

SUMMARY
The recent development of spatial omics methods has enabled single-cell profiling of the transcriptome and 3D genome organization with high spatial resolution. Expanding the repertoire of spatial omics tools, a spatially resolved single-cell epigenomics method will accelerate understanding of the spatial regulation of cell and tissue functions. Here, we report a method for spatially resolved epigenomic profiling of single cells using in-situ tagmentation and transcription followed by multiplexed imaging. We demonstrated the ability to profile histone modifications marking active promoters, putative enhancers, and silent promoters in individual cells, and generated high-resolution spatial atlas of hundreds of active promoters and putative enhancers in embryonic and adult mouse brains. Our results suggested putative promoter-enhancer pairs and enhancer hubs regulating developmentally important genes. We envision this approach will be generally applicable to spatial profiling of epigenetic modifications and DNA-binding proteins, advancing our understanding of how gene expression is spatiotemporally regulated by the epigenome.
Graphical Abstract
In Brief
An imaging-based approach is developed to enable spatially resolved single-cell epigenomic profiling with high spatial and genomic resolution.
INTRODUCTION
Spatiotemporal control of gene expression is essential for the development and function of cells and tissues. The regulatory information encoded in the epigenome, such as histone and DNA modifications, enables differential activation or repression of genes to generate different cell types during development (Allis and Jenuwein, 2016; Henikoff and Smith, 2015; Moris et al., 2016; Zhao et al., 2021; Zoghbi and Beaudet, 2016). Sequencing-based approaches have been traditionally used to profile histone and DNA modifications in an ensemble of cells. Recently, epigenetic sequencing techniques have been extended to the single-cell level to enable characterization of chromatin accessibility and epigenetic modifications in individual cells (Bartlett et al., 2021; Bartosovic et al., 2021; Buenrostro et al., 2015; Carter et al., 2019; Cusanovich et al., 2015; Gravina et al., 2016; Kaya-Okur et al., 2019; Wang et al., 2019b; Wu et al., 2021; Zhu et al., 2021).
However, the spatial context of cells is lost in sequencing-based methods that require cell dissociation. On the other hand, spatial information of epigenetic properties, such as epigenetic modifications marking active enhancers and promoters, is critical to understand how the epigenome shapes the development of cell types and control of cell states in the native context of complex tissues. For example, during embryonic brain development, morphogenic gradients and transcription factors form complex spatial patterns, giving rise to a myriad of neural progenitors destined to become different types of neurons and non-neuronal cells (Cadwell et al., 2019; Gelman et al., 2012; Hébert and Fishell, 2008; Molnár et al., 2019; O’Leary et al., 2013; Rakic, 2009). Recent evidence suggests that diverse enhancer recruitments may help generate finely delineated domains or protodomains within the developing brain, fine-tuning the broad patterns generated by transcription factors and morphogenic gradients (Pattabiraman et al., 2014; Visel et al., 2013). Progenitors from these domains then give rise to different neuronal subtypes in various brain regions (Silberberg et al., 2016). Moreover, in the adult brain, neurons from different subtypes and cortical layers exhibit different chromatin accessibilities and epigenetic modification profiles (Gray et al., 2017; Graybuck et al., 2021; Mo et al., 2015; Zhu et al., 2021). These observations highlight the need for epigenomic mapping with high spatial resolution. Tens to hundreds of thousands of epigenetic elements, such as putative enhancers, have been identified in both embryonic and adult brains (Gorkin et al., 2020; Gray et al., 2017; Graybuck et al., 2021; Mo et al., 2015; Preissl et al., 2018; Shen et al., 2012; Visel et al., 2009, 2013; Yue et al., 2014), but the spatial distributions remain unclear for most of these elements. High-resolution spatial profiling of these epigenetic elements will greatly facilitate the functional understanding of the epigenome.
The transgenic approach that delivers enhancer sequences fused to a reporter expression cassette into the animal has been used to measure the spatial patterns of thousands of putative enhancers in the embryonic mouse brain in a heroic effort that spanned more than 10 years (Kvon et al., 2020; Pattabiraman et al., 2014; Silberberg et al., 2016; Visel et al., 2007, 2013). This approach requires extensive cloning and generation of transgenic animals. In addition, mapping enhancers in a setting where the enhancer activity is shown by an adjacent reporter may not always recapitulate endogenous epigenetic activities. A spatial profiling approach that can map endogenous epigenetic activities, such as active enhancers and promoters, of individual cells in a high throughput manner is thus in demand. Moreover, it is essential that such a spatial epigenomics approach has a high genomic resolution because epigenetic elements are typically short (~1 kilobase or shorter).
Recently, spatial genomics approaches have been developed to profile the transcriptome using either imaging-based approaches (multiplexed FISH and in situ sequencing) with single cell resolution (Lein et al., 2017; Zhuang, 2021) or spatially resolved RNA capture following by sequencing (Larsson et al., 2021). The imaging-based approaches have also allowed the 3D organization of the DNA in single cells to be measured at the genome scale, imaging thousands of chromatin loci with a genomic resolution of tens of kilobases (kb) to megabases (Payne et al., 2021; Su et al., 2020; Takei et al., 2021a, 2021b). Genome-scale chromatin imaging has also been combined with protein imaging to study the spatial relationship between chromatin loci and nuclear structures, including nuclear bodies and histone marks (Su et al., 2020; Takei et al., 2021a, 2021b), but due to limited imaging resolution, it is difficult to determine whether chromatin loci that showed signal colocalization with histone marks indeed carry or are just in spatial proximity to these marks. Expansion microscopy could improve the accuracy in determining the epigenetic state of chromatin loci and has been demonstrated for imaging the histone modifications of a few genomic loci at 10-kb resolution (Woodworth et al., 2021). However, imaging the epigenetic state remains challenging for most gene regulatory elements, which are ~1kb or shorter, due to the limited image resolution and the difficulty to detect short sequences. Hence, a technique that allows epigenetic-state imaging of the chromatin in individual cells with high genomic resolution and high genomic throughput is still lacking.
Here, we developed an imaging method to measure the epigenetic modifications of chromatin in individual cells with high genomic throughput and high genomic resolution. We demonstrated the ability to image genomic loci as short as a few hundred bases, identify their epigenetic states, and map their spatial distributions. We imaged histone-modifications marking active promoters, putative enhancers, and silent promoters in individual cells and demonstrated epigenomic profiling with sub-nuclear resolution. We further used this approach to map hundreds of active promoters and putative enhancers in embryonic and adult mouse brains. Our data revealed high-resolution spatial distributions of putative enhancers and predicted previously unknown enhancer-promoter pairs and enhancer hubs for developmentally important genes in the embryonic brain.
RESULTS
Epigenomic MERFISH enables in situ spatially resolved single-cell profiling of epigenetic modifications
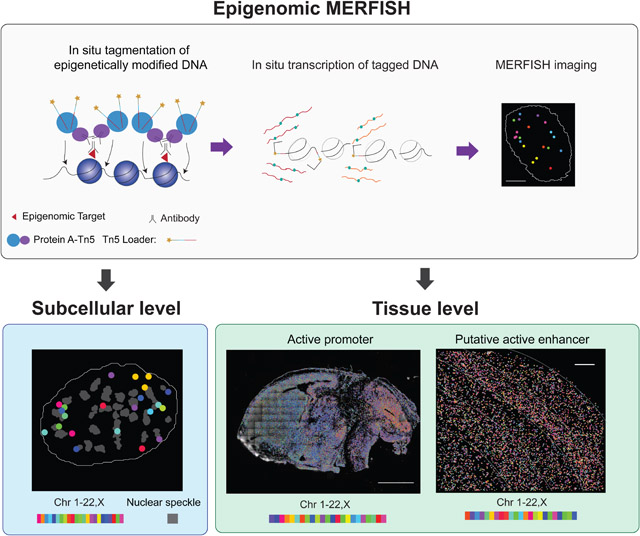
In order to image epigenomic loci in a high throughput manner, we first captured specific epigenetic modifications on the chromatin in situ and tagged the DNA with T7 promoters at or near the modification sites, followed by in situ transcription of the tagged DNA fragments to generate RNAs, and finally we detected the transcribed RNAs by multiplexed error robust fluorescence in situ hybridization (MERFISH) (Figure 1A), which allows RNA imaging at the transcriptomic scale (Chen et al., 2015). Hereafter, we referred to this method as epigenomic MERFISH.
Figure 1. Spatially resolved single-cell profiling of epigenetic modifications by epigenomic MERFISH.
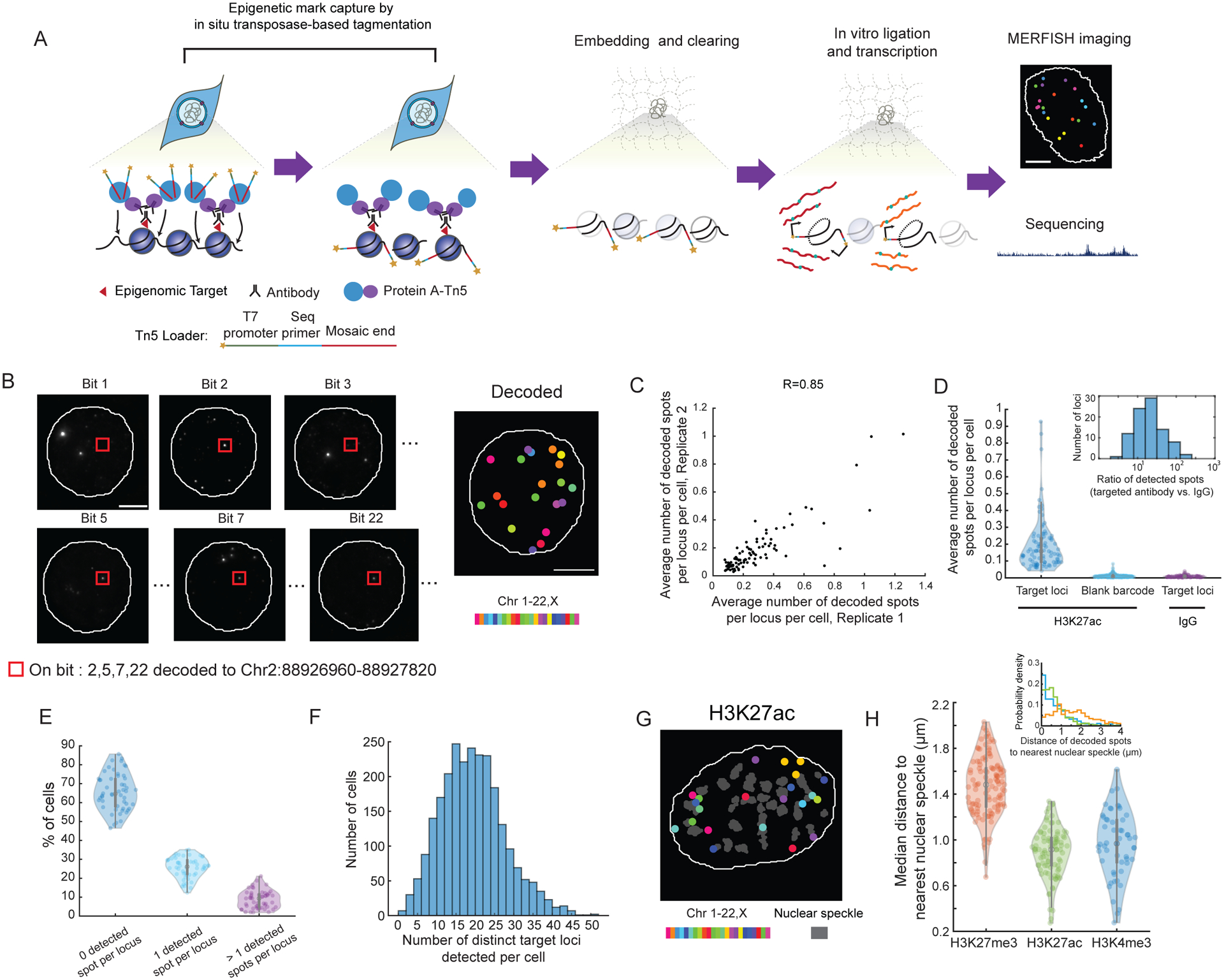
(A) Schematic of epigenomic MERFISH. Cells were treated with primary antibodies recognizing the epigenetic modifications of interest, secondary antibodies, and protein A coupled transposase (PA-Tn5) to generate DNA fragments tagged with T7 promoter and sequencing primers. The sample was embedded into polyacrylamide gel and cleared, with the tagged DNA crosslinked to the gel. The tagged DNA fragments were then transcribed into RNAs by T7 polymerase. The resulting RNA was either detected by MERFISH imaging or subjected to sequencing. Scale bar: 5 μm.
(B) Epigenomic MERFISH image of 90 target H3K27ac loci in a single cell. The images from individual bits are shown on the left. The decoded image is shown on the right, with individual spots color-coded based on the chromosomal identities of the loci. Scale bars: 5 μm.
(C) Correlation between two biological replicates of H3K27ac imaging. Each dot corresponds a single H3K27ac locus. The Pearson correlation coefficients are 0.85 between replicates 1 and 2, 0.71 between replicates 1 and 3, and 0.75 between replicates 2 and 3.
(D) Violin plots showing the average number of decoded spots per cell for each target H3K27ac locus (left) and each blank barcode (middle) when H3K27ac antibody is used to capture the epigenetic mark, as well as the average number of decoded spots per cell for each target H3K27ac locus when a control IgG is used instead (right). Each dot corresponds to a single H3K27ac locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K27ac antibody and that detected with IgG for individual H3K27ac loci. Results here are aggregates of three replicates.
(E) Violin plots showing the percentage of cells with 0, 1, or >1 detected spots for individual target H3K4me3 loci corresponding to the promoters of 52 essential genes. Each dot corresponds to a single H3K4me3 locus. For a given locus, ~35% (median percentage across 52 loci) of cells showed at least one detected spot. Results here are aggregates of three replicates.
(F) Histogram of the number of distinct H3K4me3 loci detected per cell. The median number of distinct loci detected per cell is 18, which is ~35% of the 52 total target loci. Results here are aggregates of three replicates.
(G) Decoded epigenomic MERFISH image of 90 target H3K27ac loci and nuclear speckles in a single cell. Individual colored spots correspond to detected H3K27ac loci color-coded based on the chromosomal identities of the loci. Grey shapes correspond to the segmented boundaries of nuclear speckles. Scale bar: 5 μm.
(H) Violin plots showing the median distances (across all imaged cells) between the detected H3K4me3, H3K27ac and H3K27me3 loci and the nearest nuclear speckles. Each dot corresponds to a single locus. Inset: Histograms of the spatial distances of three example H3K4me3 (blue), H3K27ac (green) and H3K27me3 (red) loci to the nearest nuclear speckles in individual cells.
See also Figure S1.
We first optimized and validated this method in cultured hTERT-RPE1 cells. To capture the epigenetic modifications, we fixed the samples and labeled the epigenetic modification of interest using antibodies (Figure 1A). The antibodies were then bound by protein A fused with transposase Tn5 (PA-Tn5), which allowed Tn5 to transpose the T7 promoters into the DNA region at or near the epigenetic modification site (Figure 1A). This procedure resulted in fragments of chromatin encompassing the epigenetic loci of interest tagged by the T7 promoters and sequencing primers at both ends, where primer tags allowed the PCR amplification and sequencing of DNA fragments, as previously done in CUT&Tag (Bartlett et al., 2021; Bartosovic et al., 2021; Harada et al., 2019; Kaya-Okur et al., 2019; Liu et al., 2020; Wang et al., 2019b; Wu et al., 2021; Zhu et al., 2021). The T7 promoter tags further allowed the DNA fragments to be transcribed into RNAs by the T7 polymerase for in situ amplification and detection. Tagmentation with the T7 promoter has also been used for signal amplification in CUT&Tag recently (Bartlett et al., 2021).
To ensure efficient and faithful capture of the epigenetic modifications in fixed cells, we screened fixation conditions using various crosslinking or precipitating fixatives and fixation durations and found that light PFA fixations with HCl treatment enabled specific transposition near the target epigenetic loci (Figure S1A and S1B).
After the tagged DNA fragments were generated, these fragments were transcribed into RNAs using the T7 RNA polymerase. This in situ transcription step amplifies a single copy of DNA fragment into many copies of RNA, which not only increases the signal of epigenomic loci to confer detection specificity, but also allows us to detect short DNA locus that would otherwise be difficult to image by FISH. To ensure efficient transcription, we embedded the sample in polyacrylamide gel and digested the sample by proteinase K to remove DNA-interacting proteins that could impede T7 transcription (Figure 1A). This embedding and clearing procedure improved T7 amplification and generated more RNAs for more efficient capture of the histone modification peaks (Figure S1B).
Finally, we used MERFISH to image the transcribed RNAs in the gel-embedded samples in a highly multiplexed manner (Chen et al., 2015; Moffitt et al., 2016b, 2016a). We used N-bit barcodes with Hamming Distance 4 (HD 4) and Hamming weight 4 (HW 4) to allow error correction, where the length the barcodes (N) was chosen based on the number of target epigenetic loci. To avoid crowdedness of the fluorescence signals in each bit such that individual loci could be clearly resolved, we assigned barcodes to the target loci in a manner such that only 3–5 loci in each chromosome was imaged in each bit (See STAR Methods for MERFISH probe design, Table S1 for oligonucleotide sequences, and Table S2 for barcode assignments).
Before performing MERFISH imaging of the target loci, we first collected and measured the transcribed RNAs in an untargeted manner by sequencing to test whether the epigenetic loci were faithfully captured. To this end, we measured the profiles of three histone modifications, H3K27ac, H3K4me3 and H3K27me3, in hTERT-RPE1 cells. H3K4me3 is a canonical marker for active promoters; H3K27ac can mark both active promoters and enhancers, and intergenic H3K27ac loci are often used to predict putative active enhancers; H3K27me3 marks silent genomic loci (Allis and Jenuwein, 2016; Sparmann and van Lohuizen, 2006). We found that the length distribution of the RNAs generated by in situ Tn5 transposition and T7 transcription was around 100–1000 bases (Figure S1C). The RNAs were subsequently reverse transcribed, PCR-amplified, and sequenced. The genome-wide profiles of H3K27ac, H3K4me3 and H3K27me3 measured using this in situ tagmentation and transcription approach agreed with those detected by ChIP-Seq and CUT&Tag, albeit with a lower peak height (Figure S1B and S1D), and the H3K27ac and H3K27me3 modifications exhibit distinct peaks (Figure S1B and S1E).
Next, we performed MERFISH imaging of the transcribed RNAs in situ to achieve spatial profiling of the epigenetic loci of interest. We first selected 90 H3K27ac-positive loci in hTERT-RPE1 cells based on the H3K27ac peaks in bulk CUT&Tag data and designed MERFISH probes targeting these loci (Table S1). The selected H3K27ac loci had a median length of ~800 bp. We measured these loci with a 24-bit, HW 4 and HD 4 code (Table S2) using 8 rounds of 3-color imaging. Among the 366 total valid barcodes, 90 were assigned to the target H3K27ac loci and the remaining ones were unassigned (referred to as blank barcodes), which allowed us to assess the mis-identification rate. The MERFISH images showed clear and decodable spots (Figure 1B). The number of decoded spots per cell for individual H3K27ac loci were reproducible between biological replicates (Figure 1C). The spots that were decoded into individual target H3K27ac loci were on average ~30 fold more abundant than the spots that were decoded into individual blank barcodes (Figure 1D), indicating a low misidentification rate. As a further control, we replaced the H3K27ac antibody with a control IgG antibody and observed a ~17-fold reduction in the number of spots decoded into the target loci (Figure 1D). Only a small fraction of loci (<2%) showed a similar number (within a factor of two) of spots detected per cell with IgG and H3K27ac antibodies, and these loci were not reproducible between replicate experiments. These results indicate that our measurements were specific to H3K27ac modifications.
We next estimated the detection rate of our epigenomic MERFISH measurements by imaging the promoters of essential genes, which should in principle be active and hence H3K4me3-positive in every cell. If our detection efficiency was 100%, we should detect at least one H3K4me3-positive spot for each promoter in every cell. We selected 52 such essential genes and designed probes targeting the ±2 kb region of their transcription starting sites (TSS) (Tables S1 and S2), which presumably covers the promoter regions of the genes. Comparisons with the blank barcode counts and with the measurements using the control IgG showed that our detection of the H3K4me3-positive loci was highly specific with a low mis-identification rate and was not contaminated by the endogenous nascent RNA signal (Figure S1F). For any given target locus, the fraction of cells exhibiting one or more detected spot is ~35% (median across target loci, Figure 1E), and in any given cell, ~35% of the target loci were detected (Figure 1F). This detection rate was higher than the detection rates of single-cell CUT&Tag (~8%) (Bartlett et al., 2021) and single-cell ATAC-seq (~5–15%) (Chen et al., 2019; Fang et al., 2021; Preissl et al., 2018).
Finally, to test whether epigenomic MERFISH can detect silent chromatin marks, we selected 115 H3K27me3-positive loci in the promoter regions (±2 kb of TSS) of genes with zero or low expression and designed MERFISH probes targeting these loci (Tables S1 and S2). Comparisons with the blank barcode counts and control IgG measurements showed that our detection of the H3K27me3 loci was also specific with a low mis-identification rate (Figure S1G).
To demonstrate the ability of epigenomic MERFISH to probe the subnuclear organization of epigenetic loci, we co-imaged nuclear speckle with the epigenetic loci in these experiments (Figure 1G). We observed that the H3K27ac and H3K4me3 loci, which mark active promoters and putative active enhancers, were in closer proximity to nuclear speckles than the H3K27me3 loci, which mark silent promoters (Figure 1H), consistent with previous findings that active chromatin has higher association rate with nuclear speckles (Chen and Belmont, 2019). These results demonstrate that epigenomic MERFISH allows for spatially resolved epigenomic profiling of chromatin with sub-nuclear resolution.
Region- or layer-specific patterns of active promoters in the mouse brain
Next, we demonstrated the spatial profiling power of epigenomic MERFISH by using it to map histone modifications in mouse brain tissues. As a proof of principle, we first imaged active promoters marked by H3K4me3 for 127 genes (Table S1), some of which are known to exhibit region-specific expression in the embryonic brain or layer-specific expression in the adult cortex. We targeted H3K4me3 ChIP-Seq peaks close to the TSS of these genes in the embryonic brain (Gorkin et al., 2020) as promoter loci (Table S2). The median length of the target loci was ~3.3 kb.
Upon further optimization of the epigenomic MERFISH protocol for tissue slices (see STAR Methods), we mapped the spatial distributions of these H3K4me3 loci in sections of the adult (Figure 2A) and embryonic day 13.5 (E13.5) (Figure 3A) mouse brain. We observed clear and decodable spot in individual cells in the brain sections (Figure S2A), and the detection of the H3K4me3 loci were highly reproducible between replicates (Figure S2B) and specific to the H3K4me3 antibody with a low mis-identification rate (Figure S2C).
Figure 2. Spatially resolved single-cell profiling of layer-specific active promoters in adult mouse cortex.
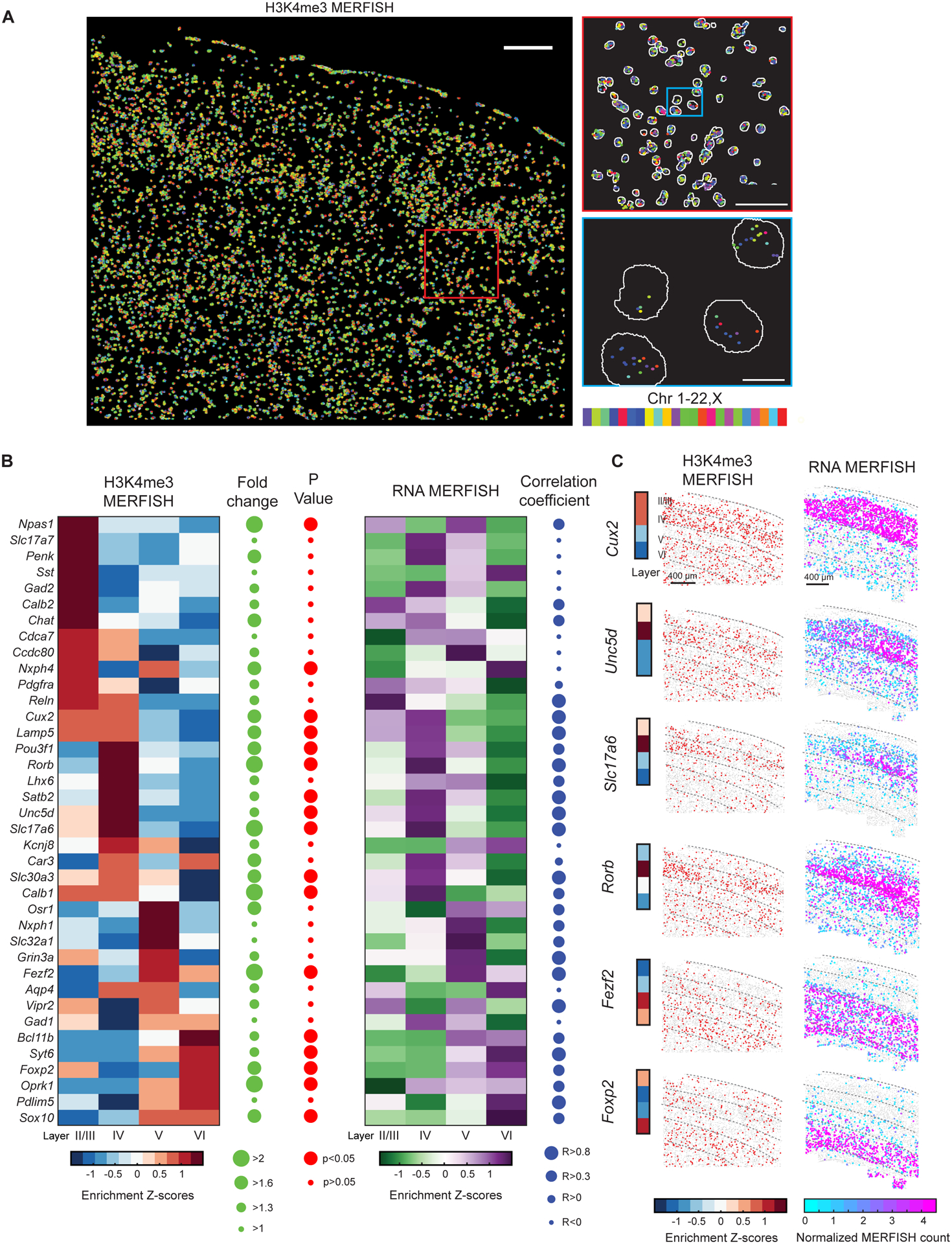
(A) Left: Epigenomic MERFISH image of 127 target H3K4me3 loci in the somatosensory cortex region of a coronal slice of an adult mouse brain. Scale bar: 200 μm. Top right: A magnified view of the red-boxed region from the left panel. Scale bar: 75 μm. Bottom right: A magnified view of the blue-boxed region from the top right panel. Segmentation of individual nuclei are shown in white and decoded spots are color-coded by the chromosomal identities of the loci. Scale bar: 10 μm.
(B) Left: Layer enrichment z-scores for the promoter H3K4me3 signals measured by epigenomic MERFISH for the indicated genes. For each promoter locus, the enrichment in a specific layer is calculated as the z-score of the fraction of cells in the layer that is H3K4me3-positive for this locus. The fold change in the fraction of H3K4me3-positive cells between the layers with the maximum and minimum enrichment (green) and the statistical significance (p-value) of the layer-specific enrichment (red) are shown on the right. Results from replicate 1 is shown here and comparison between replicates 1 and 2 is shown in Figure S3A. Right: Layer enrichment z-scores for the RNA expression level measured by RNA MERFISH for the indicated genes (Zhang et al., 2021). For each gene, the enrichment in a specific layer is calculated as the z-score of the fraction of cells in the layer that express this gene. The Pearson correlation coefficient of layer enrichment between the epigenomic MERFISH and RNA MERFISH data is shown on the right.
(C) Left: Epigenomic MERFISH images showing layer enrichment of H3K4me3 signals for the promoters of six indicated genes. Each dot in the images represent a cell and red dots represent cells with positive H3K4me3 signals. The layer enrichment heatmap on the left is reproduced from panel (B). Right: RNA MERFISH images showing the RNA expression levels for the six indicated genes in individual cells, with each cell presented as a dot. Normalized MERFISH counts is defined as RNA counts per cell divided by the imaged volume of each cell (Zhang et al., 2021). Scale bars: 400 μm.
See also Figures S2 and S3.
Figure 3. Spatially resolved profiling of active promoters in mouse embryonic brain.
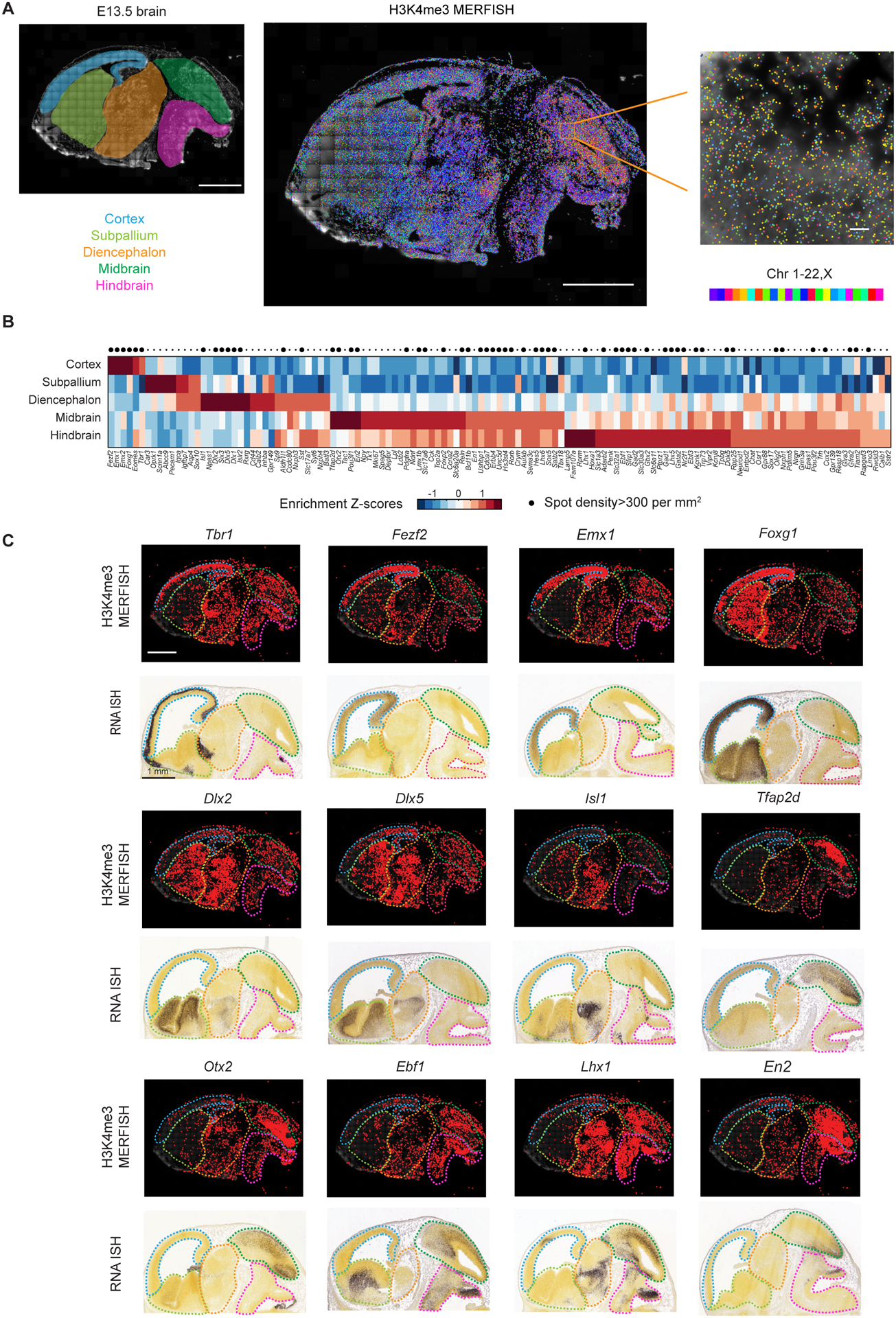
(A) Left: Schematic highlighting different brain regions, cortex, subpallium, diencephalon, midbrain, hindbrain, of an imaged sagittal slice of a E13.5 mouse brain. The background shows the DAPI signal. Scale bar: 1 mm. Middle. Epigenomic MERFISH image of 127 target H3K4me3 loci in the slice. Scale bar: 1 mm. Right: An enlarged region for orange box in the midbrain. Scale bar: 20 μm. All decoded spots are plotted on the background of the DAPI signal and are color-coded by the chromosomal identities of the loci.
(B) The brain region enrichment z-scores for each of the 127 target H3K4me3 loci in different brain regions. The brain region is segmented based on the cytological hallmarks. The z-score of the decoded spot density (spot number / DAPI-positive area) in each region is shown. Top: Large dot indicating the loci that have a H3K4me3 spot density larger than 300 per mm2.
(C) Epigenomic MERFISH images of the H3K4me3 signals of the promoters of twelve transcription factors shown in comparison with the Allen Brain RNA ISH images of the corresponding genes. Allen RNA ISH images here and in subsequent figures are taken from Allen Reference Atlas - Mouse Brain [brain atlas] available from atlas.brain-map.org (Lein et al, 2007). Scale bars: 1 mm.
See also Figure S4.
We then characterized the spatial distributions of the H3K4me3 signals of these loci in adult mouse cortex, focusing on whether the loci corresponding to the promoters of cortical-layer marker genes exhibited the expected layer-specific enrichment of H3K4me3 signals. We imaged these 127 loci in thousands (~4,200) of individual cells in the somatosensory cortex of adult mouses brains (Figure 2A), and compared the spatial patterns of the detected H3K4me3 loci with the RNA expression pattern of their corresponding genes that we recently measured by MERFISH (Zhang et al., 2021). Of the 127 H3K4me3-positive promoter loci probed here, 38 promoter loci had corresponding genes in the RNA MERFISH data, and the layer enrichment pattern that we observed for these loci and genes were largely similar between the epigenomic MERFISH and RNA MERFISH measurements (Figure 2B). For example, like RNA MERFISH signals, Cux2 and Unc5d promoter H3K4me3 signals measured by epigenomic MERFISH were enriched in layers II/III and IV, Rorb and Slc17a6 promoter H3K4me3 signals were enriched in layer IV, Fezf2 promoter H3K4me3 signal was enriched in layers V and VI, and Foxp2 promoter H3K4me3 signal was enriched in layer VI (Figure 2C). Overall, 17 of these 38 promoters showed statistically significant enrichment in specific cortical layers, and among these layer-enriched promoters, the vast majority (~80%) showed layer enrichment patterns that were both consistent between replicates and similar to the RNA expression patterns of the corresponding genes (Figure S3A). As expected, for those promoter loci that did not show statistically significant layer-specific enrichment, visual inspection of their H3K4me3 images and the RNA MERFISH images of the corresponding genes confirmed the lack of layer-specific enrichment (see examples in Figure S3B) and the z-scored layer-enrichment patterns of the H3K4me3 and RNA signals often did not appear similar with each other due to the lack of statistical significance (Figure 2B).
As an additional control, we generated null distributions of the epigenomic MERFISH signals by randomly permutating the genomic identity of the detected H3K4me3 spots among the 127 target loci (Figure S3C and S3D). The experimentally observed degree of similarity between the epigenomic MERFISH and RNA MERFISH spatial patterns was significantly higher than that observed in this randomization control (Figure S3E).
Next, we characterized how the H3K4me3 signals of the 127 target loci were distributed in the embryonic brain. To identify genes with region-specific expression, we determined the detected spot density of these 127 loci in each of the five brain regions: the cortex, subpallium, diencephalon, midbrain, and hindbrain (Figure 3A). Many loci showed enrichment of expression in specific brain regions (Figure 3B). To validate our results, we focused on those loci with reasonably high spot density (>300 spots per mm2) and compared their spatial distribution with the expression patterns of the corresponding genes reported in Allen brain in situ hybridization (ISH) atlas (Lein et al., 2007). Of the 57 loci that satisfied this spot density criterion, 46 loci have RNA ISH data of corresponding genes measured in the E13.5 brain and most of them showed spatial distributions of H3K4me3 signals that were similar to the RNA expression patterns reported in the Allen ISH atlas (Figure 3C; Figure S4A). For example, the promoters of Tbr1 and Fezf2 exhibited H3K4me3 signal enrichment within the cortex, comparable to the expression patterns of Tbr1 and Fezf2 in the RNA ISH images (Figure 3C). Canonical transcription factors for cortical development, Emx1, Emx2, and Eomes, also showed H3K4me3 signal enrichment in the cortex (Figure 3C; Figure S4A). As a telencephalon marker, Foxg1 showed expected H3K4me3 signal enrichment in the cortex and subpallium (Figure 3C). Several distal less homeodomain transcription factors (Dlx1, Dlx2 and Dlx5) showed expected H3K4me3 signal enrichment in subpallium and diencephalon (Figure 3C; Figure S4A), consistent with the knowledge that these genes are important for forebrain inhibitory neuron development (Eisenstat et al., 1999). The promoter of Isl1 showed H3K4me3 signal enrichment in the diencephalon (Figure 3C), consistent with the known expression of this gene in a subpopulation of differentiating hypothalamic neurons (Lee et al., 2016). Finally, the promoters of midbrain and/or hindbrain specific transcription factors (e.g.: Tfap2d, Otx2, Ebf1, Lhx1, and En2) (Cepeda-Nieto et al., 2005; Joyner, 1996; Rhinn et al., 1998; Wang et al., 1997) showed expected H3K4me3 signal enrichment in midbrain and/or hindbrain (Figure 3C; Figure S4A). Of the 46 loci, we classified the H3K4me3 MERFISH and RNA ISH signals as being enriched or not enriched in each of the five brain regions and found that 39 loci had the same classification for H3K4me3 MERFISH and RNA ISH signals in the majority (3 or more) of the brain regions (Figure S4A). In the randomization control where genomic identity of the detected H3K4me3 spots was randomly permutated among the 127 targeted loci, the degree of similarity between the H3K4me3 MERFISH and RNA ISH spatial patterns was much lower (Figure S4B and S4C).
Our observed good, albeit not perfect, agreement between H3K4me3 MERFISH and RNA ISH spatial patterns is consistent with the knowledge that although H3K4me3 can mark active promoters, some inactive genes can still have H3K4me3 signals at their promoters (Guenther et al., 2007) and transcription can also happen in the absence of H3K4me3 modification in some cases (Hödl and Basler, 2012). Moreover, a gene could have multiple isoforms that are controlled by different promoters (Okladnova et al., 1998), and hence a particular promoter’s H3K4me3 signal may only account for the RNA expression of the gene in a subset of the brain regions.
Overall, the observed agreement between our spatial profiling results of the H3K4me3-marked active promoters and the previously measured expression patterns of the corresponding genes further validated our epigenomic MERFISH measurements.
Layer-enrichment patterns of putative active enhancers in mouse adult cortex
Next, we applied epigenomic MERFISH to map the putative active enhancers in the brain, first aiming to reveal layer-specific enhancers by targeting genomic loci with the H3K27ac modification. When situated in regions away from the promoter sites, H3K27ac is often used as a marker for putative active enhancers (Creyghton et al., 2010; Heintzman et al., 2009; Rada-Iglesias et al., 2011). However, layer-specific bulk H3K27ac sequencing data is not readily available. Recently, layer-specific chromatin accessibility has been profiled by ATAC-seq using FACS-sorted layer-specific excitatory neurons (Gray et al., 2017; Graybuck et al., 2021). We thus used the ATAC-seq data to guide our selection of target loci, as ATAC-seq peaks that do not correspond to promoters are often considered possible candidates for enhancers. We selected 139 ATAC peaks that are >2kb away from known TSS of genes, show statistically significant signal enrichment in one cortical layer, and have non-zero reads from bulk H3K27ac ChIP-Seq data (Tables S1 and S2). The median length of these peak was ~270 bp.
We performed epigenomic MERFISH imaging of these loci, targeting the H3K27ac modification, in adult mouse sections containing the somatosensory cortex and profiled ~5,400 individual cells in this region (Figure 4A). The results were consistent between replicates and specific to the H3K27ac antibody (Figure S5A and S5B). Among 139 target loci, 35 showed a statistically significant layer-specific pattern (Figure 4B). The observation that many loci did not show layer-specific enrichment of H3K27ac signals is not surprising considering that a substantial fraction (~50%) of the ATAC-seq peaks are not overlapping with H3K27ac peaks measured by ChIP-seq (Fulco et al., 2019; Gray et al., 2017). Hence, the signal of ATAC peaks may not always reflect the H3K27ac level. Indeed, compared to those loci not exhibiting significant layer-specific enrichment, these 35 loci had a higher average H3K27ac signal (Figure S5C). The layer-enrichment patterns of these loci were largely similar to those obtained from ATAC-seq (Figure 4B), and the degree of similarity between epigenomic MERFISH and ATAC-seq patterns was significantly higher than that observed in the randomization control (Figure S5D and S5E).
Figure 4. Spatially resolved single-cell profiling of layer-specific putative active enhancers in adult mouse cortex.
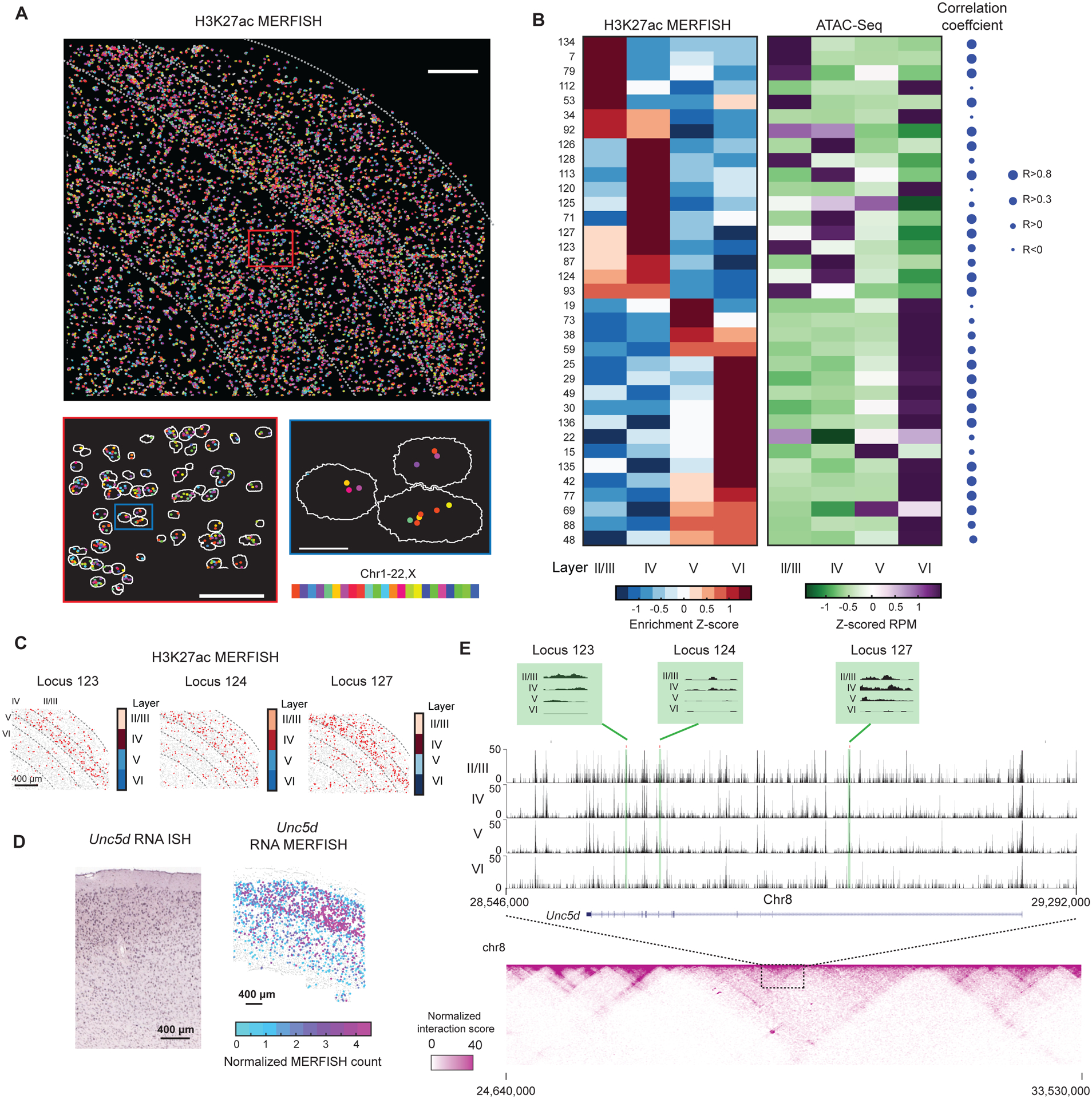
(A) Top: Epigenomic MERFISH image of 139 target H3K27ac loci in the somatosensory cortex region of a coronal slice of an adult mouse brain. Scale bar: 200 μm. Bottom left: A magnified view of the red-boxed region from the top panel. Scale bar: 75 μm. Bottom right: A magnified view of the blue-boxed region from the bottom left panel. Segmentation of individual nuclei are shown in white and decoded spots are color-coded by the chromosomal identities of the loci. Scale bar: 10 μm.
(B) Left: Layer enrichment z-scores for the H3K27ac signal measured by epigenomic MERFISH for the indicated genomic loci. Layer enrichment z-score is calculated as described in Figure 2B. Results from replicate 1 is shown and results from replicate 2 are similar. Right: The corresponding z-scored reads per million (RPM) for each of target loci from published ATAC-seq data (Gray et al., 2017). The Pearson correlation coefficients between the layer enrichment derived from epigenomic MERFISH data and ATAC seq data are shown on the right.
(C) Epigenomic MERFISH images of the H3K27ac signals for three example target loci showing enrichment in layers II/III and IV. Quantification of the layer enrichment are shown on the right (reproduced from Figure 4B). Scale bar: 400 μm.
(D) The Allen RNA ISH image (left) and RNA MERFISH image (right) of the RNA expression level of the Unc5d gene. Scale bars: 400 μm.
(E) Top: UCSC browser track of the ATAC-seq data (Gray et al., 2017) showing the location of the three target loci (loci 123, 124, 127) in the intronic regions of Unc5d. Regions marked in green are the three target loci with the green boxes above showing the enlarged version of the ATAC-seq track of the marked loci. Bottom: Hi-C map of a genomic region harboring these loci (Deng et al., 2015).
See also Figure S5.
Notably, some of these putative enhancer loci exhibited layer-specific enrichment patterns of H3K27ac signals that were similar to the spatial expression patterns of nearby genes. For example, the three putative enhancer loci (loci 123, 124 and 127) within 600 kb of the TSS of gene Unc5d showed a consistent and significant enrichment in layers II/III and IV (Figure 4C), and the Unc5d gene also showed enriched expression in layers II/III and IV (Figure 4D). The Hi-C data of the mouse brain (Deng et al., 2015) showed that these three loci and the Unc5d gene are located within the same sub-TAD (Figure 4E). These results suggest that loci 123, 124 and 127 are putative enhancers for the Unc5d gene and that the spatially profiling power of epigenomic MERFISH could help identify putative promoter-enhancer pairs.
High-resolution spatial profiling of putative active enhancers in mouse embryonic brains
We next imaged putative active enhancers marked by H3K27ac in the E13.5 embryonic mouse brain to identify spatial patterns of enhancer activity (Figure 5A). To this end, we targeted a total of 142 H3K27ac-positive loci and five loci with low H3K27ac counts as negative controls, selected based on ChIP-Seq data obtained from the embryonic brain (Gorkin et al., 2020) (Tables S1 and S2). The median length of these loci was ~2.2 kb. The five negative control loci showed a comparable number of detected spots to those of blank barcodes and ~10-fold fewer detected spots compared to the H3K27ac-positive loci, indicating a low false positive detection rate (Figure S6A). As a further validation, we compared our results with previous ChIP-Seq data obtained from E13.5 forebrain, midbrain and hindbrain (Gorkin et al., 2020) by grouping the epigenomic MERFISH signals into these three major brain regions. We observed a similar region-specific enrichment pattern to the ChIP-Seq results (Figure 5B) and the degree of similarity was significantly higher than that observed in the randomization control (Figure S6B and S6C).
Figure 5. Spatially resolved profiling of putative active enhancers in mouse embryonic brain.
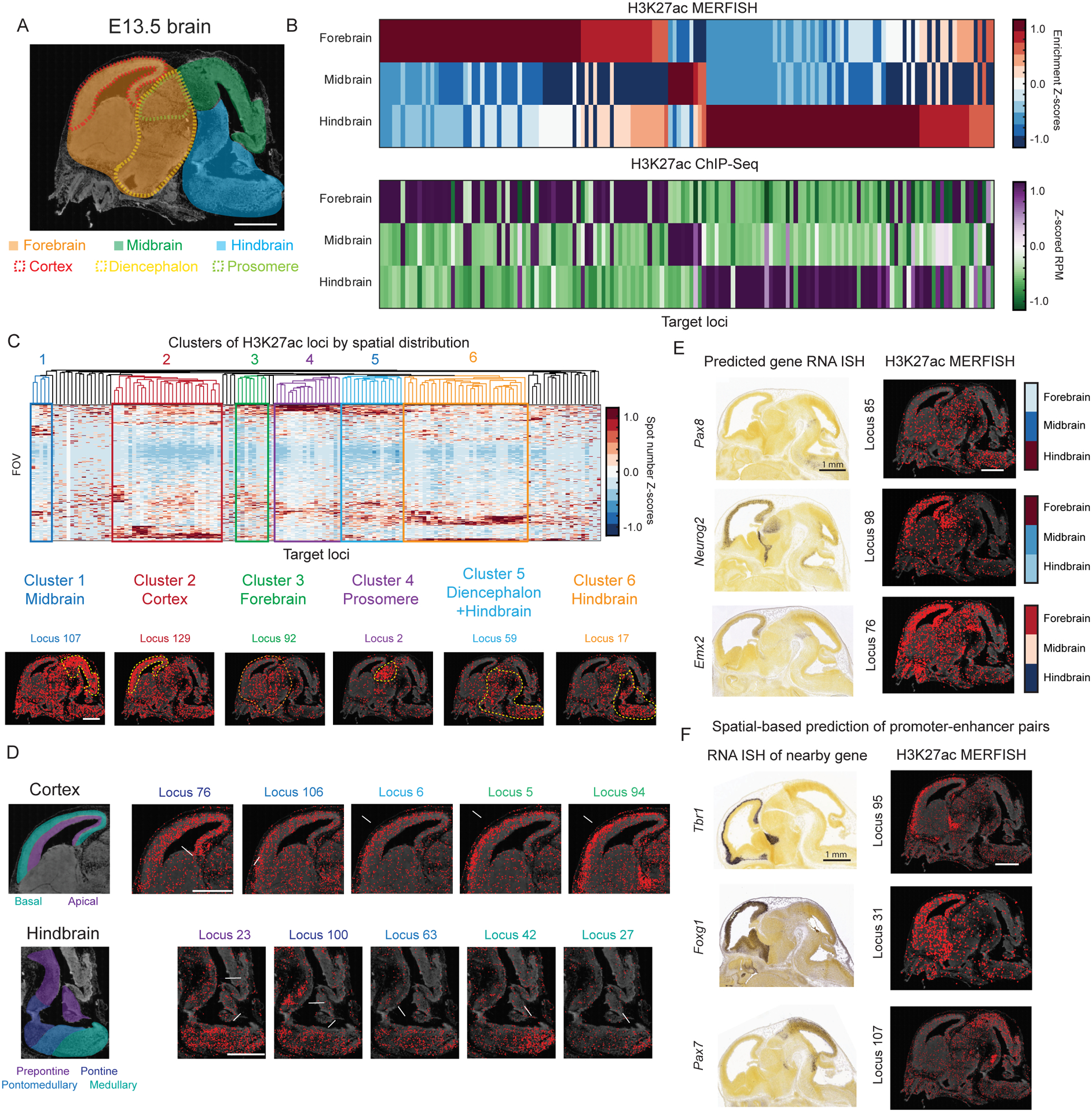
(A) Top: Schematic highlighting different brain regions (forebrain, midbrain, and hindbrain in solid color shades and cortex, diencephalon and prosomere in dotted color lines) of an imaged sagittal slice of a E13.5 mouse brain. The background shows the DAPI signal. Scale bar: 1 mm.
(B) Top: The brain region enrichment z-scores for the H3K27ac signal measured by epigenomic MERFISH for 142 target genomic loci. Brain region enrichment z-score is calculated as described in Figure 3B. Bottom: The corresponding z-scored reads per million for the target loci from published H3K27ac ChIP-seq data of the E13.5 brain (Gorkin et al., 2020).
(C) Top: Hierarchical clustering of the 142 target genomic loci based on the measured spatial distributions of the H3K27ac signals of individual loci. The spatial distribution of each locus is presented as the number of H3K27ac spots in each imaged field-of-view (FOV: 0.04 mm2) for the locus with each FOV presented as a row. Six major clusters that contains >3 loci are shown, representing six different spatial patterns (enrichment in midbrain, cortex, forebrain, prosomere, diencephalon+hindbrain, and hindbrain). Bottom: Epigenomic MERFISH images of the H3K27ac signals of six representative loci, one for each cluster. Scale bar: 1 mm.
(D) Epigenomic MERFISH images of two clusters of loci that show fine spatial distribution changes within the cortex and hindbrain. White arrows point to the region of the H3K27ac signal enrichment. Schematic shown on the left depict the regions of interest. Scale bars: 1 mm.
(E) Comparison between the spatial distributions of H3K27ac signals of three putative enhancer loci measured by epigenomic MERFISH (right) and the expression patterns of the corresponding predicted genes from the Allen RNA ISH atlas (left). Quantifications of the region-specific enrichment of the H3K27ac signals are shown on the right (reproduced from Figure 5B). Scale bars: 1 mm.
(F) Prediction of putative promoter-enhancer pairs using the H3K27ac epigenomic MERFISH data and the RNA expression pattern of the nearby genes. Epigenomic MERFISH of the H3K27ac signals of three target loci are shown on the right and Allen RNA ISH images of the corresponding nearby genes are shown on the left. Scale bars: 1 mm.
See also Figure S6.
To further explore the spatial distributions of these putative active enhancers, we performed hierarchical clustering analysis of these loci based on the spatial distributions of their H3K27ac signals. We obtained six major clusters corresponding to loci with H3K27ac signals enriched in the following six brain regions: midbrain, cortex, forebrain, prosomere, diencephalon+hindbrain, and hindbrain (Figure 5C). To understand whether each cluster of loci was potentially recognized by a specific transcription factor, we performed motif searching analysis using MEME to find transcription factor motifs enriched within these clusters of loci. We found both known motifs for specific transcription factors, including Ascl2, Rfx, Zfp652, Tcf7l2 and Sox17, Sfip1, and Sp2 motifs enriched in the midbrain, cortex, forebrain, prosomere, diencephalon+hindbrain and hindbrain clusters respectively, as well as previously unknown motifs (with top two motifs shown for each cluster in Figure S6D).
Visual inspection of the H3K27ac signals for loci within individual clusters further revealed more refined spatial patterns (Figure 5D). For example, a set of loci in the cortex cluster (loci 76, 106, 6, 5, 94) showed progressive changes in their spatial distributions from the apical to the basal side of the cortex (Figure 5D, top). Embryonic hindbrain develops into future pons, cerebellum, and medulla. Several loci in the hindbrain cluster (loci 23, 100, 63, 42, 27) showed interesting local enrichment of H3K27ac signals within different subregions of the hindbrain (Figure 5D, bottom).
For most putative enhancers, the genes that they regulate remain unknown. We posit that correlation of the enhancer-activity and gene-expression spatial patterns can help predict enhancer-gene pairs. Among the 142 putative enhancer loci that we imaged, six have previously been predicted to be putative enhancers of genes with existing ISH data in the E13.5 brain (Gorkin et al., 2020). Interestingly, three of them showed spatial patterns of H3K27ac signals that matched with the expression pattern of their predicted gene targets (Figure 5E), providing further support for these previous predictions. Such spatial correlation could also be used to generate previously unknown hypothesis of promoter-enhancer pairs. Indeed, several putative enhancer loci (for example, loci 95, 31, 107) in our measurements showed spatial distributions of H3K27ac signals that matched the spatial expression patterns of their nearest genes in the genomic space (Tbr1, Foxg1 and Pax7, respectively) (Figure 5F), suggesting potential regulation of these genes by these putative enhancers.
Putative active enhancer hubs for developmentally important genes in mouse embryonic brain
Interestingly, several of the observed spatial clusters of putative enhancer loci contained multiple loci near a common gene in the genomic space, which showed similar spatial patterns of H3K27ac activity to the expression pattern of the gene. For example, a set of ten putative enhancer loci (loci 66-75) in the prosomere cluster are within ± 300 kb genomic distance from the promoter of Tcf7l2, which are all located within the same sub-TAD (Figure 6A and 6B). All ten enhancers showed H3K27ac signals enrichment in the prosomere region, which resembled the spatial expression pattern of Tcf7l2 (Figure 6A), whereas the other gene located in the same sub-TAD (Vti1a) (Figure 6B) has a different expression pattern (enriched expression in the cortex) (Lein et al., 2007) and are known to regulate cortical development (Sokpor et al., 2021). These results suggest that these ten loci may form an enhancer hub to regulate the expression of Tcf7l2. Interestingly, when we performed motif search for these loci, seven of the ten loci were enriched for the Tcf7l2 motif (loci highlighted in green in Figure 6A, motif shown in Figure S6D). It is thus tempting to surmise that Tcf7l2, a downstream transcription factor for the developmentally important Wnt signaling, binds to its own enhancers to establish a positive feedback loop to ensure its robust expression.
Figure 6. Putative active enhancer hubs for developmentally important genes in mouse embryonic brain.
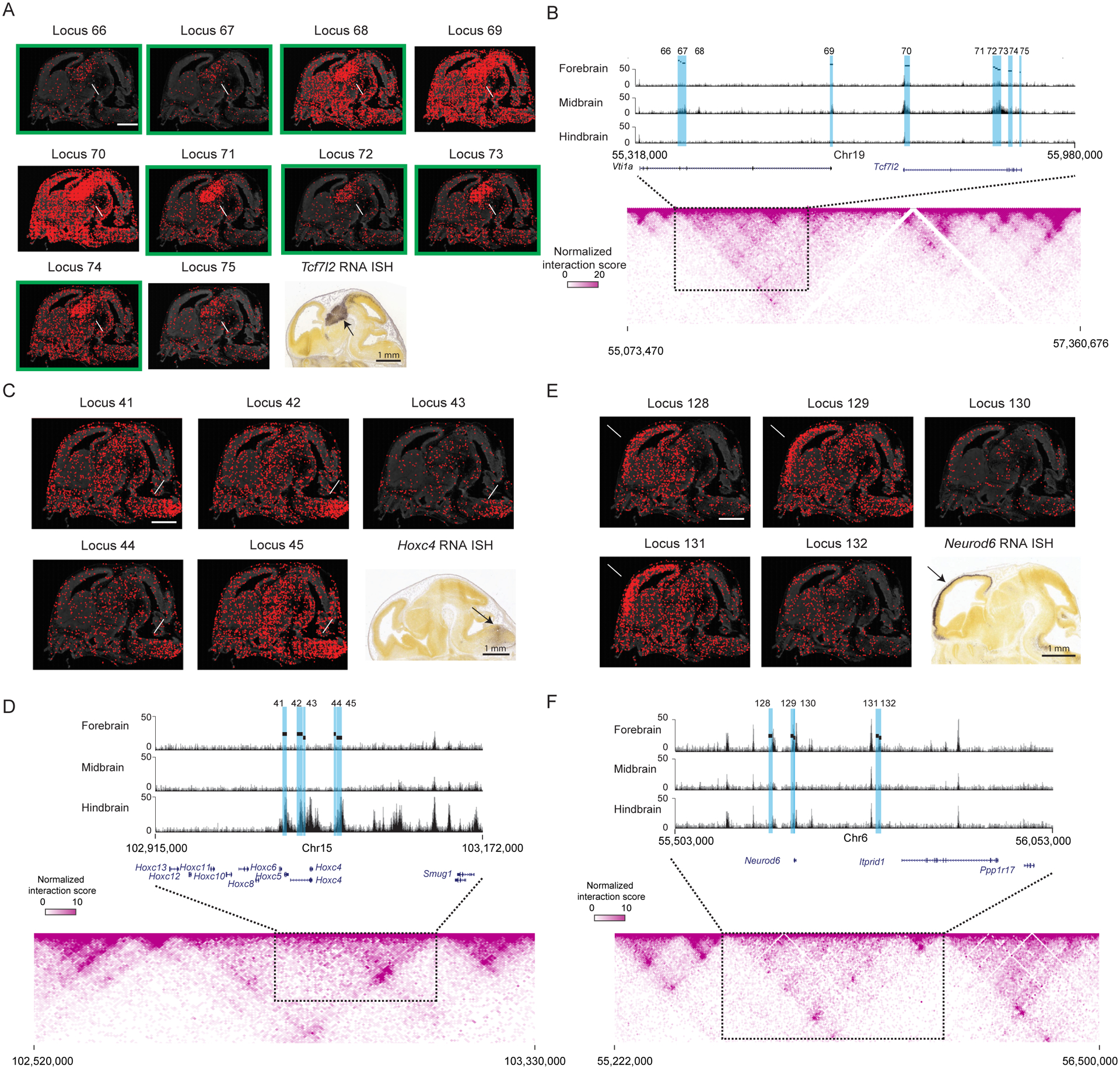
(A) Epigenomic MERFISH images of H3K27ac signals for 10 target loci in the prosomere cluster shown together with the Allen RNA ISH image of the nearby gene Tcf7l2 (bottom right). The white and black arrows respectively point to the prosomere region where the H3K27ac signals and RNA ISH signals are most enriched. Green box marking the loci that harbors a Tcf7l2 motif. Scale bars: 1 mm.
(B) Top: H3K27ac ChIP-seq track (Gorkin et al., 2020) of a region corresponding to the sub-TAD that harbors the 10 target H3K27ac loci enriched in the prosomere and the Tcf7l2 gene. Bottom: Hi-C contact map (Deng et al., 2015) of a genomic region that includes the sub-TAD and flanking regions.
(C) Epigenomic MERFISH images of H3K27ac signals of 5 target loci in the hindbrain cluster shown together with the Allen RNA ISH image of the nearby gene Hoxc4 (bottom right). Scale bars: 1 mm.
(D) As in (B) but for the 5 target loci in the hindbrain cluster.
(E) H3K27ac epigenomic MERFISH images of 5 target loci in the cortex cluster shown together with the Allen RNA ISH image of the nearby gene Neurod6 (bottom right). Scale bars: 1 mm.
(F) As in (B) but for the 5 target loci in the cortex cluster.
Similarly, we found five putative enhancer loci in the hindbrain cluster (loci 41-45) near the promoter of Hoxc4, which is known to express in the hindbrain (Figure 6C and 6D), and five putative enhancer loci in the cortex cluster (loci 128-132) near the promoter of Neurod6, which is known to express in the cortex (Figure 6E and 6F). In both cases, the putative enhancer loci resided in the same sub-TAD with the genes (Figure 6D and 6F) and exhibited spatial patterns of H3K27ac signals similar to the spatial expression pattern of the gene (Figure 6C and 6E). Like the prosomere cluster describe above, these cortex and hindbrain clusters may also form enhancer hubs to regulate the expression of the corresponding Hoxc4 (potentially some other Hoxc genes as well) and Neurod6 genes.
The phenomenon of multiple enhancers regulating the same gene and drive expression patterns that overlap in space has initially been observed in Drosophila (Hong et al., 2008) and subsequently in vertebrate system (Osterwalder et al., 2018). These clusters of dispersed enhancers, referred to as “shadow enhancers” or “redundant enhancers” could provide redundancy to ensure transcriptional robustness during development (Hong et al., 2008; Osterwalder et al., 2018). The putative enhancer hubs that we observed here may be related to these shadow or redundant enhancers. Recently, super-enhancers, clusters of enhancers that span several kb to tens of kb in the genomic space, have also been identified in various systems and suggested to play a role in regulating cell type specific genes (Hnisz et al., 2013). The putative enhancer hubs that we observed span hundreds of kb in the genomic space and hence are not typically considered super-enhancers. Whether they could assume similar functional roles as super-enhancers, for example by 3D chromatin folding, remains an open question for future studies.
DISCUSSION
In this work, we developed a method for spatially resolved single-cell epigenomic profiling. In this method, we captured the epigenetic marks of interest in situ using Tn5-based DNA tagmentation with the T7 promoter, amplified the tagged DNA fragments using in situ transcription, and then detected the resulting RNAs using MERFISH imaging. Using this epigenomic MERFISH method, we demonstrated the ability to profile epigenetic modifications on chromatin, including modifications marking active promoters, putative enhancers, and silent chromatin, in individual cells with high spatial and genomic resolution, as well as high genomic throughput. Histone modifications on genomic loci as short as a few hundred bases can be imaged, providing a genomic resolution of <1kb. The high spatial resolution afforded by imaging allowed us to determine the sub-nuclear localizations of these genomic loci and their spatial relationship with nuclear structures. In our proof-of-principle demonstrations here, we imaged histone modifications of hundreds of genomic loci. Since MERFISH allows >10,000 distinct RNAs to be imaged and identified in individual cells (Xia et al., 2019), we anticipate that the genomic throughput of epigenomic MERFISH could be further increased to allow simultaneous profiling of thousands of genomic loci with specific epigenetic modifications.
We demonstrated that epigenomic MERFISH can be applied to tissue samples. Using this approach to spatially profile two distinct histone modifications that mark active promoters and putative enhancers, we observed region-specific distributions of active promoters and putative enhancers in both adult and developing mouse brain. These measurements not only showed spatial patterns of active promoters and enhancers that are consistent with previous knowledge, but also revealed previously unknown fine spatial distributions of putative enhancers as well as putative enhancer-promoter pairs and enhancer hubs for genes involved in brain development.
Compared to sequencing-based single-cell epigenomic profiling methods that requires cell dissociation, epigenomic MERFISH retains the spatial context of cells and hence enables spatially resolved single-cell profiling of epigenetic activities in tissues. In parallel to our work, a sequencing-based spatial epigenomic profiling method (spatial-CUT&Tag) has been developed by performing CUT&Tag in situ, followed by microfluidics-assisted spatial barcoding and sequencing (Deng et al., 2022). The resolution (pixel size) of spatial CUT&Tag, limited by the microfluidic channel width to tens of microns, makes single-cell analysis challenging: only a small fraction of pixels in the spatial CUT&Tag measurements contain a single cell, whereas the vast majority of pixels have contributions from more than one cells (Deng et al., 2022). In comparison, imaging-based epigenomic MERFISH has a much higher (sub-μm) spatial resolution, which not only enables single-cell analysis but also allows sub-nuclear organization to be probed within individual cells. Epigenomic MERFISH also has its limitations. Unlike sequencing-based methods, which allow untargeted genome-wide detection of epigenetic sites, epigenomic MERFISH is a targeted approach and hence requires prior knowledge or hypothesis for selecting epigenomic loci. We do, however, anticipate that it will be possible to profile the epigenetic properties of thousands, perhaps even tens of thousands, of genomic loci simultaneously, which should mitigate this limitation.
We foresee many applications of epigenomic MERFISH. Here, we used epigenomic MERFISH to profile histone modifications that mark active promoters and enhancers or silent promoters. The in situ tagmentation by Tn5 can be applied to capture other epigenetic marks, as long as antibodies or other affinity probes for these marks exist. Thus, we anticipate that epigenomic MERFISH can be applied to study many epigenomic properties, providing spatially resolved single-cell profiling of not only histone and DNA modifications, but also the binding patterns of transcription factors and non-coding RNAs along the genomic DNA.
Recent studies demonstrated the possibility to preload PA-Tn5 with antibodies to target different epigenetic marks simultaneously (Gopalan et al., 2021). We envision that this approach can also be applied to epigenomic MERFISH, making it possible to simultaneously map multiple distinct epigenetic marks, for example marking promoter and enhancer activities simultaneously to provide a more comprehensive picture of enhancer activity and gene regulation within the cell.
As an imaging approach, we also envision that epigenomic MERFISH could be combined with different modalities of imaging-based omics measurements, such as 3D genome and transcriptome imaging, to enable simultaneous detection of the epigenetic and protein-binding profiles of chromatin, 3D organization of the chromatin, and gene expression profiles within the same cells. Such a single-cell spatial multi-omics approach promises to substantially accelerate our understanding of the mechanisms underlying transcriptional regulation and the role of gene regulation in tissue development and functions.
Limitation of the study
Although the power of epigenomic MERFISH can be further increased by combining this approach with other imaging modalities to enable single-cell multi-omic analysis, our current protocol (light fixation and HCl treatment) was designed to optimize Tn5 accessibility for DNA tagmentation and causes loss of endogenous RNA. Further protocol optimization is likely needed to ensure high-quality epigenomic and transcriptomic imaging of the same samples, either by optimizing conditions for both RNA preservation and DNA tagmentation or sequentially processing the sample for RNA and epigenomic imaging. Combined epigenomic and transcriptomic MERFISH imaging could also benefit from MERFISH probe designs that avoid crosstalk between the two signals. Currently, the detection efficiency of epigenomic MERFISH is lower than that of RNA or DNA MERFISH, which can be as high as 80-100% (Chen et al., 2015; Moffitt et al., 2016b; Su et al., 2020; Xia et al., 2019). The reduction in detection efficiency likely arises from the <100% efficiencies of antibody binding, Tn5 transposition, DNA ligation, and T7 transcription. Further optimizing these steps, such as using polyethylene glycol to increase Tn5 transposition and DNA ligation efficiencies (Picelli et al., 2014), and using more efficient polymerases or promoters to increase transcription activity (Askary et al., 2020), could potentially increase the detection efficiency of epigenomic MERFISH. In this work, we used H3K27ac to mark putative enhancers. Gene regulatory elements are often marked by multiple types of modifications (Rada-Iglesias et al., 2011), such as H3K4me1 and H3K27ac for enhancers. Simultaneous measurements of different modifications could help increase the accuracy of enhancer calling. Ultimately, validations of putative enhancers predicted based on epigenetic marks will require genetic perturbation experiments.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiaowei Zhuang (zhuang@chemistry.harvard.edu).
Materials Availability
Oligonucleotide probe sequences used for imaging can be found in Table S1. These probes or templates for making these probes can be purchased from commercial sources, as detailed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K27ac antibody | Abcam | Cat# ab4729, RRID: AB_2118291 |
| Anti-H3K4me3 antibody | ThermoFisher | Cat# 49-1005, RRID: AB_2533856 |
| Anti-H3K27me3 antibody | Cell Signaling | Cat# 9733, RRID: AB_2616029 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Cat# A-21206, RRID: AB_2535792 |
| SON DNA Binding Protein (SON) (N-Term) antibody | Antibodies-online | Cat# 7ABIN768615, RRID: AB_11207358 |
| Donkey anti-goat secondary antibody | Jackson Immuno | Cat# 705-005-003, RRID: AB_2340384 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formamide | Ambion | Cat# AM9342 |
| 20xSSC | Ambion | Cat# AM9763 |
| Triton-X | Sigma | Cat# T8787 |
| Glucose oxidase | Sigma | Cat# G2133 |
| Phusion® Hot Start Flex 2X Master Mix | New England Biolabs | Cat# M0536 |
| HiScribe™ T7 Quick High Yield RNA Synthesis Kit | New England Biolabs | Cat# E2050 |
| Maxima H Minus Reverse Transcriptase | ThermoFisher | Cat# EP0752 |
| dNTP mix | ThermoFisher | Cat# R1121 |
| Eagle’s Minimum Essential Media (EMEM) | ATCC | Cat# 30-2003 |
| Fetal Bovine Serum Plasma | Sigma | Cat# F1141 |
| 32% Paraformaldehyde | Fisher Scientific | Cat# 50-980-494 |
| MEGAscript Transcription Kit | ThermoFisher | AMB13345 |
| aminoallyl-UTP | ThermoFisher | R1091 |
| Yeast tRNA | ThermoFisher | 15401029 |
| Stellaris RNA FISH hybridization buffer | Biosearch | SMF-HB1-10 |
| PA-Tn5 | Diagenode | C01070002 |
| 1M HEPES | ThermoFisher | 15630080 |
| Spermidine | Sigma | S2626-5G |
| T4 DNA ligase | New England Biolabs | M0202L |
| T4 DNA polymerase | New England Biolabs | M0203L |
| DAPI | ThermoFisher | Cat# D1306 |
| RNase inhibitor, Murine | New England Biolabs | Cat# M0314 |
| RNasin™ Plus RNase Inhibitor | Promega | Cat# PRN2615 |
| 1M Tris, pH 8 | ThermoFisher | Cat# 15568025 |
| Catalase | Sigma | Cat# C3155 |
| 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) | Sigma | Cat# 238813 |
| tris(2-carboxyethyl)phosphi ne (TCEP) | Sigma | Cat# C4706 |
| UltraPure™ BSA (50 mg/mL) | ThermoFisher | Cat# AM2618 |
| Experimental Models: Cell lines and animals | ||
| hTERT-RPE1 | ATCC | Cat# CRL-4000 |
| P56, C57BL6 male mice | Charles River | Cat# 027 |
| Time-pregnant mice E13.5 | Charles River | Cat# 027 |
| Oligonucleotides | ||
| Primers, readout probes | Integrated DNA Technologies | See Table S1 |
| Encoding oligonucleotide probe libraries | Twist Bioscience | See Tables S1 |
| Software and Algorithms | ||
| Software for analyzing epigenomic MERFISH data | This paper | Github: https://github.com/TianLuHarvard/Code |
| Bowtie 2.1.041 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools v1.16.1 | (Li, 2011) | https://www.htslib.org/ |
| MACS 2.1.142 | (Zhang et al., 2008) | https://pypi.org/project/MACS2/ |
| IDR 2.0.243 | (Li et al., 2011) | Github: https://github.com/nboley/idr |
| Bedtools v2.30.0 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/index.html |
| diffBind v1.16.3 | (Ross-Innes et al., 2012) | https://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| featureCounts 2.0.1 | (Liao et al., 2014) | http://subread.sourceforge.net/ |
| DESeq2 1.36.0 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| deepTools v1.5.12 | (Ramírez et al., 2016) | https://deeptools.readthedocs.io/en/develop/content/list_of_tools.html |
| Deposited Data | ||
| Epigenomic MERFISH data | This paper | Zenodo: https://doi.org/10.5281/zenodo.7075964 |
| CUT&Tag data and sequencing data of in situ transcribed RNAs from tagged DNA fragments | This paper | GEO: GSE191069 |
| Hi-C data | (Deng et al., 2015) | GEO: GSE59779 |
| ATAC-seq data | (Gray et al., 2017) | GEO: GSE87548 |
| ChIP-seq data | (Gorkin et al., 2020) | ENCODE: Forebrain (ENCSR311YPF), Midbrain (ENCSR671NSS), Hindbrain (ENCSR344HHI) |
Data and Code Availability
Sequencing data reported in this work are available at NCBI GEO data repository (GSE191069). Epigenomic MERFISH data reported in this work are available at Zenodo (https://doi.org/10.5281/zenodo.7075964).
Analysis code generated in this work is available at https://github.com/TianLuHarvard/Code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
hTERT-RPE1 cells (ATCC, CRL-4000) were cultured in DMEM/F-12, GlutaMAX™ supplement (ThermoFisher, 10565042), 10% FBS (Sigma, F4135-1L) and 1% Pen/Strep (Invitrogen, 15140122) antibiotics at 37 °C. ~1 million cells were plated onto silanized coverslips one day before fixation. hTERT-RPE1 were purchased from ATCC and the identity of the cell line was authenticated by the manufacturer.
Mouse brain tissue sections
P56 male C57BL6 mice were ordered from Charles River Laboratories. The brain was dissected after euthanasia and embedded in Optimal Cutting Temperature Compound (OCT, VWR, 25608-930) and stored at −80°C. E13.5 embryonic brains were collected from timed pregnant C57BL6 mice ordered from Charles River Laboratories. The brain was dissected from the embryo and embedded in OCT and stored in −80°C. The embedded tissue was transferred to −18°C cryostat before slicing. The tissue was sliced into 10μm thick tissue slices and mounted onto silanized coverslips. The mounted tissue was left at room temperature for at least 10 mins before fixation. Animal care and experiments were carried out in accordance with NIH guidelines and were approved by the Harvard University Institutional Animal Care and Use Committee (IACUC).
METHOD DETAILS
Oligonucleotide Probe Design
Selection of target epigenetic loci
The candidate target epigenetic loci for each of the experiments were selected using the criteria as described below. Those candidate loci were subsequently filtered using criteria listed in the “Encoding probe design” section.
1. H3K27ac loci in hTERT-RPE1 cells
H3K27ac peaks were obtained by performing the CUT&Tag reaction in house following the published protocol (Kaya-Okur et al., 2019). The sequencing reads were mapped with bowtie2, followed by peak calling using MACS with q<0.10 and consistent peaks between two replicates were identified by performing irreproducible discovery rate (IDR) setting the threshold at q<0.10 (see “Sequencing Data Analysis” section for more information). These consistent peaks serve as a pool of candidate loci, from which 90 target loci were chosen for imaging.
2. H3K4me3 loci for essential genes in hTERT-RPE1 cells
Candidate H3K4me3 loci were chosen as the promoter regions (±2kb of TSS) of housekeeping genes, from which 52 highly expressed and essential genes (Hart et al., 2015) were selected for imaging to estimate the detection efficiency of epigenomic MERFISH. We further performed CUT&Tag to confirm the presence of H3K4me3 at these promoter loci, and we analyzed the CUT&Tag data similarly as described for the H3K27ac peaks above.
3. H3K27me3 loci in hTERT-RPE1 cells
The promoter regions (±2kb of TSS) of genes with zero or low expression were chosen as candidate H3K27me3 loci, from which 115 loci were selected for imaging. We further performed CUT&Tag to confirm the presence of H3K27me3 at these promoter loci, and we analyzed CUT&Tag data similarly as described for the H3K27ac peaks above.
4. H3K4me3 loci in adult mouse cortex and embryonic mouse brain
Candidate genes that potentially show layer-specific enrichment pattern in adult mouse cortex were curated using marker genes reported by Loo and coworkers (Loo et al., 2019). The gene list included marker genes for layer II-VI excitatory neurons, inhibitory neurons, microglia, endothelial cells, astrocytes, and oligodendrocytes. Candidate genes that show region-specific enrichment pattern in the embryonic brain were chosen as a list of homeodomain transcription factors with well characterized expression in the embryonic brain. From this combined list of candidates, 127 genes were selected. We then targeted the H3K4me3 ChIP-Seq peaks that are close to the TSS of these genes from E13.5 mouse brain (Gorkin et al., 2020) as active promoter loci for imaging.
5. ATAC-seq peaks in adult mouse cortex
Candidate ATAC-Seq peaks were curated from the published data (Gray et al., 2017). There were four mCherry sorted populations of cells driven by the following gene promoters: Cux2, Ntsr1, Rbp4 and Snnc1, which represent layers II/III, IV, V and VI excitatory neurons, respectively. Also, we selected peaks that are more than 2 kb away from TSS to avoid mapping the promoter associated ATAC-seq peaks. We also required that the selected ATAC-Seq peaks has >0 H3K27ac reads mapped to it, according to the bulk H3K27ac ChIP-Seq data obtained from sorted CAMKII excitatory neurons (Mo et al., 2015). Only peaks that differentially expressed in each cortical layer (differential binding performed in diffBind (Ross-Innes et al., 2012), FDR p value<0.05) were chose as candidate loci, from which 139 were selected for imaging.
5. H3K27ac ChIP-seq peaks in the embryonic mouse brain
E15.5 embryonic forebrain, midbrain, and hindbrain specific H3K27ac ChIP-seq peaks previously identified by the ENCODE consortium were merged using bedTools merge (Quinlan and Hall, 2010) to obtain a master list of forebrain, midbrain, and hindbrain peaks (Gorkin et al., 2020). The read counts for the master list of peaks were calculated using featureCount (Liao et al., 2014) with default settings. The read counts were subsequently normalized using DESeq2 norm (Love et al., 2014). Adjusted p-values were obtained using DESeq2 by doing pairwise comparison between any two brain regions. Peaks with significantly more reads in one brain region against the other two regions (fold change > 4, FDR adjusted p-value <0.05) and with normalized reads number >200 were included as candidate loci, from which 142 were selected for imaging. 132 of these loci were also significantly enriched in one brain region in E13.5. 5 loci that have less than 20 reads in H3K27ac ChIP-Seq data in forebrain, midbrain, and hindbrain were additional included as control loci. These control loci served a negative control for our epigenomic MERFISH experiment.
Encoding probe design
During epigenomic MERFISH imaging, a library of encoding probes was first added to the sample to bind to the RNAs generated by in situ T7 transcription. These encoding probes each has a 30-nt target sequence that can bind to a 30-nt target region on one of the RNAs, and 3 readout sequences that allows the encoding probes to be detected by complementary fluorescently labeled readout probes. Each distinct readout sequence corresponds to one bit in the barcode and the collection of readout sequences on an RNA determines the barcode of the RNA. For example, if the barcode reads “1” at bits 1, 3, 5, and 7 and “0” at all other bits, the collection of encoding probes on the RNA should contain readout sequences 1, 3, 5 and 7.
To design encoding probes that target human or mouse loci respectively, hg19 and mm10 genome builds were used for designing target sequences on the encoding probes. For each locus, we identify a number of 30-nt long target regions for the MERFISH encoding probes to bind. The candidate target regions were selected from a sliding window of 30 nt starting from the first nucleotide. The candidate target regions were kept if
The range of GC% is 33%−73% and the melting temperature Tm is 61°C-81°C
It doesn’t have the same 15 nt sequences within other peaks identified from the corresponding ChIP-seq/CUT&Tag data and IgG sequencing data
It doesn’t contain more than 3 consecutive dinucleotide repeats
The next candidate target region was chosen such that it can have a <4 nt overlap with the previous target region and was on the other strand of the DNA. This was repeated until the whole candidate locus was covered. This strategy allows RNA transcribed from either ends of the tagged DNA fragment to be imaged by MERFISH. After designing probes for all candidate loci, the list of candidate loci in the library were filtered by the requirement that number of encoding probes for each locus was > 1.2 - 2 probes per 100 bp in order to ensure efficient labelling of different RNA lengths.
Template library for synthesizing encoding probes were purchased from Twist Bioscience. The sequences for encoding probes are listed in Table S1.
Readout probes
Dye-labeled readout probes were purchase from Integrated DNA Technologies. In the readout probes, fluorescent dye molecules (Alexa 750, Cy5, or Atto565/Cy3B) were linked to oligonucleotide via a disulfide bond that can be cleaved by TCEP. The sequences for dye labeled readout probes are listed in Table S1.
Barcode design
The 24-bit Hamming distance 4 (HD4) and Hamming weight 4 (HW4) code, which contains 366 distinct barcodes, were adopted from La Jolla covering repository and used for MERFISH imaging in this work. The barcodes were then randomly assigned to the target genomic loci except for the requirement that for each bit, there were only 3 - 5 “on” bits (bits that read ‘1’) for each chromosome, in order to ensure that the number of spots imaged in each bit were sufficiently sparse. The barcodes are listed in Table S2.
After barcode arrangement, we next assigned the readout sequences to the encoding probes. Since the HW4 code contains four “1” bit per barcode, 4 readout sequences were assigned to each genomic locus. We required that the three readout sequences on each encoding probe correspond to three of the four readout sequences assigned to its target locus and any two adjacent encoding probes have all four readout sequences. This strategy aims to let short RNA that can only fit 2 probes have all 4 bits presented on the encoding probes.
Experimental Setup
Microscope setup for image acquisition
We used two microscope setups to perform the imaging, and the setups were as described previously (Moffitt et al., 2016a; Wang et al., 2019a). In one of the setups, a Nikon CFI Plan Apo Lambda 60× oil NA 1.4 immersion objective installed on a Nikon Ti-U microscope body was used for imaging. Illumination was provided by solid-state single-mode lasers (405 nm laser, Obis 405 nm LX 200 mW, Coherent; 488 nm laser, Genesis MX488-1000, Coherent; 560 nm laser, 2RU-VFL-P-2000-560-B1R, MPB Communications; 647 nm laser, 2RU-VFL-P-1500-647-B1R, MPB Communication; and 750 nm laser, 2RU-VFL-P-500-750-B1R, MPB Communications). Mechanical shutters were used to switch the 750 nm laser. Acousto-optic tunable filters (AOTF) were used to control the intensities of the 488 nm, 560 nm, and 647 nm lasers; the 405 nm laser was modulated by a direct digital signal. To separate the excitation illumination from the fluorescence emission, a custom dichroic (Chroma, zy405/488/561/647/752RP-UF1) and emission filter (Chroma, ZET405/488/461/647-656/752m) were used. The emission was imaged onto the Hamamatsu digital CMOS camera. During acquisition, the sample was translated using a motorized XY stage (Ludl, BioPrecision2) and kept in focus using a home-built autofocus system. A peristaltic pump (Gilson, MINIPULS 3) pulled liquid into Bioptechs FCS2 flow chamber with sample coverslips and three valves (Hamilton, MVP and HVXM 8-5) were used to select the input fluid.
In the other setup, samples were imaged on a custom-built Olympus microscope body with 60x silicone oil objective (UPLSAPO 60x S2; Olympus). Laser illumination was provided at 750, 647, 560, 488 and 405 nm with a Lumencor Celesta light system. These illumination laser wavelengths were used to excite Alexa750, Cy5 and Cy3 conjugated readout probes, Alexa-488 fiducial beads and DAPI respectively. The dichroic and emission filter were the same as described above. The rest of the imaging system was as described previously (Moffitt et al., 2016a).
Experimental Procedures and Protocols
Encoding probe synthesis
Encoding probe synthesis was as described previously (Xia et al., 2019). Briefly, the encoding probe template library was ordered from Twist Bioscience and diluted in TE buffer to about 1 ng/μL. We did qPCR (10 - 12 cycles) to amplify the oligo pools and stopped the reaction when the curve started to plateau. The amplified templates were purified and transcribed into RNAs via in situ transcription for >20 hrs at 37°C, The RNAs were reverse transcribed to ssDNAs for 1 hr at 55°C, and we then purified the DNAs via alkaline hydrolysis (to remove RNA templates), phenol-chloroform extraction (to remove proteins), and ethanol precipitation (to remove nucleotides and concentrate probes). The final concentration of the encoding probe library was about 40 μg/μL.
Imaging coverslip silanization
40-mm, round #1.5 coverslips (Bioptechs, 0420-0323-2) were first cleaned by 37.5% HCl and pure methanol for 30 mins at room temperature, washed by 70% ethanol and dried. For silanization, coverslips were covered in silanization buffer (500 mL distilled water, 1500 μL Bind-silane (Sigma, GE17-1330-01) and pH adjusted to 3.5 by glacial acetic acid) for an hour at room temperature. The coverslips were then washed with water and dried in the oven before storing in a dehumidified chamber.
Epigenomic MERFISH protocol for cell culture and tissue slices
For cell culture, the cells were fixed with 1% PFA in 1x PBS for 5 mins at room temperature and washed three times with 1× PBS. The sample was then permeabilized by 1% Triton-X for 20 mins at room temperature and washed three times with 1× PBS. Then 0.1 M HCl was added for chromatin loosening for 5 mins at room temperature and washed three times with 1× PBS. The sample was then incubated in block buffer (50% Ultrapure BSA, 1% Triton-X and 1× PBS) for one hour at room temperature and further incubated in 1:100 primary antibody (against H3K27ac, H3K4me3, or H3K27me3) for one hour at room temperature. The sample was washed three times with 1x PBS and incubated in 1:200 secondary antibody (ThermoFisher, A-21206) for one hour at room temperature. After washing three times with 1x PBS, the sample was ready for transposition.
Before the transposition, PA-Tn5 (Diagenode, C01070002) was loaded with a pair of annealed loader DNAs ordered from IDT (sequences in Table S1). The two loader DNAs were annealed at 100 μM concentration (for each loader) using the following settings at the thermocycler (95°C for 6 minutes and −5 °C per cycle for 15 cycles, each cycle lasting 1 minute). For loading PA-Tn5, 6.5 μL of annealed loader A, 6.5 μL of annealed loader B and 10 μL of PA-Tn5 were mixed and incubated at room temperature for one hour. 12.5 μL of 100% glycerol was added to the mixture and PA-Tn5 was ready for transposition and stored at −20°C.
The immunolabeled sample was incubated with 1:50 PA-Tn5 in 50 μL of PA-Tn5 binding buffer at room temperature for 1 hour to let protein A bind to antibodies. The 50 mL PA-Tn5 binding buffer was prepared by mixing 1 mL 1M HEPES (Thermo, 15630080), 3 mL 5 M NaCl, 4 μL Spermidine (Sigma S2626-5G), and 100 μl of 5% digitonin with water filled up to 50 mL. The high salt concentration in the buffer prevents the non-specific binding of the PA-Tn5 and the lack of Mg2+ in the buffer prevents the transposition of PA-Tn5 (Kaya-Okur et al., 2019). After the incubation, samples were washed with PA-Tn5 binding buffer 3 times to remove nonspecific binding. Samples were then incubated in the PA-Tn5 transposition buffer (1 mL of PA-Tn5 binding buffer with 10 μL 1M MgCl2) for 1 hr at 37°C for transposition.
After transposition, samples are washed three times with 1× PBS. For experiments that combined with nuclear speckle labels, the sample were further incubated in 1:100 SON antibody (ABIN768615) and 1:100 oligo-labeled secondary antibody in block buffer at room temperature for 1 hour, respectively. The sample was then embedded in 4% Acrylamide/Bis 19:1 gel with 1:200 Alexa-488 beads (Invitrogen) for 1 hr at room temperature. The embedded sample was digested in 2% SDS, 0.5% Triton-X and 1:100 proteinase K in 2x SSC at 37 °C for at least 16 hours. The sample was then washed 3 times with 1× PBS. Each wash was one hour at room temperature on a shaker. After wash, the sample was incubated in 50 μL nick ligation mix containing 2.5 μL of T4 DNA ligase (New England Biolabs, M0202L), 2.5 μL of T4 DNA polymerase (New England Biolabs, M0203L), 5 μL of T4 DNA ligase buffer (New England Biolabs, M0202L), 5 μL of 10 mM dNTP (New England Biolabs, N0447L) and 35 μL of water at room temperature for 40 minutes at room temperature. After wash three times with 1× PBS, the sample was incubated in transcription mix (MEGAscript Transcription Kit, ThermoFisher, AMB13345; or HiScribe T7 RNA synthesis Kit, New England Biolabs, E2040S) with aminoallyl-UTP (Thermo, R1091) at 37°C for 16-18 hours. For MEGAscript Transcription Kit, he 200 μL transcription mix contained 20 μL ATP, 15 μL UTP, 20 μL CTP, 20 μL GTP, 20 μL 10x Reaction buffer, 20 μL T7 polymerase, 10 μL RNase inhibitor and 5 μL aminoallyl-UTP. For HiScribe T7 RNA synthesis Kit, the 200 μL transcription mix contained 65 μL water, 100 μL NTP buffer mix, 20 μL T7 polymerase, 10 μL RNase inhibitor and 5 μL aminoallyl-UTP. Samples for RNA sequencing didn’t have aminoallyl-UTP. After in situ transcription, the samples were fixed in 4% PFA for 20 minutes at room temperature to crosslink the transcribed RNA via the aminoallyl group to the gel and stained with 1:30 encoding probe library in 30% hybridization buffer at 37°C overnight. The hybridization buffer contained 30% formamide (ThermoFisher, AM9342), 60% stellaris RNA FISH hybridization buffer (Biosearch, SMF-HB1-10), 10% 25 mg/mL Yeast tRNA (ThermoFisher, 15401029), and 1:100 murine RNase inhibitor. Our approach requires multiple enzymatic steps which are sensitive to freeze/thawing cycles and storage conditions. For NTP, T4 ligase buffer, dNTP, AA-UTP and pA-Tn5 loader, we recommend using fresh reagent aliquots.
For tissue slides, the protocol was similar, except that 1) the primary antibody was stained overnight at 4°C, 2) the binding of the PA-Tn5 was allowed to occur for 2 hours and 3) the transposition reaction was allowed to run overnight at 37°C.
For SON test in Figure S1, the samples were stained with SON antibodies (Antibodies-online, ABIN768615) and the PA-Tn5 was loaded with a pair of Tn5 loader DNAs (MEA and MEB) from Illumina, and the loader DNAs contained a single-stranded overhang. After transposition, the samples were washed with PBS and hybridized with 100 nM probes with a region that can target the overhang region and another region that can bind to readout probes in 10% hyb buffer (10% formamide, 80% stellaris RNA FISH hybridization buffer, 10% 25 mg/mL Yeast tRNA, and 1:100 murine RNase inhibitor) at 37°C for 1 hour. The samples were then washed in 30% formamide with 2x SSC and stained with readout probes in 10% EC buffer for imaging.
Synthesis of oligo conjugated antibodies
100 μl of 1 mg/ml donkey anti-goat secondary antibodies obtained from Jackson Immuno (705-005-003) were first incubated with 2 μl of 5 mM of DBCO-PEG5-NHS ester (Kerafast) in 1X PBS at room temperature for 1 hour. The product of the reaction was purified and resuspended in 100 μl of 1X PBS using Amicon columns according to the manufacturers’ protocol. 20 μl of 100 μM readout oligonucleotides functionalized on the 5’ end with acrydite and on the 3’ end with azide was added and the reaction was left to completion at 4°C overnight. The 5’ acrydite allows tethering of the oligonucleotides to the gel and the 3’ azide reacts with the DBCO moiety via copper-free click chemistry reaction. The final product of the reaction was purified and resuspended in 100 μl of 1X PBS using Amicon columns according to the manufacturers’ protocol. The sequences of the oligonucleotides were provided in Table S1.
RNA extraction and sequencing
The RNAs were harvested from the polyacrylamide gel using the crush and soak method. Briefly, the gel was scraped off the coverslip surface and shredded into tiny pieces before resuspension in the elution buffer (500 mM ammonium acetate and 1mM EDTA-KOH pH 8.0) with 1:100 RNase inhibitor (Promega) and rotated at room temperature for 2 - 4 hours. After 2 - 4 hours, gel pieces were removed using a 40 μm filter. RNAs in the eluate were then purified using Zymo RNA kit using the recommended protocol by Zymo. RNAs were first reverse transcribed into cDNAs using a primer specific to one mosaic end using Maxima RT kit (ThermoScientific) using the manufacturer’s protocol. The resulting cDNAs were PCR enriched using the following settings using Phusion (New England Biolabs): 1. 72°C, 5 minutes; 2. 98°C, 30 seconds; 3. 98°C, 10 seconds; 4, 63°C, 30 seconds; 5. 72°C, 45 seconds; 6. go back to step 3 for N cycles. The number of cycles (N) were first determined by running a test PCR reaction (¼ the saturation). The usual cycle number required was around 12 - 15.
Following PCR, 0.7 volume of AMPure XP beads (Beckman) was added to the PCR reaction. The mixture was incubated for 5 minutes before placing on the magnetic rack. The beads were then washed twice with 80% ethanol before they were let dry at room temperature. 20 μl of RNase free water was added for elution. The 0.7 volume ensured proper removal of the primer dimer peak. Subsequently, the PCR product was examined using DNA Tapestation for proper size distribution. Concentration of the PCR product was obtained by selecting the peak from 100 - 1000 bp. Libraries that passed the Tapestation QC were sequenced paired-ended using Novaseq SP100 or Nextseq platform.
Imaging procedure for epigenomic MERFISH
After hybridization of the encoding probes, samples are washed in 30% formamide at room temperature for 20 minutes and washed 3 times with 2x SSC before imaging. The first set of readout probes were added at 3 nM concentration in 2X SSC with 10% ethylene carbonate. The stained sample coverslips were mounted to the Bioptechs imaging chamber for imaging. Each imaging round contained three distinct steps: imaging, cleaving and hybridization. The buffer for each step was flowed into the imaging chamber via a fluidic system controlled by a custom made software (Su et al., 2020).
In the imaging step, about 2 mL of anti-photobleaching buffer was flowed into the chamber. For anti-photobleaching buffer, we used either
rPCO-PCA based buffer: 2x SSC, 5 mM 3,4-dihydroxybenzoic acid (Sigma, P5630), 2 mM (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) (Sigma, 238813), 50 μM trolox quinone, 1:500 rPCO (Oriental Yeast Company), 1:500 Murine RNase inhibitor, and 5 mM NaOH (to adjust pH to 7.0) and topped up to 50 mL with nuclease free water.
Glucose oxidase base buffer: 50 mg glucose-oxidase (Sigma, G2133), 50 mg Trolox, 300 μL catalase (Sigma, C100-500MG), 10% w/v glucose (Sigma, G8270), 5 mL 500 μM Trolox quinone and 50 μL murine RNase inhibitor and topped up to 50 mL with nuclease free water.
For cultured hTERT-RPE1 cells, 100 fields of view were imaged for each sample with 40 z-planes (step size of 200 nm) imaged per channel. For brain tissue slices, 200 - 400 fields of view were imaged for each sample with 30 z-planes (step size of 300 nm) per channel. The images were acquired at 10 Hz. After imaging, in the cleaving step, 2 mL of cleaving buffer containing 2x SSC and 50 mM TCEP (Goldbio) was flowed into the imaging chamber to cleave the dye off the readout probes and left for 12 minutes before the residual TCEP was removed by flowing in 2 mL of 2x SSC.
In the readout hybridization step, readout probes in three different colors (labeled with Alexa750, Cy5, and Atto565/Cy3B respectively) were added to the hybridization buffer (2x SSC, 10% ethylene carbonate, 200μl of 100% Triton-X) at a concentration of 3 nM for each readout probe. The readout probes were left to hybridize for 12 minutes before the unbound readout probes were washed away with a wash buffer (2x SSC, 10% ethylene carbonate). The three steps were repeated 8 times for a 24-bit imaging.
Image Analysis
Decoding of epigenomic MERFISH images
To normalize for intensity variation across different color channels, every image in a given color channel was divided by the mean-intensity image of all images in that that color channel. Images of multiple rounds were registered using Alexa 488 fiducial beads. Cell nuclei were segmented by watershed algorithm using DAPI staining as both seed and boundaries.
Epigenomic MERFISH signals from each channel in each hybridization round were identified using two spot-finding methods: In the experiments in which we segmented individual cells, the pixels in each nucleus with intensity higher than certain brightness threshold was selected. In order to connect spots detected in different z-planes, the selected pixels across different z planes were clustered by the bwareaopen function in MATLAB (Version 2021a) with the requirement that the number of pixels within a cluster should be in the range of 10-100. In order to capture clusters with relatively wide variations in spot intensity, this process was iterated using multiple brightness thresholds (from top 0.001% to top 1% of the FOV with the decrement of 0.01%). Each iteration of lowering the brightness threshold allowed the identification of additional clusters that belonged to one of the following two types: (I) dim pixel clusters that could not be recognized at higher brightness threshold in the previous iteration, and (II) larger clusters that encompassed one or more pixel clusters found in the previous iteration. Any cluster of type-I was preserved only if its total number of pixels is within the range of 10-100. If the pixel number of any cluster of type-II fell within the permissible range, it was kept; if not, it was deleted, and the smaller pixel cluster(s) found in the previous round that overlapped with this new cluster were kept instead. Each pixel cluster was then considered a spot and the x, y, and z coordinates of the spot was measured using the regionprops3 function in MATLAB (Version 2021a).
In the experiments in which we didn’t segment the cells, the 3D spot finding approach described above was computationally too slow to find the spots in the whole imaging field of view. We thus used a 2D spot finding approach first. Briefly, the 2D spots for each Z plane were first identified using the approach described above, but in 2D instead of 3D, using the regionprops function. After spot finding in every z plane, the 2D spots across all z planes were clustered by DBSCAN function in MATLAB (Version 2021a) using the distance threshold of 50 nm in the x, y plane and 300 nm in z direction and minimum spot number of 2. Each resulting spot cluster was then considered a 3D spot and the x, y, and z coordinates of the 3D spot were calculated as the mean of x, y, and z coordinates of the 2D spots in the spot clusters.
The spots identified from the two methods were further filtered by signal-to-background ratio with a threshold of 1.4. The signal-to-background ratio for a spot was defined as the intensity of the center of the spot divided by the minimal intensity of the pixels that were 500 nm away from the spot center in the xy plane.
After 3D spot finding, the spots from all bits were clustered by DBSCAN using a distance threshold of 250 nm in 3D and minimum spot number of 3. For any cluster that had 3-5 spots (i.e. a cluster that was detected in 3-5 bits), the cluster was then decoded according to the codebook allowing at most one-bit mismatch from the valid barcodes. For any cluster that had more than 5 spots, the spots within that cluster were further clustered by DBSCAN using a distance threshold of 150 nm in 3D and minimum spot number of 3 and the resulting new clusters that had 3-5 spots were decoded. The clusters that were not matching to any barcode were discarded. The final x, y, and z coordinates of the decoded spots were calculated as the mean x, y, and z coordinates of the spot across all bits. The decoded locus identity, 3D localization and barcode error of the spots are saved for further analysis.
Measurements of distance between epigenomic loci and nuclear speckles
The boundaries of nuclear speckles were determined by thresholding the intensity and area of the SON-positive areas in the immunofluorescence images of the nuclear speckles. The intensity threshold was set as top 2% of all pixels within each cell and the area thresholds for individual nuclear speckles were set to 100 pixels for lower bound and 800 pixels for upper bound. The distance of an epigenomic MERFISH spot to the nearest nuclear speckle was calculated in 3D as the minimal distance between the epigenomic spot and the SON-positive pixels.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantifying region-specific enrichment in mouse embryonic brain
The specific regions of the brain were manually segmented by comparing the DAPI staining in our images and the reference Allen brain atlas (http://developingmouse.brain-map.org/static/atlas). The total number of decoded spots within these regions were counted and divided by the DAPI positive area to calculate the spot density. The ordering of the heatmaps for imaging data in Figures 3 and 5 were done as follows: 1) The loci with maximum density in certain regions were grouped. 2) Within this group, the maximum density for those loci were ordered from largest to smallest. 3) Arrange the group in the region order as shown in the figures. All heatmaps were plotted using the Z-score of the spot density for each locus.
Quantifying layer-specific enrichment in adult mouse cortex
The layers in the mouse cortex were manually identified by comparing the DAPI staining in our images and the reference Allen brain atlas (http://mouse.brain-map.org/static/atlas). To assign each cell into each layer, we approximated the cortical layer boundaries as a set of concentric circular arcs, which matched with the manually segmented cortical layer boundaries by visual inspection. We then determined whether any given cell belongs to a cortical layer by comparing the radial position of the cell with the radii of the concentric circular arcs representing the cortical layer boundaries. The layer enrichment for a specific epigenetic activity of each locus was calculated as follows: A cell was considered H3K4me3- or H3K27ac-positive for a certain locus if at least one decoded spot for this locus was detected in the cell. For each locus, the layer enrichment in a specific layer was calculated as the z-score of the following quantity: the fraction of cells in the layer that were H3K4me3- or H3K27ac-positive for this locus. The significance of the enrichment was calculated using a chi-square test using Chi2test function in MATLAB (Version 2021a). The ordering of the heatmaps for layer enrichment in Figures 2 and 4 were done as follows: 1) The loci with maximum layer enrichment in certain layers were grouped. 2) Within each group, the maximum layer enrichment for those loci were ordered from largest to smallest. 3) Arrange the groups in the layer order as shown in the figures. All heatmaps were plotted using the Z-score of the layer enrichment for each locus.
Clustering of putative active enhancers based on spatial distribution
The putative active enhancer loci marked by H3K27ac in the embryonic mouse brain were clustered based on their spatial distributions, measured as the number of decoded spots in each field of view (FOV). Clustergram function in MATLAB (Version 2021a) was used and the linkage for clustering the FOV and enhancer loci was ‘weighted’, distance was ‘Euclidean’. To identify the main clusters with more than 3 loci, we used the linkage threshold of 22.3. The resulting six main clusters were shown in the Figure 5.
Motif enrichment analysis
Motif enrichment analysis was performed by obtaining the sequences of the loci belong to each of the six main clusters described above using bedtools getFasta (Quinlan and Hall, 2010). The sequences were uploaded to the MEME-ChIP website and motif enrichment was performed for each of the six clusters using default settings. Only the top two most significant motifs for each cluster were listed in Figure S6D.
Sequencing data analysis
Sequencing reads were aligned to the hg19 and mm10 reference sequence using Bowtie 2.1.041 (Langmead and Salzberg, 2012). Mitochondrial reads were removed. Reads were subsequently deduplicated using the rmdup option in samtools (Li, 2011). Peak calling was performed using MACS 2.1.142 (Zhang et al., 2008) using the default options outlined in the vignette with the significant value cut-off at q<0.1. For experiments with two replicates, top 100,000 reproducible peaks were sorted by the p value and selected using IDR 2.0.243 (Li et al., 2011) with cutoff of IDR ≤ 0.1 using the recommended settings. Peaks were considered differentially expressed if they have q<0.10 by DiffBind (Ross-Innes et al., 2012). The global profiles of histone marks were then plotted using deeptools (Ramírez et al., 2016) suite following the vignette centering at the peak summit.
Supplementary Material
Figure S1. The effect of fixation and clearing on epigenomic MERFISH signals and the performance of epigenomic MERFISH imaging, related to Figure 1 (A) Images comparing the SON (a marker for nuclear speckles) immunofluorescence (IF) images (left panels) in different fixation conditions (MAA: Methanol/acetic acid, 1% PFA, EGS: ethylene glycol bis(succinimidyl succinate), DSG: disuccinimidyl glutarate, 1% PFA followed by 0.1N HCl treatment) and the location of tagged DNA fragments colocalized with SON and detected by FISH (right panels). To generate tagged DNA fragments colocalized with SON, primary antibody against SON was added, followed by secondary antibody and PA-Tn5. 1% PFA with 0.1N HCl treatment gave the best colocalization between the SON immunofluorescence and tagged DNA signals. We performed screening for fixation conditions using the nuclear speckle marker as it forms distinct puncta in the nucleus, enabling rapid screening of the in situ tagmentation conditions for fixed cells. Scale bar: 10 μm.
(B) Sequencing analysis of in situ transcribed RNAs from tagged DNA fragments with the various histone modifications. The top four UCSC tracks compare the sequencing results of in situ transcribed RNAs from tagged DNA fragments with the H3K27ac modification (in situ tag. and trans.) for cleared and uncleared samples (both with 5 min PFA fixation and HCl treatment) with results obtained from H3K27ac ChIP-Seq and CUT&Tag measurement in hTERT-RPE1 cells. The sequencing result of in situ transcribed RNA from cleared sample shows more unique reads and better capture of the epigenetic profile. The middle three UCSC tracks compare the sequencing result of in situ transcribed RNAs from tagged DNA fragments with the H3K4me3 modification (in situ tag. and trans.) obtained under two fixation conditions (5 min PFA fixation, HCl treated and 10 min PFA fixation, both cleared) with results obtained H3K4me3 CUT&Tag measurements in hTERT-RPE1 cells. The sequencing result of in situ transcribed RNA from 5 min fixation with HCl treatment showed more unique reads and better capture of the epigenetic profile. The bottom two UCSC tracks compare the sequencing result of in situ transcribed RNAs from tagged DNA fragments with the H3K27me3 modification (in situ tag. and trans., 5 min PFA fixation, HCl treated, cleared) with results obtained the H3K27me3 CUT&Tag measurements in hTERT-RPE1 cells. The tracks were normalized to 1x sequencing depth.
(C) RNA Tapestation results showing the RNA length distribution of in situ transcribed RNAs from tagged DNA fragments with the H3K4me3 modification.
(D) Genome wide profiles comparing the sequencing results of in situ transcribed RNAs from tagged DNA fragments and the CUT&Tag results for the three chromatin marks (H3K27ac, H3K4me3, and H3K27me3) centered at the peak summits. Each line represents a peak in the CUT&Tag track and the same peak obtained from the sequencing result of in situ transcribed RNAs. The genomic locations of peaks are determined from the CUT&Tag data and the tracks for the in situ transcribed RNAs from the same genome regions are plotted for comparison. The scale bar shows the magnitude of the normalized peak height (1x sequencing depth). The peaks shown are significant peaks with q value < 0.1.
(E) Genome wide profiles comparing the sequencing results of in situ transcribed RNAs from tagged DNA fragments for H3K27ac and H3K27me3 experiments. Left: The genomic locations of peaks are determined from the H3K27ac data and the tracks for the H3K27me3 data from the same genome regions are plotted for comparison. The very dim signal in the H3K27me3 data indicate that the H3K27ac peaks were essentially not detected using H3K27me3 antibody. Right: The genomic locations of peaks are determined from the H3K27me3 data and the tracks for the H3K27ac data from the same genome regions are plotted for comparison. The very dim signal in the H3K27ac data indicate that the H3K27me3 peaks were essentially not detected using H3K27ac antibody.
(F) Violin plots showing the average number of decoded spots per cell for each of the 52 target H3K4me3 loci corresponding to the promoters of the 52 essential genes (left) and each blank barcode (middle) when H3K4me3 antibody is used to capture the epigenetic mark. Also shown is the violin plot of the average number of decoded spots per cell for each target H3K4me3 locus when a control IgG is used instead (right). Each dot corresponds to a single H3K4me3 locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K4me3 antibody and that detected with IgG for individual H3K4me3 loci. Results here are aggregates of three replicates. The Pearson correlation coefficients for the number of decoded spots per cell for individual H3K4me3 loci between replicates are 0.58 - 0.67. The correlation is lower than that observed for the H3K27ac measurements, as expected because the biological variation across active promoters for essential genes is relatively low. Indeed, the correlation between replicates is substantially higher for the 127 target H3K4me3 loci in the mouse cortex (see Figure S2B), where the biological variation across promoter loci is expected to be higher due to cell-type specific expression of the correspond genes.
(G) Same as (F), but for the 115 target H3K27me3 loci. Results here are aggregates of two replicates. The Pearson correlation coefficient for the number of decoded spots per cell for individual H3K27me3 loci between replicates is 0.64. The correlation is lower than that observed for the H3K27ac measurements, potentially due to an antibody specific effect. Nonetheless, the distance to nuclear speckles for individual H3K27me3 loci showed good correlation between replicates with a Pearson correlation coefficient of 0.7.
Figure S2. The performance of epigenomic MERFISH imaging of mouse brain tissues, related to Figure 2 (A) Epigenomic MERFISH image of 127 target H3K4me3 loci in a single cell. The images from individual bits are shown on the left. The decoded image is shown on the right, with individual spots color-coded based on the identities of the chromosome identity. Scale bars: 4 μm.
(B) Correlation between two biological replicates of H3K4me3 imaging. Each dot corresponds to a single H3K4me3 locus. The Pearson correlation coefficients are 0.95.
(C) Violin plots showing the average number of decoded spots per cell for each of the 127 target H3K4me3 loci (left) and each blank barcode (middle) when H3K4me3 antibody is used to capture the epigenetic mark. Also shown is the violin plot of the average number of decoded spots per cell for each target H3K4me3 locus when a control IgG is used instead (right). Each dot corresponds to a single H3K4me3 locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K4me3 antibody and that detected with IgG for individual H3K4me3 loci. Results here are aggregates of two replicates.
Figure S3. Comparisons between epigenomic MERFISH images of active promoters and the RNA MERFISH images of corresponding genes in mouse cortex and the randomization control, related to Figure 2 (A) Same as Figure 2B, but for the promoter loci that showed statistically significant enrichment in specific cortical layers in replicate 1, with results from replicate 2 for the same genes also shown for comparison. Left: The layer enrichment z-scores for the promoter H3K4me3 signals measured by epigenomic MERFISH for the indicated genes for two replicates. The Pearson correlation coefficients R between the two replicates are shown on the right. Right: The layer enrichment z-scores for the RNA expression level measured by RNA MERFISH for the corresponding genes (Zhang et al., 2021).
(B) Comparisons between epigenomic MERFISH images of example promoter loci and the RNA MERFISH images of corresponding genes that did not show statistically significant layer-specific enrichment. For each gene, the left panel show epigenomic MERFISH images of H3K4me3 signals with each dot in the images representing a cell and red dots represent cells with positive H3K4me3 signals, and the right panel shows the RNA MERFISH image of the corresponding gene (Zhang et al., 2021), with each dot showing the RNA expression level in a cell. Scale bars: 400 μm.
(C) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for three example promoter loci that show significant layer-specific enrichment. Each dot in the images represents a cell and red dots represent cells with positive H3K4me3 signals. In the randomly permutation, the spatial position of each detected spot is unchanged and the genomic identity of each detected spot is randomly selected among the 127 target H3K4me3 loci while keeping the total number of spots detected for each genomic locus the same as the experimentally observed value. Scale bar: 400 μm.
(D) Comparison of the fraction of cells that is H3K4me3-positive in each of the cortical layers between the experimentally observed values and the values derived from the randomly permutated images for the three loci shown in (C).
(E). Box plots of the Pearson correlation coefficient for the layer enrichment between the epigenomic MERFISH and RNA MERFISH data. The box plots depict the distributions of the Pearson correlation coefficients for all 38 promoter loci that has RNA MERFISH images of the corresponding genes. Red line denotes the median, box denotes the 25-75 percentile, and whiskers denote the 1.5 interquartile range. (Left) Experimentally measured results, as shown in Figure 2B. (Right) Results from the random permutation controls where the epigenomic MERFISH images are randomly permutated as described in (C). 1000 permutations were performed and the Pearson correlation coefficients from all permutations are included in the box plot.
Figure S4. Comparison between epigenomic MERFISH images of active promoters and Allen RNA ISH images of the corresponding genes in mouse embryonic brain and the randomization control, related to Figure 3 (A) Comparison between epigenomic MERFISH images of the H3K4me3 signals of the promoter loci and the Allen RNA ISH images of the corresponding genes. All 46 promoter loci that have a H3K4me3 spot density > 300 per mm2 and Allen RNA ISH images of the corresponding genes are shown here. The promoter loci are sorted based on the matching score between epigenomic MERFISH images and RNA ISH images. We segmented the imaged tissue sections into five brain regions: cortex, subpallium, diencephalon, midbrain and hindbrain and determined whether the H3K4me3-MERFISH and RNA ISH signals are enriched in each of these five regions by visual inspection. The matching score was calculated as how many regions have the same enrichment assessment (i.e, enriched or not) for H3K4me3 and RNA ISH signals. Two independent classifications were performed by two people. The results largely agreed with each other, and where the matching score differed between the two classifications, we took the average of the two scores. Scale bars: 1 mm.
(B) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for three example promoter loci that show significant enrichment in specific brain regions. The random permutation is performed among the 127 target H3K4me3 loci as described in Figure S3C. Scale bar: 1 mm.
(C) Box plots of the matching scores between epigenomic MERFISH images and RNA ISH images. The box plots depict the distributions of the matching scores derived for all 46 promoter loci shown in (A). Red line denotes the median, box denotes the 25-75 percentile, and whiskers denote the 1.5 interquartile range. Symbol denote outliers. (Left) Experimentally measured results. (Right) Results from the random permutation controls (10 permutations), where the epigenomic MERFISH images are randomly permutated as described in (B).
Figure S5. The performance of epigenomic MERFISH measurements of target H3K27ac loci in adult mouse cortex, related to Figure 4 (A) Scatter plot showing the correlation between two biological replicates of H3K27ac imaging in the adult mouse cortex. Each dot corresponds to a single H3K27ac locus. The Pearson correlation coefficient is 0.90.
(B) Violin plot showing the average number of decoded spots per cell in adult mouse cortex for each of the 139 target H3K27ac locus (left) and each blank barcode (middle) when H3K27ac antibody is used to capture the epigenetic mark. Also shown is the violin plot of the average number of decoded spots per cell for each target H3K27ac locus when a control IgG is used instead (right). Each dot corresponds to a single H3K27ac locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K27ac antibody and that detected with IgG for individual H3K27ac loci. Results here are aggregates of two replicates.
(C) Violin plots showing the H3K27ac ChIP-Seq read number for each target H3K27ac locus for loci that showed layer-specific enrichment (left) and loci that did not show significant layer-specific enrichment (right). ChIP-Seq data are taken from (Mo et al., 2015).
(D) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for two example H3K27ac loci that show significant enrichment in specific cortical layers. The random permutation is performed among the 139 target H3K27ac loci as described in Figure S3C. Scale bar: 400 μm.
(E) Box plots of the Pearson correlation coefficient for the layer enrichment between the epigenomic MERFISH and ATAC-seq data. The box plots depict the distributions of the Pearson correlation coefficients for the 35 H3K27ac loci that showed statistically significant layer-specific enrichment. Red line denotes the median, box denotes the 25-75 percentile, and whiskers denote the 1.5 interquartile range. (Left) Experimentally measured results, as shown in Figure 4B. (Right) Results from the random permutation controls (1000 permutations), where the epigenomic MERFISH images are randomly permutated as described in (D).
Figure S6. The performance of epigenomic MERFISH measurements of target H3K27ac loci in embryonic mouse brain and top sequence motifs identified for putative enhancers in the six spatial clusters shown in Figure 5, related to Figure 5 (A) Violin plot showing the number of decoded spots in the embryonic mouse brain for each of 142 target H3K27ac loci (left), each of the 5 negative control loci (middle), and each blank barcode (right).
(B) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for three example H3K27ac loci that show significant enrichment in specific brain regions. The random permutation is performed among the 142 target H3K27ac loci as described in Figure S3C. Scale bar: 1 mm.
(C) Box plots of the Pearson correlation coefficient for the brain region enrichment between the epigenomic MERFISH and ChIP-seq data. The box plots depict the distributions of the Pearson correlation coefficients for the 142 target H3K27ac loci. (Left) Experimentally measured results. (Right) Results from the random permutation controls (1000 permutations), where the epigenomic MERFISH images are randomly permutated as described in (B).
(D) The top two most enriched sequence motifs for each of the six spatial clusters of H3K27ac loci shown in Figure 5. The p-value with the percentage of locus having the motif shown in bracket for each motif were listed below.
Table S1. Sequences for all oligonucleotides used, related to all figures. The table includes the following separate list of oligonucleotide probes:
Encoding probe library for 90 target H3K27ac loci in hTERT-RPE1 cells,
Encoding probe library for 52 target H3K4me3 loci in hTERT-RPE1 cells,
Encoding probe library for 115 H3K27me3 loci in hTERT-RPE1 cells,
Encoding probe library for 127 target H3K4me3 loci in adult mouse cortex and embryonic mouse brain,
Encoding probe library for 139 target H3K27ac loci in adult mouse cortex,
Encoding probe library for 142 target H3K27ac loci and 5 negative control loci in embryonic mouse brain,
Library amplification primers for all the above encoding probe libraries,
Readout probes,
Tn5 loaders for epigenomic MERFISH experiments,
Tn5 loaders for SON tagmentation experiments,
Labeling and readout oligos for nuclear speckle imaging.
The lists for the encoding probe libraries, library amplification primers, Tn5 loaders, and labeling and readout oligos for nuclear speckle imaging include: 1) Name of each oligo, 2) Sequences of each oligo. The list for the readout probes includes 1) Bit number, 2) Readout probe name, 3) Sequence of the readout probe 4) The fluorophore for that readout probe.
Table S2. MERFISH codebooks for the target epigenomic loci, related to all figures The table includes the codebook of the six sets of target loci:
90 target H3K27ac loci in hTERT-RPE1 cells,
52 target H3K4me3 loci in hTERT-RPE1 cells,
115 target H3K27me3 loci in hTERT-RPE1 cells,
127 target H3K4me3 loci in adult mouse cortex and embryonic mouse mouse brain,
139 target H3K27ac loci in adult mouse cortex,
142 target H3K27ac loci and 5 negative control loci in embryonic mouse brain.
Each codebook contains: 1. Loci number 2. Chromosome of the loci, 3. Start genomic positions of the loci, 4. End genomic positions of the loci, (5. Name of the corresponding gene when relevant), followed by barcode of the individual loci as a series of ‘0’ (off bits) and ‘1’ (on bits). The corresponding readout probe for each bit is labeled on top.
Highlights.
Epigenomic MERFISH enables spatially resolved single-cell epigenomic profiling
Epigenomic MERFISH maps active promoters and putative enhancers in mouse brain
Epigenomic MERFISH reveals putative enhancer hubs in embryonic mouse brain
Epigenomic MERFISH images show sub-nuclear distributions of epigenetic loci
ACKNOWLEDGEMENTS
We thank Justyna Janas for providing initial aliquots of PA-Tn5. We thank Bogdan Bintu and Rongxin Fang, and other members of the Zhuang lab for helpful discussions. This work is supported in part by the National Institutes of Health (to X.Z.). T.L. is supported in part by the Harvard Biological and Biomedical Sciences Program. X.Z. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
X.Z. is an inventor of patents applied for by Harvard University related MERFISH, a co-founder and consultant of Vizgen, Inc, and a member of the Cell advisory board.
REFERENCES
- Allis CD, and Jenuwein T (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet 17, 487-500. 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Askary A, Sanchez-Guardado L, Linton JM, Chadly DM, Budde MW, Cai L, Lois C, and Elowitz MB (2020). In situ readout of DNA barcodes and single base edits facilitated by in vitro transcription. Nat. Biotechnol 38, 66-75. 10.1038/s41587-019-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DA, Dileep V, Handa T, Ohkawa Y, Kimura H, Henikoff S, and Gilbert DM (2021). High-throughput single-cell epigenomic profiling by targeted insertion of promoters (TIP-seq). J. Cell Biol 220: e202103078. 10.1083/jcb.202103078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartosovic M, Kabbe M, and Castelo-Branco G (2021). Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat. Biotechnol 39, 825-835. 10.1038/s41587-021-00869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, and Greenleaf WJ (2015). Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486-490. 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell CR, Bhaduri A, Mostajo-Radji MA, Keefe MG, and Nowakowski TJ (2019). Development and Arealization of the Cerebral Cortex. Neuron 103, 980-1004. 10.1016/j.neuron.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter B, Ku WL, Kang JY, Hu G, Perrie J, Tang Q, and Zhao K (2019). Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat. Commun 10, 3747. 10.1038/s41467-019-11559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda-Nieto AC, Pfaff SL, and Varela-Echavarría A (2005). Homeodomain transcription factors in the development of subsets of hindbrain reticulospinal neurons. Mol. Cell. Neurosci 28, 30-41. 10.1016/j.mcn.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Chen Y, and Belmont AS (2019). Genome organization around nuclear speckles. Current Opinion in Genetics & Development 55, 91-99. 10.1016/j.gde.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lareau C, Andreani T, Vinyard ME, Garcia SP, Clement K, Andrade-Navarro MA, Buenrostro JD, and Pinello L (2019). Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 20, 241. 10.1186/s13059-019-1854-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090. 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A 107, 21931-21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, and Shendure J (2015). Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910-914. 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Ma W, Ramani V, Hill A, Yang F, Ay F, Berletch JB, Blau CA, Shendure J, Duan Z, et al. (2015). Bipartite structure of the inactive mouse X chromosome. Genome Biol. 16, 152. 10.1186/s13059-015-0728-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Bartosovic M, Kukanja P, Zhang D, Liu Y, Su G, Enninful A, Bai Z, Castelo-Branco G, and Fan R (2022). Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 375, 681-686. 10.1126/science.abg7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstat DD, Liu JK, Mione M, Zhong W, Yu G, Anderson SA, Ghattas I, Puelles L, and Rubenstein JL (1999). DLX-1, DLX-2, and DLX-5 expression define distinct stages of basal forebrain differentiation. J. Comp. Neurol 414, 217-237. . [DOI] [PubMed] [Google Scholar]
- Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. (2021). Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun 12, 1337. 10.1038/s41467-021-21583-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664-1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman DM, Marín O, and Rubenstein JLR (2012). The generation of cortical interneurons. In Jasper’s Basic Mechanisms of the Epilepsies, (Oxford University Press; ), pp. 786-796. 10.1016/C2017-0-00860-1 [DOI] [Google Scholar]
- Gopalan S, Wang Y, Harper NW, Garber M, and Fazzio TG (2021). Simultaneous profiling of multiple chromatin proteins in the same cells. Mol. Cell 81, 4736-4746.e5. 10.1016/j.molcel.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, Li B, Chiou J, Wildberg A, Ding B, et al. (2020). An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744-751. 10.1038/s41586-020-2093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina S, Dong X, Yu B, and Vijg J (2016). Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biology 17. 10.1186/s13059-016-1011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray LT, Yao Z, Nguyen TN, Kim TK, Zeng H, and Tasic B (2017). Layer-specific chromatin accessibility landscapes reveal regulatory networks in adult mouse visual cortex. Elife 6: e21883. 10.7554/eLife.21883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybuck LT, Daigle TL, Sedeño-Cortés AE, Walker M, Kalmbach B, Lenz GH, Morin E, Nguyen TN, Garren E, Bendrick JL, et al. (2021). Enhancer viruses for combinatorial cell-subclass-specific labeling. Neuron 109, 1449-1464.e13. 10.1016/j.neuron.2021.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, and Young RA (2007). A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130, 77-88. 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Maehara K, Handa T, Arimura Y, Nogami J, Hayashi-Takanaka Y, Shirahige K, Kurumizaka H, Kimura H, and Ohkawa Y (2019). A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol 21, 287-296. 10.1038/s41556-018-0248-3. [DOI] [PubMed] [Google Scholar]
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515-1526. 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Hébert JM, and Fishell G (2008). The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci 9, 678-685. 10.1038/nrn2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108-112. 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, and Smith MM (2015). Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol 7, a019364. 10.1101/cshperspect.a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934-947. 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hödl M, and Basler K (2012). Transcription in the absence of histone H3.2 and H3K4 methylation. Curr. Biol 22, 2253-2257. 10.1016/j.cub.2012.10.008. [DOI] [PubMed] [Google Scholar]
- Hong J-W, Hendrix DA, and Levine MS (2008). Shadow Enhancers as a Source of Evolutionary Novelty. Science 321, 1314-1314. 10.1126/science.1160631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner AL (1996). Engrailed, Wnt and Pax genes regulate midbrain--hindbrain development. Trends Genet. 12, 15-20. 10.1016/0168-9525(96)81383-7. [DOI] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1-10. 10.1038/s41467-019-09982-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon EZ, Zhu Y, Kelman G, Novak CS, Plajzer-Frick I, Kato M, Garvin TH, Pham Q, Harrington AN, Hunter RD, et al. (2020). Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants. Cell 180, 1262-1271.e15. 10.1016/j.cell.2020.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357-359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson L, Frisén J, and Lundeberg J (2021). Spatially resolved transcriptomics adds a new dimension to genomics. Nat. Methods 18, 15-18. 10.1038/s41592-020-01038-7. [DOI] [PubMed] [Google Scholar]
- Lee B, Lee S, Lee S-K, and Lee JW (2016). The LIM-homeobox transcription factor Isl1 plays critical roles in development of multiple arcuate nucleus neurons. Development 143, 3763-3773. 10.1242/dev.133967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E, Borm LE, and Linnarsson S (2017). The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64-69. 10.1126/science.aan6827. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168-176. 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987-2993. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, and Bickel PJ (2011). Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat 5, 1752-1779. 10.1214/11-aoas466. [DOI] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923-930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu B, Xu Q, Wang Q, Feng S, Lai F, Wang P, Zheng F, Xiang Y, Wu J, Nie J, et al. (2020). The landscape of RNA Pol II binding reveals a stepwise transition during ZGA. Nature 587, 139-144. 10.1038/s41586-020-2847-y. [DOI] [PubMed] [Google Scholar]
- Loo L, Simon JM, Xing L, McCoy ES, Niehaus JK, Guo J, Anton ES, and Zylka MJ (2019). Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun 10, 134. 10.1038/s41467-018-08079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369-1384. 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Hao J, Wang G, Chen KH, Babcock HP, and Zhuang X (2016a). High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. U. S. A 113, 11046-11051. 10.1073/pnas.1612826113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt JR, Hao J, Bambah-Mukku D, Lu T, Dulac C, and Zhuang X (2016b). High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl. Acad. Sci. U. S. A 113, 14456-14461. 10.1073/pnas.1617699113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Clowry GJ, Šestan N, Alzu’bi A, Bakken T, Hevner RF, Hüppi PS, Kostović I, Rakic P, Anton ES, et al. (2019). New insights into the development of the human cerebral cortex. J. Anat 235, 432-451. 10.1111/joa.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moris N, Pina C, and Arias AM (2016). Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet 17, 693-703. 10.1038/nrg.2016.98. [DOI] [PubMed] [Google Scholar]
- Okladnova O, Syagailo YV, Tranitz M, Stöber G, Riederer P, Mössner R, and Lesch KP (1998). A promoter-associated polymorphic repeat modulates PAX-6 expression in human brain. Biochem. Biophys. Res. Commun 248, 402-405. 10.1006/bbrc.1998.8972. [DOI] [PubMed] [Google Scholar]
- O’Leary DDM, Stocker AM, and Zembrzycki A (2013). Comprehensive Developmental Neuroscience: Patterning and Cell Type Specification in the Developing CNS and PNS: Chapter 4. Area Patterning of the Mammalian Cortex (Elsevier Inc. Chapters). 10.1016/C2011-0-07685-8 [DOI] [Google Scholar]
- Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239-243. 10.1038/nature25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman K, Golonzhka O, Lindtner S, Nord AS, Taher L, Hoch R, Silberberg SN, Zhang D, Chen B, Zeng H, et al. (2014). Transcriptional regulation of enhancers active in protodomains of the developing cerebral cortex. Neuron 82, 989-1003. 10.1016/j.neuron.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne AC, Chiang ZD, Reginato PL, Mangiameli SM, Murray EM, Yao C-C, Markoulaki S, Earl AS, Labade AS, Jaenisch R, et al. (2021). In situ genome sequencing resolves DNA sequence and structure in intact biological samples. Science 371. 10.1126/science.aay3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, and Sandberg R (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033-2040. 10.1101/gr.177881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissl S, Fang R, Huang H, Zhao Y, Raviram R, Gorkin DU, Zhang Y, Sos BC, Afzal V, Dickel DE, et al. (2018). Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci 21, 432-439. 10.1038/s41593-018-0079-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841-842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279-283. 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (2009). Evolution of the neocortex: a perspective from developmental biology. Nat. Rev. Neurosci 10, 724-735. 10.1038/nrn2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160-5. 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M, Dierich A, Shawlot W, Behringer RR, Le Meur M, and Ang SL (1998). Sequential roles for Otx2 in visceral endoderm and neuroectoderm for forebrain and midbrain induction and specification. Development 125, 845-856. 10.1242/dev.125.5.845 [DOI] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389-393. 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. (2012). A map of the cis-regulatory sequences in the mouse genome. Nature 488, 116-120. 10.1038/nature11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg SN, Taher L, Lindtner S, Sandberg M, Nord AS, Vogt D, Mckinsey GL, Hoch R, Pattabiraman K, Zhang D, et al. (2016). Subpallial Enhancer Transgenic Lines: a Data and Tool Resource to Study Transcriptional Regulation of GABAergic Cell Fate. Neuron 92, 59-74. 10.1016/j.neuron.2016.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokpor G, Rosenbusch J, Kunwar AJ, Rickmann M, Tuoc T, Rizzoli SO, Tarabykin V, von Mollard GF, Krieglstein K, and Staiger JF (2021). Ablation of Vti1a/1b triggers neural progenitor pool depletion and cortical layer 5 malformation in late-embryonic mouse cortex. Neuroscience 463, 303-316. 10.1016/j.neuroscience.2021.03.021. [DOI] [PubMed] [Google Scholar]
- Sparmann A, and van Lohuizen M (2006). Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 6, 846-856. 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Su J-H, Zheng P, Kinrot SS, Bintu B, and Zhuang X (2020). Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 182, 1641-1659.e26. 10.1016/j.cell.2020.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Yun J, Zheng S, Ollikainen N, Pierson N, White J, Shah S, Thomassie J, Suo S, Eng C-HL, et al. (2021a). Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344-350. 10.1038/s41586-020-03126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei Y, Zheng S, Yun J, Shah S, Pierson N, White J, Schindler S, Tischbirek CH, Yuan G-C, and Cai L (2021b). Single-cell nuclear architecture across cell types in the mouse brain. Science 374, 586-594. 10.1126/science.abj1966. [DOI] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, and Pennacchio LA (2007). VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Research 35, D88-D92. 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854-858. 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, et al. (2013). A high-resolution enhancer atlas of the developing telencephalon. Cell 152, 895-908. 10.1016/j.cell.2012.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lu T, Emanuel G, Babcock HP, and Zhuang X (2019a). Imaging-based pooled CRISPR screening reveals regulators of lncRNA localization. Proc. Natl. Acad. Sci. U. S. A 116, 10842-10851. 10.1073/pnas.1903808116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xiong H, Ai S, Yu X, Liu Y, Zhang J, and He A (2019b). CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 76, 206-216.e7. 10.1016/j.molcel.2019.07.015. [DOI] [PubMed] [Google Scholar]
- Wang SS, Tsai RY, and Reed RR (1997). The characterization of the Olf-1/EBF-like HLH transcription factor family: implications in olfactory gene regulation and neuronal development. J. Neurosci 17, 4149-4158. 10.1523/jneurosci.17-11-04149.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodworth MA, Ng KKH, Halpern AR, Pease NA, Nguyen PHB, Kueh HY, and Vaughan JC (2021). Multiplexed single-cell profiling of chromatin states at genomic loci by expansion microscopy. Nucleic Acids Res. 10.1093/nar/gkab423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SJ, Furlan SN, Mihalas AB, Kaya-Okur HS, Feroze AH, Emerson SN, Zheng Y, Carson K, Cimino PJ, Keene CD, et al. (2021). Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat. Biotechnol 39, 819-824. 10.1038/s41587-021-00865-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia C, Fan J, Emanuel G, Hao J, and Zhuang X (2019). Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. U. S. A 116, 19490-19499. 10.1073/pnas.1912459116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355-364. 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Eichhorn SW, Zingg B, Yao Z, Cotter K, Zeng H, Dong H, and Zhuang X (2021). Spatially resolved cell atlas of the mouse primary motor cortex by MERFISH. Nature 598, 137-143. 10.1038/s41586-021-03705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Allis CD, and Wang GG (2021). The language of chromatin modification in human cancers. Nat. Rev. Cancer 21, 413-430. 10.1038/s41568-021-00357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Zhang Y, Li YE, Lucero J, Behrens MM, and Ren B (2021). Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 18, 283-292. 10.1038/s41592-021-01060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X (2021). Spatially resolved single-cell genomics and transcriptomics by imaging. Nat. Methods 18, 18-22. 10.1038/s41592-020-01037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, and Beaudet AL (2016). Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol 8. 10.1101/cshperspect.a019497 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The effect of fixation and clearing on epigenomic MERFISH signals and the performance of epigenomic MERFISH imaging, related to Figure 1 (A) Images comparing the SON (a marker for nuclear speckles) immunofluorescence (IF) images (left panels) in different fixation conditions (MAA: Methanol/acetic acid, 1% PFA, EGS: ethylene glycol bis(succinimidyl succinate), DSG: disuccinimidyl glutarate, 1% PFA followed by 0.1N HCl treatment) and the location of tagged DNA fragments colocalized with SON and detected by FISH (right panels). To generate tagged DNA fragments colocalized with SON, primary antibody against SON was added, followed by secondary antibody and PA-Tn5. 1% PFA with 0.1N HCl treatment gave the best colocalization between the SON immunofluorescence and tagged DNA signals. We performed screening for fixation conditions using the nuclear speckle marker as it forms distinct puncta in the nucleus, enabling rapid screening of the in situ tagmentation conditions for fixed cells. Scale bar: 10 μm.
(B) Sequencing analysis of in situ transcribed RNAs from tagged DNA fragments with the various histone modifications. The top four UCSC tracks compare the sequencing results of in situ transcribed RNAs from tagged DNA fragments with the H3K27ac modification (in situ tag. and trans.) for cleared and uncleared samples (both with 5 min PFA fixation and HCl treatment) with results obtained from H3K27ac ChIP-Seq and CUT&Tag measurement in hTERT-RPE1 cells. The sequencing result of in situ transcribed RNA from cleared sample shows more unique reads and better capture of the epigenetic profile. The middle three UCSC tracks compare the sequencing result of in situ transcribed RNAs from tagged DNA fragments with the H3K4me3 modification (in situ tag. and trans.) obtained under two fixation conditions (5 min PFA fixation, HCl treated and 10 min PFA fixation, both cleared) with results obtained H3K4me3 CUT&Tag measurements in hTERT-RPE1 cells. The sequencing result of in situ transcribed RNA from 5 min fixation with HCl treatment showed more unique reads and better capture of the epigenetic profile. The bottom two UCSC tracks compare the sequencing result of in situ transcribed RNAs from tagged DNA fragments with the H3K27me3 modification (in situ tag. and trans., 5 min PFA fixation, HCl treated, cleared) with results obtained the H3K27me3 CUT&Tag measurements in hTERT-RPE1 cells. The tracks were normalized to 1x sequencing depth.
(C) RNA Tapestation results showing the RNA length distribution of in situ transcribed RNAs from tagged DNA fragments with the H3K4me3 modification.
(D) Genome wide profiles comparing the sequencing results of in situ transcribed RNAs from tagged DNA fragments and the CUT&Tag results for the three chromatin marks (H3K27ac, H3K4me3, and H3K27me3) centered at the peak summits. Each line represents a peak in the CUT&Tag track and the same peak obtained from the sequencing result of in situ transcribed RNAs. The genomic locations of peaks are determined from the CUT&Tag data and the tracks for the in situ transcribed RNAs from the same genome regions are plotted for comparison. The scale bar shows the magnitude of the normalized peak height (1x sequencing depth). The peaks shown are significant peaks with q value < 0.1.
(E) Genome wide profiles comparing the sequencing results of in situ transcribed RNAs from tagged DNA fragments for H3K27ac and H3K27me3 experiments. Left: The genomic locations of peaks are determined from the H3K27ac data and the tracks for the H3K27me3 data from the same genome regions are plotted for comparison. The very dim signal in the H3K27me3 data indicate that the H3K27ac peaks were essentially not detected using H3K27me3 antibody. Right: The genomic locations of peaks are determined from the H3K27me3 data and the tracks for the H3K27ac data from the same genome regions are plotted for comparison. The very dim signal in the H3K27ac data indicate that the H3K27me3 peaks were essentially not detected using H3K27ac antibody.
(F) Violin plots showing the average number of decoded spots per cell for each of the 52 target H3K4me3 loci corresponding to the promoters of the 52 essential genes (left) and each blank barcode (middle) when H3K4me3 antibody is used to capture the epigenetic mark. Also shown is the violin plot of the average number of decoded spots per cell for each target H3K4me3 locus when a control IgG is used instead (right). Each dot corresponds to a single H3K4me3 locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K4me3 antibody and that detected with IgG for individual H3K4me3 loci. Results here are aggregates of three replicates. The Pearson correlation coefficients for the number of decoded spots per cell for individual H3K4me3 loci between replicates are 0.58 - 0.67. The correlation is lower than that observed for the H3K27ac measurements, as expected because the biological variation across active promoters for essential genes is relatively low. Indeed, the correlation between replicates is substantially higher for the 127 target H3K4me3 loci in the mouse cortex (see Figure S2B), where the biological variation across promoter loci is expected to be higher due to cell-type specific expression of the correspond genes.
(G) Same as (F), but for the 115 target H3K27me3 loci. Results here are aggregates of two replicates. The Pearson correlation coefficient for the number of decoded spots per cell for individual H3K27me3 loci between replicates is 0.64. The correlation is lower than that observed for the H3K27ac measurements, potentially due to an antibody specific effect. Nonetheless, the distance to nuclear speckles for individual H3K27me3 loci showed good correlation between replicates with a Pearson correlation coefficient of 0.7.
Figure S2. The performance of epigenomic MERFISH imaging of mouse brain tissues, related to Figure 2 (A) Epigenomic MERFISH image of 127 target H3K4me3 loci in a single cell. The images from individual bits are shown on the left. The decoded image is shown on the right, with individual spots color-coded based on the identities of the chromosome identity. Scale bars: 4 μm.
(B) Correlation between two biological replicates of H3K4me3 imaging. Each dot corresponds to a single H3K4me3 locus. The Pearson correlation coefficients are 0.95.
(C) Violin plots showing the average number of decoded spots per cell for each of the 127 target H3K4me3 loci (left) and each blank barcode (middle) when H3K4me3 antibody is used to capture the epigenetic mark. Also shown is the violin plot of the average number of decoded spots per cell for each target H3K4me3 locus when a control IgG is used instead (right). Each dot corresponds to a single H3K4me3 locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K4me3 antibody and that detected with IgG for individual H3K4me3 loci. Results here are aggregates of two replicates.
Figure S3. Comparisons between epigenomic MERFISH images of active promoters and the RNA MERFISH images of corresponding genes in mouse cortex and the randomization control, related to Figure 2 (A) Same as Figure 2B, but for the promoter loci that showed statistically significant enrichment in specific cortical layers in replicate 1, with results from replicate 2 for the same genes also shown for comparison. Left: The layer enrichment z-scores for the promoter H3K4me3 signals measured by epigenomic MERFISH for the indicated genes for two replicates. The Pearson correlation coefficients R between the two replicates are shown on the right. Right: The layer enrichment z-scores for the RNA expression level measured by RNA MERFISH for the corresponding genes (Zhang et al., 2021).
(B) Comparisons between epigenomic MERFISH images of example promoter loci and the RNA MERFISH images of corresponding genes that did not show statistically significant layer-specific enrichment. For each gene, the left panel show epigenomic MERFISH images of H3K4me3 signals with each dot in the images representing a cell and red dots represent cells with positive H3K4me3 signals, and the right panel shows the RNA MERFISH image of the corresponding gene (Zhang et al., 2021), with each dot showing the RNA expression level in a cell. Scale bars: 400 μm.
(C) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for three example promoter loci that show significant layer-specific enrichment. Each dot in the images represents a cell and red dots represent cells with positive H3K4me3 signals. In the randomly permutation, the spatial position of each detected spot is unchanged and the genomic identity of each detected spot is randomly selected among the 127 target H3K4me3 loci while keeping the total number of spots detected for each genomic locus the same as the experimentally observed value. Scale bar: 400 μm.
(D) Comparison of the fraction of cells that is H3K4me3-positive in each of the cortical layers between the experimentally observed values and the values derived from the randomly permutated images for the three loci shown in (C).
(E). Box plots of the Pearson correlation coefficient for the layer enrichment between the epigenomic MERFISH and RNA MERFISH data. The box plots depict the distributions of the Pearson correlation coefficients for all 38 promoter loci that has RNA MERFISH images of the corresponding genes. Red line denotes the median, box denotes the 25-75 percentile, and whiskers denote the 1.5 interquartile range. (Left) Experimentally measured results, as shown in Figure 2B. (Right) Results from the random permutation controls where the epigenomic MERFISH images are randomly permutated as described in (C). 1000 permutations were performed and the Pearson correlation coefficients from all permutations are included in the box plot.
Figure S4. Comparison between epigenomic MERFISH images of active promoters and Allen RNA ISH images of the corresponding genes in mouse embryonic brain and the randomization control, related to Figure 3 (A) Comparison between epigenomic MERFISH images of the H3K4me3 signals of the promoter loci and the Allen RNA ISH images of the corresponding genes. All 46 promoter loci that have a H3K4me3 spot density > 300 per mm2 and Allen RNA ISH images of the corresponding genes are shown here. The promoter loci are sorted based on the matching score between epigenomic MERFISH images and RNA ISH images. We segmented the imaged tissue sections into five brain regions: cortex, subpallium, diencephalon, midbrain and hindbrain and determined whether the H3K4me3-MERFISH and RNA ISH signals are enriched in each of these five regions by visual inspection. The matching score was calculated as how many regions have the same enrichment assessment (i.e, enriched or not) for H3K4me3 and RNA ISH signals. Two independent classifications were performed by two people. The results largely agreed with each other, and where the matching score differed between the two classifications, we took the average of the two scores. Scale bars: 1 mm.
(B) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for three example promoter loci that show significant enrichment in specific brain regions. The random permutation is performed among the 127 target H3K4me3 loci as described in Figure S3C. Scale bar: 1 mm.
(C) Box plots of the matching scores between epigenomic MERFISH images and RNA ISH images. The box plots depict the distributions of the matching scores derived for all 46 promoter loci shown in (A). Red line denotes the median, box denotes the 25-75 percentile, and whiskers denote the 1.5 interquartile range. Symbol denote outliers. (Left) Experimentally measured results. (Right) Results from the random permutation controls (10 permutations), where the epigenomic MERFISH images are randomly permutated as described in (B).
Figure S5. The performance of epigenomic MERFISH measurements of target H3K27ac loci in adult mouse cortex, related to Figure 4 (A) Scatter plot showing the correlation between two biological replicates of H3K27ac imaging in the adult mouse cortex. Each dot corresponds to a single H3K27ac locus. The Pearson correlation coefficient is 0.90.
(B) Violin plot showing the average number of decoded spots per cell in adult mouse cortex for each of the 139 target H3K27ac locus (left) and each blank barcode (middle) when H3K27ac antibody is used to capture the epigenetic mark. Also shown is the violin plot of the average number of decoded spots per cell for each target H3K27ac locus when a control IgG is used instead (right). Each dot corresponds to a single H3K27ac locus or a blank barcode. Inset: Histogram of the ratio between the number of decoded spots per cell detected with H3K27ac antibody and that detected with IgG for individual H3K27ac loci. Results here are aggregates of two replicates.
(C) Violin plots showing the H3K27ac ChIP-Seq read number for each target H3K27ac locus for loci that showed layer-specific enrichment (left) and loci that did not show significant layer-specific enrichment (right). ChIP-Seq data are taken from (Mo et al., 2015).
(D) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for two example H3K27ac loci that show significant enrichment in specific cortical layers. The random permutation is performed among the 139 target H3K27ac loci as described in Figure S3C. Scale bar: 400 μm.
(E) Box plots of the Pearson correlation coefficient for the layer enrichment between the epigenomic MERFISH and ATAC-seq data. The box plots depict the distributions of the Pearson correlation coefficients for the 35 H3K27ac loci that showed statistically significant layer-specific enrichment. Red line denotes the median, box denotes the 25-75 percentile, and whiskers denote the 1.5 interquartile range. (Left) Experimentally measured results, as shown in Figure 4B. (Right) Results from the random permutation controls (1000 permutations), where the epigenomic MERFISH images are randomly permutated as described in (D).
Figure S6. The performance of epigenomic MERFISH measurements of target H3K27ac loci in embryonic mouse brain and top sequence motifs identified for putative enhancers in the six spatial clusters shown in Figure 5, related to Figure 5 (A) Violin plot showing the number of decoded spots in the embryonic mouse brain for each of 142 target H3K27ac loci (left), each of the 5 negative control loci (middle), and each blank barcode (right).
(B) Comparison of experimentally obtained epigenomic MERFISH images and randomly permutated epigenomic MERFISH images for three example H3K27ac loci that show significant enrichment in specific brain regions. The random permutation is performed among the 142 target H3K27ac loci as described in Figure S3C. Scale bar: 1 mm.
(C) Box plots of the Pearson correlation coefficient for the brain region enrichment between the epigenomic MERFISH and ChIP-seq data. The box plots depict the distributions of the Pearson correlation coefficients for the 142 target H3K27ac loci. (Left) Experimentally measured results. (Right) Results from the random permutation controls (1000 permutations), where the epigenomic MERFISH images are randomly permutated as described in (B).
(D) The top two most enriched sequence motifs for each of the six spatial clusters of H3K27ac loci shown in Figure 5. The p-value with the percentage of locus having the motif shown in bracket for each motif were listed below.
Table S1. Sequences for all oligonucleotides used, related to all figures. The table includes the following separate list of oligonucleotide probes:
Encoding probe library for 90 target H3K27ac loci in hTERT-RPE1 cells,
Encoding probe library for 52 target H3K4me3 loci in hTERT-RPE1 cells,
Encoding probe library for 115 H3K27me3 loci in hTERT-RPE1 cells,
Encoding probe library for 127 target H3K4me3 loci in adult mouse cortex and embryonic mouse brain,
Encoding probe library for 139 target H3K27ac loci in adult mouse cortex,
Encoding probe library for 142 target H3K27ac loci and 5 negative control loci in embryonic mouse brain,
Library amplification primers for all the above encoding probe libraries,
Readout probes,
Tn5 loaders for epigenomic MERFISH experiments,
Tn5 loaders for SON tagmentation experiments,
Labeling and readout oligos for nuclear speckle imaging.
The lists for the encoding probe libraries, library amplification primers, Tn5 loaders, and labeling and readout oligos for nuclear speckle imaging include: 1) Name of each oligo, 2) Sequences of each oligo. The list for the readout probes includes 1) Bit number, 2) Readout probe name, 3) Sequence of the readout probe 4) The fluorophore for that readout probe.
Table S2. MERFISH codebooks for the target epigenomic loci, related to all figures The table includes the codebook of the six sets of target loci:
90 target H3K27ac loci in hTERT-RPE1 cells,
52 target H3K4me3 loci in hTERT-RPE1 cells,
115 target H3K27me3 loci in hTERT-RPE1 cells,
127 target H3K4me3 loci in adult mouse cortex and embryonic mouse mouse brain,
139 target H3K27ac loci in adult mouse cortex,
142 target H3K27ac loci and 5 negative control loci in embryonic mouse brain.
Each codebook contains: 1. Loci number 2. Chromosome of the loci, 3. Start genomic positions of the loci, 4. End genomic positions of the loci, (5. Name of the corresponding gene when relevant), followed by barcode of the individual loci as a series of ‘0’ (off bits) and ‘1’ (on bits). The corresponding readout probe for each bit is labeled on top.
Data Availability Statement
Sequencing data reported in this work are available at NCBI GEO data repository (GSE191069). Epigenomic MERFISH data reported in this work are available at Zenodo (https://doi.org/10.5281/zenodo.7075964).
Analysis code generated in this work is available at https://github.com/TianLuHarvard/Code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.