

Abstract
Toll-like receptors (TLRs) and CD40-related signaling pathways represent critical bridges between innate and adaptive immune responses. Here, an immunotherapy regimen that enables co-stimulation of TLR7/8- and CD40-mediated pathways is developed. TLR7/8 agonist resiquimod (R848) derived amino lipids, RAL1 and RAL2, are synthesized and formulated into RAL-derived lipid nanoparticles (RAL-LNPs). The RAL2-LNPs show efficient CD40 mRNA delivery to DCs both in vitro (90.8 ± 2.7%) and in vivo (61.3 ± 16.4%). When combined with agonistic anti-CD40 antibody, this approach can produce effective antitumor activities in mouse melanoma tumor models, thereby suppressing tumor growth, prolonging mouse survival, and establishing antitumor memory immunity. Overall, RAL2-LNPs provide a novel platform toward cancer immunotherapy by integrating innate and adaptive immunity.
Keywords: CD40, immunotherapy, lipid nanoparticles, mRNA, resiquimod
1. Introduction
Cancer immunotherapy has emerged as a revolutionizing strategy to activate the host immune system against cancer cells through numerous approaches, such as checkpoint inhibitors,[1] cancer vaccines,[2] adoptive cellular therapy,[3] monoclonal antibodies,[4] and cytokine therapy.[5] Dendritic cells (DCs) are important members of the innate and adaptive immunity. As immature DCs capture and process tumor-associated antigens (TAA) from the tumor microenvironment, they mature and migrate to the spleen or lymph nodes for antigen presentation to naïve T cells.[6] However, a variety of tumor-associated factors affect the normal myeloid cell differentiation process and block DC maturation, thereby becoming a major obstacle to T cell priming.[7] To overcome this limitation, it is important to promote the maturation process of DCs to ensure an appropriate immune response cascade.
Effective DC maturation can be induced via various pathways. For example, targeting toll-like receptors (TLRs) has been shown to enhance DC maturation in mice and humans.[8,9] The recognition of resiquimod (R848) by endosomal TLR7/8 innate immune sensor triggers the production of pro-inflammatory cytokines and chemokines through the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon regulatory factors (IRFs). Consequently, activated DCs will increase the expression of major histocompatibility (MHC) molecules and co-stimulatory molecules, which are necessary for T cell recognition and activation.[10–12] Alternatively, agonistic CD40 ligation on matured DCs is also critical in triggering adequate T-cell- and B-cell-mediated immune responses.[13] Agonistic anti-CD40 antibodies (Abs) activate CD40 pathway in vivo and effectively induce the production of cytotoxic T lymphocytes (CTLs).[14] A synergistic effect exists when combining TLR agonists with anti-CD40 Abs, leading to enhanced antigen-specific immune responses through the production of pro-inflammatory cytokines and exponential expansion of antigen-specific T cells.[15–18] In particular, concomitant delivery of TLR7 and CD40 agonists has been shown to stimulate a CD8+ T cell response 10–20-fold greater than the use of either agonist alone.[15]
Although anti-CD40 Abs have shown great antitumor potential, low CD40 expressions in tumor-infiltrating DCs present a limiting factor in eliciting CTL immune responses.[19] In vitro transcribed mRNA (IVT mRNA) resembles endogenous mRNA and holds prominent prospect for inducing high levels of protein expression.[20,21] In order for IVT mRNA to function, a safe and effective delivery system is required to protect the nucleotides from degradation and allow efficient cellular uptake.[22–24] Among the various materials used for mRNA delivery, lipid nanoparticles (LNPs) have undergone extensive investigation and have successfully entered the clinic to combat the coronavirus disease 2019 (COVID-19).[25,26] We hypothesize that the delivery of CD40 mRNAs through LNPs may be a promising strategy to transiently express CD40 proteins in DCs. This can enhance the immunotherapeutic effects of agonistic anti-CD40 antibody, thereby inducing heightened T cell response and the development of immunological memory.
To combine the TLR and CD40 signaling pathways, we designed and synthesized R848-derived amino lipids (RAL) and formulated them into lipid nanoparticles (RAL-LNPs) for CD40 mRNA delivery to DCs. Subsequently, we injected anti-CD40 Abs to boost CD40 activation on DCs and promote their maturation (Figure 1B). Our results showed that RAL2-LNPs can effectively deliver CD40 mRNA to DCs both in vitro and in vivo, thus significantly improving the tumor-specific immune response in mice. This study demonstrates the potential of integrating the innate and adaptive immunity to boost the antitumor response and develop sustained memory immunity.
Figure 1.
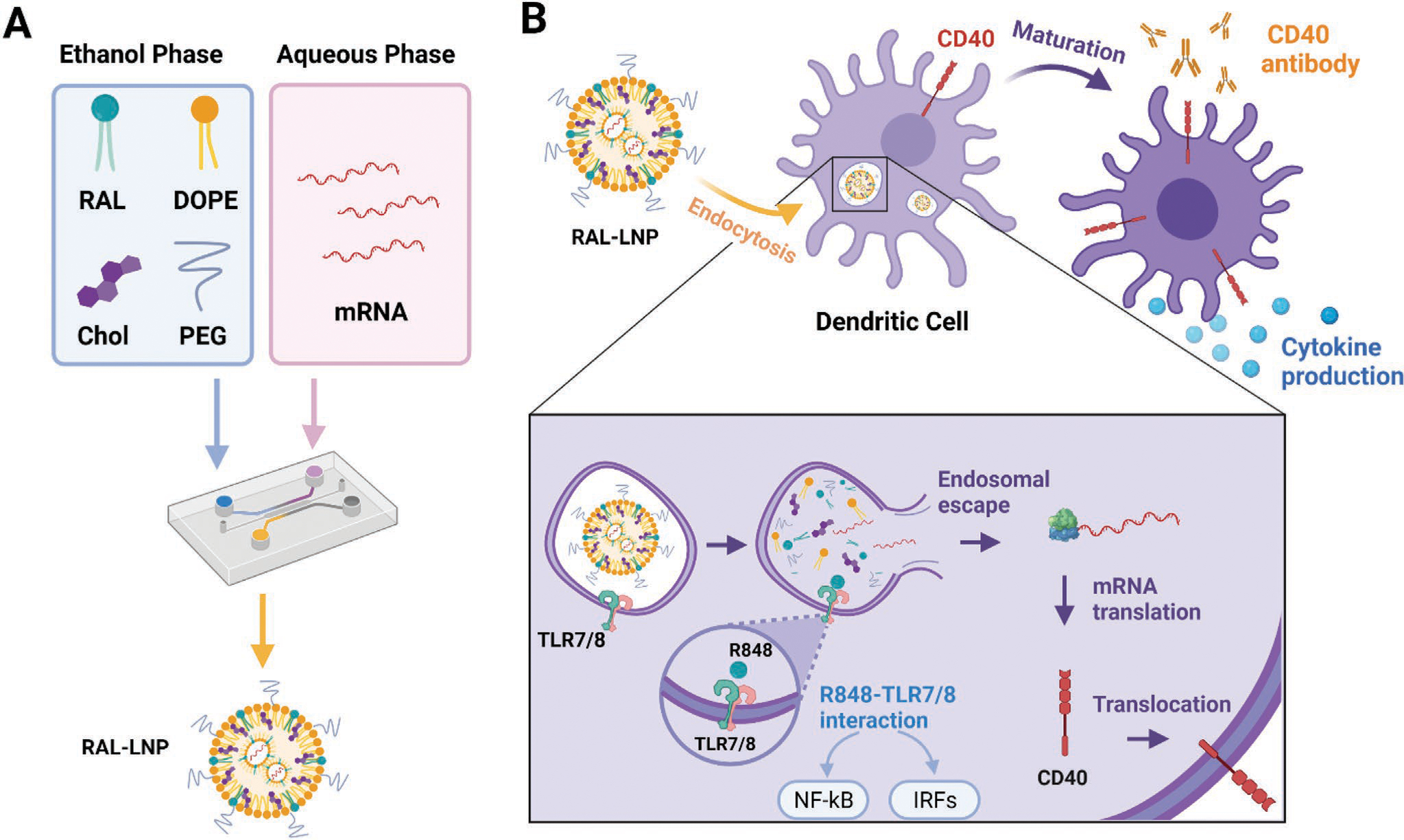
Construction of RAL-LNPs for mRNA delivery in dendritic cells (DCs). A) Illustration of RAL-LNPs components and formulation methods. B) Schematic illustration of RAL CD40-LNPs induced DC maturation. The RAL-LNP delivers CD40 mRNA to DCs, where the CD40 protein is produced in the cytoplasm and translocated onto the cell membrane. Agonistic anti-CD40 Abs activate CD40 expressed on the surface of DCs to induce subsequent antitumor immunity.
2. Results
We first synthesized amino lipid 1 and 2 for mRNA delivery, each containing two tertiary amino groups and three hydrophobic tails. The tertiary amino groups can be protonated in an acidic environment, inducing electrostatic interactions with the negatively charged phosphate groups in mRNA backbones and subsequent complication. While amino lipid 1 has linear hydrocarbon tails, amino lipid 2 has branched ester tails. Amino lipid 1 and 2 were then conjugated to the hydroxyl group on Tr-protected R848 through an ester linker to give RAL1 and RAL2, respectively (Figure 2A; Figure S1, Supporting Information). The esterification reaction was optimized by screening different coupling reagents, and Yamaguchi esterification with toluene as the solvent was found to be the optimal condition (Figure S2, Supporting Information).[27] The structures of RAL1 and RAL2 were confirmed by 1H NMR and high-resolution mass spectrometry (Supporting Information).
Figure 2.
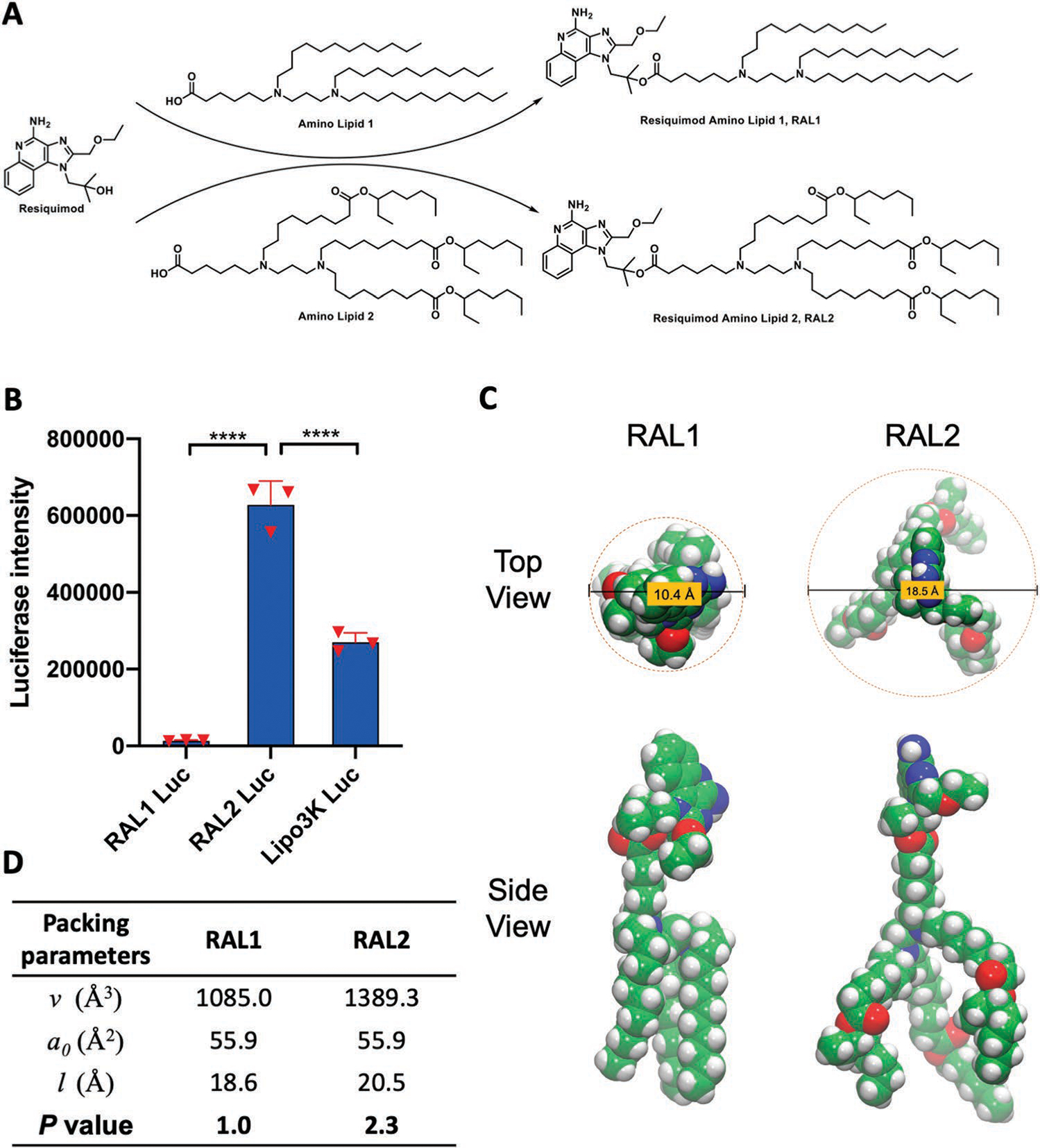
Structure and characterization of RAL-LNPs. A) The synthetic routes to RAL1 and RAL2. B) Delivery of Luc mRNA in JAWS II cells. Data in D are presented as the mean ± standard deviation (S.D.) (n = 3). C) Structural illustration of RAL1 and RAL2. D) Critical packing parameters calculated for RAL1 and RAL2 structures sampled from molecular dynamics simulation. Statistical significance in (B) was analyzed by one-way ANOVA with Dunnett’s multiple comparison test. ****p < 0.0001.
To investigate the mRNA delivery capabilities, RAL1 and RAL2 were formulated together with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), cholesterol (Chol), DMG-PEG2000 into RAL-LNPs to deliver firefly luciferase (Luc) mRNA.[28,29] The molar ratio of lipid components for the RAL-LNP formulations was RAL:DOPE:Chol:DMG-PEG2000 = 20:30:40:0.75, which was found as an appropriate molar ratio to produce robust LNP formulations and afford high mRNA delivery to cells (Figure 1A).[24,28] The RAL1 Luc-LNPs and RAL2 Luc-LNPs prepared by pipetting have a particle size ≈115 nm hydrodynamic diameter and polydispersity index (PDI) <0.2 (Figure S3A, Supporting Information). In addition, both particles are positively charged ≈20 mV, with a Luc mRNA encapsulation efficiency (EE%) of ≈87.30 ± 0.68% and 80.34 ± 2.69%, respectively (Figure S3B, Supporting Information). Next, we treated JAWS II cells, a DC cell line, with RAL-Luc LNPs at the dosage of 50 ng Luc mRNA per well in 96-well plates. The delivery efficiency of RAL2-LNPs was significantly higher than that of RAL1-LNPs and Lipofectamine 3000 (Lipo3K) (Figure 2B).
Next, we sought to understand structural factors responsible for RAL2’s high mRNA delivery efficiency by calculating its packing parameter (P). The P value derived from the geometrical shape of a single lipid molecule can predict the type of its self-assembled structure, which is a key factor governing its biological behavior and function.[30] Our calculation found that the P value of RAL2 (P = 2.3) is much higher compared to that of RAL1 (P = 1.0) (Figure 2C,D). In general, large P values (>1) that correspond to “a small head with large tails” would lead to reversed hexagonal (HII phase) or reversed spherical nanostructures, and the formation of the HII phase could in turn induce the rupture of endosomal membrane and the release of RNA payloads into the cytosol.[31,32] Taken together, our computational results suggest that RAL2 is likely to assemble into a HII phase structure that could facilitate intracellular mRNA delivery, while RAL1 forms a planar bilayer structure that is more stable in an acidic environment.[30] Thus, RAL2 was selected as a material for further characterizations and functional studies.
To investigate the efficiency of RAL2-LNPs in delivering CD40 mRNA to DCs, we treated JAWS II cells with RAL2-LNPs encapsulating CD40 mRNA (RAL2 CD40-LNPs), and quantified CD40 expression 24 h after treatment. We found that RAL2 CD40-LNPs induced 90.8 ± 2.7% CD40 expression on the JAWS II cells, which was significantly higher than that of the untreated and RAL2 Luc-LNPs control groups (Figure 3A). These results encouraged us to further characterize the physicochemical properties and internalization mechanisms of the RAL2 CD40-LNPs. The microfluidic device prepared RAL2 CD40-LNPs have a particle size of 97.5 ± 0.4 nm, which was slightly larger than that of free RAL2-LNPs (Figure 3B). In addition, RAL2 CD40-LNPs are positively charged ≈16 mV, with an mRNA EE% of 90.88 ± 0.35% (Figure 3C). Cryogenic transmission electron microscopy (cryo-TEM) image revealed the spherical morphology of RAL2 CD40-LNPs (Figure 3D). Internalization mechanisms regarding the endocytic pathway and endosomal escape of RAL2-LNPs were studied using Alexa-Fluor-647-labeled RNA. Endocytosis inhibitors 5-(N-Ethyl-N-isopropyl) amiloride (EIPA), chlorpromazine (CPZ), and methyl-β-cyclodextrin (MβCD) inhibit the macropinocytosis, clathrin, and caveolae endocytic pathways, respectively. JAWS II cells were first incubated with each endocytosis inhibitor before adding the RAL2-LNPs encapsulated with Alexa-Fluor-647-labeled RNA. We found that when incubated with MβCD, the cellular uptake of RAL2-LNPs was significantly reduced by ≈94%, indicating that caveolae-mediated endocytosis is the main pathway for JAWS II cells to uptake RAL2-LNPs (Figure 3E). The endosomal escape mechanism was examined using calcein, a cell-permeant dye that is usually entrapped in the endosomes.[33] Cells were treated with calcein alone or in combination with RAL2-LNPs. We observed diffused green-fluorescent calcein in the cells treated with calcein and RAL2-LNPs, indicating the disruption of endosomal membranes by the RAL2-LNPs. Subsequently, Alexa-Fluor-647-labeled RNA is released from endosomes (Figure 3F).
Figure 3.
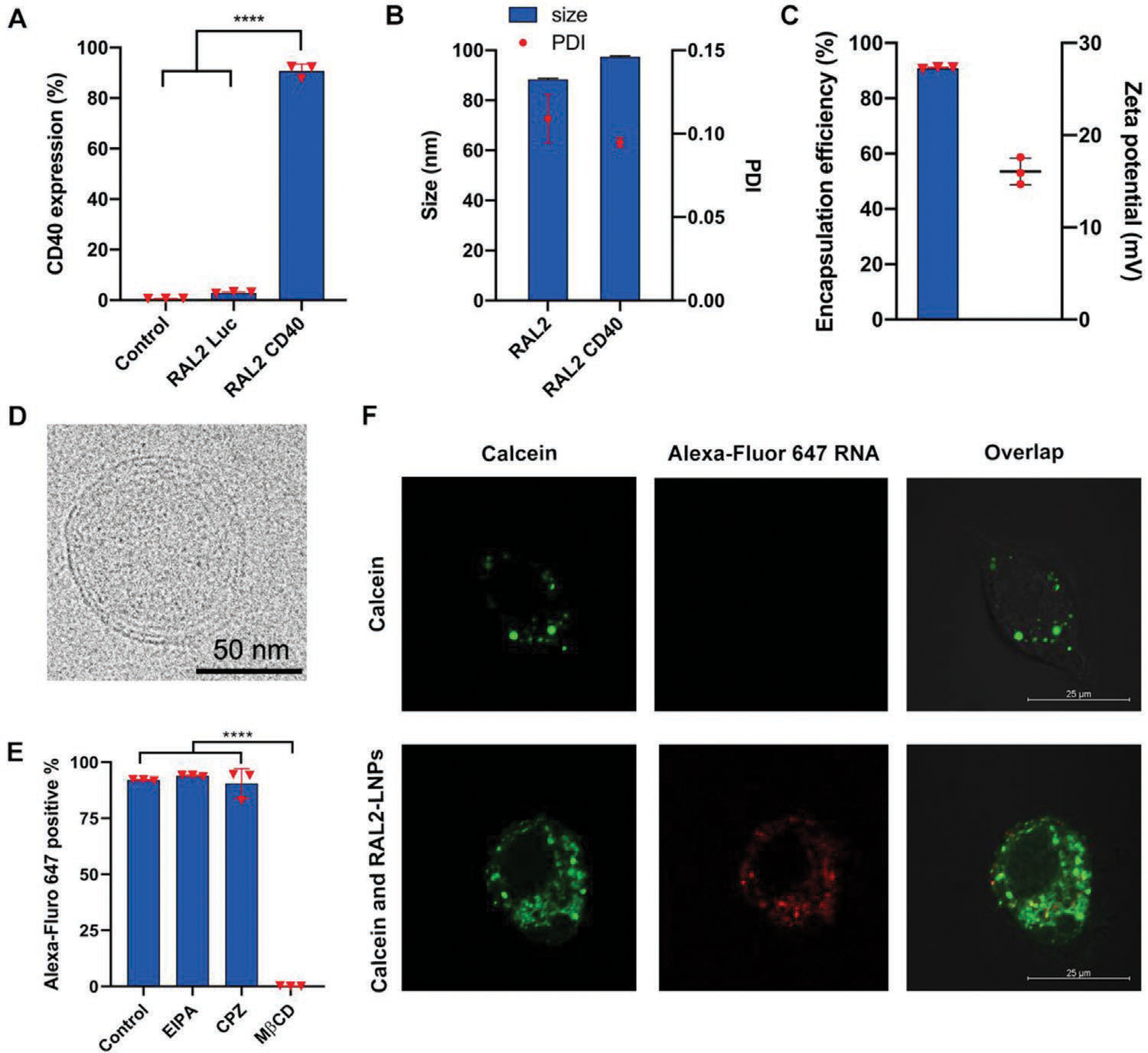
RAL2-CD40-LNP-mediated mRNA delivery in vitro. A) RAL2-LNPs-mediated CD40 mRNA delivery in JAWS II cells. B) Size and PDI of RAL2-LNPs carrying CD40 mRNAs prepared by microfluidic device. C) Encapsulation efficiency and zeta potential of RAL2-LNPs carrying CD40 mRNAs. D) Cryo-TEM image of RAL2 CD40-LNPs (scale bar = 50 nm). E) Cellular uptake of RAL2-LNPs encapsulating Alexa-Fluor-647-labeled RNAs in the presence of endocytosis inhibitors, EIPA, CPZ, and MβCD. F) Confocal images of JAWS II cells incubated with calcein alone or with RAL2-LNPs (scale bar = 25 μm). Data in (A), (B), and (E) are presented as the mean ± S.D. (n = 3). Statistical significance in (A) and (E) was analyzed by one-way ANOVA with Dunnett’s multiple comparison test. ****p < 0.0001.
To simulate the activation of DCs by RAL2-LNPs in vivo, we further studied the delivery and immune-stimulatory responses of RAL2 CD40-LNPs in murine bone marrow-derived DCs (BMDCs). As shown in Figure 4A, RAL2 CD40-LNPs significantly increased CD40 expression in BMDCs (64.3 ± 0.3%), fivefold of that of PBS (12.7 ± 0.2%) and 1.5-fold of RAL2-LNPs (38.6 ± 0.3%). The increased CD40 expression in RAL2-LNPs treated group is consistent with prior findings that R848 can induce the expression of CD40 in BMDCs.[34–36] Then we examined DC activation after co-administration of RAL2 CD40-LNPs with anti-CD40 antibodies (anti-CD40 Abs) by analyzing activation markers and cytokine production in primary DCs. We show in Figure 4B that this co-administration approach induced higher expression of DC activation markers, CD80, CD86, and MHC II. In addition, compared to the control and anti-CD40 Abs group, cytokine production of IL-12 and TNF-α in the primary DCs also significantly increased in the RAL2 CD40-LNPs + anti-CD40 Abs group (Figure 4C). Collectively, these data demonstrate that anti-CD40 Abs could work in synergy with RAL2 CD40-LNPs in triggering BMDC activation and maturation.
Figure 4.
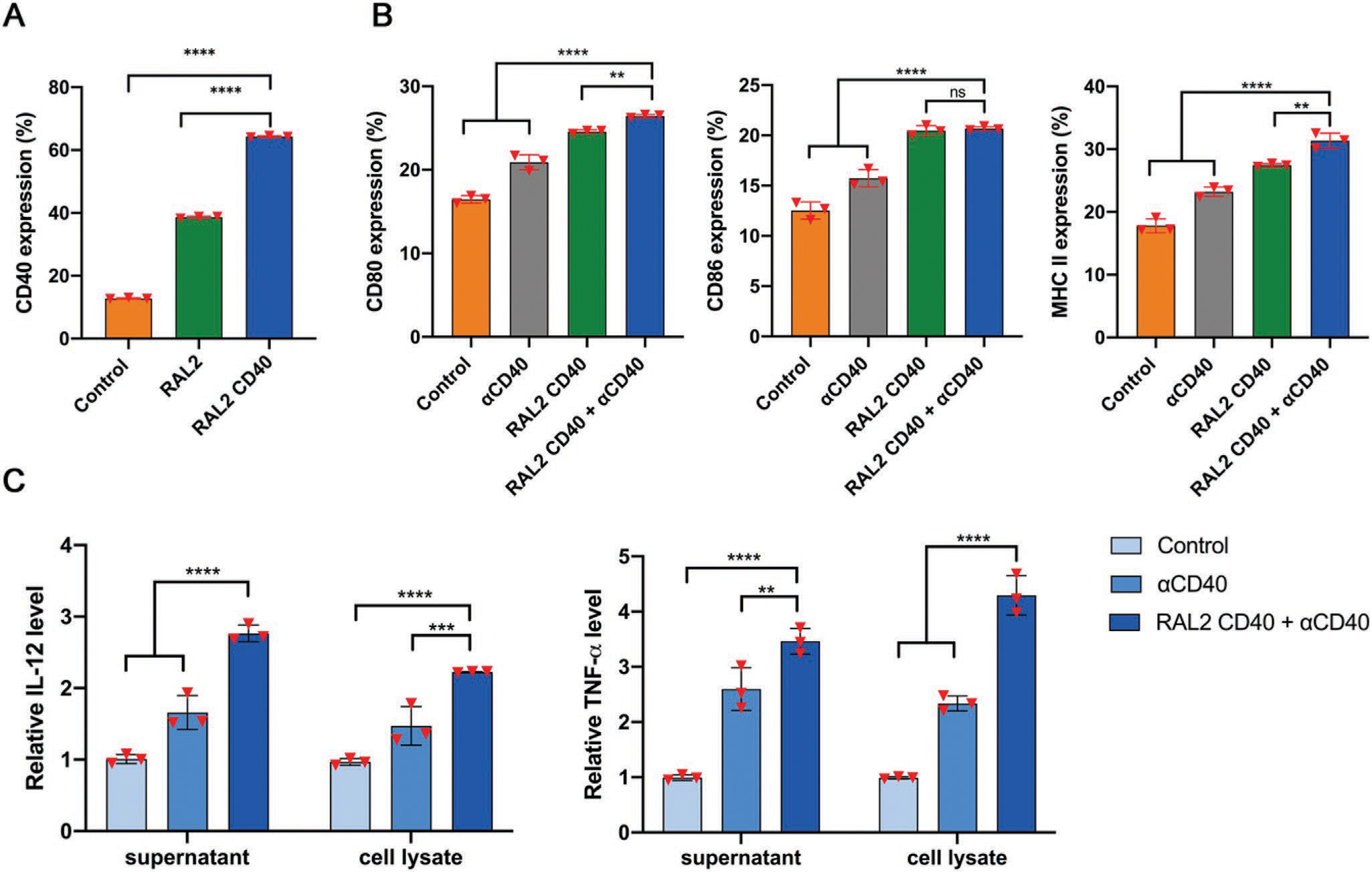
Delivery of CD40 mRNA by RAL2-LNPs mediates DC maturation and cytokine production in BMDCs ex vivo. A) RAL2 and RAL2-CD40 LNPs induced CD40 expression in BMDCs. B) Expression of DC activation markers, CD80, CD86, and MHC II on BMDCs treated with PBS, anti-CD40 Abs, RAL2 CD40-LNPs, or RAL2 CD40-LNPs + anti-CD40 Abs. C) Cytokine production of TNF-α and IL-1β from BMDCs ex vivo. Data in (A), (B), and (C) are presented as the mean ± S.D. (n = 3). Statistical significance in (A), (B), and (C) was analyzed by one-way ANOVA with Dunnett’s multiple comparison test. ns, not significant; **p < 0.01; ***p < 0.001; and ****p < 0.0001; ns, not significant.
Next, we examined the function of R848 on both mRNA delivery and DC activation using amino lipid 2 (AL2, RAL2 without R848 conjugation)-LNPs and RAL2-LNPs. While the size, PDI, and EE% are comparable between AL2 and RAL2-LNPs (Figure S4A,B, Supporting Information), RAL2 Luc-LNPs were fourfold more effective for mRNA delivery to JAWS II cells than AL2 Luc-LNPs (Figure S4C, Supporting Information). Furthermore, we studied the activation functions of AL2 CD40-LNPs and RAL2 CD40-LNPs to BMDCs. AL2 CD40-LNPs induced less CD80, CD86, and MHC II expression compared to RAL2 CD40-LNPs. These data indicate the importance of R848 conjugation on RAL2 for mRNA delivery and DC activation (Figure S4D, Supporting Information).
To assess the CD40 mRNA delivery efficacy of RAL2-LNPs in vivo, we intratumorally (i.t.) injected RAL2 CD40-LNPs or PBS in a subcutaneous B16-F10 mouse tumor model. The mice were euthanized 24 h after the injection, and tumors were harvested for further analysis. CD40 expression was quantified by flow cytometry analysis in the major immune cell populations, including CD4+ T cells, CD8+ T cells, DCs, and macrophages (gating strategy shown in Figure S5, Supporting Information). We observed significantly enhanced CD40 expression in DCs and CD8+ T cells (Figure 5A). Specifically, CD40 expression on DCs increased from 18.6 ± 4.9% (PBS treated group) to 61.3 ± 16.4% (RAL2 CD40-LNPs treated group), suggesting that RAL2-LNPs act as efficient vehicles for the delivery of CD40 mRNA in vivo.
Figure 5.
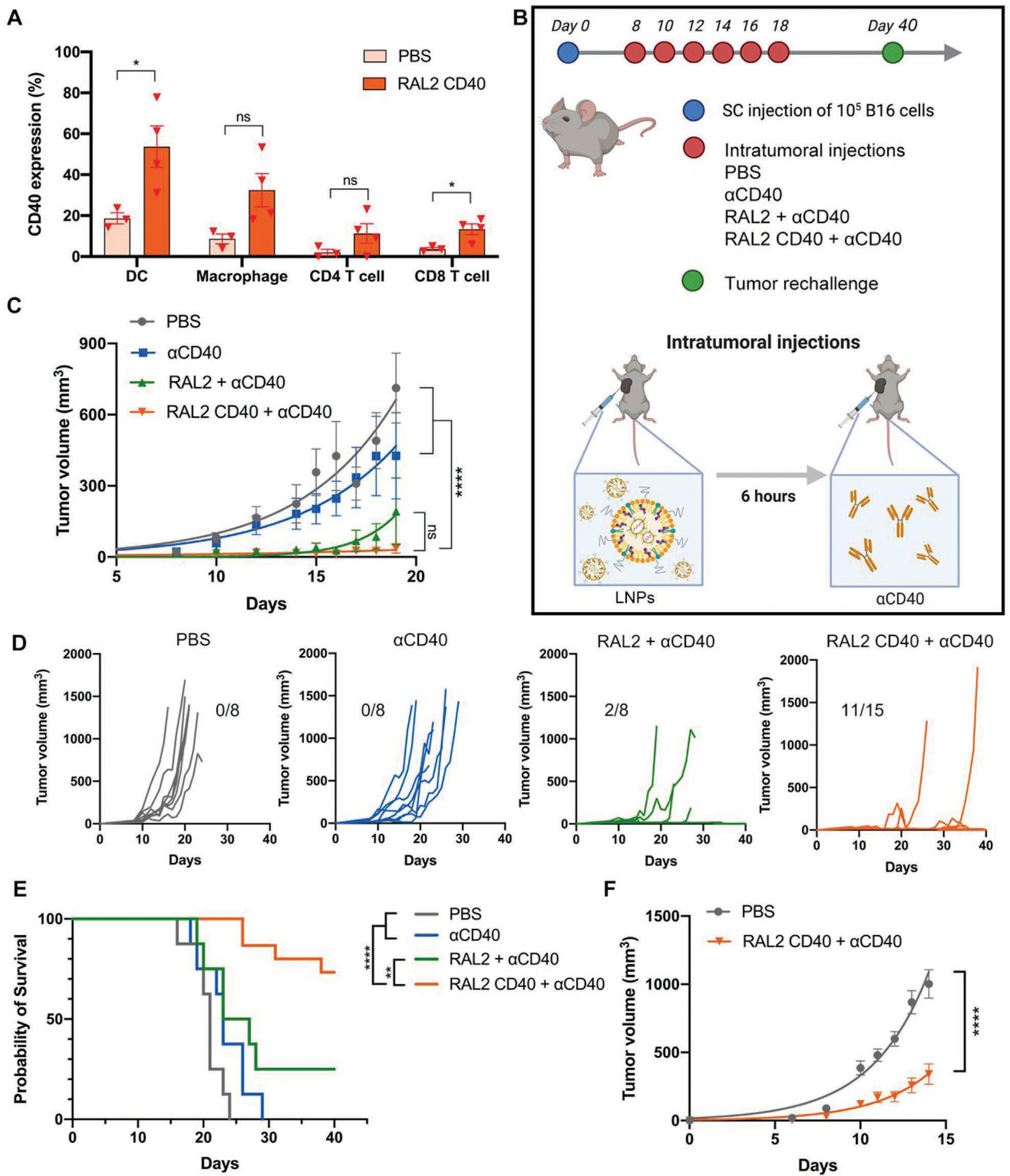
Delivery of CD40 mRNA by RAL2-LNPs inhibits B16-F10 melanoma tumor growth and induces memory immunity in mice. A) CD40 expression on the surface of dendritic cells after i.t. injection of RAL2 CD40-LNPs in subcutaneous B16-F10 tumors (n = 3,4). B) Schematic illustration of the treatment regimen in B16-F10 mouse tumor model via RAL2-LNPs delivering CD40 mRNA followed by the injection of agonistic anti-CD40 Abs (e.g., RAL2 CD40-LNPs + anti-CD40 Abs). C) Tumor volumes after six i.t. doses of PBS, anti-CD40 Abs (30 μg per mouse), RAL2-LNPs + anti-CD40 Abs, or RAL2 CD40-LNPs (10 μg mRNA per mouse) + anti-CD40 Abs. D) Tumor volumes of individual mice (n = 8–15) after tumor inoculation. E) Percentage survival after the initial tumor implantation on day 0 (n = 8–15). F) Tumor size of untreated B16-F10 tumors after tumor rechallenge on the opposite flank in cured mice on day 40 (n = 11). Data in (A), (C), and (F) are presented as the mean ± standard error of the mean (S.E.M.). Statistical significance in (A) was analyzed by unpaired two-tailed Students’ t-test. Statistical significance in (C) and (F) was analyzed by two-way ANOVA. Statistical significance in (E) was analyzed by the log-rank (Mantel–Cox) test. **p < 0.01; ****p < 0.0001; ns, not significant.
Given the efficient delivery of RAL2-LNPs to DCs both in vitro and in vivo, we tested the antitumor effects of RAL2 CD40-LNPs in B16-F10 tumor-bearing mice. Mice were treated every other day with i.t. injection of RAL2 CD40-LNPs containing 8 μg CD40 mRNA, followed by i.t. injection of 30 μg anti-CD40 Abs. A total of six treatments were conducted (Figure 5B). Four groups (n = 8–15) were included in the study: PBS, anti-CD40 Abs, RAL2-LNPs + anti-CD40 Abs, and RAL2 CD40-LNPs + anti-CD40 Abs group. Tumor growth was monitored every other day (Figure 5C,D). Compared to the other three groups, RAL2 CD40-LNPs + anti-CD40 Abs treatment showed slower tumor progression (Figure 5C) and higher survival rate (Figure 5E), which indicates that both TLR7/8 stimulation and increased CD40 expression is responsible for boosting the antitumor effect of anti-CD40 Abs. More importantly, this treatment resulted in a 70% complete tumor response. To study the potential memory immunity, we rechallenged the cured mice with B16-F10 tumor on the contralateral flank 2 weeks after no palpable tumors were observed. Compared to the naïve mice, the previously cured mice showed much slower tumor growth (Figure 5F), which indicates the induction of memory immunity against B16-F10 melanoma tumor.
Last, we evaluated the therapeutic effects of RAL2 CD40-LNPs + anti-CD40 Abs in the melanoma tumor model through systemic administration. RAL2 CD40-LNPs (30 μg mRNA) and 100 μg anti-CD40 Abs were administered to B16-F10 tumor-bearing mice through intravenous (i.v.) and intraperitoneal (i.p.) injection respectively, for a total of two treatments. As shown in Figure S6 (Supporting Information), this treatment regimen significantly slowed tumor progression and prolonged mice survival compared to the PBS group, suggesting that systemic administrations of this treatment regimen can also induce strong antitumor activities.
3. Discussion
DC-based immunotherapy is of growing attraction due to its unparalleled antigen presentation capacity, low toxicity, and potential to modulate other immune cells.[6] A number of DC-based cancer vaccines have been explored in clinical trials and revealed great therapeutic potential in patients.[37] However, due to the impaired migration of DCs to the lymph node and the transient activation state in the lymphoid tissues, subsequent adaptive immunity cannot be induced in the clinic.[16] To address this limitation, a DC-based immunotherapy that prolongs the activation state and antigen presentation abilities of DCs is needed.
In this study, RAL1 and RAL2, two novel ionizable lipids, were synthesized and formulated into RAL-LNPs with DOPE, Chol, and PEG-lipid. By comparing the in vitro Luc mRNA delivery to JAWS II cells by these two formulations, we speculate that the structure of amino lipid tails can affect mRNA delivery capacity. RAL2 with the biodegradable branched ester tails shows higher mRNA delivery efficiency compared to RAL1 with the linear hydrocarbon tails (Figure 2E). The branched-tail structure in RAL2 could increase the distance between lipids and facilitate the amine protonation process at endosomal pH, thereby allowing efficient mRNA escape from endosomes.[31,38] In addition, the ester linkers in RAL2 confers biocompatibility as they can be hydrolyzed into smaller fragments by esterase and enables rapid in vivo lipid clearance in mice following LNP administration.[39] Besides the chemical structures of RAL2, DOPE as a helper lipid is also critical for RAL2-LNPs-mediated nucleic acid delivery. The incorporation of DOPE in the LNP not only allows closer contact with the negatively charged phosphate groups of mRNA, but also improves the mRNA transfection efficiency as DOPE undergoes conformation change that facilitate endosomal escape at low pH, from a flexible bilayer to an inverse hexagonal structure.[30] We found that RAL2 Luc-LNPs without DOPE exhibited less uniformity in particle size (Figure S7A, Supporting Information) and lower mRNA EE% (Figure S7B, Supporting Information) compared to RAL2-LNPs with DOPE. Furthermore, the mRNA delivery efficiency of RAL2-LNPs with DOPE was 120-fold higher than those without DOPE in JAWS II cells (Figure S7C, Supporting Information), suggesting that DOPE is an important component in the RAL2-LNP formulations.
To promote DC maturation via TLR and CD40 co-stimulation, RAL-LNPs were used to deliver CD40 mRNA to DCs. After the RAL CD40-LNPs are taken up by the cells through the caveolae-mediated endocytosis, R848 can interact with TLR7/8 located on the endosome membrane and activate the TLR signaling pathway. On the other hand, the CD40 mRNA is translated into CD40 protein and translocated onto the DC membrane, agonistic anti-CD40 antibody is applied to allow further CD40 activation. Consistent with previous studies, the TLR and CD40 signaling act synergistically to increase cytokine production, DC maturation and MHC II mediated antigen presentation.[16,40] We found that co-stimulating the TLR7/8 and CD40 pathways demonstrated strong antitumor efficacy in the highly aggressive B16-F10 melanoma mice tumor model, with attenuated tumor growth and prolonged mice survival. One of the main tasks of cancer immunotherapy is to promote strong tumor-antigen specific immune responses. Our study showed that the combination therapy of RAL2 CD40-LNPs with anti-CD40 Abs not only induces anti-tumor immunity resulting in 70% complete tumor removal, but also inhibits the growth of the contralateral tumor. However, this immunity is not strong enough to fully eliminate tumor metastasis on the untreated side. Treatments in combination with small molecules, cytokines,[41] and checkpoint inhibitors[42,43] have been exploited to enhance CD40-mediated memory immunity, which could be a complementary strategy that merits further investigations.
Together, our results show that lipidized R848-based LNPs can serve as an effective delivery platform for CD40 mRNA. In addition, dual activation of TLR7/8 and CD40 signaling pathways initiates protective memory immunity and achieves optimal antitumor efficacy in melanoma tumor-bearing mice. We believe that with further optimization of dosage and timing, the combination of CD40 and R848 described in this work will show great promise in future clinical studies. Importantly, the coordinated stimulation of innate and adaptive signaling pathways chosen in this study only represents one of the many possible combinations. Thus, combining novel mRNA delivery platform with a wide array of immunomodulatory molecules represents a general approach toward novel antitumor drug discovery.
4. Experimental Section
Materials, Reagents, and Cells:
All chemicals and solvents were purchased from Sigma–Aldrich (St. Louis, MO, USA) unless otherwise listed. Resiquimod (R848) (InvivoGen, Cat: tlrl-r848). DOPE (Avanti Polar Lipids Inc, Cat: 850275P). DMG-PEG2000 (NOF America Corporation). JAWS ΙΙ (ATCC CRL11904) cells were purchased from ATCC (Manassas, VA, USA), and cultured in α-MEM culture medium (Thermo Fisher, Cat: 12571063) containing 20% fetal bovine serum (Invitrogen), 2 × 10−3 m L-Glutamine (Thermo Fisher, Cat: 25 030 081), 1 × 10−3 m sodium pyruvate, and 5 ng mL−1 murine GM-CSF (Sigma–Aldrich, Cat: GF026). B16-F10 cells were kind gifts from Dr. Jianhua Yu and cultured in Dulbecco’s modified Eagle medium (DMEM) culture medium containing 10% fetal bovine serum. Antibodies used for in vivo: InVivoPlus anti-mouse CD40 monoclonal antibody (BioXCell, Cat: BP0016–2). Antibodies used for FACS: anti-CD45 APC (30-F11) (Thermo Fisher, Cat: 17–0451-82), anti-CD3 PE (145–2C11) (BD Pharmingen, Cat: 553 063), anti-CD4 Pacific Blue (RM4–5) (Thermo Fisher Cat: MCD0428), anti-CD8 APC-efluor 780 (53–4.7) (Thermo Fisher, Cat: 47–0081-82), anti-CD19 Alexa-Fluor 700 (eBio1D3(1D3)) (Thermo Fisher, Cat: 56–0193-82), anti-CD11b Pacific Blue (M1/70.15) (Thermo Fisher, Cat: RM2828), anti-F4/80 PE-eFluor 610 (BM8) (Thermo Fisher, Cat: 61–4801-82), anti-CD11c PE-Cyanine7 (N418) (Thermo Fisher, Cat: 25–0114-82), anti-CD40 FITC (HM40–3) (Thermo Fisher, Cat: 11–0402-82), anti-CD80 FITC (B7–1) (Thermo Fisher, Cat: 11–0801-81), anti-CD86 PE (B7–2) (Thermo Fisher, Cat: 12–0862-81), anti-CD86 FITC (B7–2) (Thermo Fisher, Cat: 11–0862-81), and anti-MHC II APC (AF6–120.1) (Thermo Fisher, Cat: 17–5320-80). Antibodies used for ELISA: rat anti-mouse IL-12 p70 (C17–8) (Thermo Fisher, Cat: MM121), and rat anti-mouse TNFα (1F3F3D4) (Thermo Fisher, Cat: 14–7325-81), all primers were ordered from Eurofins Genomics.
CD40 mRNA Preparation:
Experimental details for mRNA sequences can be found in the Supporting Information. The cDNA sequence encoding CD40 amino acids was obtained from Ensembl (Reference sequence: ENSMUST00000017799). Linearized dsDNA sequence was ordered from IDT and incorporate into pUC19 vector through Hifi assembly using NEBuilder HiFi DNA Assembly purchased from NEB (Ipswich, MA, USA). The correct plasmid was identified by Sanger sequencing and an 867 bp long genomic DNA template was amplified by polymerase chain reaction (PCR). In vitro transcription of uncapped CD40 mRNA was synthesized using AmpliScribe T7-Flash Transcription Kit purchased from Lucigen (Middleton, WI, USA). Cap structure was added using the Vaccinia Capping System and Cap 2′-O-Methyltransferase system, both purchased from NEB (Ipswich, MA, USA). All mRNAs were purified with RNA Clean & Concentrator-25 purchased from Zymo Research (Irvine, CA, USA).
Nanoparticle Formulation Characterization:
mRNA-encapsulated LNPs were formulated with RALs and helper lipids 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), Chol, and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG2000) at a molar ratio of 20/30/40/0.75. All lipid materials were dissolved to the desired concentration in ethanol, and mRNA in citrate buffer. In vitro mRNA encapsulated LNPs were prepared by pipetting, and in vivo mRNA encapsulated LNPs were prepared by the Precision NanoSystems microfluidic device (Vancouver, BC, Canada).[33,44–47] For in vitro studies, the newly prepared mRNA encapsulated LNPs were immediately added to the cells; For in vivo studies, the mRNA encapsulated LNPs were dialyzed in PBS buffer for 80 min using Slide-A-Lyzer Dialysis Cassettes purchased from Life Technologies (Grand Island, NY, USA) before administration. The particle size and zeta potential of RAL2-LNPs were determined using Malvern Zetasizer NanoZS (Malvern, UK). The mRNA encapsulation efficacy (EE%) of the mRNA NPs were determined by Ribogreen Assay as previously described.[28]
Packing Parameter (P) Calculation:
The dimensionless packing parameter P of an amphiphilic molecule was derived from its molecular volume (v), head area (a), and tail length (l) as P = v/(la). 2D structures of both RAL1 and RAL2 molecules were converted to 3D coordinates using OpenBabel.[48] The generated 3D structures were optimized with the MMFF94 force field[49] using Avogadro[49,50] To generate more physically relevant conformations, all-atom molecular dynamics (MD) simulation was performed on a planar bilayer membrane model containing both RAL1 and RAL2 using NAMD 2.13.[51] The membrane model consists of 200 lipid molecules, 4 RAL1 and 4 RAL2. To accelerate conformational sampling, the highly mobile membrane mimetic (HMMM) model with short-tailed phospholipids and an organic solvent membrane core was used to represent the lipid bilayer.[52] The bilayer model was assembled with the CHARMM-GUI HMMM Builder.[53] The CHARMM36 lipid force field was used for lipids,[54] and the force field parameters for RAL1 and RAL2 were derived using SwissParam[55] The system was first energy-minimized using the conjugate gradient algorithm for 2000 steps, followed by heating to 300 K within 5 ns using a 1 fs time step in the constant particle number, pressure, and temperature (NPT) ensemble with a semi-isotropic pressure coupling that allows the z-axis to expand and contract independently from the x–y plane. This was followed by 100 ns of simulation with a 2 fs time step. Langevin dynamics [56] was used to control the temperature at 300 K with a damping coefficient of 2 ps−1, and the Langevin piston method [57] to keep the pressure at 1 atm. Long-range electrostatic interactions were calculated using the Particle-Mesh Ewald summation with a cut off at 12 Å.[58] Van der Waals (vdW) interactions were truncated via a force-based switching function with a switching distance of 8 Å and a cutoff distance of 12 Å. Bonds to hydrogen atoms were constrained using the SHAKE algorithm.[59] RAL1 and RAL2 conformers were collected every 10 ps from the last 10 ns MD trajectory, resulting in 4000 conformers for each molecule. These conformers were clustered with a 1.0 Å root-mean-square-deviation (RMSD) cutoff. The conformer corresponding to the center of the most populated cluster was selected for subsequent P value calculation. For each RAL1/RAL2 structure, a was measured as the cross-section area of the resiquimod group, and l was measured as the distance between the center of mass (COM) of the ester linker and the three terminal methyl groups. The vdW volume was calculated using an approximate method termed Atomic and Bond Contributions of vdW volume (VABC) as VvdW = Σall-atom-contributions − 5.92NB − 14.7RA − 3.8RNR, where NB is the number of bonds, RA is the number of aromatic rings, and RNA is the number of nonaromatic rings.[60]
In Vitro Delivery of CD40 mRNA to DCs:
To test the delivery of CD40 mRNA to JAWS II cells, FITC-labeled anti-CD40 monoclonal antibody was used to detect cell CD40 expression levels by flow cytometry. Briefly, 1 × 105 JAWS II cells were plated in a 24-well plate and incubated at 37 °C in 5% CO2 incubator for 24 h. Then cells were treated with RAL2-LNPs containing 300 ng Pseudouridine (ψ) modified mRNAs for 18 h. Afterward, the cells were washed and incubated with FITC-labeled anti-CD40 monoclonal antibody (clone HM40–3, 1:70 dilution) in cold PBS containing 1% FBS for 30 min at 4 °C. The cellular uptake was then analyzed on a BD LSR Fortessa or BD LSR II flow cytometer. To study the endocytic pathway of the RAL2-LNPs, cells were treated with different endocytosis inhibitors 30 min before adding RAL2-LNPs containing Firefly Luc mRNA and Alexa-Fluor-647-labeled RNA at a 1:1 weight ratio. After 3 h of co-incubation, the cellular uptake was analyzed on a BD LSR Fortessa or BD LSR II flow cytometer.
Endosome Escape:
A total of 3 × 104 JAWS II cells in 300 μl medium were plated in an imaging dish (Ibidi) and incubated at 37 °C in 5% CO2 incubator for 24 h. Calcein (final concentration 150 μg mL−1) was added to the cells alone or in combination with RAL2-LNPs containing Firefly Luc mRNA and Alexa-Fluor-647-labeled RNA at a 1:1 weight ratio. After 2 h of co-incubation, the cells were imaged using Nikon A1R Live Cell confocal laser scanning microscope (Melville, NY, USA) and the acquired images were analyzed with NIS-Elements AR (Version 5.20.00.)
Isolation of Monocytes from Murine Bone Marrow and Induction of Bone-Marrow-Derived DCs (BMDCs):
C57BL/6J mice were euthanized and the tibias and femurs were removed under sterile conditions. Monocytes were then isolated from the bone marrow cell suspension, suspended in RPMI-1640 medium supplemented with 1% Pen-Strep, 10% FBS, 50 ng ml−1 GM-CSF, and 50 ng mL−1 IL-4, and incubated at 37 °C in 5% CO2 incubator for 8 days to generate immature DCs. The immature DCs were then isolated using CD11c MicroBeads UltraPure, mouse kit (Miltenyi Biotec, Cat: 130–108-338) following the manufacturer’s instructions. 0.8 × 106 immature BMDCs were seeded in a 6-well plate in 1.6 mL RPMI-1640 medium supplemented with 25 ng mL−1 GM-CSF, 25 ng mL−1 IL4 for 24 h before treatment. For treatment, 125 μL of formulated RAL2 and RAL2-CD40 LNPs (RAL2-CD40 mRNA concentration: 0.01 mg mL−1) to each well and cultured for 12 h before adding anti-mouse CD40 monoclonal antibody (BioXCell, Cat: BP0016–2). The cells were cultured for an additional 12 h and stained with fluorescent labeled anti-CD40 antibodies or fluorescent labeled DC activation markers (CD80, CD86, MHCII) for 30 min at 4 °C before flow cytometry. IL-12 and TNF-α cytokine production were detected by ELISA.
Flow Cytometry Analysis of TIL Cell Populations:
Female C57BL/6J mice were subcutaneously (s.c.) inoculated with 1 × 105 B16-F10 cells in the right flank and were i.t. injected with PBS/RAL2-LNPs (containing 20 μg mRNA) after the tumor volumes reached 150 mm3. Mice were euthanized after 18 h and the tumors were harvested. A Tumor Dissociation Kit purchased from MACS (San Diego, CA, USA) was used to dissociate the tumor tissue into suspension cells. Peripheral blood stem cells (PBSCs) were isolated by gradient centrifugation using Ficoll–Paque PLUS (GE Healthcare Life Sciences, PA, USA). For T cell, APC cell, and B cell tetramer staining, 2 × 106 cells from tumor digests or purified lymphocytes were stained with corresponding antibodies for 30 min at 4 °C. The following fluorochrome-conjugate anti-mouse mAbs were used for flow cytometry: CD45-APC, CD3-PE, CD4-Pacific blue, CD8− APC-780, CD11b-Pacific blue, CD11c-PE-Cy7, F4/80-APC-Cy7, CD19-Alexa-Fluor 700, and anti-CD40-FITC. All antibodies were purchased from Invitrogen.
In Vivo Antitumor Efficacy of CD40 mRNA NPs:
All animal experiments were carried out under the guidelines for Care and Use of Laboratory Animals and were approved by IACUC of The Ohio State University (approval ID: 2014A00000106-R2). Female C57BL/6J mice were s.c. inoculated with 1 × 105 B16-F10 cells in the right flank. Tumor size was monitored using a digital caliper every 2 days and calculated as the volume (length × width × width/2). For local administrations, mice were treated every other day for a total of six treatments: i.t. injection of 50 μl RAL2- or RAL2 CD40-LNPs (RAL2-CD40 mRNA concentration: 0.2 mg ml−1) followed by 30 μg anti-CD40 antibody (FGK4.5) dissolved in PBS 6 h later. After tumors were no longer palpable, mice were rested for 2 weeks before re-challenged with the same tumor line on the opposite flank. For systematic administrations, mice were treated with 150 μL RAL2-CD40 LNPs (RAL2-CD40 mRNA concentration: 0.2 mg ml−1) through i.v. injection and 100 μg anti-CD40 antibody through i.p. injection. Tumor sizes were measured every other day. The mice were euthanized when the tumor diameter exceeded 1.5 cm.
Statistical Analysis:
No collected experimental data were excluded for the quantitative analysis. Number of repeated times in each group and definition of error bars were described in the figure legend. Statistical significance was calculated using unpaired, two-tailed Student’s t tests (two groups) or one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison test (multiple groups). Tumor volumes were analyzed using two-way ANOVA with repeated measurements, and survival curves were analyzed using the log-rank test. The P values were shown in the figures and specific statistical methods were described in the figure legends. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 were considered statistically significant. Statistical analyses were conducted on GraphPad Prism 8.
Supplementary Material
Acknowledgements
J.Y., Y.Z., and S.D. contributed equally to this work. Y.D. acknowledges the support from the Maximizing Investigators’ Research Award R35GM119679 and R35GM144117 from the National Institute of General Medical Sciences as well as the start-up fund from the College of Pharmacy at The Ohio State University. J.Y. acknowledges the support from the Professor Sylvan G. Frank Graduate Fellowship as well as the Presidential Fellowship. The authors acknowledge the use of the core facility provided by the Genomics Shared Resource (GSR) and Campus Microscopy & Imaging Facility (CMIF) at the Ohio State University. The authors acknowledge the use of biorender.com to prepare certain figures.
Footnotes
Conflict of Interest
Y.D. is a scientific advisory board member of Oncorus Inc, Arbor Biotechnologies, and FL85. Other authors have no competing interests to declare.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Jingyue Yan, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Yuebao Zhang, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Shi Du, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Xucheng Hou, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Wenqing Li, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Chunxi Zeng, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Chengxiang Zhang, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Jeffrey Cheng, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Binbin Deng, Center for Electron Microscopy and Analysis, The Ohio State University, Columbus, OH 43212, USA.
David W. McComb, Center for Electron Microscopy and Analysis, The Ohio State University, Columbus, OH 43212, USA Department of Materials Science and Engineering, The Ohio State University, Columbus, OH 43210, USA.
Weiyu Zhao, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Yonger Xue, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Diana D. Kang, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA
Xiaolin Cheng, Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA.
Yizhou Dong, Division of Pharmaceutics & Pharmacology, College of Pharmacy, The Ohio State University, Columbus, OH 43210, USA; Department of Radiation Oncology, Department of Biomedical Engineering, The Center for Clinical and Translational Science, The Comprehensive Cancer Center, Dorothy M. Davis Heart & Lung Research Institute, Center for Cancer Engineering, Center for Cancer Metabolism, Pelotonia Institute for Immune-Oncology, The Ohio State University, Columbus, OH 43210, USA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].Topalian SL, Taube JM, Anders RA, Pardoll DM, Nat. Rev. Cancer 2016, 16, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hu Z, Ott PA, Wu CJ, Nat. Rev. Immunol. 2018, 18, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME, Nat. Rev. Cancer 2008, 8, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Weiner LM, Surana R, Wang S, Nat. Rev. Immunol. 2010, 10, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Berraondo P, Sanmamed MF, Ochoa MC, Etxeberria I, Aznar MA, Pérez-Gracia JL, Rodríguez-Ruiz ME, Ponz-Sarvise M, Castañón E, Melero I, Br. J. Cancer 2019, 120, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bol KF, Schreibelt G, Gerritsen WR, de Vries IJM, Figdor CG, Clin. Cancer Res. 2016, 22, 1897. [DOI] [PubMed] [Google Scholar]
- [7].Rabinovich GA, Gabrilovich D, Sotomayor EM, Annu. Rev. Immunol. 2007, 25, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lahiri A, Das P, Chakravortty D, Vaccine 2008, 26, 6777. [DOI] [PubMed] [Google Scholar]
- [9].Tacken PJ, Zeelenberg IS, Cruz LJ, van Hout-Kuijer MA, van de Glind G, Fokkink RG, Lambeck AJA, Figdor CG, Blood 2011, 118, 6836. [DOI] [PubMed] [Google Scholar]
- [10].Nuhn L, Koker SD, Lint SV, Zhong Z, Catani JP, Combes F, Deswarte K, Li Y, Lambrecht BN, Lienenklaus S, Sanders NN, David SA, Tavernier J, Geest BGD, Adv. Mater. 2018, 30, 1803397. [DOI] [PubMed] [Google Scholar]
- [11].Schön MP, Schön M, Oncogene 2008, 27, 190. [DOI] [PubMed] [Google Scholar]
- [12].Islam MA, Rice J, Reesor E, Zope H, Tao W, Lim M, Ding J, Chen Y, Aduluso D, Zetter BR, Farokhzad OC, Shi J, Biomaterials 2021, 266, 120431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schjetne KW, Fredriksen AB, Bogen B, J. Immunol. 2007, 178, 4169. [DOI] [PubMed] [Google Scholar]
- [14].Piechutta M, Berghoff AS, ESMO Open 2019, 4, e000510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, Kedl RM, J. Exp. Med. 2004, 199, 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lapteva N, Seethammagari MR, Hanks BA, Jiang J, Levitt JM, Slawin KM, Spencer DM, Cancer Res. 2007, 67, 10528. [DOI] [PubMed] [Google Scholar]
- [17].Jain S, Chodisetti SB, Agrewala JN, PLoS One 2011, 6, e20651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ahonen CL, Wasiuk A, Fuse S, Turk MJ, Ernstoff MS, Suriawinata AA, Gorham JD, Kedl RM, Usherwood EJ, Noelle RJ, Blood 2008, 111, 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee GH, Askari A, Malietzis G, Bernardo D, Clark SK, Knight SC, Al-Hassi HO, Curr. Cancer Drug Targets 2014, 14, 610. [DOI] [PubMed] [Google Scholar]
- [20].Sahin U, Karikó K, Türeci Ö, Nat. Rev. Drug Discovery 2014, 13, 759. [DOI] [PubMed] [Google Scholar]
- [21].Pardi N, Hogan MJ, Porter FW, Weissman D, Nat. Rev. Drug Discovery 2018, 17, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hou X, Zaks T, Langer R, Dong Y, Nat. Rev. Mater. 2021, 6, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yan J, Kang DD, Dong Y, Biomater. Sci. 2021, 9, 6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li W, Zhang X, Zhang C, Yan J, Hou X, Du S, Zeng C, Zhao W, Deng B, McComb DW, Zhang Y, Kang DD, Li J, Carson WE, Dong Y, Nat. Commun. 2021, 12, 7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, et al. , N. Engl. J. Med. 2021, 384, 403.33378609 [Google Scholar]
- [26].Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Şahin U, Jansen KU, Gruber WC, Engl N J. Med. 2020, 383, 2603. [Google Scholar]
- [27].Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M, Bull. Chem. Soc. Jpn. 1979, 52, 1989. [Google Scholar]
- [28].Li B, Luo X, Deng B, Wang J, McComb DW, Shi Y, Gaensler KML, Tan X, Dunn AL, Kerlin BA, Dong Y, Nano Lett. 2015, 15, 8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang X, Zhao W, Nguyen GN, Zhang C, Zeng C, Yan J, Du S, Hou X, Li W, Jiang J, Deng B, McComb DW, Dorkin R, Shah A, Barrera L, Gregoire F, Singh M, Chen D, Sabatino DE, Dong Y, Sci. Adv. 2020, 6, eabc2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Godbey WT, in Biotechnol. Its Appl, 2nd ed. (Ed: Godbey WT), Academic Press, London, UK, 2022, pp. 287. [Google Scholar]
- [31].Buschmann MD, Carrasco MJ, Alishetty S, Paige M, Alameh MG, Weissman D, Vaccines 2021, 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Y, Sun C, Wang C, Jankovic KE, Dong Y, Chem. Rev. 2021, 121, 12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hou X, Zhang X, Zhao W, Zeng C, Deng B, McComb DW, Du S, Zhang C, Li W, Dong Y, Nat. Nanotechnol. 2020, 15, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang X, Yang Y, Jing L, Zhai W, Zhang H, Ma Q, Li C, Yan F, Cheng D, Zhang J, Ning Z, Shi H, Wang C, Zhao M, Dai J, Li Z, Ming J, Yu M, Wang H, Cheng H, Xiong H, Dong G, Front. Immunol. 2021, 12, 680068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Amiel E, Everts B, Freitas TC, King IL, Curtis JD, Pearce EL, Pearce EJ, J. Immunol. 2012, 189, 2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hargadon KM, Int. Rev. Immunol. 2016, 35, 85. [DOI] [PubMed] [Google Scholar]
- [37].Constantino J, Gomes C, Falcão A, Cruz MT, Neves BM, Transl. Res. 2016, 168, 74. [DOI] [PubMed] [Google Scholar]
- [38].Hajj KA, Ball RL, Deluty SB, Singh SR, Strelkova D, Knapp CM, Whitehead KA, Small 2019, 15, 1805097. [DOI] [PubMed] [Google Scholar]
- [39].Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, Lynn A, Bulychev A, McFadyen I, Chan J, Almarsson Ö, Stanton MG, Benenato KE, Mol. Ther. 2018, 26, 1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A, Nat. Immunol. 2005, 6, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhang M, Ju W, Yao Z, Yu P, Wei B-R, Simpson RM, Waitz R, Fassò M, Allison JP, Waldmann TA, J. Immunol. 2012, 188, 6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Singh M, Vianden C, Cantwell MJ, Dai Z, Xiao Z, Sharma M, Khong H, Jaiswal AR, Faak F, Hailemichael Y, Janssen LME, Bharadwaj U, Curran MA, Diab A, Bassett RL, Tweardy DJ, Hwu P, Overwijk WW, Nat. Commun. 2017, 8, 1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sorensen MR, Holst PJ, Steffensen MA, Christensen JP, Thomsen AR, Vaccine 2010, 28, 6757. [DOI] [PubMed] [Google Scholar]
- [44].Zhang C, Zhao W, Bian C, Hou X, Deng B, McComb DW, Chen X, Dong Y, ACS Appl. Bio Mater. 2019, 2, 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang C, Zhang X, Zhao W, Zeng C, Li W, Li B, Luo X, Li J, Jiang J, Deng B, McComb DW, Dong Y, Nano Res. 2019, 12, 855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhao W, Zhang C, Li B, Zhang X, Luo X, Zeng C, Li W, Gao M, Dong Y, Cell. Mol. Bioeng. 2018, 11, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhao P, Hou X, Yan J, Du S, Xue Y, Li W, Xiang G, Dong Y, Bioact. Mater. 2020, 5, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR, J. Cheminf. 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Halgren TA, J. Comput. Chem. 1996, 17, 490. [Google Scholar]
- [50].Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR, J. Cheminf. 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K, J. Comput. Chem. 2005, 26, 1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ohkubo YZ, Pogorelov TV, Arcario MJ, Christensen GA, Tajkhorshid E, Biophys. J. 2012, 102, 2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Qi Y, Cheng X, Lee J, Vermaas JV, Pogorelov TV, Tajkhorshid E, Park S, Klauda JB, Im W, Biophys. J. 2015, 109, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW, J. Phys. Chem. B 2010, 114, 7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zoete V, Cuendet MA, Grosdidier A, Michielin O, J. Comput. Chem. 2011, 32, 2359. [DOI] [PubMed] [Google Scholar]
- [56].Pastor RW, Brooks BR, Szabo A, Mol. Phys. 1988, 65, 1409. [Google Scholar]
- [57].Feller SE, Zhang Y, Pastor RW, Brooks BR, J. Chem. Phys. 1995, 103, 4613. [Google Scholar]
- [58].Darden T, York D, Pedersen L, J. Chem. Phys. 1993, 98, 10089. [Google Scholar]
- [59].Ryckaert J-P, Ciccotti G, Berendsen HJC, J. Comput. Phys. 1977, 23, 327. [Google Scholar]
- [60].Zhao YH, Abraham MH, Zissimos AM, J. Org. Chem. 2003, 68, 7368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.