faecalis reshapes the metabolic environment and acts as a source of fermentable amino acids for C. difficile. To validate these predictions, we measured amino acids in cells in macrocolonies. E. faecalis harbored high levels of fermentable amino acids predicted by our models to be important in metabolic cross-talk (Fig. 3a). We further observed that E. faecalis exports high levels of extracellular ornithine and depletes arginine when grown in culture (Fig. 3b). Consistent with this, mice antibiotic-depleted of enterococci harbored significantly less ornithine and enriched levels of arginine (Fig. 3c). These data suggest E. faecalis is a source of several fermentable amino acids and that ornithine and arginine are central features of metabolic remodeling. Next, we sought to determine if ornithine is cross-fed to C. difficile. Ornithine quantification from macrocolonies showed that C. difficile cells become enriched in ornithine when grown in proximity of E. faecalis, demonstrating direct cross-feeding (Fig. 3d). Ornithine is a fermentable amino acid that is important for C. difficile persistence in the gut and can be used for both oxidative and reductive fermentative pathways21. Specifically, ornithine can be catabolized by C. difficile through the proline reductase pathway to generate energy22. We utilized an activity-based chemical probe capable of monitoring proline reductase activity23 and observed a significant enhancement in proline reductase activity when C. difficile was grown in media supplemented with E. faecalis cell-free supernatants (Fig. 3e, Extended Data Fig. 6). This supports our modeling predictions and suggests that C. difficile uses E. faecalis-derived ornithine for energy.
Fig. 3. Amino acid cross-talk is central to Enterococcus-C. difficile interactions.
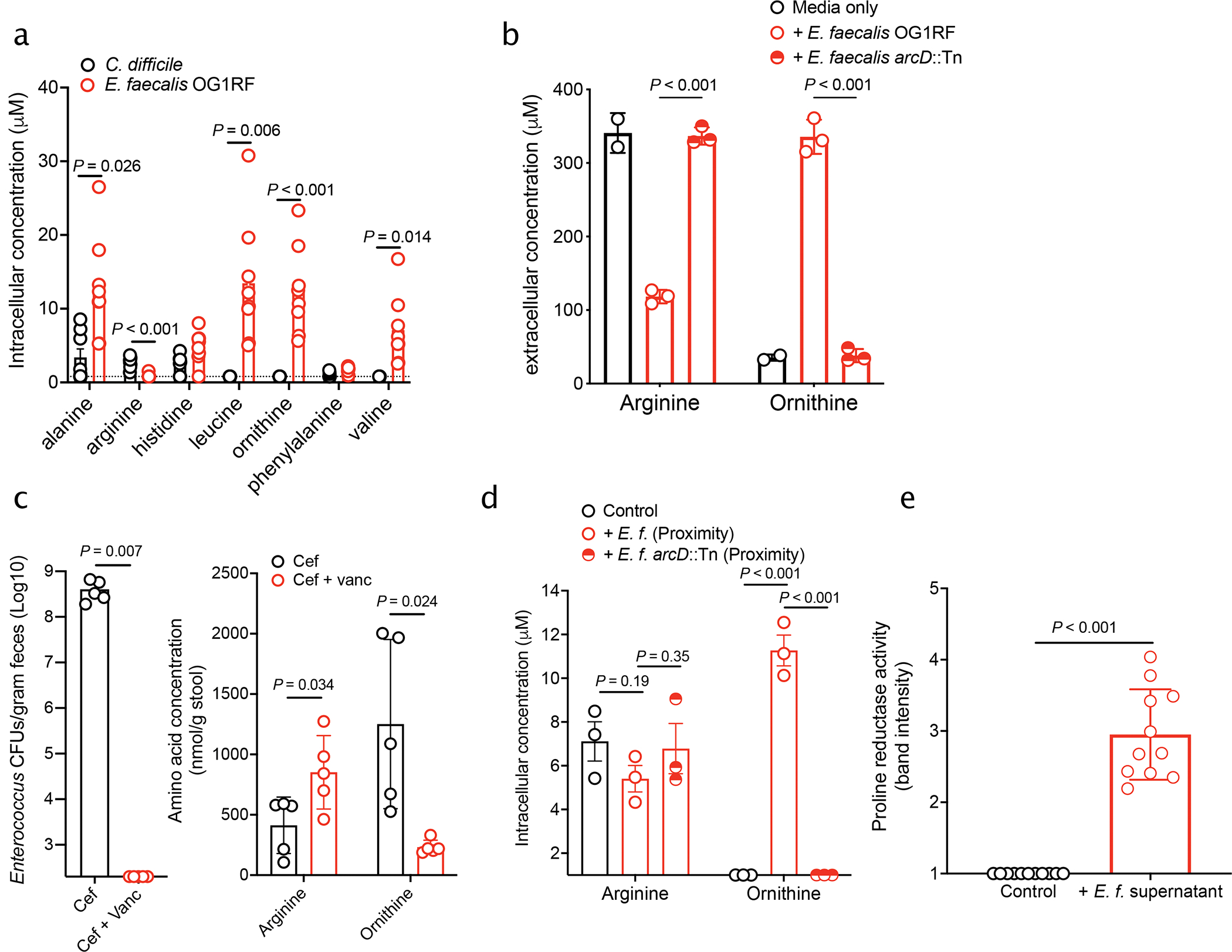
(a) Amino acid abundance measured in macrocolonies (mean ± s.d., n = 8/group, multiple two-sided t-tests with Bonferroni-Dunn method for correction for multiple comparisons, corrected P values in Supplementary Table 5). Dotted line indicated limit of detection. (b) Arginine and ornithine levels measured in cell-free supernatants of E. faecalis OG1RF or E. faecalis OG1RF arcD::Tn (mean ± s.d., n=2 media only, n = 3 +E. faecalis and +E. faecalis ArcD::Tn, multiple two-sided t-tests with Bonferroni-Dunn method for correction for multiple comparisons. OG1RF vs. arcD::Tn: Ornithine: P < 0.001; Arginine: P < 0.001). (c) Enterococcus burdens (left) and arginine and ornithine levels (right) on day of C. difficile infection quantified from stool of mice treated with cef or cef + vanc. (mean ± s.d., n = 5/group, two-sided Mann-Whitney used for Enterococcus burdens, multiple two-sided t-tests with Bonferroni-Dunn method for correction for multiple comparisons used for amino acids. Corrected P in Supplementary Table 5). (d) Arginine and ornithine levels quantified from C. difficile macrocolonies plated in proximity of E. faecalis or E. faecalis arcD::Tn macrocolonies (mean ± s.d., n = 3/group, multiple two-sided t-tests with Bonferroni-Dunn method for correction for multiple comparisons. Corrected P values in Supplementary Table 5). (e) Band intensity of fluorescently-labeled PrdA from lysates of C. difficile grown either in fresh BHIS media or in media supplemented with E. faecalis OG1RF supernatants run on SDS-PAGE (mean ± s.d., n=11/group, two-sided t-test with Welch’s correction, P<0.001).
The role of ADI pathway in cross-talk
E. faecalis catabolizes arginine for energy through the arginine deiminase (ADI) pathway. During arginine uptake, ornithine is exported through the ArcD antiporter (OG1RF_10103) (Fig. 3b). Ornithine is important in polymicrobial interactions in E. faecalis24, and our data suggest that this amino acid is central to C. difficile-E. faecalis interactions (Figs. 2e and 3). Thus, we cultured C. difficile with an E. faecalis ArcD transposon mutant (arcD::Tn), which is defective in import of arginine and export of ornithine (Fig. 3b) and unable to cross-feed C. difficile ornithine (Fig. 3d). C. difficile showed a significant growth defect when grown in supernatants from E. faecalis arcD::Tn (Extended Data Fig. 7a), supporting a role ornithine in fitness. Interestingly, enterococcal-mediated virulence enhancement in C. difficile was also abrogated when grown with E. faecalis arcD::Tn cell-free supernatants (Fig. 4a). This suggests a direct role for the enterococcal ADI system in control of C. difficile virulence.
Fig. 4. E. faecalis ADI pathway enhances C. difficile virulence.
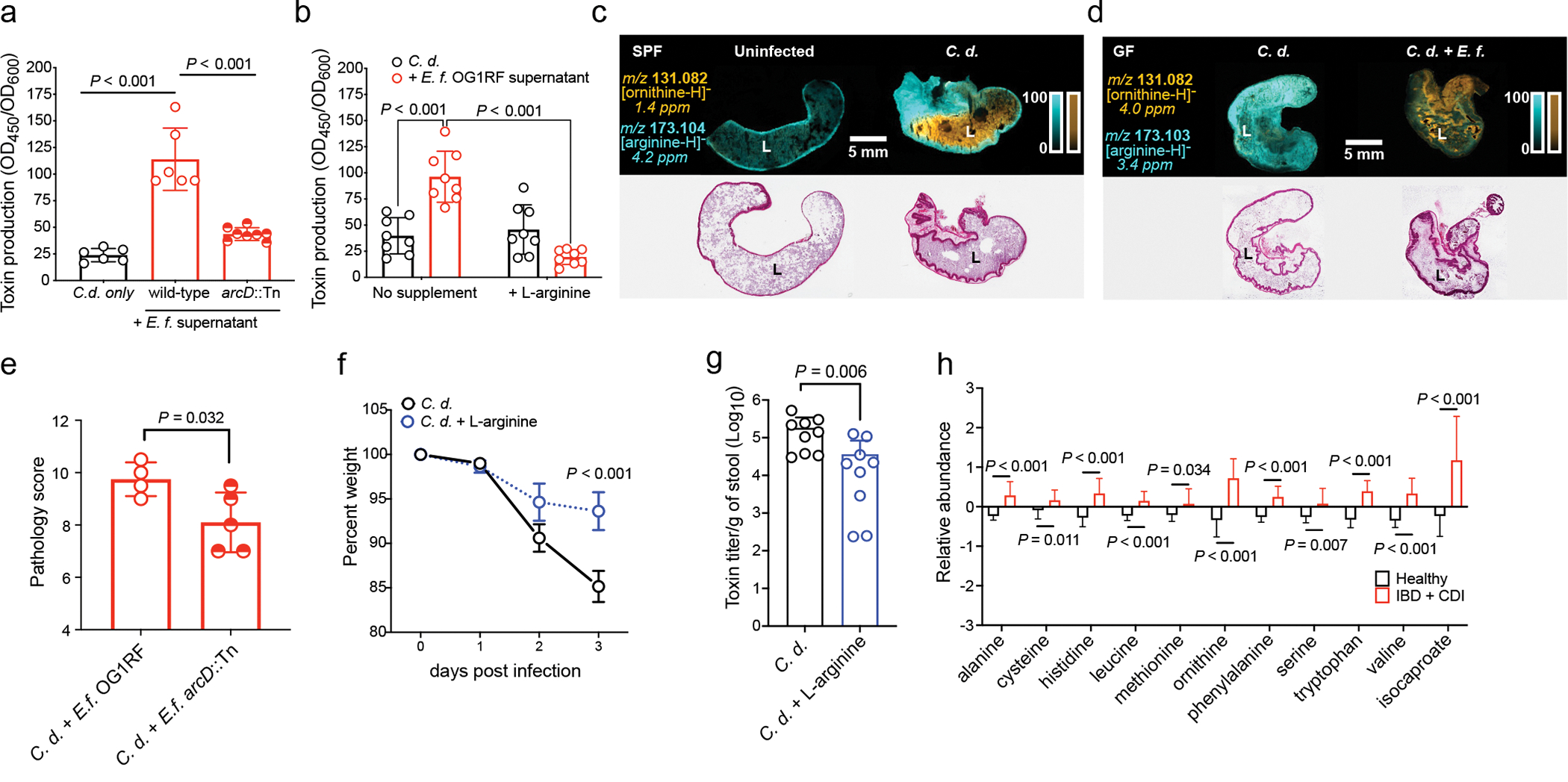
(a) C. difficile CD196 toxin production measured by ELISA (mean ± s.d., n = 6 C. d. only and OG1RF supernatant, n=8 arcD::Tn, two-sided t-tests with Welch’s correction) and (b) supplementation with L-arginine (mean ± s.d., n = 8/group, Tukey’s multiple comparison test) (C.d. = C. difficile; E.f. = E. faecalis). Corrected P values in Supplementary Table 5. (c) MALDI-IMS image of ornithine and arginine in uninfected or infected mice (3 d post-infection) (SPF) (representative of n = 5 mice) or (d) GF mice mono-infected with C. difficile CD196 or co-infected with E. faecalis OG1RF (2d post-infection) (GF) (representative of n = 4 mice). Corresponding hematoxylin and eosin stained tissue displayed. “L” = lumen. (e) Pathology score from cecum of GF mice pre-colonized with E. faecalis OG1RF (n = 4) or E. faecalis arcD::Tn (n = 5) and subsequently infected with C. difficile (mean ± s.d., two-sided t-tests with Welch’s correction, P=0.032). (f) Percent weight of mice infected with C. difficile CD196 and treated with 2% L-arginine in drinking water (mean ± s.d., n = 9/treatment group, two-way ANOVA with Bonferroni’s multiple comparison test, day 3 P<0.001). (g) C. difficile toxin titers from mice infected with C. difficile and treated with 2% L-arginine in their drinking water (mean ± s.d., n = 9/treatment group, two-sided Mann-Whitney test, P=0.006). Stool toxin titers measured by cytotoxicity. (h) Relative abundance of select amino acids and isocaproate in feces of pediatric patients with IBD and CDI (mean ± s.d., n = 20) or healthy controls (mean ± s.d., n = 19, multiple two-sided t-tests with Bonferroni-Dunn method for correction for multiple comparisons. Corrected P values in Supplementary Table 5). Metabolites shown as relative values with each value rescaled to set the median value to 1.
Analysis of a diverse collection of E. faecalis (51 genomes) and E. faecium (246 genomes) genomes25,26 showed that arcD is present in 100% of strains. Notably, all vancomycin-resistant E. faecium strains that we analyzed for MGE exchange during co-infection with C. difficile contained two copies of arcD located at different positions in the genome, despite the fact that E. faecium does not readily use arginine as an energy source27. These findings suggest that arcD is broadly conserved across clinically relevant strains of enterococci. Metagenomic analysis of adult patients with CDI28 further showed the presence of ADI systems (arc genes) across an array of taxa during infection (Extended Data Fig. 7b), including the Lachnospiraceae and Eggerthellaceae. This suggests that diverse microbiota have the capacity to impact C. difficile through the action of the ADI pathway. However, enterococcal arc genes frequently represented most arc genes in a patient, and enterococci were among the highest represented arc containing taxa during infection (Extended Data Fig. 7b–c). To determine whether arginine depletion or ornithine production controls C. difficile virulence, we grew C. difficile in cell-free supernatants from E. faecalis OG1RF that had been subsequently supplemented with L-arginine. Virulence enhancement was reversed following the addition of exogenous L-arginine (Fig. 4b). Addition of L-ornithine to rich media did not lead to increased C. difficile toxin production (Extended Data Fig. 7d), suggesting that ornithine does not have a direct role in enhancing virulence in the conditions tested. Together, these data suggest that arginine is associated with C. difficile virulence and that arginine depletion by the E. faecalis ADI system enhances C. difficile virulence29,30.
To define the role of these metabolic interactions during infection, we first performed matrix-assisted laser desorption/ionization imaging mass spectrometry (MALDI-IMS) on ceca from cefoperazone treated mice infected with C. difficile, which harbor high levels of Enterococcus (Fig. 3c). Ornithine was seen at high abundances in the lumen following infection and arginine was reciprocally localized in tissues but absent in the lumen (Fig. 4c; Extended Data Fig. 7e). Germ-free (GF) mice mono-infected with C. difficile showed an influx of arginine in the lumen and low levels of ornithine. Conversely, GF mice co-infected with C. difficile and E. faecalis were devoid of luminal arginine and had increased ornithine (Fig. 4d; Extended Data Fig. 7f, g, h). These data demonstrate a direct role for enterococci in dramatically remodeling amino acid availability in the gut. Notably, we observed that GF mice infected with C. difficile harbored increased arginine in their ceca and stool compared to uninfected GF mice (Extended Data Fig. 7f, h). This suggests that C. difficile-mediated damage to host tissue and the corresponding inflammatory response provide a nutrient source of arginine for enterococci during CDI. To test whether enterococci use arginine to thrive in the gut during CDI, we colonized conventional mice with E. faecalis OG1RF or E. faecalis arcD::Tn during CDI. E. faecalis lacking a functional ADI pathway (E. faecalis arcD::Tn) initially colonize equally as well, but showed a defect in the gut during peak of C. difficile disease (Extended Data Fig. 7i), demonstrating the importance of arginine in enterococcal fitness in the CDI gut. Next, to examine the role of the E. faecalis ArcD antiporter in promoting C. difficile disease, we pre-colonized GF mice with either E. faecalis or E. faecalis arcD:Cm. Depletion of arginine and accumulation of ornithine in the gut of GF mice was dependent on ArcD (Extended Data Fig. 7j–k) and C. difficile infected mice with E. faecalis arcD:Cm showed decreased disease pathology, demonstrating the role for these metabolites in disease outcome (Fig. 4e). Finally, to confirm the role of arginine availability in CDI, we treated mice with 2% L-arginine in drinking. Supplementation with L-arginine did not impact initial C. difficile colonization or overall burdens of enterococci, but significantly reduced toxin titers in stool and parameters of disease severity (Extended Data Fig. 8a–d, Fig. 4f–g). Together, these observations demonstrate that enterococci modulate levels of arginine and ornithine in the gut during CDI and that arginine plays a central role in C. difficile virulence. Importantly, these findings also demonstrate that modulation of enterococcal metabolism and the nutritional landscape in the gut can alter CDI.
To demonstrate the relevance of our findings in humans, we explored the fecal metabolome of children with IBD and CDI31. Consistent with our findings, patients with IBD + CDI harbored significantly higher levels of fermentable amino acids, including ornithine and leucine (Fig. 4h), and the Stickland fermentation end products (Fig. 4h). We further observed a positive correlation (Spearman’s ρ = 0.4243; n = 26) between C. difficile burdens and ornithine (Extended Data Fig. 8e), supporting the key role for ornithine in CDI. Collectively, these data suggest that enterococci and C. difficile interact during CDI through metabolic cross-talk to support increased colonization, pathogenesis, and persistence in the gut.
Discussion
Understanding factors that contribute to clinical outcomes of CDI is essential to combating this urgent public health challenge. Here we show that enterococci reshape the metabolic environment in the gut to enhance C. difficile colonization and support fitness following disease manifestation. We propose that through the action of the ADI pathway, E. faecalis enhances C. difficile fitness via ornithine exchange, while concurrently providing metabolic cues via arginine depletion to increase virulence (Extended Data Fig. 8f). The role for enterococci in enhanced C. difficile toxin production is consistent with a recent study of co-infection between C. difficile and VRE in mice32. It is important to note that mechanistic studies probing interactions between enterococci and C. difficile were only performed using E. faecalis strain OG1RF and C. difficile strain CD196. Although we present data that suggests that these interactions are widespread across clinically relevant enterococci, further studies that systematically test mechanisms of interaction between a diversity of enterococci and C. difficile strains are needed. Mechanisms by which C. difficile senses and responds to the enterococcal ADI pathway are not completely understood but our data suggest a role for nutrient availability. Future studies exploring the impact of enterococcal arginine utilization on the local microenvironment during CDI are warranted and these observations need to be extensively validated in human studies. The ability of enterococci to thrive during CDI and benefit from increased arginine availability suggests that this interaction is mutually beneficial. Although enterococci frequently dominate the microbiota during CDI and co-infection with VRE is common, it is likely that other members of the microbiota play a key role in C. difficile virulence7,33. Moreover, microbiota with ADI pathways, such as members of the Clostridia34, Lactobacillus (Extended Data Fig. 3d), Streptococcus35, and Lachnospiraceae (Extended Data Fig. 7b) may have potential to play a similar role during infection. Together, our work demonstrates the supportive role of pathogenic microbiota in the outcome of CDI and highlights the importance of integration of metabolic signals from the microbiota in pathogen virulence. These findings have implications for our understanding of the variables impacting the outcome of C. difficile infection, the risk for recurrence, and the factors impacting success of treatment.
Materials and Methods
Bacterial strains and growth conditions
Strains used in this study are listed in Supplementary Table 3. C. difficile, Enterococcus, and E. coli strains were grown at 37°C in an anaerobic chamber (85% nitrogen, 10% hydrogen, 5% carbon dioxide, Coy Lab Products) in brain-heart-infusion broth (BD Life Sciences) supplemented with 0.5% yeast extract (BD Life Sciences) and 0.1% L-cysteine (Sigma-Aldrich) (BHIS) unless otherwise stated. For bacterial growth curves, C. difficile were incubated with double orbital shaking at 37°C and OD600 was measured every 30 minutes in a BioTek Epoch 2 (BioTek; Winooski, VT, USA). For growth in presence of E. faecalis supernatants, cultures were normalized to optical density (OD) and sub-cultured (1:100) into 200μL of fresh BHIS with 70% E. faecalis supernatants in a 96-well plate sealed with a gas-permeable membrane (Breathe-Easy®) (Millipore-Sigma; Burlington, MA, USA). Sequence-defined transposon mutants used in this study were confirmed using gene specific primers36.
Quantification of C. difficile toxin production
For quantification of C. difficile toxin from culture, C. difficile was grown for 24 hours anaerobically, pelleted at 4000g for 10 minutes, and filtered through a 0.2μm filter. For quantification of toxin via ELISA, the C. difficile Tox A/B II ELISA (Meridian Bioscience) was used per manufacturer’s instruction. For quantification of C. difficile toxin titers via cytotoxicity assay, a Vero cell-rounding cytotoxicity assay was used as previously described6. Briefly, Vero cells (ATCC #CCL-81) were plated at 1 × 104 cells per well in a 96-well flat bottom cell-culture treated microtiter plate and incubated for 16–24h. C. difficile filtered supernatants were titrated in tenfold dilutions within the wells to a maximum dilution of 10−8. After an overnight incubation, Vero cell rounding was assessed under 10x magnification. Cytotoxicity data are expressed as the reciprocal value of the highest dilution that rounded 100% of the cells. For co-culture growth, toxin levels were normalized to C. difficile CFU. For cultures grown with Enterococcus cell-free supernatants, toxin levels were normalized to C. difficile OD600. When grown in the presence of Enterococcus supernatants, C. difficile was grown in 70% cell-free supernatants and 30% fresh media. The ADI pathway leads to production of ammonia and alteration in pH in some pathogens35; however, adjustment of pH in E. faecalis supernatants did not directly lead to a significant impact on toxin output in our assay. All toxin quantification assays were independently replicated at least 3 times and representative assays are shown.
Co-culture growth assays on solid media
For macrocolonies, E. faecalis and C. difficile overnight cultures were normalized based on OD600 and 10μL were plated onto BHIS + L-cysteine agar plates and grown for 7 days to produce macrocolonies. For proximity assays to measure metabolite exchange, cultures were OD normalized, and 10 μL were spotted onto BHIS + L-cysteine agar plates at a 1.5 cm distance and physical mixing of cells was avoided. Macrocolony cultures were grown for 7 days.
Animal and experimental models of C. difficile infection
Animal experiments were approved by the Animal Care and Use Committees of Vanderbilt University, the Children’s Hospital of Philadelphia, and the University of Pennsylvania Perelman School of Medicine (protocols M1700053, IAC 18–001316, 806279). For C. difficile infections in conventional facilities, 4–8-week-old C57BL/6 male mice were purchased from Jackson Laboratories and given one week to equilibrate their microbiota prior to experimentation. All experimental manipulations were performed in a biosafety level 2 laminar flow hood. Mice were housed in individual cages under the same conditions during the experiment, investigators were not blinded to treatment groups, and all mice were culture-negative for C. difficile prior to infection. For the C. difficile infection in conventional animals, mice were given antibiotics (0.5 mg/mL cefoperazone or 0.5 mg/mL cefoperazone + 1 mg/ml vancomycin) in drinking water ad libitum for 5 days followed by a 2-day recovery period and subsequent infection as previously described6,37. Mice were infected via oral gavage with 1×105 spores of C. difficile resuspended in sterile PBS. C. difficile strains CD196, M7404, and M7404 TcdA− TcdB− were used for conventional infections, as described in the text. When noted, mice were co-infected with 5 × 108 E. faecalis (wild type strain OG1RF or arcD::Tn) cells. E. faecalis cells were grown to stationary phase, washed in PBS twice prior to infection, and gavaged prior to infection with C. difficile spores. For C. difficile infection in germ-free (GF) mice, 6-week-old C57BL/6 female mice were used and mice were infected with either 1×105 spores of C. difficile (strain CD196) or C. difficile spores + 5 × 108 E. faecalis (wild type strain OG1RF or arcD::Tn) cells in PBS. Both conventional and GF mice were monitored for survival and were euthanized after reaching a terminal endpoint of appearing moribund or experiencing weight loss >20% from baseline. C. difficile and enterococcal CFUs were quantified daily from fecal samples. All samples were collected and all stool-related data are reported, unless animals were too sick to acquire a fresh stool sample. Samples were diluted and homogenized in PBS and serially plated onto taurocholate cycloserine cefoxitin fructose agar (TCCFA) for C. difficile and Bile Esculin agar for enterococci. Wild type E. faecalis OG1RF was grown on BHI agar with rifampicin (200μg/mL), and transposon mutants were grown on BHI agar with rifampicin and chloramphenicol (10μg/mL).
Histological analysis
At necropsy, ceca and colons were harvested, fixed in 10% formalin solution and embedded in paraffin. Sections were stained with hemotoxylin and eosin. Each section was given a disease score by a pathologist in a blinded fashion based on previously described criteria 37. Histological scores were reported as a cumulative score of three independent scoring criteria: inflammation, edema and epithelial cell damage.
C. difficile toxin titers from feces
C. difficile toxin cytotoxicity was determined using a Vero cell-rounding cytotoxicity as described above. Fresh fecal samples were homogenized in 1ml of sterile PBS and pelleted at 4000g for 5 minutes. The supernatants were filtered through a 0.2μm filter and titrated in tenfold dilutions within the wells to a maximum dilution of 10−8. After an overnight incubation, Vero cell rounding was assessed under 10x magnification. Cytotoxicity data are expressed as the reciprocal value of the highest dilution that rounded 100% of the cells per gram of sample.
Quantification of C. difficile and enterococci. from human stool samples
Bacterial burdens were quantified from frozen stool from the two patient cohorts as colony forming units (CFUs) per gram of stool. Samples were weighed and homogenized in sterile PBS. The samples were then serially diluted and plated on differential media. Enterococci were isolated in aerobic conditions by plating on Bile Esculin Agar (BD, 221409) and visually differentiated from streptococci by blackening around colonies from the esculetin reaction with iron salts in the media and confirmed by 16S rRNA gene sequencing. C. difficile was plated on taurocholate cycloserine cefoxitin fructose agar (TCCFA) in anaerobic conditions.
DNA isolation and 16S rRNA gene sequence analysis for bacterial identification
Genomic DNA from C. difficile and enterococci bacterial isolates were extracted from frozen pellets. Prior to extraction, the cell pellets were mechanically lysed in a PowerLyzer Homogenizer (Qiagen) with 0.1 mm glass beads. DNA was extracted using the DNeasy Blood and Tissue kit with QIAcube automation according to the manufacturer’s instructions (Qiagen). The 16S rRNA gene was amplified using universal 27F and 1492R primers. All PCR products were purified with the Monarch PCR & DNA Cleanup kit (New England Biolabs) and sequenced by Sanger DNA sequencing performed by CHOP’s Nucleic Acid PCR Core Facility. The first 50 bases and everything after 850 bases were trimmed from the 5’ end of each sequence and sequence analysis was carried out by NCBI BLASTn similarity search within the 16S Ribosomal database with an E-value cutoff of 0.01. Enterococci were identified on the parameters of highest percent identity and agreement between forward and reverse sequences.
Quantifying C. difficile survival in antibiotic treated interspecies biofilms
Biofilm protocol was adapted from Kumar et al38. OD600 normalized liquid cultures of C. difficile CD196, E. faecalis OG1RF (isogenic wildtype and noted transposon mutants)13,36, and E. coli DH5α were seeded in monoculture or in co-culture with CD196 in BHIS+ L-cysteine media in non-treated 24-well microplates. Biofilms were incubated for four days anaerobically at 37°C. All non-adherent cells were washed with PBS buffer and the cells in biofilms were treated with fresh BHIS media or fresh BHIS media + vancomycin at 5μg/mL. After overnight vancomycin treatment, all treatment media was removed from the wells and replaced with 1mL fresh BHIS. Biofilms were then scraped into solution using a sterile pipette tip. A 10-fold dilution series was prepared, and spot plated in 5μL quantities onto taurocholate cycloserine cefoxitin fructose agar (TCCFA). CFU counts were determined by counting bacterial colonies on the agar plate within the Log10 serial dilution. Biofilm assays were independently replicated at least three times and representative assays are shown in figures.
Fluorescent in situ hybridization
Fluorescent in situ hybridization (FISH) for C. difficile and Enterococcus was performed on paraffin embedded 5 μm cecal sections from C. difficile infected mouse tissues. Embedded tissues were deparaffinized using two washes with xylene, two washes with 100% ethanol, and a single wash with sterile water. Hybridization was performed overnight (16 hours) at 46°C concurrently with both fluorescently labeled oligonucleotide probes at 5 ng/ml in hybridization buffer with 35% formamide. Following hybridization, slides were washed for 40 minutes with prewarmed wash buffer at 48°C. Prior to imaging, VectaShield with DAPI was added to the slide to protect from photobleaching and a coverslip was placed prior to imaging. Imaging was performed using super resolution structured illumination microscopy (SIM) imaging at the Vanderbilt University Cell Imaging Shared Resource. Maximum intensity projections of 6.8 mm z-stacks are presented. Images were denoised using NIS Elements Artificial Intelligence Denoise.AI capabilities. Brightness and contrast were adjusted consistently for optimum visualization. HPLC purified probes for C. difficile (Cy5- CATCCTGTACTGGCTCAC)39 and Enterococcus (Cy3- CACCGCGGGTCCATCCATCA)40 with 5’ fluorophores were acquired from Integrated DNA technologies (IDT). Enterococcus was false colored green in micrographs to provide greater contrast against C. difficile cells. FISH assays were independently replicated in 5 mice and representative images are shown in figures.
RNA sequencing
Bacterial growth and RNA extraction:
OD normalized cultures of C. difficile strain CD196, E. faecalis strain OG1RF, and mixed co-cultures were grown anaerobically in biological triplicate in BHIS + L-cysteine at 37°C for 4 hours (exponential phase). For co-cultures, C. difficile was seeded at a 2:1 ratio to E. faecalis to account for differences in growth rates between species. After growth, a 1:1 solution of acetone:ethanol was added to an equal volume of the culture. Samples were stored at −80°C until used for RNA extraction, and RNA extraction was performed as previously described41. RNA was quantified and either used for RNA sequencing or quantitative PCR (qPCR) as described below.
Library construction:
RNA-seq library construction and sequencing were performed by HudsonAlpha Institute for Biotechnology (Huntsville, AL). The concentration and integrity of extracted total RNA were estimated by a Qubit 2.0 Fluorometer (Invitrogen) and an Agilent 2100 Bioanalyzer (Applied Biosystems), and 500 ng of RNA was utilized for downstream applications. rRNA was removed using the Ribo-Zero Gold (Epidemiology) kit (Illumina) according to the manufacturer’s instructions. RNA was fragmented and primed for first-strand synthesis using the NEBNext first strand synthesis module (New England BioLabs). Directional second-strand synthesis was performed using the NEBNext Ultra Directional second strand synthesis kit. Libraries were then prepared from samples using the NEBNext DNA Library Prep master mix set (Illumina) with the following slight modifications. End repair was performed and followed by polyadenylic acid (poly[A]) addition and custom adapter ligation. Ligated samples were individually barcoded with unique in-house Genomic Services Lab (GSL) primers and amplified through 12 cycles of PCR. Library quantity was assessed by a Qubit 2.0 Fluorometer, and the library quality was assessed by utilizing a DNA High Sensitivity Chip on a Caliper GX (PerkinElmer). The quantitative PCR (qPCR)-based Kapa Biosystems library quantification kit (Kapa Biosystems) was used for final accurate quantification of libraries prior to sequencing. Each library was diluted to a final concentration of 12.5 nM and pooled equimolar prior to clustering. Paired-end sequencing was performed on an Illumina HiSeq2500 sequencer (Illumina). Raw RNA sequencing data and processed data are deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE165751.
RNAseq Analysis
Single-end RNA-seq reads were trimmed of any adaptor sequences with trim-galore (version 0.6.6). The remaining reads were aligned to the C. difficile reference genome using STAR (version 2.7.6a) with the following parameters: --outSAMtype BAM SortedByCoordinate --limitBAMsortRAM 1500000000 – outSAMmapqUnique 255 –outFilterMultimapNmax 1 –alignIntronMax 1. Reads count on each gene were calculated at the same time of mapping with the parameter – quantMode GeneCounts.
Differential expression analysis between Coculture vs C. difficile alone was done using R package DESeq2 (version 1.30.0) and the Wald test. The read counts were normalized among the samples and were transformed to log2 form using the variance stabilizing transformations (VST) method. Volcano plot was drawn using the R package EnhancedVolcano (version 1.8.0). Only the differentially expressed genes (adjusted p-value < 0.01 and fold-change >= 2) were shown on the plot, AA metabolism associated genes were colored in red and only the genes with classic nomenclatures were labeled. Pathways were assigned for differentially expressed genes based on KEGG database.
Genome-scale metabolic network reconstruction analysis
Genome-scale metabolic network reconstruction (GENRE) for C. difficile R20291 (iCdR703) was used for analysis of transcriptional data from RNAseq. Although designed for C. difficile strain R20291, iCdR703 contains identical components for C. difficile strain CD19618. C. difficile GENRE (iCdR703) was accessed from (https://github.com/mjenior/Smith_etal_Enterococcus) and exchange reaction bounds were set to simulate rich BHI medium conditions. Transcriptomic read abundances were evenly subsampled between experimental replicates and each replicate was integrated individually, creating multiple unique models of context-specific metabolism for which all observed variation in structure and activity was then collapsed within experimental groups to obtain a more comprehensive model of metabolism for each. Transcriptomic data integration and context-specific model generation were performed with RIPTiDe (v3.3.6) on the default settings20. Context-specific flux sampling results were subsequently analyzed in R (v3.2.0) with the vegan42 and AUCRF43 packages. Specifically, the AUCRF package was used for random forest machine learning functions with an increment of removing 1 variable at a time during each test phase (pdel = 0) with a minimum number of variables to include in the final model of 10 (k0 = 10), and an initial seed of 906801. The iCdR703 generated metabolic network reconstruction of C. difficile that represents active metabolism in both in vitro and infection settings, as described previously18.
Quantitative PCR
RNA concentration was determined using the Synergy 2 with Gen 5 software (BioTek) and 2 μg was reverse transcribed by M-MLV reverse transcriptase (Fisher Scientific) in the presence of RNase inhibitor (Promega) and random hexamers (Promega). Reactions lacking the reverse transcriptase were used to control for DNA contamination. Newly created cDNA was diluted 1:100 and used in qRT-PCR using iQ SYBR green supermix (BIO-RAD). Amplification was achieved using a 3-step melt cure program on a CFX96 qPCR cycler (BIO-RAD). Transcript abundance was calculated using the ΔΔCT method normalized by the C. difficile rpoB gene and performed in biological triplicates. Each assay was repeated 3 independent times and a representative independent experiment is shown.
Metabolomics analyses
For targeted metabolomics analysis of amino acids in sterile, spent bacterial culture supernatants, C. difficile CD196 and E. faecalis OG1RF were grown in BHIS media, subcultured 1:100 into BHIS, and grown for 20 hours under anaerobic conditions. Cultures were then centrifuged at 4000 RPM for 10 minutes and filter sterilized with 0.22μm filters. Cell-free culture supernatants were supplemented with fresh, sterile BHIS media to generate 70% conditioned media. For targeted analyses of amino acids in stool samples, samples were weighed and homogenized in sterile PBS. Contents were sterilized with 0.22μm filters. For targeted analyses of macrocolonies, colonies were scraped from agar and cell lysates were analyzed. Analysis was performed by the Microbial Culture and Metabolomics Core of the PennCHOP Microbiome Program. Amino acids were quantified using a Waters Acquity uPLC System with an AccQ-Tag Ultra C18 1.7 μm 2.1×100mm column and a Photodiode Detector Array. Bacterial culture samples were centrifuged at 13,000g for 5 minutes, and the supernatant was transferred to a new tube and centrifuged again at 13,000g for 5 minutes. Amino acids in the supernatant were derivatized using the Waters AccQ-Tag Ultra Amino Acid Derivatization Kit (Waters Corporation, Milford, MA) and analyzed using the UPLC AAA H-Class Application Kit (Waters Corporation, Milford, MA) according to manufacturer’s instructions. Quality control checks (blanks and standards) were run every eight samples. All chemicals and reagents used were mass spectrometry grade.
Imaging mass spectrometry analyses
Bacterial colonies grown on agar as previously described to be analyzed via matrix-assisted laser desorption/ionization (MALDI) imaging mass spectrometry (IMS) were mounted onto indium tin oxide (ITO)-coated microscope slides. These slides were placed in slide boxes, shipped to the University of Florida on dry ice, and then stored at −80°C until analysis. After removal from the freezer, colony samples are dried in a desiccator at room temperature for 2–3 days prior to drying in a custom-built vacuum drying chamber (160 mtorr, room temperature, 1 hour) to remove moisture and facilitate adherence of the agar to the microscope slide. A 9-aminoacridine (9AA) MALDI matrix layer was applied using a custom-built sublimation apparatus (<50 mtorr, 180°C, 14 minutes, resulting in ~3 mg of matrix deposited on the slide)44.
Tissue samples to be analyzed via MALDI IMS were embedded in OCT, shipped to the University of Florida on dry ice, and stored at −80°C until analysis. Tissue sections were prepared at 12 μm thickness using a Leica CM 3050S Research Cryostat (Leica Biosystems, Buffalo Grove, IL, USA) (−30°C object temperature, −28°C chamber temperature) and thaw mounted onto indium tin oxide (ITO)-coated microscope slides. Samples to be compared via IMS were mounted on the same microscope slide to ensure identical sample preparation and facilitate accurate metabolite comparisons between tissue types. Slides with mounted tissue sections were then warmed to room temperature in a desiccator for ~30 minutes before application of a 1,5-diaminonapthalene MALDI matrix layer using the sublimation apparatus (<50 mtorr, 120°C, 11 minutes, resulting in ~3 mg of matrix deposited on the slide)45. A recrystallization procedure was performed following matrix application to enhance analyte extraction into the MALDI matrix layer (200 μL methanol, 60°C, 2 minutes)46.
All imaging mass spectrometry experiments were performed in negative ion mode on a 7T Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer equipped with a dynamically harmonized ParaCell (Bruker Daltonics, Billerica, MA, USA). The instrument contains an Apollo II dual MALDI/ESI source that uses a Smartbeam II Nd:YAG MALDI laser (2 kHz, 355 nm). Images were acquired at a pixel spacing of 100 μm in both the x and y dimensions using a ∼25-μm laser beam and a 90-μm Smart Walk (500 laser shots). Continuous accumulation of selected ions (CASI) was employed to improve the sensitivity of selected metabolites by setting the Q1 mass to m/z 140 and the mass window to 90 m/z47. Co-culture data were collected from m/z 100 to 1,000 using a 0.9787s time-domain transient length, resulting in a resolving power of ∼228,000 at m/z 122. Tissue data were collected from m/z 100 to 1,000 using either a 0.2447s or 0.4893s time-domain transient length, resulting in a resolving power of ∼84,000 or ~169,000 (at m/z 156), respectively. Internal calibration (quadratic fit for full spectrum data and single point correction for CASI data) was performed using matrix-related ions. Ion images were visualized using FlexImaging 5.0 (Bruker Daltonics, Billerica, MA, USA). Ion images are displayed without normalization and using interpolation. Following image acquisition, tissue sections were stained using hematoxylin and eosin (H&E), scanned using an Aperio Scanscope CS (Leica Biosystems, Buffalo Grove, IL, USA) bright field whole slide scanner, and visualized using Aperio ImageScope (Leica Biosystems, Buffalo Grove, IL, USA).
In situ labelling of C. difficile with a hydrazine probe and gel-based profiling of D-proline reductase activity
A phenylhydrazine probe was synthesized according to the literature23. Working stock solutions (0.1 mM) were prepared in H2O containing 10% dimethyl sulfoxide (DMSO) and neutralized to pH 6.0–7.0 and stored at − 80°C prior to use.
Probe treatment and gel-based analysis of probe-labelled proteins was adapted and optimized following a previous study23. Briefly, cultures were harvested (4,000g, 10 min, 4°C) and resuspended in 1 mL of growth media. Cells were treated with the hydrazine probe (1 mM) and incubated anaerobically for 30 min at 37°C. The cells were washed by centrifugation (17,000g, 3 min, 4°C) and resuspended in ice-cold deoxygenated PBS. Cell pellets were stored at − 80°C prior to use.
Pelleted C. difficile cells were resuspended in 8 M urea in PBS (400 μL), incubated on ice for 30 min and lysed with a Branson SFX250 Sonifier equipped with a 102C (3 × 10 pulses, 0.3 second on and 2 second off, 15% energy). Soluble proteomes were fractionated by centrifugation (100,000g, 30 min, 4°C) and total protein concentrations were determined using the DC protein assay (Bio-Rad Laboratories, Hercules, CA, USA) on a microplate reader (Biotek ELx808).
A freshly prepared copper(I)-catalyzed azide alkyne cycloaddition (CuAAC or “click”) reagent mixture (6 μL) containing 3 μL of 1.7 mM Tris(benzyltriazolylmethyl)amine (TBTA) in DMSO:t-BuOH (1:4 v/v), 1 μL of 50 mM CuSO4 in H2O, 1 μL of 1.25 mM rhodamine-azide (Rh-N3) in DMSO, and 1 μL of freshly prepared 50 mM tris(2-carboxyethyl) phosphine (TCEP) in H2O was added to samples (50 μL) containing the probe-labelled proteomes (1 mg/mL). After addition of the click mixture, samples were vortexed and incubated at room temperature for 1 h while rotating and quenched by addition of 17 μL of 4X sodium dodecyl sulfate (SDS) loading buffer. Probe-labelled proteins were resolved by SDS-PAGE and visualized by in-gel fluorescence scanning on a ChemiDoc MP Imaging System (Bio-Rad Laboratories, Hercules, CA, USA). D-proline reductase (0.3 mg/mL), as purified below, was incubated with the phenylhydrazine probe (3 μM, 15 min, at 37°C), conjugated to Rh-N3 and compared against C. difficile gel profiles to identify active PrdA.
Band intensities of the active (probe-labelled) PrdA subunit of D-proline reductase were quantified using ImageJ Software48 and normalized against corresponding expression profiles.
Purification of D-proline reductase from C. difficile
The purification of D-proline reductase was adapted from a previous study49. C. difficile VPI 10463 was cultivated in rich growth medium containing 20 g/L tryptone (dot scientific), 10 g/L yeast extract (Acros Organics, 451120050), 1.75 g/L K2HPO4 (Fisher), 1 μM selenite (Honeywell) and supplemented with 40 mM L-proline (Alfa Aesar) and 40 mM L-alanine (Alfa Aesar) to enhance the expression of prd at 37°C. After 24 h, cells were harvested by centrifugation (5,000g, 30 min, 4°C), flash-frozen in liquid N2 and stored at − 80°C. The cell paste (8–10 g/L culture) was resuspended (5 mL /g paste) in 50 mM Tris buffer (pH 8.4) containing 1 mM Ethylenediaminetetraacetic acid (EDTA), and 1 mM dithiothreitol (DTT) at 4°C, lysed by sonication (1 sec on and 2 sec off for 10 minutes, 40% amplitude) on Qsonica Q700 sonicator and centrifuged (30,000g, 20 min, 4°C). Stepwise ammonium sulfate fractionation (25, 40, 60 and 85% saturation) was performed to isolate D-proline reductase-containing fractions. In brief, solid ammonium sulfate (Alfa Aesar) was slowly added to the lysate stirring at 4°C to create a 25% saturated solution. After 1 h, precipitated proteins were collected by centrifugation (30,000g, 20 min, 4°C). Steps were repeated on the remaining supernatant to fractionate precipitated proteins at 40, 60 and 85% ammonium sulfate saturation. D-proline reductase was precipitated in 40–60% ammonium sulfate. The precipitate was solubilized in 50 mM Tris buffer (pH 8.4) containing 1 mM EDTA, 1 mM DTT, and 2 M ammonium sulfate and loaded onto a phenyl-Sepharose column (20 × 2.5 cm) equilibrated in the same buffer. The column was washed with equilibration buffer until absorption of the eluate at 280 nm (A280) and 260 nm (A260) was ~ 0 (~ 5 column volumes). Bound proteins were eluted by a 400 mL linear gradient of ammonium sulfate (2.0 M to 0.0 M) in 50 mM Tris buffer (pH 8.4) containing 1 mM EDTA and 1 mM DTT. Fractions containing D-proline reductase as determined by SDS-PAGE were pooled and dialyzed against 50 mM Tris buffer (pH 8.4) containing 1 mM DTT and 250 mM NaCl for 12 h at 4 °C. The protein was concentrated to ~ 4 mg/mL in 10% glycerol and flash-frozen at – 80 °C. The yield for D-proline reductase complex was 3 mg of protein per gram of cell paste. The active protein was confirmed by LC-MS/MS.
16S rRNA gene sequencing and analysis
Specimens positive for C. difficile toxin A or B by enzyme immunoassay (EIA) were captured from a cross-sectional study of human subjects (University of Pennsylvania IRB #826543). We extracted DNA from residual stool using the Powersoil kit (Qiagen), amplified the V1-V2 hypervariable region of the 16S rRNA gene using barcoded 27F and 338R primers, and performed paired-end 250bp sequencing (Illumina MiSeq). Sequences were demultiplexed and aligned with QIIME2; sequence denoising and amplicon sequence-variant (ASV) binning was performed with DADA2. Taxonomic assignment was performed by basic local alignment (BLAST) to the SILVA rRNA gene database (version 132). Linear regression relating subjects’ peripheral white blood cell (WBC) count to log base 10 Enterococcus relative abundance performed using R statistical software (version 4.1.0). Enterococcus abundance values of zero, which cannot translate to the log scale, were treated as the lowest observed Enterococcus abundance.
Identification of shared sequences present in vancomycin-resistant enterococci and C. difficile genomes
We used a previously developed approach50 to scan the genomes of clinical bacterial isolates collected from hospital-associated infections at the University of Pittsburgh Medical Center for highly similar nucleotide sequences present in the genomes of different species25. We identified nucleotide sequences >5kb that were present at 99.98% identity or greater (i.e. no more than 1 mutation per 5kb of sequence) in both VRE and C. difficile genomes. Shared sequences were annotated with Prokka51 and were compared to one another, and to publicly available genomes, with BLAST. Share sequence clusters were visualized with Cytoscape52. MGE clusters are available in Supplementary Table 4 and FASTA files available in supplementary material.
Expression of collagen adhesin in E. faecalis
The predicted coding sequence and ribosome-binding region of the CD0386-like surface adhesin was amplified from the vancomycin-resistant E. faecium isolate VRE33574 with PCR, and was cloned into the pMSP3535 expression vector using Gibson assembly53. The resulting vector was transformed into E. faecalis strain OG1RF, and transformants were selected with 15ug/mL erythromycin. Transformants were tested for their ability to form biofilms on standard and collagen-coated plates following a previously described protocol 54. Briefly, overnight cultures were diluted 100-fold into BHI broth supplemented with 0.25% glucose; 200 μL of each culture was plated into 8 replicate wells of a 96-well untreated polystyrene microtiter plate, and plates were incubated for 24 h at 37°C under static conditions. Plates were washed three times with 250 μL 1× phosphate-buffered saline (PBS) to remove unattached cells, and then wells were stained with 200 μL of 0.1% crystal violet (CV) in water. After incubation for 30 min at 4°C, stained wells were washed twice with 250 μL of 1× PBS to remove excess stain. Plates were dried, and then 250 μL of 4:1 ethanol:acetone was added to each well to solubilize the CV-stained biofilm. After incubation for 45 min at room temperature, the absorbance of the dissolved CV was measured at 595 nm using a Synergy H1 microplate reader (Biotek). Adhesin expression was induced with 25ng/mL nisin, and a transformant carrying the empty pMSP3535 vector was used as a comparison strain.
Analysis of ArcD conservation in the human microbiome
Raw reads from an adult CDI metagenomics dataset28 were aligned to all prokaryote proteins in KEGG using Diamond (v2.0.14). Reads aligned to any of the Arc operons (ArcA, ArcB, ArcC, ArcD/ArcF, and ArcR with KO ids K01478, K03758, K24446, K00926, K21827, K21828, K00611, K09065) were filtered out from the original fastq files, and the DNA sequences were then taxonomically classified using Kraken2 (v2.1.2). The normalized read counts for selected taxa were used to generate the heatmap. After reads were directly assigned a taxonomy using Kraken2, they were analyzed and visualized in R (version 3.5.2). Relative abundances were calculated by aggregating read counts at the family level and below and dividing by total number of classified reads per sample. We focused on adults with symptomatic adult CDI (n = 48). The top 10 bacterial families containing Arc-operon mapping reads are visualized in Extended Data Fig. 7b and Extended Data Figure 7c. The conservation of ArcD in enterococci was determined by pangenome analysis of previously published E. faecalis and E. faecium genomes25,26. In both cases, gene presence/absence matrices previously constructed with Roary55 were queried for arcD. Among 51 genetically diverse E. faecalis isolate genomes, arcD was found in all genomes. Among the 246 vancomycin-resistant E. faecium genomes used for shared sequence analysis, all genomes were found to encode two copies of arcD located at different positions in the genome.
Human samples
Vanderbilt University Medical Center:
Pediatric participants, ages 12 months through 18 years, were prospectively enrolled from July 2017 through December 2019 at Monroe Carell Jr. Children’s Hospital at Vanderbilt after informed parental consent and patient assent when applicable. This study was approved by the Vanderbilt Institutional Review Board (IRB# 170067). Thorough medical histories were obtained from all participants, including past hospitalizations, surgeries, and medications received 30 days prior to enrollment and confirmed by medical record review. Data were kept strictly confidential using a REDCap database (REDCap software, Vanderbilt University).56 Symptomatic C. difficile cases where children with diarrhea (unformed stools) between 12 months and 18 years of age who underwent clinical laboratory testing and tested positive for C. difficile by nucleic acid amplification-based testing (NAAT). C. difficile negative children (confirmed by NAAT) included children with cystic fibrosis, inflammatory bowel disease, and cancer. No images of human subjects are included in the figures, extended data, or supplementary materials.
Children’s Hospital of Philadelphia:
Subjects were recruited at the Children’s Hospital of Philadelphia (CHOP) from September 2015 to April 2018 and informed consent was acquired (IRB approval number 15–011817), as previously described31. Groups included healthy children (HC) and children with IBD and concurrent CDI (IBD+CDI). Healthy children were age matched to those with IBD + CDI. For inclusion in the C. difficile infected group, patients needed to 1) have an underlying diagnosis of inflammatory bowel disease 2) have loose, watery stools at least 3 times per day for >24 hours or an increase in stool frequency from baseline and 3) be positive by glutamate dehydrogenase (GDH) testing assay or PCR for the presence of the toxin A/B gene (cycle of threshold <40). Clinical manifestations of C. difficile infection ranged from mild diarrhea to fulminant colitis and shock. Patients with CDI were identified by notification of positive testing for toxigenic C. difficile by the CHOP Microbiology laboratory. Exclusion criteria included history of hemophagocytic lymphohistiocytosis or Langerhan’s cell histiocytosis or concurrent gastrointestinal infection proven by clinical testing. Prior history of C. difficile or recent antibiotic use were not exclusion criteria. Inclusion criteria for healthy controls included if they did not have a chronic diagnosis and could provide stool samples at all 3 time points. Healthy children were excluded if they had diarrhea, antibiotic use in 90 days prior to enrollment, or a family member with C. difficile enrolled into the study. Patients with CDI were included with either primary infection or recurrent infections. There were 10/23 patients in the IBD+CDI cohort who had recurrent CDI at enrollment, three of whom had a prior positive clinical test for C. difficile within three months of enrollment. Untargeted metabolomics on the stool samples from these patients was performed as previously described31. No images of human subjects are included in the figures, extended data, or supplementary materials.
The Hospital of the University of Pennsylvania:
Subjects with positive testing by enzyme immunoassay (EIA) for C. difficile toxin A or B were identified over six months of continuous screening (February – August 2017) at the Hospital of the University of Pennsylvania, with a waiver of informed consent granted by the University of Pennsylvania Institutional Review Board (IRB #826543). Fecal specimens from 50 consecutive toxin EIA+ subjects underwent 16S rRNA gene sequencing as described above, and the fecal bacterial community composition was compared to the maximum peripheral white blood cell (WBC) count within 72 hours of the index C. difficile test. Samples in which there were no detected Enterococcus reads were assigned the minimum observed value in the data. WBC data were extracted from the subjects’ electronic medical records. No images of human subjects are included in the figures, extended data, or supplementary materials.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 8 and R version 3.5.2. Specific statistical tests, replicate numbers, calculated errors and extended information for each experiment are reported in the figure legends. All data represent distinct samples unless otherwise stated (e.g. longitudinal bacterial burdens in feces).
Materials availability
All strains and materials used in this study are available upon request.
Data availability
Raw RNA sequencing data and processed data are deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE165751. MGE clusters identified in Extended Figure 2c are available in Supplementary Table 4. Conservation of arc genes performed on metagenomic reads deposited under BioProject accession number PRJNA74826228. Conservation of arc genes among enterococci performed on dataset deposited under BioProject ID: PRJNA58716125,26. All other data are available in the main text or supplementary materials.
Code availability
Code and source data for replication of C. difficile GENRE (iCdR703) analyses available at https://github.com/mjenior/Smith_etal_Enterococcus.
Extended Data
Extended Data Fig. 1. Enterococcal abundance and dynamics during CDI.
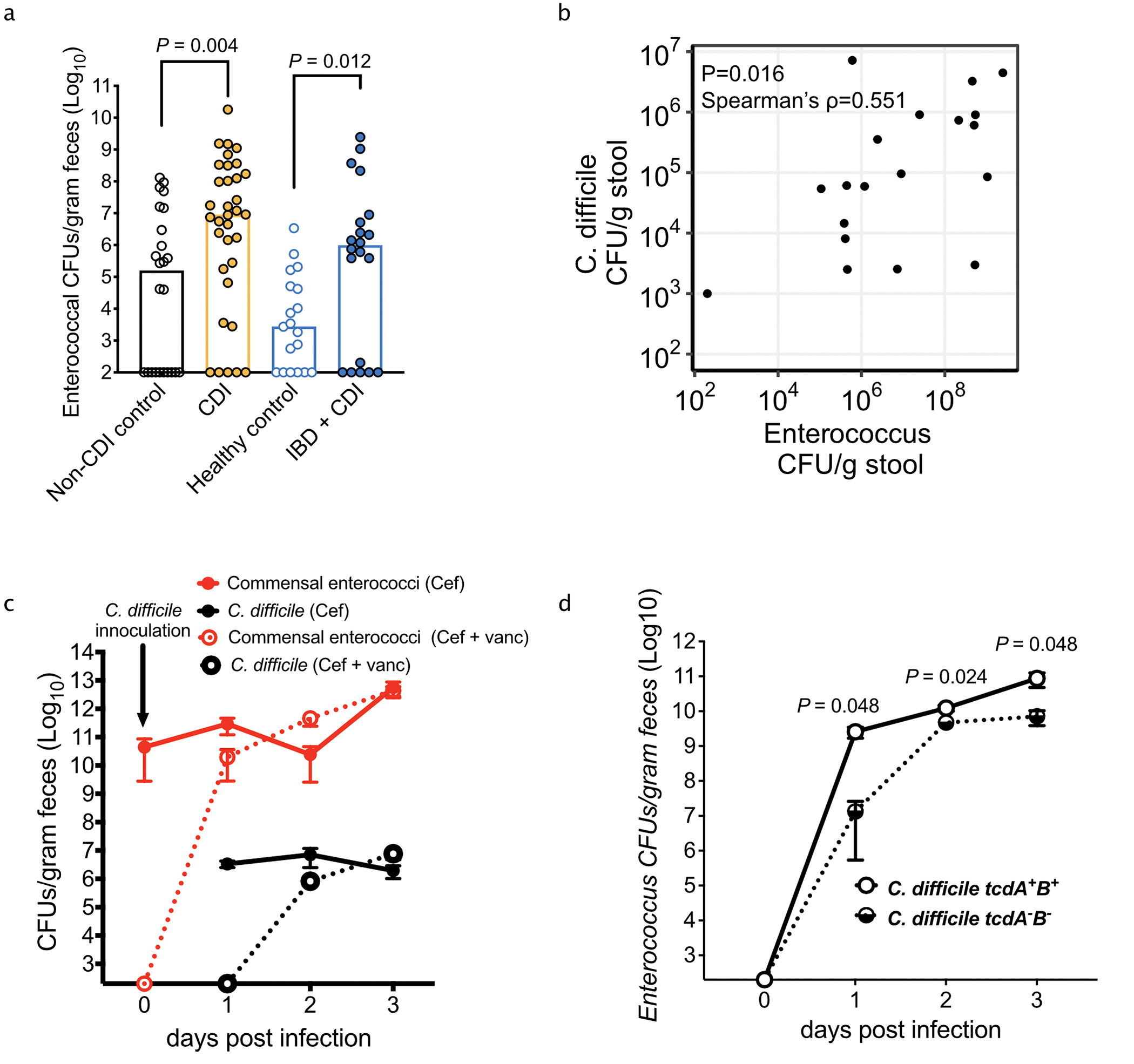
(a) Enterococcal bacterial burdens (CFUs) from pediatric patients (black and gold = Vanderbilt University (median, n = 24 for Non-CDI controls; n = 34 for CDI, two-sided Mann-Whitney, P=0.004); blue and light blue = Children’s Hospital of Philadelphia (n = 19 for healthy; n = 20 for IBD + CDI, two-sided Mann-Whitney test, P=0.012). (b) Two-sided Spearman correlation between detected C. difficile and Enterococcus burdens in pediatric patients with IBD + CDI (Spearman ρ = 0.551; n = 19). (c) Bacterial burdens quantified from mice following treatment with cefoperazone (cef) or cef + vancomycin (vanc) (mean ± s.e.m., n = 10 mice/group). (d) Enterococcal CFUs over the course of CDI. Mice were infected with a toxin producing wild type strain (M7404 TcdA+TcdB+) or a toxin-null isogenic mutant (M7404 TcdA−TcdB−) (n = 5/group) (mean ± s.e.m. two-sided Mann-Whitney with Bonferroni-Dunn method for correction for multiple comparisons, corrected P values are in Supplementary Table 5).
Extended Data Fig. 2. Biofilm formation and transfer of mobile genetic elements during interspecies interactions.
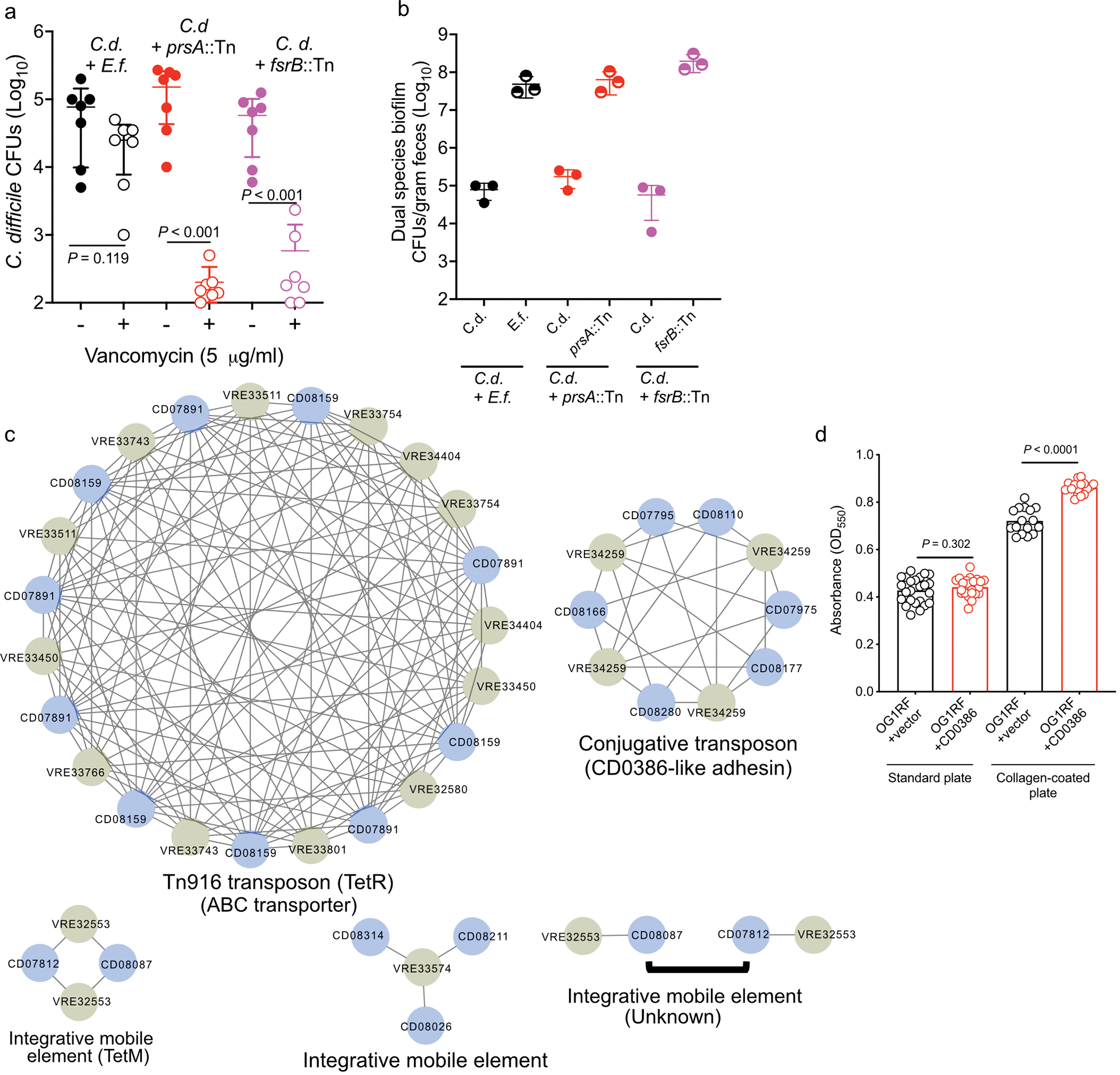
(a) Survival assay of co-culture biofilms with E. faecalis (E.f.) (P=0.119) or transposon mutants in E. faecalis genes OG1RF_11528 (fsrB::Tn) (P<0.001) and OG1RF_10423 (prsA::Tn) (P<0.001). Abundance of C. difficile in untreated (−) or vancomycin treated (+) biofilms are depicted (mean ± s.d., n=7, two-sided Mann-Whitney with Bonferroni-Dunn method for correction for multiple comparisons) (b) Abundance of C. difficile (C.d.) and E. faecalis strains (E.f., prsA::Tn, and frsB::Tn) in untreated dual species biofilms (mean ± s.d., n = 3). (c) Clusters of shared sequences detected in C. difficile (blue) and VRE (olive) genomes of clinical isolates from hospitalized patients. Lines connect sequences with at least 99.98% identity. Clusters are labeled based on mobile element type and relevant cargo, if known. Source data for each cluster can be found in Supplementary Information. (d) Biofilm formation of E. faecalis OG1RF carrying empty pMSP3535 vector or pMSP3535 carrying the CD0386-like adhesin. Biofilm formation was tested in standard (P=0.302) and collagen-coated plates (P<0.001). Crystal violet staining (OD550) values were calculated (mean ± s.d., n = 24/group for standard plates and 16/group for collagen plates, unpaired two-tailed t-tests).
Extended Data Fig. 3. Enterococcal-mediated enhancement of C. difficile toxin gene expression and production.
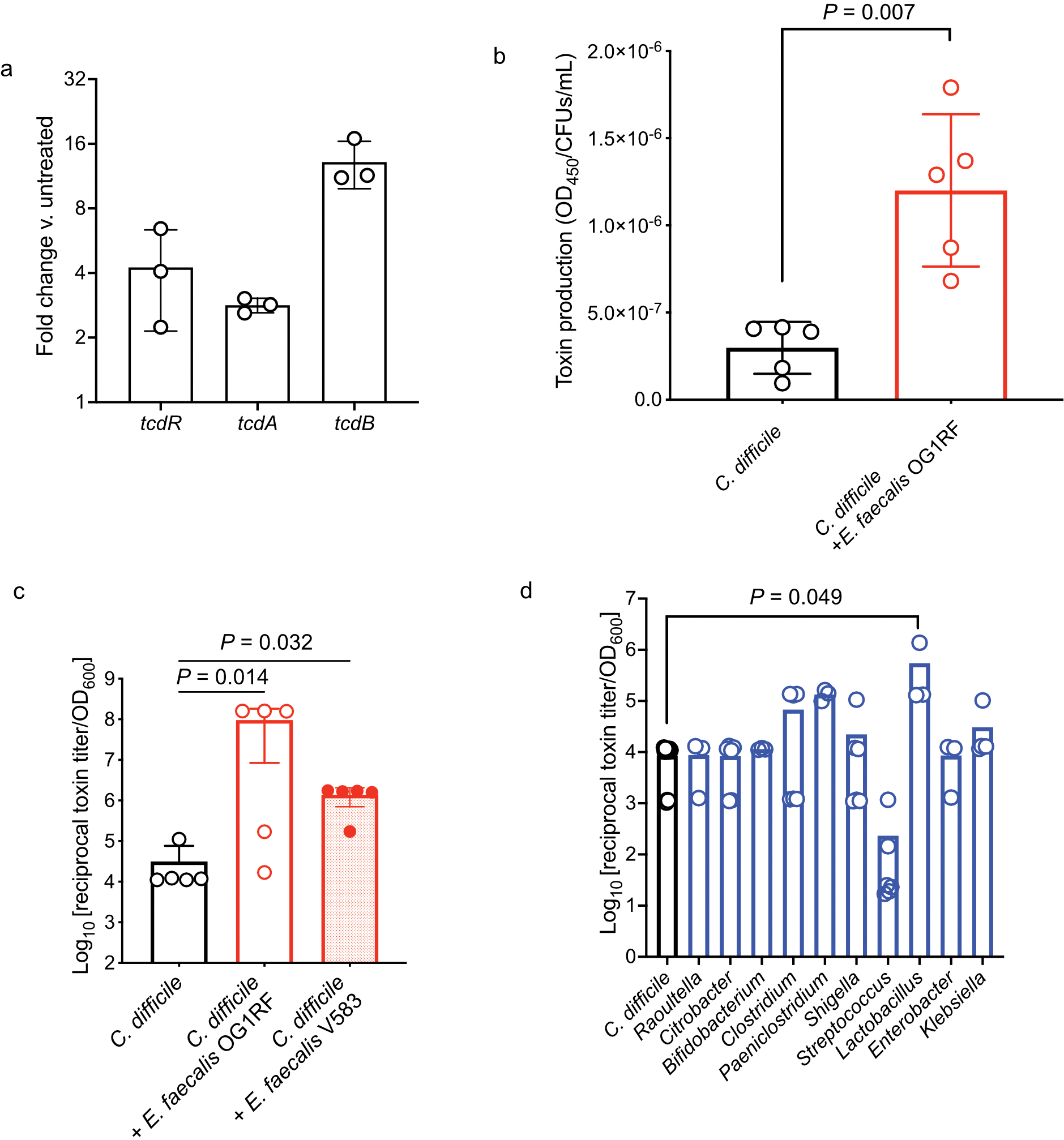
(a) Fold change of the toxin-encoding genes in C. difficile in coculture with E. faecalis versus monoculture as measured by qPCR (mean ± s.d., n=3). (b) Toxin production by C. difficile when grown in co-culture with E. faecalis as measured by ELISA. Both C. difficile and E. faecalis were grown in the same culture and differential plating was used to measure C. difficile CFUs. Toxin levels (OD450) were normalized to C. difficile CFUs in the culture to control for any difference in growth (mean ± s.d., n = 5, two-tailed t-tests with Welch’s correction, P=0.007). (c) C. difficile toxin levels measured from in vitro cultures by cytotoxicity with E. faecalis cell-free supernatants. (mean ± s.d., n = 5, Kruskal-Wallis test with Dunn’s correction for multiple comparisons, OG1RF P=0.014, V583 P=0.032). (d) C. difficile toxin production measured by cytotoxicity following introduction of cell-free supernatants from microbiota isolates cultured from human patients with CDI and IBD (mean ± s.d., n = 12 (C. difficile), 3 (Raoultella, Bifidobacterium, Enterobacter, Paeniclostridium, Lactobacillus), 5 (Klebsiella), 6 (Citrobacter, Clostridium, Shigella, Streptococcus), Kruskal-Wallis test with Dunn’s correction for multiple comparisons, Lactobacillus P=0.049). Isolates were selected to represent the spectrum of taxa cultured from these patients.
Extended Data Fig. 4. Transcriptional changes associated with C. difficile – E. faecalis interactions.
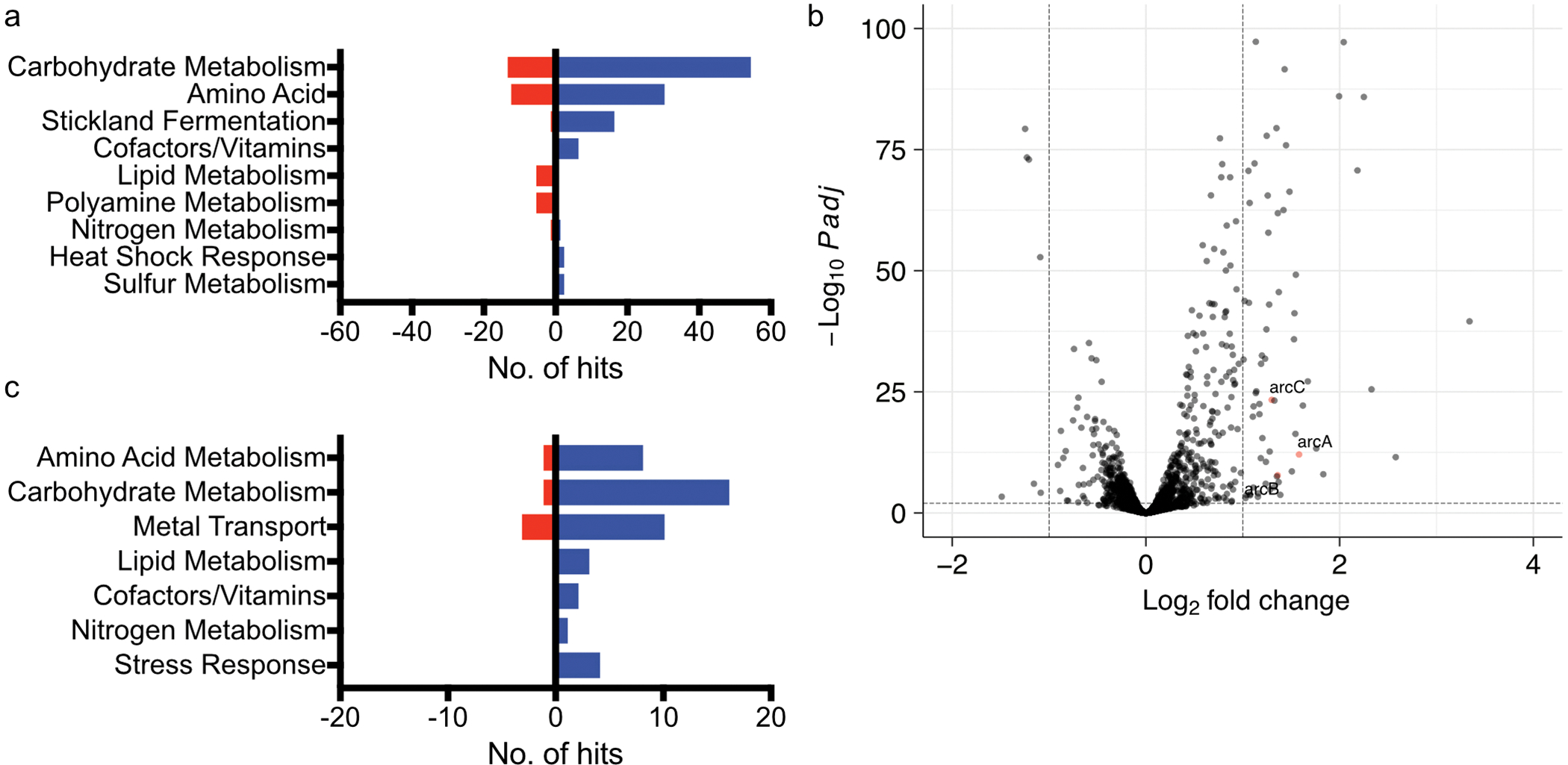
(a) Pathway analysis of C. difficile transcripts significantly altered following co-culture as measured by RNA sequencing. For pathway analyses, blue bars represent transcripts that increased in abundance and red bars represent transcripts that decreased in abundance. (b) Volcano plot showing E. faecalis transcripts significantly altered following co-culture as measured by RNA sequencing. Red points represent genes associated with amino acid metabolism. Significance determined using two-sided Wald test and corrected for multiple comparisons using the Benjamini-Hochberg method. (c) Pathway analysis of E. faecalis transcripts significantly altered following co-culture as measured by RNA sequencing.
Extended Data Fig. 5. Transcriptome-guided metabolic flux predictions using genome-scale metabolic network reconstruction for C. difficile.
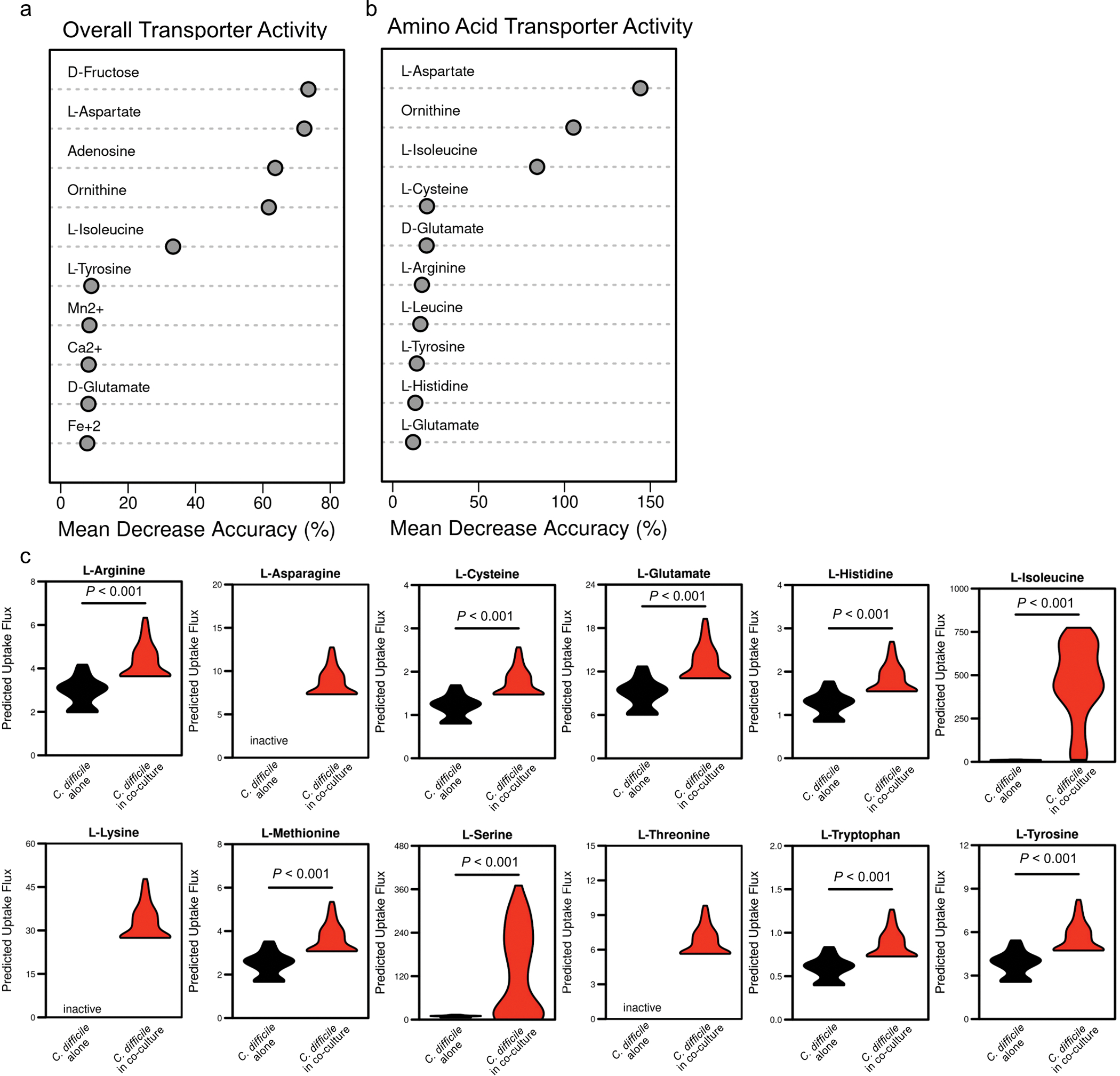
(a) AUC-Random Forest supervised machine learning results for reaction flux samples for conserved transport reactions between contexts (k = 10; OOB = 0%). (b) AUC-Random Forest supervised machine learning results for reaction flux samples for conserved amino acid transport reactions between contexts (k = 10; OOB = 0%). (c) Difference in simulated uptake of selected amino acids across context-specific models. Significance determined by two-sided Wilcoxon rank-sum test. Corrected P values in Supplementary Table 5.
Extended Data Fig. 6. In situ labelling of C. difficile with a hydrazine probe and gel-based profiling of D-proline reductase activity.
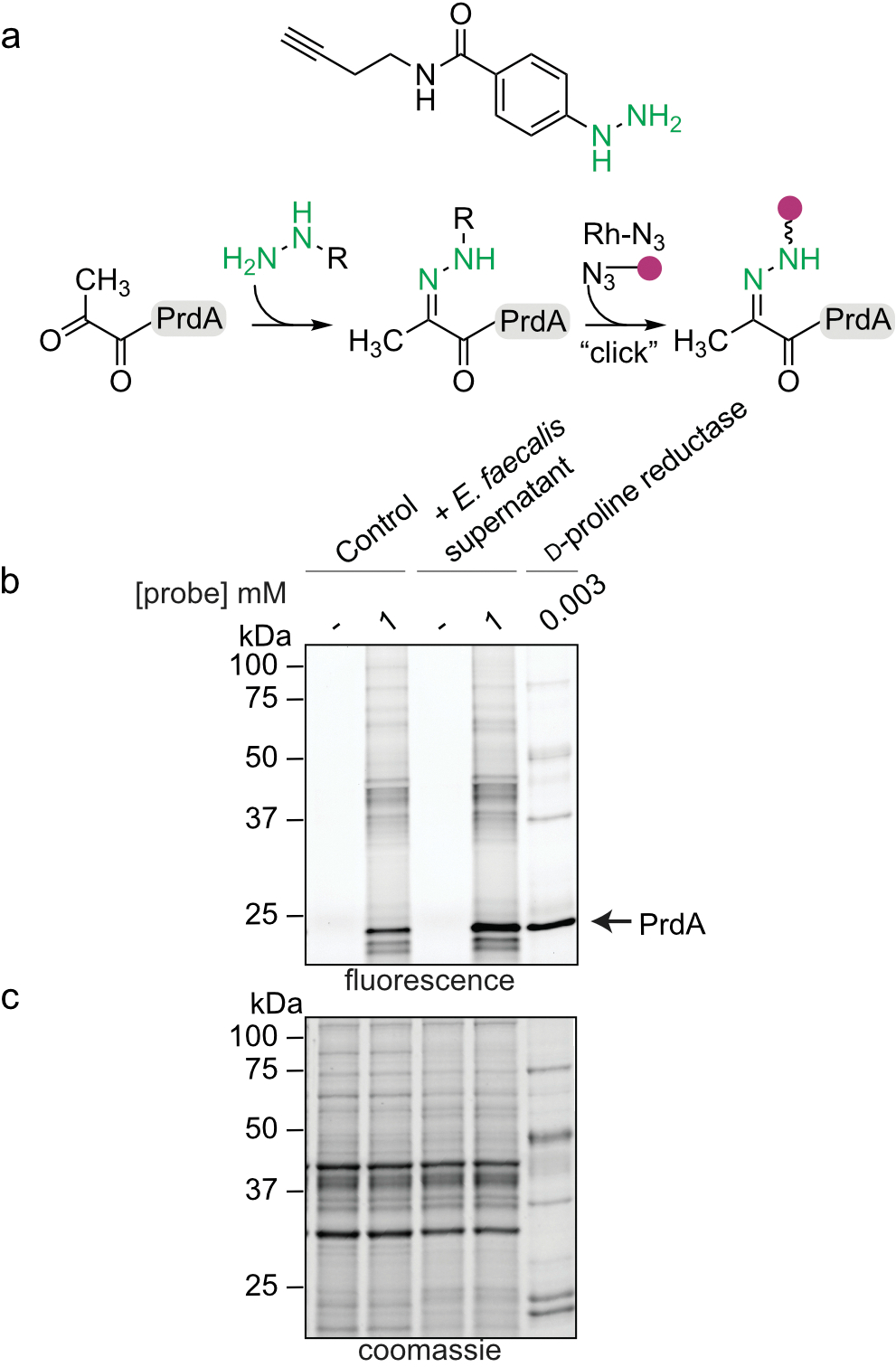
(a) Labeling schematic of hydrazine probe with PrdA of D-proline reductase. (b) Representative gel-based labelling profiles for C. difficile in the absence and presence of E. faecalis supernatant. Gel representative of three separate experiments. For Gel source data, see Supplementary Fig. 1. (c) Corresponding expression profiles after Coomassie staining.
Extended Data Fig. 7. The enterococcal ADI pathway reshapes the metabolic environment in the gut during CDI.
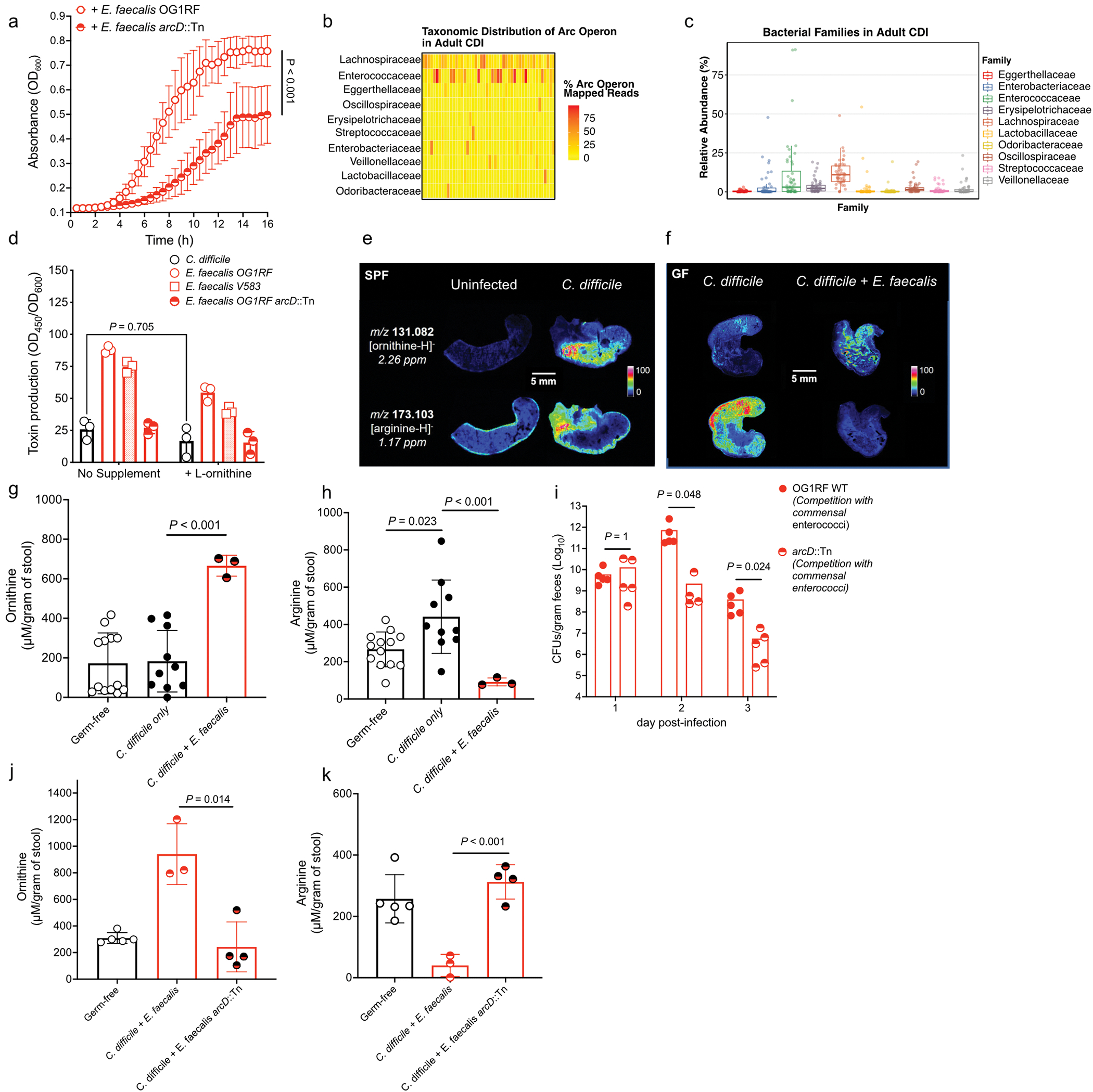
(a) C. difficile growth in presence of E. faecalis or E. faecalis arcD::Tn supernatants. (mean ± s.d., n = 8/group, two-way ANOVA, Time factor P<0.001). (b) Taxonomic distribution (at bacterial family level) of reads mapped to arc genes in adult patients (n=48) with symptomatic CDI. Each column is a subject and each row is a bacterial family. Each cell displays the percentage of reads mapped to arc genes of a specific family out of all arc mapped reads. (c) Relative abundance of the top 10 arc operon containing bacterial families in each adult patient symptomatically infected with C. difficile (n=48, lower and upper hinges correspond to the first (25%) and third (75%) quartiles. The upper and lower whiskers extend from the hinge to the largest value no further than 1.5*IQR. Data beyond the whiskers are plotted individually). (d) Toxin production of C. difficile following introduction of supernatants from E. faecalis and addition of exogenous L-ornithine measured by ELISA (mean ± s.d., n = 3, Tukey’s multiple comparisons test, C. difficile +/− ornithine P=0.705). (e) MALDI-IMS image of uninfected or infected mice (3d post-infection) (SPF) (representative of n = 5 mice) or (f) GF mice mono-infected with C. difficile CD196 or co-infected with E. faecalis OG1RF (2d post-infection) (representative of n = 4 mice). Individual heatmaps of arginine and ornithine. (g) Ornithine levels in stool measured by targeted metabolomics in GF mice infected with C. difficile only (mean ± s.d., n = 10) or C. difficile + E. faecalis OG1RF (mean ± s.d., n = 3, two-sided t-tests with Welch’s correction, P<0.001). Metabolomics were performed on GF mice prior to infection (GF group, n = 13). (h) Arginine levels in stool of GF mice infected with C. difficile only (mean ± s.d., n = 10, two-sided t-test with Welch’s correction, P=0.023) or C. difficile + E. faecalis OG1RF (mean ± s.d., n = 3, two-sided t-test with Welch’s correction, P<0.001). Stool metabolomics performed prior to infection (GF group, n = 13). (i) CFU of E. faecalis OG1RF (wild type) or E. faecalis arcD::Tn during CDI. Each strain introduced prior to CDI and naturally competed with endogenous enterococci (n = 5/group) (mean ± s.e.m. two-sided Mann-Whitney test with Bonferroni-Dunn method for correction for multiple comparisons, day 2 P=0.048, day 3 P=0.024). (j) Ornithine (P=0.014) and (k) arginine (P<0.001) levels in stool measured by targeted metabolomics in GF mice infected with C. difficile. Mice pre-colonized for 1 day with E. faecalis (n = 3) or E. faecalis arcD::Tn (n = 4) (mean ± s.d.. two-sided t-tests with Welch’s correction). Metabolomics performed on GF mice prior to infection (GF group, n = 5).
Extended Data Fig. 8. Arginine supplementation decreases C. difficile pathogenesis in mice.
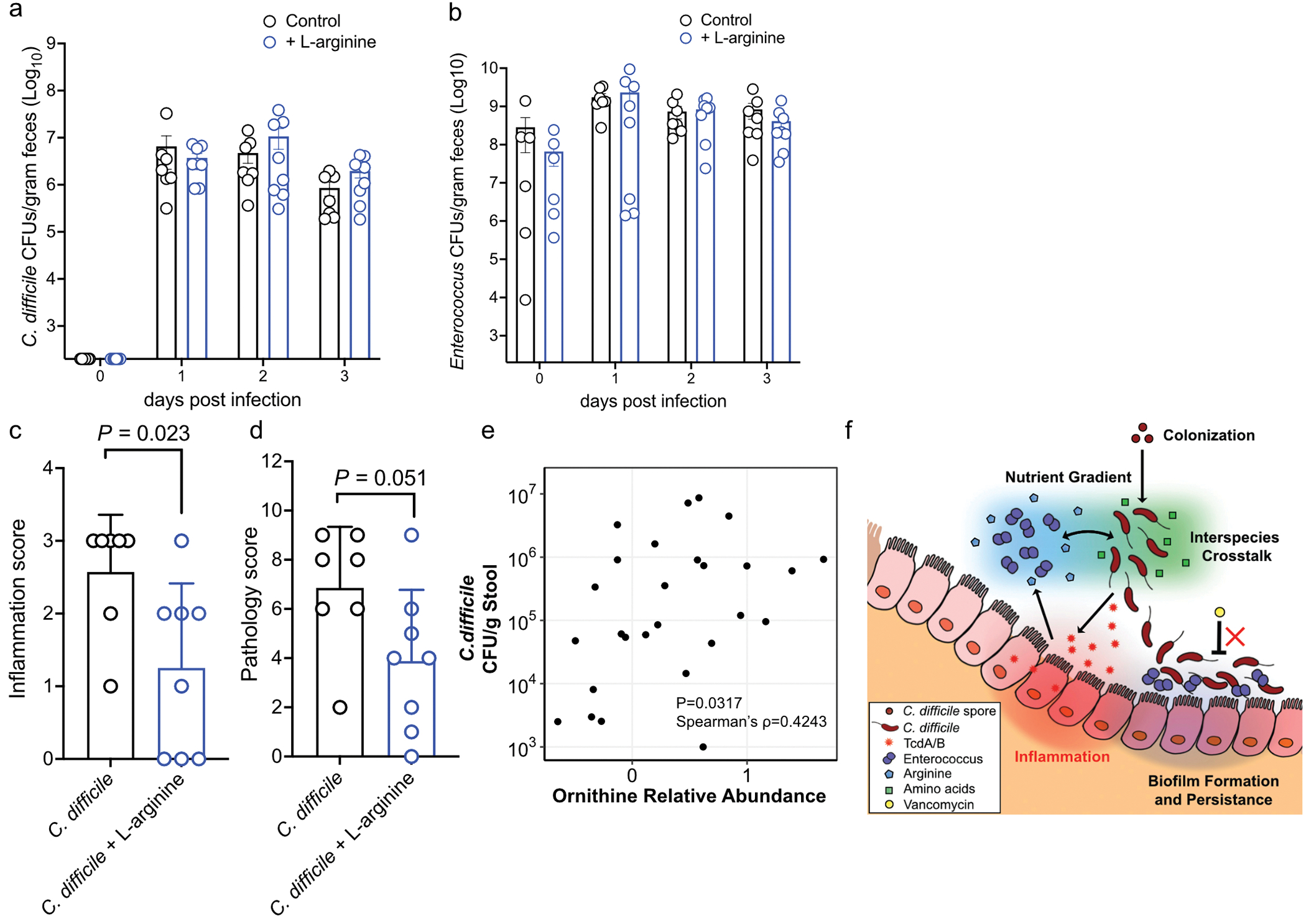
(a) C. difficile and (b) Enterococcus burdens quantified from mice following cefoperazone treatment and subsequent infection. Mice were treated with 2% L-arginine in drinking water starting 2 days prior to infection and subsequently during the course of infection (mean ± s.d.. n = 7 for control, n = 8 for L-arginine treated; Mann-Whitney with Bonferroni-Dunn method for correction for multiple comparisons for each comparison). (c) Inflammation score (P=0.023) and (d) cumulative pathology score (P=0.051) measured 3 days post-infection for control (n = 7) and L-arginine treated (n = 8) mice (mean ± s.d.. two-sided t-tests with Welch’s correction). (e) Spearman correlation between ornithine abundance in stool and C. difficile burdens in pediatric patients with IBD and CDI with detectable C. difficile based on culture (two-sided Spearman’s ρ = 0.4243; n = 26). (f) Proposed model of multifaceted cooperative interactions between enterococci and C. difficile during infection.
Supplementary Material
Acknowledgments:
We gratefully acknowledge Drs. Lee Harrison and Jane Marsh for sharing genome sequence data and VRE isolates from the EDS-HAT study (R01AI127472). We thank the Children’s Hospital of Philadelphia Junior Faculty Breakfast Club and the Children’s Hospital of Philadelphia MICRO group for feedback on the manuscript. We thank all members of the Zackular Lab for providing feedback and support throughout this endeavor. We thank the Children’s Hospital of Philadelphia Biostatistics and Data Management Core for statistical consulting and support. We thank Dr. Elliot Friedman and the Penn Microbial Culture and Metabolomics Core for their support and expertise. We also thank Drs. Kyle Bittinger and Ceylan Tanes for thoughtful discussions on analyses detailed in this study.
Funding:
National Institutes of Health grant K22AI7220 (JPZ)
National Institutes of Health grant R35GM138369 (JPZ)
National Institutes of Health grant R01AI138581 (EPS)
National Institutes of Health grant R01AI145992 (EPS)
Children’s Hospital of Philadelphia Junior Faculty Pilot Grant (JPZ)
Cell and Molecular Biology Training Grant T32GM07229 (ABS)
UVA TransUniversity Microbiome Initiative Pilot Grant (MLJ, JAP)
National Institutes of Health grant R01AT010253 (JAP)
Chemical and Biology Interface Training Grant 5T32GM071339–15 (KAB)
National Institutes of Health grant R01HD090061 (JAG)
National Institutes of Health grant UL1TR000445 (MRN)
National Institutes of Health grant K23 AI121485 (BJK)
Centers for Disease Control and Prevention grant BAA 200–2016-91937 (BJK)
National Institutes of Health grant 1DP1DA051620 (MLM)
Commonwealth Universal Research Enhancement program grant SAP#4100068710 (RNB)
Footnotes
Competing interests: Authors declare that they have no competing interests.
References
- 1.Abbas A & Zackular JP Microbe-microbe interactions during Clostridioides difficile infection. Curr Opin Microbiol 53, 19–25, doi: 10.1016/j.mib.2020.01.016 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buffie CG et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208, doi: 10.1038/nature13828 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lessa FC, Winston LG, McDonald LC & Team, E. I. P. C. d. S. Burden of Clostridium difficile infection in the United States. N Engl J Med 372, 2369–2370, doi: 10.1056/NEJMc1505190 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schubert AM et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio 5, e01021–01014, doi: 10.1128/mBio.01021-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auchtung JM, Preisner EC, Collins J, Lerma AI & Britton RA Identification of Simplified Microbial Communities That Inhibit Clostridioides difficile Infection through Dilution/Extinction. mSphere 5, doi: 10.1128/mSphere.00387-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zackular JP et al. Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection. Nat Med 22, 1330–1334, doi: 10.1038/nm.4174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomkovich S, Stough JMA, Bishop L & Schloss PD The Initial Gut Microbiota and Response to Antibiotic Perturbation Influence Clostridioides difficile Clearance in Mice. mSphere 5, doi: 10.1128/mSphere.00869-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berkell M et al. Microbiota-based markers predictive of development of Clostridioides difficile infection. Nat Commun 12, 2241, doi: 10.1038/s41467-021-22302-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antharam VC et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol 51, 2884–2892, doi: 10.1128/JCM.00845-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poduval RD, Kamath RP, Corpuz M, Norkus EP & Pitchumoni CS Clostridium difficile and vancomycin-resistant Enterococcus: the new nosocomial alliance. Am J Gastroenterol 95, 3513–3515, doi: 10.1111/j.1572-0241.2000.03291.x (2000). [DOI] [PubMed] [Google Scholar]
- 11.Ubeda C et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120, 4332–4341, doi: 10.1172/JCI43918 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taur Y et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 55, 905–914, doi: 10.1093/cid/cis580 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willett JLE et al. Comparative Biofilm Assays Using Enterococcus faecalis OG1RF Identify New Determinants of Biofilm Formation. mBio 12, e0101121, doi: 10.1128/mBio.01011-21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willett JLE, Robertson EB & Dunny GM The Phosphatase Bph and Peptidyl-Prolyl Isomerase PrsA Are Required for Gelatinase Expression and Activity in Enterococcus faecalis. J Bacteriol 204, e0012922, doi: 10.1128/jb.00129-22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee IPA, Eldakar OT, Gogarten JP & Andam CP Bacterial cooperation through horizontal gene transfer. Trends Ecol Evol, doi: 10.1016/j.tree.2021.11.006 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Roberts AP & Mullany P Tn916-like genetic elements: a diverse group of modular mobile elements conferring antibiotic resistance. FEMS Microbiol Rev 35, 856–871, doi: 10.1111/j.1574-6976.2011.00283.x (2011). [DOI] [PubMed] [Google Scholar]
- 17.Chambers CJ, Roberts AK, Shone CC & Acharya KR Structure and function of a Clostridium difficile sortase enzyme. Sci Rep 5, 9449, doi: 10.1038/srep09449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jenior ML et al. Novel Drivers of Virulence in Clostridioides difficile Identified via Context-Specific Metabolic Network Analysis. mSystems 6, e0091921, doi: 10.1128/mSystems.00919-21 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang X, Lloyd CJ & Palsson BO Reconstructing organisms in silico: genome-scale models and their emerging applications. Nat Rev Microbiol, doi: 10.1038/s41579-020-00440-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenior ML, Moutinho TJ, Dougherty BV & Papin JA Transcriptome-guided parsimonious flux analysis improves predictions with metabolic networks in complex environments. PLoS Comput Biol 16, e1007099, doi: 10.1371/journal.pcbi.1007099 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pruss KM et al. Oxidative ornithine metabolism supports non-inflammatory C. difficile colonization. Nat Metab 4, 19–28, doi: 10.1038/s42255-021-00506-4 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barker HA Amino acid degradation by anaerobic bacteria. Annu Rev Biochem 50, 23–40, doi: 10.1146/annurev.bi.50.070181.000323 (1981). [DOI] [PubMed] [Google Scholar]
- 23.Matthews ML et al. Chemoproteomic profiling and discovery of protein electrophiles in human cells. Nat Chem 9, 234–243, doi: 10.1038/nchem.2645 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keogh D et al. Enterococcal Metabolite Cues Facilitate Interspecies Niche Modulation and Polymicrobial Infection. Cell Host Microbe 20, 493–503, doi: 10.1016/j.chom.2016.09.004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundermann AJ et al. Whole Genome Sequencing Surveillance and Machine Learning of the Electronic Health Record for Enhanced Healthcare Outbreak Detection. Clin Infect Dis, doi: 10.1093/cid/ciab946 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryan NC et al. Genomic and Functional Characterization of Enterococcus faecalis Isolates Recovered From the International Space Station and Their Potential for Pathogenicity. Front Microbiol 11, 515319, doi: 10.3389/fmicb.2020.515319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deibel RH Utilization of arginine as an energy source for the growth of Streptococcus faecalis. J Bacteriol 87, 988–992, doi: 10.1128/jb.87.5.988-992.1964 (1964). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fishbein SR et al. Multi-omics investigation of Clostridioides difficile-colonized patients reveals pathogen and commensal correlates of C. difficile pathogenesis. Elife 11, doi: 10.7554/eLife.72801 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karasawa T, Maegawa T, Nojiri T, Yamakawa K & Nakamura S Effect of arginine on toxin production by Clostridium difficile in defined medium. Microbiol Immunol 41, 581–585, doi: 10.1111/j.1348-0421.1997.tb01895.x (1997). [DOI] [PubMed] [Google Scholar]
- 30.Fredrick CM, Lin G & Johnson EA Regulation of Botulinum Neurotoxin Synthesis and Toxin Complex Formation by Arginine and Glucose in Clostridium botulinum ATCC 3502. Appl Environ Microbiol 83, doi: 10.1128/AEM.00642-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bushman FD et al. Multi-omic Analysis of the Interaction between Clostridioides difficile Infection and Pediatric Inflammatory Bowel Disease. Cell Host Microbe, doi: 10.1016/j.chom.2020.07.020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keith JW et al. Impact of Antibiotic-Resistant Bacteria on Immune Activation and Clostridioides difficile Infection in the Mouse Intestine. Infect Immun 88, doi: 10.1128/IAI.00362-19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesniak NA et al. The Gut Bacterial Community Potentiates Clostridioides difficile Infection Severity. mBio 13, e0118322, doi: 10.1128/mbio.01183-22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Girinathan BP et al. In vivo commensal control of Clostridioides difficile virulence. Cell Host Microbe 29, 1693–1708.e1697, doi: 10.1016/j.chom.2021.09.007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirose Y et al. Streptococcus pyogenes upregulates arginine catabolism to exert its pathogenesis on the skin surface. Cell Rep 34, 108924, doi: 10.1016/j.celrep.2021.108924 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 36.Dale JL et al. Comprehensive Functional Analysis of the Enterococcus faecalis Core Genome Using an Ordered, Sequence-Defined Collection of Insertional Mutations in Strain OG1RF. mSystems 3, doi: 10.1128/mSystems.00062-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theriot CM et al. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2, 326–334, doi: 10.4161/gmic.19142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar L, Cox CR & Sarkar SK Matrix metalloprotease-1 inhibits and disrupts Enterococcus faecalis biofilms. PLoS One 14, e0210218, doi: 10.1371/journal.pone.0210218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bloedt K, Riecker M, Poppert S & Wellinghausen N Evaluation of new selective culture media and a rapid fluorescence in situ hybridization assay for identification of Clostridium difficile from stool samples. J Med Microbiol 58, 874–877, doi: 10.1099/jmm.0.009811-0 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Wellinghausen N, Bartel M, Essig A & Poppert S Rapid identification of clinically relevant Enterococcus species by fluorescence in situ hybridization. J Clin Microbiol 45, 3424–3426, doi: 10.1128/JCM.00861-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knippel RJ et al. Heme sensing and detoxification by HatRT contributes to pathogenesis during Clostridium difficile infection. PLoS Pathog 14, e1007486, doi: 10.1371/journal.ppat.1007486 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixon P VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927–930, doi: 10.1111/j.1654-1103.2003.tb02228.x (2003). [DOI] [Google Scholar]
- 43.Calle ML, Urrea V, Boulesteix AL & Malats N AUC-RF: a new strategy for genomic profiling with random forest. Hum Hered 72, 121–132, doi: 10.1159/000330778 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Hankin JA, Barkley RM & Murphy RC Sublimation as a method of matrix application for mass spectrometric imaging. J Am Soc Mass Spectrom 18, 1646–1652, doi: 10.1016/j.jasms.2007.06.010 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas A, Charbonneau JL, Fournaise E & Chaurand P Sublimation of new matrix candidates for high spatial resolution imaging mass spectrometry of lipids: enhanced information in both positive and negative polarities after 1,5-diaminonapthalene deposition. Anal Chem 84, 2048–2054, doi: 10.1021/ac2033547 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Yang J & Caprioli RM Matrix sublimation/recrystallization for imaging proteins by mass spectrometry at high spatial resolution. Anal Chem 83, 5728–5734, doi: 10.1021/ac200998a (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prentice BM et al. Dynamic Range Expansion by Gas-Phase Ion Fractionation and Enrichment for Imaging Mass Spectrometry. Anal Chem 92, 13092–13100, doi: 10.1021/acs.analchem.0c02121 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675, doi: 10.1038/nmeth.2089 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson S, Calos M, Myers A & Self WT Analysis of proline reduction in the nosocomial pathogen Clostridium difficile. J Bacteriol 188, 8487–8495, doi: 10.1128/JB.01370-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans DR et al. Systematic detection of horizontal gene transfer across genera among multidrug-resistant bacteria in a single hospital. Elife 9, doi: 10.7554/eLife.53886 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069, doi: 10.1093/bioinformatics/btu153 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504, doi: 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bryan EM, Bae T, Kleerebezem M & Dunny GM Improved vectors for nisin-controlled expression in gram-positive bacteria. Plasmid 44, 183–190, doi: 10.1006/plas.2000.1484 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Chilambi GS et al. Evolution of vancomycin-resistant Enterococcus faecium during colonization and infection in immunocompromised pediatric patients. Proc Natl Acad Sci U S A 117, 11703–11714, doi: 10.1073/pnas.1917130117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Page AJ et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693, doi: 10.1093/bioinformatics/btv421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Obeid JS et al. Procurement of shared data instruments for Research Electronic Data Capture (REDCap). Journal of biomedical informatics 46, 259–265, doi: 10.1016/j.jbi.2012.10.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stabler RA et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10, R102, doi: 10.1186/gb-2009-10-9-r102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carter GP et al. Defining the Roles of TcdA and TcdB in Localized Gastrointestinal Disease, Systemic Organ Damage, and the Host Response during Clostridium difficile Infections. mBio 6, e00551, doi: 10.1128/mBio.00551-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanahan D Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166, 557–580, doi: 10.1016/s0022-2836(83)80284-8 (1983). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RNA sequencing data and processed data are deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE165751. MGE clusters identified in Extended Figure 2c are available in Supplementary Table 4. Conservation of arc genes performed on metagenomic reads deposited under BioProject accession number PRJNA74826228. Conservation of arc genes among enterococci performed on dataset deposited under BioProject ID: PRJNA58716125,26. All other data are available in the main text or supplementary materials.