

Abstract
Background and Objectives
Identifying protein targets that provide cognitive reserve is a strategy to prevent and treat Alzheimer disease and Alzheimer disease related dementias (AD/ADRD). Previous studies using bulk human brain tissue reported 12 proteins associated with cognitive reserve. This study examined whether the same proteins from induced neurons (iNs) are associated with cognitive reserve of their human donors.
Methods
Induced pluripotent stem cell (iPSC) lines were generated from cryopreserved peripheral blood mononuclear cells of older adults who were autopsied as part of the Religious Orders Study or Rush Memory and Aging Project. Neurons were induced from iPSCs using a standard neurogenin2 protocol. Tandem mass tag proteomics analyses were conducted on iNs day 21. Cognitive reserve of their human donors was measured as person-specific slopes of cognitive change not accounted for by common neuropathologies.
Results
The 53 human donors died at a mean age of 91 years, all were non-Latino White, and 36 (67.9%) were female. Eighteen were diagnosed with Alzheimer dementia proximate to death, and 34 had pathologic AD diagnosis at autopsy. Approximately 60% of the donors had above-average cognitive reserve such that their cognition declined slower than an average person with comparable burdens of neuropathologies. Eight of the 12 candidate proteins were quantified in iNs proteomics analyses. Higher adenylate kinase 4 (AK4) expression in iNs was associated with lower cognitive reserve, consistent with the previous report for brain AK4 expression.
Discussion
By replicating cortical protein associations with cognitive reserve in human iNs, these data provide a valuable molecular readout for studying complex clinical phenotypes such as cognitive reserve in a dish.
Neuritic plaques and neurofibrillary tangles are the neuropathologic hallmarks of Alzheimer disease (AD).1,2 Targeting these misfolded proteins that define the biology of AD may facilitate pharmacologic treatment of the disease. However, anti-β amyloid (Aβ) agent and drug discovery have yielded limited success in clinical trials.3 One contributing factor is that while AD is the leading driver of Alzheimer disease and Alzheimer disease related dementias (AD/ADRD), the latter is a highly complex and polygenic brain disorder that is attributable to not only AD and other neurodegenerative and cerebrovascular conditions that commonly coexist in aging brain4 but also cognitive reserve, that is, the brain's ability to maintain cognition in the presence of neuropathologic insults.5 The interplay between the accumulation of multiple brain pathologies and cognitive reserve suggests that AD-specific therapeutics alone may not be sufficient against the onset and progression of AD/ADRD.6 Identifying targets that provide cognitive reserve offers an alternative strategy and has the potential to offset the detrimental effects of any and all combinations of common brain pathologies.7
Research on protein targets involved in cognitive reserve has only recently emerged. In 2 separate studies using proteomics data from bulk brain tissue from hundreds of community-dwelling old adults,8,9 we used residual cognitive decline (i.e., person-specific rates of cognitive change not accounted for by AD and other common non-AD brain pathologies) as a proxy for cognitive reserve and reported 12 cortical proteins associated with residual cognitive decline. For older adults with comparable burdens of brain pathologies, we showed that the rates of cognitive decline varied widely depending on the levels of these proteins. These findings raise the possibility that perturbation of these candidate proteins may affect the progression of AD/ADRD, above and beyond accumulating brain pathologies.
We recently generated induced pluripotent stem cell (iPSC) lines from 53 autopsied older adults from the Religious Orders Study or Rush Memory and Aging Project (ROSMAP) and differentiated these lines into cortical neuronal fate, that is, induced neurons (iNs). We showed that Aβ and tau production in iNs was associated with cognition and neuritic plaques and neurofibrillary tangles in brain.10 In this study, we extended that work by interrogating proteins in iNs in relation to cognitive reserve of their human donors. Specifically, we targeted proteins that were previously associated with cognitive reserve from proteomics analyses of bulk neocortical tissue. We found that AK4 from human iPSC-derived iNs was associated with cognitive reserve of their human donors. These data provide a valuable molecular readout for studying the complex clinical phenotype such as cognitive reserve in a dish. Furthermore, the approach offers a potentially powerful pathway to personalized medicine for neurodegenerative diseases by leveraging person-specific differences in polygenic risk.
Methods
Study Design
The study aims to investigate whether proteins associated with cognitive reserve identified using bulk brain tissue are replicable in human iNs. To this end, 53 iPSC lines were generated from cryopreserved peripheral blood mononuclear cells (PBMCs) of autopsied older adults. Neurons were induced from iPSCs using a standard neurogenin2 protocol. Tandem mass tag proteomics analyses were conducted on iNs day 21. Cognitive reserve of their human donors was measured using residual cognitive decline. Regression analyses examined the associations of target proteins from iNs with residual cognitive decline of their human donors.
Participants Selected for iPSC Lines
The 53 iPSC lines were generated from older adults who participated in the ROSMAP. The ROSMAP were clinicopathologic cohort studies of aging and dementia. Community-dwelling older adults were enrolled without known dementia, and all agreed to annual detailed clinical evaluations and brain donation after death.11 Most agreed to annual collection of blood, with cryopreserved PBMCs collected annually beginning in 2008.
Participants selected for iPSC line generation were all deceased and underwent brain autopsy with neuropathologic assessments. They represent a broad spectrum of cognitive status proximate to death. Specifically, of these 53 participants, 31 had no cognitive impairment (NCI), 3 had mild cognitive impairment (MCI), and 19 had dementia of whom 18 had Alzheimer dementia. The median Mini-Mental Status score proximate to death was 27, with an interquartile range between 14 and 28.
Clinical and Neuropathologic Assessments
Participants underwent annual detailed clinical evaluations, including a comprehensive cognitive testing battery that assessed 5 relatively dissociable cognitive domains. Raw scores for the individual tests were standardized using baseline mean and SD of the entire ROSMAP cohorts. The resulting z scores were averaged to obtain a global cognitive score, and higher scores indicate better cognition. Domain-specific composite scores were computed similarly.12
A uniform postmortem evaluation systematically assessed burdens of AD, other non-AD degeneration, that is, Lewy bodies, limbic predominant age-related TDP-43 encephalopathy (LATE), hippocampal sclerosis, and cerebrovascular conditions of macroscopic infarcts and microinfarcts, cerebral amyloid angiopathy, atherosclerosis, and arteriolosclerosis. Molecular-specific immunohistochemistry assessed the burdens of Aβ and paired helical filament (PHF)tau tangles in 8 predetermined brain regions. Sections stained using amyloid and phosphorylated tau antibodies were imaged for quantitative analysis.13 Aβ deposition was quantified as percentage area positive for amyloid, and PHFtau tangles were quantified as cortical density of phosphorylated tangles per squared millimeter. For Aβ load, and separately PHFtau tangle density, region-specific measures were square root transformed to alleviate skewness and then averaged across the brain regions to obtain a summary score. The presence of Lewy bodies in the neocortex was determined using immunohistochemistry with an α-synuclein antibody.14 LATE was quantified with a phosphorylated monoclonal TAR5P-1D3 anti-TDP43 antibody and summarized into 4 stages (0: absent of TDP43, 1: TDP43 localized to the amygdala, 2: TDP43 extended to the hippocampus and/or entorhinal cortex, and 3: TDP43 further extended to neocortex).15 The presence of hippocampal sclerosis was determined using hematoxylin and eosin staining. The presence of chronic macroscopic infarcts and microinfarcts were recorded, and cerebral amyloid angiopathy, atherosclerosis, and arteriolosclerosis were assessed using 4-level semiquantitative measures (i.e., none, mild, moderate, and severe), as previously described.16-19
Residual Cognitive Decline as a Proxy for Cognitive Reserve
On average, participants were followed up for 9 years before death (range: 1–21), and annual cognitive scores captured the history of cognitive decline, the clinical hallmark of AD/ADRD. We used residual cognitive decline as a proxy for cognitive reserve. The metric assesses the extent to which person-specific rates of cognitive change deviate from the decline predicted by common neuropathologies. In brief, we fit a linear mixed-effects model with annual cognitive scores as the longitudinal outcome. The model consists of 2 components, that is, the fixed effects and random effects. The fixed effects component quantified the effects of the abovementioned 9 neuropathologies on the slope of cognitive change. By fixed effects alone, individuals with the same burden of neuropathologies would have the same rate of decline. In addition, the random effects component, specifically random slope, quantifies how person-specific slope of change deviates from the decline predicted by neuropathologies, hence the name residual cognitive decline. Individuals with positive random slopes have slower than expected decline, given the burden of neuropathologies, indicating a high reserve. By contrast, individuals with negative random slopes have faster than expected decline, indicating a low reserve. The approach has been used in this and other groups to quantify reserve or resilience.5,8,9,20-22
Generation of iPSC Lines and iNs
Technical details on the generation of iPSC lines and iNs are previously described.10 In brief, the iPSC lines were generated using cryopreserved PBMCs from autopsied ROSMAP participants. PBMC samples were expanded, and cells were primed. Primed samples were seeded for reprogramming using CytoTuneTM-iPS Sendai Reprogramming v2.0 kit. Reprogrammed cells were identified with live cell surface staining using the iPSC marker Tra-1-60. Colonies were isolated from successfully reprogrammed cell lines, manually picked, and then expanded through automation on the NYSCF Global Stem Cell Array platform.23 All iPSC lines underwent rigorous quality control procedures that include testing for sterility, mycoplasma, karyotyping, and pluripotency.
Neurons were induced from iPSCs following a neurogenin2 direct induction protocol.24 The iPSCs were plated in mTeSR1 media for viral transduction. Transduced cells were dissociated and plated in StemFlex medium for differentiation. The medium was changed to KSR with doxycycline on day 1, 1:1 KSR:N2B with puromycin on day 2, and N2B with 1:100 B27 on day 3. Cells were cultured starting on day 4 in NBM media with 1:50 B27 and BDNF, GDNF, and CNTF. The analyses in this work use culture at day 21 postinduction. Notably, more than 98% of cells in iN cultures express neuronal markers, and further testing reveals a robust and consistent differentiation across cell lines.10
Tandem Mass Tag Proteomics Analysis of iNs
Details on tandem mass tag proteomics analysis of iNs are provided as eMethods (links.lww.com/WNL/C243). In brief, day 21 iNs were twice-washed and flash-frozen before being lysed in 100 μL of lysis buffer. Protein concentrations were determined by the bicinchoninic acid method. Tandem mass tag labeling and high pH fractionation were performed.25-27 Mass spectrometry was conducted with a high-field asymmetric waveform ion mobility spectrometry Pro equipped with Orbitrap Eclipse (Thermo Scientific, Waltham, MA). Raw data were analyzed using the Proteome Discoverer Suite (version 2.3, Thermo Scientific). MS/MS spectra were searched against the UniProtKB human proteome database. Following spectral assignment, peptides were assembled into proteins. Postquantification quality control procedures included normalization to a Global Internal Standard, removal of peptides with excessive missing, and imputation with k-nearest neighbor algorithm. A ComBat algorithm was used to remove variance induced by cell harvest batch. The final iN proteomics data file included 94,162 peptides with 11,508 unique proteins.
We previously identified 12 cortical proteins associated with residual cognitive decline using bulk brain tissue. Eight of the 12 proteins were detected and passed filters in the iNs proteomics analyses, including neuritin, alpha-actinin-4, ubiquitin-like modifier-activating enzyme 1, small glutamine-rich tetratricopeptide repeat-containing protein beta, complexin-1, endophilin-A2 (SH3GL1), adenylate kinase 4 (AK4), and inositol-tetrakisphosphate 1-kinase. Statistical analyses were restricted to these 8 proteins.
Statistical Analysis
The Student t test and Pearson or Spearman correlations examined the relationship between the iNs proteins and the donors' clinical and pathologic characteristics. To investigate the associations of iNs proteins with residual cognitive decline, we fit parallel linear regression models with person-specific random slopes for change in global cognition as the continuous outcome and each of the 8 protein expressions the predictor. We repeated the analyses for 5 cognitive domains to investigate potential domain-specific signals. To minimize testing burden, the primary analysis examined the protein expressions by averaging peptide expression levels. In supporting analyses, for signals associated with residual cognitive decline, we further examined the correlation structure of corresponding peptides to determine whether the associations were driven by specific sequences. Statistical analysis was performed using SAS/STAT software, version 15.2 and R program, version 3.6.3. Correction for multiple testing was addressed by using the Benjamini-Hochberg procedure that controls the false discovery rate at an α level of 0.05.
Standard Protocol Approvals, Registrations, and Patient Consents
The ROSMAP studies were approved by an institutional review board of Rush University Medical Center. Each participant signed an informed consent, an Anatomical Gift Act for organ donation, and a separate repository consent allowing for sharing and repurposing of data and biospecimens.
Data Availability
Data used in this study are available on request through the Rush Alzheimer Disease Center Resource Sharing Hub at https://www.radc.rush.edu/. Cell lines are available on request at the same web link.
Results
Characteristics of Human Donors
The 53 older adults died at a mean age of 91 years, all were non-Latino White, 36 (67.9%) were female, and the mean years of education was 16 years (Table 1). At autopsy, 34 (64.2%) met modified National Institute on Aging Reagan criteria for pathologic AD, 5 (9.4%) had neocortical Lewy bodies, 6 (11.3%) had hippocampal sclerosis, and 12 (22.6%) had TDP-43 pathology that extended beyond the amygdala. Chronic macroscopic infarcts or microinfarcts were present in 20 (37.7%) individuals. In addition, 15 (28.3%) had moderate to severe amyloid angiopathy, 12 (22.6%) had moderate to severe atherosclerosis, and 13 (24.5%) had moderate to severe arteriolosclerosis. Furthermore, 34 (64.2%) had more than 1 pathologic diagnosis, 13 (24.5%) had 1 pathologic diagnosis, and only 6 (11.3%) did not meet pathologic criteria for any disease (Figure 1).
Table 1.
Demographic and Clinicopathologic Characteristics of Human Donors
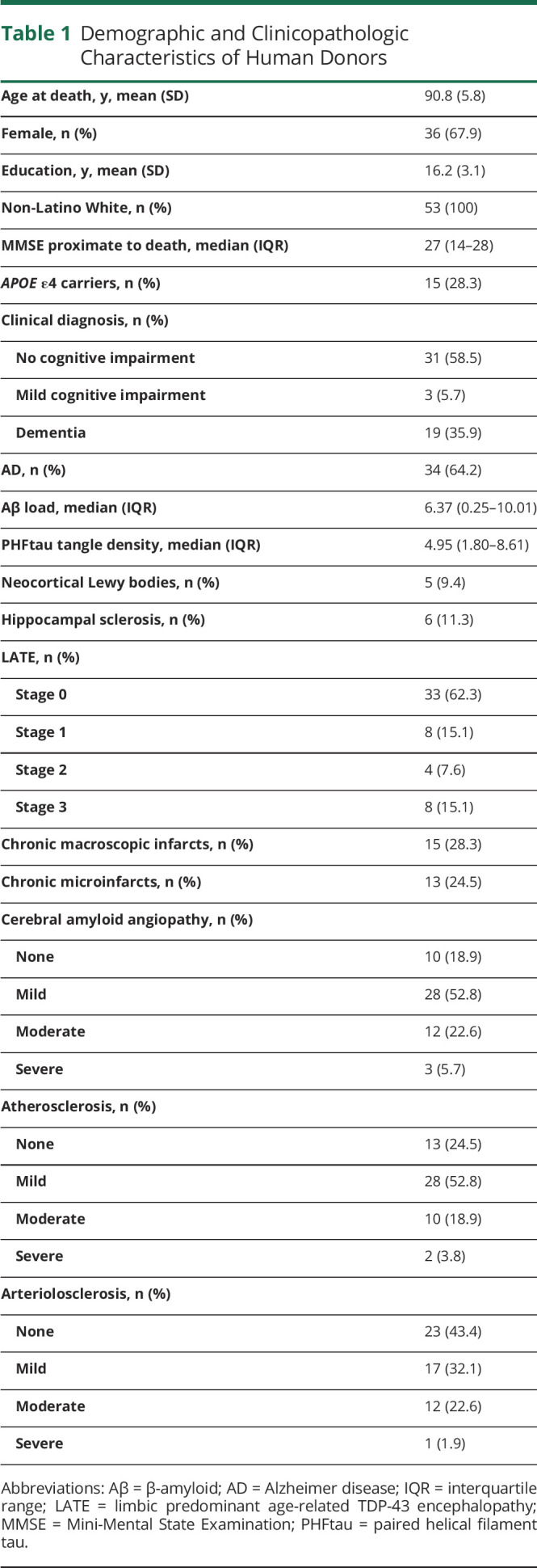
Figure 1. Burdens of Age-Related Neuropathologies.

The figure illustrates the burdens of neuropathologies in the human donors. Each column represents person-specific combination of pathologies present in the brain of individual donor case. The colors of the columns differentiate the clinical diagnosis proximate to death, that is, light blue columns are cases of no cognitive impairment, blue columns are cases of mild cognitive impairment, and dark blue columns are cases of dementia. The rows from bottom to top represent the presence of AD, neocortical Lewy bodies, TDP43 pathology beyond the amygdala, hippocampal sclerosis, macroscopic infarcts, microinfarcts, moderate-to-severe amyloid angiopathy, moderate-to-severe atherosclerosis, and moderate-to-severe arteriolosclerosis. AD = Alzheimer disease; LATE = limbic predominant age-related TDP-43 encephalopathy.
iN Proteins and Clinical and Pathologic Characteristics of Human Donors
Compared with the neuropathologic assessment where 34 of the 53 human donors had pathologic AD, only 19 were diagnosed with dementia. Almost half (16 of 34) of older adults diagnosed with AD at autopsy did not manifest dementia, of which 13 had NCI and the remaining 3 had MCI proximate to death.
First, we examined the extent to which the 8 iN protein expressions differed by dementia status of the donor cases (Figure 2). In this sample, 5 of the 8 proteins were differentially expressed between human donors with and without dementia, of which AK4 survived the correction for multiple testing. Specifically, the mean AK4 level was lower in those without dementia. Consistent results were observed for longitudinal change in cognition over the years before death (eFigure 1, links.lww.com/WNL/C243).
Figure 2. iN Proteins by Dementia Status.

The figure illustrates the difference in iN protein expressions by dementia status. The figure consists of 8 panels, each representing 1 specific candidate protein. Each panel is a violin plot that compares the iN protein expression level between the donors with and without dementia. The colored outer shapes show the distributions of protein expression (colored area), and the inner boxplots show the median, interquartile range, and potential outliers. The p values at the top were derived from the Student t tests. ACTN4 = alpha-actinin-4; AK4 = adenylate kinase 4; CPLX1 = complexin-1; iN = induced neuron; ITPK1 = inositol-tetrakisphosphate 1-kinase; NRN1 = neuritin; SGTB = small glutamine-rich tetratricopeptide repeat-containing protein beta; SH3GL1 = endophilin-A2; UBA1 = ubiquitin-like modifier-activating enzyme 1.
Next, we examined the associations of the iN proteins with individual neuropathologic indices assessed at autopsies (Figure 3). Overall, we did not observe strong correlations between proteins from iNs and Aβ load or PHFtau tangle density of their human donors. One isolated signal for Aβ was the SH3GL1 protein, in which higher SH3GL1 expression was correlated with lower Aβ load. After the correction for multiple testing, none of the iN proteins showed differential expression for non-AD neurodegenerative (i.e., the presence of cortical Lewy bodies, advanced stages [2 or 3] for LATE, and the presence of hippocampal sclerosis) or cerebrovascular conditions (i.e., the presence of macroscopic infarcts and microinfarcts, moderate or severe amyloid angiopathy, atherosclerosis, and arteriolosclerosis).
Figure 3. iN Proteins and Age-Related Neuropathologies.

The figure illustrates the associations of iN protein expressions with common age-related neuropathologic indices. Results are presented as −log10(p) such that higher the value, more significant the association. Red color represents a positive association, and blue represents a negative association. Vertical dotted line is the reference cutoff representing p = 0.00625 (i.e., 0.05/8). ACTN4 = alpha-actinin-4; AK4 = adenylate kinase 4; CPLX1 = complexin-1; iN = induced neuron; ITPK1 = inositol-tetrakisphosphate 1-kinase; NRN1 = neuritin; SGTB = small glutamine-rich tetratricopeptide repeat-containing protein beta; SH3GL1 = endophilin-A2; UBA1 = ubiquitin-like modifier-activating enzyme 1.
iN Proteins and Cognitive Reserve
By leveraging data from annual cognitive assessments for up to 21 years before death and comprehensive neuropathologic evaluations at autopsy, we estimated person-specific residual cognitive slopes as a proxy for cognitive reserve. Of these 53 human donors, 33 had residual slopes of cognitive change slower than the mean, whereas 20 had faster residual slopes. This suggests that approximately 60% of the donors had above-average cognitive reserve such that their cognition showed no decline or even improvement after accounting for the effects of known neuropathologies.
We regressed person-specific residual slopes on each of the 8 iN protein expressions (Figure 4). We found that higher AK4 expression was associated with more negative residual slopes of cognitive decline, suggesting that human donors with higher cognitive reserve have lower AK4 levels in iNs. The results for other proteins were not significant. Next, we conducted 2 supporting analyses. First, we examined whether our observed AK4 association with residual cognitive slopes was driven by specific AK4 sequences. Five AK4 peptides were quantified from the iN proteomics analysis (eFigure 2, links.lww.com/WNL/C243). A principal component analysis revealed that the abundance of these AK4 sequences were highly correlated, such that the first principal component explained more than 85% of the total variance, indicating that the results are unlikely to differ by AK4 sequences. Second, we examined whether the iN protein associations with residual cognitive decline differed for 5 cognitive domains. The domain-specific analyses showed that overall these associations were consistent (Figure 5). The AK4 protein remained the strongest signal and was inversely associated with the residual cognitive slope for episodic memory and semantic memory.
Figure 4. iN Proteins and Residual Cognitive Decline.

The figure illustrates the associations of iN protein expressions with residual cognitive decline. The figure consists of 8 panels, each representing 1 specific candidate protein. Each panel is a scatter plot. Candidate protein expression is on the x-axis, and the measure of residual cognitive decline is on the y-axis. Individual data points were overplotted with a regression line and the 95% confidence band. The dashed blue line represents the reference for no association. If the reference line is entirely covered by the 95% confidence band, it indicates that the association did not reach a nominal significance. Corresponding p values are shown at the top. ACTN4 = alpha-actinin-4; AK4 = adenylate kinase 4; CPLX1 = complexin-1; iN = induced neuron; ITPK1 = inositol-tetrakisphosphate 1-kinase; NRN1 = neuritin; SGTB = small glutamine-rich tetratricopeptide repeat-containing protein beta; SH3GL1 = endophilin-A2; UBA1 = ubiquitin-like modifier-activating enzyme 1.
Figure 5. iN Proteins and Domain-Specific Residual Cognitive Decline.

The figure illustrates the associations of iN protein expressions with domain-specific residual cognitive decline. Candidate proteins are on the x-axis, and measures for domain-specific residual cognitive declines are on the y-axis. Red tile represents a positive association (i.e., higher expression is associated with slower residual decline), and blue tile represents a negative association (i.e., higher expression is associated with faster residual decline). The size of black dot inside the tile signifies the strength of an association. ACTN4 = alpha-actinin-4; AK4 = adenylate kinase 4; CPLX1 = complexin-1; iN = induced neuron; ITPK1 = inositol-tetrakisphosphate 1-kinase; NRN1 = neuritin; SGTB = small glutamine-rich tetratricopeptide repeat-containing protein beta; SH3GL1 = endophilin-A2; UBA1 = ubiquitin-like modifier-activating enzyme 1.
Discussion
In this study, we investigated the extent to which proteins in postmortem neocortical tissue related to cognitive reserve are conserved in iNs. Consistent with the finding in bulk human brain tissue, we showed that higher AK4 in induced neuronal culture was strongly associated with faster residual cognitive decline, that is, lower cognitive reserve, in their respective human donors.
Accumulating evidence from clinicopathologic studies demonstrates that AD/ADRD is a complex function of numerous common brain pathologies with additive effects.4,28,29 Furthermore, not all old adults with neuropathologic conditions manifest cognitive impairment or dementia.30,31 Indeed, we previously identified many proteins associated with slower or faster cognitive decline after adjusting for the effects of common brain pathologies.8,9,32 Together, this suggests that targeting any individual pathology will have a small effect on overall disease burden. With the public health imperative to prevent AD/ADRD, it seems inconceivable that we can use numerous biomarkers to target the myriad different combinations of brain pathologies and offer multiple drug cocktails to millions of people in the United States at risk for cognitive impairment. By contrast, targets for cognitive reserve act on mechanisms agnostic to any and all combinations of neuropathologic insults and could offset the effects of these pathologies on cognitive impairment.
Replicating protein signals from proteomic analysis of bulk brain tissue in stem cell–derived neurons offers an opportunity to conduct drug screening in an ex vivo model that captures person-specific differences of their respective human donors. Of the 8 candidate proteins investigated in this work, we robustly replicate 1 protein signal, namely AK4, in relation to residual cognitive decline. While association does not directly infer causality, several lines of evidence in this work provide strong support for a potential causal relationship. Following the seminal work by Bradford-Hill,33 there are a series of guidelines for assessing causality in epidemiology. Among the most important ones are strength of association, consistency of association, specificity of association, and temporal sequence of association.34 In this study, the observed association of AK4 and cognitive reserve survived the most stringent correction for multiple testing, in support of strength. The observed association is also consistent with a previous report using protein data from bulk human brain tissue in a much larger sample, in support of consistency. Furthermore, the association was initially detected by a Bayesian network model of a 390-gene messenger RNA expression module.32 Finally, the unique data from iPSC technology provide further evidence for specificity and temporality. Distinct from aged bulk brain tissue where protein differences can be attributable to both genetic and environmental factors, iPSC-derived neurons are stripped of the epigenome and are devoid of the environmental exposures traditionally associated with cognitive reserve (e.g., education, cognitive activity, and social activity). We are measuring AK4 production in early embryonic age neurons. Consequently, the observed protein association is primarily, if not exclusively, hardcoded in the genomic background, which places the protein upstream of cognition. In totality, the evidence strongly suggests that neuronal AK4 exerts effects on cognitive reserve.
AK4 belongs to a family of adenylate kinase isozymes that are involved in cellular energy homeostasis. The protein is localized in the mitochondrial matrix and is highly expressed in multiple organs including the kidney, liver, heart, and brain.35 AK4 has consistently been implicated as a stress-response protein critical to cell survival and proliferation. Previous studies reported that AK4 gene and protein expressions were elevated in cultured cells under hypoxic condition.36-38 An in vivo study of rat liver also revealed an upregulation of AK4 in response to oxidative stress induced by various hepatotoxicants.39 There are reports of the protein's involvement in inflammation.40 Further functional studies suggest that AK4 knockdown using shRNA suppresses cell growth, while AK4 overexpression protects cells from H2O2-induced cell death.41 An earlier trial on deferoxamine for hepatocellular carcinoma observed an upregulation of AK4 during the drug administration, and the follow-up experiments revealed that AK4 knockdown cells showed increased ATP production and greater sensitivity to hypoxia and the anticancer drug.42 These data together indicate that AK4 may serve as a potential therapeutic target. The mechanism through which AK4 acts on change in cognition is still unclear. Postmortem neuroimaging data link AK4 to microstructural changes in brains.43 AK4 in bulk human cortical tissue was associated with transverse relaxation rate (R2) on postmortem MRI, which mediates half of the protein association with cognitive decline. Of note, slower R2 in white matter is reflective of greater water content, which can result from hypoxia-driven demyelination.44 Future work is warranted to elucidate the function of AK4 in AD/ADRD.
We did not detect significant associations for any other candidate proteins. One possibility is the small sample size. While not reaching the statistical significance, the 2 proteins, that is, complexin-1 and SH3GL1, showed moderate correlations with residual slopes of cognitive change. Besides sample size, an alternative explanation for lack of association in iNs is that traditional proteomics analysis from bulk brain tissue does not differentiate expressions between cell types, and the previously reported associations could be driven by non-neuronal CNS cells such as astrocytes, microglia, and endothelial cells. Of note, the human iPSC lines that we developed can be readily differentiated into other cell types to further test this hypothesis. In addition, because the neurogenin2-induced day 21 iN culture system is a highly controlled model of cortical neuron, potential environmental effects on cognitive reserve are lost during the process of reprogramming. As such, when an association is reproduced between aged tissue and iNs, there is a strong implication that the effect is neuronal and that it is hardcoded in the genetics. On the contrary, if a protein association was responding to environmental factors, the signal would not be expected to show up in iNs.
Finally, interrogating protein associations in a human culture system offers a unique opportunity to probe the protein molecules for druggability. While omicwide analyses are crucial for high-throughput screening and nomination for potential targets for complex human diseases, establishing druggability requires in vitro experiments. The iPSC experimental system provides an important tool for functional validations and protein perturbations. In addition, it could capture the genomic underpinnings that are most responsive to protein perturbations. Using cultured astrocytes and induced human neurons, we previously demonstrated that target genes perturbation altered Aβ42 levels in induced astrocytes.32 Furthermore, the human iPSC-based experimental system creates a controlled and manipulable platform that links multilevel omics data from cell culture to clinicopathologic characteristics from their human donors. As such, it has the potential of creating cell culture profiles that are predictive of complex human phenotypes, such as cognitive reserve. Ultimately, we hope that it would allow us to perturb these phenotypes in a dish and identify therapeutic targets for AD/ADRD. Because the model system strips the epigenome, the results are presumably based on person-specific differences in polygenic risk. Indeed, in our previous study, we found that an AD polygenic risk score (PRS) was associated with Aβ and tau produced in iNs. Because a PRS can be generated in living humans, the approach offers a potentially powerful approach to personalized medicine.
Strengths of this work include our unique set of iPSC lines, which opens a new avenue for identifying and optimizing much needed therapeutic targets for complex AD/ADRD phenotypes. While the study may seem at first to be small with a sample size of 53, a viable path to personalized medicine will of necessity need to be robust with modest sample sizes. Our finding is robust and survived correction for multiple comparisons. Human donors were deeply phenotyped and had undergone uniform detailed clinical and neuropathologic evaluations. Multiomics data from postmortem brain tissue were leveraged to assess the congruence of the data from induced human neurons. Limitations are noted. Our uniform standard neuropathologic assessments may not completely capture the pathologic burdens of the autopsied. All the cell lines were generated using biospecimens from donors of European ancestry, and the current results may not be applicable to minority populations. While these donor cases selected for iPSC lines shared similar demographics as the overall ROSMAP participants who had died and undergone brain autopsy, there are differences in clinical diagnoses and neuropathologic burdens (eTable 1, links.lww.com/WNL/C243). Furthermore, ROSMAP are voluntary cohorts that require brain donation, and participants on average were older with high education, and a majority were female individuals. As a result, they may not be representative of the general aging population. The small sample size and the voluntary nature of the ROSMAP cohorts limit the generalizability of findings in this work.
Acknowledgment
The authors acknowledge the contributions from all the ROSMAP participants and investigators and staff at the Rush Alzheimer Disease Center.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- AD/ADRD
Alzheimer disease and Alzheimer disease related dementias
- AK4
adenylate kinase 4
- iN
induced neuron
- iPSC
induced pluripotent stem cell
- LATE
limbic predominant age-related TDP-43 encephalopathy
- MCI
mild cognitive impairment
- NCI
no cognitive impairment
- PBMC
peripheral blood mononuclear cell
- PHFtau
paired helical filament tau
- PRS
polygenic risk score
- ROSMAP
Religious Orders Study or Rush Memory and Aging Project
- SH3GL1
endophilin-A2
Appendix. Authors

Study Funding
This study was supported by NIH grants R01AG055909, R01NS117446, R01AG15819, R01AG17917, P30AG10161, P30AG072975, R01AG067482, and U01AG61356.
Disclosure
J.A. Schneider, P.L. De Jager, T.L. Young-Pearse, and D.A. Bennett report grants from National Institute on Aging. All other authors declare no relevant competing interests. Go to Neurology.org/N for full disclosures.
References
- 1.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6(4):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66(2):200-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson RS, Nag S, Boyle PA, et al. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology. 2013;80(13):1202-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings J. The National Institute on Aging—Alzheimer's Association framework on Alzheimer's disease: application to clinical trials. Alzheimers Dement. 2019;15(1):172-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett DA. Mixed pathologies and neural reserve: implications of complexity for Alzheimer disease drug discovery. PLoS Med. 2017;14(3):e1002256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu L, Tasaki S, Schneider JA, et al. Cortical proteins associated with cognitive resilience in community-dwelling older persons. JAMA Psychiatry. 2020;77(11):1172-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu L, Petyuk VA, Gaiteri C, et al. Targeted brain proteomics uncover multiple pathways to Alzheimer's dementia. Ann Neurol. 2018;84(1):78-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lagomarsino VN, Pearse RV II, Liu L, et al. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron. 2021;109(21):3402-3420.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis. 2018;64(s1):S161-S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyle PA, Wang T, Yu L, et al. To what degree is late life cognitive decline driven by age-related neuropathologies? Brain. 2021;144(7):2166-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapasi A, Leurgans SE, Arvanitakis Z, Barnes LL, Bennett DA, Schneider JA. Aβ (amyloid beta) and tau tangle pathology modifies the association between small vessel disease and cortical microinfarcts. Stroke. 2021;52(3):1012-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197-2204. [DOI] [PubMed] [Google Scholar]
- 15.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol. 2015;77(6):942-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider JA, Wilson RS, Cochran EJ, et al. Relation of cerebral infarctions to dementia and cognitive function in older persons. Neurology. 2003;60(7):1082-1088. [DOI] [PubMed] [Google Scholar]
- 17.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011;42(3):722-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer's dementia attributed to age-related neuropathologies. Ann Neurol. 2019;85(1):114-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol. 2016;15(9):934-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu L, Boyle PA, Segawa E, et al. Residual decline in cognition after adjustment for common neuropathologic conditions. Neuropsychology. 2015;29(3):335-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain. 2020;143(8):2561-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White CC, Yang HS, Yu L, et al. Identification of genes associated with dissociation of cognitive performance and neuropathological burden: multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med. 2017;14(4):e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paull D, Sevilla A, Zhou H, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. 2015;12(9):885-892. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Pak C, Han Y, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78(5):785-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ping L, Duong DM, Yin L, et al. Global quantitative analysis of the human brain proteome in Alzheimer's and Parkinson's disease. Sci Data. 2018;5:180036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higginbotham L, Ping L, Dammer EB, et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer's disease. Sci Adv. 2020;6(43):eaaz9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ping L, Kundinger SR, Duong DM, et al. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer's disease. Sci Data. 2020;7(1):315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehta RI, Schneider JA. What is ‘Alzheimer's disease’? The neuropathological heterogeneity of clinically defined Alzheimer's dementia. Curr Opin Neurol. 2021;34(2):237-245. [DOI] [PubMed] [Google Scholar]
- 29.White LR, Edland SD, Hemmy LS, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology. 2016;86(11):1000-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837-1844. [DOI] [PubMed] [Google Scholar]
- 31.Stephan BCM, Hunter S, Harris D, et al. The neuropathological profile of mild cognitive impairment (MCI): a systematic review. Mol Psychiatry. 2012;17(11):1056-1076. [DOI] [PubMed] [Google Scholar]
- 32.Mostafavi S, Gaiteri C, Sullivan SE, et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat Neurosci. 2018;21(6):811-819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill AB. The Environment and Disease: Association or Causation? Sage Publications, 1965. [Google Scholar]
- 34.Wakeford R. Association and Causation in Epidemiology–Half a Century since the Publication of Bradford Hill's Interpretational Guidance. SAGE Publications Sage UK, 2015:4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noma T, Fujisawa K, Yamashiro Y, et al. Structure and expression of human mitochondrial adenylate kinase targeted to the mitochondrial matrix. Biochem J. 2001;358(pt 1):225-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen L, Fink T, Ebbesen P, Zachar V. Temporal transcriptome of mouse ATDC5 chondroprogenitors differentiating under hypoxic conditions. Exp Cell Res. 2006;312(10):1727-1744. [DOI] [PubMed] [Google Scholar]
- 37.Kong F, Binas B, Moon JH, Kang SS, Kim HJ. Differential expression of adenylate kinase 4 in the context of disparate stress response strategies of HEK293 and HepG2 cells. Arch Biochem Biophys. 2013;533(1-2):11-17. [DOI] [PubMed] [Google Scholar]
- 38.Wujak M, Veith C, Wu C-Y, et al. Adenylate kinase 4—a key regulator of proliferation and metabolic shift in human pulmonary arterial smooth muscle cells via akt and HIF-1α signaling pathways. Int J Mol Sci. 2021;22(19):10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto T, Kikkawa R, Yamada H, Horii I. Investigation of proteomic biomarkers in in vivo hepatotoxicity study of rat liver: toxicity differentiation in hepatotoxicants. J Toxicol Sci. 2006;31(1):49-60. [DOI] [PubMed] [Google Scholar]
- 40.Chin WY, He CY, Chow TW, Yu QY, Lai LC, Miaw SC. Adenylate kinase 4 promotes inflammatory gene expression via Hif1α and AMPK in macrophages. Front Immunol. 2021;12:630318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu R, Ström AL, Zhai J, et al. Enzymatically inactive adenylate kinase 4 interacts with mitochondrial ADP/ATP translocase. Int J Biochem Cell Biol. 2009;41(6):1371-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujisawa K, Terai S, Takami T, et al. Modulation of anti-cancer drug sensitivity through the regulation of mitochondrial activity by adenylate kinase 4. J Exp Clin Cancer Res. 2016;35:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim N, Yu L, Dawe R, et al. Microstructural changes in the brain mediate the association of AK4, IGFBP5, HSPB2, and ITPK1 with cognitive decline. Neurobiol Aging. 2019;84:17-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dawe RJ, Yu L, Leurgans SE, et al. Postmortem MRI: a novel window into the neurobiology of late life cognitive decline. Neurobiol Aging. 2016;45:169-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used in this study are available on request through the Rush Alzheimer Disease Center Resource Sharing Hub at https://www.radc.rush.edu/. Cell lines are available on request at the same web link.