

Abstract
We examined the characteristics of individuals with biomarker evidence of tauopathy but without β-amyloid (Aβ) (A-T+) in relation to individuals with (A+T+) and without (A-T-) evidence of Alzheimer’s disease (AD). We included 561 participants with Aβ and tau PET from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). We compared A-T- (n = 316), A-T+ (n = 63), and A+T+ (n = 182) individuals on demographics, amyloid, tau, hippocampal volumes, and cognition. A-T+ individuals were low on apolipoprotein E ɛ4 prevalence (17%) and had no evidence of subtly elevated brain Aβ within the negative range. The severity of tau deposition, hippocampal atrophy, and cognitive dysfunction in the A-T+ group was intermediate between A-T- and A+T+ (all p < 0.001). Tau uptake patterns in A-T+ individuals were heterogeneous, but approximately 29% showed tau deposition in the medial temporal lobe only, consistent with primary age-related tauopathy and an additional 32% showed a pattern consistent with AD. A-T+ individuals also share other features that are characteristic of AD such as cognitive impairment and neurodegeneration, but this group is heterogeneous and likely reflects more than one disorder.
Keywords: Tauopathies, Tau, Positron emission tomography, Primary age-related tauopathy, 18F-flortaupir
1. Introduction
In the biomarker-based framework with a dichotomous assessment of β-amyloid (Aβ, A), pathological tau (T), and neurodegeneration (N) (Jack et al., 2018a; Jack et al., 2016a), people with abnormal brain Aβ (A+) are defined as being on the Alzheimer’s continuum regardless of clinical symptoms. This scheme has been useful for the investigation of disease mechanisms and the prediction of outcomes in cognitively normal and impaired people (Jack et al., 2017a; van Maurik et al., 2019; Yu et al., 2019).
However, relatively little is known about A-T+ individuals who are outside the Alzheimer’s continuum with normal Aβ levels but who have either abnormal tau alone (A-T+N-) or both abnormal tau and neurodegeneration (A-T+N+). The A-T+ individuals, therefore, include two likely overlapping categories: suspected non-Alzheimer’s pathophysiology (SNAP) (Jack et al., 2012a), a heterogeneous biomarker-defined category defined by abnormal neurodegeneration in the absence of Aβ and has evolved to alternatively include abnormal tau (Jack et al., 2018a); and primary age-related tauopathy (PART), which is a neuropathological diagnosis characterized as mild to moderate tau deposition in the absence of Aβ pathology (Crary et al., 2014; Jack et al., 2016b). In contrast to tauopathies with only one form of tau, PART includes both 3R and 4R forms of tau, and deposition is focused in the medial temporal lobe but may extend to lateral temporal and cingulate regions. PART is clinically heterogeneous, including individuals without impairment as well as a cognitively impaired group previously referred to as tangle-only dementia (Yamada, 2003). Other substrates of T+ SNAP could be frontotemporal lobar degeneration or other primary tauopathies, while non-tau related conditions (T-N+) may include vascular brain injury, limbic-predominant age-related TDP-43 encephalopathy (LATE) (Nelson et al., 2019), or dementia with Lewy bodies.
Positive and negative thresholds are a critical and controversial aspect of the characterization of these groups. The definition of these thresholds is specific to the modality (cerebrospinal fluid [CSF], positron emission tomography [PET], plasma) used to define amyloid and tau as well as the specific methodological and analytical tools used for quantification. Nonetheless, studies using a variety of modalities and methodologies have converged on estimates that about 20–30% of individuals in research series are normal on amyloid with abnormal tau or neurodegeneration (SNAP) (Jack et al., 2016b), of which a small, but significant proportion (around 10–15%) reflects tauopathy without Aβ (A-T+) (Altomare et al., 2019; Dodich et al., 2020; Jack et al., 2017a; Yu et al., 2019) regardless of N status.
We performed this study in order to define the characteristics of A-T+ individuals, to qualitatively characterize the tau uptake pattern of these individuals, and to compare them on several quantitative characteristics to individuals with no evidence of any Alzheimer’s disease (AD) pathology (A-T-), to individuals with AD (A+T+).
2. Methods
2.1. Participants
We obtained the data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (ida.loni.usc.edu) and downloaded the data of the present study in November 2019.
Six hundred eighty-five participants who underwent 18F-flortaucipir (FTP) PET and Aβ PET (18F-florbetaben [FBB] or 18F-florbetapir [FBP]) within 1 year were initially included. Groups were comprised of those who were cognitively normal (n = 391, 47 of whom had subjective memory complaints), and those diagnosed with mild cognitive impairment (MCI, n = 226) or AD dementia (n = 68) according to the standard ADNI protocol (Thomas et al., 2019); the latter two groups were combined in our results as cognitively impaired. According to threshold values (described below), we divided participants into 4 AT groups based only on Aβ and tau PET biomarkers. We excluded 118 A+T- participants, leaving 69 A-T+ subjects as individuals of interest and 316 A-T- and 182 A+T+ as comparison groups. A+T- individuals were excluded because they represent a similar category as A+T+ individuals in that they are on the AD pathway, but their T-status provides minimal information about tau in relation to the A-T+ group (Guo et al., 2020). All amyloid images in the A-T+ group were then reviewed by a nuclear medicine physician (K.P.). Six amyloid scans quantitatively categorized as negative were read as positive because of focal regions of tracer accumulation (5) or generalized tracer accumulation that was sub-threshold (1) (See A-T+ PET Visual Review below). These individuals were excluded, resulting in 63 subjects in the A-T+ group. All subjects had comprehensive data related to demographics, medical history, neuropsychological assessments, magnetic resonance imaging, CSF and blood biomarkers, and Apolipoprotein E (APOE) genotyping as defined by the ADNI protocol.
2.2. Standard protocol approvals, registrations, and patient consents
The ADNI study was approved by the Institutional Review Boards at each of the participating institutions, written informed consent was obtained for all study participants, and all data were deidentified. All ADNI data are available at adni.loni.usc.edu with documentation on the ADNI website (www.adni-info.org) including the inclusion/exclusion criteria (ADNI protocol - http://adni.loni.usc.edu/wp-content/uploads/2010/09/ADNI_GeneralProceduresManual.pdf).
2.3. Aβ PET processing and thresholds
Processing methods for ADNI FBB, FBP (Landau and Jagust, 2015; Landau et al., 2013), and FTP PET data (Landau and Jagust, 2016; Maass et al., 2017; Maass et al., 2018) have been previously described. Full details of the image acquisition (http://adni.loni.ucla.edu/wp-content/uploads/2010/05/ADNI2_PET_Tech_Manual_0142011.pdf) and processing steps (http://adni.loni.usc.edu/methods/pet-analysis/pre-processing/) can be found online. Aβ PET analyses for standardized uptake value ratio (SUVR) used a whole cerebellar reference region and a composite cortical region, all with FreeSurfer v5.3 segmentation of a contemporary coregistered MRI, to define regional tracer retention (Jagust et al., 2015). Thresholds for Aβ PET positivity were defined as composite FBP SUVR≥ 1.11 or FBB SUVR ≥1.08 as described on the ADNI website (ADNI_UCBERKELEY_AV45_Methods_12.03.15.pdf and UCBerkeley_FBB_Methods_04.11.19.pdf). We obtained centiloid values as described in the Centiloid project (Klunk et al., 2015), with the following conversion equations: Centiloid = (196.9 × SUVRFBP) – 196.03 and Centiloid = (159.08 × SUVRFBB) – 151.65 (ADNI_Centiloid_Methods_Instruction_20181113.pdf).
2.4. Hippocampal volume
Hippocampal volume was assessed via FreeSurfer v5.3 segmentation and was adjusted for intracranial volume by regressing out the association between hippocampal volume and intracranial volume in ADNI cognitively normal individuals (Guo et al., 2020; Jack et al., 2012b).
2.5. Tau PET processing and thresholds
FTP PET analyses used similarly defined regions of interest (ROIs, not partial volume corrected) intensity normalized using the inferior cerebellar grey matter as a reference region to create SUVRs. We used bilateral FreeSurfer defined ROIs that approximated Braak pathological staging as previously described (Braak and Braak, 1991, 1995; Maass et al., 2017). Braak 1: entorhinal cortex; Braak 34 (inferolateral temporal): parahippocampal, fusiform, lingual, amygdala, middle temporal, cingulate, insula, inferior temporal, and temporal pole regions; Braak 56 (extratemporal neocortical): superior frontal, orbitofrontal, frontal pole, middle frontal, Broca’s area, lateral occipital, supramarginal, inferior parietal, superior parietal, precuneus, superior temporal, bank of superior temporal sulcus, transverse temporal, pericalcarine, postcentral, cuneus, precentral, and paracentral regions (UCBERLELEY_AV1451_Methods_Aug2018.pdf). Braak 2 (hippocampus) was excluded due to the vulnerability of this region to off-target binding in the choroid plexus (Maass et al., 2017). We also examined FTP retention in a temporal metaROI which included entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal ROIs (Jack et al., 2017b).
Our criteria for abnormal tau focused on entorhinal and inferolateral temporal regions, following the working criteria of PART (Crary et al., 2014). We used previously defined ROC analyses to define SUVR thresholds for entorhinal (Braak 1; 1.21, Supplementary Fig. 1) and inferolateral temporal (Braak 34; 1.23, Supplementary Fig. 2) regions that best differentiated Aβ+ impaired from Aβ-unimpaired individuals (Guo et al., 2020). Abnormal tau (T+) status was defined as exceeding these thresholds for either Braak 1 or Braak 34 regions or both. Therefore T+ status could be due to abnormal entorhinal tau, abnormal inferolateral temporal tau, or abnormal tau that spans entorhinal and inferolateral temporal regions.
2.6. A-T+ PET visual review
Each amyloid and tau PET scan of individuals the A-T+ group was reviewed visually in order to (1) confirm that the amyloid PET scan was truly negative, (2) determine whether there was an AD-like pattern of tau binding seen in AD (medial temporal FTP signal that may extend into inferolateral temporal regions and/or into other neocortical regions) or (3) determine whether the scan showed other clinically important types of tau deposition patterns.
A grayscale MRI was overlayed with the coregistered PET image (coregistration carried out in SPM). The PET image was displayed using the ‘Jet’ colormap (matplotlib library in Python), alpha=70%, thresholded from 0.0 to 2.3. We used the Freesurfer anatomical atlas as a reference point for each of the slices. Amyloid PET visual review criteria and findings are summarized above (2.1). All authors reviewed all FTP PET scans in the A-T+ group in a consensus conference, and a nuclear medicine physician (K.P) categorized them as fitting into one of four patterns: (1) “Negative” or “false-positive” quantification, characterized by elevation of FTP signals outside of brain areas like skull base or off-target binding, (2) “medial temporal binding” showing mild to moderate elevation of FTP signal predominantly in bilateral medial temporal regions compatible with Braak stage 1, (3) “AD-like” showing bilateral medial temporal FTP signal with extension to inferolateral temporal lobes compatible with Braak stages 1–4 (Braak et al., 2006; Braak and Braak, 1991; Cho et al., 2016; Schwarz et al., 2016) or in some cases including further extension to frontal and parietal lobes (4) “Atypical” or “non-AD” demonstrating FTP signal not following this typical tau distribution of AD. Pattern 2 was considered consistent with normal aging or PART, and pattern 3 was considered consistent with PART or AD (Sonni et al., 2020). To define the topographical distribution, we ranked the highest SUVR region for each participant in both A-T+ and A+T+ groups and used this information to create a frequency bar chart and brain maps that demonstrated the proportion of subjects in whom each ROI was the region of highest tracer accumulation.
2.7. Cognitive tests
To evaluate cognition we compared the composite memory score (Crane et al., 2012) (ADNI_Methods_UWMPSYCHSUM_20160112.pdf), the composite executive function score (Gibbons et al., 2012), the Alzheimer’s Disease Assessment Scale-Cognitive subscale (ADAS-cog) score (Mohs et al., 1983), and the Preclinical Alzheimer Cognitive Composite (PACC) (Donohue et al., 2014) between groups. The composite memory score consisted of the auditory verbal learning test, the word list learning and recognition components of ADAS-cog, word recall items of Mini-Mental State Examination (MMSE), and logical memory I from the Wechsler Memory Test-Revised. The composite executive function score comprised the digit symbol substitution and digit span backward tests, trail making test parts A and B, animal and vegetable category fluency, digit cancellation, and the clock drawing test. The PACC consisted of logical memory delayed recall, MMSE total score, digit symbol, and ADAS-cog word recall.
2.8. Statistical analysis
We applied a Chi-square test for categorical variables, the Kruskal-Wallis test for non-normally distributed continuous variables, and the analysis of variance for normally distributed ones to identify differences between the 3 groups. Analysis of covariance (ANCOVA) was adjusted for age, sex, education, or clinical diagnosis as covariates with Bonferroni-corrected posthoc tests. Statistical analyses were performed using SPSS version 20.0 (IBM, Armonk, NY, USA) with a value for statistical significance of p < 0.05.
3. Results
3.1. Subject characteristics
Table 1 shows the characteristics of all 561 participants. The A-T+ group accounted for 9.2% of all initial participants (N = 685). The A-T+ group was older than the A-T- group and more educated than the A+T+ group. The percentage of APOE ɛ4 carriers in the A-T+ group (17.0%) was similar to the A-T- group (22.8%) but lower than the A+T+ group (64.2%). Individuals in the A-T+ group were intermediate between the two other groups in the prevalence of cognitive impairment. The A-T+ group had a higher proportion of individuals with hyperlipidemia than both other groups, but they did not differ on other cardiovascular risk factors. The A-T+ group did not differ from the others on the geriatric depression scale, but had lower neuropsychiatric inventory scores than the A+T+ subjects. Hippocampal volume was intermediate to the 2 other groups, and significantly smaller than only the A-T- group. The proportion of subjects studied with FBP and FBB was similar across groups (FBP, A-T- 60.2% vs. A-T+ 65.1% vs. A+T+ 64.8%) and the A-T+ subjects showed no evidence of even minimal amyloid elevation, with similar centiloid and CSF Aβ measurements between A-T+ and A-T-. CSF Ptau was not elevated in the A-T+ participants compared to the A-T- group, but it was lower than the A+T+ group. In general CSF ratios were more similar to A-T- than A+T+, although the Aβ42/Ptau ratio was statistically intermediate to the 2 other groups.
Table 1.
Characteristics of 561 participants by AT group
| AT_group | A-T- (n = 316) |
A-T+ (n = 63) |
A+T+ (n = 182) |
p1 | p2 | p3 |
|---|---|---|---|---|---|---|
| Age, year | 72.0 ± 7.2 | 75.8 ± 7.6 | 75.6 ± 7.8 | <0.001a, b | ||
| Female | 177 (56.0) | 26 (41.3) | 93 (51.1) | 0.087 | ||
| Education, year | 16.6 ± 2.5 | 17.1 ± 2.5 | 16.1 ± 2.5 | 0.011c | ||
| Diagnosis | <0.001 | |||||
| NL | 230 (72.8) | 35 (55.6) | 50 (27.5) | |||
| CI | 86 (27.2) | 28 (44.4) | 132 (72.5) | |||
| APOEε4_carrierd | 63 (22.8) | 9 (17.0) | 88 (64.2) | <0.001b, c | ||
| Diabetes | 41 (13.0) | 10 (14.5) | 22 (12.1) | 0.743 | ||
| Hypertension | 123 (38.9) | 32 (50.8) | 85 (46.7) | 0.095 | ||
| Hyperlipidemia | 77 (24.4) | 27 (42.9) | 47 (25.8) | 0.010a, c | ||
| Cholesterole,mg/dl | 193.5 ± 30.8 | 204.1 ± 36.3 | 191.6 ± 48.2 | 0.411 | ||
| MMSE | 28.9 ± 1.3 | 28.6 ± 1.9 | 26.0 ± 4.0 | <0.001b, c | <0.001b, c | |
| GDS | 1.1 ± 1.5 | 1.6 ± 2.0 | 1.7 ± 1.8 | 0.001b | 0.003b | |
| NPI | 2.2 ± 5.1 | 2.9 ± 6.7 | 5.5 ± 8.3 | <0.001b, c | <0.001b, c | |
| Hippocampal_Volume, ml | 5809.7 ± 1018.6 | 5035.6 ± 1547.4 | 4727.5 ± 1226.7 | <0.001a, b | <0.001a, b | |
| Amyloid PET | ||||||
| Centiloid | 4.23 ± 9.93 | 1.90 ± 11.14 | 82.92 ± 35.08 | <0.001b, c | <0.001b, c | |
| CSF, pg/ml | ||||||
| Ptauf | 18.17 ± 6.28 | 20.75 ± 7.62 | 35.79 ± 17.66 | <0.001b, c | <0.001b, c | |
| Aβ42f | 1158.75 ± 844.23 | 1049.16 ± 1035.94 | 486.41 ± 416.34 | <0.001b, c | <0.001b, c | |
| Aβ42/Ptauf | 87.27 ± 24.80 | 79.40 ± 22.67 | 23.57 ± 16.42 | <0.001a, b, c | <0.001b, c | |
| Aβ42/Aβ40g | 0.08 ± 0.02 | 0.08 ± 0.02 | 0.03 ± 0.01 | <0.001b, c | <0.001b, c | |
| Ptau/Aβ40g | 0.0010 ± 0.0002 | 0.0011 ± 0.0003 | 0.0020 ± 0.0007 | <0.001b, c | <0.001b, c | |
p1, ANOVA, Kruskal-Wallis, or chi-square; p2, ANCOVA adjusted by age, sex, and diagnosis; p3, ANCOVA adjusted by age, sex, and education. Tukey posthoc analysis was conducted in p1 and Bonferroni posthoc analysis was conducted in p2 and p3.
Key: APOE, apolipoprotein E; Aβ, beta-amyloid; CI, cognitive impairment; CSF, cerebrospinal fluid; Ptau, hyperphosphorylated tau; GDS, geriatric depression scale; NL, normal cognition; MMSE, mini-mental state examination; NPI, neuropsychiatric inventory; PET, positron emission tomography; SUVR, standard uptake value ratio.
p < 0.05 between A-T- and A-T+
p < 0.05 between A-T- and A+T+
p < 0.05 between A-T+ and A+T+
Missing data 95
Missing data 372
Missing data 156
Missing data 287. Data are shown as mean ± SD or number (%).
3.2. FTP-PET results
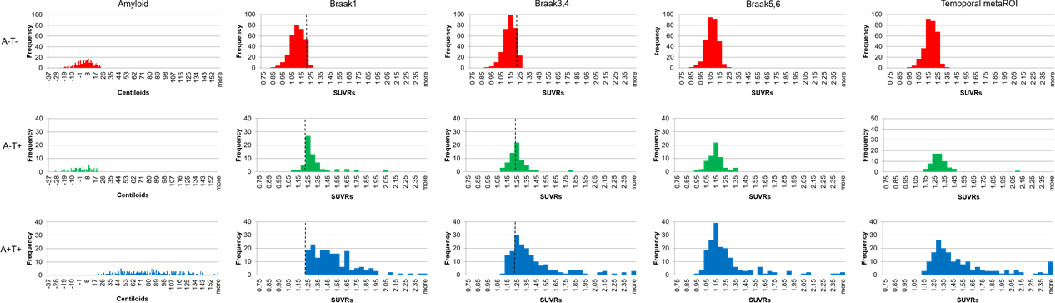
As shown in Fig. 1, the A-T+ group demonstrated intermediate FTP levels in each Braak stage among the 3 groups after adjusting for age, sex, and clinical diagnosis. Frequency histograms of centiloids and tau PET regions of interest illustrate the expected relationships, with tau distributions of A-T+ individuals that are intermediate to A-T- and A+T+ individuals (Supplementary Fig. 3). Examples of visually-identified patterns of FTP binding in the A-T+ group are provided in Fig. 2. Seventeen A-T+ individuals (27.0%) were categorized as showing no significant cortical uptake (negative or false positive pattern) (Fig. 2A), 18 (28.6%) as a pattern with bilateral medial temporal binding (Fig. 2B), 20 (31.7%) showing AD-like pattern (Fig. 2C and 2D), and the remaining 8 (12.7%) included 1 atypical AD-like pattern (Fig. 2E) and 7 non-AD patterns (Fig. 2F and 2G). These unusual patterns included a highly asymmetric focal uptake case suggestive of logopenic variant primary progressive aphasia but without Aβ deposition (Fig. 2E) and several other cases that appeared to be primary tauopathies including a pattern suggestive of the microtubule-associated protein tau (MAPT) R406W mutation (Fig. 2F) or other frontotemporal lobar degeneration (Fig. 2G).
Fig. 1.
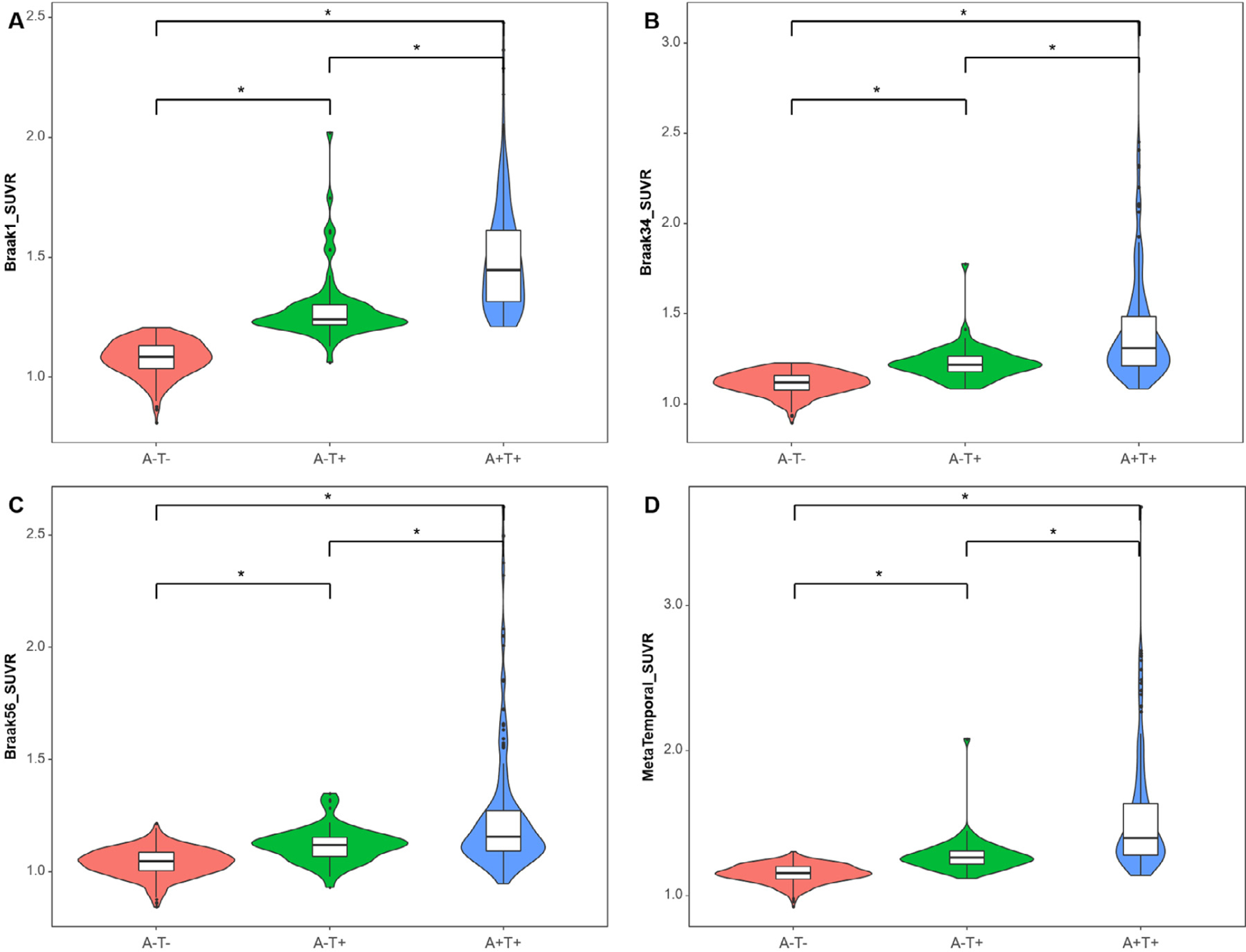
Tau levels by Braak stage among 316 A-T- individuals, 63 A-T+, and 182 A+T+ individuals. Violin plots and Boxplots represent tau SUVR in grouped regions of interest corresponding to Braak 1 (A), Braak 34 (B), Braak 56 (C), and Metatemporal ROI (D) by AT group. The Y-axis is a tau SUVR value in each Braak stage. Regardless of Braak stages, the A-T+ group shows an intermediate level of FTP SUVR between A-T- and A+T+ groups. Asterisk (∗) represents a statistically significant p-value (p < 0.001) in the analysis of covariance adjusted by age, sex, and diagnosis with Bonferroni correction. Abbreviations: FTP, 18F-flortaucipir; ROI, region of interests; SUVR, standard uptake value ratio.
Fig. 2.
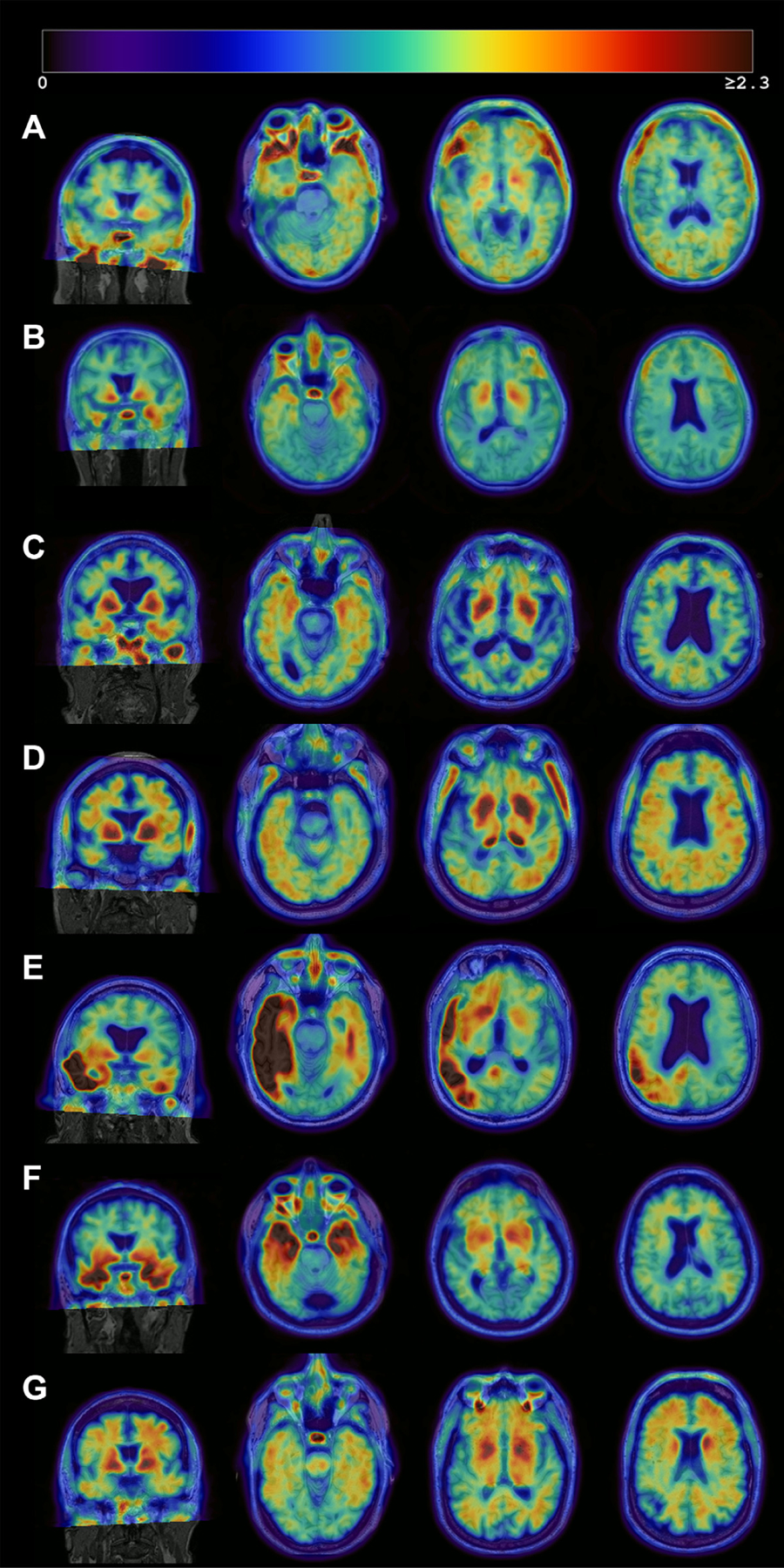
Heterogeneous patterns of FTP binding in the A-T+ group. Each image consists of an MRI (grayscale) with the co-registered PET image overlaid. The images represent Negative or false-positive pattern (A), bilateral medial temporal binding pattern (B) or AD-like pattern (C, D), and atypical (E) or non-AD pattern (F, G). The right hemisphere is displayed on the right side. (A) No significant cortical binding on visual assessment. Likely false-positive quantification due to partial volume effect in the inferior temporal lobe, high off-target signal in the base of the skull, and non-specific signal in the white matter (68/female, MCI, MMSE 28). (B) Mild bilateral medial temporal FTP binding which could reflect PART (61/male, dementia, MMSE 23), (C) Mild bilateral medial and lateral temporal tracer retention which could represent PART or early AD (86/male, MCI, MMSE 28). (D) Mild lateral temporal, parietal, and frontal tracer retention in a pattern suggestive of AD (75/male, Normal, MMSE 26), (E) Intense asymmetric left-dominant parieto-temporal binding, highly suggestive of lvPPA due to AD despite amyloid negativity (71/male, Normal, MMSE 29), and (F) Intense binding in bilateral temporal poles, suggestive of MAPT mutation (68/male, MCI, MMSE 23) and (G) Mild to moderate tracer binding predominantly in the frontal white matter, suggestive of non-AD pathology (possible primary tauopathy) (68/male, MCI, MMSE 29). Abbreviations: AD, Alzheimer’s disease; FTP, 18F-flortaucipir; lvPPA, logopenic variant primary progressive aphasia; MAPT, microtubule-associated protein tau MCI, mild cognitive impairment; MMSE, mini-mental state examination; PART, primary age-related tauopathy.
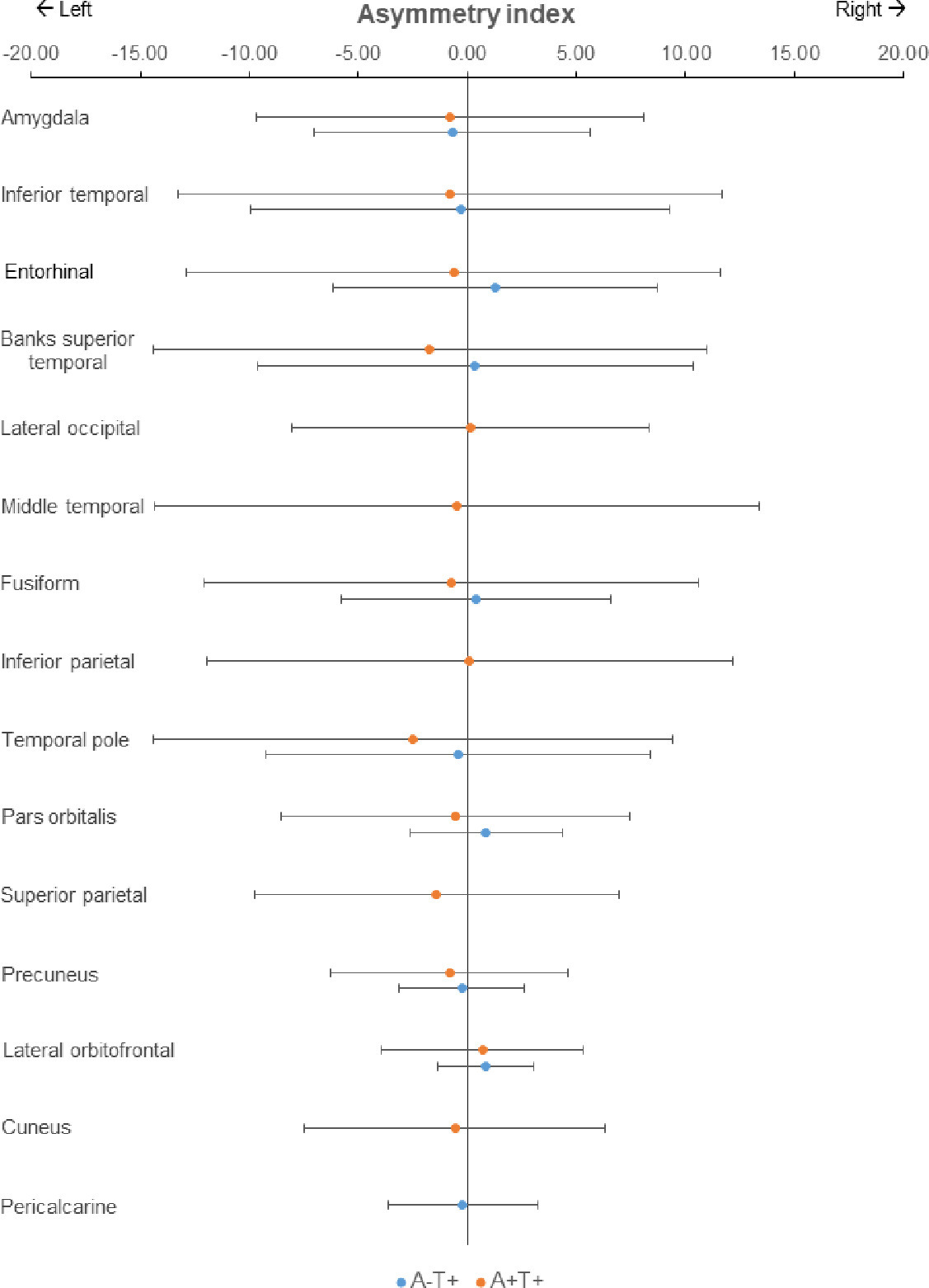
Across subjects, given that the asymmetry index of tau SUVRs in the regions that showed the highest SUVR uptake, there was no asymmetry (Suppl. Fig. 4). Fig. 3(A) displays the order of regions most frequently observed as the highest areas of tau accumulation in the brain, both left and right combined, and Fig. 3(B) represents the topography separately for left and right. Bilateral amygdala was the highest accumulation region in both A-T+ and A+T+ groups (Fig. 3A and 3B). The patterns of FTP deposition were similar between the two groups, although bilateral inferior temporal lobes were the second-highest region in A+T+ and entorhinal cortex was the second-highest in A-T+. Otherwise, both groups showed high levels in the medial, inferior, and lateral temporal lobes as well as orbitofrontal lobes and amygdala, but the A+T+ group also showed high uptake in medial and lateral parietal lobes and lateral frontal lobes (Fig. 3C and D).
Fig. 3.
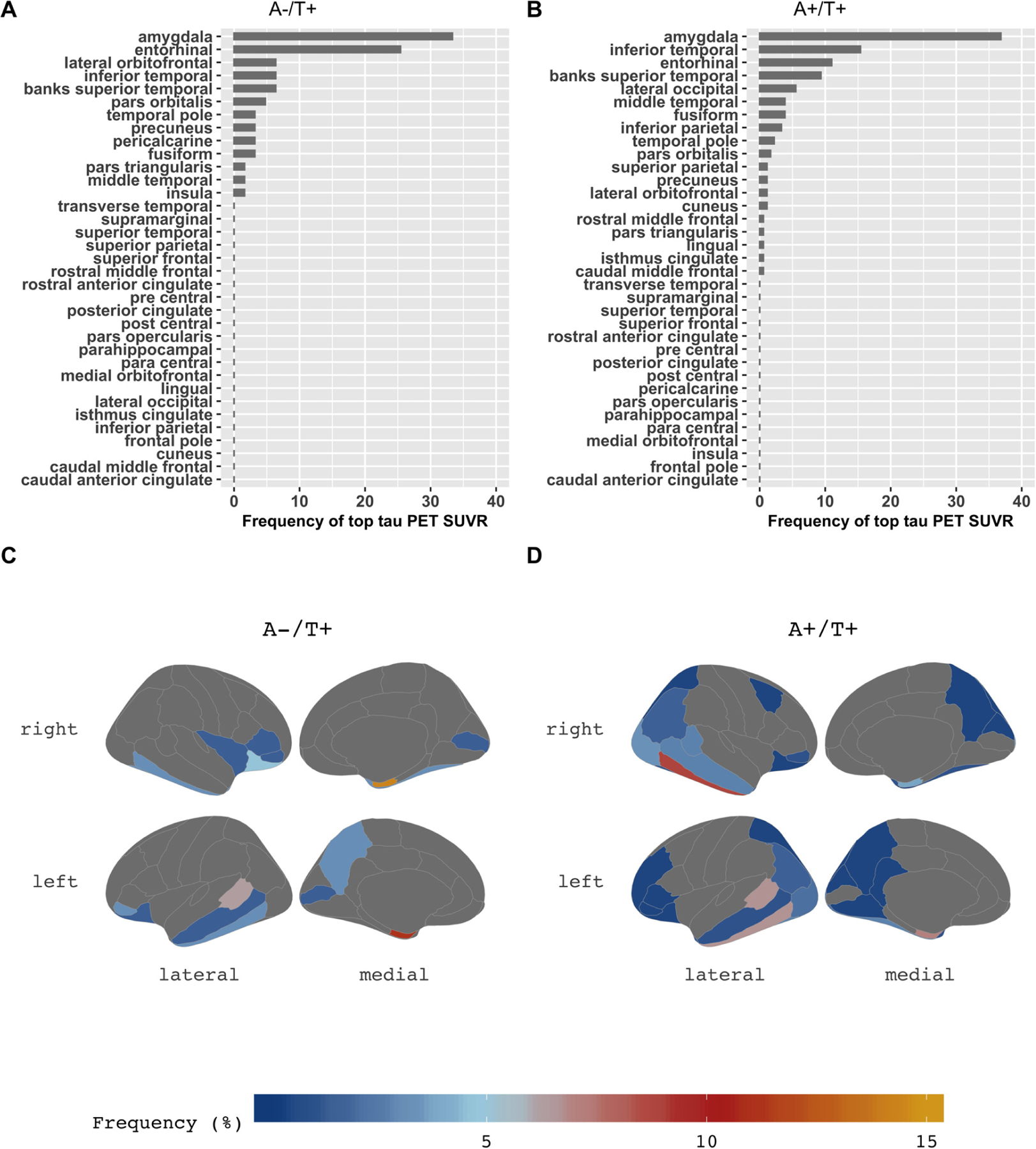
The topography of tau deposition. In the bar charts of the A-T+ group (A) and the A+T+ group (B), the X-axis is the percentage of frequency that each region appeared as the highest tau SUVR (averaged across hemispheres). Detailed asymmetry index in the regions of each group were presented in Suppl. Fig. 4. Amygdala is the highest area of tau deposition in both the A-T+ group and the A+T+ group. The brain maps (C, D) demonstrate the distribution of tau deposition according to the frequency coded in the color bar below. Compared to the A-T+ group (C), the A+T+ group (D) shows a more widespread distribution of tau deposition extending to parietal, frontal, and occipital lobes, but similar patterns showing amygdala, temporal and orbitofrontal lobes. Abbreviations: SUVR, standard uptake value ratio.
3.3. Cognition
Results of ANCOVAs adjusting for age, sex, and education revealed that cognitive performance of the A-T+ group was intermediate to the other two groups on the executive function and memory composites, ADAS-cog, and PACC. Performance of the A-T+ group on executive function and ADAS-cog was significantly lower than A+T+ individuals but did not differ from A-T- individuals, but A-T+ individuals were significantly different from both other groups on memory (p < 0.001) (Fig. 4A) and PACC (< 0.001) (Fig. 4D).
Fig. 4.
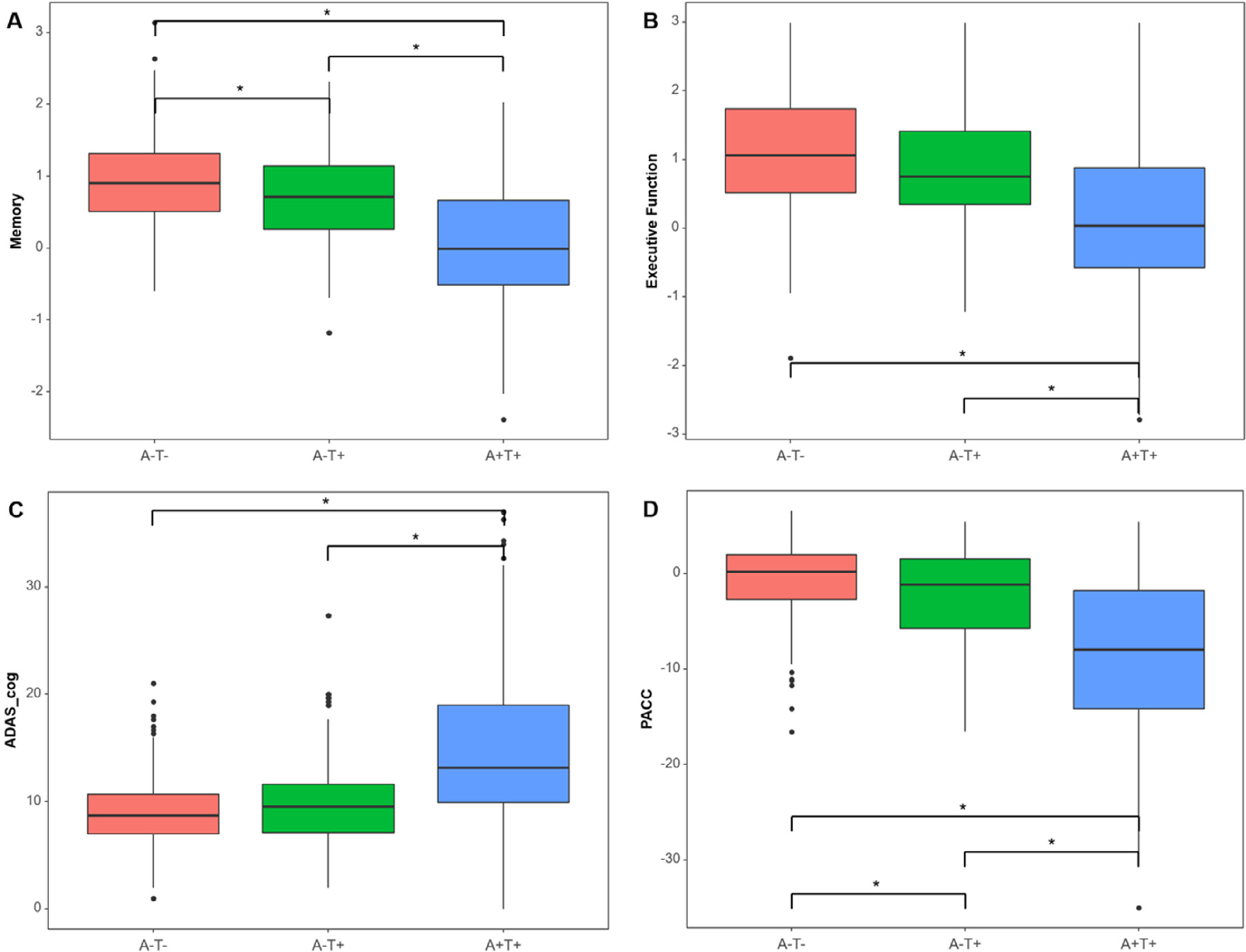
Cognitive profile comparisons among 3 groups. Boxplots represent (A) memory composite score, (B) executive function composite score, (C) ADAS-cog, and (D) PACC by AT group. The Y-axis is a normalized value (z-scores) of each cognitive measure. The cognition of the A-T+ group is intermediate between the 3 groups. Asterisk (∗) indicates a statistically significant p-value (p < 0.001) in the analysis of covariance adjusted by age, sex, and education with Bonferroni correction. Abbreviations: ADAS-cog, Alzheimer’s disease assessment scale-cognitive subscale; PACC, preclinical Alzheimer cognitive composite.
4. Discussion
Individuals with abnormal tau but normal Aβ comprised about 10% of impaired and unimpaired participants, which was similar to prior studies (Altomare et al., 2019; Dodich et al., 2020). However, these individuals are heterogeneous and no single explanation accounts for their prevalence. First, about 9% of the initially characterized 69 A-T+ subjects were subsequently classified as A+ by visual read and excluded from further analyses, consistent with the likelihood that a proportion of A-T+ individuals are not truly amyloid negative. While this could suggest that subthreshold levels of Aβ exist throughout the A-T+ group, this explanation seems unlikely since we excluded individuals with visually positive Aβ PET, and our resulting A-T+ group had normal Aβ PET and CSF measurements that did not differ from A-T- subjects. It is possible that these individuals might nevertheless be unusually susceptible to Aβ requiring only low levels to initiate a toxic tau cascade. Visual inspection of A-T+ tau PET scans indicated that 60.3 % of the A-T+ group had mild bilateral medial temporal binding or an AD-like pattern that is suggestive of PART. In addition to this predominant pattern, there were a number of other atypical patterns including possible primary tauopathies.
The topography of quantitatively-assessed tau deposition was similar in the A-T+ and A+T+ groups. In both groups, the amygdala was a region in the brain with the highest uptake in the majority of participants; the entorhinal cortex and inferior temporal cortex were also frequently observed as the highest regions. This pattern, with some variation, has been previously reported in both normal aging and AD (Adams et al., 2019; Cho et al., 2016; Lowe et al., 2018). The primary difference between the A-T+ and A+T+ groups was the wider spread of tau throughout neocortex in the latter subjects. A minority of A-T+ individuals had maximal tau regions in frontal or posterior regions, but this was an atypical pattern, and these extra-temporal tau elevations likely occurred in individuals with elevated diffuse tau throughout the brain. Together, these findings suggests that regardless of amyloid status, tau is usually highest in a consistent set of medial temporal regions, even if the overall amount of tau or spread beyond those medial temporal regions is variable.
Although PART is a neuropathologically-defined phenomenon, our biomarker-based characterization of PART largely agrees with pathology work. For example, the A-T+ group in this study, though heterogeneous, had tau deposition that was intermediate to A-T+ and A+T+ (AD) individuals, consistent with pathology studies showing relatively restricted tau deposition in PART compared to AD (Besser et al., 2017). We also found that A-T+ had a low prevalence of APOE ε4 carriers, consistent with previous PART research (Bell et al., 2019; Crary et al., 2014; Kim et al., 2019; Teylan et al., 2019; Teylan et al., 2020; Zhang et al., 2019). With respect to hippocampal volume and cognition, A-T+ again demonstrated an intermediate state between A-T- and A+T+. The relatively better cognitive performance in A-T+ compared with A+T+ individuals agrees with previous reports of relatively preserved cognition in neuropathologically-defined PART (Bell et al., 2019; Besser et al., 2017; Crary, 2016; Crary et al., 2014; Jellinger et al., 2015) as does relatively preserved hippocampal volume in A-T+ than A+T+ (Josephs et al., 2017; Quintas-Neves et al., 2019).
The wider distribution of tau in the A+T+ subjects is consistent with other data suggesting that Aβ facilitates tau spread (Hanseeuw et al., 2019; Jack et al., 2018b). Whether A-T+ individuals may ultimately evolve into A+T+, and thus AD, is unknown since the current analyses are only cross-sectional. However, it is clear from pathology studies that many PART individuals with intermediate levels of tau do not develop AD during life (Bell et al., 2019; Besser et al., 2017; Crary, 2016; Crary et al., 2014; Jellinger et al., 2015) supporting arguments that PART should be considered a distinct category from AD.
Imaging studies offer the potential advantage over pathological examinations of PART in that we can observe whether such individuals become amyloid positive over time. However, based on their very low current levels of brain Aβ seen with PET, the progression of amyloid pathology seems unlikely. Moreover, recent studies have shown that A-T+ individuals convert to AD or another dementia at a much lower rate than A+T+ individuals (Eckerström et al., 2021), supporting the idea that the A-T+ group is a distinct disease entity different from Alzheimer’s continuum.
Because PART is a neuropathologic-based entity, and the ability to image tau in vivo is relatively recent, the biomarker-based concept of SNAP has been used more frequently in imaging studies. Although SNAP was originally conceived as abnormal neurodegeneration in the absence of Aβ, as in vivo tau markers have become more accessible, the concept of SNAP has transitioned to inclusion of abnormal neurodegeneration and/or tau (N+ and/or T+) in the absence of Aβ (Jack et al., 2018a). While neurodegeneration is closely linked to tau, they do not always agree, and PART as a tau-focused concept may constitute a subset of SNAP which is more broadly defined. Here, we focused on PART in particular but examined the full range of neurodegeneration (hippocampal volume) in A-T+ individuals without restricting the sample to individuals with abnormal hippocampal atrophy. Characteristics of PART overlap with, but are not the same as, characteristics of SNAP. For example, one study showed temporal tau in SNAP did not differ from A-N- individuals (Mormino et al., 2016). On the other hand, slower progression of cognition or slower atrophy of hippocampal volume in SNAP than those in A+ groups has been reported in previous longitudinal studies (Burnham et al., 2016; Caroli et al., 2015; Chung et al., 2017; Mormino et al., 2016; Zhao et al., 2018), which is in agreement with the findings reported here for A-T+ individuals. Furthermore, we found that 44% of the A-T+ individuals were cognitively impaired (compared with 72% of A+T+ and 27% of A-T- individuals), consistent with an Aβ−independent role for tauopathy in the neurodegenerative process of pathologic brain aging (Josephs et al., 2017) and with the idea that disease progression is accelerated when tauopathy is combined with amyloidosis (Altomare et al., 2019; Bilgel et al., 2018; Fortea et al., 2014; Pascoal et al., 2017; Soldan et al., 2019; Yu et al., 2019; Zhao et al., 2018).
The present study has several limitations. First, FTP binds predominantly to paired helical filaments typically observed in AD. Because of this, we are unable to report on A-T+ individuals who may harbor non-AD forms of tau, providing an incomplete picture of this biomarker category. Second, we note that PART is a neuropathologic diagnosis, so we do not consider A-T+ status defined here as interchangeable with PART because of the possibility of false positivity on tau PET, or tauopathies among A-T+ individuals that may be identified pathologically. Third, relying on a statistical threshold for tau positivity can introduce misclassification. However, this is a routine research practice, and we attempted to minimize this by reviewing all images in the A-T+ group. Fourth, the study employed a cross-sectional approach to data analysis, which cannot determine causality or long-term change. Once longitudinal data becomes available for this sample, it will be important to examine the trajectories of A-T+ individuals. Fifth, we did not obtain information about MAPT, progranulin gene, or other mutations that are associated with elevated FTP uptake. Finally, while we excluded regions such as the hippocampus which are known to be influenced by off-target binding in the neighboring choroid plexus, neuropathological validation is ultimately required to understand the basis of PET images, and PART itself remains a neuropathological diagnosis. The autopsy-imaging studies reported to date do not indicate that FTP PET detects PART (Fleisher et al., 2020; Lowe et al., 2020). However, there are relatively limited data, and no imaging-pathology studies have specifically focused on PART (Table 1).
5. Conclusions
Although PART is a pathological entity that has not been examined extensively during life, biomarker studies are beginning to determine the extent to which PART may represent “normal” versus “pathological” aging. Our findings suggest that A-T+ individuals span both of these categories: while tau positivity in the absence of amyloid is associated with healthy aging for some individuals, in other individuals it is associated with several features that are common to AD, such as tau extending beyond the medial temporal lobe, cognitive symptoms, and hippocampal atrophy.
In summary, our investigation of A-T+ individuals suggests that they are a heterogeneous group, with the majority sharing features of AD that nevertheless occur in the clear absence of Aβ. Whether these individuals reflect PART, an early stage of AD, or an entirely different entity will depend on pathological validation and longitudinal observation. It seems most likely that regional vulnerability to tau is a shared feature among older people, which produces modest cognitive impairment and neurodegeneration on its own but may become more malignant in the presence of Aβ.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by grant U01 AG024904 from the National Institute on Aging. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; Bio-Clinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute, University of Southern California. ADNI data are disseminated by the Laboratory for NeuroImaging at the University of Southern California.
Footnotes
Disclosure statement
Dr. Yoon, Dr. Guo, Dr. Provost, Korman D, and Ward TJ report no disclosures relevant to the manuscript. Dr. Landau has served as a consultant to NeuroVison and Cortexyme. Dr. Jagust has served as a consultant to Genentech, Biogen, Novartis, CuraSen, Bioclinica, and Grifols.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neurobiolaging.2021.09.019.
CRediT authorship contribution statement
Bora Yoon: Conceptualization, Methodology, Formal analysis, Writing – original draft, Writing – original draft. Tengfei Guo: Formal analysis, Software, Visualization. Karine Provost: Formal analysis, Writing – review & editing. Deniz Korman: Data curation, Formal analysis, Software. Tyler J. Ward: Data curation, Formal analysis, Visualization. Susan M. Landau: Investigation, Conceptualization, Methodology, Supervision, Writing – review & editing. William J Jagust: Investigation, Conceptualization, Methodology, Supervision, Resources, Writing – review & editing.
References
- Adams JN, Maass A, Harrison TM, Baker SL, Jagust WJ, 2019. Cortical tau deposition follows patterns of entorhinal functional connectivity in aging. Elife 8, e49132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altomare D, de Wilde A, Ossenkoppele R, Pelkmans W, Bouwman F, Groot C, van Maurik I, Zwan M, Yaqub M, Barkhof F, van Berckel BN, Teunissen CE, Frisoni GB, Scheltens P, van der Flier WM, 2019. Applying the ATN scheme in a memory clinic population. Neurology 93, e1635. [DOI] [PubMed] [Google Scholar]
- Bell WR, An Y, Kageyama Y, English C, Rudow GL, Pletnikova O, Thambisetty M, O’Brien R, Moghekar AR, Albert MS, Rabins PV, Resnick SM, Troncoso JC, 2019. Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. Alzheimers Dement 15 (1), 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser LM, Crary JF, Mock C, Kukull WA, 2017. Comparison of symptomatic and asymptomatic persons with primary age-related tauopathy. Neurology 89 (16), 1707–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgel M, An Y, Helphrey J, Elkins W, Gomez G, Wong DF, Davatzikos C, Ferrucci L, Resnick SM, 2018. Effects of amyloid pathology and neurodegeneration on cognitive change in cognitively normal adults. Brain 141 (8), 2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K, 2006. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112 (4), 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82 (4), 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1995. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16 (3), 278–284 271–278; discussion. [DOI] [PubMed] [Google Scholar]
- Burnham SC, Bourgeat P, Dore V, Savage G, Brown B, Laws S, Maruff P, Salvado O, Ames D, Martins RN, Masters CL, Rowe CC, Villemagne VL, 2016. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer’s disease pathophysiology (SNAP) or Alzheimer’s disease pathology: a longitudinal study. Lancet Neurol 15 (10), 1044–1053. [DOI] [PubMed] [Google Scholar]
- Caroli A, Prestia A, Galluzzi S, Ferrari C, van der Flier WM, Ossenkoppele R, Van Berckel B, Barkhof F, Teunissen C, Wall AE, Carter SF, Scholl M, Choo IH, Grimmer T, Redolfi A, Nordberg A, Scheltens P, Drzezga A, Frisoni GB, 2015. Mild cognitive impairment with suspected nonamyloid pathology (SNAP): Prediction of progression. Neurology 84 (5), 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Choi JY, Hwang MS, Kim YJ, Lee HM, Lee HS, Lee JH, Ryu YH, Lee MS, Lyoo CH, 2016. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 80 (2), 247–258. [DOI] [PubMed] [Google Scholar]
- Chung JK, Plitman E, Nakajima S, Caravaggio F, Iwata Y, Gerretsen P, Kim J, Takeuchi H, Shinagawa S, Patel R, Chakravarty MM, Graff-Guerrero A, 2017. Hippocampal and Clinical Trajectories of Mild Cognitive Impairment with Suspected Non-Alzheimer’s Disease Pathology. J Alzheimers Dis 58 (3), 747–762. [DOI] [PubMed] [Google Scholar]
- Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, Jones RN, Mukherjee S, Curtis SM, Harvey D, Weiner M, Mungas D, 2012. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 6 (4), 502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary JF, 2016. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine 1 (6), 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT, 2014. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128 (6), 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodich A, Mendes A, Assal F, Chicherio C, Rakotomiaramanana B, Andryszak P, Festari C, Ribaldi F, Scheffler M, Schibli R, Schwarz AJ, Zekry D, Lövblad K-O, Boccardi M, Unschuld PG, Gold G, Frisoni GB, Garibotto V, 2020. The A/T/N model applied through imaging biomarkers in a memory clinic. Eur J Nuclear Med Mol Imaging 47 (2), 247–255. [DOI] [PubMed] [Google Scholar]
- Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, Weiner M, Aisen PSfor the Australian Imaging, B., Ageing, L.F.S.o., Initiative, t.A.s.D.N., Study, t.A.s.D.C., 2014. The Preclinical Alzheimer Cognitive Composite: Measuring Amyloid-Related Decline. JAMA Neurol 71 (8), 961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerström C, Svensson J, Kettunen P, Jonsson M, Eckerström M, 2021. Evaluation of the ATN model in a longitudinal memory clinic sample with different underlying disorders. Alzheimer’s & Dementia: Diagnosis. Assess Dis Monit 13 (1), e12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Pontecorvo MJ, Devous MD Sr., Lu M, Arora AK, Truocchio SP, Aldea P, Flitter M, Locascio T, Devine M, Siderowf A, Beach TG, Montine TJ, Serrano GE, Curtis C, Perrin A, Salloway S, Daniel M, Wellman C, Joshi AD, Irwin DJ, Lowe VJ, Seeley WW, Ikonomovic MD, Masdeu JC, Kennedy I, Harris T, Navitsky M, Southekal S, Mintun MA, 2020. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol 77, 829–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortea J, Vilaplana E, Alcolea D, Carmona-Iragui M, Sanchez-Saudinos MB, Sala I, Anton-Aguirre S, Gonzalez S, Medrano S, Pegueroles J, Morenas E, Clarimon J, Blesa R, Lleo A, 2014. Cerebrospinal fluid beta-amyloid and phospho-tau biomarker interactions affecting brain structure in preclinical Alzheimer disease. Ann Neurol 76 (2), 223–230. [DOI] [PubMed] [Google Scholar]
- Gibbons LE, Carle AC, Mackin RS, Harvey D, Mukherjee S, Insel P, Curtis SM, Mungas D, Crane PK, 2012. A composite score for executive functioning, validated in Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav 6 (4), 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Korman D, Baker SL, Landau SM, Jagust WJ, 2020. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in alzheimer’s disease pathophysiology. Biol Psychiatry 89 (8), 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, Buckley RF, Papp KV, Amariglio RA, Dewachter I, Ivanoiu A, Huijbers W, Hedden T, Marshall GA, Chhatwal JP, Rentz DM, Sperling RA, Johnson K, 2019. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol 76, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R, 2018a. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14 (4), 535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B, 2016a. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87 (5), 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Chetelat G, Dickson D, Fagan AM, Frisoni GB, Jagust W, Mormino EC, Petersen RC, Sperling RA, van der Flier WM, Villemagne VL, Visser PJ, Vos SJ, 2016b. Suspected non-Alzheimer disease pathophysiology–concept and controversy. Nat Rev Neurol 12 (2), 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, Kantarci K, Gunter JL, Senjem ML, Ivnik RJ, Roberts RO, Rocca WA, Boeve BF, Petersen RC, 2012a. An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol 71 (6), 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, Kantarci K, Gunter JL, Senjem ML, Ivnik RJ, Roberts RO, Rocca WA, Boeve BF, Petersen RC, 2012b. An operational approach to National Institute on Aging–Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol 71 (6), 765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Schwarz CG, Lowe VJ, Senjem ML, Vemuri P, Weigand SD, Therneau TM, Knopman DS, Gunter JL, Jones DT, Graff-Radford J, Kantarci K, Roberts RO, Mielke MM, Machulda MM, Petersen RC, 2018b. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 141 (5), 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, Senjem ML, Gunter JL, Rocca WA, Petersen RC, 2017a. Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol 16 (6), 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, Machulda MM, Mielke MM, Roberts RO, Vemuri P, Reyes DA, Petersen RC, 2017b. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement 13 (3), 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM, Koeppe RA, Reiman EM, Chen K, Mathis CA, Price JC, Foster NL, Wang AY, 2015. The Alzheimer’s Disease Neuroimaging Initiative 2 PET Core: 2015. Alzheimer’s Dementia 11 (7), 757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, Dickson DW, Hof PR, Hyman BT, Jack CR Jr., Jicha GA, Knopman DS, Kovacs GG, Mackenzie IR, Masliah E, Montine TJ, Nelson PT, Schmitt F, Schneider JA, Serrano-Pozo A, Thal DR, Toledo JB, Trojanowski JQ, Troncoso JC, Vonsattel JP, Wisniewski T, 2015. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 129 (5), 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, Weigand SD, Boeve BF, Kantarci K, Petrucelli L, Lowe VJ, Jack CR Jr., Petersen RC, Parisi JE, Dickson DW, 2017. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 133 (5), 705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Kim HS, Choi SM, Kim BC, Lee MC, Lee KH, Lee JH, 2019. Primary Age-Related Tauopathy: An Elderly Brain Pathology Frequently Encountered during Autopsy. J Pathol Transl Med 53 (3), 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD Sr., Jagust WJ, Johnson KA, Mathis CA, Minhas D, Pontecorvo MJ, Rowe CC, Skovronsky DM, Mintun MA, 2015. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 11 (1), 1–15. e11–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau S, Jagust W, 2015. Florbetapir Processing Methods http://adni.loni.usc.edu/
- Landau S, Jagust W, 2016. Flortaucipir (AV-1451) Processing Methods http://adni.loni.usc.edu/.
- Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ, Mintun MA, 2013. Amyloid-beta imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 54 (1), 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Lundt ES, Albertson SM, Min HK, Fang P, Przybelski SA, Senjem ML, Schwarz CG, Kantarci K, Boeve B, Jones DT, Reichard RR, Tranovich JF, Hanna Al-Shaikh FS, Knopman DS, Jack CR Jr., Dickson DW, Petersen RC, Murray ME, 2020. Tau-positron emission tomography correlates with neuropathology findings. Alzheimers Dement 16 (3), 561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, Kantarci K, Jones DT, Vemuri P, Graff-Radford J, Schwarz CG, Machulda MM, Mielke MM, Roberts RO, Knopman DS, Petersen RC, Jack CR Jr., 2018. Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141 (1), 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Landau S, Baker SL, Horng A, Lockhart SN, La Joie R, Rabinovici GD, Jagust WJ, 2017. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage 157, 448–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass A, Lockhart SN, Harrison TM, Bell RK, Mellinger T, Swinnerton K, Baker SL, Rabinovici GD, Jagust WJ, 2018. Entorhinal Tau Pathology, Episodic Memory Decline, and Neurodegeneration in Aging. J Neurosci 38 (3), 530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohs RC, Rosen WG, Davis KL, 1983. The Alzheimer’s disease assessment scale: an instrument for assessing treatment efficacy. Psychopharmacol Bull 19 (3), 448–450. [PubMed] [Google Scholar]
- Mormino EC, Papp KV, Rentz DM, Schultz AP, LaPoint M, Amariglio R, Hanseeuw B, Marshall GA, Hedden T, Johnson KA, Sperling RA, 2016. Heterogeneity in Suspected Non–Alzheimer Disease Pathophysiology Among Clinically Normal Older Individuals. JAMA Neurol 73 (10), 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, Rademakers R, Alafuzoff I, Attems J, Brayne C, Coyle-Gilchrist ITS, Chui HC, Fardo DW, Flanagan ME, Halliday G, Hokkanen SRK, Hunter S, Jicha GA, Katsumata Y, Kawas CH, Keene CD, Kovacs GG, Kukull WA, Levey AI, Makkinejad N, Montine TJ, Murayama S, Murray ME, Nag S, Rissman RA, Seeley WW, Sperling RA, White Iii CL, Yu L, Schneider JA, 2019. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain 142 (6), 1503–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal TA, Mathotaarachchi S, Shin M, Benedet AL, Mohades S, Wang S, Beaudry T, Kang MS, Soucy JP, Labbe A, Gauthier S, Rosa-Neto P, 2017. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimers Dement 13 (6), 644–653. [DOI] [PubMed] [Google Scholar]
- Quintas-Neves M, Teylan MA, Besser L, Soares-Fernandes J, Mock CN, Kukull WA, Crary JF, Oliveira TG, 2019. Magnetic resonance imaging brain atrophy assessment in primary age-related tauopathy (PART). Acta Neuropathol Commun 7 (1), 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, Joshi AD, Devous MD Sr, Mintun MS, 2016. Regional profiles of the candidate tau PET ligand 18 F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139 (5), 1539–1550. [DOI] [PubMed] [Google Scholar]
- Soldan A, Pettigrew C, Fagan AM, Schindler SE, Moghekar A, Fowler C, Li Q-X, Collins SJ, Carlsson C, Asthana S, 2019. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology 92 (14), e1567–e1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonni I, Lesman Segev OH, Baker SL, Iaccarino L, Korman D, Rabinovici GD, Jagust WJ, Landau SM, La Joie R for the Alzheimer’s Disease Neuroimaging, I., 2020., 2021. Evaluation of a visual interpretation method for tau-PET with 18F-flortaucipir. Alzheimer’s & Dementia: Diagnosis. Assess Dis Monitoring 12 (1), e12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teylan M, Besser LM, Crary JF, Mock C, Gauthreaux K, Thomas NM, Chen Y-C, Kukull WA, 2019. Clinical diagnoses among individuals with primary age-related tauopathy versus Alzheimer’s neuropathology. Lab Invest 99, 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teylan M, Mock C, Gauthreaux K, Chen YC, Chan KCG, Hassenstab J, Besser LM, Kukull WA, Crary JF, 2020. Cognitive trajectory in mild cognitive impairment due to primary age-related tauopathy. Brain 143, 611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KR, Edmonds EC, Eppig JS, Wong CG, Weigand AJ, Bangen KJ, Jak AJ, Delano-Wood L, Galasko DR, Salmon DP, Edland SD, Bondi MW, 2019. MCI-to-normal reversion using neuropsychological criteria in the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 15 (10), 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Maurik IS, Vos SJ, Bos I, Bouwman FH, Teunissen CE, Scheltens P, Barkhof F, Frolich L, Kornhuber J, Wiltfang J, Maier W, Peters O, Ruther E, Nobili F, Frisoni GB, Spiru L, Freund-Levi Y, Wallin AK, Hampel H, Soininen H, Tsolaki M, Verhey F, Kloszewska I, Mecocci P, Vellas B, Lovestone S, Galluzzi S, Herukka SK, Santana I, Baldeiras I, de Mendonca A, Silva D, Chetelat G, Egret S, Palmqvist S, Hansson O, Visser PJ, Berkhof J, 2021. van der Flier WM, 2019. Biomarker-based prognosis for people with mild cognitive impairment (ABIDE): a modelling study. Lancet Neurol 18 (11), 1034–1044. [DOI] [PubMed] [Google Scholar]
- Yamada M, 2003. Senile dementia of the neurofibrillary tangle type (tangle-only dementia): neuropathological criteria and clinical guidelines for diagnosis. Neuropathology 23 (4), 311–317. [DOI] [PubMed] [Google Scholar]
- Yu J-T, Li J-Q, Suckling J, Feng L, Pan A, Wang Y-J, Song B, Zhu S-L, Li D-H, Wang H-F, 2019. Frequency and longitudinal clinical outcomes of Alzheimer’s AT (N) biomarker profiles: A longitudinal study. Alzheimer’s Dementia 15 (9), 1208–1217. [DOI] [PubMed] [Google Scholar]
- Zhang X, Sun B, Wang X, Lu H, Shao F, Rozemuller AJM, Liang H, Liu C, Chen J, Huang M, Zhu K, 2019. Phosphorylated TDP-43 Staging of Primary Age-Related Tauopathy. Neurosci Bull 35 (2), 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tudorascu DL, Lopez OL, Cohen AD, Mathis CA, Aizenstein HJ, Price JC, Kuller LH, Kamboh MI, DeKosky ST, Klunk WE, Snitz BE, 2018. Amyloid β Deposition and Suspected Non-Alzheimer Pathophysiology and Cognitive Decline Patterns for 12 Years in Oldest Old Participants Without Dementia. JAMA Neurol 75 (1), 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.