

Summary
How the homeostatic drive for sleep accumulates over time and is released remains poorly understood. In Drosophila, we previously identified the R5 ellipsoid body (EB) neurons as putative sleep drive neurons1 and recently described a mechanism by which astrocytes signal to these cells to convey sleep need2. Here, we examine the mechanisms acting downstream of the R5 neurons to promote sleep. EM connectome data demonstrate that R5 neurons project to EPG neurons3. Broad thermogenetic activation of EPG neurons promotes sleep, while inhibiting these cells reduces homeostatic sleep rebound. Perforated patch-clamp recordings reveal that EPG neurons exhibit elevated spontaneous firing following sleep deprivation, which likely depends on an increase in extrinsic excitatory inputs. Our data suggest that cholinergic R5 neurons participate in the homeostatic regulation of sleep, and epistasis experiments indicate that the R5 neurons act upstream of EPG neurons to promote sleep. Finally, we show that the physical and functional connectivity between the R5 and EPG neurons increases with greater sleep need. Importantly, dual patch-clamp recordings demonstrate that activating R5 neurons induces cholinergic-dependent excitatory post-synaptic responses in EPG neurons. Moreover, sleep loss triggers an increase in the amplitude of these responses, as well as in the proportion of EPG neurons that respond. Together, our data support a model whereby sleep drive strengthens the functional connectivity between R5 and EPG neurons, triggering sleep when a sufficient number of EPG neurons are activated. This process could enable the proper timing of the accumulation and release of sleep drive.
eTOC Blurb
The circuit mechanisms underlying the homeostatic regulation of sleep remain enigmatic. Ho et al. find that sleep deprivation induces plastic changes in a homeostatic sleep circuit, which potentiates signaling to a downstream sleep-promoting circuit. They propose that this process mediates transmission of homeostatic sleep drive in Drosophila.
Results and Discussion
EPG neurons promote sleep and contribute to sleep homeostasis
We recently delineated a pathway signaling sleep need from astrocytes to the ellipsoid body (EB) R5 sleep drive neurons in Drosophila2. To further elucidate the circuit mechanisms underlying the homeostatic regulation of sleep, we sought to identify neuronal groups that act downstream of the R5 neurons in this process. The fly hemibrain connectome data demonstrate that R5 neurons synapse with EPG (“compass”) neurons, shown to be involved in navigation3–6. As expected, a split-GAL4 driver for R5 neurons (R30G03-AD, R58H05-DBD) driving expression of DenMark7 and synaptotagmin-GFP8 (syt-GFP) labelled synaptic terminals in the EB ring, while R19G02-GAL4>UAS-DenMark, UAS-syt-GFP flies (labelling EPG neurons) demonstrated both synaptic terminal and dendritic labeling throughout the EB ring and additional synaptic terminal staining in the protocerebral bridge (PB) (Figures S1A and S1B). Intriguingly, we previously performed a large-scale screen for GAL4 lines affecting sleep behavior, and R19G02-GAL4 (Figures 1A and S1C) was identified as a driver inducing increased sleep during neural activation with dTRPA11. We repeated those findings and obtained similar results with thermogenetic activation of an additional split-GAL4 line (SS50574) that also drives restricted expression in EPG neurons (Figures 1B, 1C, S1D, and S1E). The sleep induced by heat treatment of R19G02-GAL4>UAS-dTRPA1 or SS50574>UAS-dTRPA1 flies was more consolidated with longer sleep bout duration and reduced sleep bout number, compared to controls (Table S1).
Figure 1. A sleep-promoting role for EPG neurons.
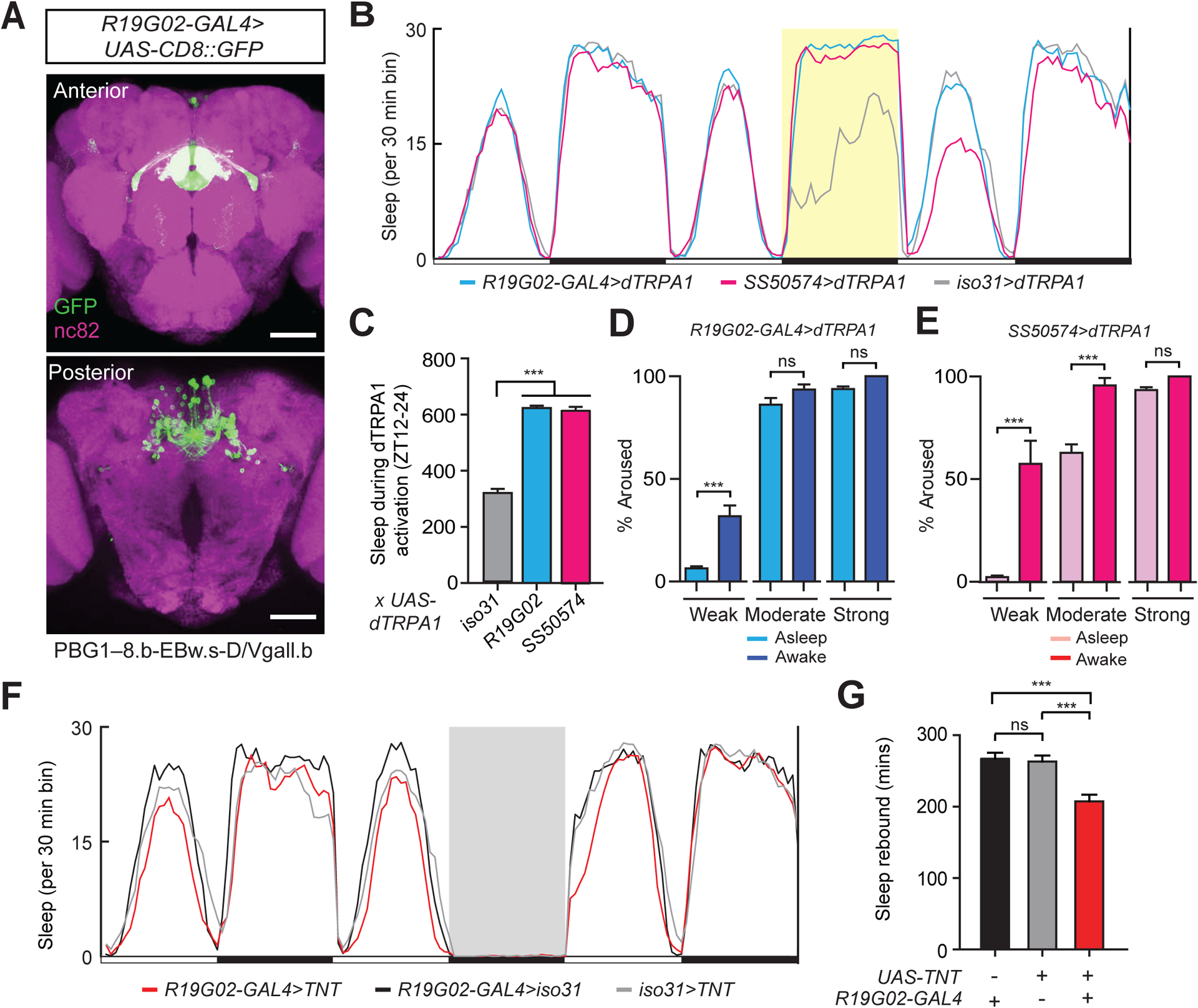
(A) Immunostaining of EPG neurons. Whole-mount brain immunostaining of an R19G02-GAL4>UAS-CD8::GFP animal with anti-GFP (green) and anti-Bruchpilot (BRP, nc82, magenta) antibody staining. Maximal intensity projection of the central brain is shown. Scale bar indicates 50 μm.
(B) Sleep profiles of R19G02-GAL4>dTRPA1 (blue), SS50574>dTRPA1 (magenta), and iso31>dTRPA1 (gray) flies. Sleep time plotted in 30 min bins. White and black bars indicate 12 hr light and dark periods, respectively. The period of 12hr dTRPA1 activation at 29°C is indicated with a yellow background. Data are from same animals as in (C).
(C) Sleep amount during 12h dTRPA1 activation for iso31>dTRPA1 (n=96), R19G02-GAL4>dTRPA1 (n=96), and SS50574>dTRPA1 (n=93) flies. Mean ± SEM is shown; one way ANOVA with Tukey’s post-hoc test.
(D and E) Arousal threshold is increased during EPG neuron activation. The percentage of R19G02-GAL4>dTRPA1 (D) or SS50574-GAL4>dTRPA1 (E) flies undergoing 12h dTRPA1 activation at 29°C that were asleep (light blue or light magenta) or awake (dark blue or dark magenta) aroused by weak (0.2 g, n=116 and 152), moderate (0.25 g, n=155 and 171), and strong (0.35 g, n=139 and 140) mechanical rotational stimuli at ZT16, ZT18, ZT20, and ZT22. Mean ± SEM is shown; Student’s t test for each stimulus condition, with Bonferroni correction.
(F) Sleep profiles of R19G02-GAL4>UAS-TNT (red), R19G02-GAL4>iso31 (black), and iso31>UAS-TNT (gray) flies. Sleep time plotted in 30 min bins. White and black bars indicate 12 hr light and dark periods, respectively. The period of sleep deprivation via mechanical shaking is indicated with a gray background. Data are from same animals as in (G).
(G) Rebound sleep following deprivation from ZT0-6 for R19G02-GAL4>iso31 (n=62), iso31>UAS-TNT (n=90), and R19G02-GAL4>UAS-TNT (n=81) flies. Mean ± SEM is shown; one way ANOVA with Tukey’s post hoc test. In this and in subsequent figures *, **, ***, and ns denote P<0.05, P<0.01, P<0.001, and not significant, respectively. See also Figure S1 and Table S1.
To address whether the quiescent behavior seen with activation of these drivers simply represented locomotor inactivity, we performed arousal threshold experiments. As shown in Figures 1D and 1E, flies during these consolidated quiescent states displayed a significant increase in arousal threshold to submaximal mechanical stimulation, compared to awake flies. Because EPG neurons function in navigation, we next asked whether thermogenetic activation of EPG neurons caused locomotor impairment. First, R19G02-GAL4>UAS-dTRPA1 or SS50574>UAS-dTRPA1 flies were subjected to 2 hr heat treatment at 29°C, and climbing ability was assessed. Climbing ability was similar for these genotypes compared to controls (Figure S1F). Second, we analyzed peak activity data following strong mechanical stimuli from our arousal threshold experiments at 29°C. The peak activity of SS50574>UAS-dTRPA1 flies was similar to controls, while it was reduced for R19G02-GAL4>UAS-dTRPA1 flies (Figure S1G). Together, these data argue that activation of EPG neurons promotes sleep and not behavioral quiescence due to locomotor inactivity or impairment.
To further confirm that the increased sleep phenotype maps to EPG neurons, we tested additional GAL4 drivers that label these cells (R15C03-GAL4 and R60D05-GAL4)5,9,10. Unexpectedly, thermogenetic activation using these drivers did not enhance sleep (Figure S1I). We hypothesized that these phenotypic differences could arise from 1) differences in driver strength, 2) a requirement for a distinct subset, or 3) a need for a sufficient number of EPG cells. To address issues related to driver strength, we repeated these experiments using a stronger effector (20XUAS-dTRPA1). However, under these conditions, sleep was still not induced using the R60D05-GAL4 and R15C03-GAL4 drivers (Table S1). Interestingly, examination of Multi-Color Flp Out (MCFO)11 data from the HHMI Janelia imaging database12 suggests relatively limited expression in EPG neurons for the R15C03-GAL4 driver, compared to R19G02-GAL4 (R60D05-GAL4 was not included in this MCFO analysis). Thus, we next combined the R60D05-GAL4 + R15C03-GAL4 drivers to perform thermogenetic activation, which resulted in increased sleep amount (Figures S1H and S1I and Table S1). To examine the relationship between EPG cell number and the ability of a given driver to promote sleep, we performed cell counting analyses. The two EPG driver lines that do not promote sleep (R15C03-GAL4 and R60D05-GAL4) label the fewest presumed EPG neurons, whereas R19G02-GAL4, SS50574, and R15C03-GAL4+R60D05-GAL4 label higher numbers of these cells (Figure S1J). These data suggest that activation of a sufficient number of EPG neurons is required to promote sleep.
We next asked whether EPG neurons are required for baseline sleep or homeostatic regulation of sleep. To address this question, we expressed tetanus toxin in EPG neurons (R19G02-GAL4>UAS-TNT) and measured sleep under baseline conditions and following mechanical sleep deprivation (SD). Inhibiting synaptic transmission of EPG neurons did not affect baseline daily sleep amount or consolidation (Table S1), but significantly reduced “rebound sleep” after 12 hr SD (Figures 1F and 1G). Together, these findings suggest that the EPG neurons are sleep-promoting and contribute to the homeostatic regulation of sleep.
Sleep need increases EPG neuron activity via extrinsic inputs
We next investigated whether EPG neuron activity is altered with increased sleep need. We performed perforated patch-clamp recordings of EPG neurons following either 12 hr of mechanical SD or undisturbed sleep (control) (Figure 2A). These experiments revealed a ~2-fold increase in spontaneous firing rate of EPG neurons in SD vs control animals (Figures 2B and 2C). This increase in spontaneous spiking of EPG neurons following SD was also reflected by a reduced interspike interval and an increase in instantaneous action potential (AP) firing rate (Figures S2A and S2B). These data suggest that sleep loss induces an increase in EPG neuron activity.
Figure 2. EPG neuron activity is increased with greater sleep need.
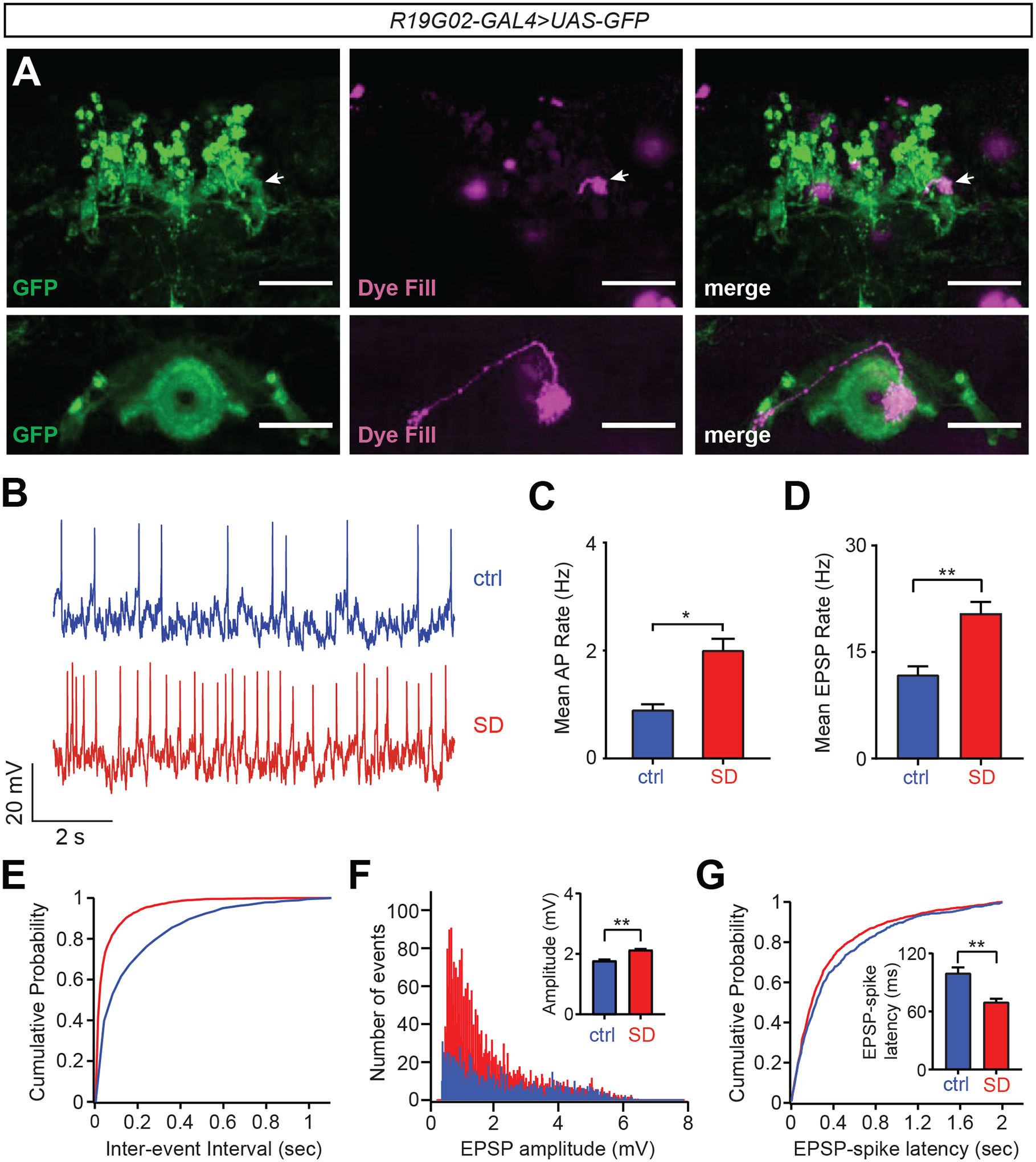
(A) Whole-mount brain immunostaining of R19G02-GAL4>UAS-CD4::tdGFP animal with anti-GFP (green) and single cell dye fill (biocytin, magenta) following electrophysiological measurements demonstrating labeling of EPG projections. Arrows point to dye-filled terminal overlapping GFP signal in the PB. Scale bar, 50 um.
B) Representative traces of membrane potentials of EPG neurons from R19G02-GAL4>UAS-CD4::tdGFP flies at ZT0-ZT3 in the presence and absence of 12 hr sleep deprivation (SD).
(C and D) Mean spontaneous action potential (AP) frequency (C) or mean spontaneous excitatory postsynaptic potential (sEPSP) frequency (D) of EPG neurons (R19G02-GAL4>UAS-CD4::tdGFP) at ZT0-3 (blue, n=5) and ZT0-3 with SD (red, n=4) flies; Student’s t-test.
(E) Cumulative probability plot of spontaneous excitatory postsynaptic potential (sEPSP) frequency from EPG neurons following SD (red) vs control without SD (blue).
(F) Distribution and quantification (inset) of sEPSP peak amplitude from EPG neurons in the presence (red) and absence (blue) of SD; Student’s t-test.
(G) Cumulative probability plot and quantification (inset) of EPSP-spike latencies for EPG neurons in ctrl (blue) vs SD (red) flies; Student’s t-test. Data in panels (C–G) are from the same flies. See also Figure S2.
We conducted additional analyses to address whether this increase in EPG activity following SD was due to changes in extrinsic inputs or intrinsic excitability. There was no change in resting membrane potential (RMP) or AP threshold in EPG neurons following SD (Figures S2C and S2D). In addition, measurement of evoked spiking frequency following current injection did not reveal a significant increase in overall intrinsic excitability of EPG neurons after sleep loss, compared to controls (although EPG spiking frequency was greater specifically with 100 pA current injections) (Figure S2E). These findings suggest that changes in extrinsic input, rather than intrinsic excitability, drive the increase in spontaneous firing of EPG neurons following sleep loss. To further address this possibility, we examined spontaneous excitatory post-synaptic potentials (EPSPs) in EPG neurons. As shown in Figures 2D and 2E, spontaneous EPSP frequency was greater in these cells after SD, compared to controls, suggesting an increase in excitatory synaptic inputs under these conditions. To assess for possible synaptic potentiation following SD, we examined EPSP amplitude and EPSP-spike coupling13. There was a significant increase in EPSP amplitude in EPG neurons under conditions of greater sleep need (Figures 2F and S2F). In addition, we observed a reduction in EPSP-spike latency in EPG cells following SD (Figures 2G and S2G), suggesting greater EPSP-spike coupling14. Together, these findings suggest that EPG neuron activity is increased with greater homeostatic sleep need, which is largely mediated by an increase in excitatory synaptic inputs onto these cells.
R5 neurons act upstream of EPG neurons to promote sleep
What is the presynaptic source of SD-triggered excitatory input to the EPG neurons? We hypothesized that the R5 neurons activate EPG neurons to signal sleep drive and induce sleep. However, the predominant mode of signaling within the EB ring is thought to be inhibitory (i.e., GABAergic)15,16. Thus, we first characterized the neurotransmitter identity of R5 neurons by performing immunostaining using antibodies against GABA, ChAT, and VGlut (Figure 3A). As expected, GABA neurotransmitter expression was observed throughout the ellipsoid body ring. Regarding R5 neurons specifically, colocalization of both anti-GABA and anti-ChAT signal with GFP signal was observed in R5 neuron axons (R30G03-AD,R58H05-DBD>UAS-CD8::GFP). In contrast, no vGluT signal was detected in the EB ring (Figure 3A). Next, to quantify the proportion of R5 neurons expressing different neurotransmitters, we performed double labeling experiments using R58H05-QF2>QUAS-tdTomato with anti-GABA antibody or ChAT- and VGluT-GAL4 MIMIC lines17 driving expression of Stinger-GFP. These analyses suggested that ~30% of R5 neurons are GABA+ (6.8 out of 22.8 per brain) and that ~30% of R5 neurons are ChAT+ (6.4 out of 20.4 neurons per brain). No overlap of R5 neurons was seen with neurons labeled by the VGluT MIMIC line (Figures 3B and 3C). Because we suspected that excitatory signals from R5 neurons act on EPG neurons to promote the homeostatic regulation of sleep, we performed ChAT knockdown in R5 neurons and assessed sleep before and after SD. We first confirmed that the UAS-ChAT-miR line could be used to knockdown ChAT expression (8411 ± 1266 arbitrary units (A.U.) for MB247-GAL4>+ (n=6), 8413 ± 770 A.U. for +>UAS-ChAT-miR (n=6), and 2212 ± 682 A.U. for MB247-GAL4>UAS-ChAT-miR (n=6) flies, P<0.001, one-way ANOVA with post-hoc Tukey’s test) (Figure S3A). Compared to controls, R58H05-GAL4>UAS-ChAT-miR flies exhibited a subtle reduction in rebound sleep after SD, but no change in baseline daily sleep (Figures 3D–3F).
Figure 3. R5 neurons act upstream of EPG neurons to promote sleep.
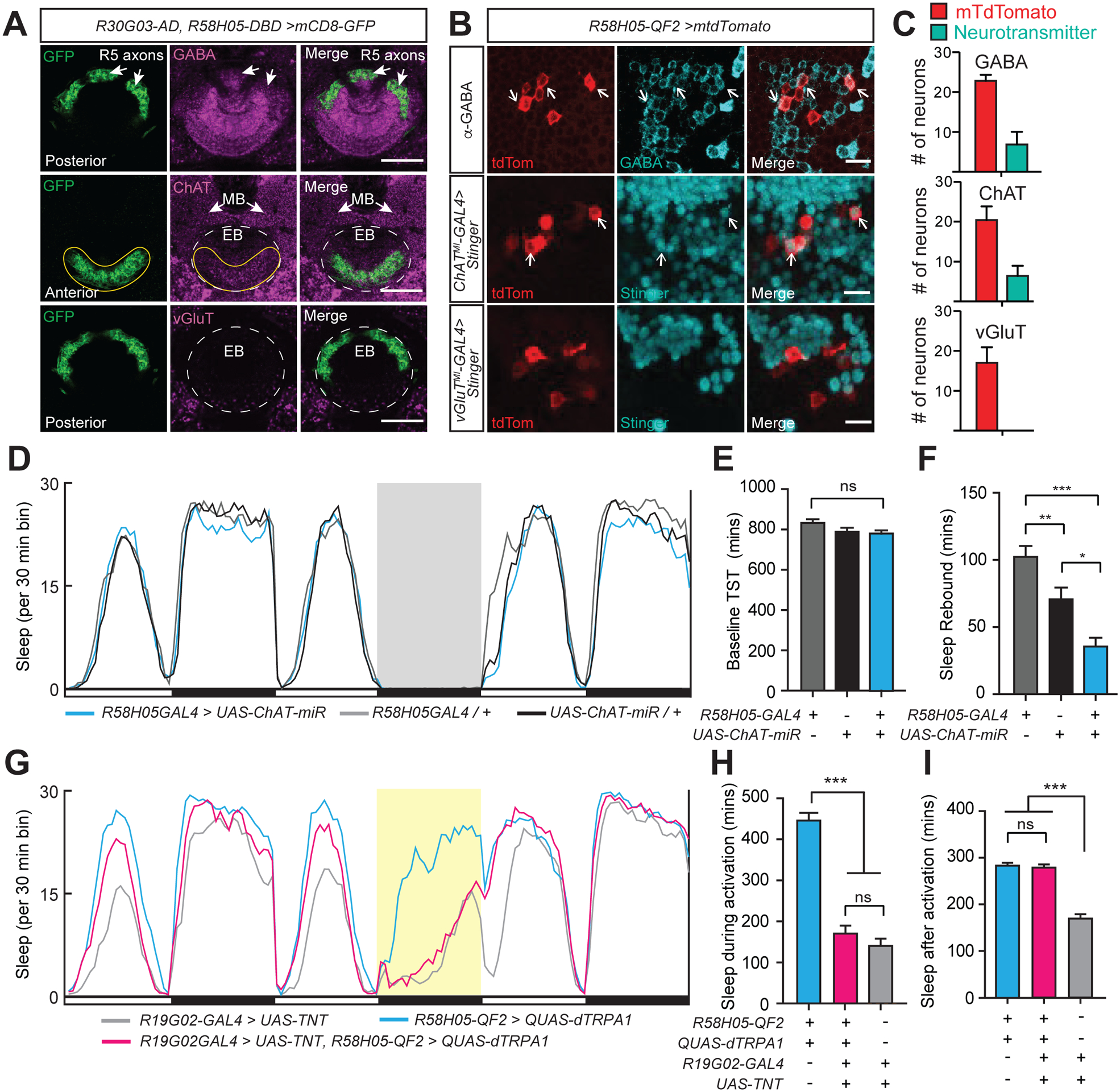
(A) Neurotransmitter staining of R5 ring neurons in an R30G03-AD, R58H05-DBD>UAS-mCD8::GFP animal. Whole-mount brain immunostaining showing anti-GFP (green), either anti-GABA, anti-ChAT, or anti-VGluT (magenta), and merged signals. Top row, posterior aspect, arrows indicate co-localization of R5 axons with GABA signal. Middle row, anterior aspect, ChAT staining colocalization with GFP signal is indicated (yellow solid line), while the location of EB ring is shown with white dashed lines. Arrows point to mushroom body (MB) lobes. Bottom, posterior aspect, no detectable overlap between vGLUT expression and GFP. EB ring is outlined with white dashed lines. Scale bars indicate 20 μm.
(B) Co-staining of R5 neurons and neurotransmitter-labeled cell bodies. R5 neurons are labeled with R58H05-QF2-7>mtdTomato (anti-dsRed, red). Neurotransmitter expression of cell bodies is labeled either by anti-GABA (top row) antibody (anti-GABA, green) or MIMIC line insertions for ChAT (middle row, ChATMI-GAL4>Stinger) or vGluT (bottom row, vGluTMI-GAL4>Stinger) driving Stinger (anti-GFP, green) expression. Arrows indicate R5 cell bodies which show co-labeling with neurotransmitter. Representative images are shown. Scale bar indicates 10 μm.
(C) Neuronal cell counts of R5 neurons and co-labeling with GABA, ChAT, and vGluT. In R58H05-QF2-7>mtdTomato animals stained with anti-GABA (n=6), an average of 6.8 out of 22.8 R5 neurons per brain were co-stained with GABA. For R58H05-QF2-7>mtdTomato animals with ChATMI-GAL4>Stinger (n=8), an average of 6.4 out of 20.4 R5 neurons per brain were cholinergic. No R5 neurons in R58H05-QF2-7>mtdTomato, vGluTMI-GAL4>Stinger animals (n=8) were observed to have vGluT co-labeling. Mean ±SEM is shown. These data are quantified from imaging experiments in Figure 3B.
(D) Sleep profiles of R58H05-GAL4>UAS-ChAT-miR (blue), R58H05GAL4>iso31 (gray), and iso31>UAS-ChAT-mir (black) flies. Sleep time plotted in 30 min bins. White and black bars indicate 12 hr light and dark periods, respectively. The period of sleep deprivation via mechanical shaking is indicated with a gray background. Data are from same flies as in (E) and (F).
(E and F) Baseline total sleep time (TST) (E) and rebound sleep from ZT0-6 (F) for R58H05GAL4>iso31 (n=64), iso31>UAS-ChAT-miR (n=60), and R58H05-GAL4>UAS-ChAT-miR (n=60). Mean ± SEM is shown; one way ANOVA with Tukey’s post hoc test.
(G) Sleep profiles of R58H05-QF2-A>QUAS-dTRPA1; R19G02-GAL4>UAS-TNT (magenta), R19G02-GAL4>UAS-TNT (gray), and R58H05-QF2-A>QUAS-dTRPA1 (blue) flies. Sleep time plotted in 30 min bins. White and black bars indicate 12 hr light and dark periods, respectively. The period of dTRPA1 activation at 29°C from ZT12-24 is indicated with a yellow background. Data are from same flies as in (H) and (I).
(H and I) Sleep during (H) and after (I, ZT0-6) dTRPA1 activation for R58H05-QF2-A>QUAS-dTRPA1 (n=48), R58H05-QF2-A>QUAS-dTRPA1; R19G02-GAL4>UAS-TNT (n=53), and R19G02-GAL4>UAS-TNT (n=56) flies. Mean ± SEM is shown; one way ANOVA with Tukey’s post hoc test. See also Figure S3.
We previously demonstrated that activation of R5 neurons promotes sleep both during and after the activation1,2. Recent work has found that some GAL4 drivers labeling R5 neurons also express in peripheral pickpocket-expressing (ppk) neurons18. Ppk+ nociceptive neurons are found in the periphery on legs and in the fly abdominal body wall and have been shown to promote arousal and subsequent “rebound” sleep19,20. We confirmed that R30G03-GAL4 exhibits expression in peripheral neurons in the legs and abdominal body wall, while R69F08-GAL4 and R46C03-GAL4 labels peripheral neurons in the abdominal body wall (Figure S3B). In contrast, R58H05-GAL4 did not drive detectable expression in peripheral neurons in the legs or abdomen, as was also the case for 2 split-GAL4 drivers (R58H05-AD, R46C03-DBD and R30G03-AD, R58H05-DBD) (Figure S3C and S3D). Thermogenetic activation of these drivers, as well as R58H05-GAL4 combined with 2 copies of ppk-GAL80, recapitulated the increase in sleep during and after heat treatment (Figures S3E–S3J).
To investigate a role for EPG neurons acting downstream of R5 neurons in promoting sleep, we performed epistasis experiments. To do this, we first characterized an R58H05-QF2 line that we generated and found minimal to no labeling of peripheral neurons, expression in the R5 ring with minimal expression in the VNC, and that it induced sleep during and after thermogenetic activation (Figures S3C, S3D, S3F, and S3G). Inhibiting synaptic transmission from EPG neurons markedly suppressed the increased sleep induced by thermogenetic activation of R5 neurons, whereas the sleep amount after activation was not affected (Figures 3G–3I). These findings support a model whereby R5 neurons signal to EPG neurons to promote sleep.
Sleep pressure triggers increased morphological and functional connectivity between R5 and EPG neurons
We previously showed that R5 neurons undergo plastic changes (as measured by an increase in the active zone marker Bruchpilot/BRP) following sleep loss1. Thus, we postulated that R5 neuron plasticity may enhance their physical and functional connectivity with EPG neurons. First, we asked whether R5 neurons exhibit morphological changes following SD. We performed single cell dye-fills of R5 neurons using biocytin in the presence and absence of 12 hr SD. Following SD, R5 neurons exhibited a greater number of presynaptic bouton-like puncta, as well as increased thickness of the ring structure in the anterior-to-posterior axis, compared to controls. Importantly, 12 hr recovery sleep reversed these morphological changes (Figures 4A–4E). To examine whether the physical connectivity between the R5 and EPG neurons is influenced by sleep need, GFP Reconstitution Across Synaptic Partners (GRASP)21 experiments were performed in the presence and absence of SD. While GRASP signal was detected in both 12 hr SD and baseline sleep conditions, the relative GRASP signal between R5 and EPG neurons was significantly increased following SD (Figures 4F–4H). These data suggest that sleep loss triggers structural plasticity in R5 neurons and strengthens physical connectivity between R5 and EPG neurons.
Figure 4. Functional Connectivity between R5 and EPG Neurons increases with Greater Sleep Need.
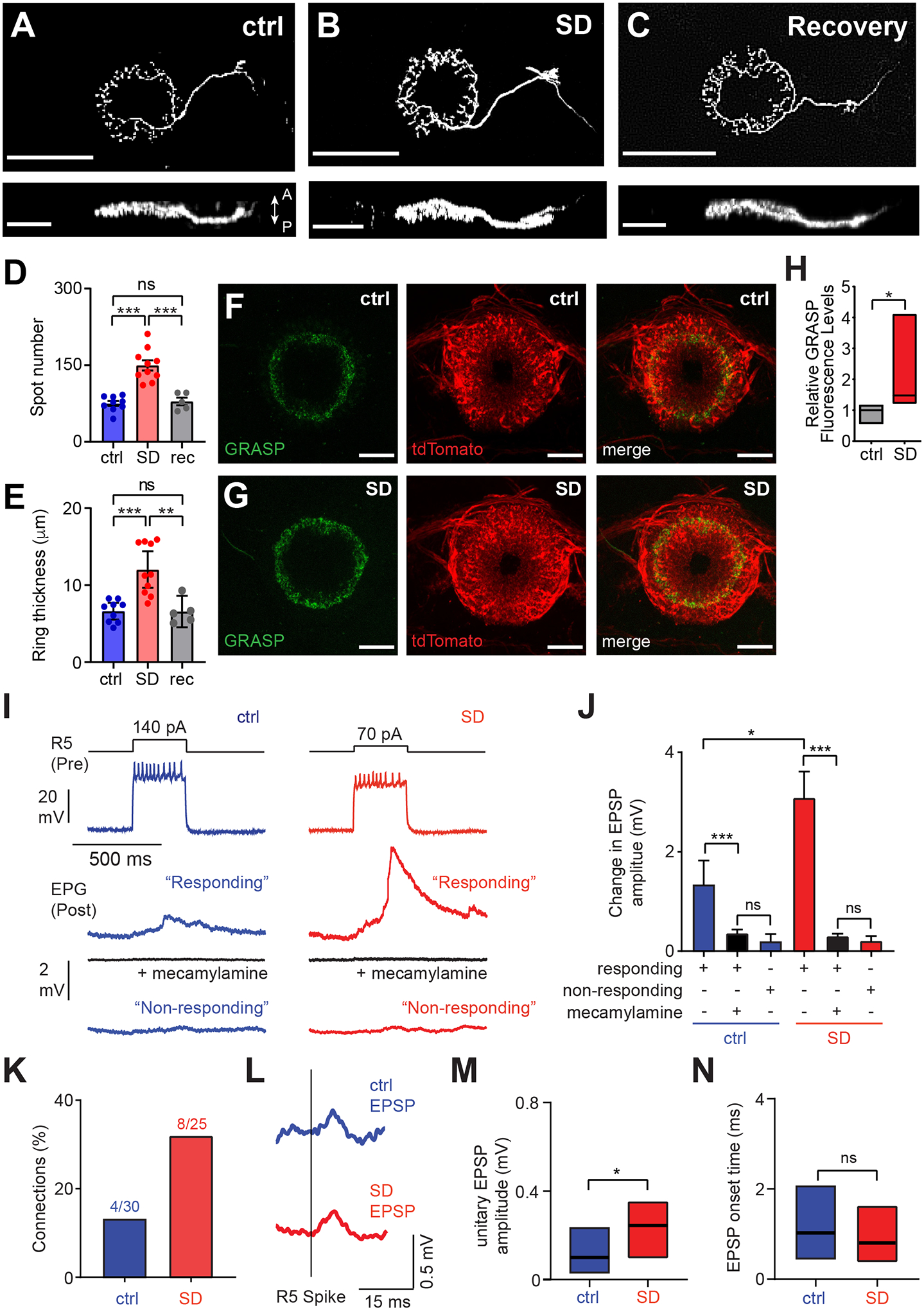
(A–C) Representative single cell dye-fill images of R5 neurons in R58H05-GAL4>UAS-CD4::tdGFP animals after baseline sleep at ZT0-3 (A, ctrl), mechanical sleep deprivation at (SD) from ZT12-24 (B), or 12 hr recovery after SD (C). Upper and lower panels provide anterior/posterior and dorsal/ventral views, respectively. Scale bars denote 50 μm and 25 μm in upper and lower panels, respectively. Arrows indicate anterior (A) and posterior (P) directions.
(D and E) Number of spots in the ring region (D) and anterior-posterior ring thickness (E) of R5 neurons in control (blue, n=9), SD (red, n=10), and 12 hr recovery (gray, rec, n=5) animals, as described in (A–C); one-way ANOVA with post-hoc Tukey’s test.
(F and G) Representative EB ring GRASP, tdTomato, and merged signals for R5 and EPG neurons in R30G03-AD, R58H05-DBD>UAS-syb-spGFP1-10; R19G02-LexA>LexAop-CD4-spGFP11, LexAop-tdTomato animals with (G) and without SD (F). Scale bar denotes 20 μm.
(H) Relative GRASP fluorescence levels (GFP/tdTomato) for the flies described in Figures (F) and (G) in the absence (gray, n=4) and presence of 12 hr SD (red, n=7); Mann-Whitney U test. Simplified box plots show 25th, 50th, and 75th percentiles.
(I) Traces from paired recordings of R5 (presynaptic, Pre) and EPG (postsynaptic, Post) neurons from R19G02-GAL4, R58H05-GAL4>UAS-CD4::tdGFP flies in the absence (ctrl, blue) or presence (SD, red) of SD from ZT12-24. Activation of R5 neurons elicited small but detectable depolarization responses in paired EPG neurons at baseline, whereas concomitant SD triggered a greater response in paired EPG cells. Top: Representative R5 membrane voltage in response to a depolarizing current injection (140 pA and 70 pA for ctrl and SD conditions, respectively). Middle: Averaged traces of simultaneously recorded EPG membrane voltages from cells exhibiting a “response”, without or with 50 μM mecamylamine (black). Bottom: Averaged traces of simultaneously recorded EPG membrane voltage showing “no response.”
(J) Mean EPG membrane voltage change in the paired recordings described in (I), for “responding” cells in the presence and absence of mecamylamine and “non-responding” cells in animals at ZT0-3 following baseline sleep (ctrl, n=30) or 12 hr SD (SD, n=25); two-way ANOVA with post hoc Tukey’s test.
(K) Frequency of synaptic connections identified between R5 and EPG pairs, in the presence (red) or absence (blue) of SD.
(L) R5 spike-triggered average of putative unitary EPG EPSPs in the presence (red) or absence (blue) of SD. Timing of R5 spikes is shown.
(M and N) Amplitude (M) and onset time (defined as a response reaching 10% of EPSP peak) (N) of R5 spike-triggered EPG EPSPs; Mann-Whitney U tests. Simplified box plots show 25th, 50th, and 75th percentiles. Data shown in panels (I)-(N) are from the same flies. See also Figure S4.
To assess whether these morphological changes produce a functional increase in synaptic strength, we performed dual patch-clamp recordings of R5 and EPG neurons after 12 hrs of SD or baseline sleep. Because the cell bodies of these two groups of neurons are on opposite sides of the fly brain, we developed a novel electrophysiological preparation to conduct these studies, where the brain is mounted vertically (Figure S4A). Owing to their increased excitability following sleep loss1, smaller currents were injected into R5 neurons in SD brains to yield levels of firing similar to controls (Figure 4I). EPG spike-triggered EPSP amplitude was significantly increased following SD, compared to controls. Furthermore, in control brains, spike-triggered EPSPs in EPG neurons were observed in only a minority (4/30) of recordings. In contrast, after SD, a greater proportion of brains exhibited a postsynaptic response (8/25) (Figures 4I–4K). To confirm that the EPG membrane potential (Vm) changes were dependent on R5 firing, we compared Vm changes triggered by R5 spikes with those occurring at random. As expected, Vm changes triggered by R5 spikes were significantly larger than background Vm fluctuations, and this difference was greater after SD (Figures S4B–S4D).
We next asked whether cholinergic R5 neurons signal to EPG neurons in our dual patch-clamp recordings. EPG responses to R5 firing were essentially abolished upon application of mecamylamine, a nicotinic acetylcholine receptor antagonist (Figures 4I and 4J), suggesting that cholinergic signaling from R5 neurons triggers excitatory post-synaptic responses in EPG neurons. We next examined unitary EPSPs from EPG neurons following individual R5 spikes. The amplitude of these unitary EPSPs was increased following SD, compared to controls (Figures 4L and 4M), further demonstrating potentiation of R5-EPG synapses with greater sleep need. To assess whether the connection between R5 and EPG is monosynaptic, we quantified EPSP onset times from unitary EPG EPSPs triggered by R5 spikes. The EPSP onset time (~1 ms) was comparable to that reported for monosynaptic connections in the Drosophila antennal lobe22,23 and was not altered by changes in sleep need (Figure 4N). These data demonstrate that sleep loss induces an increase in the likelihood and strength of EPG postsynaptic responses following R5 firing.
Although previous studies have suggested that most EB ring neuron signaling is GABAergic15,16, cholinergic EB ring subpopulations have been described24. Our findings suggest that cholinergic R5 neurons signal to EPG neurons to promote sleep. However, given the modest effects on sleep associated with R5 ChAT knockdown, additional signaling mechanisms likely play a role in this process. Our data further show that sleep loss enhances the anatomical and functional connectivity between R5 and EPG neurons, which appears to be driven primarily by presynaptic plasticity in the R5 neurons. Synaptic plasticity triggered by homeostatic drive is unusual25,26,27 and may promote persistence of homeostatically-generated behaviors under conditions of significant internal need. Interestingly, while inhibiting EPG neurotransmission largely blocks the sleep-promoting effects of activating R5 neurons, it does not impact the persistent sleep following this activation. This finding suggests that multiple, redundant mechanisms may underlie the sleep persistence phenotype seen with R5 activation.
EPG neurons have been shown to encode directional heading5,6,9,10,28. How does one reconcile a role for these neurons in navigation with a potential role in homeostatic sleep regulation? We find that a large number of EPG neurons must be activated to promote sleep behavior. In addition, our dual patch-clamp recordings reveal that, not only does the synaptic strength between R5 and EPG neurons increase with greater sleep need, but also the proportion of these functional connections. Thus, we speculate that the R5/EPG synapse acts as a “gate” for transmission of homeostatic sleep drive. In this model, under baseline conditions, EPG neurons function as compass neurons and receive broad, inhibitory inputs29,30 and weak and infrequent excitatory R5 inputs. Following sleep deprivation, plasticity of R5 neurons is induced1,31, enhancing R5→EPG excitatory signaling. Moreover, sleep loss promotes synchronization of R5 neuron activity32, which could further strengthen and coordinate sleep need-dependent R5 signaling to EPG neurons. These changes could lead to activation of a greater number of EPG neurons and promote recovery sleep.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for data, resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Mark N. Wu (marknwu@jhmi.edu).
Materials Availability
Transgenic Drosophila strains generated for this manuscript are available upon request to the lead contact.
Data and Code Availability
Raw data, analysis files, and images generated for this manuscript are available upon request to the lead contact.
Jupyter notebooks are available upon request from the lead contact. Additional code for this manuscript is available on Github (https://github.com/margiezilla/Ho_etal_2022).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Adult female Drosophila melanogaster of age 4–10 days old were used for all experiments. Flies were reared on standard food containing molasses, cornmeal, and yeast under a 12:12 h LD cycle at 25°C, with the exception of crosses utilizing dTRPA1 (in which case flies were reared at 23°C).
METHOD DETAILS
Transgenic Fly Strains
All GMR GAL4 and LexA lines were obtained from the Bloomington Drosophila Stock Center36,37. Other flies were obtained as described in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GABA | Sigma | Cat# A2052 RRID: AB_477652 |
| Mouse monoclonal anti-ChAT | Developmental Studies Hybridoma Bank | Cat# 4B1 RRID: AB_528122 |
| Rabbit polyclonal anti-vGluT | Daniels et al.33 | RRID: AB_2567386, |
| Chicken polyclonal anti-GFP | Invitrogen | Cat# A10262 RRID: AB_2534023 |
| Rabbit polyclonal anti-dsRed | Clontech | Cat # 632496 RRID: AB_10013483 |
| Mouse monoclonal anti-BRP | Developmental Studies Hybridoma Bank | Cat# nc82 RRID: AB_2314866 |
| Alexa Fluor 488 Goat anti-Chicken IgY | Invitrogen | Cat# A11039 RRID: AB_2534096 |
| Alexa Fluor 568 Goat anti-Mouse IgG | Invitrogen | Cat# A11031 RRID: AB_144696 |
| Alexa Fluor 647 Goat anti-Mouse IgG | Invitrogen | Cat# A21235 RRID: AB_2535804 |
| Alexa Fluor 568 Goat anti-Rabbit IgG | Invitrogen | Cat# A11011 RRID: AB_143157 |
| Rabbit polyclonal anti-GFP | Thermo Fisher | Cat# A11122 RRID: AB_221569 |
| Alexa Fluor 488 Goat anti-Rabbit IgG | Thermo Fisher | Cat# A27034 RRID: AB_2536097 |
| Alexa Fluor 568-conjugated streptavidin | Thermo Fisher | Cat# S11226 |
| Bacterial and virus strains | ||
| N/A | ||
| Biological samples | ||
| N/A | ||
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase | Sigma | Cat# C5138 |
| Protease XIV | Sigma | Cat# P5147 |
| Dispase | Sigma | Cat# D4693 |
| Escin | Santa Cruz Biotechnology | Cat# 6805-41-0 |
| Mecamylamine | Sigma | Cat# M9020 |
| Critical commercial assays | ||
| N/A | ||
| Deposited data | ||
| N/A | ||
| Experimental models: Cell lines | ||
| N/A | ||
| Experimental models: Organisms/strains | ||
| iso31 | Bloomington Drosophila Stock Center | BDSC# 5905 |
| R19G02-GAL4 | Bloomington Drosophila Stock Center | BDSC# 48860 |
| SS50574 (VT017491-AD, VT043927-DBD) | Janelia | SS50574 |
| R60D05-GAL4 | Bloomington Drosophila Stock Center | BDSC# 39247 |
| R15C03-GAL4 | Bloomington Drosophila Stock Center | BDSC# 47865 |
| R58H05-GAL4 | Bloomington Drosophila Stock Center | BDSC# 39198 |
| R46C03-GAL4 | Bloomington Drosophila Stock Center | BDSC# 50258 |
| R30G03-GAL4 | Bloomington Drosophila Stock Center | BDSC# 49646 |
| R69F08-GAL4 | Bloomington Drosophila Stock Center | BDSC# 39499 |
| R58H05-QF2 #7 | Blum et al.2 | |
| R58H05-QF2 #A | Wu Lab | |
| R58H05-AD (86Fb), R46C03-DBD (vk27) | Blum et al.2 | |
| R30G03-AD (86Fb), R58H05-DBD (vk27) | Liu et al.1 | |
| R19G02-LexA | Bloomington Drosophila Stock Center | BDSC# 52550 |
| UAS-ChAT-miR1-A | Wu Lab | |
| UAS-mCD8-GFP | Bloomington Drosophila Stock Center | BDSC# 5137 |
| UAS-dTRPA1 | Bloomington Drosophila Stock Center | BDSC# 26263 |
| UAS-TNT | Bloomington Drosophila Stock Center | BDSC# 28838 |
| UAS-CD4-tdGFP | Bloomington Drosophila Stock Center | BDSC# 35836 |
| UAS-mtdTomato | Bloomington Drosophila Stock Center | BDSC# 30124 |
| ChaTMI04508-GAL4 | Bloomington Drosophila Stock Center Diao et al.17 | BDSC# 60317 |
| vGluTMI04979-GAL4 | Bloomington Drosophila Stock Center. Diao et al.17 | BDSC# 60312 |
| UAS-Stinger | Bloomington Drosophila Stock Center | BDSC# 84277 |
| QUAS-dTRPA1-7 | Gift of C. Potter | |
| UAS-spGFP1-10 | Bloomington Drosophila Stock Center | BDSC# 93016 |
| lexAop-CD4-spGFP11 | Bloomington Drosophila Stock Center | BDSC# 93019 |
| LexAop-tdTomato | Bloomington Drosophila Stock Center | BDSC# 77138 |
| UAS-syt-GFP, UAS-DenMark | Bloomington Drosophila Stock Center | BDSC# 33065 |
| 10XUAS-syn21-GFP-p10 (attP2) pJFRC81 | Gift of G. Rubin | Pfeiffer et al.34 |
| 20XUAS-IVS-dTRPA1 (attP18) pJFRC124 | Gift of G. Rubin | |
| ppk-Gal80 | Gift of W. Joiner | |
| MB247-GAL4 | Bloomington Drosophila Stock Center | BDSC# 50742 |
| 20XUAS-6XGFP | Bloomington Drosophila Stock Center | BDSC# 52261 |
| 10XQUAS-6XGFP | Gift of C. Potter | |
| Oligonucleotides | ||
| Oligo 5’
ATAGAATTCCGCCAGATCTTTTAAAGTCCACAACTCATCAAGGAAAAT GAAAGTCAAAGTTGGCAGCTTACTTAAACTTAATCACAGCCTTTAAT GTACCGCTGGCTGGGAACTTTCATTAAGTTAATATACCATATCTAATTAAAGT TCCCAGCCAGCGGTGTACCTAAAGTGCCTAACATCATTATTTAATTTTTTTTTTTTT TGGCACACGAATAACCATGCCGTTTTGGATCTTTTAAAGTCCACAACTCATCAAGGAAAATGAAAGTCAAAGTTGGCAGCTTAC TTAAACTTAATCACAGCCTTTAATGTAGATGCACGAGCTGTTCAAC GATAAGTTAATATACCATATCTATCTTTGAACAGCTCGTGCATCTGTACCTAAA GTGCCTAACATCATTATTTAATTTTTTTTTTTTTTGGCACACGAATAACCATGCCGTTTTGGATCCGTGCGGCCGCAAC 3’ |
GeneArt | UAS-ChaT-miR |
| Recombinant DNA | ||
| pUAST | Brand and Perrimon35 | |
| Software and algorithms | ||
| FIJI | Open Source |
https://imagej.net/software/fiji/ RRID: SCR_002285 |
| MATLAB | Mathworks | RRID: SCR_001622 |
| Custom MATLAB Scripts for sleep analysis | Blum et al.2 | https://github.com/marknwulab/Blum-et-al.-2020 |
| Custom Python code in Jupyter Notebook | Wu Lab | https://github.com/margiezilla/Ho_etal_2022 |
| Prism 7 | Graphpad | RRID: SCR_002798 |
| DAMFileScan311 | Trikinetics | N/A |
| DAMFileScan113 | Trikinetics | N/A |
| Zen Black | Zeiss | RRID:SCR_013672 |
| Jupyter Notebook | https://jupyter.org/ | RRID:SCR_018315 |
| Python Programming Language | http://www.python.org/ | RRID:SCR_008394 |
| Other | ||
| Dental Wax | GC Corp | Cat# 27B2X00008000016 |
| DAM2 Activity Monitors | Trikinetics | Cat# DAM2 |
| LC-4 Light Controller | Trikinetics | Cat# LC4 |
| PSIU9 Power Supply Interface | Trikinetics | Cat# PSIU9 |
| Vortexer with Vortex Mounting Plate | Trikinetics | Cat# TVOR-120 |
| Laser-cut Adaptor Plate for Vortexer | Wu Lab | https://github.com/marknwulab/Blum-et-al.-2020 |
| DVX-2500 Digital Multi-tube Vortexer | VWR | DVX-2500 |
Molecular Biology
The ChAT microRNA (miR) was constructed as previously described38. For UAS-ChaT-miR, two 22-mers in exon 4 (5’-ACCGCTGGCTGGGAACTTTAAT-3’) and exon 7 (5’-AGATGCACGAGCTGTTCAAAGA-3’) were used to create the two hairpin loops. The miR sequence was synthesized in vitro (GeneArt) and subcloned into pUAST using EcoRI and NotI. The UAS-ChAT-miR1-A line was generated via random P-element mediated insertion into iso31 flies and screened for viability and general health. The R58H05-QF2-7 line was as described in Blum et al., 20212. The R58H05-QF2-A line was generated by re-injecting the construct into iso31 flies using P-element mediated random insertion and was selected to avoid expression in peripheral tissues. The R58H05-p65AD and R46C03-GAL4 DBD lines were generated as described in Blum et al., 20212.
Immunostaining
Neurotransmitter immunostaining was performed as previously described16. The following primary antibodies were used: rabbit-anti-GABA (1:500, Sigma, A2052, RRID: AB_477652), mouse-anti-ChAT (1:100, DSHB 4B1, RRID: AB_528122), and rabbit-anti-vGluT (1:5000, RRID: AB_2567386). Secondary antibodies were raised in goat against rabbit, chicken and mouse antisera (Invitrogen) and conjugated to Alexa Fluor 488 (1:1000), Alexa Fluor 555 (1:1000) or Alexa Fluor 647 (1:300). Antibodies were prepared in blocking buffer with 0.02% NaN3, and primary antibodies were reused several times. Whole mount brain immunostaining for other antigens was performed as previously described16. The following primary antibodies were used: chicken-anti-GFP (1:1000, Invitrogen A10262, RRID:AB_2534023), rabbit-anti-dsRed (1:1000, Clontech, 632496, RRID: AB_10013483) and mouse-anti-BRP (1:10, DSHB nc82, RRID: AB_2314866). Secondary antibodies were raised in goat against chicken, rabbit, or mouse antisera (Invitrogen) and conjugated to Alexa Fluor 488 (1:1000), or Alexa Fluor 568 (1:1000) or Alexa Fluor 647 (1:300). For counting putative EPG cells labeled by various drivers in Figure S1J, single or combined transgenic driver lines were used to express mCD8::GFP, and immunostaining was performed as described. GFP+ cells with expected projections to the PB were counted as EPG cells. In addition, in cases where their projections could not be clearly visualized, GFP+ cells of similar size immediately surrounding those cells were counted as putative EPG neurons. To quantify ChAT knockdown in Figure S3A, a single Z-slice (1 μm) that captured the thickest portion of the mushroom body γ-lobe was chosen for analysis. Using ImageJ, a region of interest (ROI) was drawn around a single γ-lobe per brain, and the average fluorescence intensity was measured.
Behavioral measurements
Strains used in behavioral experiments were outcrossed at least four times to iso31 background (RRID: BDSC_5905), and 5–8 day old female flies were used for all behavioral experiments. Sleep behavior was measured using the Drosophila Activity Monitoring system (Trikinetics) and established metrics as previously described39. Sleep parameters were analyzed using custom MATLAB scripts2, and additional post-processing analysis was performed with Python using Jupyter notebook (https://github.com/margiezilla/Ho_etal_2022). Flies were loaded in sucrose locomotor tubes (5% sucrose, 2% agar) for all behavioral experiments. For UAS-dTRPA1 experiments, adult flies were raised at 23°C. 1 day of baseline sleep was recorded at 22°C and then the temperature was raised to 29°C during ZT12-24 during the second night. The temperature was returned to 22°C starting at ZT0 the following day. For arousal threshold measurements during dTRPA1 activation at 29C, flies were subjected to stimuli of increasing intensity and duration using a digital vortexer (500RPM for 1s (0.2g), 2s (0.25g), and 4s (0.35g) at ZT16, ZT18, ZT20 and ZT22). Asleep flies were defined as flies not moving for the 5 minutes (no beam crosses detected) prior to stimulus, whereas awake flies were those observed to be moving. Aroused flies were identified as flies moving within the 3 min interval following the stimulus. For sleep deprivation experiments, adult flies were raised at 25°C and assayed at 25°C. Sleep deprivation was performed by mechanical stimulation, by shaking 2–3 sec/minute in a digital vortexer (VWR DVX-2500 Multi-tube vortexer) at 1300–2000 RPM from ZT12-24. Homeostatic sleep rebound from the following 6 hours (ZT0-6) was then recorded and compared to previous daytime sleep to calculate rebound sleep. Only flies exhibiting ≥95% sleep deprivation were used in analyses. Locomotor climbing performance following thermogenetic activation of EPG neurons was performed essentially as previously described40,41. 10–12 flies (5 days old) were exposed to 29°C for 2 hrs (ZT0-2) and then tapped to the bottom of a clean plastic vial with another vial placed above. The proportion of flies climbing above a 10 cm mark was measured after 10 s. To assess peak locomotor activity during thermogenetic activation of EPG neurons, we measured the maximum number of beam crossings within 1 min for the 5 min window immediately following the strong shaking stimuli shown in Figures 1D and 1E.
Single cell dye labeling of R5 neurons.
Single cell dye labeling of R5 neurons was performed by iontophoretic injection of biocytin for at least 15 min in order to quantify the puncta number and ring thickness of an R5 neuron. After iontophoretic injection of biocytin, the brain was fixed in 4% paraformaldehyde in Phosphate Buffered Saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.7 mM KH2PO4, pH 7.4) overnight at 4°C. After washing for 1 hr in several changes of PBST (0.3% Triton X-100 in PBS) at room temperature, the brain was incubated with rabbit anti-GFP antibodies (Thermo Fisher A11122, 1:200) for 16–40 hrs on a shaker at 4°C, followed by incubation with fluorescent Alexa Fluor 488 anti-rabbit (Thermo Fisher A27034, 1:1000) secondary antibodies and Alexa 568-conjugated streptavidin (Thermo Fisher S11226, 1:100) for 24–40 hrs on a shaker at 4°C. After a 1 hr wash, samples were cleared in 70% glycerol in PBS for 5 min at room temperature and then mounted in Vectashield (Vector Labs). Brains were then imaged using a confocal imaging system (LSM-700; Carl Zeiss). Serial optical sections were acquired at 0.7–1.0 μm intervals.
To quantify spot number and ring thickness of dye-injected R5 neurons, we used Imaris image analysis software (Imaris x64 9.7.1, Oxford instruments). To measure spot number, we first manually measured representative spot diameter and applied this variable as the “Estimated Diameter.” We selected “quality” as the filter type for our analysis. We then manually tuned the quality parameter until every punctate region of interest on the R5 axonal ring was covered by a spot. Typically, only a single class (“Class A”) was generated based on the average distance to 3 nearest neighbors, but if the algorithms returned more than one class (e.g. “Class A” and “Class B”), only class A was selected for analysis because other classes were below the distance threshold (i.e, too close together). Finally, we manually removed spots outside the R5 ring structure and selected “Total Number of Spots” as the output.
To measure anterior-posterior R5 ring thickness, we used the “Measurement Points” function in Imaris. We selected the thickest area of the structure and quantified anterior-posterior thickness with measurement points on the anterior and posterior surfaces. To ensure the only difference in measurement was on the z-axis, we synchronized the x- and y-axis of the two points
Connectivity using GRASP
GRASP21 was used to assess physical connectivity between R5 and EPG neurons. The R30G03-p65AD; R58H05-DBD and R19G02-LexA were used to drive expression of UAS-CD4-spGFP1-10 in R5 neurons and LexAop-CD4-spGFP11 and LexAop-tdTomato in EPG neurons, respectively. These flies were subjected to mechanical SD or undisturbed (non-SD) from ZT12-24 as described above. The brains of SD and non-SD control flies were quickly dissected in cold AHLS (2 mM CaCl2, 108 mM NaCl, 5 mM KCl, 8.2 mM MgCl2, 4 mM NaHCO3, 1 mM NaH2PO4, 5 mM HEPES, 10 mM Sucrose, 5 mM Trehalose, pH 7.5) and native GFP fluorescence was imaged with confocal microscopy (Zeiss LSM700) using a 63x oil immersion objective. Relative quantification of GRASP fluorescence levels was performed by measuring GFP signal against tdTomato. Stacks containing the EB region labeled by tdTomato signal were projected into images by using the “average intensity” function in ImageJ. The ROI was also determined by tdTomato signal labeling the EB neuropil. The “Analyze -> Measure” function of ImageJ was used to quantify the GFP and tdTomato fluorescence intensity in the ROI.
Electrophysiological recordings
The procedures were performed as previously described42. Sleep deprivation was performed using mechanical stimulation with a multi-tube digital vortexer (VWR VX-2500) at 5 s/min from ZT12-ZT24, and flies with >90% sleep loss during this period were used for electrophysiological recordings. Recordings were performed at ZT0-3.
ex vivo preparation
Flies were chilled on ice for anesthesia and immobilized on a dissecting chamber following isolation of the head. The brain was exposed by opening the head capsule and dissected in a Drosophila physiological saline solution (101 mM NaCl, 3 mM KCl, 1 mM CaCl2, 4 mM MgCl2, 1.25 mM NaH2PO4, 20.7 mM NaHCO3, and 5 mM glucose; pH 7.2). The tracheae and the intracranial muscles were removed. To better visualize the recording site and increase the likelihood of a successful recording, the glial sheath surrounding the brain was focally and carefully removed after treating with an enzymatic cocktail, collagenase (0.2 mg/mL), protease XIV (0.4 mg/mL), and dispase (0.6 mg/mL), at 22°C for 1–2 min. The surface of the cell body was briefly cleaned with a small stream of saline that was pressure-ejected from a large-diameter pipette under visualization of a dissecting microscope. For dual recordings of R5 and EPG neurons, brains were mounted vertically to allow electrode access from both anterior and posterior sides. Brains were vertically immobilized on the bottom of the recording chamber, by placing dental wax (GC Corp., 27B2X00008000016) in front of and behind the brain.
Perforated patch-clamp recordings
Glass pipettes (8–12 MΩ) were fashioned from borosilicate capillaries with a Flaming/Brown puller (P-1000; Sutter Instrument). A final concentration of 50 μM escin (Santa Cruz Biotechnology) was added fresh into the internal pipette solution (102 mM potassium gluconate, 0.085 mM CaCl2, 0.94 mM EGTA, 8.5 mM HEPES, 4 mM Mg-ATP, 0.5mM Na-GTP, 17 mM NaCl; pH7.2). Because escin is light-sensitive, filling syringes were wrapped with aluminum foil. Pipette tips were dipped briefly for 1 s or less into a small container with escin-free internal pipette solution, and then were back-filled with the escin-containing solution from the filling syringe. 50 μM mecamylamine was used to isolate the cells from excitatory synaptic inputs for the experiments with pharmacological blockade. Perforated patches could develop spontaneously over time (usually ~1–8 min) without any suction pulse applied in the pipette. Single electrode recordings from EPG neurons were acquired with an Axopatch 200B amplifier (Molecular Devices), and dual recordings from R5 and EPG neurons were performed using two patch-clamp amplifiers–an Axopatch 200B amplifier and a Model 2400 amplifier with 100 MΩ headstage (A-M systems). The voltage signals were sampled at 20 kHz and lowpass filtered at 2 kHz.
Electrophysiology data analysis
To quantify spontaneous excitatory postsynaptic potentials (sEPSPs) from EPG neurons under single electrode current-clamp recording configuration, a median filter with a time constant of 3 ms was applied to the unfiltered membrane potential. Then, background noise power was computed based on root mean square values from the all-points amplitude in each dataset. These computations were used to define noise-rejection criteria to decide signal threshold27. Once sEPSPs were sorted, their amplitude was then defined as the difference between the maximum/minimum potential of each sEPSP. EPSP-spike coupling was defined as temporal distance between adjacent presynaptic R5 action potential (AP, preceding) and EPG EPSP (following) epochs. To examine the functional connectivity between R5 and EPG neurons under dual electrode current-clamp recording configuration, depolarizing current injections were applied to R5 cells to evoke spike trains, and the membrane potentials of EPG were simultaneously recorded. The change in EPG membrane voltage was calculated by subtracting the mean membrane potential before current injection from the mean membrane potential during the period of current injection. We carefully modulated the amount of injected current in every experiment so that we could induce a similar number of action potentials between R5 with and without sleep deprivation. This is particularly important, because the membrane excitability of R5 is increased after sleep deprivation1. Within the “responding” paired recordings, we sorted a 30 ms time window of membrane voltage traces for EPG centered on a simultaneously measured R5 spike to assess for a monosynaptic interaction between R5 and EPG neurons. By performing visual inspection, the sorted EPG membrane voltages were further categorized into detectable or undetectable unitary EPSPs based on the presence of a clear depolarization (typically >0.2 mV), as well as monotonic rise time kinetics and phase-locked latency indicating monosynaptic excitation. The sorted EPG membrane voltages categorized as detectable were quantified in their amplitudes of peak depolarization. To exclude the possibility that detected depolarization could be coincidentally observed in the absence of R5 spike events, we also quantified randomly chosen 30 ms membrane voltage traces of EPG and confirmed that there was no detectable depolarization. The monosynaptic EPSP onset time43 was defined as the time from the peak of R5 AP to the time reaching 10% of the peak depolarization after R5 AP. All analyses were performed using MATLAB (The MathWorks).
Quantification and Statistical Analysis
Statistical analyses were performed with Prism (GraphPad). For comparisons of two groups of normally distributed data, Student’s t tests were performed. For multiple comparisons of normally-distributed data, one-way or two-way ANOVAs followed by Tukey’s post-hoc tests were performed. For multiple comparison of non-normally distributed data, Kruskal-Wallis test followed by Dunn’s post-hoc test was performed.
Supplementary Material
Highlights.
Broad activation of EPG compass neurons promotes sleep in Drosophila
EPG activity increases with sleep need and facilitates homeostatic sleep rebound
R5 sleep drive neurons act upstream of EPG neurons
Sleep loss strengthens R5/EPG connectivity to enhance transmission of sleep drive
Acknowledgments
We thank T. Wolff, W. Joiner, V. Jayaraman, and G. Rubin (HHMI Janelia) for kindly sharing reagents. We thank A. Hutson, Y. Zhang, and Y. Xie for assistance with Imaris imaging and I.D. Blum for assistance with sleep behavior analysis. We thank the Bloomington Stock Center (supported by NIH grant P40OD018537) and the Vienna Drosophila Stock Center (www.vdrc.at) for fly stocks. This work was supported by NINDS Center Grant P30 BNS050274 for use of the Core Machine Shop and the Multiphoton Imaging Core, ERC Starting Grant 758580 (S.L.), the Howard Hughes Medical Institute (A.L.K.), and NIH grants K99NS117654 (M.C.W.H.), K99NS101065 (M.T.), F31NS117175 (M.B.), R01NS100792 (M.N.W.), and R35NS122181 (M.N.W.).
Inclusion and Diversity
We support inclusive, diverse, and equitable conduct of research.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Liu S, Liu Q, Tabuchi M, and Wu MN (2016). Sleep Drive Is Encoded by Neural Plastic Changes in a Dedicated Circuit. Cell 165, 1347–1360. 10.1016/j.cell.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blum ID, Keles MF, Baz ES, Han E, Park K, Luu S, Issa H, Brown M, Ho MCW, Tabuchi M, et al. (2021). Astroglial Calcium Signaling Encodes Sleep Need in Drosophila. Current biology : CB 31, 150–162 e157. 10.1016/j.cub.2020.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scheffer LK, Xu CS, Januszewski M, Lu Z, Takemura SY, Hayworth KJ, Huang GB, Shinomiya K, Maitlin-Shepard J, Berg S, et al. (2020). A connectome and analysis of the adult Drosophila central brain. Elife 9. 10.7554/eLife.57443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hulse BK, Haberkern H, Franconville R, Turner-Evans DB, Takemura SY, Wolff T, Noorman M, Dreher M, Dan C, Parekh R, et al. (2021). A connectome of the Drosophila central complex reveals network motifs suitable for flexible navigation and context-dependent action selection. Elife 10. 10.7554/eLife.66039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seelig JD, and Jayaraman V (2015). Neural dynamics for landmark orientation and angular path integration. Nature 521, 186–191. 10.1038/nature14446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green J, Adachi A, Shah KK, Hirokawa JD, Magani PS, and Maimon G (2017). A neural circuit architecture for angular integration in Drosophila. Nature 546, 101–106. 10.1038/nature22343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicolai LJ, Ramaekers A, Raemaekers T, Drozdzecki A, Mauss AS, Yan J, Landgraf M, Annaert W, and Hassan BA (2010). Genetically encoded dendritic marker sheds light on neuronal connectivity in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 107, 20553–20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang YQ, Rodesch CK, and Broadie K (2002). Living synaptic vesicle marker: synaptotagmin-GFP. Genesis 34, 142–145. 10.1002/gene.10144. [DOI] [PubMed] [Google Scholar]
- 9.Wolff T, Iyer NA, and Rubin GM (2015). Neuroarchitecture and neuroanatomy of the Drosophila central complex: A GAL4-based dissection of protocerebral bridge neurons and circuits. The Journal of comparative neurology 523, 997–1037. 10.1002/cne.23705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolff T, and Rubin GM (2018). Neuroarchitecture of the Drosophila central complex: A catalog of nodulus and asymmetrical body neurons and a revision of the protocerebral bridge catalog. The Journal of comparative neurology 526, 2585–2611. 10.1002/cne.24512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nern A, Pfeiffer BD, and Rubin GM (2015). Optimized tools for multicolor stochastic labeling reveal diverse stereotyped cell arrangements in the fly visual system. Proceedings of the National Academy of Sciences of the United States of America 112, E2967–2976. 10.1073/pnas.1506763112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meissner GW, Dorman Z, Nern A, Forster K, Gibney T, Jeter J, Johnson L, He Y, Lee K, Melton B, et al. (2020). An image resource of subdivided Drosophila GAL4-driver expression patterns for neuron-level searches. bioRxiv. 10.1101/2020.05.29.080473. [DOI] [Google Scholar]
- 13.Konig P, Engel AK, and Singer W (1996). Integrator or coincidence detector? The role of the cortical neuron revisited. Trends in neurosciences 19, 130–137. 10.1016/s0166-2236(96)80019-1. [DOI] [PubMed] [Google Scholar]
- 14.Zsiros V, and Hestrin S (2005). Background synaptic conductance and precision of EPSP-spike coupling at pyramidal cells. Journal of neurophysiology 93, 3248–3256. 10.1152/jn.01027.2004. [DOI] [PubMed] [Google Scholar]
- 15.Kahsai L, Carlsson MA, Winther AM, and Nassel DR (2012). Distribution of metabotropic receptors of serotonin, dopamine, GABA, glutamate, and short neuropeptide F in the central complex of Drosophila. Neuroscience 208, 11–26. 10.1016/j.neuroscience.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Xie X, Tabuchi M, Brown MP, Mitchell SP, Wu MN, and Kolodkin AL (2017). The laminar organization of the Drosophila ellipsoid body is semaphorin-dependent and prevents the formation of ectopic synaptic connections. Elife 6. 10.7554/eLife.25328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diao F, Ironfield H, Luan H, Diao F, Shropshire WC, Ewer J, Marr E, Potter CJ, Landgraf M, and White BH (2015). Plug-and-play genetic access to Drosophila cell types using exchangeable exon cassettes. Cell reports 10, 1410–1421. 10.1016/j.celrep.2015.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Satterfield LK, De J, Wu M, Qiu T, and Joiner WJ (2022). Inputs to the sleep homeostat originate outside the brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 10.1523/JNEUROSCI.2113-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimono K, Fujimoto A, Tsuyama T, Yamamoto-Kochi M, Sato M, Hattori Y, Sugimura K, Usui T, Kimura K, and Uemura T (2009). Multidendritic sensory neurons in the adult Drosophila abdomen: origins, dendritic morphology, and segment- and age-dependent programmed cell death. Neural Dev 4, 37. 10.1186/1749-8104-4-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seidner G, Robinson JE, Wu M, Worden K, Masek P, Roberts SW, Keene AC, and Joiner WJ (2015). Identification of Neurons with a Privileged Role in Sleep Homeostasis in Drosophila melanogaster. Current biology : CB 25, 2928–2938. 10.1016/j.cub.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gordon MD, and Scott K (2009). Motor control in a Drosophila taste circuit. Neuron 61, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazama H, and Wilson RI (2008). Homeostatic matching and nonlinear amplification at identified central synapses. Neuron 58, 401–413. 10.1016/j.neuron.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Zhang W, Qiao W, Hu A, and Wang Z (2010). Functional connectivity and selective odor responses of excitatory local interneurons in Drosophila antennal lobe. Neuron 67, 1021–1033. 10.1016/j.neuron.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 24.Martin-Pena A, Acebes A, Rodriguez JR, Chevalier V, Casas-Tinto S, Triphan T, Strauss R, and Ferrus A (2014). Cell types and coincident synapses in the ellipsoid body of Drosophila. The European journal of neuroscience 39, 1586–1601. 10.1111/ejn.12537. [DOI] [PubMed] [Google Scholar]
- 25.Kong D, Dagon Y, Campbell JN, Guo Y, Yang Z, Yi X, Aryal P, Wellenstein K, Kahn BB, Sabatini BL, and Lowell BB (2016). A Postsynaptic AMPK-->p21-Activated Kinase Pathway Drives Fasting-Induced Synaptic Plasticity in AgRP Neurons. Neuron 91, 25–33. 10.1016/j.neuron.2016.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu T, Kong D, Shah BP, Ye C, Koda S, Saunders A, Ding JB, Yang Z, Sabatini BL, and Lowell BB (2012). Fasting activation of AgRP neurons requires NMDA receptors and involves spinogenesis and increased excitatory tone. Neuron 73, 511–522. 10.1016/j.neuron.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, Tabuchi M, Liu S, Kodama L, Horiuchi W, Daniels J, Chiu L, Baldoni D, and Wu MN (2017). Branch-specific plasticity of a bifunctional dopamine circuit encodes protein hunger. Science 356, 534–539. 10.1126/science.aal3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seelig JD, and Jayaraman V (2013). Feature detection and orientation tuning in the Drosophila central complex. Nature 503, 262–266. 10.1038/nature12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flores-Valle A, Goncalves PJ, and Seelig JD (2021). Integration of sleep homeostasis and navigation in Drosophila. PLoS Comput Biol 17, e1009088. 10.1371/journal.pcbi.1009088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SS, Rouault H, Druckmann S, and Jayaraman V (2017). Ring attractor dynamics in the Drosophila central brain. Science 356, 849–853. 10.1126/science.aal4835. [DOI] [PubMed] [Google Scholar]
- 31.Huang S, Piao C, Beuschel CB, Gotz T, and Sigrist SJ (2020). Presynaptic Active Zone Plasticity Encodes Sleep Need in Drosophila. Current biology : CB 30, 1077–1091 e1075. 10.1016/j.cub.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 32.Raccuglia D, Huang S, Ender A, Heim MM, Laber D, Suarez-Grimalt R, Liotta A, Sigrist SJ, Geiger JRP, and Owald D (2019). Network-Specific Synchronization of Electrical Slow-Wave Oscillations Regulates Sleep Drive in Drosophila. Current biology : CB 29, 3611–3621 e3613. 10.1016/j.cub.2019.08.070. [DOI] [PubMed] [Google Scholar]
- 33.Daniels RW, Gelfand MF, Collins CA, and DiAntonio A (2008). Visualizing glutamatergic cell bodies and synapses in Drosophila larval and adult CNS. J Comp Neurol 508, 131–152. [DOI] [PubMed] [Google Scholar]
- 34.Pfeiffer BD, Truman JW, and Rubin GM (2012). Using translational enhancers to increase transgene expression in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 109, 6626–6631. 10.1073/pnas.1204520109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brand AH and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer BD, Jenett A, Hammonds AS, Ngo TT, Misra S, Murphy C, Scully A, Carlson JW, Wan KH, Laverty TR, et al. (2008). Tools for neuroanatomy and neurogenetics in Drosophila. Proceedings of the National Academy of Sciences of the United States of America 105, 9715–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenett A, Rubin GM, Ngo TT, Shepherd D, Murphy C, Dionne H, Pfeiffer BD, Cavallaro A, Hall D, Jeter J, et al. (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell reports 2, 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CH, Huang H, Ward CM, Su JT, Schaeffer LV, Guo M, and Hay BA (2007). A synthetic maternal-effect selfish genetic element drives population replacement in Drosophila. Science 316, 597–600. [DOI] [PubMed] [Google Scholar]
- 39.Shaw PJ, Cirelli C, Greenspan RJ, and Tononi G (2000). Correlates of sleep and waking in Drosophila melanogaster. Science 287, 1834–1837. [DOI] [PubMed] [Google Scholar]
- 40.Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FA, Gubb DC, and Lomas DA (2005). Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 132, 123–135. [DOI] [PubMed] [Google Scholar]
- 41.Woolums BM, McCray BA, Sung H, Tabuchi M, Sullivan JM, Ruppell KT, Yang Y, Mamah C, Aisenberg WH, Saavedra-Rivera PC, et al. (2020). TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII-dependent elevation of intracellular Ca(2). Nat Commun 11, 2679. 10.1038/s41467-020-16411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabuchi M, Monaco JD, Duan G, Bell B, Liu S, Liu Q, Zhang K, and Wu MN (2018). Clock-Generated Temporal Codes Determine Synaptic Plasticity to Control Sleep. Cell 175, 1213–1227 e1218. 10.1016/j.cell.2018.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson RI, Turner GC, and Laurent G (2004). Transformation of olfactory representations in the Drosophila antennal lobe. Science 303, 366–370. 10.1126/science.1090782. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data, analysis files, and images generated for this manuscript are available upon request to the lead contact.
Jupyter notebooks are available upon request from the lead contact. Additional code for this manuscript is available on Github (https://github.com/margiezilla/Ho_etal_2022).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.