

Abstract
Cancer affects more than 19 million people and is the second leading cause of death in the world. One of the principal strategies used in cancer therapy is the inhibition of topoisomerase II, involved in the survival of cells. Side effects and adverse reactions limit the use of topoisomerase II inhibitors; hence, research is focused on discovering novel compounds that can inhibit topoisomerase II and have a safer toxicological profile. Marine organisms are a source of secondary metabolites with different pharmacological properties including anticancer activity. The objective of this review is to present and discuss the pharmacological potential of marine-derived compounds whose antitumor activity is mediated by topoisomerase II inhibition. Several compounds derived from sponges, fungi, bacteria, ascidians, and other marine sources have been demonstrated to inhibit topoisomerase II. However, some studies only report docking interactions, whereas others do not fully explain the mechanisms of topoisomerase II inhibition. Further in vitro and in vivo studies are needed, as well as a careful toxicological profile evaluation with a focus on cancer cell selectivity.
Keywords: topoisomerase II, cancer chemotherapy, marine compounds, sponges, marine fungi, marine bacteria, marine invertebrates
1. Introduction
Cancer is the second leading cause of death in the world after cardiovascular diseases, affecting an estimated 19 million people and causing approximately 10 million deaths in 2020 [1].
Chemotherapy represents the main anticancer therapeutic approach. Nowadays, the principal clinically employed anticancer drugs are natural products, or their structural analogs [2,3,4,5,6]. However, several factors limit their effectiveness: (i) their efficacy is inversely proportional to disease progression; (ii) occurrence of chemoresistance; (iii) severe toxicity caused by lack of selectivity against cancer cells [7,8]. For this reason, the discovery of anticancer agents characterized by an improved pharmaco-toxicological profile remains a major aim of pharmacological research.
One of the principal targets of drugs used in chemotherapy to stop the aberrant proliferation of cancer cells is topoisomerase (topo) II [9].
Topo is a class of nuclear enzymes essential for cell survival. They regulate the topology of DNA and are involved in replication, transcription, proliferation, and chromosome segregation during the cell cycle. Vertebrates express two different isoforms of topo II–α and β–and although they possess 70% sequence homology and show similar enzyme activity, they are expressed and regulated differently [10].
The mechanism of action of topo II is the temporary break of both DNA strands to allow supercoil relaxation and the physiological cellular process.
Specifically, topo II acts on both strands of DNA, being capable of removing knots or tangles from the entire DNA duplex. In fact, the cut that is occasioned in a specific region of DNA (Gate-segment) allows another DNA duplex (Transport-segment) to be crossed throughout this break, unwinding the DNA. Topo II generates a covalent interaction–called cleavage complex–with the newly cut G-segment [9]. In particular, the catalytic cycle of topo II is composed of: (i) binding DNA segments (G- and T-); (ii) flexing of the G-segment in the presence of metals ions; (iii) formation of the cleavage complex by a nucleophilic attack occasioned by tyrosine residues present in the catalytic site of the enzyme; (iv) closing the gate to constrain the T-segment to pass through G-segment; (v) ligation of the G-segment and release of the T-segment, and (vi) releasing of the G-segment mediated by ATP hydrolysis and arrangement of the enzyme for a new catalytic cycle (Figure 1) [9].
Figure 1.

Catalytic cycle of topo II. Binding DNA segments ➀; flexing of the G-segment in the presence of metals ions ➁; formation of the cleavage complex ➂; closing the gate to constrain the T-segment to pass through the G-segment ➃; ligation of the G-segment ➄; release of the T-segment ➅; release of the G-segment ➆; enzyme ready for a new catalytic cycle ➇.
Thus, the inhibition of topo activity allows the blocking of the cell cycle and then conduces to cell death [11]. Topo II-mediated DNA breakage is a critical step for cell survival and must be finely regulated to avoid a possible fragmentation of the entire genome [9]. In a healthy cell, there is fine control of the formation of cleavage complexes, which are short-lived and reversible. Topo II inhibitors are compounds capable of modulating the formation of cleavable complexes and altering this equilibrium.
There are two different mechanisms described for topo II inhibition: (i) poisoning or (ii) catalytic inhibition. Poisoning is the main mechanism and acts on the stabilization of the cleavable complex, leading to maintaining the permanent breakage of DNA. Indeed, when the levels of cleavable complexes become high, they cannot be repaired by topo II, thus becoming irreversible DNA lesions that activate different signaling pathways and result in cell death by apoptosis [12]. On the other hand, catalytic inhibition implies that the inhibitor prevents the formation of the cleavage complex. If the amount of cleavage complexes is poor, the DNA relaxation is impeded, the daughter chromosomes remain entangled, and segregation is not possible during mitotic replication. In addition, in this case, apoptotic cell death is activated [9].
The stabilization of the cleavage complex mediated by topo II poisons or the blocking of its catalytic activity by topo II catalytic inhibitors are two opposite processes that both lead to cell death by induction of apoptosis.
One of the most important classes of anticancer drugs targeting topo II is anthracyclines, extracted from Streptomyces genus bacteria. The most used anthracycline is doxorubicin (DOXO), as well as its epimer epirubicin [13] and its derived valrubicin [13] that act as topo II poisons [14]. However, it has been described that the use of DOXO leads to important side effects, such as cardiomyopathy [15].
Other drugs, derived from a natural source, act as topo II poisons: for instance, etoposide (ETO) and its analog teniposide, two podophyllotoxins obtained from the herbaceous plant of the Podophyllum genus [16], and resveratrol [17], an important polyphenol found in several vegetable sources. Examples of natural topo II catalytic inhibitors are tryptanthrin, obtained from Candida lipolytica yeast or from several plant genera as Clanthe, Isatis, Wrightia, Couroupota [18]; fisetin [19] and myricetin [20], flavonoids present in several fruits; or daurinol [21], a triterpene isolated from Haplophyllum dauricum.
For several years, the research of new molecules with anticancer activity has also been centered on marine sources such as sponges, fungi, bacteria, etc. [22]. In fact, due to some unfavorable environmental conditions, such as salinity, temperature and pressure alterations, competition for free soil, etc., the marine organisms had to develop several adaptative mechanisms, which are mediated by the secondary metabolites [23]. Secondary metabolites exhibit a wide range of biological effects and pharmaceutical activities. However, their discovery and characterization are limited by the low quantities that are achievable from these organisms. Despite this limitation, several marine-derived compounds possess very interesting antitumor potential [24].
The purpose of this study was to investigate compounds derived from marine sources–specifically sponges, fungi, bacteria, ascidians, echinoderms, and marine microalgae, with proven antitumor activity mediated by topo II inhibition–through a systematic review. In July 2022, a literature search was conducted using the public databases Pubmed Scopus, and Web of Science. The search strategy used free descriptors and terms, limiting articles to the human topo II and the English language, regardless of publication year. The search retrieved all relevant articles related to marine-derived compounds, topo II inhibition, and antitumor activity. In vivo and in vitro studies are included.
2. Topo II Inhibitors from Marine Sponges
2.1. Neoamphimedine and Amphimedine
The alkaloids neoamphimedine (neo) (Figure 2a) and its regioisomer amphimedine (Figure 2b) are two pyridoacridines isolated from Xestospongia sp.
Figure 2.

Chemical structure of neoamphimedine ((a), CAS number: 221456-55-9) and amphimedine ((b), CAS number: 86047-14-5).
Neo was highly cytotoxic in several tumor cell lines [25,26]. In addition, neo was equally cytotoxic in wild-type A2780 ovarian cancer cells and in multidrug-resistant (MDR)-expressing A2780AD cell line (Table 1). Of note, taxol, DOXO, and amsacrine (m-AMSA) had a 15-, 33-, and 8-fold lower cytotoxicity than neo [25]. In vivo, the administration of neo (12.5–50 mg/kg for 19 days) to Balb/c nu/nu mice bearing HCT-116 and KB xenograft reduced tumor growth (Table 1) and displayed the same efficacy as ETO [25].
Table 1.
Sponge-derived inhibitors of topo II.
| Compound | Sponge Species (Sp.) | Topo II Inhibitory Activity | Antitumor Effect(s) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Assay | IC50/IC90 a or Range of Concentrations Tested | Outcomes | Experimental Model | Cytotoxic Activity (IC50 b) | Other Antitumor Mechanism(s) |
|||
| (1′R,5′S,6′S)-2-(3′,5′-dibromo-1′,6′-dihydroxy-4′-oxocyclohex-2′-enyl) acetonitrile |
Pseudoceratina sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | 2.5–10 μg/mL | ↓ DNA relaxation in presence of topo IIα | K562 | IC50 (72 h): 1.4 μg/mL | Apoptosis (↑ cleaved PARP, ↑ cleaved caspase-3, ↑ XIAP protein expression) DNA damage (↑ p-ATM, ↑ p-ATR, ↑ γH2A.X protein expression, ↑ p-BRCA, ↑ p-Chk2 protein expression) Oxidative stress (↑ ROS) ↓ IKK/NFκB pathway ↑ PI3K/Akt pathway |
[35] |
| HeLa | IC50 (72 h): 4.8 μg/mL | |||||||
| MCF-7 | IC50 (72 h):1.9 μg/mL | |||||||
| MDA-MB-231 | IC50 (72 h): 5.5 μg/mL | |||||||
| 2-tetraprenil-1,4-benzoquinone | Carteriospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 0.43 μg/mL | ↓ DNA relaxation and formation of supercoiled DNA products in presence of topo IIα | Molt-4 | IC50 (72 h): 0.34 μg/mL | Apoptosis (↓ MMP) | [74] |
| K562 | IC50 b (72 h): 0.70 μg/mL | |||||||
| HL-60 | IC50 (72 h): 0.42 μg/mL | |||||||
| U937 | IC50 (72 h): 0.65 μg/mL | |||||||
| Sup-T1 | IC50 (72 h): 0.33 μg/mL | |||||||
| Ca9-22 | IC50 (72 h): 0.97 μg/mL | |||||||
| Cal-27 | IC50 (72 h): 0.51 μg/mL | |||||||
| LNCaP | IC50 (72 h): >20 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 15.41 μg/mL | |||||||
| T-47D | IC50 (72 h): 1.06 μg/mL | |||||||
| Immunodeficient athymic mice bearing Molt-4 xenograft | ↓ Tumor growth (1.14 μg/g/day, for 33 days) | |||||||
| 10-acetylirciformonin B | Ircinia sp. | Cell-free DNA cleavage assay using an enzyme-mediated supercoiled pHOT1 plasmid DNA and human topo II | 1.5–6.0 μM | ↓ DNA relaxation and formation of supercoiled DNA products in the presence of topo IIα | HL-60 | Not indicated | Apoptosis (↓ MMP; ↓ Bcl-2, ↓ Bcl-xL, ↓ XIAP, ↓ survinin, ↑ BAX, ↑ cleaved PARP protein expression, ↑ cyt c release) ↓ p-Akt, ↓ p-PTEN, ↓ Src, ↓ HKII, ↓ PKM 2, ↑ p-ERK, ↑ p-38, ↑ p-JNK, ↑ p-GSK 3β protein expression Oxidative stress (↑ ROS) |
[57] |
| Western Blotting on HL-60 cells | 3.0 μM | ↓ Topo IIα protein expression | ||||||
| 12β-(3′β-hydroxybutanoyloxy)-20,24-dimethyl-24-oxo-scalara- 16-en-25-al | Carteriospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 1.98 μg/mL | ↓ DNA relaxation and formation of supercoiled DNA products in presence of topo IIα | K562 | IC50 (72 h): 0.01 μg/mL | [74] | |
| Molt-4 | IC50 (72 h): 0.01 μg/mL | Apoptosis (↓ MMP; ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ cleaved PARP protein expression) ↑ DNA damage (↑ γH2AX protein expression, ↑ DNA DSBs) Oxidative stress (↑ ROS) ↑ ER stress (↑ Ca2+ release; ↑ IRE 1α, ↑ Bip, ↑ CHOP, ↑ Grp94, ↑ Hsp70, ↑ ATF6, ↓ PERK protein expression) ↓ Hsp90 (↓ Akt, ↓ p70S6k, ↓ NFκB, ↓ Raf-1, ↓ p-GSK3β, ↓ XIAP, ↓ MDM 2 ↓ Rb2, ↓ CDK4 ↓Cyclin D3, ↓ HIF 1 ↓ HSF1; ↑ Hsp70 protein expression) |
||||||
| HL-60 | IC50 (72 h): 0.01 μg/mL | |||||||
| U937 | IC50 (72 h): 0.01 μg/mL | |||||||
| Sup-T1 | IC50 (72 h): 0.13 μg/mL | |||||||
| Ca9-22 | IC50 (72 h): 0.10 μg/mL | |||||||
| Cal-27 | IC50 (72 h): 0.56 μg/mL | |||||||
| LNCaP | IC50 (72 h): 13.87 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 2.33 μg/mL | |||||||
| T-47D | IC50 (72 h): 2.19 μg/mL | |||||||
| 12β-(3′β-hydroxypentanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al | Carteriospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 0.37 μg/mL | ↓ DNA relaxation | K562 | IC50 (72 h): 0.35 μg/mL | [74] | |
| Molt-4 | IC50 (72 h): 0.30 μg/mL | Apoptosis (↓ MMP) DNA damage (↑ γH2AX protein expression) |
||||||
| HL-60 | IC50 (72 h): 0.22 μg/mL | |||||||
| U937 | IC50 (72 h): 0.61 μg/mL | |||||||
| Sup-T1 | IC50 (72 h): 0.42 μg/mL | |||||||
| Ca9-22 | IC50 (72 h): 1.48 μg/mL | |||||||
| Cal-27 | IC50 (72 h): 3.17 μg/mL | |||||||
| LNCaP | IC50 (72 h): >20 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 1.71 μg/mL | |||||||
| T-47D | IC50 (72 h): 1.87 μg/mL | |||||||
| 14-methoxy-xestoquinone | Xestopongia sp. | Cell-free DNA relaxation assay with supercoiled pBR322 DNA plasmid and topo II of drosophila | IC90: 143 μM | ↓ DNA relaxation | HCT-116 | IC50 (18 + 72 h): 28 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): 4.3 μM | |||||||
| 14-chloro-15-hydroxyxestoquinone | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 110 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): 33 μM | [77] | |
| Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 135 μM | ↓ DNA relaxation | CHO xrs-6 c | IC50 (18 + 72 h): 27 μM | ||||
| 15-methoxy-xestoquinone | Xestopongia sp. | Cell-free DNA relaxation assay with supercoiled pBR322 DNA and Topo II of drosophila | IC90: 143 μM | ↓ DNA relaxation | HCT-116 | IC50 (18 + 72 h): 28 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): 4.3 μM | |||||||
| 15-chloro-14-hydroxyxestoquinone | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 110 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): 33 μM | [77] | |
| Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 135 μM | ↓ DNA relaxation | CHO xrs-6 c | IC50 (18 + 72 h): 27 μM | ||||
| 24R, 25S-Manoalide | Luffariella sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 1.18 μM | ↓ DNA relaxation | Molt-4 | IC50 (72 h): 0.82 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-8, ↑ cleaved caspase-9 protein expression) DNA damage (↑ p-ATM, ↑ p-Chk2, ↑ γH2AX protein expression, ↑ DSBs) Oxidative stress (↑ ROS) |
[61] |
| K562 | IC50 (72 h): 7.67 μM | |||||||
| Sup-T1 | IC50 (72 h): 1.35 μM | |||||||
| U937 | IC50 (72 h): 1.56 μM | |||||||
| Adociaquinone A | Xestopongia sp. | Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 118 μM | ↓ DNA relaxation | HCT-116 | IC50 (18 + 72 h): 24 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): 78 μM | |||||||
| Adociaquinone B | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: <11 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): 21 μM | [77] | |
| Cell-free DNA relaxation assay with supercoiled pBR322 DNA and topo II of drosophila | IC90: 78 μM | ↓ DNA relaxation | CHO xrs-6 c | IC50 (18 + 72 h): 23 μM | ||||
| KSDS assay | / | Formation of enzyme-DNA cleavable complex | ||||||
| Aeroplysinin 1 | Pseudoceratina sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 1.37 μM | ↓ DNA relaxation | Molt-4 | IC50 (72 h): 0.12 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α, ↓ HO-1, ↑ catalase, ↑ MnSOD, ↓ NOX4, ↑ NOX2 protein expression) ↑ Hsp70 protein expression ↓ EGFR, ↓ p-EGFR, ↓ β-catenin protein expression |
[36] |
| Western blotting on Molt-4 cells | 0.1–0.4 μM | ↓ Topo IIα protein expression | K562 | IC50 (72 h): 0.54 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α protein expression) ↓ β-catenin protein expression |
|||
| Western blotting on PC-3 cells | 0.8–3.2 μM | ↓ Topo IIα protein expression | PC-3 | IC50 (72 h): 0.58 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP ↓ Bcl-2, ↓ p-mTOR protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α, ↓ HO-1, ↑ catalase, ↑ MnSOD, ↑ NOX4, ↓ NOX2 protein expression) ↓ Hsp90, ↑ Hsp70 protein expression ↓ Colony formation ↓ Cell migration ↓ EMT ↓ EGFR, ↓ p-EGFR, ↓ β-catenin protein expression |
|||
| Du145 | IC50 (72 h): 0.33 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↓ p-Akt, ↓ XIAP ↓ Bcl-2, ↓ p-mTOR protein expression) Oxidative stress (↑ ROS; ↓ HIF-1 α, ↓ HO-1, ↑ catalase, ↑ MnSOD, ↓ NOX4, ↑ NOX2 protein expression) ↓ Hsp90 protein expression ↓ colony formation ↓ cell migration ↓ EMT |
||||||
| CCD966S d | IC50 (72 h): 1.54 μM | |||||||
| NR8383 d | IC50 (72 h): 6.77 μM | |||||||
| Bastadin-14 | Psammaplysilla purpurea | Not indicated | IC50: 2 μg/mL | ↓ Topo IIα protein activity | A-549 | IC50: 2 μg/mL | [79] | |
| HT-29 | ||||||||
| P388 e | ||||||||
| CV-1 d | IC50: 2.5 μg/mL | |||||||
| Batzelline A | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (88.45 μM) | ↓ DNA decatenation (58%) | Panc-1 | IC50 (72 h): >17.68 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (88.45 μM) | DNA intercalation (18%) | AsPC-1 | IC50 (72 h): >17.68 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): >17.68 μM | |||||||
| Mia PaCa2 | ||||||||
| Vero d | ||||||||
| Batzelline B | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (93.02 μM) | ↓ DNA decatenation (63%) | Panc-1 | IC50 (72 h): >18.61μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (93.02 μM) | DNA intercalation (21%) | AsPC-1 | IC50 (72 h): >18.61μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): >18.61μM | |||||||
| Mia PaCa2 | ||||||||
| Vero d | ||||||||
| Elenic acid | Plakinastrella sp. | Not indicated | IC50: 0.1 μg/mL | ↓ Topo IIα activity | P388 e | IC50 (time not indicated): 5 μg/mL | [80] | |
| A-549 | ||||||||
| MEL-28 | ||||||||
| Halenaquinone | Petrosia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | IC50: 0.0055 μg/mL (0.017 μM) | ↓ DNA relaxation | Molt-4 | IC50 (24 h): 0.61 μg/mL IC50 (72 h): 0.18 μg/mL |
Apoptosis (↓ MMP; ↑ c-PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-7, ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ Bax, ↑ cyt c, ↓ Bcl-2, ↓ Bid protein expression) ↓ p-Akt, ↓ p-PTEN, ↓ p-GSK3β, ↓ p-PDK1 and ↓ HKII protein expression Oxidative stress (↑ ROS) Inhibition of HDAC activity (↑ acetyl-H3, ↑ acetyl-H3K18 protein expression) |
[75] |
| Western Blotting on Molt-4 cells | 1.25 μg/mL | ↓ Topo IIα protein expression | Immunodeficient athymic mice bearing Molt-4 xenograft | ↓ Tumor growth, ↓ tumor weight, ↓ tumor volume (1μg/g/day for 30 days) | ||||
| K562 | IC50 (72 h): 0.48 μg/mL | Apoptosis (↑ cleaved PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-7 protein expression) | ||||||
| MDA-MB-231 | IC50 (72 h): 8 μg/mL | |||||||
| DLD-1 | IC50 (72 h): 6.76 μg/mL | |||||||
| Heteronemin | Hippospongia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA and human topo II | 2.56–40.9 μM | ↓ DNA relaxation and formation of supercoiled DNA products in the presence of topo IIα | LNCaP | IC50 (24 h) 1.4 μM IC50 (48 h): 0.8 μM IC50 (72 h): 0.4 μM |
Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3 protein expression) Oxidative stress (↑ ROS) ↑ ER stress (↑ Ca2+ release, ↑ IRE 1α, ↑ Bip, ↑ CHOP, ↑ Hsp70, ↓ ATF6, ↓ PERK protein expression) ↓ Hsp90, ↓ IRAK1, ↓ p-Akt, ↓ XIAP, ↓ Rb2, ↓ HDAC1, ↓ PCNA ↓ CDK4, ↓ p-STAT3, ↑ Hsp70 protein expression Autophagy (↑ LC3-II protein expression) |
[62] |
| Western blotting on LnCaP cells | 0.64–2.56 μM | ↓ Topo IIα protein expression | PC-3 | IC50 (24 h): 2.7 μM | Apoptosis | |||
| Immunodeficient athymic mice bearing LNcaP xenograft | IC50 (24 h): 7 μM | ↓ Tumor size, ↓ tumor growth (1 mg/Kg b.w.; for 29 days) | ||||||
| Hippospongic acid A | Hippospongia sp. | Cell-free DNA relaxation assay using supercoiled pUC19 DNA plasmid and topo II | IC50: 15 μM | ↓ DNA relaxation | NUGC-3 | IC50 (time not indictaed): 9.5 μM | ↓ Cell cycle in phases G1 and G2/M Apoptosis (↑ DNA fragmentation) |
[55] |
| Isobatzelline A | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (88.73 μM) | ↓ DNA decatenation (36%) | Panc-1 | IC50 (72 h): 9.37 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (88.73 μM) | DNA intercalation (54%) | AsPC-1 | IC50 (72 h): 1.74 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 2.39 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 4.34 μM | |||||||
| Verod | IC50 (72 h): >17.75 μM | |||||||
| Isobatzelline C | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (106.38 μM) | ↓ DNA decatenation (27%) | Panc-1 | IC50 (72 h): 9.99 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (106.38 μM) | DNA intercalation (56%) | AsPC-1 | IC50 (72 h): 1.72 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 1.31 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 2.34 μM | |||||||
| Verod | IC50 (72 h): >21.28 μM | |||||||
| Isobatzelline D | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (89.61 μM) | ↓ DNA decatenation (26%) | Panc-1 | IC50 (72 h): 5.72 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (89.61 μM) | DNA intercalation (47%) | AsPC-1 | IC50 (72 h): 1.48 μM | ↓ cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 1.48 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 2.67 μM | |||||||
| Verod | IC50 (72 h): 15.70 μM | |||||||
| Isobatzelline E | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (107.30 μM) | ↓ DNA decatenation (95%) | Panc-1 | IC50 (72 h): >21.46 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (107.30 μM) | DNA intercalation (27%) | AsPC-1 | IC50 (72 h): >21.46 μM | ↓ Cell cycle in phase G2 | |||
| BxPC-3 | IC50 (72 h): >21.46 μM | |||||||
| Mia PaCa2 | ||||||||
| Verod | ||||||||
| Makaluvamine A |
Zyzzya cf. marsailis |
Cell-free decatenation reaction of kinetoplast DNA | IC90: 41 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 1.3 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 2.1 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 0.41 μM | ||||
| Neutral filter elution assay | Not indicated | Production of cleavable complexes (strand scission factor = 1.38) | Balb/C nu/nu athymic mice bearing OVCAR-3 xenograft | ↓ Tumor mass: T/C: 62% (0.5 mg/kg for 4 weeks) | ||||
| Analysis of absorbance spectra of calf thymus DNA | Not indicated | 53% absorption hypochromism | ||||||
|
Zyzzya
fuliginosa |
Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid | 91 mM | 17% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Makaluvamine B |
Zyzzya cf. marsailis |
Cell-free decatenation reaction of kinetoplast DNA | IC90: 500 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): >50 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 181 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 13.49 μM | ||||
| Makaluvamine C |
Zyzzya cf. marsailis |
Cell-free decatenation reaction of kinetoplast DNA | IC90: 420 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 36.2 μM | / | [49] |
| in vitro cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 1.2 μM | Topo II-mediated cleavage of plasmid DNA | ||||||
| Analysis of absorbance spectra of calf thymus DNA | Not indicated | 66% absorption hypochromism | CHO xrs-6 c | IC50 (time not indicated): 5.4 μM | ||||
| Balb/C nu/nu athymic mice bearing OVCAR-3 xenograft | ↓ Tumor mass: T/C: 48% (5 mg/kg for 4 weeks) | |||||||
| Immune competent mice inoculated with P388 e cells | ↑ MLS: 18% (5 mg/kg for 4 weeks) | |||||||
|
Zyzzya
fuliginosa |
Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and human topo II | 91 mM | 16% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Cell-free cleavage assay of pUC 19 radiolabeled DNA with human topo II | 33–466 μM | Cleavage of pUC 19 DNA at nucleoside A329 Formation of cleavable complex |
||||||
| Makaluvamine D |
Zyzzya cf. marsailis |
Cell-free decatenation reaction of kinetoplast DNA | IC90: 320 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 17.1 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 52 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 14 μM | ||||
|
Zyzzya
fuliginosa |
Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 5% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| in vitro cell-free cleavage assay of pUC 19 radiolabeled DNA with human topo II | 33–466 μM | Cleavage of pUC 19 DNA at nucleoside A329 Formation of cleavable complex |
||||||
| Makaluvamine E |
Zyzzya cf. marsailis |
Cell-free decatenation reaction of kinetoplast DNA | IC90: 310 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 1.2 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 15 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 1.7 μM | ||||
| Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 22% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Cell-free cleavage assay of pUC 19 radiolabeled DNA with human topo II | 33–466 μM | Cleavage of pUC 19 DNA at nucleoside A329 Formation of cleavable complex |
||||||
| Makaluvamine F |
Zyzzya cf. marsailis |
Cell-free decatenation reaction of kinetoplast DNA | IC90: 25 μM | ↓ DNA relaxation | HCT-116 | IC50 (time not indicated): 0.17 μM | [49] | |
| Cell-free DNA cleavage assay with supercoiled pBR322 DNA plasmid | IC50: 1.1 μM | Topo II-mediated cleavage of plasmid DNA | CHO xrs-6 c | IC50 (time not indicated): 0.08 μM | ||||
| Makaluvamine H |
Zyzzya
fuliginosa |
Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 33% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | Athymic nude mice bearing KB xenograft | Not indicated | ↓ Tumor growth (22 mg/kg, days 1, 4, and 8 for 28 days) | [51] |
| Makaluvamine I |
Zyzzya
fuliginosa |
Cell-free DNA cleavage assay with radiolabeled and supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 61% of Topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | CHO xrs-6 c | IC50 (time not indicated): 0.4 μM | [51] | |
| CHO AA8 c | IC50 (time not indicated): 2 μM | |||||||
| Athymic nude mice bearing KB xenograft | ↓ Tumor growth (11 mg/kg, days 1, 4, and 8 for 28 days) | |||||||
| Makaluvamine N |
Zyzzya
fuliginosa |
Cell-free DNA relaxation assay using a supercoiled pBR322 DNA plasmid and human topo II | / | ↓ Topo II unwinding (>90% at 5 μg/mL) | HCT-116 | IC50 (72 h): 0.6 μg/mL | [46] | |
|
Zyzzya
fuliginosa |
Cell-free DNA cleavage assay with radiolabeled supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 26% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | ||||
| Makaluvamine V | Zyzzya fuliginosa | Cell-free DNA cleavage assay with radiolabeled supercoiled rf M13 mp19 DNA plasmid and topo II | 91 mM | 2% of topo II-mediated cleavage of plasmid DNA (compared to 100% of etoposide) | [51] | |||
| Manoalide 25-acetals |
Hirtios erecta | Not indicated | IC50: 25 μM | ↓ DNA-unknotting activity of calf thymus DNA topo II | CDF1 mice inoculated with P388 e cells | T/C: 150% (1 μg/g) | [60] | |
| Neoamphidemine | Xestospongia sp. | Cell-free decatenation reaction of kinetoplast DNA | 0.5–30 μM | ↓ DNA decatenation | HEK-293 | IC50 (72 h): 0.8 μM | [29] | |
| Malachite green assay with supercoiled DNA, recombinant human topoIIα | 0–10 μM | Competitive inhibition of topo II-mediated ATP hydrolysis | HEK293-Metnase | IC50 (72 h): 0.5 μM | ||||
| Molecular docking | / | Bind to the ATPase site of topo II | ||||||
| Not indicated | Not indicated | Catenation of DNA to high molecular weight complex | CHO AA8 c | IC50 (time not indicated): 2 μg/mL | [27] | |||
| Not indicated | Not indicated | 3% of topo II-mediated DNA cleavage | ||||||
| Cell-free DNA cleavage assay using radiolabeled and supercoiled rf M13 mp19 DNA and human topo II | Not indicated | Catenation of DNA to high molecular weight complex | HCT-116 | IC50 (72 h): 4.5 μM | [25] | |||
| 50 μM | 8.9% of topo II-mediated DNA cleavage | |||||||
| Transmission electron microscopy analysis | Not indicated | Catenation of DNA | SK-mel-5 | IC50 (72 h): 7.6 μM | ||||
| DNA filter-binding assay | 100–600 μM | Catenation of DNA through DNA aggregation | KB | IC50 (72 h): 6 μM | ||||
| MCF-7 | IC50 (72 h): 1.8 μM | |||||||
| A2780 | IC50 (72 h): 0.9 μM | |||||||
| A2780AD | IC50 (72 h): 0.83 μM | |||||||
| CHO AA8 c | IC50 (72 h): 2.5 μM | |||||||
| CHO xrs-6 c | IC50 (72 h): 1.6 μM | |||||||
| Balb/c nu/nu mice bearing HCT-116 xenograft | ↓ Tumor growth (12.5, 25, and 50 mg/kg for 19 days) | |||||||
| Balb/c nu/nu mice bearing KB xenograft | ↓ Tumor growth (50 mg/kg for 19 days) | |||||||
| Popolohuanone E | Dysidea sp. | Not indicated | IC50: 400 μM | ↓ Topo II activity |
P388 e | IC50: 20 μg/mL | [81] | |
| HT-29 | IC50: >20 μg/mL | |||||||
| A549 | IC50: 2.5 μg/mL | |||||||
| CV-1 d | IC50: >20 μg/mL | |||||||
| Secobatzelline A | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (98.04 μM) | ↓ DNA decatenation (61%) | Panc-1 | IC50 (72 h): 10.39 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (98.04 μM) | DNA intercalation (34%) | AsPC-1 | IC50 (72 h): 3.62 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): 4.10 μM | |||||||
| Mia PaCa2 | IC50 (72 h): 5.62 μM | |||||||
| Verod | IC50 (72 h): 14.03 μM | |||||||
| Secobatzelline B | Batzella sp. | Cell-free decatenation reaction of kinetoplast DNA | 25 μg/mL (97.66 μM) | ↓ DNA decatenation (13%) | Panc-1 | IC50 (72 h): 17.38 μM | [54] | |
| Ethidium bromide displacement fluorescence assay | 25 μg/mL (97.66 μM) | DNA intercalation (17%) | AsPC-1 | IC50 (72 h): >19.531 μM | ↓ Cell cycle in phase S | |||
| BxPC-3 | IC50 (72 h): >19.53 μM | |||||||
| Mia PaCa2 | ||||||||
| Verod | ||||||||
| Secoadociaquinone A | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 113 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): >143 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): >247 μM | |||||||
| Secoadociaquinone B | Xestopongia sp. | Cell-free decatenation reaction of kinetoplast DNA | IC90: 113 μM | ↓ DNA decatenation | HCT-116 | IC50 (18 + 72 h): >143 μM | [77] | |
| CHO xrs-6 c | IC50 (18 + 72 h): >247 μM | |||||||
| Xestoquinone | Petrosia sp. | Cell-free DNA cleavage assay using an enzyme-mediated negatively-supercoiled pHOT1 plasmid DNA and topo II | IC50: 0.094 μM | ↓ DNA relaxation |
Molt-4 | IC50 (24 h): 2.95 μM | Apoptosis (↓ MMP; ↑ cleaved PARP, ↑ cleaved caspase-3, ↑ cleaved caspase-7, ↑ cleaved caspase-8, ↑ cleaved caspase-9, ↑ Bax, ↑ Bak, ↑ cyt c, ↑ Fas, ↑ TRADD, ↓ Bcl-2, ↓ Bid, ↓ XIAP protein expression ↑ ER stress (↑ Ca2+ release; ↑ CHOP, ↓ IRE 1α, ↓ PERK protein expression) ↓ HDAC activity (↓ HDAC1, ↓ HDAC3, ↓ HDAC4, ↓ HDAC6, ↓ HDAC7, ↓ HDAC8 protein expression) DNA damage (↑ p-Chk1, ↑ p-Chk2, ↑ γH2AX protein expression) Oxidative stress (↑ ROS) Interaction with Hsp90 |
[76] |
| Immunodeficient athymic mice bearing Molt-4 xenograft | ↓ Tumor growth, ↓ tumor weight, ↓ tumor volume (1μg/g/day for 50 days) ↓ HDAC1, ↓ HDAC3, ↓ HDAC8 protein expression |
|||||||
| Western Blotting on Molt-4 and K562 cells | 7.84 μM | ↓ Topo IIα protein expression | K562 | IC50 (24 h): 6.22 μM | ||||
| Sup-T1 | IC50 (24 h): 8.58 μM | |||||||
| U937 | IC50 (24 h): 11.12 μM | |||||||
| NR8383 d | IC50 (24 h): >30 μM | |||||||
↑: upregulation/induction; ↓: downregulation/inhibition; a: concentration that inhibits 50% or 90% of topo II activity; b: concentration that inhibits 50% of cell viability; c: Chinese hamster ovary (CHO) double strand break repair-deficient cells; d: non tumor cells; e: murine cancer cells; A2780AD: A2780 multidrug resistant cells; Akt: protein kinase B; ATF6: activating transcription factor 6; ATM: ataxia telangiectasia mutated; ATR: ATM and RAD3-related; Bax: BCL2-associated X protein; Bcl-2: B-cell lymphoma 2; Ca2+: calcium; CDK4: cyclin-dependent kinase 4; Chk1: checkpoint kinase 1; Chk2: checkpoint kinase 2; Cyt c: cytochrome c; CHOP: C/EBP homologous protein; DSBs: double-strand breaks; EGFR: epidermal growth factor receptor; EMT: epithelial-mesenchymal transition; ER: endoplasmic reticulum; ERK: extracellular signal-regulated kinase; GSK 3β: glycogen synthase kinase-3 β; GRP4: glucose-regulated protein 94; H3: histone H3; HDAC: histone deacetylase; HK: hexokinase; HIF 1: hypoxia-inducible factor 1; HIF-1α: hypoxia-inducible factor 1-alpha; HO-1: heme oxygenase 1; HSF1: heat shock transcription factor 1; Hsp70: heat shock protein 70; Hsp90: heat shock protein 90; IRAK1: interleukin-1 receptor-associated kinase 1; IRE 1α: inositol-requiring enzyme 1α; JNK: Jun N-terminal kinase; KSDS: potassium sodium dodecyl sulfate; LC3-II: microtubule-associated proteins 1A/1B light chain 3B, LC3-phosphatidylethanolamine conjugate; mTOR: mammalian target of rapamycin; MDM 2: murine double minute 2; MLS: median life span; MMP: mitochondrial membrane permeabilization; MnSOD: manganese superoxide dismutase; NFκB: nuclear factor kappa B; NOX: NADPH oxidase; p-: phosphorylated; PARP: poly (ADP-ribose) polymerase; PCNA: proliferating cell nuclear antigen; PDK1: 3-phosphoinositide-dependent kinase 1; PERK: protein kinase RNA-like endoplasmic reticulum kinase; PKM2: pyruvate kinase muscle isozyme M2; PTEN: phosphatase and tensin homolog; Raf-1: V-raf-1 murine leukemia viral oncogene homolog 1; ROS: reactive oxygen species; Src: proto-oncogene tyrosine-protein kinase; STAT3: signal transducers and activators of transcription 3; T/C: ratio between the tumor volume in the treated (T) group and in the untreated control (C) group; TRADD: Fas-associated death domain protein; XIAP: X-linked inhibitor of apoptosis protein. Human breast cancer cell lines: MCF-7; MDA-MB-231; T-47D. Human cervical cancer cell line: HeLa. Human colon cancer cell lines: DLD-1; HCT-116; HT-29. Human epithelial carcinoma cell line: KB. Human gastric cancer cell line: NUGC-3. Human leukemia cell lines: MOLT-4; K562; HL-60. Human lymphoma cell lines: U937; Sup-T1. Human lung cancer cell line: A549. Human melanoma cell lines: MEL-28; SK-mel-5. Human ovarian cancer cell lines: OVCAR-3; A2780. Human oral carcinoma cell lines: Ca9-22; Cal-27. Human pancreatic cancer cell lines: Panc-1; AsPC-1; BxPC-3; Mia PaCa2. Human prostate cancer cell lines: LnCap; PC-3; Du-145.
Focusing on neo as a topo II inhibitor, early studies showed that this metabolite did not act as a topo II poison. Firstly, neo slightly cleaved DNA through the formation of cleavage complexes (Table 1) [25,27]; secondly, it did not cause a more pronounced cytotoxicity on Chinese hamster ovary (CHO) xrs-6 cells [double strand breaks (DSBs) repair-deficient] compared to CHO AA8 cells (DSBs repair-competent) [25], a typical behavior of topo II poisons [28]. Instead, neo inhibited the catalytic activity of topo II through the topo II-mediated catenation of DNA (both supercoiled and relaxed), only in the presence of active topo II, and promoted the aggregation of DNA into high molecular weight complexes, resulting from neither protein–DNA aggregation nor chemical cross-linking to DNA [25,27]. The regioisomer amphimedine did not exhibit any of these effects [25,27]. More recently, Ponder et al. reported that neo (0.5–30 μM) suppressed topo IIα-mediated DNA decatenation and competitively inhibited topo IIα-dependent ATP hydrolysis by binding to the ATPase site of the enzyme, with a binding energy equal to −61.8 kcal/mol, as reported in computational docking studies [29]. This in silico approach also elucidated that the lack of activity of amphidemine was due to the different position of the carbonyl moiety, which prevented the binding to the ATPase site of the enzyme. In the same study, the authors analyzed the topo IIα inhibitory activity of neo in the presence of metnase, a DNA repair protein that facilitates the reaction of DNA decatenation and contributes to the development of resistance against topo II inhibitors such as DOXO and ETO [30,31]. Neo maintained and exhibited even greater topo IIα inhibitory activity, as observed in human embryonic kidney 293 (HEK293) cells that over-express metnase compared to wild-type HEK293 cells [29]. Those results suggest that (i) the binding affinity of neo for topo IIα is higher when topo IIα interacts with metnase, and (ii) neo seems to elude the metnase-based mechanism of resistance.
The neo capability to act as a topo IIα ATP-competitive inhibitor was associated with the reversion of the epithelial–mesenchymal transition (EMT), as shown in a multicellular tumor spheroid model (MCTS) of colon cancer cells (SW620) [32]. EMT is a process that occurs when epithelial cells lose their characteristics and assume a mesenchymal phenotype. EMT boosts the metastatic potential of tumor cells, enabling them to get through the extracellular matrix, get into the bloodstream, and then proliferate in a distinct tissue [33]. Aberrant T-cell factor (TCF) transcription and β-catenin are involved in the Wnt signaling pathway, which actively participates in the EMT process. Topo IIα has a key role in the Wnt signaling pathway: it interacts with β-catenin, TCF4, Wnt response elements (WREs), and promoters of downstream target genes of TCF (c-Myc, vimentin, and axis inhibition protein 2). Acting as a topo IIα ATP-competitive inhibitor, neo reduced the topo II-dependent TCF transcription, both in vitro (colorectal cancer MCTS cells; 10 μM, 72 h of treatment) and in vivo (SW620 xenografted athymic nude mice; 5 mg/kg, once a week for 22 days) [32]. In SW620 MCTS, neo also prevented the binding of topo IIα and TCF4 to WREs and promoters and reverted EMT through (i) the downregulation of the protein expression of mesenchymal markers (vimentin, Slug, zinc-finger E-box binding homeobox 1, and c-Myc) and (ii) the upregulation of epithelial ones (zonula occludens-1 and E-cadherin) [32]. Overall, neo, as a topo IIα inhibitor, downregulates the transcriptional activity of the β-catenin/TCF4 nuclear complex, which can be considered an interesting target for the types of cancers–such as colon cancer–in which the Wnt pathway largely contributes to the carcinogenic process [34].
2.2. Aeroplysinin-1 and Its Oxidized Derivative
The brominated isoxazoline alkaloid aeroplysinin-1 (apl-1) (Figure 3a) and its oxidized derivative [DT; (1′R,5′S,6′S)-2-(3′,5′-dibromo-1′,6′-dihydroxy-4′-oxocyclohex-2′-enyl) acetonitrile] (Figure 3b) were isolated from the marine sponge Pseudoceratina sp. [35,36].
Figure 3.

Chemical structure of aeroplysinin-1 ((a), CAS number: 28656-91-9) and its oxidized derivative ((b), CAS number: 294208-35-8).
DT was cytotoxic on different tumor cell lines. Additionally, DT had a selective cytotoxic effect on tumor cells, since the cell viability of rat alveolar macrophage NR8383 cells was more than 80% after exposure to the highest tested concentration of the compound [35]. In the same study, DT (0.01–10 μg/mL) was found to inhibit topo IIα using a cell-free DNA cleavage assay with an enzyme-mediated negatively supercoiled pHOT1 plasmid DNA. In the presence of topo IIα, DT at low concentrations (0.01, 0.1, and 1 µg/mL) caused DNA relaxation, and at high concentrations (2.5, 5, and 10 µg/mL) blocked DNA relaxation. This means that DT interferes with the topo IIα catalytic cycle [35]. However, the compound did not generate linear DNA [35], which is associated with the stabilization of topo II-DNA cleavage complex typical of topo II poisons [37].
The link between the inhibition of topo IIα and the apoptotic activity of DT is controversial. DT increased the apoptotic fraction of K562 cells at concentrations of 2.5, 5.0, and 10 μg/mL. Moreover, the compound at 0.5 and 1.0 μg/mL activated caspase-3 (Casp-3) and cleaved poly (ADP-ribose) polymerase (PARP), while at 5 μg/mL it decreased Casp-3 activity and PARP cleavage. DT also induced the phosphorylation of various DNA damage-related proteins, including H2A histone family member X (H2A.X), ataxia telangiectasia mutated (ATM), breast cancer gene (BRCA), and ataxia-telangiectasia rad3-related (ATR) in the same concentration-dependent manner. Additionally, while 2.5 μg/mL of DT increased intracellular reactive oxygen species (ROS) levels in a time-dependent manner (0–60 min), at 5 μg/mL, ROS levels rose up to 30 min and then gradually decreased time-dependently [35]. This could possibly explain the lower activation of Casp-3 and the lower phosphorylation of DNA damage-related proteins in cells treated with DT 5 μg/mL. At the same time, the pre-treatment of cells with the ROS scavenger N-acetyl cysteine (NAC) inhibited the apoptotic activity and the protein expression of phosphorylated H2A.X (γ-H2A.X) induced by DT at 5 μg/mL [35]. This result points out that, although inhibition of topo IIα is associated with the activation of DNA damage-related proteins, overproduction of ROS also contributes to increase DNA damage and seems to be the major pro-apoptotic trigger. ROS-induced apoptosis by DT has been found to involve the IKK (IκB kinases)/NFκB (nuclear factor kappa B) and PI3K (phosphatidylinositol 3-kinase)/Akt signaling pathways, as demonstrated by the reduced expression of IKK/NFκB-related proteins and the increased phosphorylation of Akt [35]. Given that the continuous activation of IKK/NF-κB pathway promotes tumorigenesis [38], its inhibition by DT could be considered an additional mechanism of its antitumor effect. However, Akt activation is associated with tumor aggressiveness and drug resistance [39]. Hence, further investigation should be carried out to clearly understand the effects of DT resulting from the activation of Akt.
Regarding apl-1, Shih and colleagues explored its antitumor activity on leukemic and prostatic cancer cell lines, focusing also on its ability to inhibit topo II. Apl-1 was highly cytotoxic (Table 1) and induced apoptosis through the dysregulation of the oxidative balance, as demonstrated by the excess of ROS and NOX (active nicotinamide adenine dinucleotide phosphate oxidase) production [36]. In addition, apl-1 reduced the activity of the PI3K/Akt/mTOR (mammalian target of rapamycin) pathway, a mechanism associated with an antitumor activity [40]. Moreover, apl-1 inhibited the relaxation of supercoiled DNA, showing an IC50 (concentration that inhibited the 50% of DNA relaxation) value of 1.37 μM (Table 1). As DT, apl-1 did not generate linear DNA [36], meaning that it could not stabilize the DNA cleavage complex. A further study determined that apl-1, despite increasing phosphorylation of H2A.X, did not produce DNA single strand breaks (SSBs) and DSBs, and did not increase the number of nuclear γ-H2A.X foci [41]. All these findings show that apl-1, in contrast to its oxidized derivative, acts as a topo IIα catalytic inhibitor, without inducing DNA damage.
Apl-1 inhibited the protein expression of heat shock protein 90 (Hsp90) in PC-3 and Du145 prostate cancer cells, making it a dual target inhibitor [36]. Hsp90 chaperon ensures the stability, integrity, shape, and function of critical oncogenic proteins (also called Hsp90 client proteins), which play critical roles in signal transduction, cell proliferation and survival, cell-cycle progression and apoptosis, as well as invasion, tumor angiogenesis, and metastasis [42]. Other marine topo II inhibitors, in addition to apl-1, possess this dual inhibitory activity of topo II and Hsp90, as discussed in the next sections. This is probably due to the similar ATPase domain structures of topo II and Hsp90 [43]. Other studies found that apl-1 inhibited the Wnt/β-catenin pathway through the proteasomal degradation of β-catenin [44] and the epidermal growth factor (EGF)-dependent proliferation of breast cancer cells (MCF-7 and ZR-75-1), probably by blocking the phosphorylation of EGF receptor [45]. Moving toward the later stages of the carcinogenic process, apl-1 showed antimetastatic and antiangiogenic effects: in PC-3 and Du145 cells, it inhibited cell migration and colony formation, and suppressed the EMT process induced by the transforming growth factor-β1 (TGF-β1) [36].
Overall, apl-1 exerted a marked antitumor activity in different tumor cell models and modulated multiple targets. Despite this, conflicting results are reported regarding its selective activity toward cancer cells. In normal rat macrophage cells (NR8383) and normal human skin cells (CCD966SK), the IC50, calculated for its cytotoxic effects, was almost 4− and 17−fold higher, respectively, than the average IC50 calculated for tumor cells (0.39 μM) [36]. However, apl-1 induced apoptosis and blocked cell-cycle progression indiscriminately in leukemia (THP-1 and NOMO-1) cells and in bovine aortic endothelial cells [41]. Thus, the toxicological profile of apl-1 needs more in-depth studies.
2.3. Makaluvamines
Another type of alkaloids produced by sponges are pyrroloiminoquinones, which include makaluvamines and batzellines.
Makaluvamines (Figure 4) were isolated from sponges mainly belonging to the Zyzza genus. In the 1990s, these compounds were the subject of intensive studies to evaluate their antitumor activity. All makaluvamines (A-V) exhibited a marked cytotoxic activity. [46,47,48]. In addition, makaluvamine A and C reduced the tumor mass of human ovarian carcinoma OVCAR3-xenograft in Balb/c nu/nu athymic mice (Table 1) in vivo [49].
Figure 4.

Chemical structure of makaluvamines A-V. Makaluvamine A (CAS number: 146555-78-4), makaluvamine B, C (CAS number: not available), makaluvamine D (CAS number: 146555-81-9), makaluvamine E (CAS number: 146555-82-0), makaluvamine F (CAS number: 146555-83-1), makaluvamine G (CAS number: 152273-69-3), makaluvamine H (CAS number 174232-34-9), makaluvamine I (CAS number: 138087-43-1), makaluvamine J (CAS number:174232-35-0), makaluvamine K (CAS number: 174232-36-1), makaluvamine L (CAS number: 174232-37-2), makaluvamine M (CAS number: 174232-41-8), makaluvamine N (CAS number: 187964-02-9), makaluvamine V (CAS number: 227103-87-9).
Regarding the ability of makaluvamines to inhibit topo II, the results are somewhat ambiguous: makaluvamine G did not inhibit topoisomerase II; for the other makaluvamines, there are conflicting data on whether they act as topo II catalytic inhibitors or poisons. Makaluvamine N inhibited more than 90% of the relaxation of supercoiled pBR322 DNA at 5.0 μg/mL [46,49], while makaluvamines A-F modulated topo II-mediated decatenation of kinetoplast DNA (kDNA) differently [49,50]. Overall, makaluvamine B was inactive, while makaluvamine A and F were the most effective, exhibiting IC90 (concentration that inhibits 90% of kDNA decatenation) values of 41 μM and 25 μM, respectively [49]. Later, Matsumoto et al. demonstrated that different makaluvamines promoted the formation of cleavable complex. Makaluvamine C, D, and E (33–466 μM) cleaved radiolabeled pUC 19 DNA in the presence of human topo II in a concentration-dependent manner, although they showed fewer and weaker cleavage sites than ETO and mitoxantrone. In addition, when also testing other makaluvamines at 91 mM using a cell-free cleavage assay with radiolabeled rf M13 mp 19 plasmid DNA, they found that makaluvamine I and H were the most efficient in inducing topo II-mediated cleavage of plasmid DNA, showing a 61% and 33% of cleavage, respectively, compared to the 100% of ETO, at the same tested concentration (Table 1). In both assays, makaluvamine D and E exhibited a comparable behavior, i.e., a weak and marked formation of cleavable complex, respectively, whereas makaluvamine C was more efficient in cleaving plasmid DNA than radiolabeled pUC 19 DNA [51]. Overall, this latter study points out that makaluvamines may act as topo II poisons. In support of this hypothesis, there are various data. Firstly, makaluvamine A intercalated into DNA and induced DNA DSBs in the neutral filter elution assay, which measures the formation of protein-linked DNA DSBs, compatible with the generation of DNA cleavable complex. The effect was comparable to that of the known DNA intercalating topo II poison m-AMSA [49]. Similar findings were reported for makaluvamine C [50]. Secondly, the most active makaluvamines (A and F) were much more cytotoxic in CHO xrs-6 cells compared to CHO BR1 cells (DSBs repair-competent): they exhibited a hypersensitive factor (HF, i.e., the ratio of IC50 on xrs-6 to that on BR1 cells) equal to 9 (for makaluvamine A) and 6 (for makaluvamine F), and thus equal to or higher than that of m-AMSA (HF = 6) [49]. Similarly, makaluvamine I showed a 5-fold lower IC50 in xrs-6 cells (0.4 μM) compared to AA8 DNA repair-competent cells (2 μM) [51]. This evidence shows a typical behavior of DNA intercalating topo II poisons. Overall, it is very likely that some makaluvamines have the formation of cleavable complexes as their predominant mechanism and thus act as a poison. However, the lack of extensive studies does not allow to clearly identify the mechanism of topo II inhibition of the different compounds. In addition, further experiments on their activity on in vitro or in vivo models are needed to identify their potential use as anticancer agents.
Recently, different makaluvamine analogs as well as a hybrid derived from makaluvamine A and ellipticine have been found to inhibit the catalytic activity of topo II and block DNA relaxation [52,53]. However, the hybrid derivative was equally cytotoxic on both prostate cancer cells and normal fibroblasts, thus demonstrating a non-selective activity toward tumor cells [53].
2.4. Batzellines
Batzellines are a group of alkaloids isolated from the marine sponge Batzella sp. (Figure 5), structurally linked to other marine substances such as makaluvamines and discorhabdins.
Figure 5.

Chemical structure of batzellines. Batzelline A (CAS number: 123064-89-1), batzelline B (CAS number: 123064-90-4), isobatzelline A (CAS number: 133401-01-1), isobatzelline C (CAS number: 133401-03-3), isobatzelline D (CAS number: 133401-04-4), isobatzelline E (CAS number: 437980-21-7), secobatzelline A (CAS number: 247590-59-6), secobatzelline B (CAS number: 247590-60-9).
Among them, isobatzelline A, isobatzelline C, isobatzelline D, and secobatzelline A were highly cytotoxic on a panel of pancreatic cancer cell lines (Table 1). Surprisingly, cytotoxic activity was found to be inversely proportional to the inhibition of topo II-mediated DNA decatenation [54]. Isobatzelline E and batzelline B, which are not among the most cytotoxic, inhibited 95% and the 63%, respectively, of DNA decatenation at 25 μg/mL; at the same concentration, isobatzellines A, C, and D, which are the most cytotoxic, inhibited 36%, 27%, and 26% of topo II-mediated DNA decatenation, respectively. These latter significantly intercalated into DNA, while the most potent topo II inhibitor isobatzelline E was the less potent DNA-intercalating compound [54]. This different behavior seems to influence the mechanism by which batzellines interfere with cell-cycle progression in a different way. In fact, only the most potent topo II inhibitor isobatzelline E blocked cells in the G2 phase of the cell cycle, whereas all the others, characterized by a less pronounced inhibitory activity on topo II and a greater ability to intercalate into DNA, blocked cell-cycle progression in the S phase [54]. Overall, these results indicate that batzellines cytotoxicity relies upon both topo II inhibition and DNA-intercalation, and that the more batzellines intercalate into the DNA, the greater the cytotoxicity of the specific compound [54]. Bearing in mind the close similarity with makaluvamines and, especially, the marked ability of isobatzellins A, C, D to intercalate with DNA, more in-depth studies should be carried out to assess whether batzellines induce DNA damage and act as topo II poisons by promoting the formation of DNA cleavable complex.
2.5. Hippospongic Acid A
Hippospongic acid A (HA-A) is a triterpene isolated from the marine sponge Hippospongia sp.
Both the natural enantiomer (R)-HA-A (Figure 6a) and the racemate (±)-HA-A (Figure 6b), which consists of the natural stereoisomer [(R)-HA-A] and the unnatural one [(S)-HA-A], dose-dependently inhibited both human and yeast topo II relaxation activity, showing an IC50 value of 15 μM. Inhibition of topo I has also been observed, although with a higher IC50 value (25 μM), together with the inhibition of DNA polymerases within 2-fold higher IC50 values [55]. (R)-HA-A and (±)-HA-A at 10 μM blocked cell-cycle progression in both G1 and G2/M phases, and induced apoptosis in NUGC-3 human gastric cancer cells. The G1-phase arrest was probably due to the inhibition of DNA polymerases, while the G2/M-phase block was mainly due to the inhibition of topoisomerases [55]. Based on these results, it seems likely that several mechanisms, namely inhibition of topo I, topo II, and DNA polymerases, are involved in the compound’s antitumor activity rather than the exclusive inhibition of topo II.
Figure 6.

Chemical structure of (R)-HA-A ((a) CAS number: not available) and (±)-HA-A ((b) CAS number: 183381-06-8).
2.6. 10-Acetylirciformonin B
10-Acetylirciformonin B (10AB) (Figure 7) is a furanoterpenoid derivative isolated with other terpenoid-derived metabolites from the marine sponge Ircinia sp. [56].
Figure 7.

Chemical structure of 10-Acetylirciformonin B (CAS number: 1334233-11-2).
Among all the isolated compounds, 10AB was the most cytotoxic (Table 1). Interestingly, it seems to exert a selective cytotoxic effect for cancer cells: in HL-60 cells, 10AB at 6.0 μM induced 80% apoptosis; in rat alveolar NR8383macrophages, it suppressed cell viability by 18.3% [57]. A previous study reported that in HL-60 cells 10AB induced Casp-dependent apoptosis and promoted the formation of DNA DSBs, accompanied by the phosphorylation of H2A.X and checkpoint kinase 2 (Chk2), two markers of nuclear DNA damage [58]. A more recent study showed that 10AB-induced DNA damage may be related to its ability to inhibit topo IIα catalytic activity: 10AB (1.5, 3.0, 6.0, and 12.0 μM) inhibited DNA relaxation without producing linear DNA (like the topo IIα poison ETO), and at 3 μM decreased the protein expression of topo IIα in HL-60 cells. All these findings indicate that 10AB could act as a DNA damaging agent and compromise the topo IIα catalytic cycle, leading to apoptotic cell death [57]. In this regard, in HL-60 cells 10AB (1.5, 3.0, and 6.0 μM) disrupted MMP (mitochondrial membrane potential) and reduced the protein expression of anti-apoptotic proteins (Bcl-2 and Bcl-X) as well as of other proteins involved in the apoptotic process, as X-linked inhibitor of apoptosis protein (XIAP) and survivin. 10AB also generated ROS, activated the mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinase (ERK) pathway, and inhibited the PI3K/PTEN/Akt/mTOR signaling pathway [57]. Akt transcriptionally regulates the expression of hexokinase II (HK-II) [59]. HKs are enzymes that catalyze the phosphorylation of glucose, i.e., the first step of glycolysis, and are upregulated in many tumors characterized by a high glycolytic activity. Moreover, HK-II has a pro-survival activity and protects mitochondria against mitochondrial apoptotic cell death by interfering with anti- and pro-apoptotic proteins and decreasing ROS generation [59]. Thus, downregulation of HK allows the shift of cancer cells’ metabolism to oxidative phosphorylation and increases ROS levels, which leads to cell death. The demonstrated ability of 10AB to downregulate p-Akt protein expression may lead to the downregulation of HK-II. This means that 10AB-induced apoptosis seems to be mediated by topo IIα inhibition and oxidative stress, as well as the perturbation of metabolic and cell survival pathways.
2.7. Manoalide-Like Sesterterpenoids
In 1994, Kobayashi et al. isolated four sesterterpenes from the sponge Hyrtios erecta [60]. Among them, manoalide 25-acetals (Figure 8) inhibited the DNA-unknotting activity of calf thymus topo II, showing an IC50 value of about 25 μM. In addition, it exhibited antitumor activity on CDF1 mice inoculated whit P388 leukemia cells, with a T/C% score (the ratio between the tumor volume in the treated group and in the untreated control group) of 150% at 1 mg/kg (Table 1) [60].
Figure 8.

Chemical structure of manoalide 25-acetals (CAS number: not available).
More recently, 10 manoalide-like sesterterpenoids (Figure 9) were isolated from Luffariella sp. sponge: 24R,25R-luffariellin A (L1), 24R,25S-luffariellin A (L2), 24R-O-Methyl-25R-luffariellin A (L3), 24R-O-Methyl-25S-luffariellin A (L4), 24S-O-Methyl-25S-luffariellin A (L5), 24R,25R-manoalide (M6), 24R,25S-manoalide (M7), 24R-O-Methyl-25R-manoalide (M8), 24R-O-Methyl-25S-manoalide (M9), and 24R,25S-thorectolide (T10) [61].
Figure 9.

Chemical structure of the manoalide-like sesterterpenoids. M1-M5 (CAS numbers not available), M6 (CAS number: 2328074-79-7), M7-M9 (CAS numbers not available), T10 (CAS number not available).
All the derivates were tested on multiple leukemia cell lines (Table 1). The compounds L2, L4, M7, and M9, bearing a 24R, 25S configuration, were the most effective, thus assuming that the cytotoxic activity was configuration-dependent [61]. The administration of M7 to immunodeficient athymic mice (1 μg/kg every day for 33 days) reduced the tumor growth of Molt-4 xenograft by about 66%, without affecting body weight [61].
M7 has been shown to act as a catalytic inhibitor of topo IIα. Moreover, it inhibited DNA relaxation with an IC50 value of 1.18 μM and promoted the formation of supercoiled DNA products in the presence of topo IIα [61]. Compared to manoalide 25-acetals, the inhibitory activity of M7 toward topo II was greatly higher, although purified topo II from two different organisms were used: human for M7 [61] and calf thymus for manoalide 25-acetals [60]. The topo IIα catalytic inhibitor activity was associated with DNA damage, as demonstrated by its ability to promote the phosphorylation of ATM, Chk2, and H2A.X and to induce DNA DSBs at 0.75 μM in Molt-4 cells. M7-induced DNA damage has been found to activate apoptotic cell death, as indicated by and the activation of Casp-3, -8, and -9, the disruption of MMP, and the cleavage of PARP [61].
2.8. Heteronemin
Another marine sesterterpenoid-type product, heteronemin (Figure 10), was separated from the Hippospongia sp. sponge [62].
Figure 10.
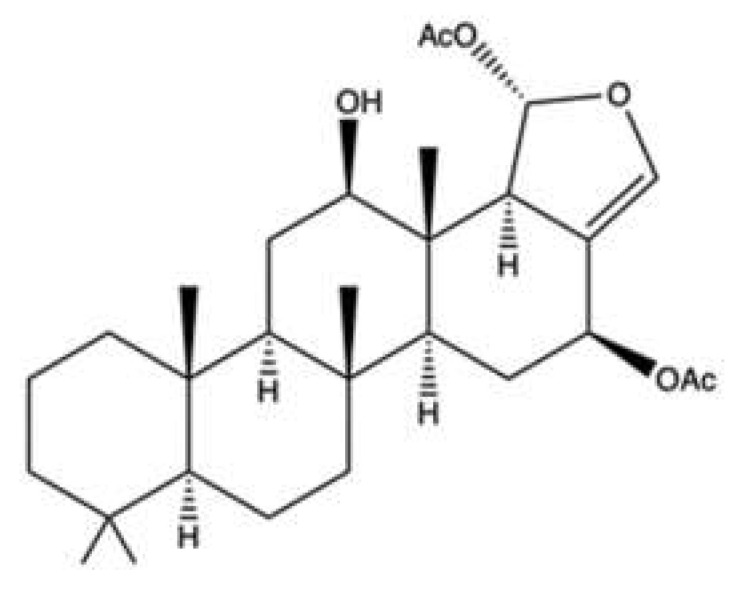
Chemical structure of heteronemin (CAS number: 62008-04-2).
Heteronemin was able to induce apoptosis as well as inhibit the proliferation of different cancer cell lines [63,64]. Interestingly, in hepatocellular carcinoma HA22T and HA59T cells, heteronemin induced both apoptosis and ferroptosis [65], a non-apoptotic programmed cell death mechanism characterized by the iron-dependent accumulation of lipid ROS [66]. Due to the well-known occurrence of multi-drug resistance caused by the deregulation of apoptosis [67], the evidence that heteronemin is a ferroptosis inducer is very interesting.
Deepening the molecular mechanisms involved in heteronemin’s cytotoxicity in prostate cancer cells, Lee et al. found that it induced both autophagy and apoptosis [62]. Autophagy promotes either cell survival or cell death in a context- and cell-dependent manner [68]. Autophagy induced by heteronemin seems to possess a cytoprotective effect rather than a pro-apoptotic one [62]. Indeed, heteronemin (1.28 and 2.56 μM) activated LC3-B II (LC3-phosphatidylethanolamine conjugate), a marker of autophagy, but at 5.12 μM, when apoptosis was markedly induced, autophagy was blocked. Moreover, the pre-treatment with two autophagy inhibitors (3-methyladenine and chloroquine) raised the percentage of LNCaP apoptotic cells [62]. Similarly, in A498 renal carcinoma cells, the inhibition of autophagy increased the pro-apoptotic activity of heteronemin [69].
The marine sesterterpene completely inhibited DNA relaxation in the cell-free DNA cleavage assay and reduced topo IIα protein expression in LNCaP cells, which resulted in the block of the total catalytic activity of the enzyme. Heteronemin did not produce linear DNA, thus assuming its inability to stabilize DNA-topo II cleavable complex [62].
Mechanisms other than the inhibition of topo II are possibly involved in the antitumor activity of heteronemin.
Heteronemin suppressed the expression of Hsp90 and that of its client proteins, thus being able to modulate the expression of oncogenic proteins and transcription factors involved in tumorigenesis [62]. Moreover, it blocked NF-κB activation via proteasome inhibition in K562 cells [70] and the activation of ERK1/2 and STAT3 in breast cancer cells [63,64]. In LnCaP cells, heteronemin (1.28–5.12 μM) disrupted MMP, fostering mitochondrial dysfunction. Due to the overproduction of ROS and Ca2+ release, heteronemin promoted oxidative and endoplasmic reticulum (ER) stress, therefore triggering the unfolded protein response (UPR) signaling network to re-establish ER homeostasis [62]. Oxidative and ER stress results from the activation of protein tyrosine phosphatases (PTPs) [62]. PTPs modulate the levels of cellular protein tyrosine phosphorylation and control cell growth, differentiation, survival, and death. PTPs exert both tumor-suppressive and oncogenic functions in a context-dependent manner [71]. Pre-treatment of LnCaP with a PTP inhibitor reduced heteronemin-induced ROS generation and ER stress, thus demonstrating that in this experimental setting, PTPs exhibits a tumor-suppressive mechanism and participates in the antitumor activity of heteronemin [62].
Oxidative stress was also involved in the heteronemin-induced anticancer effects in Molt-4 cells. In this cell line, it enhanced γ-H2A.X protein expression, probably due to apoptosis rather than DNA damage occurrence. Indeed, although γ-H2A.X is the most sensitive biomarker of DNA damage, its measure by ELISA and/or immunoblotting allows to evaluate the total H2A.X protein levels in a sample, but apoptotic cells with pan-nuclear H2A.X expression cannot be differentiated from surviving cells, which may alter H2A.X quantification. In contrast, the fluorescent microscopic quantification of foci is the most sensitive approach and can distinguish between pan-nuclear staining and foci formation [72]. The increased γ-H2A.X protein expression induced by heteronemin in Molt-4 cells was demonstrated by using Western Blot, as for all the other sponge-derived topo II inhibitors, and, unlike other studies, the expression of other DNA damage-related proteins was not evaluated. Thus, it is not clear whether heteronemin induces DNA damage in this experimental model.
In vivo, heteronemin inhibited the growth of Molt-4 and LnCaP xenograft in Balb/c nude mice and in immunodeficient athymic mice, respectively, treated with 0.31 μg/g (three times a week for 24 days) and 1 mg/kg (every day for 29 days) of heteronemin [62,73].
2.9. Scalarane Sesterterpenoids
A dual inhibitory effect on topo II and Hsp90 was also reported for two new scalarane sesterterpenoids (SS) [12β-(3′β-hydroxybutanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al (SS1, Figure 11a) and 12β-(3′β-hydroxypentanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al (SS2, Figure 11b)], and a tetraprenyltoluquinol-related metabolite (2-tetraprenil-1,4-benzoquinone, TPL, Figure 11c), all isolated from the sponge Carteriospongia sp. [74].
Figure 11.

Chemical structure of 12β-(3′β-hydroxybutanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al (a), 12β-(3′β-hydroxypentanoyloxy)-20,24-dimethyl-24-oxo-scalara-16-en-25-al (b), and 2-tetraprenil-1,4-benzoquinone ((c) CAS numbers not available).
SS1, SS2, and TPL were cytotoxic on many tumor cell lines [74] (Table 1). All three compounds inhibited DNA relaxation, reaching almost 100% inhibition at the highest tested concentration (20 μg/mL). There was no information regarding the production of linear DNA [74]. Topo II inhibition was associated with DNA damage: SS1 (0.0625–0.25 μg/mL) increased the protein expression of γ-H2A.X and, at 0.0625 μg/mL; it also induced DNA DSBs in Molt-4 cells [74]. Although SS2 enhanced γ-H2A.X protein expression, it is difficult to associate this event exclusively with DNA damage since neither other marker of DNA damage nor the formation of DSBs have been evaluated. SS1, like heteronemin [62], promoted ROS generation and ER stress and induced mitochondrial apoptosis [74]. In addition, SS1 shared with heteronemin the ability to inhibit Hsp90 protein expression and that of its client proteins [74]. Although Lai and colleagues investigated SS1 more deeply than TPL, the latter was also tested in a Molt-4 cells xenograft animal model, showing that its daily administration (1.14 μg/g) for 33 days inhibited almost 50% of xenograft tumor growth in male immunodeficient athymic mice [74]. Authors justified their choice to only test TPL in vivo by the small amount they were able to isolate for the other two compounds. However, considering the marked antitumor activity of SS1, a possible in vivo study of this compound should be considered as well.
2.10. Polycyclic Quinone-Type Metabolites
The two polycyclic quinone-type compounds halenaquinone [75] (Figure 12a) and xestoquinone [76] (Figure 12b), which differ for a carbonyl group, were isolated from the sponge Petrosia sp.
Figure 12.

Chemical structure of halenaquinone ((a), CAS number: 86690-14-4) and xestoquinone ((b), CAS number: 97743-96-9).
Halenaquinone and xestoquinone exhibited a comparable cytotoxic activity [75,76]. In vivo, the administration of halenaquinone (1 μg/g for 30 days) and xestoquinone (1 μg/g for 50 days) suppressed the growth of Molt-4 xenograft in immunodeficient athymic mice, without affecting body weight (Table 1) [75,76].
Both compounds strongly inhibited either the topo II-catalyzed DNA relaxation and the protein expression of topo IIα in Molt-4 [75,76] and K562 cells [76]. For DNA relaxation, xestoquinone showed an IC50 value of 0.094 μM [76], and halenaquinone showed an IC50 about 5.5-fold lower (0.017 μM) [75]. These results indicate that they act as potent catalytic inhibitors of topo II. However, they did not form DNA-topo II cleavage complex, since no linear DNA was observed in the cell-free DNA relaxation assay [75,76]. Additionally, molecular docking studies reported that xestoquinone was capable of binding topo II with a docking score of −26.9, although a similar or even a lower value was observed for topo I (−24.0) and Hsp90 (−15.5) [76]. These results demonstrate that the compound can bind to multiple targets. Xestoquinone (7.84 μM) treatment of Molt-4 cells markedly increased the expression of multiple DNA damage markers (p-Chk1, p-Chk2, and γ-H2A.X), pointing out that its topo II catalytic activity inhibition induced DNA damage [76]. No markers of DNA damage were evaluated for the congener halenaquinone. Nonetheless, given the close similarities in the antitumor mechanisms of both compounds, it cannot be excluded that congener halenaquinone was a topo II catalytic inhibitor. In fact, both compounds have been shown to inhibit the activity of histone deacetylase (HDAC) in vitro [75,76] and in a Molt-4 xenograft mouse in vivo model [76]. This is not so surprising, as several studies report that topo II and HDAC mutually modulate their activity [43]. In addition to this, ROS overproduction [75,76], induction of ER stress, and binding to protein Hsp90 [76] recorded for both compounds led to apoptosis. Notably, the two polycyclic quinone-type metabolites promoted both apoptotic pathways as the disruption of MMP, decrease in anti-apoptotic proteins (Bcl-2, Bcl-X, Bid), increase in pro-apoptotic ones (Bax, Bak) (all markers of intrinsic apoptosis), and activation of Casp-8 and -9 (markers of extrinsic apoptosis) were observed in Molt-4 and K562 cells [75,76].
Alongside halenaquinone and xestoquinone, other polycyclic quinone-type metabolites were isolated from the sponge Xestospongia sp. [77]. All studied compounds inhibited topo II (Table 1). Among those, adociaquinone B (Figure 13) was the most potent with an IC90 (the concentration inducing the 90% of inhibition) < 11 μM and 78 μM for DNA decatenation and relaxation, respectively. In contrast to xestoquinone and halenaquinone, adociaquinone B was a non-intercalating DNA topo II poison. In fact, it strongly promoted the formation of the enzyme-DNA cleavable complex to the same extent as mitoxantrone, a known topo II poison [78]. However, in contrast to mitoxantrone, adociaquinone B did not intercalate into DNA since it was not able to displace ethidium bromide from calf thymus DNA [77]. Secoadociaquinone A and B, two other Xestospongia sp. metabolites, inhibited topo II activity in the cell-free DNA decatenation assay without exhibiting cytotoxicity since they were unable to permeate cell membranes. Thus, it is not sufficient to test the inhibitory activity of topo II only on cell-free systems, as very often the physicochemical properties of the tested compounds prevent their entry into cells and consequently a possible interaction with intracellular targets, such as topo II [77].
Figure 13.

Chemical structure of adociaquinone B (CAS number: 113831-00-8).
3. Topo II Inhibitors from Marine Fungi and Bacteria
3.1. Leptosin F
Leptosin F (LEP, Figure 14) is an indole derivative containing sulphur that is derived from the fungus Leptoshaeria sp., which grows on the marine alga Sargassum tortile [82].
Figure 14.

Chemical structure of leptosin F (CAS number: not available).
Yanagihara and colleagues demonstrated that LEP potently inhibited the growth of RPMI-8402 T cell acute lymphoblastic leukemia cells–more powerfully than ETO and with an IC50 value in the nM range–and induced apoptosis [82]. A pro-apoptotic effect has also been reported for LEP in normal human embryo kidney cells (293 cell line), where it activated Casp-3 at doses as low as 1 to 10 µM [82]. These results could indicate that LEP does not act selectively against cancer cells, but rather on all rapidly proliferating cells.
The in vitro kDNA decatenation assay revealed its ability to inhibit topo II [82]. Gel electrophoresis of the kDNA after decatenation assay showed that LEP did not act as a catalytic inhibitor of topo II, as the authors instead stated. Further studies would be necessary to define the exact mechanism of interaction between LEP and the enzyme. Moreover, since the compound concentration required to exert cytotoxic activity on RPMI-8402 cells was extremely lower (nM range) than that required to inhibit topo II (µM range), the cytotoxicity of LEP at the cellular level might involve other pathways in addition to the inhibition of topo II.
3.2. Pericosine A
Pericosine A (PA, Figure 15) is a metabolite produced by a strain of Periconia byssoides OUPS-N133, a marine fungus originally separated from the sea hare Aplysia kurodai [83].
Figure 15.

Chemical structure of pericosine A (CAS number: 200335-68-8).
Some studies reported the ability of PA to induce growth inhibition on different cancer cell lines [83,84] (Table 2). Furthermore, in mice inoculated with P388 leukemic cells, PA increased the median survival days compared to vehicle (13.0 versus 10.7 days) (Table 2). In the same study, the authors reported that PA at 100–300 mM inhibited topo II and at 449 μM inhibited the epidermal growth factor receptor (EGFR) by 40−70%. Since PA seems to exert its inhibitory effects on topo II at very high concentrations, it is unlikely that this mechanism of action was responsible for its in vitro and in vivo antitumor effects. The inhibition of EGFR, a protein kinase known to promote cell proliferation and counteract apoptosis [85], could be a more plausible mechanism [83]. The lack of important information on its antitumor activity in vitro and in vivo does not permit a clear characterization of the anticancer activity of PA. Therefore, further experiments should be conducted to fully understand the potential usefulness of PA in the oncological area.
Table 2.
Topo II inhibitors derived from marine fungi and bacteria.
| Compound | Source | Topo II Inhibitory Activity | Antitumor Effect(s) | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Assay | Concentration(s) Tested or IC50 | Outcomes | Experimental Model | Cytotoxic Activity | Other Antitumor Mechanism(s) | |||
| 2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol | Bacterium Streptomyces sp. VITJS8 | Molecular docking | HepG2 | IC50 (16 h): 250 µg/mL | Apoptosis (caspase-9, caspase-8, caspase-3 cleavage, regulation of Bcl-2 family proteins, cell shrinkage, chromatin condensation, apoptotic bodies, DNA fragmentation, incomplete nuclear membrane) ↓ Cell growth (cell cycle arrest: ↑ cells in S and G2/M phases, ↓ cells in G0/G1 phase) |
[94] | ||
| 3-hydroxyholyrine A | Bacterium Streptomyces strain OUCMDZ-3118 | Cell free DNA relaxation assay using supercoiled pBR322 DNA plasmid and topo II α | Not indicated | A-549 | IC50 (48 h): 0.51 µM | [97] | ||
| MCF-7 | IC50 (48 h): 5.0 µM | |||||||
| K562 | IC50 (48 h): 7.2 µM | |||||||
| AGS | IC50 (48 h): 1.7 µM | Apoptosis (↓ survivin protein expression) DNA damage (↑ γ-H2AX protein expression) |
||||||
| MKN45 | IC50 (48 h): 4.3 µM | Apoptosis (↓ survivin protein expression) | ||||||
| Aspergiolide A | Fungus Aspergillus glaucus |
A-549 | IC50 (24 h): 0.13 µM | [88] | ||||
| HL-60 | IC50 (72 h): 0.28 µM | |||||||
| BEL-7402 | IC50 (24 h): 7.5 µM | |||||||
| P388 f | IC50 (72 h): 35.0 µM | |||||||
| Spectrofluorimetric decatenation reaction of kinetoplast DNA | 10–100 µM | ↓ Topo II activity | HeLa | IC50 (72 h): 2.37–7.07 µM | [87] | |||
| SMMC-7721 | ||||||||
| SGC-7901 | ||||||||
| MCF-7 | ||||||||
| MDA-MB-468 | ||||||||
| U251 | ||||||||
| A431 | ||||||||
| SK-OV-3 | ||||||||
| BxPC-3 | ||||||||
| 786-O | ||||||||
| BEL-7402 | IC50 (72 h): 2.37–7.07 µM | Apoptosis (procaspase-3, procaspase-8, procaspase-9 and PARP cleavage, ↑ Bax protein expression, ↓ Bcl-2 protein expression) DNA damage (↑ γ-H2AX protein expression) |
||||||
| KN mice inoculated with H22 f cells | ↓ Tumor growth (5, 15, 45 mg/kg i.p.) | |||||||
| Nude mice bearing BEL-7402 xenografts | ↓ Tumor volume (7, 14, 28 mg/kg/day i.p. for 21 days) | |||||||
| Jadomycin DS | Bacterium Streptomyces venezuelae ISP5230 | WaterLOGSY NMR spectroscopy | Interaction with topo IIβ | [91] | ||||
| Leptosin F | Fungus Lestoshaeria sp. |
Cell-free decatenation reaction of kinetoplast st DNA | 10–30 µM | ↓ Topo II activity | RPMI8402 | IC50 (72 h): 8.2 nM | Apoptosis (↑ caspase-3 activity, ↑ DNA degradation) | [82] |
| 293 d | Apoptosis (caspase-3 activation Akt inactivation) |
|||||||
| P388 f | ED50: 0.056 µg/cm3 | [98] | ||||||
| Marinactinone B | Bacterium Marinactinospora thermotolerans | Cell free relaxation assay using supercoiled pBV220 DNA plasmid and topo II from rat liver cells | 607 µM | ↓ Topo II activity | SW1990 | IC50 (72 h): 99 µM | [86] | |
| Pericosine A | Fungus Periconia byssoides | 100–300 mM | ↓ Topo II activity | P388 f | ED50: 0.1 µg/mL | [83] | ||
| HBC-4 | Log GI50/M: -4.76 | |||||||
| BSY-1 | Log GI50/M: -4.75 | |||||||
| HBC-5 | Log GI50/M: -5.22 | |||||||
| MCF-7 | Log GI50/M: -4.66 | |||||||
| MDA-MB-231 | Log GI50/M: -4.74 | |||||||
| U-251 | Log GI50/M: -4.76 | |||||||
| SF-268 | Log GI50/M: -4.72 | |||||||
| SF-295 | Log GI50/M: -4.62 | |||||||
| SF-539 | Log GI50/M: -4.71 | |||||||
| SNB-75 | Log GI50/M: -7.27 | |||||||
| SNB-78 | Log GI50/M: -4.71 | |||||||
| HCC2998 | Log GI50/M: -4.75 | |||||||
| KM-12 | Log GI50/M: -4.73 | |||||||
| HT-29 | Log GI50/M: -4.70 | |||||||
| WiDr | Log GI50/M: -4.64 | |||||||
| HCT-15 | Log GI50/M: -4.77 | |||||||
| HCT-116 | Log GI50/M: -4.75 | |||||||
| NCI-H23 | Log GI50/M: -4.78 | |||||||
| NCI-H226 | Log GI50/M: -4.80 | |||||||
| NCI-H522 | Log GI50/M: -4.95 | |||||||
| NCI-H460 | Log GI50/M: -4.72 | |||||||
| A-549 | Log GI50/M: -4.61 | |||||||
| DMS273 | Log GI50/M: -4.68 | |||||||
| DMS114 | Log GI50/M: -4.82 | |||||||
| LOX-IMVI | Log GI50/M: -4.72 | |||||||
| OVCAR-3 | Log GI50/M: -4.85 | |||||||
| OVCAR-4 | Log GI50/M: -4.68 | |||||||
| OVCAR-5 | Log GI50/M: -4.79 | |||||||
| OVCAR-8 | Log GI50/M: -4.78 | |||||||
| SK-OV-3 | Log GI50/M: -4.76 | |||||||
| RXF-631L | Log GI50/M: -4.73 | |||||||
| ACHN | Log GI50/M: -4.72 | |||||||
| St-4 | Log GI50/M: -4.65 | |||||||
| MKN1 | Log GI50/M: -4.78 | |||||||
| MKN7 | Log GI50/M: -4.70 | |||||||
| MKN28 | Log GI50/M: -4.72 | |||||||
| MKN45 | Log GI50/M: -4.75 | |||||||
| MKN74 | Log GI50/M: -4.69 | |||||||
| Mice inoculated with P388 f cells | ↑ Survival days compared to controls (25 mg/kg administered i.p. on day 1 and 5) | |||||||
| Streptomyces sp. VITJS4 strain crude extract | Bacterium Streptomyces sp. VITJS4 | Molecular docking | HeLa | IC50 (time not indicated): 50 µg/mL | Apoptosis DNA damage (stained nuclei with round morphology, chromatin condensation, DNA fragmentation) |
[95] | ||
| HepG2 | IC50 (time not indicated): 50 µg/mL | |||||||
| Sulochrin | Fungus Aspergillus falconensis | Molecular docking | L5178Y f | IC50 (24 h): 5.1 µM | [96] | |||
| MDA-MB-231 | No cytotoxic activity | ↓ Cell migration | ||||||
↑: upregulation/induction; ↓: downregulation/inhibition; d non-transformed cells; f murine cancer cells; Akt: protein kinase B; Bax: Bcl-2 associated X protein; Bcl-2: B-cell lymphoma 2; ED50: dose effective in 50% of treated subjects; γ-H2AX: phosphorylated H2A histone family member X; IC50: concentration that inhibits 50% of the investigated activity; i.p.: intraperitoneal; Log GI50: logarithm of concentration that inhibits 50% of cell growth; PARP: poly (ADP-ribose) polymerase. Human breast cancer cell lines: MCF-7; MDA-MB-231; MDA-MB-468; BSY-1. Human cervical cancer cell line: HeLa. Human colon cancer cell lines: HCT-116; HT-29; HCT2998; KM-12; WiDr; HCT-15. Human epidermoid cancer cell line: A431. Human gastric cancer cell line: AGS; MKN45; SGC-7901; St-4; MKN1; MKN7; MKN28; MKN74. Human glioma cell lines: U251. Human glioblastoma cell lines: SF-268; SF-295; SF-539; SNB-75; SNB-78. Human hepatic cancer cell lines: HepG2; BEL-7402; SMMC-7721. Human leukemia cell lines: K562; HL-60; RPMI8402. Human lung cancer cell line: A549; NCI-H23; NCI-H226; NCI-H522; NCI-H460; DMS273; DMS114. Human melanoma cell lines: LOX-IMVI. Human ovarian cancer cell lines: SK-OV-3; OVCAR-3; OVCAR-4; OVCAR-5; OVCAR-8. Human pancreatic cancer cell lines: BxPC-3; SW1990. Human renal cancer cell lines: 786-O; RXF-6312; ACHN.
3.3. Marinactinone B
Marinactinone B (MB, Figure 16) is a γ-pyrone derivate isolated from the bacterial strain Marinactinospora thermotolerans SCSIO 00606, found in the sediments of the northern South China Sea [86].
Figure 16.

Chemical structure of marinactinone B (CAS number: 1344677-16-2).
MB was evaluated for its anticancer activity against breast (MCF-7), pancreatic (SW1990), hepatic (HepG2 and SMCC-7721), lung (NCI-H460), and cervical (HeLa) cancer cell lines. It exhibited cytotoxicity at medium-elevated concentration values only against SW1990 (99 µM) and SMCC-7721 (45 µM) cell lines. It was also a very weak inhibitor of topo II with an IC50 value of 607 µM [86]. With such a high IC50 value, MB is not a promising compound per se. However, given its interaction with topo II, MB could constitute the basis for the development of analogues with antitumor activity.
3.4. Aspergiolide A
Aspergiolide A (ASP, Figure 17) is an anthracycline [87] isolated from Aspergillus glaucus, which was obtained from the marine sediment around mangrove roots harvested in the Chinese province of Fujian [88].
Figure 17.

Chemical structure of aspergiolide A (CAS number: 915160-58-6).
ASP was cytotoxic on different human and murine cancer cell lines (Table 2) [88].
Wang et al. have delved into the antitumor efficacy of ASP in vitro and in vivo. The compound induced Casp-dependent apoptosis as early as 12 h after treatment [87]. In addition, ASP increased γ-H2A.X protein expression. Considering its anthracyclinic structure, it has been hypothesized that the inhibition of topo II could be involved in its apoptotic activty. The kDNA decatenation assay demonstrated that ASP inhibited the enzyme in a fashion comparable to DOXO. The results of in vivo experiments in H22 hepatoma-bearing mice and on BEL-7402 cancer xenografts (Table 2) corroborated the in vitro findings. ASP reduced tumor volume dose-dependently in H22 mice and showed comparable activity to that of DOXO (2 mg/kg). In BEL-7402 xenografts, ASP showed significantly milder activity than DOXO. Interestingly, in both in vivo models, ASP altered mice body weight considerably less than DOXO, suggesting less toxicity than the benchmark anthracycline [87]. The study also investigated the pharmacokinetic profile of ASP, which has been shown to distribute throughout the body in a perfusion- and blood-flow-dependent manner, and was able to concentrate in tumor tissues. Additionally, ASP penetrated the blood brain barrier. No clinical signs of toxicity or organs morphological changes were found in mice treated with the maximal tolerable dose of ASP (more than 400 mg/kg) [87], which is considerably higher than the dose necessary to produce the antitumor effects. The genotoxic potential of ASP was also evaluated via the in vivo bone marrow erythrocyte micronucleus assay. The number of micronuclei produced following treatment with ASP was comparable to the negative control, suggesting that ASP was not genotoxic [87].
Anthracyclines are proven to cause significant cardiotoxicity and electrocardiogram abnormalities including long QT syndrome, a potentially lethal condition induced by several drugs [89]. Long QT syndrome has been found to be caused by the blockade of hERG (human ether-a-go-go-related gene), a gene codifying the pore-forming subunit of the potassium channels, which are relevant for cardiac repolarization [90]. Thus, Li et al. investigated the in vitro inhibitory rates of ASP on the hERG current. The resulting values indicated that ASP was unable to inhibit the hERG channel, and hence it is unlikely to produce cardiotoxicity through this mechanism [87].
On the whole, the studies reported above identify ASP as an attractive candidate in the oncological area. However, further studies will be necessary to clarify whether the effects of the compound can be attributed to topo II inhibition.
3.5. Jadomycin DS
Jadomycin DS (JAD, Figure 18) is a polyketide produced by the bacterium Streptomyces venezuelae ISP5230 under stress conditions [91].
Figure 18.

Chemical structure of jadomycin DS (CAS number: not available).
JAD shares three common features with ETO and DOXO: (i) a lactone ring, (ii) a quinone moiety, and (iii) a copper-mediated DNA cleavage activity. To estimate the molecular interactions of JAD, binding studies were conducted using a nuclear magnetic resonance spectroscopy (NMR) method that allows the identification of molecules capable of binding a ligand-protein with binding affinity (KD) in the μM−mM range [92,93]. JAD bound topo IIβ. However, the overall KD for JAD-topo IIβ complex was equal to 9.4 mM, suggesting that the bond formed between JAD and topo IIβ is weak [91]. The high binding constant between the compound and topo IIβ does not depict JAD as an attractive anti-cancer drug. Moreover, JAD interacted unselectively with several unrelated enzymes including serum albumin [91], making it difficult to determine its actual mode of action and severely compromise its hypothetic in vivo application.
3.6. 2R-Acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol
2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol (2RA, Figure 19) is a sesquiterpene derived from Streptomyces sp. VITJS8 found in Indian marine soil [94].
Figure 19.

Chemical structure of 2R-acetoxymethyl-1,3,3-trimethyl-4t-(3-methyl-2-buten-1-yl)-1t-cyclohexanol (CAS number: not available).
2RA was cytotoxic [94], blocked the cell cycle in the G2/M phase, and triggered Casp-dependent apoptosis in HepG2 cells. To determine whether 2RA was able to interact with human topo IIα, a molecular docking study was performed, demonstrating that 2RA was able to bind to the active receptor pocket with a binding energy of −7.84 kJ/mol [94]. In addition, an increased formation of hydrogen bonds in the protein–ligand complex was recorded compared to the protein, indicating that the protein–ligand complex had a higher binding affinity and stability than the protein [94]. However, in vitro studies should be conducted to demonstrate that 2RA is a topo II α inhibitor.
3.7. Streptomyces sp. VITJS4 Ethyl Acetate Crude Extract
Streptomyces sp. VITJS4 bacterial strain was isolated from the marine environment in Tamil Nadu, India [95]. VITJS4 ethyl acetate crude extract exerted cytotoxic effects against HepG2 and HeLa cancer cells with identical IC50 values of 50 μg/mL and induction of apoptosis. Hence, this would suggest a cell line-independent mechanism of action [95]. Gas chromatography–mass spectrum analysis (GC–MS) identified a phthalate derivative, namely 1, 2-benzenedicarboxylic acid, mono- (2-ethylhexyl) ester, as the major bioactive metabolite among the 52 bioactive compounds of the ethyl acetate extract, which is probably responsible for the activity observed on the two human cancer cell lines. Molecular docking analysis was conducted to assess the interaction between the compound and topo IIα. What emerged is the formation of bonds at the active pocket of protein with a binding energy of −5.87 kJ/mol [95].
3.8. Sulochrin
Sulochrin (Figure 20) is a benzophenone derivative isolated from Aspergillus falconensis after cultivating it on a solid rice medium containing 3.5% of (NH4)2SO4 [96].
Figure 20.
Chemical structure of sulochrin (CAS number: 519-57-3).
Sulochrin was cytotoxic on L5178Y murine lymphoma cell line with an IC50 value of 5.1 µM [96]. The compound was not cytotoxic on MDA-MB-231 human breast cancer cells; however, at a concentration of 70 µM, it dramatically reduced cell migration [96]. Molecular docking studies indicated the interaction of sulochrin with topo II. With a free binding energy of −12.11 kcal/mol, the compound showed a robust stability through the formation of several stable bonds within the active sites, comparable to that exerted by DOXO (−16.28 kcal/mol). Molecular docking studies also demonstrated the capacity of the compound to even bind within the active sites of two further enzymes: the cyclin-dependent kinase 2 (CDK2) involved in cell-cycle progression, and the matrix metalloproteinase 13 (MMP-13) involved in the EMT process, with moderate free binding energies [96].
3.9. 3-Hydroxyholyrine A
3-hydroxyholyrine A (3HA, Figure 21) is an indolocarbazole produced by the marine-derived bacterium Streptomyces strain OUCMDZ-3118 in the presence of 5-hydroxy-L-tryptophan [97].
Figure 21.

Chemical structure of 3-hydroxyholyrine A (CAS number: 2226941-28-0).
3HA exerted cytotoxic effects on many tumor cell lines (Table 2) and reduced the expression of the antiapoptotic protein survivin more potently than ETO in MKN45 cells [97]. In supercoiled plasmid DNA relaxation assay, 3HA potently inhibited the activity of topo IIα enzyme at 1.0, 5.0, and 10.0 μM. Of note, 3HA exhibited an inhibitory activity at concentrations lower than ETO (50 μM). The inhibition of topo IIα resulted in DNA damage, as demonstrated by the concentration-dependent increase in the expression of γ-H2A.X.
4. Topo II Inhibitors Derived from Ascidians, Echinoderms and Marine Microalgae
4.1. Wakayin
Wakayin (Figure 22) is a pyrroloiminoquinone alkaloid isolated from an ascidian, commonly called sea squirt, belonging to the species Clavelina [99].
Figure 22.

Chemical structure of wakayin (CAS number: 134781-25-2).
In early studies evaluating its activity, wakayin induced cytotoxic effects on the human colon HCT-116 cancer cell line with an IC50 value of 0.5 μg/mL. On the same cell line, it inhibited topo II enzyme at a concentration of 250 μM [99]. Moreover, wakayin exhibited a higher cytotoxicity on DSBs repair-deficient CHO xrs-6 cells than on DSBs repair-proficient CHO BR1 cells. Their IC50 ratio was indeed 9.8, higher than that of ETO corresponding to 7.0. Those results clearly indicate DSB induction as a mechanism involved in the cytotoxicity of wakayin [100]. Taking into account this evidence and the planar quinonic structure of wakayin, it was hypothesized and then demonstrated that wakayin inhibited the decatenation of kDNA in a concentration-dependent manner in the range of 40 to 133 μg/mL [100]. However, the difference between the concentration inhibiting the purified enzyme (40–133 μg/mL) and the concentration exerting the cytotoxic effects (0.5 μg/mL) suggests that other mechanisms, not just topo II inhibition, could contribute to wakayin-induced DNA damage.
4.2. Ascididemin
Ascididemin (ASC, Figure 23) is a pyridoacridine alkaloid isolated from the mediterranean ascidian Cystodytes dellechiajei collected near the Balearic Islands [101] as well as from Okinawan ascidian Didemnum sp., from Kerama Islands [102].
Figure 23.

Chemical structure of ascididemin (CAS number: 114622-04-7).
It has been reported that ASC was 10-fold more cytotoxic in CHO xrs-6 (DSBs repair deficient) than in CHO BR1 (DSBs repair proficient) cells, while exhibiting identical toxicity in CHO-BR1 (SSB repair-proficient) and CHO-EM9 (SSB repair-deficient) cells, raising the hypothesis that DSBs were involved in its in vitro anticancer activity [103]. Moreover, ASC was cytotoxic on human leukemia, colon, and breast cancer cell lines [102]. Cytotoxicity elicited by ASC (Table 3) was related to the induction of Casp-dependent apoptosis, even at the lowest concentrations [102,104]. Meanwhile, it inhibited the growth of the non-malignant African green monkey kidney cell line BSC-1, revealing a lack of selectivity against cancer cells [103].
Table 3.
Topo II inhibitors derived from ascidians, echinoderms, and marine microalgae.
| Compounds | Source | Topo II Inhibitory Activity | Antitumor Effect(s) | Ref | ||||
|---|---|---|---|---|---|---|---|---|
| Assay | Concentration(s) Tested or IC50 | Outcomes | Experimental Model | Cytotoxic Activity |
Other Antitumor Mechanism(s) |
|||
| Ascididemin | Ascidians Cystodytes dellechiajei and Didemnum sp. |
L1210 d | IC50 (time not indicated): 0.39 µg/mL | ↑ Ca2+ release in sarcoplasmic reticulum | [102] | |||
| P388 d | IC50 (time not indicated): 0.4 µM | [103] | ||||||
| HCT-116 | IC50 (time not indicated): 0.3 µM | |||||||
| MCF-7 | IC50 (time not indicated): 0.3 µM | |||||||
| CHO xrs-6 b | IC50 (time not indicated): 0.03 µM | |||||||
| CHO EM9 c | IC50 (time not indicated): 0.3 µM | |||||||
| IC50: 30 µM | ↓ Topo II activity | [105] | ||||||
| Cell free DNA relaxation assay using supercoiled pKMp27 DNA plasmid and topo II α | 0.5–50 µM | Weak ↓ topo II activity | HL-60 | IC50 a (72 h): 0.48 µM | Apoptosis (caspase-3 activation, PARP cleavage, DNA fragmentation) Cell cycle inhibition (↑ cells in sub-G1 phase, ↓ cells in G1 and G2 phases) |
[104] | ||
| Cell-free DNA cleavage assay using radiolabeled pKMp27 DNA plasmid | 25–100 µM | Stimulation of DNA cleavage Stabilization of DNA–topoisomerase II covalent complexes |
HL-60/MX2 e | IC50 a (72 h): 0.65 µM | Cell cycle inhibition (↑ cells in sub-G1 phase, ↓ cells in G1 and G2 phases) | |||
| Cell-free DNA cleavage assay using radiolabeled and supercoiled rf M13 mp19 DNA | 91 µM | ↓ Topo II activity | CHO AA8 | IC50 (72 h): 3.1 µM | [106] | |||
| CHO EM9 c | IC50 (72 h): 0.4 µM | |||||||
| CHO xrs-6 b | IC50 (72 h): 0.7 µM | |||||||
| HCT-116 | IC50 (72 h): 0.1 µM | |||||||
| KB | IC50 (72 h): 0.6 µM | |||||||
| P388 d | IC50 (time not indicated): 0.4 µM | [115] | ||||||
| Bengacarboline | Ascidian Didemnum sp. |
32 µM | ↓ Topo II activity | [116] | ||||
| A-549 | IC50 (72 h): 7.1 µM | [117] | ||||||
| BxPC3 | IC50 (72 h): 10.0 µM | |||||||
| LoVo | IC50 (72 h): 9.9 µM | |||||||
| MCF-7 | IC50 (72 h): 8.6 µM | |||||||
| Echinoside A | Echinoderm Holothuria nobilis | Cell-free decatenation reaction of kinetoplast DNA with topo IIα | 1–125 µM | ↓ DNA decatenation | K562 | IC50 (time not indicated): 5.42 µM | [109] | |
| K562/A02 e | IC50 (time not indicated): 5.31 µM | |||||||
| MCF-7 | IC50 (time not indicated): 1.32 µM | |||||||
| MCF-7/ADRe | IC50 (time not indicated): 1.26 µM | |||||||
| KB | IC50 (time not indicated): 2.78 µM | |||||||
| KB/VCRe | IC50 (time not indicated): 3.29 µM | |||||||
| In vivo complexes of the enzyme (ICE) bioassay | 0.5–1 μM | Formation of stable cleavage complexes | HL-60 | Not indicated | Apoptosis (pro-caspase-3 and pro-caspase-8 cleavage, PARP degradation) DNA damage (↑ γ-H2AX protein expression) |
|||
| Cell-free DNA relaxation assay using supercoiled pBR322 plasmid DNA and topo IIα | 5–125 µM | ↓ DNA relaxation | KM mice inoculated with S-180 d cells | ↓ Tumor growth (1.5, 3.0 mg/kg/day for 7 days i.v.) |
||||
| Cell-free DNA unwinding assay using supercoiled pBR322 plasmid DNA and topo IIα | 5–125 µM | No intercalation in the DNA | KM mice inoculated with H22 d cells | ↓ Tumor growth (1.5, 3.0, 6.0 mg/kg/day for 7 days i.v) | ||||
| Ethidium bromide displacement fluorescence assay with human topo IIα | 1–125 µM | No intercalation in the DNA | Nude mice bearing human prostate carcinoma PC-3 xenografts | ↓ Tumor growth (2.6, 5.2 mg/kg/week for 4 weeks i.v.) | ||||
| Cell-free topo IIα –mediated DNA religation assay using supercoiled pBR322 plasmid DNA | ↑ Topo IIα-dependent DSBs formation | |||||||
| Cell free ATP hydrolysis assay with human topo IIα assay | 100 µM | No alteration of the ATPase activity of topo IIα | Nude mice bearing human prostate carcinoma PC-3 xenografts |
↓ Tumor growth (2.6, 5.2 mg/kg/week for 4 weeks i.v.) | ||||
| Eusynstyelamide B | Ascidian Didemnum candidum | MDA-MB-231 | IC50 (72 h): 4.95 µM | Apoptosis (PARP cleavage) Cell cycle inhibition (↑ cells in G2/M phase, ↓ cells in G0/G1 phase) |
[112] | |||
| LNCaP | IC50 (72 h): 5.0 µM | Cell cycle inhibition (↑ cells in G2/M phase, ↓ S phase progression, ↓ CDK1, CCNB1, ↓ CDC25A, ↓ CDC2 gene expression, ↑ CDKN1A gene expression, ↑ p21CIP1/WAF1, ↓ total CDC2 protein expression DNA damage induction (↑ γH2AX foci, ↓ expression of MKI67, ↑ GADD45A, GADD45G ↑ CHK2 phosphorylation |
[113] | |||||
| Ethidium bromide displacement fluorescence assay with topo II | 6.25–50 µM | No intercalation | MDA-MB-231 | Cell cycle inhibition (↑ cells in G2/M phase, ↓ cells in G0/G1 phase, ↑ CDKN1A gene expression ↑ p21CIP1/WAF1 ↓ CCNB1 gene expression, ↑ total CDC2 protein, ↑ p53, ↑ total p53 expression) DNA damage (↑ γH2AX foci, ↑ CHK2 phosphorylation ↑ CHK1 phosphorylation) |
||||
| DNA melting temperature analysis | 6.25–50 µM | No direct interaction with DNA | ||||||
| Decatenation reaction of kinetoplast DNA with topo II | 25–100 µM | ↓ Decatenation of kDNA | ||||||
| GA3Pl+ | Microalga Gymnodinium sp. A3 | Cell-free decatenation reaction of kinetoplast DNA with recombinant human topo IIα | IC50: 0.017 μg/mL | ↓ Topo IIα activity | HBC-4 | GI50 (time not indicated): 5.2 µg/mL | [108] | |
| Cell-free DNA cleavage assay using supercoiled pT2GN plasmid DNA and recombinant human topo IIα |
IC50 0.048 μg/mL | ↓ Topo IIα activity |
BSY-1 | GI50 (time not indicated): 0.67 µg/mL | ||||
| HBC-5 | GI50 (time not indicated): 6.2 µg/mL | |||||||
| MCF-7 | GI50 (time not indicated): 2.9 µg/mL | |||||||
| MDA-MB-231 | GI50 (time not indicated): 1.5 µg/mL | |||||||
| U251 | GI50 (time not indicated): 2.0 µg/mL | |||||||
| SF-268 | GI50 (time not indicated): 4.7 µg/mL | |||||||
| SF-295 | GI50 (time not indicated): 2.7 µg/mL | |||||||
| SF-539 | GI50 (time not indicated): 1.8 µg/mL | |||||||
| SNB-75 | GI50 (time not indicated): 1.5 µg/mL | |||||||
| SNB-78 | GI50 (time not indicated): 2.4 µg/mL | |||||||
| HCC2998 | GI50 (time not indicated): 2.3 µg/mL | |||||||
| KM-12 | GI50 (time not indicated): 3.7 µg/mL | |||||||
| HT-29 | GI50 (time not indicated): 3.6 µg/mL | |||||||
| WiDr | GI50 (time not indicated): 3.1 µg/mL | |||||||
| HCT-15 | GI50 (time not indicated): 3.4 µg/mL | |||||||
| HCT-116 | GI50 (time not indicated): 3.7 µg/mL | |||||||
| NCI-H23 | GI50 (time not indicated): 2.8 µg/mL | |||||||
| NCI-H226 | GI50 (time not indicated): 2.2 µg/mL | |||||||
| NCI-H522 | GI50 (time not indicated): 1.3 µg/mL | |||||||
| NCI-H460 | GI50 (time not indicated): 3.8 µg/mL | |||||||
| A-549 | GI50 (time not indicated): 11 µg/mL | |||||||
| DMS273 | GI50 (time not indicated): 2.0 µg/mL | |||||||
| DMS114 | GI50 (time not indicated): 2.7 µg/mL | |||||||
| LOX-IMVI | GI50 (time not indicated): 6.3 µg/mL | |||||||
| OVCAR-3 | GI50 (time not indicated): 2.2 µg/mL | |||||||
| OVCAR-4 | GI50 (time not indicated): 3.2 µg/mL | |||||||
| OVCAR-5 | GI50 (time not indicated): 6.8 µg/mL | |||||||
| OVCAR-8 | GI50 (time not indicated): 4.1 µg/mL | |||||||
| SK-OV-3 | GI50 (time not indicated): 8.1 µg/mL | |||||||
| RXF-631L | GI50 (time not indicated): 9.1 µg/mL | |||||||
| ACHN | GI50 (time not indicated): 8.3 µg/mL | |||||||
| St-4 | GI50 (time not indicated): 8.4 µg/mL | |||||||
| MKN1 | GI50 (time not indicated): 3.0 µg/mL | |||||||
| MKN7 | GI50 (time not indicated): 5.9 µg/mL | |||||||
| MKN28 | GI50 (time not indicated): 7.0 µg/mL | |||||||
| MKN45 | GI50 (time not indicated) 2.9 µg/mL | |||||||
| MKN74 | GI50 (time not indicated): 4.6 µg/mL | |||||||
| GA3Pl- | Microalga Gymnodinium sp. A3 | Decatenation reaction of kinetoplast DNA with recombinant human topo IIα | IC50 0.015 μg/mL | ↓ Topo IIα activity | ||||
| Cell-free DNA cleavage assay using supercoiled pT2GN plasmid DNA and recombinant human topo IIα | IC50 0.052 μg/mL | |||||||
| Meridine | Ascidian Amphicarpa meridiana |
Decatenation reaction of kinetoplast DNA | IC50 3 µM | ↓ Topo II activity | P388 d | IC50 (72 h): 0.08 µM | [118] | |
| A-549 | IC50 (72 h): 0.08 µM | |||||||
| HT-29 | IC50 (72 h): 0.84 µM | |||||||
| MEL-28 | IC50 (72 h): 0.08 µM | |||||||
| 75 µM | ↓ Topo II activity | [105] | ||||||
| Wakayin | Ascidian Clavelina sp. |
HCT-116 | IC50 (time not indicated): 0.5 µg/mL | [99] | ||||
| Decatenation reaction of kinetoplast DNA with avian topo II | 40–133 µg/mL | ↓ Topo II activity | CHO BR1 | IC50 (72 h): 3.05 µg/mL | [100] | |||
| CHO xrs-6 b | IC50 (72 h): 0.31 µg/mL | |||||||
↑: upregulation/induction; ↓: downregulation/inhibition; a: concentration that inhibits 50% of the proliferation b: DNA-double strand break repair-deficient cells; c: DNA-single strand break repair-deficient cells; d: murine cancer cells; e: multidrug resistant cancer cells; Ca2+: calcium; CCBN1: cyclin B1; CDC2: cell division cycle 2; CDC25A: cell division cycle 25 homolog A; CDK1: cyclin dependent kinase 1; CDKN1A: cyclin dependent kinase inhibitor 1A; CHK1: checkpoint kinase 1; CHK2: checkpoint kinase 2; CHO: Chinese hamster ovary; DSBs: double-strand breaks; γ-H2AX: phosphorylated H2A histone family member X; GADD45A: growth arrest and DNA damage inducible alpha; GADD45G: growth arrest and DNA damage inducible gamma; GI50 concentration that inhibits 50% of cell growth; IC50: concentration that inhibits 50% of the investigated activity; i.v.: intravenous; MKI67: marker of proliferation Ki-67; p21CIP1/WAF1: cyclin dependent kinase inhibitor 1; PARP: poly (ADP-ribose) polymerase. Human breast cancer cell lines: MCF-7; MDA-MB-231; BSY-1. Human colon cancer cell lines: HCT-116; HT-29; HCT2998; KM-12; WiDr; HCT-15. Human gastric cancer cell line: MKN45; St-4; MKN1; MKN7; MKN28; MKN74. Human glioma cell lines: U251. Human glioblastoma cell lines: SF-268; SF-295; SF-539; SNB-75; SNB-78. Human leukemia cell lines: K562; HL-60; Human lung cancer cell line: A549; NCI-H23; NCI-H226; NCI-H522; NCI-H460; DMS273; DMS114. Human melanoma cell lines: LOX-IMVI; MEL-28. Human ovarian cancer cell lines: SK-OV-3; OVCAR-3; OVCAR-4; OVCAR-5; OVCAR-8. Human pancreatic cancer cell lines: BxPC-3. Human renal cancer cell lines: RXF-6312; ACHN. Human prostate cancer cell lines: PC-3; LNCaP.
ASC was shown to inhibit topo II activity at a concentration equal to 30 μM [105]. Nearly 10 years later, Dassonneville and colleagues evaluated its interaction with topo II and demonstrated that this compound can (i) inhibit DNA ligation after it has been cleaved by topo II, and (ii) stimulate DNA cleavage with most cleavage sites having a C on the side of the cleaved bond [104]. Based on these results, ASC could be defined as a site-specific topo II poison for the purified enzyme, although its activity appeared to be inferior compared to the positive control ETO [104]. However, the capability of ASC to function as a topo II poison was not demonstrated in cellular assays. Indeed, comparing the cytotoxic activity of ASC on human leukemia cells sensitive (HL-60) or resistant (HL-60/MX2) to mitoxantrone, ASC was cytotoxic with similar IC50 values (0.48 μM for HL-60 and 0.65 μM for HL-60/MX2) [104]. Matsumoto and coworkers performed a cell-free assay to clarify the mechanism of action of ASC. The results proved that ASC was able to cleave the DNA in a concentration- and time-dependent manner, even in the absence of topo II. Moreover, experimental results demonstrated (i) the generation of ROS, (ii) that antioxidants treatment protected against DNA cleavage, and (iii) that cells deficient in ROS-induced damage repair system were more susceptible to ASC. On the whole, those results suggest that ROS production is involved in the cytotoxicity of ASC [106]. The production of ROS could be due to the direct reduction of ASC iminoquinone heterocyclic ring to semiquinone, with production of H2O2 [106]. Considering the potential of ASC to intercalate in DNA, it is probable that ROS production occurs in proximity of the nucleic acid, thereby producing DNA damage [106].
4.3. Gymnodinium sp. A3 Acidic Polysaccharide
The OKU-1 strain of the marine microalga, Gymnodinium sp. A3 (GA3), was first discovered in the water of the Seto Inland Sea (Japan). GA3 generates an extracellular acidic polysaccharide, which is a D-galactan sulfate associated with L-(+)-lactic acid, denominated GA3P [107].
Umemura and coworkers evaluated different GA3P formulations bearing high (>80%) and low (<20%) lactic acid percentage (GA3Pl+ and GA3Pl−, respectively) [108]. Both preparations of GA3P inhibited kDNA decatenation with similar IC50 values (0.048 μg/mL for GA3P+ and 0.052 μg/mL for GA3P−), proving that GA3P was a topo II inhibitor and that lactic acid percentage had no impact on topo II inhibition [108]. Gel electrophoresis of pT2GN plasmid DNA revealed that GA3P+ did not induce the accumulation of cleavable complexes and acted as a catalytic inhibitor. Furthermore, the analysis of plasmid DNA showed that GA3P+, when simultaneously added to teniposide, inhibited the stabilization of teniposide-induced cleavable complexes [108].
In a large panel of cells, the polysaccharide slightly inhibited cell proliferation with GI50 values ranging from 0.67 to 11 μg/mL [108]. However, no further cellular assays were undertaken to elucidate the cytotoxic activity or the possible death mechanism exerted by the compound. Despite evidence showing that GA3P+ was a topo II catalytic inhibitor, its chemical profile and high molecular weight can hamper its entry into the nucleus and its interaction with DNA or topo II. Certainly, further studies will be required to clarify the mechanism of action of GA3P against cancer cells.
4.4. Echinoside A
Echinoside A (ECH, Figure 24) is a saponin isolated from the sea cucumber Holothuria nobilis (Selenka), an echinoderm retrieved from the sea ground of the Dongshan Island (P. R. China) [109].
Figure 24.

Chemical structure of echinoside A (CAS number: 75410-53-6).
ECH exerted a broad-spectrum anticancer activity against a panel of 26 human and murine cancer cell lines with very similar IC50 ranging from 1.0 to 6.0 μM [109]. Fluorescent TUNEL staining of ECH-treated HL-60 cells and DNA fragmentation indicated that the observed cytotoxicity resulted from Casp-dependent apoptosis. The potent effects observed in cancer cells were confirmed by in vivo experiments on animal cancer models (Table 3).
An extensive and comprehensive set of in vitro experiments with topo IIα enzyme was conducted to investigate its topo II inhibitor activity. The results indicate that ECH effectively reduced the pBR322 plasmid DNA relaxation and suppressed kDNA decatenation [109]. An assay with top IIα extracted from HL-60 cells proved that ECH 0.5 μM induced the formation of stable cleavage complexes, which is a common mechanism for topo II poisons, along with intercalation in DNA. However, two different experiments (Table 3) reported that ECH was a non-intercalative agent, even at high concentrations [109]. The activity of ECH toward topo IIα-DNA binding was evaluated using a fluorescence anisotropy assay, which revealed that ECH inhibited the binding between the enzyme and DNA. Molecular docking studies clarified that ECH, through its sugar moiety, established strong hydrogen bonds with the DNA binding site of topo IIα, working as a catalytic inhibitor that competes with DNA for the substrate [109].
Further studies explored the effects of ECH on the cleavage/religation equilibrium using a cell-free assay. ECH produced an increase in DNA cleavage and enhanced DSBs formation, without significant effects on religation [109]. The ability of ECH to promote DNA cleavage without affecting DNA ligation makes it similar to topo II poisons such as ellipticin, genistein, and quinolones [110,111], which act with the same mechanism. However, ECH has been found to possess the peculiar characteristics of (i) blocking the noncovalent binding of topo IIα to DNA by competing with DNA for the DNA-binding domain of the enzyme, and (ii) hindering topo IIα-mediated pre-strand passage cleavage/religation equilibrium. Taken together, the studies presented above suggest that ECH is a potent non-intercalative topo II inhibitor with a peculiar mechanism of action. It acts as a topoisomerase poison (stabilization of cleavable complexes and induction of DSBs) and a catalytic inhibitor (inhibition on the topo II-DNA binding, interference with the pre-strand passage cleavage/religation equilibrium). Due to these characteristics, it constitutes a promising starting point for the development of anticancer drugs based on topo II inhibition
4.5. Eusynstyelamide B
Eusynstyelamide B (EUB, Figure 25) is a bis-indole alkaloid extracted from the marine ascidian Didemnum candidum found in the Great Barrier Reef [112].
Figure 25.

Chemical structure of eusynstyelamide B (CAS number: not available).
EUB was able to induce cytotoxicity in breast MDA-MB-231 and LNCaP prostate cancer cells [112,113]. Table 3 reports the differences in gene and protein expression between MDA-MB-231 and LNCaP cell lines, emphasizing the cell line-specific mechanisms of EUB. The COMET assay and the quantitative evaluation of γ-H2A.X foci supported the production of DNA damage via DSBs in both cell lines.
With the aim to investigate whether the observed DNA damage derived from the direct interaction of EUB with DNA, a displacement assay and a DNA melting temperature analysis were performed. Both demonstrated that EUB did not directly interact with DNA but instead acted as a topo II poison [113]. EUB was also highly cytotoxic in two non-transformed cell lines (NFF primary human neonatal foreskin fibroblasts and RWPE-1 epithelial prostate cell line), with IC50 values even lower than that reported on tumor cell lines. NFF and RWPE-1 cells are highly proliferating and express high levels of topo IIα [114]. This means that the effects of EUB were not specific for cancer cells. Further in vitro and in vivo studies have to be performed to assess the safety profile of EUB.
5. Conclusions
Of the compounds discussed in this review, only a few acts as topo II poisons (adociaquinone B and EUB) and as catalytic inhibitors (neo and apl-1). Several others exhibit topo II inhibitory activity but, due to the paucity of experimental evidence, their mode of inhibition has not been elucidated, making it difficult to establish their mechanism of action.
Although topo II inhibitors, particularly topo II poisons, are successfully used as anticancer agents, the occurrence of drug resistance and severe side effects, such as cardiotoxicity and the development of secondary malignancies, limit their use [43]. An approach to overcome these limitations could be the use of dual inhibitors. Multiple marine-derived compounds described in this review such as 25-acetals manoalide, xestoquinone, HA-A, and M7, inhibit both topo I and topo II [55,60,61,76], while for others, topo II inhibitory activity is accompanied by the inhibition of Hsp90 [36,62,74] or HDAC [75,76]. The resulting advantages are manifold. Simultaneous inhibition of topo I and topo II could reduce the possible onset of resistance. The same advantage can be achieved by inhibiting topo II and Hsp90 [43]. Concerning topo II and HDAC inhibition, HDAC inhibition-mediated histone hyperacetylation increases chromatin decondensation and DNA accessibility. These effects may promote topo II binding and enhance topo II inhibiting activity [43]. Among the marine compounds presented in this review, heteronemin is the most interesting. Indeed, its cytotoxic activity was highly multimechanistic, with inhibition of the catalytic activities of both topo I and topo II and inhibition of Hsp90, associated with oxidative and ER stress. However, the dual inhibitors are often compounds with a high molecular weight [119], which could limit their druggability and their safety profile as well as indicate that their pharmacokinetics should be thoroughly explored
Another issue to consider is the ability of topo II inhibitors to cause DNA lesions that, if not repaired or not cytotoxic, could lead to chromosome aberrations and secondary malignancies such as leukemias [120]. Although topo II catalytic inhibitors are usually associated with no or limited direct DNA damage [121], some marine-derived topo II catalytic inhibitors presented in this review induce DNA DSBs and/or increase the protein expression of DNA damage-related proteins. Thus, it would be of great relevance to clarify whether their genotoxicity results from their topo II catalytic inhibition or involves different mechanisms. A further concern related to the toxicological profile is the lack of selectivity toward cancer cells exhibited by some marine compounds, which prompts more extensive studies on non-transformed cells to assess the safety of such molecules.
Lastly, some marine compounds exhibited a strong binding affinity for topo II, demonstrated through molecular docking studies. Among those, the most interesting are neo, ECH, and sulochrin, which are characterized by a binding energy of -61.8, -39.21, and -12.11 kcal/mol, respectively. However, in some cases, this interaction has not been confirmed by cellular assays, making it difficult to know whether topo II binding leads to the actual inhibition of the enzyme activity. Thus, at least DNA decatenation and/or relaxation assays are necessary to confirm their topo II inhibitory activity. These cell-free assays certainly provide early indications of the effective inhibition of topo II. However, they may not be sufficient because, as shown for secoadociaquinone A and B and GA3P [77,108], their inhibitory activity on the purified enzyme does not necessarily lead to the inhibition of topo II at the cellular level.
In conclusion, in this review, we reported current studies on marine-derived compounds targeting topo II, highlighted their pharmacological potential, and discussed their toxicological issues.
Author Contributions
Conceptualization, C.F.; methodology, G.G., V.P. and I.C.-C.; data curation, G.G., V.P. and I.C.-C.; writing—original draft preparation, G.G., V.P. and I.C.-C.; writing—review and editing, G.A., C.S. and C.F.; supervision, C.F. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
I.C.-C. was supported by the VII Program of Inner Initiative for Research and Transfer of the University of Seville (VIIPPIT-2022-III.2) and by a postdoctoral fellowship from the Andalusian Government Ministry of Economy, Knowledge, Business, and University (DOC_00587/2020).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Global Cancer Observatory. [(accessed on 13 September 2022)]. Available online: https://gco.iarc.fr/
- 2.Dias D.A., Urban S., Roessner U. A Historical Overview of Natural Products in Drug Discovery. Metabolites. 2012;2:303–336. doi: 10.3390/metabo2020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fimognari C., Lenzi M., Ferruzzi L., Turrini E., Scartezzini P., Poli F., Gotti R., Guerrini A., Carulli G., Ottaviano V., et al. Mitochondrial Pathway Mediates the Antileukemic Effects of Hemidesmus Indicus, a Promising Botanical Drug. PLoS ONE. 2011;6:e21544. doi: 10.1371/journal.pone.0021544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lenzi M., Malaguti M., Cocchi V., Hrelia S., Hrelia P. Castanea sativa Mill. Bark Extract Exhibits Chemopreventive Properties Triggering Extrinsic Apoptotic Pathway in Jurkat Cells. BMC Complement. Altern. Med. 2017;17:251. doi: 10.1186/s12906-017-1756-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenzi M., Cocchi V., Malaguti M., Barbalace M.C., Marchionni S., Hrelia S., Hrelia P. 6-(Methylsulfonyl) Hexyl Isothiocyanate as Potential Chemopreventive Agent: Molecular and Cellular Profile in Leukaemia Cell Lines. Oncotarget. 2017;8:111697–111714. doi: 10.18632/oncotarget.22902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Newman D.J., Cragg G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 7.Scotti L., Bezerra Mendonca F.J., Ribeiro F.F., Tavares J.F., da Silva M.S., Barbosa Filho J.M., Scotti M.T. Natural Product Inhibitors of Topoisomerases: Review and Docking Study. Curr. Protein Pept. Sci. 2018;19:275–291. doi: 10.2174/1389203718666170111114442. [DOI] [PubMed] [Google Scholar]
- 8.Schirrmacher V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment. Int. J. Oncol. 2019;54:407–419. doi: 10.3892/ijo.2018.4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vann K.R., Oviatt A.A., Osheroff N. Topoisomerase II Poisons: Converting Essential Enzymes into Molecular Scissors. Biochemistry. 2021;60:1630–1641. doi: 10.1021/acs.biochem.1c00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vélez-Cruz R., Osheroff N. Encyclopedia of Biological Chemistry. Elsevier; Amsterdam, The Netherlands: 2004. DNA Topoisomerases: Type II; pp. 806–811. [Google Scholar]
- 11.Buzun K., Bielawska A., Bielawski K., Gornowicz A. DNA Topoisomerases as Molecular Targets for Anticancer Drugs. J. Enzyme Inhib. Med. Chem. 2020;35:1781–1799. doi: 10.1080/14756366.2020.1821676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bax B.D., Murshudov G., Maxwell A., Germe T. DNA Topoisomerase Inhibitors: Trapping a DNA-Cleaving Machine in Motion. J. Mol. Biol. 2019;431:3427–3449. doi: 10.1016/j.jmb.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ormrod D., Holm K., Goa K., Spencer C. Epirubicin: A Review of Its Efficacy as Adjuvant Therapy and in the Treatment of Metastatic Disease in Breast Cancer. Drugs Aging. 1999;15:389–416. doi: 10.2165/00002512-199915050-00006. [DOI] [PubMed] [Google Scholar]
- 14.Prajapati D., Parihar L., Sharma P., Sharma S. A Review on Doxorubicn Induced Cardiotoxicity and Its Molecular Mechanism. Int. J. Pharm. Sci. 2020;11:2521–2527. [Google Scholar]
- 15.Singal P.K., Iliskovic N. Doxorubicin-Induced Cardiomyopathy. N. Engl. J. Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 16.Holthuis J.J.M. Etoposide and Teniposide: Bioanalysis, Metabolism and Clinical Pharmacokinetics. Pharm. Weekbl. 1988;10:101–116. doi: 10.1007/BF01959294. [DOI] [PubMed] [Google Scholar]
- 17.Lee J.H., Wendorff T.J., Berger J.M. Resveratrol: A Novel Type of Topoisomerase II Inhibitor. J. Biol. Chem. 2017;292:21011–21022. doi: 10.1074/jbc.M117.810580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaur R., Manjal S.K., Rawal R.K., Kumar K. Recent Synthetic and Medicinal Perspectives of Tryptanthrin. Bioorg. Med. Chem. 2017;25:4533–4552. doi: 10.1016/j.bmc.2017.07.003. [DOI] [PubMed] [Google Scholar]
- 19.Grynkiewicz G., Demchuk O.M. New Perspectives for Fisetin. Front. Chem. 2019;7:697. doi: 10.3389/fchem.2019.00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semwal D., Semwal R., Combrinck S., Viljoen A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrients. 2016;8:90. doi: 10.3390/nu8020090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang K., Oh S.H., Yun J.H., Jho E.H., Kang J.-H., Batsuren D., Tunsag J., Park K.H., Kim M., Nho C.W. A Novel Topoisomerase Inhibitor, Daurinol, Suppresses Growth of HCT116 Cells with Low Hematological Toxicity Compared to Etoposide. Neoplasia. 2011;13 doi: 10.1593/neo.11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saeed A.F.U.H., Su J., Ouyang S. Marine-Derived Drugs: Recent Advances in Cancer Therapy and Immune Signaling. Biomed. Pharmacother. 2021;134:111091. doi: 10.1016/j.biopha.2020.111091. [DOI] [PubMed] [Google Scholar]
- 23.Demain A.L., Fang A. The Natural Functions of Secondary Metabolites. In: Fiechter A., editor. History of Modern Biotechnology I. Volume 69. Advances in Biochemical Engineering/Biotechnology; Springer; Berlin/Heidelberg, Germany: 2000. pp. 1–39. [DOI] [PubMed] [Google Scholar]
- 24.Molinski T.F., Dalisay D.S., Lievens S.L., Saludes J.P. Drug Development from Marine Natural Products. Nat. Rev. Drug Discov. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 25.Marshall K.M., Matsumoto S.S., Holden J.A., Concepción G.P., Tasdemir D., Ireland C.M., Barrows L.R. The Anti-Neoplastic and Novel Topoisomerase II-Mediated Cytotoxicity of Neoamphimedine, a Marine Pyridoacridine. Biochem. Pharmacol. 2003;66:447–458. doi: 10.1016/S0006-2952(03)00209-0. [DOI] [PubMed] [Google Scholar]
- 26.Li L., Abraham A.D., Zhou Q., Ali H., O’Brien J.V., Hamill B.D., Arcaroli J.J., Messersmith W.A., LaBarbera D.V. An Improved High Yield Total Synthesis and Cytotoxicity Study of the Marine Alkaloid Neoamphimedine: An ATP-Competitive Inhibitor of Topoisomerase IIα and Potent Anticancer Agent. Mar. Drugs. 2014;12:4833–4850. doi: 10.3390/md12094833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Guzman F.S., Carte B., Troupe N., Faulkner D.J., Harper M.K., Concepcion G.P., Mangalindan G.C., Matsumoto S.S., Barrows L.R., Ireland C.M. Neoamphimedine: A New Pyridoacridine Topoisomerase II Inhibitor Which Catenates DNA. J. Org. Chem. 1999;64:1400–1402. doi: 10.1021/jo982047x. [DOI] [Google Scholar]
- 28.Jeggo P.A., Caldecott K., Pidsley S., Banks G.R. Sensitivity of Chinese Hamster Ovary Mutants Defective in DNA Double Strand Break Repair to Topoisomerase II Inhibitors. Cancer Res. 1989;49:7057–7063. [PubMed] [Google Scholar]
- 29.Ponder J., Yoo B.H., Abraham A.D., Li Q., Ashley A.K., Amerin C.L., Zhou Q., Reid B.G., Reigan P., Hromas R., et al. Neoamphimedine Circumvents Metnase-Enhanced DNA Topoisomerase IIα Activity through ATP-Competitive Inhibition. Mar. Drugs. 2011;9:2397–2408. doi: 10.3390/md9112397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wray J., Williamson E.A., Royce M., Shaheen M., Beck B.D., Lee S.-H., Nickoloff J.A., Hromas R. Metnase Mediates Resistance to Topoisomerase II Inhibitors in Breast Cancer Cells. PLoS ONE. 2009;4:e5323. doi: 10.1371/journal.pone.0005323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wray J., Williamson E.A., Sheema S., Lee S.-H., Libby E., Willman C.L., Nickoloff J.A., Hromas R. Metnase Mediates Chromosome Decatenation in Acute Leukemia Cells. Blood. 2009;114:1852–1858. doi: 10.1182/blood-2008-08-175760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Q., Abraham A.D., Li L., Babalmorad A., Bagby S., Arcaroli J.J., Hansen R.J., Valeriote F.A., Gustafson D.L., Schaack J., et al. Topoisomerase IIα Mediates TCF-Dependent Epithelial-Mesenchymal Transition in Colon Cancer. Oncogene. 2016;35:4990–4999. doi: 10.1038/onc.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Georgakopoulos-Soares I., Chartoumpekis D.V., Kyriazopoulou V., Zaravinos A. EMT Factors and Metabolic Pathways in Cancer. Front. Oncol. 2020;10:499. doi: 10.3389/fonc.2020.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bian J., Dannappel M., Wan C., Firestein R. Transcriptional Regulation of Wnt/β-Catenin Pathway in Colorectal Cancer. Cells. 2020;9:2125. doi: 10.3390/cells9092125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su J.-H., Chen Y.-C., El-Shazly M., Du Y.-C., Su C.-W., Tsao C.-W., Liu L.-L., Chou Y., Chang W.-B., Su Y.-D., et al. Towards the Small and the Beautiful: A Small Dibromotyrosine Derivative from Pseudoceratina sp. Sponge Exhibits Potent Apoptotic Effect through Targeting IKK/NFκB Signaling Pathway. Mar. Drugs. 2013;11:3168–3185. doi: 10.3390/md11093168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shih S.-P., Lu M.-C., El-Shazly M., Lin Y.-H., Chen C.-L., Yu S.S.-F., Liu Y.-C. The Antileukemic and Anti-Prostatic Effect of Aeroplysinin-1 Is Mediated through ROS-Induced Apoptosis via NOX Activation and Inhibition of HIF-1a Activity. Life. 2022;12:687. doi: 10.3390/life12050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vos S.M., Tretter E.M., Schmidt B.H., Berger J.M. All Tangled up: How Cells Direct, Manage and Exploit Topoisomerase Function. Nat. Rev. Mol. Cell Biol. 2011;12:827–841. doi: 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Ignazio L., Batie M., Rocha S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-ΚB. Biomedicines. 2017;5:21. doi: 10.3390/biomedicines5020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nitulescu G., Van De Venter M., Nitulescu G., Ungurianu A., Juzenas P., Peng Q., Olaru O., Grădinaru D., Tsatsakis A., Tsoukalas D., et al. The Akt Pathway in Oncology Therapy and beyond (Review) Int. J. Oncol. 2018;53:2319–2331. doi: 10.3892/ijo.2018.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Song M., Bode A.M., Dong Z., Lee M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019;79:1019–1031. doi: 10.1158/0008-5472.CAN-18-2738. [DOI] [PubMed] [Google Scholar]
- 41.Stuhldreier F., Kassel S., Schumacher L., Wesselborg S., Proksch P., Fritz G. Pleiotropic Effects of Spongean Alkaloids on Mechanisms of Cell Death, Cell Cycle Progression and DNA Damage Response (DDR) of Acute Myeloid Leukemia (AML) Cells. Cancer Lett. 2015;361:39–48. doi: 10.1016/j.canlet.2015.02.030. [DOI] [PubMed] [Google Scholar]
- 42.Yun C.W., Kim H.J., Lim J.H., Lee S.H. Heat Shock Proteins: Agents of Cancer Development and Therapeutic Targets in Anti-Cancer Therapy. Cells. 2019;9:60. doi: 10.3390/cells9010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skok Ž., Zidar N., Kikelj D., Ilaš J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J. Med. Chem. 2020;63:884–904. doi: 10.1021/acs.jmedchem.9b00726. [DOI] [PubMed] [Google Scholar]
- 44.Park S., Kim J.-H., Kim J.E., Song G.-Y., Zhou W., Goh S.-H., Na M., Oh S. Cytotoxic Activity of Aeroplysinin-1 against Colon Cancer Cells by Promoting β-Catenin Degradation. Food Chem. Toxicol. 2016;93:66–72. doi: 10.1016/j.fct.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 45.Kreuter M.-H., Leake R.E., Rinaldi F., Müller-Klieser W., Maidhof A., Müller W.E.G., Schröder H.C. Inhibition of Intrinsic Protein Tyrosine Kinase Activity of EGF-Receptor Kinase Complex from Human Breast Cancer Cells by the Marine Sponge Metabolite (+)-Aeroplysinin-1. Comp. Biochem. Physiol. B Biochem. 1990;97:151–158. doi: 10.1016/0305-0491(90)90194-X. [DOI] [PubMed] [Google Scholar]
- 46.Venables D.A., Concepción G.P., Matsumoto S.S., Barrows L.R., Ireland C.M. Makaluvamine N: A New Pyrroloiminoquinone from Zyzzya Fuliginosa. J. Nat. Prod. 1997;60:408–410. doi: 10.1021/np9607262. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt E.W., Harper M.K., Faulkner D.J. Makaluvamines H-M and Damirone C from the Pohnpeian Sponge Zyzzya Fuliginosa. J. Nat. Prod. 1995;58:1861–1867. doi: 10.1021/np50126a008. [DOI] [PubMed] [Google Scholar]
- 48.Carney J.R., Scheuer P.J., Kelly-Borges M. Makaluvamine G, a Cytotoxic Pigment from an an Indonesian Sponge Histodermella sp. Tetrahedron. 1993;49:8483–8486. doi: 10.1016/S0040-4020(01)96256-8. [DOI] [Google Scholar]
- 49.Radisky D.C., Radisky E.S., Barrows L.R., Copp B.R., Kramer R.A., Ireland C.M. Novel Cytotoxic Topoisomerase II Inhibiting Pyrroloiminoquinones from Fijian Sponges of the Genus Zyzzya. J. Am. Chem. Soc. 1993;115:1632–1638. doi: 10.1021/ja00058a003. [DOI] [Google Scholar]
- 50.Barrows L.R., Radisky D.C., Copp B.R., Swaffar D.S., Kramer R.A., Warters R.L., Ireland C.M. Makaluvamines, Marine Natural Products, Are Active Anti-Cancer Agents and DNA Topo II Inhibitors. Anticancer Drug Des. 1993;8:333–347. [PubMed] [Google Scholar]
- 51.Matsumoto S.S., Haughey H.M., Schmehl D.M., Venables D.A., Ireland C.M., Holden J.A., Barrows L.R. Makaluvamines Vary in Ability to Induce Dose-Dependent DNA Cleavage via Topoisomerase II Interaction. Anticancer Drugs. 1999;10:39–45. doi: 10.1097/00001813-199901000-00006. [DOI] [PubMed] [Google Scholar]
- 52.Shinkre B.A., Raisch K.P., Fan L., Velu S.E. Analogs of the Marine Alkaloid Makaluvamines: Synthesis, Topoisomerase II Inhibition, and Anticancer Activity. Bioorg. Med. Chem. Lett. 2007;17:2890–2893. doi: 10.1016/j.bmcl.2007.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boucle S., Melin C., Clastre M., Guillard J. Design, Synthesis and Evaluation of New Marine Alkaloid-Derived Pentacyclic Structures with Anti-Tumoral Potency. Mar. Drugs. 2015;13:655–665. doi: 10.3390/md13010655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guzmán E.A., Johnson J.D., Carrier M.K., Meyer C.I., Pitts T.P., Gunasekera S.P., Wright A.E. Selective Cytotoxic Activity of the Marine-Derived Batzelline Compounds against Pancreatic Cancer Cell Lines. Anticancer Drugs. 2009;20:149–155. doi: 10.1097/CAD.0b013e32831fa39e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mizushina Y. Molecular Action Mode of Hippospongic Acid A, an Inhibitor of Gastrulation of Starfish Embryos. J. Biochem. 2003;133:541–552. doi: 10.1093/jb/mvg070. [DOI] [PubMed] [Google Scholar]
- 56.Su J.-H., Tseng S.-W., Lu M.-C., Liu L.-L., Chou Y., Sung P.-J. Cytotoxic C21 and C22 Terpenoid-Derived Metabolites from the Sponge Ircinia sp. J. Nat. Prod. 2011;74:2005–2009. doi: 10.1021/np2004209. [DOI] [PubMed] [Google Scholar]
- 57.Shih H.-C., El-Shazly M., Juan Y.-S., Chang C.-Y., Su J.-H., Chen Y.-C., Shih S.-P., Chen H.-M., Wu Y.-C., Lu M.-C. Cracking the Cytotoxicity Code: Apoptotic Induction of 10-Acetylirciformonin B Is Mediated through ROS Generation and Mitochondrial Dysfunction. Mar. Drugs. 2014;12:3072–3090. doi: 10.3390/md12053072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su J.-H., Chang W.-B., Chen H.-M., El-Shazly M., Du Y.-C., Kung T.-H., Chen Y.-C., Sung P.-J., Ho Y.-S., Kuo F.-W., et al. 10-Acetylirciformonin B, a Sponge Furanoterpenoid, Induces DNA Damage and Apoptosis in Leukemia Cells. Molecules. 2012;17:11839–11848. doi: 10.3390/molecules171011839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts D.J., Miyamoto S. Hexokinase II Integrates Energy Metabolism and Cellular Protection: Akting on Mitochondria and TORCing to Autophagy. Cell Death Differ. 2015;22:248–257. doi: 10.1038/cdd.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kobayashi M., Okamoto T., Hayashi K., Yokoyama N., Sasaki T., Kitagawa I. Marine Natural Products. XXXII. Absolute Configurations of C-4 of the Manoalide Family, Biologically Active Sesterterpenes from the Marine Sponge Hyrtios Erecta. Chem. Pharm. Bull. 1994;42:265–270. doi: 10.1248/cpb.42.265. [DOI] [PubMed] [Google Scholar]
- 61.Lai K.-H., Peng B.-R., Hsu Y.-M., El-Shazly M., Du Y.-C., Lu M.-C., Su J.-H., Liu Y.-C. The Configuration-Dependent Anti-Leukemic Effect of Manoalide Stereoisomers: Reignite Research Interest in These Sponge-Derived Sesterterpenoids. Bioorg. Chem. 2021;114:105150. doi: 10.1016/j.bioorg.2021.105150. [DOI] [PubMed] [Google Scholar]
- 62.Lee M.-G., Liu Y.-C., Lee Y.-L., El-Shazly M., Lai K.-H., Shih S.-P., Ke S.-C., Hong M.-C., Du Y.-C., Yang J.-C., et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Mar. Drugs. 2018;16:E204. doi: 10.3390/md16060204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang Y.-C.S.H., Li Z.-L., Huang T.-Y., Su K.-W., Lin C.-Y., Huang C.-H., Chen H.-Y., Lu M.-C., Huang H.-M., Lee S.-Y., et al. Effect of Estrogen on Heteronemin-Induced Anti-Proliferative Effect in Breast Cancer Cells with Different Estrogen Receptor Status. Front. Cell Dev. Biol. 2021;9:688607. doi: 10.3389/fcell.2021.688607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chung C.-C., Huang T.-Y., Chu H.-R., De Luca R., Candelotti E., Huang C.-H., Yang Y.-C.S.H., Incerpi S., Pedersen J.Z., Lin C.-Y., et al. Heteronemin and Tetrac Derivatives Suppress Non-Small Cell Lung Cancer Growth via ERK1/2 Inhibition. Food Chem. Toxicol. 2022;161:112850. doi: 10.1016/j.fct.2022.112850. [DOI] [PubMed] [Google Scholar]
- 65.Chang W.-T., Bow Y.-D., Fu P.-J., Li C.-Y., Wu C.-Y., Chang Y.-H., Teng Y.-N., Li R.-N., Lu M.-C., Liu Y.-C., et al. A Marine Terpenoid, Heteronemin, Induces Both the Apoptosis and Ferroptosis of Hepatocellular Carcinoma Cells and Involves the ROS and MAPK Pathways. Oxid. Med. Cell Longev. 2021;2021:7689045. doi: 10.1155/2021/7689045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu T., Ding W., Ji X., Ao X., Liu Y., Yu W., Wang J. Molecular Mechanisms of Ferroptosis and Its Role in Cancer Therapy. J. Cell Mol. Med. 2019;23:4900–4912. doi: 10.1111/jcmm.14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neophytou C.M., Trougakos I.P., Erin N., Papageorgis P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers. 2021;13:4363. doi: 10.3390/cancers13174363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan X., Zhou R., Ma Z. Autophagy—Cell Survival and Death. In: Qin Z.-H., editor. Autophagy: Biology and Diseases. Volume 1206. Springer; Singapore: 2019. pp. 667–696. Advances in Experimental Medicine and Biology. [DOI] [PubMed] [Google Scholar]
- 69.Wu S.-Y., Sung P.-J., Chang Y.-L., Pan S.-L., Teng C.-M. Heteronemin, a Spongean Sesterterpene, Induces Cell Apoptosis and Autophagy in Human Renal Carcinoma Cells. BioMed Res. Int. 2015;2015:738241. doi: 10.1155/2015/738241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schumacher M., Cerella C., Eifes S., Chateauvieux S., Morceau F., Jaspars M., Dicato M., Diederich M. Heteronemin, a Spongean Sesterterpene, Inhibits TNFα-Induced NF-ΚB Activation through Proteasome Inhibition and Induces Apoptotic Cell Death. Biochem. Pharmacol. 2010;79:610–622. doi: 10.1016/j.bcp.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 71.Nunes-Xavier C.E., Mingo J., López J.I., Pulido R. The Role of Protein Tyrosine Phosphatases in Prostate Cancer Biology. Biochim. Biophys. Acta Mol. Cell Res. 2019;1866:102–113. doi: 10.1016/j.bbamcr.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 72.Bonner W.M., Redon C.E., Dickey J.S., Nakamura A.J., Sedelnikova O.A., Solier S., Pommier Y. ΓH2AX and Cancer. Nat. Rev. Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saikia M., Retnakumari A.P., Anwar S., Anto N.P., Mittal R., Shah S., Pillai K.S., Balachandran V.S., Peter V., Thomas R., et al. Heteronemin, a Marine Natural Product, Sensitizes Acute Myeloid Leukemia Cells towards Cytarabine Chemotherapy by Regulating Farnesylation of Ras. Oncotarget. 2018;9:18115–18127. doi: 10.18632/oncotarget.24771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lai K.-H., Liu Y.-C., Su J.-H., El-Shazly M., Wu C.-F., Du Y.-C., Hsu Y.-M., Yang J.-C., Weng M.-K., Chou C.-H., et al. Antileukemic Scalarane Sesterterpenoids and Meroditerpenoid from Carteriospongia (Phyllospongia) sp., Induce Apoptosis via Dual Inhibitory Effects on Topoisomerase II and Hsp90. Sci. Rep. 2016;6:36170. doi: 10.1038/srep36170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shih S.-P., Lee M.-G., El-Shazly M., Juan Y.-S., Wen Z.-H., Du Y.-C., Su J.-H., Sung P.-J., Chen Y.-C., Yang J.-C., et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs. 2015;13:3132–3153. doi: 10.3390/md13053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang K.-C., Lu M.-C., Hsu K.-C., El-Shazly M., Shih S.-P., Lien S.-T., Kuo F.-W., Yang S.-C., Chen C.-L., Yang Y.-C.S.H. The Antileukemic Effect of Xestoquinone, a Marine-Derived Polycyclic Quinone-Type Metabolite, Is Mediated through ROS-Induced Inhibition of HSP-90. Molecules. 2021;26:7037. doi: 10.3390/molecules26227037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Concepcion G.P., Foderaro T.A., Eldredge G.S., Lobkovsky E., Clardy J., Barrows L.R., Ireland C.M. Topoisomerase II-Mediated DNA Cleavage by Adocia- and Xestoquinones from the Philippine Sponge Xestospongia sp. J. Med. Chem. 1995;38:4503–4507. doi: 10.1021/jm00022a016. [DOI] [PubMed] [Google Scholar]
- 78.Evison B.J., Sleebs B.E., Watson K.G., Phillips D.R., Cutts S.M. Mitoxantrone, More than Just Another Topoisomerase II Poison. Med. Res. Rev. 2016;36:248–299. doi: 10.1002/med.21364. [DOI] [PubMed] [Google Scholar]
- 79.Carney J.R., Scheuer P.J., Kelly-Borges M. A New Bastadin from the Sponge (i) Psammaplysilla Purpurea (i) J. Nat. Prod. 1993;56:153–157. doi: 10.1021/np50091a025. [DOI] [PubMed] [Google Scholar]
- 80.Juagdan E.G., Kalidindi R.S., Scheuer P.J., Kelly-Borges M. Elenic Acid, an Inhibitor of Topoisomerase II, from a Sponge, Plakinastrella sp. Tetrahedron Lett. 1995;36:2905–2908. doi: 10.1016/0040-4039(95)00432-C. [DOI] [Google Scholar]
- 81.Carney J.R., Scheuer P.J. Popolohuanone E, a Topoisomerase-II Inhibitor with Selective Lung Tumor Cytotoxicity from the Pohnpei Sponge Dysidea sp. Tetrahedron Lett. 1993;34:3727–3730. doi: 10.1016/S0040-4039(00)79211-2. [DOI] [Google Scholar]
- 82.Yanagihara M., Sasaki-Takahashi N., Sugahara T., Yamamoto S., Shinomi M., Yamashita I., Hayashida M., Yamanoha B., Numata A., Yamori T., et al. Leptosins Isolated from Marine Fungus Leptoshaeria Species Inhibit DNA Topoisomerases I and/or II and Induce Apoptosis by Inactivation of Akt/Protein Kinase B. Cancer Sci. 2005;96:816–824. doi: 10.1111/j.1349-7006.2005.00117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamada T., Iritani M., Ohishi H., Tanaka K., Minoura K., Doi M., Numata A. Pericosines, Antitumour Metabolites from the Sea Hare-Derived Fungus PEriconia Byssoides. Structures and Biological Activities. Org. Biomol. Chem. 2007;5:3979. doi: 10.1039/b713060k. [DOI] [PubMed] [Google Scholar]
- 84.Usami Y., Ichikawa H., Arimoto M. Synthetic Efforts for Stereo Structure Determination of Cytotoxic Marine Natural Product Pericosines as Metabolites of Periconia sp. from Sea Hare. Int. J. Mol. Sci. 2008;9:401–421. doi: 10.3390/ijms9030401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wee P., Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers. 2017;9:E52. doi: 10.3390/cancers9050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang F., Tian X., Huang C., Li Q., Zhang S. Marinactinones A-C, New γ-Pyrones from Marine Actinomycete Marinactinospora Thermotolerans SCSIO 00606. J. Antibiot. 2011;64:189–192. doi: 10.1038/ja.2010.153. [DOI] [PubMed] [Google Scholar]
- 87.Li J., Wang Y., Qi X., Li D., Zhu T., Mo X. Anticancer Efficacy and Absorption, Distribution, Metabolism, and Toxicity Studies of Aspergiolide A in Early Drug Development. Drug Des. Dev. Ther. 2014;2014:1965–1977. doi: 10.2147/DDDT.S64989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du L., Zhu T., Fang Y., Liu H., Gu Q., Zhu W. Aspergiolide A, a Novel Anthraquinone Derivative with Naphtho[1,2,3-de]Chromene-2,7-Dione Skeleton Isolated from a Marine-Derived Fungus Aspergillus Glaucus. Tetrahedron. 2007;63:1085–1088. doi: 10.1016/j.tet.2006.11.074. [DOI] [Google Scholar]
- 89.Ducroq J., Moha ou Maati H., Guilbot S., Dilly S., Laemmel E., Pons-Himbert C., Faivre J., Bois P., Stücker O., Le Grand M. Dexrazoxane Protects the Heart from Acute Doxorubicin-Induced QT Prolongation: A Key Role for IKs. Br. J. Pharmacol. 2010;159:93–101. doi: 10.1111/j.1476-5381.2009.00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Witchel H.J. Drug-Induced HERG Block and Long QT Syndrome: HERG, Drugs, and LQTS. Cardiovasc. Ther. 2011;29:251–259. doi: 10.1111/j.1755-5922.2010.00154.x. [DOI] [PubMed] [Google Scholar]
- 91.Martinez-Farina C.F., McCormick N., Robertson A.W., Clement H., Jee A., Ampaw A., Chan N.-L., Syvitski R.T., Jakeman D.L. Investigations into the Binding of Jadomycin DS to Human Topoisomerase IIβ by WaterLOGSY NMR Spectroscopy. Org. Biomol. Chem. 2015;13:10324–10327. doi: 10.1039/C5OB01508A. [DOI] [PubMed] [Google Scholar]
- 92.Raingeval C., Cala O., Brion B., Le Borgne M., Hubbard R.E., Krimm I. 1D NMR WaterLOGSY as an Efficient Method for Fragment-Based Lead Discovery. J. Enzyme Inhib. Med. Chem. 2019;34:1218–1225. doi: 10.1080/14756366.2019.1636235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang R., Leung I.K.H. Methods in Enzymology. Volume 615. Elsevier; Amsterdam, The Netherlands: 2019. Protein–Small Molecule Interactions by WaterLOGSY; pp. 477–500. [DOI] [PubMed] [Google Scholar]
- 94.Naine S.J., Devi C.S., Mohanasrinivasan V., Doss C.G.P., Kumar D.T. Binding and Molecular Dynamic Studies of Sesquiterpenes (2R-Acetoxymethyl-1,3,3-Trimethyl-4t-(3-Methyl-2-Buten-1-Yl)-1t-Cyclohexanol) Derived from Marine Streptomyces sp. VITJS8 as Potential Anticancer Agent. Appl. Microbiol. Biotechnol. 2016;100:2869–2882. doi: 10.1007/s00253-015-7156-2. [DOI] [PubMed] [Google Scholar]
- 95.Jemimah Naine S., Subathra Devi C., Mohanasrinivasan V., George Priya Doss C. Bioactivity of Marine Streptomyces sp. VITJS4: Interactions of Cytotoxic Phthalate Derivatives with Human Topoisomerase II α: An in Silico Molecular Docking Analysis. Interdiscip. Sci. 2018;10:261–270. doi: 10.1007/s12539-016-0187-2. [DOI] [PubMed] [Google Scholar]
- 96.El-Kashef D.H., Youssef F.S., Reimche I., Teusch N., Müller W.E.G., Lin W., Frank M., Liu Z., Proksch P. Polyketides from the Marine-Derived Fungus Aspergillus Falconensis: In Silico and in Vitro Cytotoxicity Studies. Bioorg. Med. Chem. 2021;29:115883. doi: 10.1016/j.bmc.2020.115883. [DOI] [PubMed] [Google Scholar]
- 97.Wang C., Monger A., Wang L., Fu P., Piyachaturawat P., Chairoungdua A., Zhu W. Precursor-Directed Generation of Indolocarbazoles with Topoisomerase IIα Inhibitory Activity. Mar. Drugs. 2018;16:E168. doi: 10.3390/md16050168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Takahashi C., Numata A., Ito Y., Matsumura E., Araki H., Iwaki H., Kushida K. Leptosins, Antitumour Metabolites of a Fungus Isolated from a Marine Alga. J. Chem. Soc. Perkin Trans. 1. 1994;13:1859–1864. doi: 10.1039/p19940001859. [DOI] [Google Scholar]
- 99.Copp B.R., Ireland C.M., Barrows L.R. Wakayin: A Novel Cytotoxic Pyrroloiminoquinone Alkaloid from the Ascidian Clavelina Species. J. Org. Chem. 1991;56:4596–4597. doi: 10.1021/jo00015a005. [DOI] [Google Scholar]
- 100.Swaffar D.S., Ireland C.M., Barrows L.R. A Rapid Mechanism-Based Screen to Detect Potential Anti-Cancer Agents. Anti-Cancer Drugs. 1994;5:15–23. doi: 10.1097/00001813-199402000-00003. [DOI] [PubMed] [Google Scholar]
- 101.Bonnard I., Bontemps N., Lahmy S., Banaigs B., Combaut G., Francisco C., Colson P., Houssier C., Waring M.J., Bailly C. Binding to DNA and Cytotoxic Evaluation of Ascididemin, the Major Alkaloid from the Mediterranean Ascidian Cystodytes Dellechiajei. Anticancer Drug Des. 1995;10:333–346. [PubMed] [Google Scholar]
- 102.Kobayash J., Cheng J., Nakamura H., Ohizumi Y., Hirata Y., Sasaki T., Ohta T., Nozoe S. Ascididemin, a Novel Pentacyclic Aromatic Alkaloid with Potent Antileukemic Activity from the Okinawan Tunicate Didemnum sp. Tetrahedron Lett. 1988;29:1177–1180. doi: 10.1016/S0040-4039(00)86681-2. [DOI] [Google Scholar]
- 103.Lindsay B.S., Barrows L.R., Copp B.R. Structural Requirements for Biological Activity of the Marine Alkaloid Ascididemin. Bioorg. Med. Chem. Lett. 1995;5:739–742. doi: 10.1016/0960-894X(95)00106-4. [DOI] [Google Scholar]
- 104.Dassonneville L., Wattez N., Baldeyrou B., Mahieu C., Lansiaux A., Banaigs B., Bonnard I., Bailly C. Inhibition of Topoisomerase II by the Marine Alkaloid Ascididemin and Induction of Apoptosis in Leukemia Cells. Biochem. Pharmacol. 2000;60:527–537. doi: 10.1016/S0006-2952(00)00351-8. [DOI] [PubMed] [Google Scholar]
- 105.Schmitz F.J., DeGuzman F.S., Choi Y.-H., Bilayet Hossain B., Rizvi S.K., van der Helm D. Biologically Active Compounds from Marine Organisms. Pure Appl. Chem. 1990;62:1393–1396. doi: 10.1351/pac199062071393. [DOI] [Google Scholar]
- 106.Matsumoto S.S., Biggs J., Copp B.R., Holden J.A., Barrows L.R. Mechanism of Ascididemin-Induced Cytotoxicity. Chem. Res. Toxicol. 2003;16:113–122. doi: 10.1021/tx025618w. [DOI] [PubMed] [Google Scholar]
- 107.Hasui M., Matsuda M., Yoshimatsu S., Okutani K. Production of a Lactate-Associated Galactan Sulfate by a Dinoflagellate Gymnodinium A3. Fish. Sci. 1995;61:321–326. doi: 10.2331/fishsci.61.321. [DOI] [Google Scholar]
- 108.Umemura K., Yanase K., Suzuki M., Okutani K., Yamori T., Andoh T. Inhibition of DNA Topoisomerases I and II, and Growth Inhibition of Human Cancer Cell Lines by a Marine Microalgal Polysaccharide. Biochem. Pharmacol. 2003;66:481–487. doi: 10.1016/S0006-2952(03)00281-8. [DOI] [PubMed] [Google Scholar]
- 109.Li M., Miao Z.-H., Chen Z., Chen Q., Gui M., Lin L.-P., Sun P., Yi Y.-H., Ding J. Echinoside A, a New Marine-Derived Anticancer Saponin, Targets Topoisomerase2α by Unique Interference with Its DNA Binding and Catalytic Cycle. Ann. Oncol. 2010;21:597–607. doi: 10.1093/annonc/mdp335. [DOI] [PubMed] [Google Scholar]
- 110.Fortune J.M., Osheroff N. Topoisomerase II as a Target for Anticancer Drugs: When Enzymes Stop Being Nice. Prog. Nucleic. Acid Res. Mol. Biol. 2000;64:221–253. doi: 10.1016/s0079-6603(00)64006-0. [DOI] [PubMed] [Google Scholar]
- 111.Robinson M.J., Corbett A.H., Osheroff N. Effects of Topoisomerase II-Targeted Drugs on Enzyme-Mediated DNA Cleavage and ATP Hydrolysis: Evidence for Distinct Drug Interaction Domains on Topoisomerase II. Biochemistry. 1993;32:3638–3643. doi: 10.1021/bi00065a016. [DOI] [PubMed] [Google Scholar]
- 112.Liberio M.S., Sadowski M.C., Nelson C.C., Davis R.A. Identification of Eusynstyelamide B as a Potent Cell Cycle Inhibitor Following the Generation and Screening of an Ascidian-Derived Extract Library Using a Real Time Cell Analyzer. Mar. Drugs. 2014;12:5222–5239. doi: 10.3390/md12105222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liberio M.S., Sadowski M.C., Davis R.A., Rockstroh A., Vasireddy R., Lehman M.L., Nelson C.C. The Ascidian Natural Product Eusynstyelamide B Is a Novel Topoisomerase II Poison That Induces DNA Damage and Growth Arrest in Prostate and Breast Cancer Cells. Oncotarget. 2015;6:43944–43963. doi: 10.18632/oncotarget.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heck M.M., Earnshaw W.C. Topoisomerase II: A Specific Marker for Cell Proliferation. J. Cell Biol. 1986;103:2569–2581. doi: 10.1083/jcb.103.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lindsay B.S., Christiansen H.C., Copp B.R. Structural Studies of Cytotoxic Marine Alkaloids: Synthesis of Novel Ring-E Analogues of Ascididemin and Their in Vitro and in Vivo Biological Evaluation. Tetrahedron. 2000;56:497–505. doi: 10.1016/S0040-4020(99)01038-8. [DOI] [Google Scholar]
- 116.Foderaro T.A., Barrows L.R., Lassota P., Ireland C.M. Bengacarboline, a New β-Carboline from a Marine Ascidian Didemnum sp. J. Org. Chem. 1997;62:6064–6065. doi: 10.1021/jo962422q. [DOI] [Google Scholar]
- 117.Pouilhès A., Kouklovsky C., Langlois Y., Baltaze J.-P., Vispé S., Annereau J.-P., Barret J.-M., Kruczynski A., Bailly C. Synthesis and Biological Evaluation of Bengacarboline Derivatives. Bioorg. Med. Chem. Lett. 2008;18:1212–1216. doi: 10.1016/j.bmcl.2007.11.113. [DOI] [PubMed] [Google Scholar]
- 118.Angel de la Fuente J., Jesús Martín M., del Mar Blanco M., Pascual-Alfonso E., Avendaño C., Carlos Menéndez J. A C-Ring Regioisomer of the Marine Alkaloid Meridine Exhibits Selective in Vitro Cytotoxicity for Solid Tumours. Bioorg. Med. Chem. 2001;9:1807–1814. doi: 10.1016/S0968-0896(01)00078-5. [DOI] [PubMed] [Google Scholar]
- 119.Seo Y.H. Dual Inhibitors Against Topoisomerases and Histone Deacetylases. J. Cancer Prev. 2015;20:85–91. doi: 10.15430/JCP.2015.20.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McClendon A.K., Osheroff N. DNA Topoisomerase II, Genotoxicity, and Cancer. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007;623:83–97. doi: 10.1016/j.mrfmmm.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Matias-Barrios V.M., Radaeva M., Song Y., Alperstein Z., Lee A.R., Schmitt V., Lee J., Ban F., Xie N., Qi J., et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front. Oncol. 2021;10:633142. doi: 10.3389/fonc.2020.633142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.