

Abstract
Fatty acid binding protein (FABP4) inhibitors are of synthetic and therapeutic interest and ongoing clinical studies indicate that they may be a promise for the treatment of cancer, as well as other diseases. As part of a broader research effort to develop more effective FABP4 inhibitors, we sought to identify new structures through a two-step computing assisted molecular design based on the established scaffold of a co-crystallized ligand. Novel and potent FABP4 inhibitors have been developed using this approach and herein we report the synthesis, biological evaluation and molecular docking of the 4-amino and 4-ureido pyridazinone-based series.
Keywords: fatty acid binding protein, FABP4, FABP4is, FABP4 inhibitors, pyridazinone, computing assisted molecular design
1. Introduction
Fatty acids (FAs) are long carbon chain organic carboxylic acids responsible for different actions in the human organism [1,2]. Their chronic high concentration in circulation leads to various disorders [3,4], including atherosclerosis [5], diabetes [6] and obesity [7]. Considering that their chemical structure is characterized by high lipophilicity, FAs are insoluble in water, and their trafficking into the body requires specific carriers such as the fatty acid-binding proteins (FABPs). [8]. Since their discovery, FABPs have been classified into different families based on their localization in the human body, such as A-FABP (adipocyte), B-FABP (brain), E-FABP (epidermal), H-FABP (muscle and heart), I-FABP (intestinal), Il-FABP (ileal), L-FABP (liver), M-FABP (myelin), and T-FABP (testis). FABP4 (aP2 or A-FABP) is the subtype expressed in adipocytes [9], and the research into small molecule inhibitors for such protein initially started when it was reported that knockout animal models of FABP4 produced protective effects against the development of insulin resistance [10], as well as several pathological events linked to the metabolic syndrome and atherosclerosis [11,12,13]. Interestingly, pharmacological approaches with small molecules that inhibit the normal function of the protein are also valid in this regard, demonstrating similar results as the genetic procedures by mimicking the phenotype of FABP4-deficient mice [14]. This family of transporter proteins also has a role in cancer progression [15], and it was discovered that non-physiological expressions of FABPs are present in some of the most common cancers such as renal cell carcinoma, bladder and prostate, as well as other types of cancer cells [16,17,18]. It was recently discovered that FABP4 promotes the metastasis and invasion of colon cancer and that the treatment with a classical small molecule inhibitor (BMS309403) weakened the migration and invasion of colon cancer cells [19]. FABP4 leads also to abnormal metastasis patterns in ovarian cancer, and recent findings demonstrate that the protein is responsible for the disease’s aggressivity, contributing to poor prognosis in this tumor [20]. Moreover, the transporter has also been shown to play a role in accelerating glioblastoma cell growth [21]. All these recent findings related to cancer research proved that FABP4 targeting may represent an effective and promising therapeutic strategy against oncological conditions, in addition to the established effects on metabolic and cardiovascular diseases.
Recently, a variety of effective FABP4 inhibitors (FABP4i) have been developed, but unfortunately, none of them is currently in the clinical research phases [14,22]. Computer-aided drug design represents a promising and effective tool for the identification of molecular hits as FABP4i [23,24,25,26,27]. In line with our recent interest in the development of new antitumor compounds and the identification of novel bioactive heterocycles [28,29,30,31,32], herein we report the design, synthesis and in vitro characterization of 4-amino and 4-ureido pyridazinone-based series of FABP4i inspired by the scaffold hopping of an established ligand co-crystallized within the protein.
2. Results and Discussion
2.1. Heterocyclic Small-Molecule Design
To generate a novel series of FABP4 inhibitors we have exploited a two-step computing assisted molecular design. As shown in Figure 1, in the first step of the drug-design process we focused on the search for bioisosteric-replacements/scaffold hopping of the pyrimidine scaffold of the co-crystallyzed ligand (2-[(2-oxo-2-piperidin-1 -ylethyl)sulfanyl]-6-(trifluoromethyl)pyrimidin-4-ol; pdbID: 1TOU). Our bioisosteric replacement analysis led to the selection of three nitrogen-containing heterocyclic frameworks, i.e., pyridazinones, pyridines and benzo[d]thiazole (see Supplementary Materials). Considering the synthetic accessibility of pyridazinone-based molecules and that pyridazinone was not investigated earlier as a scaffold to access FABP4 inhibitors, we envisaged to use this heterocycle to carry out automated ligand growing experiments inside the FABP4 cavity, as described in the Section 3, leading to 52 target molecules. The compounds were then synthesized and screened against FABP4 and the chemical structures are reported in Table 1 and Table 2. Both the scaffold hopping and the ligand growing experiments were conducted using Spark (https://www.cresset-group.com/products/spark/ accessed on 15 June 2022) [33].
Figure 1.

Schematic representation of the computer assisted design of the 4-amino and 4-ureido pyridazinones.
Table 1.
4-NH2-pyridazinones synthesized and screened against FABP4.
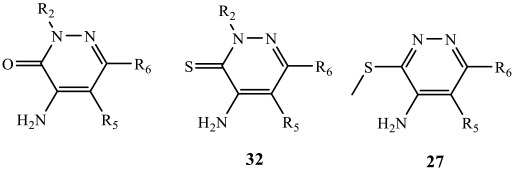
| |||
|---|---|---|---|
| Comp. | R2 | R5 | R6 |
| 4a | H | CONHPh | Ph |
| 4b | H | CONHnC3H7 | Ph |
| 5a | C2H5 | CONHPh | Ph |
| 5b | C2H5 | CONHnC3H7 | Ph |
| 6 | H | CONH2 | Ph |
| 7 | H | CN | Ph |
| 16 | Ph | CONH2 | CH3 |
| 17 | Ph | CN | CH3 |
| 18 | Ph | COCH3 | H |
| 21 | cC6H11 | CONH2 | Ph |
| 22 | cC6H11 | CN | Ph |
| 24d | iC3H7 | H | Ph |
| 24e | nC3H7 | H | Ph |
| 24f | nC4H9 | H | Ph |
| 27 | - | H | Ph |
| 32 | CH3 | H | Ph |
| 37a | H | H | 3-thienyl |
| 37c | H | H | cC6H11 |
| 37d | H | H | iC3H7 |
| 38a | CH3 | H | 3-thienyl |
| 38b | CH3 | H | cC6H11 |
| 38c | CH3 | H | iC3H7 |
| 38d | CH3 | H | CH2-Ph |
| 42a | CH3 | H | 2-(OH)-Ph |
| 42b | CH3 | H | 4-(NH2)-Ph |
| 44 | CH3 | H | 4-(NHCOCH3)-Ph |
| 48 | CH3 | H | 2-pyridinyl |
| 54 | Ph | H | CH3 |
| 57 | Ph | pyrazole | CH3 |
Table 2.
4-Amino and 4-ureido pyridazinones synthesized and screened against FABP4.
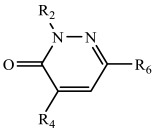
| |||
|---|---|---|---|
| Comp. | R2 | R4 | R6 |
| 25a | CH3 | NHCONH2 | Ph |
| 25b | cC6H11 | NHCONH2 | Ph |
| 25c | C2H5 | NHCONH2 | Ph |
| 25d | iC3H7 | NHCONH2 | Ph |
| 25e | nC3H7 | NHCONH2 | Ph |
| 25f | nC4H9 | NHCONH2 | Ph |
| 28 | H | NHCONH2 | Ph |
| 29a | CH3 | NHCOCH3 | Ph |
| 29b | CH3 | NHCOC2H5 | Ph |
| 29c | CH3 | NHCOiC3H7 | Ph |
| 29d | CH3 | NHCOnC3H7 | Ph |
| 30a | CH3 | NH-(3-CN)-Ph | Ph |
| 30b | CH3 | NH-(2-CN)-Ph | Ph |
| 31a | CH3 | NH-(3-CONH2)-Ph | Ph |
| 31b | CH3 | NH-(2-CONH2)-Ph | Ph |
| 35 | CH3 |
|
Ph |
| 39a | CH3 | NHCONH2 | cC6H11 |
| 39b | CH3 | NHCONH2 | iC3H7 |
| 40 | H | NHCONH2 | 2-(OH)-Ph |
| 43 | CH3 | NHCONH2 | 2-(OH)-Ph |
| 49 | CH3 | NHCONH2 | 2-pyridinyl |
| 51 | CH3 | CONH2 | Ph |
| 55 | Ph | NHCONH2 | CH3 |
2.2. Chemistry
The synthetic procedures carried out to obtain the target compounds containing the pyridazinone scaffold are reported in Scheme 1, Scheme 2, Scheme 3, Scheme 4, Scheme 5, Scheme 6, Scheme 7, Scheme 8 and Scheme 9. The structures were confirmed on the basis of analytical and spectral data. Scheme 1 shows the synthetic pathway affording the final compounds 4a,b, 5a,b, 6 and 7. Intermediate 2 [34] was obtained starting from isoxazole-pyridazinone 1, synthesized by adopting previously reported protocols [29,30,31,32] and using methanol and triethylamine for opening the isoxazole nucleus. The subsequent hydrolysis (acid 3 [35]) and acylation with thionyl chloride, triethylamine and appropriate amine led to final compounds 4a,b. Products 5a,b were obtained from alkylation reaction of 4a,b with ethyl bromide in standard conditions. The opening of isoxazole core of the starting material 1 with 33% NH4OH afforded to amide 6 [28] which, by subsequent dehydration with POCl3, led to compound 7. The synthesis of final compounds 16–18 is reported in Scheme 2. The reaction between sodium salt of diketone 8 with the commercially available ethyl chloro(hydroximino)acetate 9 in ethanol led to a mixture of isomers 10 and 11 [36] that were cyclized to isoxazole-pyridazinone 12 and 13 using phenylhydrazine and PPA. After chromatographic separation, the latter were subjected to a series of reactions to obtain the compounds 16–18. The treatment of intermediate 13 with ammonium formate and Pd/C provided compound 18, while the treatment of 12 with methanol and triethylamine led to pyridazinone 14. Intermediate 14 was first hydrolyzed to acid (15), then converted to amide (16) and finally treated with POCl3 to obtain the cyano derivative 17. The final compounds 21 and 22 were obtained through a procedure similar to that shown in Scheme 1 for amide derivative 6 and cyano derivative 7, using intermediate 20 as the starting material, which was obtained by reaction of cyclohexyl hydrazine and PPA with isoxazole 19 [34] (see Scheme 3). Scheme 4 reports the synthesis of the pyridazinone-based derivatives of type 24 and 25 (unsubstituted at position 5), compound 28 and the thio-derivative 27. Intermediate 23 [37] was reacted with the appropriate brominated alkylating agent in presence of potassium carbonate and dry DMF to afford 24a–f derivatives (24a, [38]; 24c, [34]). The formation of urea derivatives of type 25 was carried out using sodium acetate and triphosgene in dry THF at reflux, and then treated with ammonia. The urea 28 was directly obtained from intermediate 23 using the same conditions used for compound type 25. The transformation of the carbonyl (C=O) in thiocarbonyl group (C=S) was carried out using the Lawesson’s reagent in toluene (26) and the subsequently alkylation with methyl iodide in standard condition led to the thio derivative 27. In Scheme 5 and Scheme 6 are reported the synthetic procedures of other un-substituted pyridazinones at position 5, but bearing different groups/functions at position 4 and 6. In particular, Scheme 5 depicts the synthetic pathways for compounds with a phenyl ring at position 6 and a methyl group at N-2, while different substituents are introduced at position 4. Starting from compound 24a [38] (Scheme 4), the amino group at position 4 was acylated using the suitable anhydride in pyridine in a sealed/pressure vessel to obtain the final compounds 29a–d. Moreover, the same amino group was also subjected to a coupling reaction using the appropriate R-phenylboronic acid in presence of copper (II) acetate and triethylamine to furnish the derivatives 30a,b and 33. The substituent R on the phenyl at position 4 was further elaborated. The m/o-CN group of compounds 30a,b was converted into m/o-CONH2 (compounds 31a,b, respectively) with 80% sulfuric acid under reflux. The 4-carbethoxy function in product 33 was firstly hydrolyzed to acid 34, converted into the corresponding acid chloride with thionyl chloride and then acylated with 1-acetylpiperazine (compound 35). Lastly, the carbonyl group of intermediate 24a was converted in thiocarbonyl (32) using the same procedure discussed in Scheme 4. In Scheme 6 are depicted pyridazinone-based derivatives with a methyl group or a hydrogen at N-2, an amino group or urea functionality at position 4, but bearing different groups e/o functions (e.g., R-phenyl, alkyl, cycloalkyl) at position 6. Starting from commercially available intermediates 36a–f, the introduction of an amino group at position 4 with hydrazine hydrate at high temperature led to compounds 37a–e (37e, [38]) and the subsequent alkylation with methyl iodide provided products 38a–d. The derivatives 39a,b and 40 were obtained from reaction with triphosgene and ammonia in the same conditions reported in Scheme 4, starting from 38b,c and 37e, respectively. The direct alkylation of the intermediates 36e and 36f afforded the corresponding N-methyl derivatives 41a,b (41a, [39]), which were subsequently converted into compounds 42a,b through the same reaction used to obtain 37a–e. In particular, the reaction conditions used to introduce an amino group at position 4 led also to the reduction in the nitro group in compound 42b. The latter was subjected to acylation reaction with acetyl chloride to obtain product 44. Instead, intermediate 42a was subjected to triphosgene treatment to obtain the urea derivative 43. Scheme 7 reports the synthesis of final compounds 48 and 49. Intermediate 47 was obtained starting from isoxazole 45, previously synthesized by us [34] by cyclization reaction with methyl hydrazine (46) and subsequent opening of the isoxazole ring with ammonium formate and palladium on carbon. The deacetylation (on 47) with 48% bromic acid at high temperature led to compound 48, which was subsequently treated with triphosgene and ammonia to obtain the urea derivative 49. Compound 51 was obtained through alkylation reaction using standard conditions [40], but starting from product 50 [41] (Scheme 8). Lastly, the final compound 55 (Scheme 9) was obtained starting from intermediate 52 [42], through the same reactions of isoxazole nucleus opening (53 [42]), deacetylation (54 [43]) and formation of the urea function. In Scheme 9 is also illustrated the treatment of 52 with dimethylformamide dimethyl acetal to generate intermediate 56, which was subsequently converted into compound 57 using hydrazine hydrate.
Scheme 1.

Reagents and conditions: (a) Et3N, CH3OH, 60 °C, 2 h; (b) 6N NaOH, EtOH, reflux, 30 min; (c) (i) SOCl2, Et3N, r.t., 30 min; (ii) R-NH2, anhydrous THF, r.t., 2 h; (d) CH3CH2Br, K2CO3, anhydrous DMF, reflux, 30–90 min; (e) 33% NH4OH, C5H11N, 60 °C, 90 min; (f) POCl3, 60 °C, 2h.
Scheme 2.

Reagents and conditions: (a) anhydrous EtOH, 0 °C, 1h; (b) phenylhydrazine, PPA, EtOH, 70 °C, 30 min; (c) CH3OH, Et3N, 60 °C, 2h; (d) NaOH, EtOH, reflux, 30 min; (e) (i) SOCl2, Et3N, reflux, 30 min.; (ii) 33% NH4OH, anhydrous THF, r.t., 15 min; (f) POCl3, 60 °C, 2h; (g) HCOONH4, 10% Pd/C, EtOH, reflux, 2 h.
Scheme 3.

Reagents and conditions: (a) cyclohexylhydrazine, PPA, EtOH, 70 °C, 30 min; (b) 33% NH3, piperidine, 60 °C, 90 min; (c) POCl3, 60 °C, 2h.
Scheme 4.

Reagents and conditions: (a) suitable R-Br, K2CO3, anhydrous DMF, reflux, 1–4 h; (b) (i) dry THF, CH3COONa, 0 °C then triphosgene, reflux, 2 h; (ii) NH3 33%, 0 °C, 1 h; (c) Lawesson’s reagent, anhydrous toluene, reflux, 5 h.
Scheme 5.

Reagents and conditions: (a) suitable (R-CO2)O, anhydrous C6H5N, closed tube, 140 °C, 5 h; (b) 2/3-cyanophenylboronic acid (for 30a,b) or 4-ethoxycarbonylphenylboronic acid (for 33), Cu(Ac)2, Et3N, dry CH2Cl2, r.t., 12 h; (c) H2SO4 80%, 80 °C, 4 h; (d) Lawesson’s reagent, anhydrous toluene, reflux, 10 h; (e) NaOH 6N, EtOH 96%, reflux, 1 h; (f) (i) SOCl2, Et3N (catalytic), reflux, 1 h; (ii) anhydrous THF, 1-acetylpiperazine, 0 °C then r.t., 1 h.
Scheme 6.

Reagents and conditions: (a) NH2NH4·H2O, sealed/pressure vessel, 180 °C, 12 h; (b) CH3I, K2CO3, anhydrous DMF, 80 °C, 2–4 h; (c) (i) anhydrous THF, CH3COONa, 0 °C then triphosgene, reflux, 2 h; (ii) NH4OH 33%, 0 °C, 1 h; (d) ClCOCH3, anhydrous THF, 0 °C, then r.t., 20 min.
Scheme 7.

Reagents and conditions: (a) CH3(NH)NH2, EtOH 96%, r.t., 2 h; (b) HCOONH4, Pd/C, EtOH 96%, reflux, 2h; (c) HBr 48%, sealed/pressure vessel, 130 °C, 3 h; (d) (i) anhydrous THF, CH3COONa, 0 °C then triphosgene, reflux, 2 h; (ii) 33% NH4OH, 0 °C, 1 h.
Scheme 8.

Reagents and conditions: (a) CH3I, K2CO3, anhydrous DMF, 80 °C, 2 h.
Scheme 9.

Reagents and conditions: (a) HCOONH4, Pd/C 10%, EtOH 96%, reflux, 2 h; (b) HBr 48%, sealed/pressure vessel, 130 °C, 3 h; (c) (i) anhydrous THF, CH3COONa, 0 °C then triphosgene, reflux, 2 h; (ii) NH4OH 33%, 0 °C, 1 h; (d) DMF-DMA, 90 °C, 1 h; (e) NH2NH4·H2O, anhydrous EtOH, 70 °C, 10 h.
Based on the analytical and spectral data (proton and carbon NMR) and mass spectrometry (MS), all the new compounds confirmed the predicted chemical structures, as well as satisfactory results in terms of formulation and purity (See Section 3; in Supporting Information are reported representative examples of analytical characterization data of the compounds processed to FABP4 inhibition assay in vitro). Reversed phase liquid chromatography was used to perform a qualitative analysis of the dataset’s purity. The formation of the products was monitored by UV absorbance at wavelengths of 281 nm and 254 nm. The retention times range was from 6 to 17 min (See Section 3 and Supporting Information). The overall feature of mass spectra (LC-MS) of this series of pyridazinone-derivatives is the presence of a predominant peak corresponding to the molecular ion [M + H]+ (See Section 3 and Supporting Information).
2.3. FABP4 Inhibition Evaluation
FABP4 inhibitory activity was assessed by measuring the decrease in fluorescent signal of a detection reagent (DR) when displaced by a strong FABP4 ligand. Specifically, the DR exhibits an increased fluorescence intensity when bound to FABP4. Therefore, any effective ligand of the protein, which binds to the same binding pocket and can displace the DR, determines a reduction in the fluorescence read-out. The new molecular series was screened in a two-step procedure. Firstly, a single concentration of 5 µM was used to gain an estimation of the overall inhibitory effect of all the molecules. Subsequently, only the compounds that were able to reduce the fluorescence reading of at least 95% were further evaluated by measuring the IC50 values (µM), which were lastly compared with the activity of the arachidonic acid (i.e., FABP4 established ligand). The single point displacement results are reported in Figure 2. Based on the data of the first screening, 10 molecules were selected as most effective compounds—i.e., able to reduce the fluorescence of the DR to at least 95%, for which the IC50 (µM) was calculated. Arachidonic acid was used as a positive control, resulting with an IC50 of 3.42 µM. The IC50 values of our set of compounds are reported in Table 3. Compound 25a demonstrated a potent inhibitory activity, with an IC50 value (i.e., 2.97 µM) lower than the reference arachidonic acid.
Figure 2.

Single point displacement experiment for selected compounds.
Table 3.
Measured IC50 values for selected compounds.
| Compounds | IC50 (µM) |
|---|---|
| Arachidonic acid | 3.42 ± 0.54 |
| 4b | 8.27 ± 0.20 |
| 25a | 2.97 ± 0.26 |
| 30b | 23.18 ± 0.52 |
| 22 | 15.23 ± 0.76 |
| 25c | >50 |
| 35 | >50 |
| 25e | >50 |
| 54 | >50 |
| 55 | >50 |
| 27 | >50 |
2.4. Molecular Modelling Studies
Since the first apo-FABP crystal structure was published in 1992, many other holo-FABP structures with a variety of ligands have been solved. The hydrophobic pocket side chains engage a hydrogen bond to the carboxylate of FAs toward several amino acids. Moreover, a network of water molecules may be involved in mediating these interactions. The docking experiments of the molecular series compounds were conducted on the most active compounds 4b, 25a, 30b, and 22. Figure 3 shows the 2D binding interactions for the molecules, while Figure 4 displays the predicted poses inside the binding pocket of FABP4. All the compounds are able to engage several interactions with relevant residues in the binding pocket, such as R126 and Y128, as well as R106. R126 can interact with both the carbonyls of the most potent compound 25a, that also interacts directly with Y128 and, through the network of water molecules, with S53. The 4b is well allocated inside the binding pocket and is engaging a strong H-bond interaction with R126. Differently, compound 22 is not suitably allocated inside the pocket to generate appropriate binding with R126 and Y128 and most of the stabilizing interactions are due to pi–pi stacking with A75, F16 and M20. Lastly, the -CN group of 30b results responsible of the stabilizing interaction with R126 and Y128, that are likely to account for the lower activity of the compound, as determined by the lower binding interaction for this group with the residues.
Figure 3.

(a) 2D interaction between 4b and FABP4. (b) 2D interaction between 25a and FABP4. (c) 2D interaction between 30b and FABP4. (d) 2D interaction between 22 and FABP4.
Figure 4.

Docked poses inside FABP4 of molecules 4b (green), 25a (blue), 30b (dark yellow) and 22 (light red).
3. Experimental Section
3.1. General Remarks
All the chemical reagents were purchased from Merk and Sigma Aldrich of reagent grade and were used without any further purification. Extracts were dried over Na2SO4 and the solvents were removed under reduced pressure. All reactions were monitored by thin-layer chromatography (TLC) using commercial plates (Merck) pre-coated with silica gel 60 F-254. Visualization was performed by UV fluorescence (λmax = 254 nm) or by staining with iodine or potassium permanganate. Chromatographic separations were performed on silica gel columns by gravity (Kieselgel 40, 0.063–0.200 mm; Merck) or flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. When reactions were performed in anhydrous conditions, the mixtures were maintained under nitrogen atmosphere. Compounds were named following IUPAC rules as applied by Beilstein-Institut AutoNom 2000 (4.01.305) or CA Index Name. All melting points were determined on a microscope hot stage Büchi apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were obtained on a Bruker AVANCE 400 spectrometer at 400 MHz and 100 MHz, respectively, using 5 mm i.d. glass tubes. Chemical shifts (δ) values are expressed as parts per million (ppm) using DMSO (d6) (2.50 for proton and 39.52 for carbon), methanol (d4) (3.31 for proton and 49.00 for carbon) or CDCl3 (7.26 for proton and 77.16 for carbon) as solvents. The coupling constants (J) are reported in Hz. The following splitting patterns are identified: s, singlet; d, doublet; t, triplet; m, multiplet; or any combination of these e.g., dd, dt, etc. Analytical reversed-phase high performance liquid chromatography (reversed-phase HPLC) was conducted out on HP 1050 instrument (Agilent Technologies, Waldbronn, Germany) to ascertain the chromatographic purity of compounds. The system includes a quaternary pump, an autosampler, and a Kontron DEG 104 degasser (Kontron, Tokyo, Japan). A C18 column, Zorbax,80 Å, 3.5 μm, 2.1 × 100 mm was used with a total run time of 30 min. The mobile phase is composed of 0.1% Trifluoro acetic acid (TFA) in Milli-Q H2O and Acetonitrile (can) at a flow rate of 0.3 mL/min with an injection volume of 10–30 μL [44]. The compounds were detected at 281 nm and 254 nm UV wavelengths. The values of the retention times (tR) are given in minutes. Mass spectrometry (LC-MS) experiments were performed on all the samples. The stock solutions (1 mg/mL in MeOH) where diluted with 0.1% HCOOH in MeOH/H2O (50:50) to a final concentration of 50 µg/mL prior to analysis. The instrument used consisted of a Thermo Accela LC system interfaced to a Thermo TSQ Access triple quadrupole mass spectrometer with a HESI source. The data were processed with Xcalibur software (version 2.0). An amount of 10 µL of sample was analyzed in flow injection, with a flow rate of 0.2 mL/min of mobile phase 0.1% HCOOC in MeOH/H2O (50:50). Parameters used for the analysis in positive ion mode were: spray voltage 3500 V; vaporizer temperature 300 °C; sheath gas pressure 50 au; capillary temperature 350 °C; capillary offset 35.
3.2. Chemistry
3.2.1. General Procedure for Compounds 4a,b
A mixture of 3 (0.35 mmol) [35], a catalytic amount of Et3N (0.1 mL) and SOCl2 (9.35 mmol) was stirred at room temperature for 30 min. Then the excess of SOCl2 was removed in vacuo and the residue oil was dissolved in cold anhydrous THF (1 mL). To this suspension, the appropriate amine (0.75 mmol) was added and the mixture was stirred at room temperature for 2 h. After cooling, cold water was added (2–5 mL) and the suspension was extracted with CH2Cl2 (3 × 15 mL); the solvent was evaporated under vacuum to afford the desired final compounds, which were purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 as eluent (4a), or by crystallization from ethanol (4b).
5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid phenylamide (4a)
Yield = 40%; mp = 228–229 °C (EtOH). Light brown solid, 1H NMR (400 MHz, DMSO-d6) δ 6.57 (s, 2H, NH2), 7.02 (t, 1H, J = 7.4 Hz, ArCONH), 7.23 (t, 2H, J = 7.8 Hz, Ar), 7.32 (d, 2H, J = 7.3 Hz, Ar), 7.39 (d, 2H, J = 8.0 Hz, Ar), 7.47–7.49 (m, 2H, Ar), 10.04 (s, 1H, CONH2), 12.88 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ163.75, 156.35, 155.43, 145.54, 141.87, 138.61, 128.43, 128.24, 127.93, 123.86, 119.95, 109.88. MS-ESI for C17H14N4O2 (Calcd, 306.11), [M + H]+ at m/z 306.96, tR = 11.825. Anal. Calcd for C17H14N4O2: C, 66.66; H, 4.61; N, 18.29. Found C, 66.92; H, 4.63; N, 18.36.
5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid propylamide (4b)
Yield = 35%; mp = 228–230 °C (EtOH). Yellow coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 0.57–0.61 (m, 2H, CH3), 1.18 (dp, 2H, J = 14.2, 7.2 Hz, CH2), 2.94 (q, 2H, J = 6.8 Hz, NHCH2), 6.31 (s, 2H, NH2), 7.36 (dt, 2H, J = 4.5, 1.6 Hz, Ar), 7.44 (dq, 2H, J = 6.4, 1.7 Hz, Ar), 7.98 (t, 1H, J = 5.8 Hz, Ar), 12.79 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ 164.94, 156.30, 145.52, 141.61, 137.07, 128.17, 127.90, 127.80, 110.26, 40.55, 21.51, 11.24. MS-ESI for C14H16N4O2 (Calcd, 272.13), [M + H]+ at m/z 273.02, tR = 9.970. Anal. Calcd for C14H16N4O2: C, 61.75; H, 5.92; N, 20.58. Found C, 61.99; H, 5.94; N, 20.66.
3.2.2. General Procedure for Compounds 5a,b
A mixture of 4a,b (0.43 mmol), K2CO3 (0.86 mmol) and 0.50 mmol of ethyl bromide in anhydrous DMF (2 mL) was refluxed for 30–90 min. After cooling, the mixture was diluted with cold water (15 mL) and compound 5a was recovered by filtration under vacuum. For compound 5b the suspension was extracted with CH2Cl2 (3 × 15 mL) and the solvent was evaporated in vacuo. The crude products were purified by crystallization from ethanol.
5-Amino-1-ethyl-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid phenylamide (5a)
Yield = 90%; mp = 172–173 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.42 (t, 3H, CH3CH2, J = 7.2 Hz), 4.26 (q, 2H, CH3CH2, J = 7.2 Hz), 6.70 (exch br s, 1H, CONH), 6.93 (d, 2H, Ar, J = 8.0 Hz), 7.04 (t, 1H, Ar, J = 8.0 Hz), 7.20 (t, 2H, Ar, J = 8.0 Hz), 7.49–7.54 (m, 3H, Ar), 7.55–7.60 (m, 2H, Ar). Anal. Calcd for C19H18N4O2: C, 68.25; H, 5.43; N, 16.76. Found C, 68.41; H, 5.44; N, 16.72.
5-Amino-1-ethyl-6-oxo-3-phenyl-1,6-dihydro-pyridazine-4-carboxylic acid propylamide (5b)
Yield = 80%; mp = 141–143 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 0.64 (t, 3H, NH-CH2CH2CH3, J = 7.2 Hz), 1.15 (sex, 2H, NH-CH3CH2CH2, J = 7.6 Hz), 1.43 (t, 3H, N-CH2CH3, J = 7.2 Hz), 3.05 (q, 2H, NH-CH2CH2CH3, J = 7.2 Hz), 4.25 (q, 2H, N-CH2CH3, J = 7.2 Hz), 5.02 (exch br s, 1H, CONHCH2), 6.95 (exch br s, 2H, NH2), 7.45–7.51 (m, 5H, Ar). Anal. Calcd for C16H20N4O2: C, 63.98; H, 6.71; N, 18.65. Found C, 63.83; H, 6.70; N, 18.70.
3.2.3. 5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid amide (6)
A mixture of isoxazolopyridazinone 1 (0.94 mmol) [29], 2 mL of 33% NH3 and a catalytic amount of piperidine was stirred at 60 °C for 90 min in a sealed/pressure vessel. After cooling the precipitate was recovered by suction and recrystallized with diethyl ether. Yield = 46%; mp > 300 °C (Et2O). Light brown solid, 1H NMR (400 MHz, DMSO-d6) δ 6.38 (s, 2H, NH2), 7.38 (dd, 3H, Ar, J = 5.0, 2.1 Hz), 7.47–7.49 (m, 2H, Ar), 12.79 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ 167.25, 156.28, 145.36, 141.66, 137.17, 128.17, 127.97, 127.83, 109.75. MS-ESI for C11H10N4O2 (Calcd, 230.08), [M + H]+ at m/z 230.95, tR = 6.091. Anal. Calcd for C11H10N4O2: C, 57.39; H, 4.38; N, 24.34. Found C, 57.16; H, 4.36; N, 24.24.
3.2.4. 5-Amino-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carbonitrile (7)
A suspension of 6 (0.40 mmol) in POCl3 (8 mmol) was stirred at 60 °C for 1–2 h. After cooling, the reaction mixture was treated with cold water (15 mL) and the suspension was extracted with CH2Cl2 (3 × 15 mL). The organic solvent was evaporated to afford the desired final compound which was purified by crystallized from diethyl ether. Yield = 48%; mp = 287–289 °C (Et2O). Yellow coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 6.78 (s, 1H, NH2), 7.48 (tt, 3H, J = 3.9, 2.4 Hz, Ar), 7.57–7.61 (m, 2H, Ar), 12.98 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ154.47, 154.06, 149.95, 145.72, 135.21, 129.28, 128.29, 128.13, 115.68, 113.42. MS-ESI for C11H8N4O (Calcd, 212.07), [M + H]+ at m/z 212.89, tR = 10.234. Anal. Calcd for C11H8N4O: C, 62.26; H, 3.80; N, 26.40. Found C, 62.01; H, 3.78; N, 26.29.
3.2.5. 4-Acetyl-isoxazole-3-carboxylic acid ethyl ester (10)
To a cooled (−5 °C) and stirred suspension of 8 (9.9 mmol) in anhydrous ethanol, a solution of ethyl chloro(hydroximino)acetate 9 (6.6 mmol) in the same solvent (11 mL) was added dropwise. The solvent was evaporated in vacuo, cold water was added (10 mL) and the suspension was extracted with CH2Cl2 (3 × 15 mL). A mixture of isoxazoles 10 and 11 [36] was obtained and they were separated by flash column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield = 15%; oil. 1H NMR (400 MHz, CDCl3) δ 1.45 (t, 3H, CH2CH3, J = 7.2 Hz), 2.55 (s, 3H, COCH3), 4.51 (q, 2H, CH2CH3, J = 7.2 Hz), 8.95 (s, 1H, Ar). Anal. Calcd for C8H9NO4: C, 52.46; H, 4.95; N, 7.65. Found C, 52.33; H, 4.94; N, 7.67.
3.2.6. General Procedure for Compounds 12 and 13
To a cooled and stirred mixture of isoxazoles 10 or 11 (6.56 mmol) and 2.5 g of PPA (25 mmol) in 2 mL of anhydrous EtOH, 7.87 mmol of phenylhydrazine were added. The reaction was carried out at 70 °C for 30 min. After cooling the solvent was evaporated under vacuum, cold water was added (10 mL) and the suspension was extracted with CH2Cl2 (3 × 15 mL). Evaporation of the solvent afforded the desired compounds.
4-Methyl-6-phenyl-6H-isoxazolo [3,4-d]pyridazin-7-one (12)
Yield = 90%; mp = 200–201 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.54 (s, 3H, CH3), 7.38 (t, 1H, Ar, J = 7.6 Hz), 7.45–7.50 (m, 2H, Ar), 7.55–7.60 (m, 2H, Ar), 9.22 (s, 1H, C=CH). Anal. Calcd for C12H9N3O2: C, 63.43; H, 3.99; N, 18.49. Found C, 63.58; H, 4.00; N, 18.44.
3-Methyl-6-phenyl-6H-isoxazolo [3,4-d]pyridazin-7-one (13)
Yield = 80%; mp = 188–190 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.88 (s, 3H, CH3), 7.42 (t, 1H, Ar, J = 7.4 Hz), 7.51 (t, 2H, Ar, J = 7.8 Hz), 7.60 (d, 2H, Ar, J = 7.6 Hz), 8.16 (s, 1H, N=CH). Anal. Calcd for C12H9N3O2: C, 63.43; H, 3.99; N, 18.49. Found C, 63.55; H, 3.99; N, 18.46.
5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carboxylic acid methyl ester (14)
A mixture of 12 (6.21 mmol) and Et3N (0.8 mL) in 2 mL of CH3OH was heated at 60 °C for 2 h. After cooling, ice water (20 mL) was added and the suspension was extracted with CH2Cl2 (3 × 15 mL). Then the solvent was evaporated in vacuo to afford compound 14 which was purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 80%; mp = 91–93 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.53 (s, 3H, CH3), 3.95 (s, 3H, COOCH3), 7.39 (t, 1H, Ar, J = 7.4 Hz), 7.49 (t, 2H, Ar, J = 8.4 Hz), 7.65 (d, 2H, Ar, J = 8.4 Hz), 8.16 (exch br s, 2H, NH2). Anal. Calcd for C13H13N3O3: C, 60.22; H, 5.05; N, 16.21. Found C, 60.08; H, 5.06; N, 16.26.
3.2.7. 5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carboxylic acid (15)
A mixture of 14 (2.12 mmol), ethanol (3 mL) and 6N NaOH (2 mL) was stirred at reflux for 30 min. After cooling, the solvent was evaporated under vacuum, cold water was added (2–3 mL) and the mixture was acidified with 6N HCl. The precipitate was recovered by vacuum filtration and crystallized from cyclohexane. Yield = 90%; mp = 214–216 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 2.37 (s, 3H, CH3), 7.39 (t, 1H, Ar, J = 7.2 Hz), 7.46 (t, 2H, Ar, J = 8.0 Hz), 7.54 (d, 2H, Ar, J = 8.0 Hz), 8.25 (exch br s, 2H, NH2). Anal. Calcd for C12H11N3O3: C, 58.77; H, 4.52; N, 17.13. Found C, 58.61; H, 4.51; N, 17.16.
3.2.8. 5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carboxylic acid amide (16)
A mixture of 15 (1.88 mmol), a catalytic amount of Et3N (0.1 mL) and SOCl2 (51 mmol) was refluxed for 30 min. After cooling, the excess of SOCl2 was removed in vacuo and the residue oil was dissolved in cold dry THF (1 mL). To this suspension a solution of 33% NH3 (2 mL) in 1.5 mL of dry THF was added and the mixture was stirred at room temperature for 15 min. After evaporation of the solvent, the mixture was diluted with cold water (20 mL) and the precipitate obtained was filtered and crystallized from ethanol. Yield = 80%; mp = 247–249 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 2.21 (s, 3H, CH3), 7.35–7.40 (m, 1H, Ar), 7.45–7.51 (m, 5H, Ar), 7.65 (s, 1H, NH2), 7.88 (s, 1H, CONH2). 13C NMR (100 MHz, DMSO-d6) δ 167.39, 155.53, 143.12, 141.99, 141.63, 128.82, 127.99, 125.85, 110.42, 20.24. MS-ESI for C12H12N4O2 (Calcd, 244.10), [M + H]+ at m/z 244.95, tR = 7.921. Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94. Found C, 59.24; H, 4.97; N, 23.03.
3.2.9. 5-Amino-3-methyl-6-oxo-1-phenyl-1,6-dihydropyridazine-4-carbonitrile (17)
Compound 17 was obtained starting from compound 16, through the same procedure described for 7. After dilution with cold water, the precipitate was recovered by filtration under vacuum and the solid obtained was purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 20%; mp = 201–203 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.26 (s, 3H, CH3), 7.38 (ddd, 1H, J = 7.7, 5.5, 3.6 Hz, Ar), 7.42–7.51 (m, 4H, Ar). 13C NMR (100 MHz, DMSO-d6) δ 153.37, 149.50, 144.17, 141.60, 130.03, 127.68, 125.90, 115.14, 100.83, 20.47. MS-ESI for C12H10N4O (Calcd, 226.08), 226.96 m/z [M + H]+, 435.11 m/z [2M+H-H2O]+, 451.07 m/z [2M-H2+H]+. tR =11.385. Anal. Calcd for C12H10N4O: C, 63.71; H, 4.46; N, 24.76. Found C, 63.96; H, 4.48; N, 24.85.
3.2.10. 5-Acetyl-4-amino-2-phenylpyridazin-3(2H)-one (18)
Intermediate 13 (1.01 mmol) was suspended in 3.5 mL of EtOH, then 6.08 mmol of HCOONH4 and 40 mg of 10% Pd/C were added. The mixture was refluxed for 2 h and after cooling, CH2Cl2 (5 mL) was added. The solution was stirred for 5 min, then the catalyst was filtered off and the solvent was evaporated in vacuo to furnish desiderd compound 18. Yield = 98%; mp = 181–183 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.60 (s, 3H, COCH3), 6.95 (exch br s, 1H, NH2), 7.42 (t, 1H, Ar, J = 7.6 Hz), 7.51 (t, 2H, Ar, J = 7.6 Hz), 7.64 (d, 2H, Ar, J = 7.6 Hz), 8.13 (s, 1H, C6-H), 9.15 (exch br s, 1H, NH2). Anal. Calcd for C12H11N3O2: C, 62.87; H, 4.84; N, 18.33. Found C, 62.69; H, 4.83; N, 18.28.
3.2.11. 6-Cyclohexyl-4-phenyl-6H-isoxazolo [3,4-d]pyridazin-7-one (20)
Compound 20 was obtained starting from 19 [34] adopting the general procedure described for compounds 12 and 13, but using cyclohexyl hydrazine as reagent. The mixture was heated at 70 °C for 5 h. After dilution with ice-water, the precipitate was recovered by filtration under vacuum and crystallized from ethanol. Yield = 45%; mp = 211–213 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.27–1.33 (m, 1H, C6H11), 1.45–1.51 (m, 3H, C6H11), 1.75–1.80 (m, 1H, C6H11), 1.85–1.95 (m, 5H, C6H11), 5.05–5.10 (m, 1H, C6H11), 7.50–7.60 (m, 3H, Ar), 7.85 (d, 2H, Ar, J = 7.2 Hz), 9.30 (s, 1H, isoxazole). Anal. Calcd for C17H17N3O2: C, 69.14; H, 5.80; N, 14.23. Found C, 69.33; H, 4.82; N, 18.28.
3.2.12. 5-Amino-1-cyclohexyl-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carboxylic acid amide (21)
A mixture of 20 (0.64 mmol) and 33% NH3 was stirred at 120 °C for 3 h in a sealed/pressure vessel. After cooling, ice-water was added and the suspension was extracted with CH2Cl2 (3 × 15 mL). Evaporation of the solvent afforded the desired final compound. Yield = 20%; mp = 125–128 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 1.20–1.25 (m, 1H, C6H11), 1.35–1.48 (m, 2H, C6H11), 1.60–1.65 (m, 2H, C6H11), 1.70–1.88 (m, 5H, C6H11), 4.77–4.82 (m, 1H, C6H11), 6.55 (exch br s, 2H, NH2), 7.25–7.31 (m, 3H, Ar), 7.44 (d, 2H, Ar, J = 7.6 Hz), 8.50 (exch br s, 2H, CONH2). Anal. Calcd for C17H20N4O2: C, 65.37; H, 6.45; N, 17.94. Found C, 65.52; H, 6.46; N, 17.99.
3.2.13. 5-Amino-1-cyclohexyl-6-oxo-3-phenyl-1,6-dihydropyridazine-4-carbonitrile (22)
Compound 22 was obtained starting from compound 21, through the same procedure described for 7 and 17. After dilution with cold water, the precipitate was recovered by suction and the solid was purified by crystallization from etanol. Yield = 90%; mp = 170–172 °C (EtOH). Yellow coloured solid, 1H NMR (400 MHz, CDCl3) δ 1.23 (qt, 1H, J = 13.2, 3.3 Hz, CH2-cyclohexane), 1.46 (ttt, 2H, J = 13.8, 7.8, 3.0 Hz, CH2-cyclohexane), 1.71 (dt, 1H, J = 13.3, 3.4 Hz, CH2-cyclohexane), 1.84–1.91 (m, 6H, J = 4.4, 3.5 Hz, CH2-cyclohexane), 4.84–4.93 (m, 1H, CH2-cyclohexane), 7.45–7.50 (m, 3H, Ar), 7.72–7.74 (m, 2H, Ar). 13C NMR (100 MHz, CDCl3) δ 152.65, 148.93, 144.34, 135.02, 129.91, 128.75, 128.20, 114.83, 85.25, 58.19, 30.99, 25.60. MS-ESI for C17H18N4O (Calcd, 294.15), [M + H]+ at m/z 294.99, [M + ACN + H]+ at m/z 336.01, tR = 17.509. Anal. Calcd for C17H18N4O: C, 69.37; H, 6.16; N, 19.03. Found C, 69.09; H, 6.13; N, 18.95.
3.2.14. General Procedure for 24b, 24d-f
A mixture of 23 [37] (0.80 mmol), K2CO3 (1.60 mmol) and 0.96–1.44 mmol of the appropriate alkyl or cycloalkyl bromide in anhydrous DMF (1 mL) was refluxed for 2–4 h. After cooling, the mixture was diluted with cold water (20 mL) and extracted with CH2Cl2 (3 × 15 mL). Evaporation of the solvent afforded the desired final compounds which were purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 (for 24b,e,f) or 1:2 (for 24d) as eluent.
4-Amino-2-cyclohexyl-6-phenylpyridazin-3(2H)-one (24b)
Yield = 21%; mp = 120–124 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.20–1.35 (m, 1H, C6H11), 1.40–1.50 (m, 2H, C6H11), 1.70–1.80 (m, 1H, C6H11), 1.90–2.05 (m, 6H, C6H11), 4.85–5.10 (m, 3H, 1H C6H11 + 2H NH2), 6.75 (s, 1H, -CH pyridaz.), 7.35–7.50 (m, 3H, Ar), 7.70 (d, 2H, Ar, J = 7.6 Hz). Anal. Calcd for C16H19N3O: C, 71.35; H, 7.11; N, 15.60. Found C, 71.52; H, 7.10; N, 15.56.
4-Amino-2-isopropyl-6-phenylpyridazin-3(2H)-one (24d)
Yield = 85%; mp = 122–124 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.45 (d, 6H, CH(CH3)2, J = 6.8 Hz), 4.97 (exch br s, 2H, NH2), 5.41 (quin, 1H, CH(CH3)2, J = 6.8 Hz), 6.75 (s, 1H, -CH pyridaz.), 7.40–7.50 (m, 3H, Ar), 7.81 (d, 2H, Ar, J = 7.6 Hz). Anal. Calcd for C13H15N3O: C, 68.10; H, 6.59; N, 18.33 Found C, 68.31; H, 6.60; N, 18.29.
4-Amino-6-phenyl-2-propylpyridazin-3(2H)-one (24e)
Yield = 83%; mp = 79–81 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.02 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.93 (sex, 2H, CH2CH2CH3, J = 7.2 Hz), 4.23 (t, 2H, CH2CH2CH3, J = 7.2 Hz), 4.99 (exch br s, 2H, NH2), 6.73 (s, 1H, -CH pyridaz.), 7.38–7.50 (m, 3H, Ar), 7.78 (d, 2H, Ar, J = 8.0 Hz). Anal. Calcd for C13H15N3O: C, 68.10; H, 6.59; N, 18.33 Found C, 68.28; H, 6.60; N, 18.31.
4-Amino-2-butyl-6-phenylpyridazin-3(2H)-one (24f)
Yield = 94%; mp = 67–69 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.00 (t, 3H, CH2CH2CH2CH3, J = 7.2 Hz), 1.45 (m, 2H, CH2CH2CH2CH3), 1.87 (m, 2H, CH2CH2CH2CH3), 4.26 (t, 2H, CH2CH2CH2CH3, J = 7.2 Hz), 4.99 (exch br s, 2H, NH2), 6.75 (s, 1H, -CH pyridaz.), 7.38–7.50 (m, 3H, Ar), 7.76 (d, 2H, Ar, J = 8.0 Hz). Anal. Calcd for C14H17N3O: C, 69.11; H, 7.04; N, 17.27 Found C, 69.29; H, 7.03; N, 17.32.
3.2.15. General Procedure for Compounds 25a–f
To a cooled (0 °C) and stirred suspension of the appropriate pyridazinone 24a–f (0.65 mmol) in anhydrous THF (1–3 mL), anhydrous sodium acetate (1.55 mmol) and triphosgene (2.26 mmol) were added. The mixture was stirred for 10 min at room temperature and refluxed for 2 h. Then, the suspension was cooled to 0 °C and 1 mL of 33% NH3 was added and the mixture was stirred for 30–90 min at room temperature. After evaporation of the solvent, ice/cold water was added (15 mL) and the precipitate obtained was recovered by filtration under vacuum and purified by crystallization from ethanol to obtain the pure samples of 25a–f.
(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25a)
Yield = 65%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.77 (s, 3H, CH3), 7.42–7.51 (m, 3H, Ar), 7.73–7.76 (m, 2H, Ar), 8.35 (s, 1H, Ar), 8.98 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.69, 155.11, 145.12, 137.92, 135.75, 129.39, 129.09, 126.02, 106.62, 20.93. MS-ESI for C12H12N4O2 (Calcd, 244.10), [M + H]+ at m/z 244.95, tR = 11.531. Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94 Found C, 59.24; H, 4.97; N, 23.03.
(2-Cyclohexyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25b)
Yield = 35%; mp = 261–263 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.18–1.31 (m, 1H, C6H11), 1.40–1.51 (m, 2H, C6H11), 1.64–1.72 (m, 1H, C6H11), 1.70–1.90 (m, 6H, C6H11), 4.87 (m, 1H, C6H11), 6.80 (exch br s, 2H, NH2), 7.45–7.55 (m, 3H, Ar), 7.79 (d, 2H, Ar, J = 7.6 Hz), 8.37 (s, 1H, -CH pyridaz.), 8.96 (exch br s, 1H, NHCONH2). Anal. Calcd for C17H20N4O2: C, 65.37; H, 6.45; N, 17.94. Found C, 65.18; H, 6.46; N, 17.91.
(2-Ethyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25c)
Yield = 25%; mp = 270–271 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 1.33 (t, 3H, J = 7.1 Hz, NCH2CH3), 4.20 (q, 2H, J = 7.2 Hz, NCH2), 6.70 (exch br s, 2H, NHCONH2) 7.47 (dt, 3H, J = 13.1, 7.1 Hz, Ar), 7.73 (dd, 2H, J = 23.5, 7.6 Hz, Ar), 8.34 (s, 1H, Ar), 8.98 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.36, 154.51, 150.89, 145.06, 137.97, 135.75, 128.88, 125.84, 106.29, 47.00, 13.39. MS-ESI for C13H14N4O2 (Calcd, 258.11), [M + H]+ at m/z 259.02, 215.90 m/z [M-CONH2 + H]+. tR = 12.090. Anal. Calcd for C13H14N4O2: C, 60.45; H, 5.46; N, 21.69. Found C, 60.21; H, 5.44; N, 21.60.
(2-Isopropyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25d)
Yield = 68%; mp = 260–263 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 1.36 (d, 6H, J = 6.7 Hz, NCH(CH3)2), 5.24 (q, 1H, J = 6.6 Hz, NCH), 7.47 (dt, 2H, J = 15.9, 7.2 Hz, Ar), 7.67–7.79 (m, 3H, Ar), 8.34 (s, 1H, Ar), 8.95 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.69, 154.31, 144.75, 137.69, 136.13, 132.04, 129.30, 129.08, 127.97, 125.91, 105.93, 49.71, 20.96. MS-ESI for C14H16N4O2 (Calcd, 272.13), [M + H]+ at m/z 272.95, tR = 14.248. Anal. Calcd for C14H16N4O2: C, 61.75; H, 5.92; N, 20.58. Found C, 61.99; H, 5.94; N, 20.66.
(3-Oxo-6-phenyl-2-propyl-2,3-dihydro-pyridazin-4-yl)urea (25e)
Yield = 60%; mp = 273–275 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 0.89 (td, 3H, J = 7.4, 2.7 Hz, NCH2CH2CH3), 1.79 (q, 2H, J = 7.3 Hz, NCH2CH2), 4.13 (t, 2H, J = 7.1 Hz, NCH2), 7.42–7.51 (m, 2H, Ar), 7.69 (d, 1H, J = 5.6 Hz, Ar), 7.73–7.76 (m, 2H, Ar), 8.33 (s, 1H, Ar), 8.96 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.68, 154.88, 145.06, 137.94, 135.87, 132.03, 129.36, 129.07, 128.06, 126.03, 106.37, 53.19, 21.39, 11.13. MS-ESI for C14H16N4O2 (Calcd, 272.13), [M + H]+ at m/z 272.95, 229.90 m/z [M-CONH2 + H]+. tR = 14.037. Anal. Calcd for C14H16N4O2: C, 61.75; H, 5.92; N, 20.58. Found C, 61.99; H, 5.94; N, 20.66.
(2-Butyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (25f)
Yield = 85%; mp = 265–267 °C (EtOH). White solid, 1H NMR (400 MHz, DMSO-d6 + D2O) δ 0.88 (t, 3H, J = 7.4 Hz, CH3), 1.27 (q, 2H, J = 7.3 Hz, CH2CH3), 1.73 (q, 2H, J = 7.2 Hz, CH2CH2CH3), 4.13–4.16 (m, 2H, CH2ArN), 7.41–7.51 (m, 3H, Ar), 7.73–7.75 (m, 2H, Ar), 8.08 (s, 1H, NHCONH2), 8.36 (s, 1H, Ar), 9.10 (s, 2H, CONH2). 13C NMR (100 MHz, DMSO-d6) δ 154.80, 154.60, 137.56, 135.69, 129.20, 128.94, 127.78, 125.79, 118.03, 106.44, 51.11, 29.92, 19.24, 13.53. MS-ESI for C15H18N4O2 (Calcd, 286.14), [M + H]+ at m/z 286.94, tR = 31.162. Anal. Calcd for C15H18N4O2: C, 62.92; H, 6.34; N, 19.57. Found C, 62.66; H, 6.31; N, 19.49.
3.2.16. 4-Amino-6-phenylpyridazine-3(2H)-thione (26)
A mixture of 23 [37] (0.86 mmol) and Lawesson’s reagent (1.71 mmol) in anhydrous toluene (2–3 mL) was heated at 90 °C for 5 h. After cooling the solvent was evaporated under vacuum, cold water was added (10 mL) and the mixture was extracted with CH2Cl2 (3 × 15 mL). Evaporation of the solvent afforded 26 which was purified by flash column chromatography using CH2Cl2/CH3OH 10:1 as eluent. Yield = 63%; mp = 175–178 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 5.77 (exch br s, 2H, NH2), 6.80 (s, 1H, -CH pyridaz.), 7.45–7.55 (m, 3H, Ar), 7.75–7.81 (m, 2H, Ar). Anal. Calcd for C10H9N3S: C, 59.09; H, 4.46; N, 20.67. Found C, 59.23; H, 4.45; N, 20.62.
3.2.17. 3-Methylsulfanyl-6-phenyl-pyridazin-4-ylamine (27)
Compound 27 was obtained, starting from compound 26, through the general procedure described for 24b and 24d–f. After dilution with cold water, the precipitate was recovered by suction and purified by crystallization. Yield = 40%; mp = 168–170 °C (Cyclohexane). Greenish colour solid, 1H NMR (400 MHz, DMSO-d6) δ 2.64 (s, 3H, SCH3), 6.27 (exch br s, 2H, NH2), 7.00 (s, 1H, Ar), 7.46 (dt, 3H, ArH, J = 12.6, 6.9 Hz), 7.90–7.93 (m, 2H, Ar). 13C NMR (100 MHz, DMSO-d6) δ155.31, 147.71, 144.50, 137.20, 129.38, 129.02, 126.51, 102.80, 12.72. MS-ESI for C11H11N3S (Calcd, 217.07), [M + H]+ at m/z 217.86, tR = 9.922. Anal. Calcd for C11H11N3S: C, 60.80; H, 5.10; N, 19.34. Found C, 60.56; H, 5.08; N, 19.26.
3.2.18. (3-Oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)urea (28)
Compound 28 was obtained, starting from 23 [37], through the same procedure described for 25a–f. Yield = 85%; mp >300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 7.41–7.50 (m, 3H, Ar), 7.72–7.75 (m, 2H, Ar), 8.33 (s, 1H, Ar), 8.94 (s, 1H, NHCONH2), 13.21 (s, 1H, ArNH). 13C NMR (100 MHz, DMSO-d6) δ156.00, 155.33, 145.54, 139.57, 138.27, 135.77, 134.50, 128.87, 125.70, 106.97. MS-ESI for C11H10N4O2 (Calcd, 230.08), [M + H]+ at m/z 230.88, t = 10.042. Anal. Calcd for C11H10N4O2: C, 57.39; H, 4.38; N, 24.34. Found C, 57.62; H, 4.39; N, 24.44.
3.2.19. General Procedure for Compounds 29a–d
A mixture of 24a [38] (0.39 mmol) and the appropriate R-anhydride (13.1 mmol) in 1 mL of pyridine was heated at 140 °C for 5 h in a sealed/pressure vessel. After cooling, ice/cold water was added (50 mL), the precipitate was recovered by filtration under vacuum and purified by crystallization from ethanol to obtain the desired compounds.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)acetamide (29a)
Yield = 90%; mp = 211–212 °C (EtOH). Brownish black coloured solid, 1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H, CH3CONH), 3.91 (s, 3H, CH3ArN), 7.42–7.48 (m, 3H, Ar), 7.80–7.83 (m, 2H, Ar), 8.61 (s, 1H, ArH). 13C NMR (100 MHz, CDCl3) δ 196.96, 155.66, 146.61, 135.61, 135.59, 129.59, 128.97, 126.46, 110.82, 40.91, 24.98. MS-ESI for C13H13N3O2 (Calcd, 243.10), [M + H]+ at m/z 243.90, tR = 13.311. Anal. Calcd for C13H13N3O2: C, 64.19; H, 5.39; N, 17.27. Found C, 64.45; H, 5.41; N, 17.34.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)propionamide (29b)
Yield = 93%; mp = 210–211 °C (EtOH). Ash coloured solid, 1H NMR (400 MHz, CDCl3) δ 1.26 (td, 3H, J = 7.5, 1.1 Hz, CH3CH2CONH), 2.49–2.55 (m, 2H, CH2CONH), 3.92 (s, 3H, CH3ArN), 7.41–7.47 (m, 3H, Ar), 7.81–7.84 (m, 2H, Ar), 8.65 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 173.77, 155.73, 146.62, 135.63, 135.60, 129.58, 128.95, 126.43, 110.76, 40.90, 31.00, 9.24. MS-ESI for C14H15N3O2 (Calcd, 257.12), [M + H]+ at m/z 257.90, tR = 14.604. Anal. Calcd for C14H15N3O2: C, 65.36; H, 5.88; N, 16.33. Found C, 65.10; H, 5.90; N, 16.39.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)isobutyramide (29c)
Yield = 95%; mp = 146–148 °C (EtOH). Brown coloured solid, 1H NMR (400 MHz, CDCl3) δ 1.28 (dd, 6H, J = 6.9, 1.2 Hz, (CH3)2CHCONH), 2.64–2.71 (m, 1H, CHCONH), 3.92 (s, 3H, CH3ArN), 7.41–7.46 (m, 3H, Ar), 7.81–7.85 (m, 2H, Ar), 8.66 (s, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 177.10, 155.82, 146.61, 135.71, 135.59, 129.57, 128.94, 126.42, 110.83, 40.87, 36.95, 19.45. MS-ESI for C15H17N3O2 (Calcd, 271.13), [M + H]+ at m/z 272.04, tR = 16.829. Anal. Calcd for C15H17N3O2: C, 66.40; H, 6.32; N, 15.49. Found C, 66.66; H, 6.34; N, 15.55.
N-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-yl)butyramide (29d)
Yield = 92%; mp = 187–189 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 1.05 (t, 3H, CH2CH2CH3, J = 7.2 Hz), 1.80 (sex, 2H, CH2CH2CH3, J = 7.2 Hz), 2.49 (q, 2H, CH2CH2CH3, J = 7.2 Hz), 3.94 (s, 3H, N-CH3), 7.45–7.50 (m, 3H, Ar), 7.85 (d, 2H, Ar, J = 7.6 Hz), 8.63 (exch br s, 1H, NH), 8.68 (s, 1H, -CH pyridaz.). Anal. Calcd for C15H17N3O2: C, 66.40; H, 6.32; N, 15.49. Found C, 66.25; H, 6.31; N, 15.44.
3.2.20. General procedure for compounds 30a,b and 33
A mixture of compound 24a [38] (0.79 mmol), the appropriate R-phenylboronic acid (0.79 mmol), copper acetate (1.19 mmol) and triethylamine (1.59 mmol) in CH2Cl2 (5 mL) was stirred at room temperature for 3–12 h. After evaporation of the solvent, ethyl acetate was added (15–20 mL) and the solution was extracted first with 33% NH3 (3 × 5 mL) and then with water (2 × 5 mL). The organic layer was evaporated under vacuum and the residue was purified by crystallization from ethanol.
3.2.21. 3-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzonitrile (30a)
Yield = 60%; mp = 234–235 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 3.80 (s, 3H, CH3), 7.23 (s, 1H, Ar), 7.42–7.48 (m, 3H, Ar), 7.55–7.59 (m, 2H, Ar), 7.80 (dd, 3H, J = 8.0, 1.8 Hz, ArCN), 7.86 (d, 1H, J = 1.8 Hz, ArCN), 9.03 (exch br s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ 193.78, 157.05, 151.81, 140.85, 136.62, 129.18, 126.83, 111.09, 100.21, 23.94. MS-ESI for C18H14N4O (Calcd, 302.12), [M + H]+ at m/z 302.90, [M + ACN + H]+ at m/z 343.92, tR = 16.247. Anal. Calcd for C18H14N4O: C, 71.51; H, 4.67; N, 18.53. Found C, 71.22; H, 4.65; N, 18.45.
3.2.22. 2-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzonitrile (30b)
Yield = 32%; mp = 178–180 °C (EtOH). White coloured solid, 1H NMR (400 MHz, CDCl3) δ 3.95 (s, 3H, CH3), 7.12 (s, 1H, Ar), 7.41–7.47 (m, 3H, ArCN), 7.55 (d, 1H, J = 8.3 Hz, ArCN), 7.65 (td, 1H, J = 7.8, 1.6 Hz, Ar), 7.72 (ddd, 3H, J = 7.6, 3.6, 1.7 Hz, Ar), 7.96 (exch br s, 1H, NH). 13C NMR (100 MHz, CDCl3) δ 147.91, 138.56, 13.22, 129.42, 128.97, 126.44, 124.63, 121.19, 114.68, 100.94, 40.59. MS-ESI for C18H14N4O (Calcd, 302.12), [M + H]+ at m/z 302.97, [M + ACN + H]+ at m/z 344.06, tR = 16.180. Anal. Calcd for C18H14N4O: C, 71.51; H, 4.67; N, 18.53. Found C, 71.22; H, 4.65; N, 18.45.
3.2.23. 4-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzoic acid ethyl ester (33)
Yield = 80%; mp = 171–172 °C (Cyclohexane). 1H NMR (400 MHz, CDCl3) 1.42 (t, 3H, CH2CH3, J = 7.2 Hz), 3.96 (s, 3H, CH3), 4.41 (q, 2H, CH2CH3, J = 7.2 Hz), 7.30–7.40 (m, 3H, 2H Ar + CH pyridaz.), 7.45–7.50 (m, 3H, Ar), 7.77 (d, 2H, Ar, J = 8.8 Hz), 7.90 (exch br s, 1H, NH), 8.12 (d, 2H, Ar, J = 8.8 Hz). Anal. Calcd for C20H19N3O3: C, 68.75; H, 5.48; N, 12.03. Found C, 68.58; H, 5.47; N, 12.06.
3.2.24. General Procedure for Compounds 31a,b
A mixture of appropriate pyridazin-benzonitrile 30a or 30b (0.165 mmol) and 80% H2SO4 (2 mL) was stirred at 80 °C for 4 h. After cooling, ice/cold water (2–3 mL) was slowly added, the precipitate obtained was recovered by filtration under vacuum and purified by crystallization.
3.2.25. 3-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzamide (31a)
Yield = 93%; mp = 214–216 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H, CH3), 7.13 (s, 1H, -CH pyridaz.), 7.40–7.50 (m, 5H, 4H Ar + 1H CONH2), 7.60 (d, 1H, Ar, J = 9.2 Hz), 7.64 (d, 1H, Ar, J = 7.2 Hz), 7.76 (d, 2H, Ar, J = 8.0 Hz), 7.91 (s, 1H, Ar), 8.02 (exch br s, 1H, CONH2), 8.93 (exch br s, 1H, NH). Anal. Calcd for C18H16N4O2: C, 67.49; H, 5.03; N, 17.49. Found C, 67.36; H, 5.04; N, 17.53.
3.2.26. 2-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzamide (31b)
Yield = 95%; mp = 140–142 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 3.80 (s, 3H, CH3), 7.15 (t, 1H, Ar, J = 7.6 Hz), 7.38 (s, 1H, -CH pyridaz.), 7.43–7.50 (m, 3H, Ar), 7.57 (t, 1H, Ar, J = 7.6 Hz), 7.66 (exch br s, 1H, CONH2), 7.77 (t, 2H, Ar, J = 9.2 Hz), 7.84 (d, 2H, Ar, J = 6.8 Hz), 8.17 (exch br s, 1H, CONH2), 10.68 (exch br s, 1H, NH). Anal. Calcd for C18H16N4O2: C, 67.49; H, 5.03; N, 17.49. Found C, 67.36; H, 5.04; N, 17.53.
3.2.27. 4-Amino-2-methyl-6-phenylpyridazine-3(2H)-thione (32)
Compound 32 was obtained, starting from compound 24a [38], through the same procedure described for 26. In this case, the mixture was refluxed for 10 h. After cooling, ice/cold water was added. The precipitate was recovered by suction and purified by flash column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 85%; mp = 134–135 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 4.38 (s, 3H, CH3), 5.90 (exch br s, 2H, NH2), 6.78 (s, 1H, -CH pyridaz.), 7.45–7.50 (m, 3H, Ar), 7.80–7.85 (m, 2H, Ar). Anal. Calcd for C11H11N3S: C, 60.80; H, 5.10; N, 19.34. Found C, 60.97; H, 5.11; N, 19.30.
3.2.28. 4-(2-Methyl-3-oxo-6-phenyl-2,3-dihydro-pyridazin-4-ylamino)benzoic acid (34)
Compound 34 was obtained through the general procedure described for 15. After cooling, the mixture was acidified with 6N HCl and the final product was filtered off to obtain the desired compound. Yield = 90%; mp = 280–281 °C (Diethyl ether). 1H NMR (400 MHz, DMSO-d6) δ 3.82 (s, 3H, CH3), 7.37 (s, 1H, -CH pyridaz.), 7.40–7.50 (m, 3H, Ar), 7.58 (d, 2H, Ar, J = 8.8 Hz), 7.84 (d, 2H, Ar, J = 8.4 Hz), 7.95 (d, 2H, Ar, J = 8.4 Hz), 9.16 (exch br s, 1H, NH), 12.78 (exch br s, 1H, OH). Anal. Calcd for C18H15N3O2: C, 67.28; H, 4.71; N, 13.08. Found C, 67.44; H, 4.71; N, 13.05.
3.2.29. 4-[4-(4-Acetyl-piperazine-1-carbonyl)-phenylamino]-2-methyl-6-phenylpyridazin-3(2H)-one (35)
Compound 35 was obtained starting from 34 through the same procedure described for 4a,b. In this case the mixture was stirred at room temperature for 40 min. After cooling, THF was removed in vacuo and cold water was added (10 mL). The crude precipitate was recovered by filtration under vacuum and purified by crystallization. Yield = 94%; mp = 213–215 °C (Cyclohexane). Ligrownown solid, 1H NMR (400 MHz, CDCl3) δ 2.14 (s, 3H, CH3CONH), 3.94 (s, 3H, CH3), 3.60 (d, 8H, J = 46.0 Hz, 2 × NCH2CH2N), 7.22 (s, 1H, Ar), 7.33 (d, 2H, J = 7.9 Hz, Ar), 7.47 (dd, 6H, J = 24.3, 7.9 Hz, Ar), 7.71–7.80 (m, 2H, NH + Ar). 13C NMR (100 MHz, CDCl3) δ 170.15, 169.37, 156.24, 146.00, 140.96, 139.58, 136.38, 130.72, 129.39, 129.32, 128.96, 126.42, 120.74, 99.58, 40.75, 21.55. MS-ESI for C24H25N5O3 (Calcd, 431.20), [M + H]+ at m/z 432.10, [M + Na]+ at m/z 454.08, tR = 13.347. Anal. Calcd for C24H25N5O3: C, 66.81; H, 5.84; N, 16.23. Found C, 66.54; H, 5.82; N, 16.16.
3.2.30. General procedure for compounds 37a–d and 42a,b
A suspension of appropriate pyridazinone 36a–d (1.29 mmol), commercially available, and hydrazine hydrate (48 mmol) was stirred in a sealed/pressure vessel at 180–200 °C for 6–12 h. After cooling, ice-cold water was added (15 mL) and the precipitate obtained was recovered by filtration under vacuum to obtain the desired compounds 37a–d. To obtain compounds 42a,b we adopted the same procedure, using 41a,b (41a, [39]) as starting materials.
3.2.31. 4-Amino-6-thiophen-3-yl-pyridazin-3(2H)-one (37a)
Yield = 52%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 6.40 (exch br s, 2H, NH2), 6.68 (s, 1H, -CH pyridaz.), 7.45 (dd, 1H, thiophene, J1 = 1.2 Hz and J2 = 4.8 Hz), 7.60 (dd, 1H, thiophene, J1 = 2.8 Hz and J2 = 4.8 Hz), 7.81 (ds, 1H, thiophene, J = 1.2 Hz), 12.54 (exch br s, 1H, NH). Anal. Calcd for C8H7N3OS: C, 49.73; H, 3.65; N, 21.75. Found C, 49.61; H, 3.65; N, 21.69.
3.2.32. 4-Amino-6-cyclohexylpyridazin-3(2H)-one (37b)
Yield = 46%; mp = 284–287 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.20–1.39 (m, 5H, C6H11), 1.72–1.83 (m, 5H, C6H11), 2.30–2.35 (m, 1H, C6H11), 6.14 (s, 1H, CH pyridaz.), 6.18 (exch br s, 2H, NH2), 12.24 (exch br s, 1H, NH). Anal. Calcd for C10H15N3O: C, 62.15; H, 7.82; N, 21.74. Found C, 62.29; H, 7.80; N, 21.79.
3.2.33. 4-Amino-6-isopropylpyridazin-3(2H)-one (37c)
Yield = 40%; mp = 246–248 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.11 (d, 6H, CH(CH3)2, J = 7.2 Hz), 2.67 (m, 1H, CH(CH3)2), 6.16 (s, 1H, -CH pyridaz.), 6.18 (exch br s, 2H, NH2), 12.23 (exch br s, 1H, NH). Anal. Calcd for C7H11N3O: C, 54.89; H, 7.24; N, 27.43. Found C, 54.76; H, 7.23; N, 27.51.
3.2.34. 4-Amino-6-benzylpyridazin-3(2H)-one (37d)
Yield = 42%; mp = 247–250 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.70 (s, 2H, CH2Ph), 6.04 (s, 1H, -CH pyridaz.), 6.22 (exch br s, 2H, NH2), 7.20–7.40 (m, 5H, Ar), 12.31 (exch br s, 1H, NH). Anal. Calcd for C11H11N3O: C, 65.66; H, 5.51; N, 20.88. Found C, 65.84; H, 5.50; N, 20.83.
3.2.35. 4-Amino-6-(2-hydroxyphenyl)-2-methylpyridazin-3(2H)-one (42a)
Yield = 58%; mp = 212–213 °C (EtOH). White coloured solid, 1H NMR (400 MHz, CDCl3) δ 3.85 (s, 3H, CH3), 6.84 (s, 1H, Ar), 6.92 (td, 1H, J = 7.7, 1.2 Hz, Ar), 7.00–7.05 (m, 1H, Ar), 7.29 (td, 1H, J = 8.3, 7.8, 1.6 Hz, Ar), 7.58–7.50 (m, 1H, Ar). 13C NMR (100 MHz, CDCl3) δ 157.81, 131.24, 126.35, 119.47, 118.30, 98.93, 29.87. MS-ESI for C11H11N3O2 (Calcd, 217.08), [M + H]+ at m/z 217.93, tR = 11.693. Anal. Calcd for C11H11N3O2: C, 60.82; H, 5.10; N, 19.34. Found C, 60.57; H, 5.08; N, 19.26.
3.2.36. 4-Amino-6-(4-aminophenyl)-2-methylpyridazin-3(2H)-one (42b)
Yield = 55%; mp = 208–209 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 3.65 (s, 3H, N-CH3), 5.35 (exch br s, 2H, NH2), 6.33 (exch br s, 2H, Ph-NH2), 6.59 (d, 2H, Ar, J = 8.0 Hz), 6.63 (s, 1H, -CH pyridaz.), 7.42 (d, 2H, Ar, J = 8.0). Anal. Calcd for C11H11N4O: C, 61.10; H, 5.59; N, 25.91. Found C, C, 61.27; H, 5.58; N, 25.85.
3.2.37. General Procedure for Compounds 38a–d
A mixture of the appropriate pyridazinone 37a–d (0.67 mmol), K2CO3 (1.34 mmol) and CH3I (1.01 mmol) in anhydrous DMF (1.5 mL) was stirred at 80 °C for 1–4 h. After cooling, the mixture was diluted with cold water (15 mL) and compound 38a was recovered by suction and crystallized from ethanol. For compounds 38b–d the suspension was extracted with CH2Cl2 (3 × 15 mL) and the solvent was evaporated in vacuo. The final compounds were purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 (for 38b,d) or CH2Cl2/CH3OH 9.5:0.5 (for 38c) as eluents.
3.2.38. 4-Amino-2-methyl-6-thiophen-3-yl-pyridazin-3(2H)-one (38a)
Yield = 62%; mp = 178–179 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.67 (s, 3H, N-CH3), 6.50 (exch br s, 2H, NH2), 6.68 (s, 1H, -CH pyridaz.), 7.47 (d, 1H, thiophene, J = 4.8 Hz), 7.61 (m, 1H, thiophene), 7.84 (s, 1H, thiophene). Anal. Calcd for C9H9N3OS: C, 52.16; H, 4.38; N, 20.27. Found C, 52.05; H, 4.37; N, 20.22.
3.2.39. 4-Amino-6-cyclohexyl-2-methylpyridazin-3(2H)-one (38b)
Yield = 58%; oil. 1H NMR (400 MHz, CDCl3) δ 1.30–1.43 (m, 5H, C6H11), 1.68–1.92 (m, 5H, C6H11), 2.40 (m, 1H, C6H11), 3.75 (s, 3H, N-CH3), 4.91 (exch br s, 2H, NH2), 6.21 (s, 1H, -CH pyridaz.). Anal. Calcd for C11H17N3O: C, 63.74; H, 8.27; N, 20.27. Found C, 63.87; H, 8.29; N, 20.23.
3.2.40. 4-Amino-6-isopropyl-2-methyl-2H-pyridazin-3-one (38c)
Yield = 49%; oil. 1H NMR (400 MHz, CDCl3) δ 1.18 (d, 6H, CH(CH3)2, J = 7.2 Hz), 2.75 (m, 1H, CH(CH3)2), 3.74 (s, 3H, N-CH3), 4.96 (exch br s, 2H, NH2), 6.21 (s, 1H, -CH pyridaz.). Anal. Calcd for C8H13N3O: C, 57.46; H, 7.84; N, 25.13. Found C, 57.58; H, 7.82; N, 25.07.
3.2.41. 4-Amino-6-benzyl-2-methylpyridazin-3(2H)-one (38d)
Yield = 48%; mp = 104–108 °C (EtOH). 1H NMR (400 MHz, CDCl3) δ 3.80 (s, 3H, N-CH3), 3.82 (s, 2H, CH2Ph), 4.81 (exch br s, 2H, NH2), 6.07 (s, 1H, -CH pyridaz.), 7.22–7.35 (m, 5H, Ar). Anal. Calcd for C12H13N3O: C, 66.96; H, 6.09; N, 19.52. Found C, 66.83; H, 5.50; N, 20.83.
3.2.42. General Procedure for Compounds 39a,b, 40 and 43
Compounds 39a,b, 40 and 43 were obtained starting from 38b,c, 37e and 42a, respectively, through the same procedure described for compound 25a–f.
3.2.43. (6-Cyclohexyl-2-methyl-3-oxo-2,3-dihydropyridazin-4-yl)urea (39a)
Yield = 66%; mp = 251–254 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.30–1.40 (m, 5H, C6H11), 1.70–1.85 (m, 5H, C6H11), 2.45 (m, 1H, C6H11), 3.64 (s, 3H, N-CH3), 6.74 (exch br s, 2H, CONH2), 7.79 (s, 1H, -CH pyridaz.), 8.84 (exch br s, 1H, NHCO). Anal. Calcd for C12H18N4O2: C, 57.58; H, 7.25; N, 22.38. Found C, 57.41; H, 7.23; N, 22.43.
3.2.44. (6-Isopropyl-2-methyl-3-oxo-2,3-dihydropyridazin-4-yl)urea (39b)
Yield = 60%; mp = 248–251 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.15 (d, 6H, CH(CH3)2, J = 6.8 Hz), 2.80 (m, 1H, CH(CH3)2), 3.65 (s, 3H, N-CH3), 6.74 (exch br s, 2H, CONH2), 7.81 (s, 1H, -CH pyridaz.), 8.85 (exch br s, 1H, CONH). Anal. Calcd for C9H14N4O2: C, 51.42; H, 6.71; N, 26.65. Found C, 51.31; H, 6,70; N, 26.61.
3.2.45. [6-(2-Hydroxy-phenyl)-3-oxo-2,3-dihydro-pyridazin-4-yl]-urea (40)
Yield = 85%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 6.75 (exch br s, 2H, CONH2), 6.90–6.95 (m, 2H, Ar), 7.27 (t, 1H, Ar, J = 8.4 Hz), 7.45 (dd, 1H, Ar, J1 = 1.2 Hz and J2 = 8.0 Hz), 8.39 (s, 1H, -CH pyridaz.), 8.92 (exch br s, 1H, NHCO), 10.43 (exch br s, 1H, OH), 13.18 (exch br s, 1H, NH). Anal. Calcd for C11H10N4O3: C, 53.66; H, 4.09; N, 22.75. Found C, 53.51; H, 4,08; N, 22.81.
3.2.46. [6-(2-Hydroxyphenyl)-2-methyl-3-oxo-2,3-dihydropyridazin-4-yl]-urea (43)
Yield = 95%; mp = 278–280 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 3.76 (s, 3H, N-CH3), 6.79 (exch br s, 2H, NH2), 6.88–6.95 (m, 2H, Ar), 7.27 (t, 1H, Ar, J = 7.2 Hz), 7.44 (d, 1H, Ar, J = 6.8 Hz), 8.37 (s, 1H, -CH pyridaz.), 8.93 (exch br s, 1H, NHCO), 10.17 (exch br s, 1H, OH). Anal. Calcd for C12H12N4O3: C, 55.38; H, 4.65; N, 21.53. Found C, 55.49; H, 4,65; N, 21,49.
3.2.47. N-[4-(5-Amino-1-methyl-6-oxo-1,6-dihydropyridazin-3-yl)-phenyl]-acetamide (44)
To a cooled (0 °C) and stirred solution of 42b (0.93 mmol) in anhydrous THF (2–3 mL), 1.02 mmol of acetyl chloride was added and the mixture was stirred at room temperature for 20 min. After dilution with cold water (20–30 mL), the precipitate was recovered by filtration under vacuum and purified by crystallization. Yield = 92%; mp = 270–272 °C (Cyclohexane). 1H NMR (400 MHz, DMSO-d6) δ 2.06 (s, 3H, COCH3), 3.69 (s, 3H, N-CH3), 6.48 (exch br s, 2H, NH2), 6.71 (s, 1H, -CH pyridaz.), 7.60–7.70 (m, 4H, Ar), 8.80 (exch br s, 1H, NHCO). Anal. Calcd for C13H14N4O2: C, 60.45; H, 5.46; N, 21.69. Found C, 60.58; H, 5.45; N, 21.63.
3.2.48. 3,6-Dimethyl-4-pyridin-2-yl-isoxazolo [3,4-d]pyridazin-7(6H)-one (46)
To a cooled (0–4 °C) solution of 45 [34] (0.38 mmol) in EtOH (2–3 mL), methylhydrazine (1.30 mmol) was added and the mixture was stirred at room temperature for 90 min. The precipitate was recovered by filtration under vacuum to obtain the desired compound. Yield = 84%; mp = 154–155 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 2.94 (s, 3H, C3-CH3), 3.74 (s, 3H, N-CH3), 7.57 (t, 1H, Ar, J = 5.2 Hz), 7.95–8.05 (m, 2H, Ar), 8.76 (d, 1H, Ar, J = 5.2 Hz). Anal. Calcd for C12H10N4O2: C, 59.50; H, 4.16; N, 23.13. Found C, 59.66; H, 4.16; N, 23.20.
3.2.49. 5-Acetyl-4-amino-2-methyl-6-pyridin-2-yl-pyridazin-3(2H)-one (47)
A mixture of 46 (0.82 mmol), 10% Pd/C (20 mg) and ammonium formate (4.9 mmol) in EtOH (5 mL), was refluxed for 2 h. After addition of CH2Cl2 (4–5 mL) and filtration of charcoal, evaporation of the solvent afforded the product 47. Yield = 65%; mp = 201–203 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 1.89 (s, 3H, COCH3), 3.73 (s, 3H, N-CH3), 7.23 (exch br s, 2H, NH2), 7.44–7.50 (m, 1H, Ar), 7.89 (d, 1H, Ar, J = 7.2 Hz), 7.96 (t, 1H, Ar, J = 7.2 Hz), 8.57 (d, 1H, Ar, J = 4.4 Hz). Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94. Found C, 59.18; H, 4.96; N, 22.99.
3.2.50. 4-Amino-2-methyl-6-pyridin-2-yl-pyridazin-3(2H)-one (48)
A suspension of 47 (0.53 mmol) in 1 mL of 48% HBr was stirred in a sealed/pressure vessel at 130 °C for 3 h. After cooling ice-cold water was added and the precipitate was recovered by filtration under vacuum to obtain the desired product 48. Yield = 65%; mp = 294–295 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.75 (s, 3H, N-CH3), 7.24 (s, 1H, -CH pyridaz.), 7.51 (m, 1H, Ar), 8.00 (t, 1H, Ar, J = 7.2 Hz), 8.12 (d, 1H, Ar, J = 8.0 Hz), 8.66 (d, 1H, Ar, J = 4.4 Hz). Anal. Calcd for C10H10N4O: C, 59.40; H, 4.98; N, 27.71. Found C, 59.51; H, 4.99; N, 27.75.
3.2.51. (2-Methyl-3-oxo-6-pyridin-2-yl-2,3-dihydropyridazin-4-yl)-urea (49)
Compound 49 was obtained starting from 48, through the same procedure described for compounds 25a–f, 39a,b, 40 and 43. Yield = 15%; mp > 300 °C (EtOH). 1H NMR (400 MHz, DMSO-d6) δ 3.82 (s, 3H, N-CH3), 6.80 (exch br s, 2H, NH2), 7.46 (m, 1H, Ar), 7.92 (t, 1H, Ar, J = 7.6 Hz), 8.10 (d, 1H, Ar, J = 7.6 Hz), 8.67 (d, 1H, Ar, J = 4.8 Hz), 8.83 (s, 1H, -CH pyridaz.), 8.97 (exch br s, 1H, NHCO). Anal. Calcd for C11H11N5O2: C, 53.87; H, 4.52; N, 28.56. Found C, 53.78; H, 5.00; N, 27.71.
3.2.52. 2-Methyl-3-oxo-6-phenyl-2,3-dihydropyridazine-4-carboxamide (51)
Compound 51 was obtained starting from 50 [41], through the same procedure described for compounds 38a–d. The compound was purified by crystallization from diethyl ether. Yield = 55%; mp = 215–217 °C (Et2O). Light brown solid, 1H NMR (400 MHz, CDCl3) δ 3.99 (s, 3H, CH3), 5.98 (exch br s, 1H, CONH2), 7.43–7.51 (m, 3H, Ar), 7.84–7.88 (m, 2H, Ar), 8.72 (s, 1H, Ar), 9.41 (exch br s, 1H, CONH2). 13C NMR (100 MHz, CDCl3) δ 163.58, 160.21, 145.33, 134.16, 132.64, 130.02, 129.23, 129.04, 126.15, 41.53. MS-ESI for C12H11N3O2 (Calcd, 229.08), [M + H]+ at m/z 229.90, tR = 12.205. Anal. Calcd for C12H11N3O2: C, 62.87; H, 4.84; N, 18.33. Found C, 62.62; H, 4.82; N, 18.26.
3.2.53. 1-(6-Methyl-3-oxo-2-phenyl-2,3-dihydropyridazin-4-yl)urea (55)
Compound 55 was obtained strating from 54 [43], through the same procedure for the formation of urea described for compounds 25a–f, 39a,b, 40 and 43. Yield = 95%; mp = 288–290 °C (EtOH). White coloured solid, 1H NMR (400 MHz, DMSO-d6) δ 2.25 (s, 3H, CH3), 7.40–7.42 (m, 1H, Ar), 7.47 (d, 2H, J = 8.2 Hz, Ar), 7.50–7.54 (m, 2H, Ar), 7.78 (s, 1H, Ar), 8.91 (s, 1H, NHCONH2). 13C NMR (100 MHz, DMSO-d6) δ 155.56, 154.89, 146.52, 141.98, 138.04, 128.86, 128.12, 125.97, 109.55, 21.48. MS-ESI for C12H12N4O2 (Calcd, 244.10), [M + H]+ at m/z 244.88, tR = 10.100. Anal. Calcd for C12H12N4O2: C, 59.01; H, 4.95; N, 22.94. Found C, 59.25; H, 4.97; N, 23.03.
3.2.54. (E)-3-(2-(Dimethylamino)vinyl)-4-methyl-6-phenylisoxazolo [3,4-d]pyridazin-7(6H)-one (56)
A mixture of 52 (1.04 mmol) [42] in 2.5 mL of DMF-DMA was hetaed at 90–100 °C for 1 h. After cooling, ice/cold water was added (15 mL) and the precipitate obtained was recovered by filtration under vacuum to obtained the pure desired compound. Yield = 90%; mp = 224–226 °C dec. (Cyclohexane). 1H NMR (400 MHz, CDCl3) δ 2.50 (s, 3H, CH3), 3.00–3.20 (m, 6H, N(CH3)2), 5.25 (d, 1H, CH=CH-N, J = 10.0 Hz), 7.30–7.35 (m, 1H, Ar), 7.45–7.50 (m, 2H, Ar), 7.59–7.64 (m, 3H, 1H CH=CH-N + 2H Ar). Anal. Calcd for C16H16N4O2: C, 64.85; H, 5.44; N, 18.91. Found C, 65.10; H, 5.46; N, 18.98.
3.2.55. 4-Amino-6-methyl-2-phenyl-5-(1H-pyrazol-5-yl)pyridazin-3(2H)-one (57)
A mixture of intermediate 56 (0.81 mmol) and 1 mL of hydrazine hydrate (excess) in 2 mL of abs. EtOH was hetaed at 70 °C for 10 h. After cooling, ice/cold water was added (15 mL). The precipitate obtained was recovered by filtration under vacum and purified by crystallization from ethanol. Yield = 65%; mp = 119–121 °C. (Cyclohexane). Yellow coloured solid, 1H NMR (400 MHz, Methanol-d4) δ 2.31 (s, 3H, CH3), 6.58 (exch br s, 2H, NH2), 7.43 (t, 1H, J = 7.3 Hz, Ar), 7.52 (t, 3H, J = 7.6 Hz, Ar), 7.58 (d, 2H, J = 7.6 Hz, Ar), 7.83 (s, 1H, NH). 13C NMR (100 MHz, Methanol-d4) δ 174.64, 147.92, 143.30, 129.88, 129.32, 127.14, 106.95, 24.30. MS-ESI for C14H13N5O (Calcd, 267.11), [M + H]+ at m/z 267.98. tR = 10.882. Anal. Calcd for C14H13N5O: C, 62.91; H, 4.90; N, 26.20. Found C, 62.66; H, 4.88; N, 26.09.
3.3. Molecular Modeling and Biological Data
The 2D chemical structures were built using Marvin Sketch and all the structures were subjected to molecular mechanics energy minimization using the MMFF94 force field present in the same software [45]. The 3D geometry of all compounds was then optimized using the PM3 Hamiltonian [46], as implemented in MOPAC 2016 package assuming a pH of 7.0 [47]. Once built and optimized, all structures were used in the bioisostere replacement tool Spark 10.4.0. Five hundred compounds were generated for the substitution (50 best compounds reported in the Supplementary Materials). The isosteric replacement was performed using the same 178,558 fragments for each part; in particular, the fragments derive from ChEMBL and Zinc databases with a protocol already reported and validated [27,48,49]. Ligand growing experiments were performed in the selected pyridazinone structure using an already reported protocol [50]. Docking calculations were made using AutoDock with the default docking parameters and a validated protocol [51,52]. The setup was done with YASARA [47]. The Lamarckian genetic algorithm implemented in AutoDock was used for the calculations. The ligand-centered maps were generated by AutoGrid with a spacing of 0.375 Å and dimensions that encompass all atoms extending 5 Å from the surface of the ligand. All of the parameters were inserted at their default settings. The X-ray crystal structures of the co-crystal FABP4/(2-[(2-oxo-2-piperidin-1-ylethyl)sulfanyl]-6-(trifluoromethyl)pyrimidin-4-ol) (PDBid: 1TOU) was downloaded from the Protein Data Bank (www.rcsb.org accessed on 15 June 2022).
3.4. FABP Inhibitory Activity Assays
To analyze the inhibitory activity of FABP4 ligands, a displacement assay was utilized as described by the Cayman’s instruction, FABP4 Inhibitor/Ligand Screening Assay Kit, Item 10,010,231 (see Supplementary Materials for additional details). The samples of compounds for activity determination were prepared as a stock solution (1 mM) in DMSO. On the day of activity assay, the compounds were all diluted in phosphate buffer solution (PBS, pH 7.4) to different concentrations (100, 50, 10, 5, 2, 1, and 0 µM). Appropriate concentrations of DMSO in PBS were used as control. The detection reagent (FABP Assay Detection Reagent, Item 10010376) was used as provided by the Cayman’s kit. The diluted Detection Reagent probe was mixed with FABP4 protein present in the kit and incubated for 10 min at room temperature. Compounds were then added and equilibrated for another 10 min. Lastly, the fluorescence signal was recorded at 470 nm (i.e., emission, with the excitation fixed at 370 nm) with a CytoFluor® Series 4000 Fluorescence Multi-Well Plate Reader. The IC50 was calculated as indicated in the kit booklet of FABP4 Inhibitor/Ligand Screening Assay Kit (Item No. 10010231) Cayman chemicals, as follows: 1) calculate the average fluorescence of each sample; 2) calculate the background corrected fluorescence (BCF) by subtracting the blank; 3) divide the BCF of each sample by the maximum BCF and multiply by 100% (this is the value in percent fluorescence units, i.e., % FU); 4) plot the % FU values against the concentration of inhibitor/ligand used; 5) find the concentration of inhibitor/ligand that corresponds to 50% FU, to determine IC50 values.
4. Conclusions
We have identified novel 4-amino and 4-ureido pyridazinone-based FABP4 inhibitors whose design was directed by computing assisted molecular design of bioisosteric-replacements/scaffold hopping of the pyrimidine skeleton of the co-crystallyzed ligand 1TOU. Selected compounds have been synthesized and tested for their ability to inhibit FABP4. Among the new series, ten compounds were further evaluated on the basis of their inhibitory activity on FABP4 established via a single point displacement assay. In particular, 4b, 25a, 30b and 22 exhibited high FABP4 inhibitory activity with IC50 in the low micromolar range. The results demonstrated that compound 25a was the most potent analogue in terms of displacement of the arachidonic acid, with an IC50 value of 2.97 μM, which is lower than the IC50 of the positive control (3.42 µM). Docking experiments, conducted with the most active compounds 4b, 25a, 30b, 22, confirmed the ability of these molecules to interact with several amino acid residues present inside the FABP4 binding pocket, with the stronger interaction exhibited by compound 25a. This result is in agreement with the higher activity recorded in vitro for 25a, in comparison to the other 4-amino and 4-ureido pyridazinone-based analogues developed in this study.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ph15111335/s1, 1H NMRs of selected compounds; 13C NMRs of selected compounds; Mass spectra of selected compounds; HPLC/UV chromatograms of selected compounds; 50 ‘best-fit’ compounds generated with scaffold hopping replacement; Info on the FABP4 inhibitor assay kit; Averaged data as Background corrected fluorescence for IC50 measured compounds.
Author Contributions
Conceptualization, L.C., G.F. and A.C.; methodology, L.C., G.F., D.M., R.R.d.O.S., F.M. and C.V.; software, G.F. and C.Z.; formal analysis, L.C., G.F., D.M., R.R.d.O.S., F.M., C.V. and A.C.; resources, G.F, C.Z., M.P.G., A.C.; data curation, L.C., G.F., D.M., R.R.d.O.S., F.M.; writing—original draft preparation, L.C., G.F.,C.Z. and D.M.; writing—review and editing, L.C., G.F., A.C.; supervision, M.P.G. and A.C.; project administration, M.P.G. and A.C.; funding acquisition, M.P.G. and A.C. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research has received funding for a scholarship to R.R.d.O.S from the Coordination for the Improvement of Higher Education Personnel—Brazil (CAPES-PRINT, funding number 88887.570120/2020-00).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Das U.N. Essential Fatty acids—A review. Curr. Pharm. Biotechnol. 2006;7:467–482. doi: 10.2174/138920106779116856. [DOI] [PubMed] [Google Scholar]
- 2.Furuhashi M., Hotamisligil G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boden G. Free fatty acids (FFA), a link between obesity and insulin resistance. Front. Biosci. 1998;3:d169–d175. doi: 10.2741/A272. [DOI] [PubMed] [Google Scholar]
- 4.Hotamisligil G.S., Bernlohr D.A. Metabolic functions of FABPs--mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015;11:592–605. doi: 10.1038/nrendo.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boden G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am. 2008;37:635–646. doi: 10.1016/j.ecl.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeFronzo R.A. Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int. J. Clin. Pract. Suppl. 2004;58:9–21. doi: 10.1111/j.1368-504X.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- 7.Sheth S.G., Gordon F.D., Chopra S. Nonalcoholic steatohepatitis. Ann. Intern. Med. 1997;126:137–145. doi: 10.7326/0003-4819-126-2-199701150-00008. [DOI] [PubMed] [Google Scholar]
- 8.Storch J., Thumser A.E. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta. 2000;1486:28–44. doi: 10.1016/S1388-1981(00)00046-9. [DOI] [PubMed] [Google Scholar]
- 9.Queipo-Ortuno M.I., Escote X., Ceperuelo-Mallafre V., Garrido-Sanchez L., Miranda M., Clemente-Postigo M., Perez-Perez R., Peral B., Cardona F., Fernandez-Real J.M., et al. FABP4 dynamics in obesity: Discrepancies in adipose tissue and liver expression regarding circulating plasma levels. PLoS ONE. 2012;7:e48605. doi: 10.1371/journal.pone.0048605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Syamsunarno M.R., Iso T., Hanaoka H., Yamaguchi A., Obokata M., Koitabashi N., Goto K., Hishiki T., Nagahata Y., Matsui H., et al. A critical role of fatty acid binding protein 4 and 5 (FABP4/5) in the systemic response to fasting. PLoS ONE. 2013;8:e79386. doi: 10.1371/journal.pone.0079386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson B.R., Mazurkiewicz-Munoz A.M., Suttles J., Carter-Su C., Bernlohr D.A. Interaction of Adipocyte Fatty Acid-binding Protein (AFABP) and JAK2. J. Biol. Chem. 2009;284:13473–13480. doi: 10.1074/jbc.M900075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adida A., Spener F. Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor gamma agonists in cultured cell. BBA-Mol. Cell Biol. 2006;1761:172–181. doi: 10.1016/j.bbalip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 13.Fu Y., Luo L., Luo N., Garvey W.T. Lipid metabolism mediated by adipocyte lipid binding protein (ALBP/aP2) gene expression in human THP-1 macrophages. Atherosclerosis. 2006;188:102–111. doi: 10.1016/j.atherosclerosis.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 14.Floresta G., Pistarà V., Amata E., Dichiara M., Marrazzo A., Prezzavento O., Rescifina A. Adipocyte fatty acid binding protein 4 (FABP4) inhibitors. A comprehensive systematic review. Eur. J. Med. Chem. 2017;138:854–873. doi: 10.1016/j.ejmech.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 15.Nieman K.M., Kenny H.A., Penicka C.V., Ladanyi A., Buell-Gutbrod R., Zillhardt M.R., Romero I.L., Carey M.S., Mills G.B., Hotamisligil G.S., et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolle A., Suhail S., Jung M., Jung K., Stephan C. Fatty acid binding proteins (FABPs) in prostate, bladder and kidney cancer cell lines and the use of IL-FABP as survival predictor in patients with renal cell carcinoma. BMC Cancer. 2011;11:302. doi: 10.1186/1471-2407-11-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uehara H., Takahashi T., Oha M., Ogawa H., Izumi K. Exogenous fatty acid binding protein 4 promotes human prostate cancer cell progression. Int. J. Cancer. 2014;135:2558–2568. doi: 10.1002/ijc.28903. [DOI] [PubMed] [Google Scholar]
- 18.Yang A., Zhang H., Sun Y., Wang Y., Yang X., Yang X., Zhang H., Guo W., Zhu G., Tian J., et al. Modulation of FABP4 hypomethylation by DNMT1 and its inverse interaction with miR-148a/152 in the placenta of preeclamptic rats and HTR-8 cells. Placenta. 2016;46:49–62. doi: 10.1016/j.placenta.2016.08.086. [DOI] [PubMed] [Google Scholar]
- 19.Tian W., Zhang W., Zhang Y., Zhu T., Hua Y., Li H., Zhang Q., Xia M. FABP4 promotes invasion and metastasis of colon cancer by regulating fatty acid transport. Cancer Cell Int. 2020;20:512. doi: 10.1186/s12935-020-01582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gharpure K.M., Pradeep S., Sans M., Rupaimoole R., Ivan C., Wu S.Y., Bayraktar E., Nagaraja A.S., Mangala L.S., Zhang X.N., et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018;9 doi: 10.1038/s41467-018-04987-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H.Y., Lv B.B., Bi Y.H. FABP4 accelerates glioblastoma cell growth and metastasis through Wnt10b signalling. Eur. Rev. Med. Pharmacol. Sci. 2018;22:7807–7818. doi: 10.26355/eurrev_201811_16405. [DOI] [PubMed] [Google Scholar]
- 22.Floresta G., Patamia V., Zagni C., Rescifina A. Adipocyte fatty acid binding protein 4 (FABP4) inhibitors. An update from 2017 to early 2022. Eur. J. Med. Chem. 2022;240:114604. doi: 10.1016/j.ejmech.2022.114604. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Law W.K., Hu J.S., Lin H.Q., Ip T.M., Wan D.C. Discovery of FDA-approved drugs as inhibitors of fatty acid binding protein 4 using molecular docking screening. J. Chem. Inf. Model. 2014;54:3046–3050. doi: 10.1021/ci500503b. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Y., Nie T., Zhang Y., Song M., Li K., Ding M., Ding K., Wu D., Xu Y. The discovery of novel and selective fatty acid binding protein 4 inhibitors by virtual screening and biological evaluation. Bioorg. Med. Chem. 2016;24:4310–4317. doi: 10.1016/j.bmc.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 25.Floresta G., Cilibrizzi A., Abbate V., Spampinato A., Zagni C., Rescifina A. FABP4 inhibitors 3D-QSAR model and isosteric replacement of BMS309403 datasets. Data Brief. 2019;22:471–483. doi: 10.1016/j.dib.2018.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Floresta G., Gentile D., Perrini G., Patamia V., Rescifina A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Mar. Drugs. 2019;17:624. doi: 10.3390/md17110624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Floresta G., Cilibrizzi A., Abbate V., Spampinato A., Zagni C., Rescifina A. 3D-QSAR assisted identification of FABP4 inhibitors: An effective scaffold hopping analysis/QSAR evaluation. Bioorg. Chem. 2019;84:276–284. doi: 10.1016/j.bioorg.2018.11.045. [DOI] [PubMed] [Google Scholar]
- 28.Crocetti L., Floresta G., Nazir S., Vergelli C., Bhogal A., Biancalani C., Cesari N., Giovannoni M.P., Cilibrizzi A. Synthesis and inverse virtual screening of new bi-cyclic structures towards cancer-relevant cellular targets. Struct Chem. 2022;33:769–793. doi: 10.1007/s11224-022-01889-0. [DOI] [Google Scholar]
- 29.Floresta G., Crocetti L., Giovannoni M.P., Biagini P., Cilibrizzi A. Repurposing strategies on pyridazinone-based series by pharmacophore- and structure-driven screening. J. Enzime Inhib. Med. Chem. 2020;35:1137–1144. doi: 10.1080/14756366.2020.1760261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giovannoni M.P., Vergelli C., Cilibrizzi A., Crocetti L., Biancalani C., Graziano A., Dal Piaz V., Loza M.I., Cadavid M.I., Díaz J.L., et al. Pyrazolo[1′,5′:1,6]pyrimido[4,5-d]pyridazin-4(3H)-ones as selective human A1 adenosine receptor ligands. Bioorg. Med. Chem. 2010;18:7890–7899. doi: 10.1016/j.bmc.2010.09.043. [DOI] [PubMed] [Google Scholar]
- 31.Biagini P., Biancalani C., Graziano A., Cesaria N., Giovannoni M.P., Cilibrizzi A., Dal Piaz V., Vergelli C., Crocetti L., Delcanale M., et al. Functionalized pyrazoles and pyrazolo[3,4-d]pyridazinones: Synthesis and evaluation of their phosphodiesterase 4 inhibitory activity. Bioorg. Med. Chem. 2010;18:3506–3517. doi: 10.1016/j.bmc.2010.03.066. [DOI] [PubMed] [Google Scholar]
- 32.Giovannoni M.P., Ciciani G., Cilibrizzi A., Crocetti L., Daniele S., Di Cesare Mannelli L., Ghelardini C., Giacomelli C., Guerrini G., Martini C., et al. Further studies on pyrazolo[1′,5′:1,6]pyrimido[4,5-d]pyridazin-4(3H)-ones as potent and selective human A1 adenosine receptor antagonists. Eur. J. Med. Chem. 2015;89:32–41. doi: 10.1016/j.ejmech.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 33.Cheeseright T., Mackey M., Rose S., Vinter A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006;46:665–676. doi: 10.1021/ci050357s. [DOI] [PubMed] [Google Scholar]
- 34.Dal Piaz V., Aguilar I.N., Buil Albero M.A., Garrido R.Y., Giovannoni M.P., Gracia F.J., Lumeras A.W., Vergelli C. Preparation of pyridazin-3(2H)-ones and their use as PDE4 inhibitors. PCT WO2005049581A120050602. 2005 June 2;
- 35.Aguilar Izquierdo N., Carrascal Riera M., Dal Piaz V., Gracia Ferrer J., Lumeras Amador W., Masdeu Margalef M., Warrellow G. Preparation of pyridazin-3(2H)-one derivatives as PDE4 inhibitors for the treatment of pathological diseases. PCT WO2005123693A120051229. 2005 December 29;
- 36.Biancalani C., Giovannoni M.P., Pieretti S., Cesari N., Graziano A., Vergelli C., Cilibrizzi A., Di Gianuario A., Colucci M., Mangano G., et al. Further Studies on Arylpiperazinyl Alkyl Pyridazinones: Discovery of an Exceptionally Potent, Orally Active, Antinociceptive Agent in Thermally Induced Pain. J. Med. Chem. 2009;52:7397–7409. doi: 10.1021/jm900458r. [DOI] [PubMed] [Google Scholar]
- 37.Sircar I. Synthesis of 4-amino-6-phenyl-3(2H)-pyridazinones: A general procedure. J. Heterocycl. Chem. 1983;20:1473–1476. doi: 10.1002/jhet.5570200607. [DOI] [Google Scholar]
- 38.Coates W.J., McKillop A. Preparation of 4-amino-3(2H)-pyridazinones by direct amination of 3(2H)-pyridazinones with hydrazine. Heterocycles. 1989;29:1077–1090. doi: 10.3987/COM-89-4836. [DOI] [Google Scholar]
- 39.Hubbard R.D., Wang L., Park C.H., Sun C., Mcdaniel K.F., Pratt J.K., Soltwedel T.N., Wendt M.D., Holms J.H., Liu D. Preparation of pyridinone and pyridazinone derivatives for the treatment of inflammatory diseases, diabetes, obesity, cancer, and AIDS. U.S. Patent US 20130331382A120131212. 2013 December 12;
- 40.Vergelli C., Schepetkin I.A., Ciciani G., Cilibrizzi A., Crocetti L., Giovannoni M.P., Guerrini G., Iacovone A., Kirpotina L.N., Khlebnikov A.I., et al. 2-Arylacetamido-4-phenylamino-5-substituted pyridazinones as formyl peptide receptors agonists. Bioorg. Med. Chem. 2016;24:2530–2543. doi: 10.1016/j.bmc.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wermuth C.G., Schlewer G., Bourguignon J.J., Maghioros G., Bouchet M.J., Moire C., Kan J.P., Worms P., Biziere K. 3-Aminopyridazine derivatives with atypical antidepressant, serotonergic and dopaminergic activities. J. Med. Chem. 1989;32:528–537. doi: 10.1021/jm00123a004. [DOI] [PubMed] [Google Scholar]
- 42.Vergelli C., Giovannoni M.P., Pieretti S., Giannuario A.D., Piaz V.D., Biagini P., Biancalani C., Graziano A., Cesari N. 4-Amino-5-vinyl-3(2H)-pyridazinones and analogues as potent antinociceptive agents: Synthesis, SARs, and preliminary studies on the mechanism of action. Bioorg. Med. Chem. 2007;15:5563–5575. doi: 10.1016/j.bmc.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 43.Cilibrizzi A., Quinn M.T., Kirpotina L.N., Schepetkin I.A., Holderness J., Ye R.D., Rabiet M.J., Biancalani C., Cesari N., Graziano A., et al. 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones: A novel class of small-molecule agonists for formyl peptide receptors. J. Med. Chem. 2009;52:5044–5057. doi: 10.1021/jm900592h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Memdouh S., Gavrilović I., Ng K., Cowan D., Abbate V. Advances in the detection of growth hormone releasing hormone synthetic analogs. Drug Test. Anal. 2021;13:1871–1887. doi: 10.1002/dta.3183. [DOI] [PubMed] [Google Scholar]
- 45.Cheng A., Best S.A., Merz K.M., Jr., Reynolds C.H. GB/SA water model for the Merck molecular force field (MMFF) J. Mol. Graph. Model. 2000;18:273–282. doi: 10.1016/S1093-3263(00)00038-3. [DOI] [PubMed] [Google Scholar]
- 46.Stewart J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004;10:155–164. doi: 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- 47.Stewart J.J. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990;4:1–105. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 48.Floresta G., Pittalà V., Sorrenti V., Romeo G., Salerno L., Rescifina A. Development of new HO-1 inhibitors by a thorough scaffold-hopping analysis. Bioorg. Chem. 2018;81:334–339. doi: 10.1016/j.bioorg.2018.08.023. [DOI] [PubMed] [Google Scholar]
- 49.Floresta G., Amata E., Dichiara M., Marrazzo A., Salerno L., Romeo G., Prezzavento O., Pittalà V., Rescifina A. Identification of Potentially Potent Heme Oxygenase 1 Inhibitors through 3D-QSAR Coupled to Scaffold-Hopping Analysis. ChemMedChem. 2018;13:1336–1342. doi: 10.1002/cmdc.201800176. [DOI] [PubMed] [Google Scholar]
- 50.Floresta G., Fallica A.N., Salerno L., Sorrenti V., Pittalà V., Rescifina A. Growing the molecular architecture of imidazole-like ligands in HO-1 complexes. Bioorg. Chem. 2021;117:105428. doi: 10.1016/j.bioorg.2021.105428. [DOI] [PubMed] [Google Scholar]
- 51.Varrica M.G., Zagni C., Mineo P.G., Floresta G., Monciino G., Pistarà V., Abbadessa A., Nicosia A., Castilho R.M., Amata E., et al. DNA intercalators based on (1,10-phenanthrolin-2-yl)isoxazolidin-5-yl core with better growth inhibition and selectivity than cisplatin upon head and neck squamous cells carcinoma. Eur. J. Med. Chem. 2018;143:583–590. doi: 10.1016/j.ejmech.2017.11.067. [DOI] [PubMed] [Google Scholar]
- 52.Zagni C., Citarella A., Oussama M., Rescifina A., Maugeri A., Navarra M., Scala A., Piperno A., Micale N. Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker. Int. J. Mol. Sci. 2019;20:945. doi: 10.3390/ijms20040945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is contained within the article or supplementary material.