

Abstract
Activated effector T cells induce pro-inflammatory responses in rheumatoid arthritis (RA) which then lead to inflammation of the joints. In this report, we demonstrate that polymeric nanoparticles with alpha keto-glutarate (aKG) in their polymer backbone (termed as paKG NPs) modulate T cell responses in vitro and in vivo. Impressively, a low dose of only three administrations of methotrexate, a clinically and chronically administered drug for RA, in conjunction with two doses of paKG NPs, reversed arthritis symptoms in collagen-induced arthritis (CIA) mice. This was further followed by significant decreases in pro-inflammatory antigen-specific T helper type 17 (TH17) responses and a significant increase in anti-inflammatory regulatory T cell (TREG) responses when CIA treated splenic cells were isolated and re-exposed to the CIA self-antigen. Overall, this study supports the concurrent and short term, low dose of paKG NPs and methotrexate for the reversal of RA symptoms.
Keywords: immunoengineering, methotrexate, polymer nanoparticle, rheumatoid arthritis
Graphical Abstract
1. Introduction
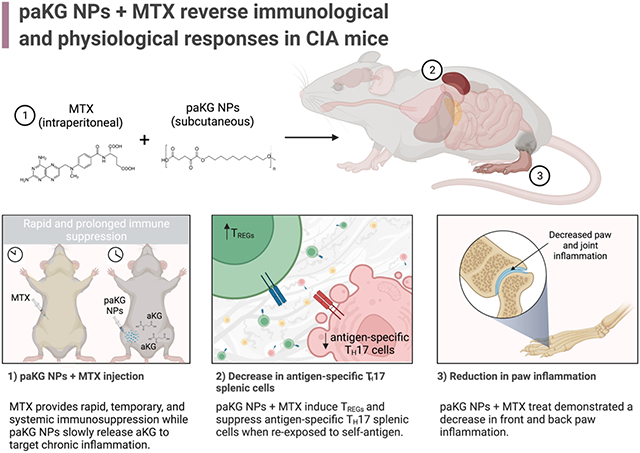
Immunoengineering approaches have made a great impact on generating techniques for anti-inflammatory applications 1-7. Immunoengineering via delivery of metabolites is an emerging area of research, which can have great implications in treatment of various autoimmune diseases and disorders associated with inflammation 8-10. We previously demonstrated that glycolytic inhibition of innate immune cells, such as dendritic cells (DCs), along with the delivery of a self-antigen, via anti-inflammatory alphaketo-glutarate (aKG) based microparticles, can lead to antigen-specific immune suppression in collagen-induced arthritis (CIA) mice 8,11. Although, this process is highly effective in reducing symptoms of rheumatoid arthritis (RA) in CIA mice, a rapid reduction of symptoms in clinic is highly desirable. Methotrexate (MTX), a clinically used RA regimen, is administered to patients for extended periods of time. This treatment temporarily alleviates systemic RA symptoms by non-specifically suppressing pro-inflammatory effector T cell function while enhancing anti-inflammatory regulatory T cell (TREG) function 12-14. Unfortunately, the short half-life of MTX 15, and its inability to induce increased immunosuppressive antigen-specific T cell responses does not lead to long-term disease resolution. Therefore, in this study, aKG-based polymeric NPs (paKG NPs) were administered to aid in the resolution of prolonged inflammation, by a sustained release of the aKG metabolite, while MTX was delivered to provide a temporary yet rapid and potent immune response for alleviation of symptoms (Figure 1a).
Figure 1: Characterization of paKG NPs for the treatment of collagen-induced arthritis (CIA) in mice.

(a) Graphical summary of in vivo study design and results (created using biorender.com). (b) On the day of nanoparticle synthesis, dynamic light scattering data demonstrates the average diameter of nanoparticles to be 191.3 ± 16.2 nm (n = 3; avg ± SEM). The measurement was executed in DI H2O. (c) Scanning electron microscope images of paKG NPs with a gold sputter coating of nm (nanoparticles were gold coated only for the purposes of imaging) demonstrated a smooth and spherical surface morphology the day of synthesis (scale bar = 500 nm; magnification = 100,000x).
2. Materials and Methods
Polymer synthesis
The paKG polymer was formulated using previously described techniques 8,16. Briefly, an equimolar ratio of alpha-ketoglutarate and 1,10-decanediol were placed under a vacuum in a round bottom flask and was continuously stirred for 2 hrs at 130°C. Methanol was used to precipitate the generated polymer. Any remaining methanol was removed using a rotary evaporator.
Nanoparticle Generation
The nanoparticles were synthesized using previously described emulsion methodologies 8,16,17. Briefly, 50 mg of the alpha-ketoglutarate polymer was dissolved in 4 mL of chloroform and added to 25 mL of 4% polyvinyl alcohol (PVA). This mixture was sonicated at 50% amplitude for 2 mins using a 700 Watts probe sonicator (12.7 mm probe size (220-B) and 3.2 mm microtip size) and then transferred to a beaker with 40 mL of 2% PVA. The contents were stirred continuously at 600 rpm for 3 hrs to allow the chloroform to evaporate. The particles were then centrifuged at 10,000 rpm for 5 mins and washed with DI H2O for a total of three times. The nanoparticles were then resuspended in deionized water (DI H2O), placed in −80°C overnight, and lyophilized for 24-48 hrs. The nanoparticles were then stored at −20°C until use.
Scanning Electron Microscopy
The nanoparticles were imaged using a scanning electron microscope focused ion bean – Nova 200 at Erying Materials Center at Arizona State University. The scanning electron microscopy samples were gold sputter coated for 180 seconds to have a coating thickness of approximately 15 nm. Nanoparticles were only gold sputter coated for the purposes of scanning electron microscopy imaging.
Dynamic Light Scattering
The average diameter of the nanoparticles was observed by dynamic light scattering (Zetasizer Nano, Cambridge, UK). The DLS samples for size and zeta potential were prepared by resuspending paKG NPs in DI H20 and placing the samples in a bath sonicator for 5 minutes for particle dispersion.
In vitro T cell culture
The splenocytes from C57BL/6 mice were isolated by placing the spleen atop of a 40 μm cell strainer and gently pressing a pestle against the spleen to separate the splenocytes. The effluent was centrifuged at 300 RFC for 5 mins. Once the supernatant was removed, 3 mL of 1x red blood cell lysis buffer was added for 5 mins at 4°C. The splenocytes were then centrifuged at 300 RFC for 5 mins. The supernatant was discarded and the cells were resuspended in RPMI-1640 Medium with L-glutamine (VWR, Radnor, PA), 10% fetal bovine serum, 1% sodium pyruvate (VWR, Radnor, PA), 1% non-essential amino acids (VWR, Radnor, PA), and 1% penicillin–streptomycin (VWR, Radnor, PA). The splenocytes were plated at 250,000 cells per well and were treated 2 hrs after the cells were seeded. The final concentration in each well of each treatment was as follows: 0.1 mg/mL of paKG nanoparticles, 1 mg/mL of methotrexate and 2.5 μg/mL of concanavalin A (positive control). No treatment was used as a negative control. The treated splenocytes were cultured for 48-72 hrs.
Flow cytometry
Immunofluorescence antibodies were used as purchased. The 0.1% flow staining buffer was formulated by adding 0.1% bovine serum albumin (VWR, Radnor, PA), 2mM Na2EDTA (VWR, Radnor, PA) and 0.01% NaN3 (VWR, Radnor, PA) in DI H2O. Flow cytometry was completed by following the manufacturer's guidance of the Attune NXT Flow cytometer (ThermoFisher Scientific, Waltham, MA, USA) in Arizona State University’s flow cytometry core. The following immunofluorescence antibodies were utilized in the flow cytometry studies: MHC-Tetramer: - I-A(q) bovine collagen II 271-285 GEPGIAGFKGEQGPK (NIH Tetramer Core Facility), CD4 (RM4-5, 566407, BD Biosciences), CD8 (53-6.7, 564983, BD Bioscience), CD25 (PC61, 552880, BD Biosciences), CD44 (IM7, 566200, BD Biosciences), Tbet (4B10, 644835, BioLegend), GATA3 (L50-823, 565449, BD Biosciences), RORyT (Q31-378, 564722, BD Biosciences), Foxp3 (FJK-16s, 48-5773-82, Invitrogen) and Ki67 (SolA15, 11-5698-82, Invitrogen).
Enzyme-linked immunosorbent assay (ELISA) analysis on in vitro T cell culture supernatant
The extracellular supernatant of the in vitro treated T cell culture was analyzed using a DuoSet ELISA development system for the quantification of IL-10 (DY417-05) and IFNγ (DY485-05) as per the manufacturer’s instructions. The absorbance was measured using a plate spectrometer (Speedmax M2e).
CIA induction
All animal experiments completed within protocol number: 19-1712R were approved by Arizona State University’s Institutional Animal Care and Use Committee. Eight- to ten-week-old male DBA/1J (Jackson Laboratories) mice were placed in a restrainer and injected subcutaneously one-third of the way down the tail with a complete Freund’s adjuvant (CFA) + bovine collagen type II (bc2) emulsion on day 0, and on day 21, a booster of incomplete Freund’s adjuvant (IFA) + bc2 was injected subcutaneously one-third of the way down the tail. Additionally, on day 28 mice were intraperitoneally (IP) injected with 10 μg of lipopolysaccharide (LPS) to synchronize and trigger CIA onset.
Paw weights and scores
CIA mice weights and paw photos (for arthritic scores) were collected on day 0 and 1-3 times a week starting from day 21 to the end of study, day 57. In order to fully encompass the variability and severity of paw swelling within the CIA model, arthritic scores were completed using a previously described method 8 where paw inflammation was scored on a scale of 0 to 6. A score of 3 or more signified moderate-severe arthritis. Briefly, scores were equated by assessing the amount of swelling and/or redness in the back left digits and then assigning a point of 0 (no swelling), 1 (mild), 2 (moderate) or 3 (severe). Then, the amount of swelling and/or redness in the back left mid-paw was assessed and assigned a point of 0 (no swelling), 1 (mild), 2 (moderate), or 3 (severe). The same was completed for the back right digits and mid-paw. Then the summation of the four points were given their respective score which can be located in Table 1. The front and back paws were scored separately. Scores were assigned by the first author in an unblinded manner.
Table 1:
Point and scoring strategy for arthritis in mice.
| Points | Score |
|---|---|
| 0 | 0 |
| 1 | 1 |
| 2 | 2 |
| 3-4 | 3 |
| 5-7 | 4 |
| 8-10 | 5 |
| 11-12 | 6 |
CIA treatment studies
CIA mice treated with paKG nanoparticles were subcutaneously injected with 0.5 mg of nanoparticles in sterile 1x phosphate-buffered saline (PBS - 50 μL18) in the right and left hind limb on days 35 and 39. One mouse received a total of 1 mg of particles on each treatment day for a total of 2 mg of particles in the entirety of the study. The particles were utilized in vivo within 30 days of synthesis. No treatment and No CIA mice were administered sterile 1x PBS (50 μL), following the same procedure as paKG nanoparticle treated mice. CIA mice treated with methotrexate were injected IP with 2.5 mg/kg of methotrexate in 50 μL sterile 1x PBS on days 35, 37 and 39. A power of 0.8 and an alpha of 0.05 were utilized to determine group sizes. CIA mice were sacrificed on day 57 and the blood was obtained for serum cytokine analysis. The cervical lymph nodes and spleen were collected for in vivo, and ex vivo flow cytometry studies, and the knee was harvested for histological analysis.
Blood serum cytokine measurement
Blood serum cytokine analysis was performed using a custom ProcartaPlex mouse cytokine multiplex Luminex assay as specified by the manufacturer for the following targets: IL-10, IL-21, IL-1β, IL-4, IL-5, GM-CSF, TNFα, and IL-17A (CTLA-8). Readings were measured on the Luminex™ 200™ Instrument System (ThermoFisher Scientific).
Ex vivo recall reaction
At the completion of CIA studies, the spleen of CIA mice was extracted for recall reaction experiments. Plates were coated with anti-CD3 (0.5 μg/mL) and incubated for 2 hrs at 37°C. The isolated splenocytes were then incubated in 3 mL of 1x red blood cell lysis buffer for 5 mins at 4°C, centrifuged at 300 RFC for 5 mins, and, after discarding the supernatant, were resuspended in RPMI-1640 Medium with L-glutamine (VWR, Radnor, PA), 10% fetal bovine serum, and 1% penicillin–streptomycin (VWR, Radnor, PA). The anti-CD3 coating on the plates were then discarded and washed with 1x PBS twice. Approximately 106 splenocytes/mL were seeded in the presence of bc2 (10 μg/mL). For splenocyte stimulation, anti-CD28 (0.5 μg/mL) and IL-2 (10 ng/mL) were added. ELISA, antibody staining and flow cytometry were done 48-72 hrs after culturing the splenocytes in 37°C.
Histopathologic evaluation
The right knee was extracted, and the surrounding muscles and tissues were removed. The knee was then placed in 4% paraformaldehyde overnight in 4°C and then kept in 30% sucrose for 72 hrs in 4°C. The knees were then decalcified using 0.05 M ethylenediaminetetraacetic acid and were incubated in 4°C for an additional 72 hrs. Once the knees were decalcified, they were embedded and frozen in optimal cutting temperature compound. The center of the knee was sectioned with a tissue thickness of 20 μm and stained with hematoxylin and eosin.
Statistical analysis
Data are expressed as average ± standard error mean. Comparison between two groups was performed using two-tailed Student t-test (Microsoft, Excel). For multiple comparison’s one-way ANOVA was used and followed by unpaired t with Welch’s correction (GraphPad Prism Software 6.0, San Diego, CA). In all cases, p-values of 0.05 or less were considered statistically significant.
3. Results and discussion
To generate paKG NPs, polymers of aKG and 1,10 decanediol were generated using condensation reaction. Nanoparticles were then synthesized from these polymers by utilizing emulsion-evaporation techniques. As determined by dynamic light scattering (DLS), the average diameter of the paKG NPs on the day of synthesis was 191.3 ± 16.2 nm (n = 3; avg ± SEM) (Figure 1b) and the zeta potential was −8.2 ± 1.7 mV (n = 3; avg ± SEM). Moreover, the particles had smooth surface morphology on the day of synthesis as observed by scanning electron microscopy (SEM) (Figure 1c). Additionally, to test the stability of the paKG NPs, the average diameter and the zeta potential of the particles was assessed 21 days post synthesis. As determined by DLS, the average diameter of the particles 3 weeks post synthesis was determined to be 273.7 ± 62.6 nm (n = 3; avg ± SEM) (Figure S1a) and the zeta potential was −16.3 ± 10.9 mV (n = 3; avg ± SEM). These data demonstrate a slight increase in the average diameter of the particles and a slight decrease in the zeta potential over 21 days. Nonetheless, the paKG NPs remained smooth and continued to demonstrate a spherical morphology (Figure S1b). Our prior work found the paKG polymer to have a number-average molecular weight of 15.3 – 23.9 kDa 16. We have previously demonstrated that paKG polymeric particles can release aKG in a sustained manner, and therefore it is expected that paKG NPs, will be able to release the aKG metabolite effectively 8,16.
To test if paKG NPs can modulate T cell function, splenocytes were isolated and cultured with paKG NPs, in the presence or absence of MTX, with no treatment as negative control and concanavalin A (ConA) as the positive control. The cells were then stained for CD4, CD8, CD44, CD25, Tbet, GATA3, RORyt and Foxp3 to identify changes in T helper (TH - CD4+) cell and cytotoxic T (TC - CD8+) cell responses (Figure S2a). In the absence of inflammation (ConA), it was observed that paKG NPs, with and without MTX, slightly increased activation (CD44+) in the anti-inflammatory TREG population (CD4+CD25+Foxp3+) when being compared to MTX alone, although not significantly (Figure S2b). Notably, paKG NPs with and without MTX upregulated TH2 (CD4+GATA3+) responses (anti-inflammatory responses in the context of RA) 19 while no treatment and MTX demonstrated significantly lower TH2 frequencies (Figure 2a). TH2 cells are known to produce the cytokine interleukin 4 (IL-4), which has shown to play a role in polarizing macrophages with a destructive phenotype in RA (M1 - classically activate macrophages) toward a phenotype involved in tissue regeneration and remodeling (M2 – alternatively activated macrophages) 20. Importantly, paKG NPs + MTX exhibited a significant decrease in the activation of pro-inflammatory TH1 (CD4+Tbet+ - Figure 2b) and TH17 (CD4+RORyT+ - Figure 2c) cell types. Therefore, paKG NPs with and without MTX were capable of preventing the activation of these cell types, in turn disabling their function 21.
Figure 2: In vitro cellular responses to paKG NPs + MTX.

(a-h) Flow cytometry of in vitro splenic T cell responses treated with a final well concentration of 1 mg/mL of methotrexate and/or 0.1 mg/mL of paKG nanoparticles and cultured in 37°C for 48-72 hrs (negative control = No treatment), (a-c) in the absence and, (d-g) presence of a final well concentration of 2.5 μg/mL of Concanavalin A (ConA – used to mimic inflammation in vitro) (n = 4-6; avg ± SEM; * = p ≤ 0.05; One-Way ANOVA, Brown-Forsythe and Welch ANOVA tests, unpaired t with Welch’s correction).
Nevertheless, assessing the influence of these formulations on T cells in the presence of inflammation (e.g. RA-associated inflammation) will provide an understanding on how effective these formulations are in modulating pro-inflammatory T cell mediated responses within RA. Therefore, the T cell activator, ConA, was added 22. Notably, in the presence of ConA, MTX significantly increased the frequency of TREGs as compared to all other groups. Interestingly paKG NPs + MTX demonstrated significantly lowered TREG frequencies as compared to no treatment and MTX (Figure S2c). However, when assessing the activated TREG population in the presence of ConA, it was observed that MTX decreased this population, while paKG NPs with and without MTX slightly increased this frequency as compared to MTX alone, although not significantly (Figure S2d). This data suggests that paKG NPs may be able to prevent the decrease in activated TREGs in the presence of inflammation. Furthermore, paKG NPs and paKG NPs + MTX significantly increased total TH2 responses when being compared to ConA and MTX (Figure 2d). Furthermore, paKG NPs + MTX, paKG NPs alone and MTX alone significantly increased the activated TH2/activate TH1 ratio, whereas ConA alone demonstrated lower frequencies (Figure 2e). Therefore, this data suggests that the separate and combined delivery of MTX and paKG NPs can lead to a decrease in TH1 function, which is observed in Figure 2f). Additionally, MTX and paKG NPs with and without MTX, significantly decreased TH17 activation (Figure 2g) which was not observed in the ConA alone group. Therefore, in the presence of inflammation, paKG NPs + MTX increased the total TH2 population while decreasing TH1 and TH17 activation. The decrease in TH1 function in response to the paKG NPs + MTX was further supported by an observed significant decrease in the extracellular production of interferon-gamma (IFNγ), a pro-inflammatory cytokine produced by TH1 cells 23, when comparing the supernatant of the cultured splenocytes treated with paKG NPs + MTX to the supernatant of the cultured splenocytes treated with ConA (Figure S2e). Extracellular IFNγ levels in the in vitro supernatant of splenocyte cultures was not significantly different between paKG NPs + ConA and ConA only treated groups. This suggests that aKG released from paKG NPs in 72 hrs by itself was not sufficient to reduce IFNγ levels in the presence of ConA. Furthermore, this data also suggests that the presence of MTX is required for reducing the IFNγ levels in this short-term experiment. Additionally, paKG NPs + MTX treatment group was not significantly different than MTX alone treatment, suggesting the importance of MTX for short-term prevention of IFNγ release in vitro. However, the extracellular production of IL-10, an anti-inflammatory cytokine 24, was undetectable in the supernatant of all treated and untreated splenocyte cultures. Thus, the combinatorial delivery of paKG NPs and MTX holds promise in reducing pro-inflammatory T cell responses in diseases where pro-inflammatory TH1 and TH17 cells are implicated, such as in RA 25.
Next, to test if the paKG NPs can modulate inflammation within joints, a CIA mouse model was utilized. Briefly, paKG NPs were injected twice on days 35 and 39, whereas MTX was injected thrice on days 35, 37 and 39. Day 35 was chosen for treatment as the mice presented moderate-severe inflammation in the paws on day 35. Based on our prior work 8, it was not expected for the day 35 NP injection site to clear before the day 39 NP injections. Furthermore, upon euthanasia on day 57, the injection site showed that the particles were still present (Figure S3). No treatment, MTX alone, paKG NPs alone, and no CIA induction were utilized as controls for this study (Figure 3a). The in vivo dosing of the NP concentration was determined based on the 0.1 mg/mL concentration of paKG NPs delivered to cultured splenocytes in vitro in Figure 2a-h. Given that mice on average weigh approximately 20 g and that 0.1 mg/mL is equivalent to 0.1 mg/g, 20 g multiplied by 0.1 mg equates to 2 mg. Therefore, the in vitro culture conditions of 0.1 mg/mL of paKG NPs assisted in determining that the administration of 2 mg of paKG NPs during the entirety of the study would be suitable for in vivo studies. Furthermore, the in vitro degradation profile of paKG microparticles (MPs), which demonstrate a controlled and sustained release of aKG for 30 days, but not complete degradation within 30 days 8,16 further supported the delivery of 2 mg of paKG NPs for the entirety of the study. Although, we expect the NPs to degrade faster than MPs. Arthritis scores and weight of the mice was measured on day 0 and 1-3 times a week from day 21 to the end of the study on day 57. If a constant score was observed for one week, then the disease was considered to be non-worsening or resolved. It was observed that mice without CIA induction significantly gained weight as observed by the difference between the weight at the end of the study on day 57 (one No CIA mouse was sacrificed at an earlier timepoint on day 50) and day 0, the beginning of the study. On the other hand, the control mice groups of MTX, paKG NPs alone, and no treatment either lost weight as compared to day 0 or maintained a similar weight to day 0. The paKG NPs + MTX group was the only treated group that had a significant increase in weight on day 57 as compared to day 0 (Figure 3b).
Figure 3: paKG NPs in conjunction with methotrexate (MTX) reduces inflammation in collagen-induced arthritis (CIA) mice.

(a) Study design for inducing CIA and treatment. (b) Change in weight of mice after paKG NP + MTX treatment demonstrated this as the only treatment group to have a significant increase in weight at the end of the study as compared to day 0. Red line represents the cut off for weight loss (n = 3-4; avg ± SEM; * = p ≤ 0.05 when comparing end of study weight to day 0 within same group; two-tailed unpaired Student’s t-test). (c,d) Arthritis scores of (c) front and (d) back paws of paKG NPs + MTX mice significantly decreased as compared to no treatment on days 40, 49, 54 and 57 (No CIA: n = 3, all other groups: n = 6; avg ± SEM; * = p ≤ 0.05 as compared to No treatment; two-tailed unpaired Student’s t-test; dotted black line represents treatment start day on day 35).
The inflammation in mice was quantified by utilizing a point and scoring system based on the observed swelling and/or redness in the digits and mid-paw (Figure S4). The point and scoring strategy are shown in Table 1. The scoring for the front and back paws were done separately. It was observed that the front paw arthritis score of mice treated with the controls of MTX alone or paKG NPs alone were not significantly different than the no treatment control for all days. On the other hand, the mice treated with paKG NPs + MTX had significantly decreased front paw arthritis scores as compared to no treatment on days 40, 49, 54 and 57 (Figure 3c). Although the paKG NPs were delivered subcutaneously in the hind limbs, similar to paKG MPs, the NPs may have trafficked from the injection site to the lymph nodes for a systemic modulation 8. Therefore, we expect the NPs to also be trafficked and/or freely drained from the injection site to the front-paw draining lymph nodes for a modulation of responses within the front paws 26. Similar to the front paws, the back paw arthritis score of mice treated with paKG NPs alone and MTX alone were not significantly different to no treatment. However, the back paw arthritis score was significantly decreased in mice treated with paKG NPs + MTX as compared to no treatment on days 40, 49, 54 and 57. A back paw arthritis score of 1 or less was observed starting on day 49 and continued till the end of the study (Figure 3d). Whereas, in our prior work, a back paw arthritic score of 1 was not observed until day 63 8. Notably, a significant overall difference between the curves in Figure 3c and 3d was demonstrated when an ordinary two-way ANOVA, uncorrected Fisher’s LSD, with individual variances computed for each comparison was preformed. Since we utilized a CIA mouse model, and given that collagen is present in all joints, it is expected that arthritis was induced in all joints within this mouse model 27,28. Therefore, these data suggest that the combination of paKG NPs and MTX may reduce inflammation in all major joints in CIA mice. Notably, RA in humans is a systemic and progressive disease where effector T cells induce inflammation progressively in several joints 29-31. It is important to highlight that the rebound in paw scores after day 40 may be occurring due to the MTX that is administered on days 35, 37, 39. Although, MTX is potent, it has a short half-life and therefore clears the body quickly. However, the short but potent effects of MTX are acting while the slower yet more long-term effects of paKG NPs begins to take place as CIA onset is occurring. Therefore, the effects of the paKG NPs may not be apparent until day 49. The dosing may be able to be further optimized to balance the short and long-term effects by either prolonging the short-term effect by administering 3 additional doses of MTX on days 41, 43 and 45 or by expediating the degradation and release of aKG from the NPs by generating porous particles 32. Nevertheless, a formulation that reduces inflammation within joints for long-periods of time (>1 week in mice) can be highly beneficial.
Several drugs that are utilized in clinic, including MTX, target T cells and prevent their pro-inflammatory functions 33,34. Therefore, in this study we investigated if the combination of paKG NPs and MTX can modulate the function of T cells in vivo. Specifically, mice were euthanized on day 57, and T cell responses were assessed in the cervical lymph nodes (CLNs) and in a portion of the spleen of treated and untreated mice. Interestingly, mice treated with paKG NPs + MTX demonstrated significantly lower TC17 (Figure S5a) and activated TH1 cell types (Figure S5b) in the spleen as compared to mice treated with MTX alone. Furthermore, a significant increase in bc2-specific Th2 cells were observed in the CLNs of mice treated with paKG NPs + MTX as compared to mice treated with paKG NPs, MTX, and untreated mice (Figure S5c). Therefore, suggesting that the delivery of paKG NPs + MTX was more effective in inducing favorable Th2 type of immune responses for the treatment of RA as compared to the clinically approved MTX treatment.
Furthermore, to understand how T cells from each treatment group would respond in the presence of the CIA self-antigen, bovine collagen type II (bc2), splenocytes obtained from a portion of the spleen from mice on day 57 were cultured with bc2. This recall response provides the ability to gauge how T cells would respond when being reintroduced to collagen in vivo, for example in joint tissue. Moreover, to further enhance the understanding of these responses when they occur in vivo, the cells were cultured with anti-CD3, anti-CD28, and IL-2 for the additional analysis of activated and stimulated naïve and memory T cell populations in the presence of bc2. The cells were then stained with a bc2-specific MHC-II tetramer, CD4, CD8, CD44, CD25, Tbet, GATA3, RORyt, Foxp3 and Ki67 to identify changes in TH cell responses (Figure S2a). It was observed that paKG NPs and paKG NPs + MTX treated mice had a decreased frequency of activated TH cells (Figure 4a) and a decreased frequency of proliferation (Ki67+) within these activated TH cells (Figure 4b) as compared to untreated mice. These data suggest that when exposed to the antigen, the frequency of activated TH cells will increase in the no treatment group and lead to higher inflammation in mice, as observed from the arthritis score (Figure 4c,d). Additionally, the frequency of TH cells that responded to bc2 were also antigen-specific (bc2+), with higher responses in no treatment as compared to paKG NPs and paKG NPs + MTX (Figure 4c). Notably, mice treated with paKG NPs + MTX had a significantly higher frequency of TREGs (Figure 4d), as compared to the no treatment control when re-exposed to bc2. Although TREGs only marginally increased in the paKG NPs + MTX group, TREGs play a crucial role in counteracting RA progression 35. Therefore, since TREGs are involved in decreasing disease severity, and given that paKG NPs + MTX treated mice displayed recovery in weight (Figure 3b) and front and back paw scores (Figure 3c,d) on day 57, it is expected for the TREGs to decrease and normalize to homeostatic measurements as RA associated symptoms resolve. Notably, at day 57, mice treated with paKG NPs alone or MTX alone treated mice did not display a recovery in weight (Figure 3b) or front and back paw scores (Figure 3c,d) and also did not have a significant increase in TREGs as compared to the no treatment control when responding to bc2 (Figure 4d). These data strongly suggest that the combinatorial delivery of paKG NPs + MTX will lead to the generation of immunosuppressive T cells in the presence of the antigen. Interestingly, the paKG NPs and paKG NPs + MTX treated mice demonstrated a downregulation of bc2-specific TH17 (Figure 4e) and activated bc2-specific TH17 (Figure 4f) when the splenocytes were exposed to the bc2 antigen as compared to the no treatment control. TH17 cells are known to play an important part in propagating autoimmune diseases such as RA 36, and therefore, the ability of paKG NPs + MTX to reduce these cell populations after exposure to bc2 is highly encouraging. Moreover, unlike untreated mice, bc2 exposed splenocytes from mice treated with paKG NPs + MTX were able to decrease the frequency of activated TH1 cells (Figure 4g). Lastly, bc2 exposed splenocytes from mice treated with paKG NPs alone, and paKG NPs + MTX were able to decrease the frequency of activated TC1 (Figure 4h) cells as compared to untreated mice. TH1 cells are responsible for promoting inflammation by releasing interferon gamma, whereas TC1 cells are responsible for directly killing cells and further inducing inflammation 37,38. Therefore, a decrease in the frequency of these populations after exposure to bc2 suggests that paKG NPs + MTX can reduce inflammation inducing T cells in an antigen dependent manner. Interestingly, T cells exposed to paKG NPs and paKG NPs + MTX in Figure 2a-h and Figures 4a-4c, 4e, 4f and 4h demonstrate similar responses. However, paKG NP treated mice did not display improvements in physiological outputs such as weight (Figure 3b) and front and back paw scores (Figure 3c,d) as observed in paKG NPs + MTX treated mice. Additionally, unlike splenic T cells in the presence of bc2 from paKG NP treated mice, splenic T cells from paKG NPs + MTX treated mice, which were cultured with bc2, demonstrated significantly higher TREGs (Figure 4d) and significantly lower activated TH1 populations (Figure 4g) as compared to splenic T cells from untreated mice. Thus, demonstrating a difference in T cell behavior between paKG NP treated and paKG NP + MTX treated mice. However, blood serum cytokine levels did not demonstrate significance differences between the TH2 produced cytokine IL-5 (Figure S5d) and pro-inflammatory cytokine TNFα (Figure S5e). Additionally, blood serum cytokine levels of IL-10, IL-21, IL-1β, IL-4, GM-CSF and IL-17A (CTLA-8) were undetectable in all CIA groups. Nonetheless, the combinatorial delivery of paKG NPs + MTX may potentially be improving physiological symptoms through the upregulation in TREGs (Figure 4d) and downregulation in activated TH1 cells (Figure 4g). Combination index or synergy between paKG NPs and MTX was not determined in this work and will be tested in future experiments39. Furthermore, the H&E staining of the knee section suggest that the cellular infiltration in the tissue of the knee was decrease as compared to the no treatment group in mice treated with paKG NPs + MTX group (Figure S6).
Figure 4: paKG NPs along with methotrexate (MTX) have an additive effect in decreasing pro-inflammatory splenic recall responses against bovine collagen 2 (bc2).

End of study ex vivo flow cytometry of splenic CIA T cell responses cultured with anti-CD3, anti-CD28, IL-2 and bc2 (a) Activated T helper cells, (b) Proliferating activated T helper cells, (c) bc2-specific T helper cells, (d) Regulatory T cells, (e) bc2-specific T helper type 17 cells, (f) Activated bc2-specific T helper type 17 cells, (g) Activated T helper type 1 cells, (h) Activated cytotoxic T cells (n = 4 mice; n = 2 replicates; avg ± SEM; * = p ≤ 0.05 as compared to No treatment; two-tailed unpaired Student’s t-test).
Furthermore, it is important to note that the resemblance of the CIA mouse model to human RA is one reason why the CIA mouse model is the most widely used RA mouse model 40. However, even with the utmost care during CIA induction, variability in disease can occur which might explain the bimodular effect within certain T cell frequencies in the ex vivo recall reaction. To circumvent biases, mice were randomized into treatment groups on day 35 and each treatment group had to meet the criteria of having an overall average back paw score of approximately 3, which is observed on day 35 (Figure 3d). Thus, promoting equality in each treatment group. Nonetheless, 18 days after the last treatment day, on day 39, splenic T cells treated with paKG NPs + MTX induced favorable T cell responses, such as suppressing activated TH1 cell types and increasing TREG frequencies, when re-exposed to the bc2 antigen. Therefore, suggesting that the combinatorial delivery of paKG NPs + MTX can have effects lasting 18 days after treatment administration. Overall, these data suggest that the short term effect of MTX, due to its potent yet short half-life, and the long-term effect of paKG particles, as observed by a sustained release of aKG 8,16, are required to achieve a prolonged reduction in physiological and immunological responses.
4. Conclusions
In conclusion, only CIA mice treated with the combinatorial short and low dose of paKG NPs and MTX demonstrated a reduction in systemic symptoms, as observed by a normalization in weight and a decrease in front and back paw inflammation. Furthermore, this decrease in inflammation is driven by a downregulation of TH17 bc2-specific antigen responses and an enhancement of TH2 type of T cell responses. Overall, a short and low dose of paKG NPs and MTX is a potential method of providing long-term alleviation of RA symptoms.
Supplementary Material
Acknowledgements
The authors would like to acknowledge the Flow Cytometry Core, the Regenerative Medicine Imaging Facility for histopathological services, the FEI at Erying Materials Center, and the Department of Animal Care and Technologies at Arizona State University. The authors are also grateful for the support of the National Institute of Health (Grant Nos. 1R01AR078343-01, 1R01GM144966-01 and 1 R01 AI155907-01).
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of DACT at Arizona State University and approved by the Animal Ethics Committee of IACUC at ASU.
Abbreviations:
- RA
rheumatoid arthritis
- aKG
alphaketo-glutarate
- paKG NPs
poly alphaketo-glutarate nanoparticles
- MP
microparticle
- CIA
collagen-induced arthritis
- TH
T helper cell
- TREG
regulatory T cells
- TC
cytotoxic T cell
- DCs
dendritic cells
- DLS
dynamic light scatter
- SEM
scanning electron microscopy
- IFNγ
interferon-gamma
- MTX
methotrexate
- PVA
polyvinyl alcohol
- DI H2O
deionized water
- CLN
cervical lymph node
- CFA
complete Freund’s adjuvant
- IFA
incomplete Freund’s adjuvant
- bc2
bovine collagen type II
- LPS
lipopolysaccharide
- ELISA
Enzyme-linked immunosorbent assay
- PBS
phosphate-buffered saline
- IP
intraperitoneally
- ConA
concanavalin A
- IL-4
interleukin 4
Footnotes
Conflicts of interest
Abhinav P. Acharya is affiliated with a start-up company, Immunometabolix, LLC. The M20-015L “Metabolic reprogramming of immune for Autoimmune disorders Prevention/Reversal” have Abhinav P. Acharya and Joslyn L. Mangal as authors, which is under option by ImmunoMetabolix, LLC. There are no other conflicts to declare.
References
- 1.Le T, Aguilar B, Mangal JL and Acharya AP, Bioengineering and Translational Medicine, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klimak M, Nims RJ, Pferdehirt L, Collins KH, Harasymowicz NS, Oswald SJ, Setton LA and Guilak F, Acta Biomaterialia, 2021, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner AB, Lee SKC, Woods EC and Acharya AP, BioMed Research International, 2013. [Google Scholar]
- 4.Fisher JD, Acharya AP and Little SR, Clinical Immunology, 2015, 160, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ratay ML, Balmert SC, Acharya AP, Greene AC, Meyyappan T and Little SR, Scientific Reports, , DOI: 10.1038/s41598-017-17869-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher J, Balmert S, Zhang W, Schweizer R, Schnider J, Komatsu C, Dong L, Erbas V, Unadkat J, Aral A, Acharya A, Kulahci Y, Turnquist H, Thomson A, Solari M, Gorantla V and Little S, Proc Natl Acad Sci U S A. [Google Scholar]
- 7.Fisher JD, Zhang W, Balmert SC, Aral AM, Acharya AP, Kulahci Y, Li J, Turnquist HR, Thomson AW, Solari MG, Gorantla VS and Little SR, Science Advances, , DOI: 10.1126/sciadv.aax8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mangal JL, Inamdar S, Le T, Shi X, Curtis M, Gu H and Acharya AP, Biomaterials, , DOI: 10.1016/j.biomaterials.2021.121079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mangal JL, Handlos JL, Esrafili A, Inamdar S, McMillian S, Wankhede M, Gottardi R and Acharya AP, Cancers, 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mangal JL, Basu N, Wu H-JJ and Acharya AP, Immunometabolism, , DOI: 10.20900/immunometab20210032. [DOI] [Google Scholar]
- 11.Liu S, Yang J and Wu Z, Mediators of Inflammation, 2021, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guggino G, Giardina A, Ferrante A, Giardina G, Schinocca C, Sireci G, Dieli F, Ciccia F and Triolo G, Rheumatology International, , DOI: 10.1007/s00296-014-3030-2. [DOI] [PubMed] [Google Scholar]
- 13.Ćalasan MB, Vastert SJ, Scholman RC, Verweij F, Klein M, Wulffraat NM, Prakken BJ and van Wijk F, Rheumatology (United Kingdom), , DOI: 10.1093/rheumatology/kev101. [DOI] [PubMed] [Google Scholar]
- 14.Cribbs AP, Kennedy A, Penn H, Amjadi P, Green P, Read JE, Brennan F, Gregory B and Williams RO, Arthritis and Rheumatology, , DOI: 10.1002/art.39031. [DOI] [PubMed] [Google Scholar]
- 15.Bannwarth B, Péhourcq F, Schaeverbeke T and Dehais J, Clinical Pharmacokinetics, 1996, 30. [DOI] [PubMed] [Google Scholar]
- 16.Mangal JL, Inamdar S, Yang Y, Dutta S, Wankhede M, Shi X, Gu H, Green M, Rege K, Curtis M and Acharya AP, Journal of Materials Chemistry B, , DOI: 10.1039/d0tb00790k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ratay ML, Glowacki AJ, Balmert SC, Acharya AP, Polat J, Andrews LP, Fedorchak MV, Schuman JS, Vignali DAA and Little SR, Journal of Controlled Release, , DOI: 10.1016/j.jconrel.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langer SW, Sehested M and Buhl Jensen P, Annals of Oncology, , DOI: 10.1023/A:1011163823321. [DOI] [PubMed] [Google Scholar]
- 19.Van Roon JAG and Bijlsma JWJ, Annals of the Rheumatic Diseases, 2002, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Hsu HC and Mountz JD, Current Rheumatology Reports, 2012, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alberts B, Johnson A and Lewis J, in Molecular Biology of the Cell, 2002. [Google Scholar]
- 22.Hegyi B, Kudlik G, Monostori É and Uher F, Biochemical and Biophysical Research Communications, , DOI: 10.1016/j.bbrc.2012.01.150. [DOI] [PubMed] [Google Scholar]
- 23.Tau GZ, Von Der Weid T, Lu B, Cowan S, Kvatyuk M, Pernis A, Cattoretti G, Braunstein NS, Coffman RL and Rothman PB, Journal of Experimental Medicine, , DOI: 10.1084/jem.192.7.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couper KN, Blount DG and Riley EM, The Journal of Immunology, , DOI: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 25.Li G, Kolan SS, Guo S, Marciniak K, Kolan P, Malachin G, Grimolizzi F, Haraldsen G and Skålhegg BS, Frontiers in Immunology, , DOI: 10.3389/fimmu.2021.689057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manolova V, Flace A, Bauer M, Schwarz K, Saudan P and Bachmann MF, European Journal of Immunology, , DOI: 10.1002/eji.200737984. [DOI] [PubMed] [Google Scholar]
- 27.Brand DD, Latham KA and Rosloniec EF, Nature Protocols, , DOI: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 28.Marinova-Mutafchieva L, Williams RO, Mason LJ, Mauri C, Feldmann M and Maini RN, Clinical and Experimental Immunology, , DOI: 10.1046/j.1365-2249.1997.2901181.x. [DOI] [PubMed] [Google Scholar]
- 29.Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker J-C, Hagerty DT and Moreland LW, New England Journal of Medicine, , DOI: 10.1056/nejmoa035075. [DOI] [Google Scholar]
- 30.Langdon K and Haleagrahara N, International Reviews of Immunology, 2018, 37. [DOI] [PubMed] [Google Scholar]
- 31.Malemud CJ, Cells, 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hadipour Moghaddam SP, Mohammadpour R and Ghandehari H, Journal of Controlled Release, , DOI: 10.1016/j.jconrel.2019.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Genestier L, Paillot R, Fournel S, Ferraro C, Miossec P and Revillard JP, Journal of Clinical Investigation, , DOI: 10.1172/JCI2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davignon J-L, Rauwel B, Degboé Y, Constantin A, Boyer J-F, Kruglov A and Cantagrel A, Arthritis Research & Therapy, , DOI: 10.1186/s13075-018-1725-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Esensten JH, Wofsy D and Bluestone JA, Nature Reviews Rheumatology, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kato H and Fox DA, Clinical and Translational Science, 2010, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruschpler P and Stiehl P, Cellular and molecular biology (Noisy-le-Grand, France: ). [PubMed] [Google Scholar]
- 38.Isomäki P, Luukkainen R, Lassila O, Toivanen P and Punnonen J, Immunology, , DOI: 10.1046/j.1365-2567.1999.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou T-C, Synergy, 2018, 7, 49–50. [Google Scholar]
- 40.Luan J, Hu Z, Cheng J, Zhang R, Yang P, Guo H, Nan G, Guo N and Gou X, Experimental and Therapeutic Medicine, , DOI: 10.3892/etm.2021.10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.