

Summary
Chronic pain remains challenging to treat, despite numerous reports of its pathogenesis, including neuronal plasticity in the spinal dorsal horn (SDH). We hypothesized that understanding plasticity only at a specific time point after peripheral nerve injury (PNI) is insufficient to solve chronic pain. Here, we analyzed the temporal changes in synaptic transmission and astrocyte-neuron interactions in SDH after PNI. We found that synaptic transmission in the SDH after PNI changed in a time-dependent manner, which was accompanied by astrocyte proliferation and loss of inhibitory and excitatory neurons. Furthermore, neuronal loss was accompanied by necroptosis. Short-term inhibition of astrocytes after PNI suppressed these physiological and morphological changes and long-term pain-related behaviors. These results are the first to demonstrate that the inhibition of astrocyte proliferation after PNI contributes to the long-term regulation of plasticity and of necroptosis development in the SDH.
Subject areas: Molecular biology, Neuroscience, Cell biology
Graphical abstract
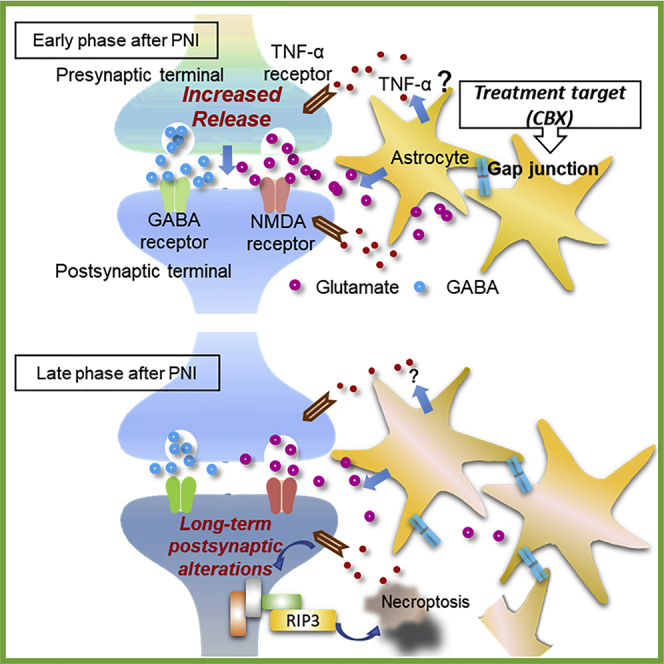
Highlights
-
•
Synaptic transmission in SDH after PNI is altered over time
-
•
Astrocyte proliferation induces neuronal loss with necroptosis in the SDH
-
•
Astrocyte regulation prevents transition to chronic pain
Molecular biology; Neuroscience; Cell biology
Introduction
Peripheral nerve injury (PNI) leads to plasticity and large-scale reorganization of neural circuits in the central nervous system (CNS), which in part leads to chronic pain.1,2 In somatosensory pathways, plasticity in chronic pain is the result of reorganization of the spinal cord or multiple brain regions because of PNI.3 The spinal dorsal horn (SDH), especially the substantia gelatinosa (SG), has several reports of plasticity, as it is the central terminal of primary afferents and a key area for pain processing.
In the SDH, structural plasticity has been reported as glial cell proliferation4,5,6 and neuronal degeneration and/or loss.7,8 Recently, a relationship between chronic pain and a pathway that leads to necroptosis, which is not apoptosis but controlled necroptotic cell death, has also been reported.9 On the other hand, abnormal sensation (allodynia and hyperalgesia), expansion of the receptive field (RF), enhancement of excitatory synaptic transmission represented by long-term potentiation (LTP), and disinhibition of inhibitory synaptic transmission are considered to be the actual manifestation of functional plasticity.10 However, how PNI induces plasticity in the SDH leading to chronic pain, and whether functional plasticity is a consequence of structural plasticity, remains largely unknown. This is because most previous studies have been cross-sectional at specific time points after PNI.11,12,13
Changes in neuronal activity after PNI in SDH are regulated to some extent by glial cells.14 TNFα and IL-1β derived from glial cells are also involved in the induction of LTP, which is considered pathogenesis of chronic pain.15 In particular, astrocytes are well known to be activated following microglia after PNI and are involved in the maintenance of chronic pain.5 Therefore, we focused on the interaction between astrocytes and neurons to elucidate the establishment process of functional and structural plasticity of SDH to understand the pathogenesis of chronic pain.
For this goal, we performed in vivo whole-cell patch clamp recordings from SDH neurons after chronic constriction injury (CCI),4,16 a model of chronic pain in rats. Although RF expansion and allodynia were observed from the early post-PNI, the synaptic responses in SDH were significantly different between the early and late post-PNI. Specifically, the pathophysiology in the late post-PNI period, that is, when chronic pain was established, was mainly because of changes in the sensitivity of postsynaptic receptors rather than presynaptic excitability enhancement. We also showed directly for the first time in vivo that activation of NMDARs, which is essential for the induction of LTP in SDH, is caused by PNI. The possibility of neuronal degeneration was suggested by electrophysiological studies, we analyzed morphologically the process of neuronal degeneration or loss because of astrocyte-neuron interaction induced by PNI. We demonstrated that both the number of excitatory and inhibitory SDH neurons decreased after PNI, in parallel with astrocyte proliferation. As astrocyte activation increases the TNFα production that induces LTP,15 and TNFα induces necroptosis, a type of necrosis,17 we hypothesized that neuronal loss might be accompanied by necroptosis. The results showed that neuronal loss is accompanied by necroptosis and that inhibition of astrocyte proliferation for only one week after PNI leads to long-term suppression of necroptosis and RF expansion and allodynia.
Results
Functional changes in response to RF stimuli after PNI are time-dependent
To identify the time-dependent functional changes in SDH neurons after PNI, the in vivo patch-clamp recording was used to examine the changes in the RF size and the response to noxious and innoxious stimuli; The CCI of the sciatic nerve was used for chronic pain model4,16 (Figure 1A). After CCI, rats showed a marked reduction in the ipsilateral hind paw withdrawal threshold, and this increase in the mechanical stimulus response threshold persisted for more than 1 month (Figure S1). In subsequent experiments, comparisons were performed between Sham, CCI-E (early stage after CCI, 7 to 14 days after CCI), and CCI-L (late stage after CCI, 21 to 28 days after CCI) rats.
Figure 1.

Post-PNI functional changes in the SDH
(A) Schematic diagram of in vivo patch-clamp recording from SG neurons at the L4 or L5 level. The ipsilateral hindlimb was stimulated with a brush (touch stimulus) or toothed forceps (pinch stimulus).
(B and C) RF map based on evoked EPSCs in response to the touch stimulus. The mean RF area was 0.42 ± 0.09 cm2 (n = 13 cells from 5 rats) in the Sham rats, 1.05 ± 0.16 cm2 (n = 19 cells from 10 rats) in the CCI-E rats, and 1.15 ± 0.14 cm2 (n = 18 cells from 8 rats) in the CCI-L rats. (Sham versus CCI-E: ∗∗p = 0.0090, versus CCI-L: ∗∗p = 0.027, One-way ANOVA).
(D) Representative chart recordings of the AUCs evoked by touch and pinch stimuli of the RF in Sham, CCI-E, and CCI-L rats.
(E) Comparison of AUC to touch stimulus. Sham: 1.15 ± 0.39 × 105 ΔpA/5 s, n = 13 cell from 5 rats; CCI-E: 3.48 ± 0.54 × 105 ΔpA/5 s, n = 23 cells from 12 rats, ∗p = 0.0342; CCI-L: 4.18 ± 1.05 × 105 ΔpA/5 s, n = 13 cells from 7 rats, ∗∗p = 0.0133. One-way ANOVA.
(F) Comparison of AUC to pinch stimulus. Sham: 1.19 ± 0.48 × 105 ΔpA/5 s, n = 10 cells from 5 rats; CCI-E: 4.23 ± 0.73 × 105 ΔpA/5 s, n = 23 cells from 12 rats, ∗p = 0.0188; CCI-L: 2.41 ± 0.79 × 105 ΔpA/5 s, n = 15 cells from 7 rats, p = 0.4861. One-way ANOVA. n.s., not significant.
(G) SG neurons were classified into 4 types according to their response to RF stimuli in in vivo patch-clamp recordings. Bars above the traces indicate the duration of the stimulation (5 s).
(H) Relative proportions of the 4 types of neurons among Sham, CCI-E, and CCI rats. The numbers in the graphs indicate the percentages. Sample sizes are indicated in parentheses. ∗p = 0.0489, Chi-squared test.
In vivo patch-clamp recordings were performed from SG neurons (50–150 μm from the dorsal surface of the spinal cord), which are the central terminals of primary afferents and a key area for pain processing.18,19 Although RF reorganization is an established characteristic of post-PNI plasticity,20,21 there are no reports of post-PNI RF mapping over time from the SDH. We identified the RF by marking the response sites to the touch stimulus by analyzing EPSCs in voltage-clamp mode. Innoxious stimuli to the shaved RF skin elicited a barrage of EPSCs in almost all neurons, consistent with the results of previous studies.22,23 The RF size in response to innoxious stimuli was significantly expanded in both CCI-E and CCI-L rats compared to Sham rats, but the degree of expansion did not differ between CCI-E and CCI-L (Figures 1B and 1C). The excitability (glutamatergic response) of SG neurons to stimuli was quantified by measuring AUC (Figure 1D), and the excitability to innoxious or noxious stimuli was compared between Sham and CCI rats. The innoxious stimulus-induced AUC increased in CCI-E and CCI-L rats compared to Sham rats (Figure 1E). Conversely, noxious stimulus-induced AUC increased only in CCI-E rats, and there was no significant difference between CCI-L and Sham rats (Figure 1F).
SDH neurons can be classified into several categories according to their response patterns to stimuli.12,24 According to the response pattern of EPSCs to noxious and innoxious stimuli applied to the hindlimb, we were able to classify them into four types: responding only to touch (low threshold [LT]), only to pinch (high threshold [HT]), to both pinch and touch (multi-receptive [MR]), and non-responding (NR) neurons (Figures 1G and 1H). We compared changes in the properties of the four neuron types between Sham and CCI rats (CCI-E and CCI-L). All cells, including NR neurons, were judged to be neurons, because we could confirm spontaneous EPSCs (sEPSCs) caused by glutamate release from presynaptic nerve terminals. The relative proportion of neurons was significantly different between Sham and CCI rats (both CCI-E and CCI-L) (Figure 1H). In both CCI rat types, the population of LT neurons decreased and the population of MR neurons increased compared to those in Sham rats. Of interest, the population of NR neurons also increased in CCI rats (Figure 1H). These findings indicated that SG neuron properties were altered after PNI. The decrease in LT neurons and the increase in MR neurons may be explained by the alteration of synaptic pathways because of disinhibition or loss of neurons. The increase of NR neurons also suggests the possibility of neuronal degeneration in the SDH.
These results indicate that functional changes in SDH neurons, such as RF expansion changes and altered neuronal response patterns to stimuli, persist early and long post-PNI. However, in the long term, neuronal excitability to noxious stimuli returned to the same level as in Sham rats, whereas responses to innoxious stimuli remained abnormally enhanced. This suggests that the neuronal pathway to innoxious stimuli remains pathologically degenerated during chronic pain.
Post-PNI mechanical allodynia is mediated by NMDA receptors
SDH EPSCs are predominantly mediated by non-NMDA receptors,18 and NMDA receptors are almost silent under physiological condition.23 NMDA receptors have been shown to elicit allodynia and/or hyperalgesia associated with increased excitatory neurotransmitter from terminals of primary afferents after PNI13,25,26 but no direct reports have been shown in vivo. To clarify that post-PNI mechanical allodynia is mediated by NMDA receptors, we analyzed evoked EPSCs under administration of antagonists of AMPA, GABA, and glycine receptors (Figures 2A and 2B). Contrarily to that in Sham rats, the touch stimulus-evoked EPSCs in CCI rats was not suppressed by AMPA, GABA, or glycine receptor antagonism. Furthermore, this induced EPSCs were completely abolished by NMDA receptor antagonist AP5. In other words, the response to the innoxious stimulus in CCI rats was mediated by NMDA receptors (Figure 2C). Of interest, the pinch stimulus-evoked EPSCs was abolished both in Sham and CCI rats, indicating that noxious stimuli were mainly transmitted by AMPA receptor-mediated excitatory synaptic responses even in CCI rats. This showed directly, for the first time, that post-PNI mechanical stimuli-elicited allodynia is an NMDA receptor-mediated response.
Figure 2.

PNI-induced tactile allodynia is mediated via NMDA receptors
To analyze only NMDA receptor-mediated responses, the RF was stimulated under bicuculine (GABA receptor antagonist; 20 μM), strychnine (glycine receptor antagonist; 2 μM), CNQX (AMPA receptor antagonist; 20 μM) perfusion. Consequently, all responses were abolished under AP5 (NMDA receptor antagonist; 50 μM) perfusion.
(A–C) Representative chart recordings under antagonist administration in Sham, CCI-E, and CCI-L rats, respectively.
(D) Comparison of touch-evoked AUCs via only NMDAR between Sham, CCI-E, and CCI-L rats and confirmation of the loss of NMDAR-mediated responses by AP5 perfusion. In Sham rats, touch-evoked AUC via only NMDAR was almost not observed, whereas it was significantly increased in CCI-L rats. AUC - Sham: 0.11 ± 0.08 × 105 ΔpA/5 s; CCI-E: 1.09 ± 0.30 × 105 ΔpA/5 s, p = 0.055; CCI-L: 1.23 ± 0.34 × 105 ΔpA/5 s, †p = 0.0235. NMDAR-mediated evoked EPSCs were abolished by AP5 perfusion. AUC - CCI-E post-AP5: 0.041 ± 0.017 × 105 ΔpA/5 s, ∗p = 0.0221 (versus CCI-E pre AP5); CCI-L post-AP5: 0.042 ± 0.020 × 105 ΔpA/5 s, ∗p = 0.0410 (versus CCI-L pre AP5).
(E) NMDAR-mediated pinch-induced AUC was almost not observed in either Sham, CCI-E, or CCI-L rats. Pinch-induced AUC was not altered by AP5 administration. AUC - Sham: 0.04 ± 0.02 × 105 ΔpA/5 s; CCI-E: 0.22 ± 0.13 × 105 ΔpA/5 s, p = 0.4653; CCI-L: 0.26 ± 0.15 × 105 ΔpA/5 s, p = 0.3351. NMDAR-mediated evoked EPSCs were abolished by AP5 perfusion. AUC - CCI-E post-AP5: 0.071 ± 0.052 × 105 ΔpA/5 s, p = 0.3896 (versus CCI-E pre AP5); CCI-L post-AP5: 0.038 ± 0.024 × 105 ΔpA/5 s, p = 0.2245 (versus CCI-L pre AP5). Significant differences were assessed using the one-way ANOVA and two-sided unpaired Student’s t test. n.s., not significant.
Presynaptic hyperexcitability after PNI is transient, but postsynaptic excitability is maintained
Changes in synaptic input strength via changes in excitatory neurotransmitter (mainly glutamate) release and receptor sensitivity are thought to be key mechanisms that induce chronic pain transition. Previous studies using electrophysiological techniques have reported that glutamate release frequency (EPSC frequency) in the SDH increases after PNI,27 whereas others have reported no difference from naive animals.13 This difference may be because of the inconsistent post-PNI analysis period in each study. Recently, it has become clear that different molecular biological mechanisms are involved in the development of chronic pain in the early- and late-phase after PNI.4,27 However, many animal studies investigating the mechanisms of chronic pain-related neuronal plasticity in the SDH were conducted 1–2 weeks after nerve injury.6,7,8
To elucidate the temporal post-PNI changes in pre- and postsynaptic excitatory neurotransmission in the SDH, we compared sEPSCs over time. Increased mean sEPSC frequency was observed only in CCI-E rats (Figures 3A and 3B), whereas an increase in mean amplitude was observed in both CCI-E and CCI-L rats (Figures 3A and 3E). These results are partially consistent with previous studies showing that excitability of SDH neurons increased after PNI.4,12 In a more detailed temporal post-PNI analysis, we found that sEPSC frequency significantly increased on postoperative days 5 and 7 (Figure 3C), and that sEPSC amplitude was significantly increased on postoperative days 9, 14, and 21 (Figure 3F), both of which returned to control levels by day 28.
Figure 3.

Time-dependent sEPSCs and mEPSCs after PNI
(A) Representative examples of sEPSCs and mEPSCs (under perfusion of TTX) in SG neurons from Sham and CCI rats (holding potential = −70 mV).
(B) Summarised frequency data of sEPSCs. Sham: 15.76 ± 4.85 Hz, n = 12 cells from 5 rats; CCI-E: 37.28 ± 5.75 Hz, n = 17 cells from 12 rats, ∗∗p = 0.0061; CCI-L: 23.20 ± 2.78 Hz, n = 17 cells from 8 rats, p = 0.2835.
(C) Time-dependent data of frequency of sEPSCs after PNI compared to those in Sham rats. Sham: 15.76 ± 4.85 Hz, n = 12 cells from 5 rats; CCI day 5: 67.71 ± 8.86 Hz, n = 5 cells from 4 rats, ∗∗∗p < 0.001; day 7: 63.67 ± 10.44 Hz, n = 5 cells from 4 rats, ∗∗∗p < 0.001; day 9: 27.13 ± 6.29 Hz, n = 6 cells from 4 rats, p = 0.5740; day 14: 25.45 ± 4.79 Hz, n = 6 cells from 4 rats, p = 0.7237; day 21: 21.57 ± 5.04 Hz, n = 6 cells from 5 rats, p = 0.9622; day 28: 23.34 ± 3.74 Hz, n = 10 cells from 7 rats, p = 0.7929.
(D) Summarised frequency data of mEPSCs. Sham: 10.25 ± 2.2 Hz, n = 7 cells from 4 rats; CCI-E: 24.56 ± 2.56 Hz, n = 7 cells from four rats, ∗∗∗p = 0.0004; CCI-L: 15.45 ± 1.84 Hz, n = 5 cells from 4 rats, p = 0.0832.
(E) Summarised amplitude data of sEPSCs. Sham: 96.7 ± 22.63 pA, n = 12 cells from 5 rats; CCI-E: 300.0 ± 54.88 pA, n = 17 cells from 12 rats, ∗∗p = 0.0069; CCI-L: 262.7 ± 40.13 Hz, n = 17 cells from 8 rats, ∗p = 0.0290.
(F) Time-dependent data of amplitude of sEPSCs after PNI compared to those in Sham rats. Sham: 96.7 ± 22.63 pA, n = 12 cells from 5 rats; CCI day 5: 54.01 ± 7.49 pA, n = 5 cells from 4 rats, p = 0.9880; day 7: 74.54 ± 31.25 pA, n = 5 cells from 4 rats, p = 0.9996; day 9: 328.1 ± 19.15 pA, n = 6 cells from 4 rats, ∗∗p = 0.0093; day 14: 459.7 ± 112.1 pA, n = 6 cells from 4 rats, ∗∗∗p < 0.001; day 21: 377.1 ± 88.39 pA, n = 6 cells from 5 rats, ∗∗p = 0.0012; day 28: 200.1 ± 34.32 pA, n = 10 cells from 7 rats, p = 0.3653.
(G) Summarised amplitude data of mEPSCs. Sham: 32.68 ± 10.87 pA, n = 7 cells from 4 rats; CCI-E: 124.0 ± 15.98 pA, n = 7 cells from 4 rats, ∗∗∗p = 0.0002; CCI-L: 98.60 ± 11.21 Hz, n = 6 cells from 4 rats, ∗∗p = 0.0038. Significant differences were assessed using one-way ANOVA followed by Dunnett’s post hoc test. n.s., not significant.
We further recorded mEPSCs in the presence of tetrodotoxin (TTX, 0.5 μM). Postsynaptic excitatory neuronal activity was significantly reduced by TTX perfusion in Sham, CCI-E, and CCI-L rats (Figures 3A and S1). The mEPSC frequency was significantly increased in CCI-E rats compared to Sham rats, but not in CCI-L rats (Figure 3D). In contrast, mEPSC amplitude increased significantly in both CCI-E and CCI-L rats (Figure 3G). These results indicated that postsynaptic neurons are exposed to excessive glutamate release immediately after PNI, but that over time the spontaneous glutamate release returns to control levels with postsynaptic excitatory facilitation, suggesting that the presynaptic hyperexcitability is a transient phenomenon, while postsynaptic excitability is maintained.
Inhibitory postsynaptic dysfunction after PNI is maintained early after PNI
Apart from enhanced excitatory synaptic transmission, inhibitory neuronal system dysfunction is also reported to be an important mechanism for the establishment of chronic pain. GABAergic or glycinergic inhibitory interneurons are densely distributed in the SDH, and their loss of function (disinhibition) has been proposed as one of the mechanisms of chronic pain.26,28 However, the claim that disinhibition is because of GABAergic interneurons after PNI7 is controversial.28 First, we analyzed the temporal changes in spontaneous inhibitory postsynaptic currents (sIPSCs) and mIPSCs after PNI in SDH to investigate the presence of disinhibition (Figure S2).
The sIPSC and mIPSC frequencies did not change in either CCI-E or CCI-L rats compared with those in Sham rats (Figures 4A, 4B, and 4D). A more detailed analysis showed that the frequencies were rather higher than those in Sham rats on days 5 and 14 post-PNI (Figure 4C). Conversely, sIPSC and mIPSC amplitudes decreased from the early post-PNI phase and dramatically decreased in the late post-PNI phase (Figures 4A, 4E, and 4G). Notably, mIPSC decay time increased (Figures 4H and 4I) in CCI-E and CCI-L rats. These results suggest that PNI did not alter inhibitory neurotransmitter (GABA and/or glycine) release, but altered the function of postsynaptic receptors.
Figure 4.

Time-dependent sIPSCs and mIPSCs after PNI
(A) Representative examples of sIPSCs and mIPSCs (under perfusion of TTX) in SG neurons from Sham and CCI rats (holding potential = 0 mV).
(B) Summarized frequency data of sIPSCs. Sham: 23.22 ± 3.92 Hz, n = 10 cells from 5 rats; CCI-E: 28.68 ± 2.88 Hz, n = 20 cells from 12 rats, p = 0.4955; CCI-L: 34.78 ± 4.77 Hz, n = 13 cells from 8 rats, p = 0.1030.
(C) Time-dependent frequency of sIPSCs after PNI compared to those from Sham rats. Sham: 23.22 ± 3.92 Hz, n = 10 cells from 5 rats; CCI day 5: 43.80 ± 3.43 Hz, n = 6 cells from 4 rats, ∗p = 0.0130; day 7: 25.86 ± 4.10 Hz, n = 7 cells from 4 rats, p = 0.9964; day 9: 19.29 ± 1.37 Hz, n = 7 cells from 4 rats, p = 0. 9767; day 14: 42.92 ± 3.97 Hz, n = 6 cells from 5 rats, ∗p = 0.0188; day 21: 30.93 ± 3.97 Hz, n = 6 cells from 5 rats, p = 0.7176; day 28; 38.09 ± 5.84 Hz, n = 7 cells from 5 rats, p = 0.0912.
(D) Summarized frequency data of mIPSCs. Sham: 8.07 ± 1.38 Hz, n = 8 cells from 5 rats; CCI-E: 6.87 ± 1.97 Hz, n = 6 cells from 4 rats, p = 0.8811; CCI-L: 9.16 ± 2.29 Hz, n = 5 cells from 4 rats, p = 0.9113.
(E) Summarized amplitude data of sIPSCs. Sham: 259.9 ± 30.07 pA, n = 10 cells from 5 rats; CCI-E: 119.7 ± 13.73 pA, n = 20 cells from 12 rats, ∗∗∗p < 0.001; CCI-L: 44.90 ± 7.12 Hz, n = 13 cells from 8 rats, ∗∗∗p < 0.001.
(F) Time-dependent amplitude of sIPSCs after PNI compared to those from Sham rats. Sham: 259.9 ± 4.90 pA, n = 10 cells from 5 rats; CCI day 5: 258.7 ± 25.34 pA, n = 6 cells from 4 rats, p > 0.9999; day 7: 86.98 ± 11.76 pA, n = 7 cells from 4 rats, ∗∗∗p < 0.001; day 9: 133.6 ± 16.92 pA, n = 7 cells from 4 rats, ∗∗p = 0.0013; day 14: 141.5 ± 37.47 pA, n = 6 cells from 5 rats,∗p = 0.0045; day 21: 63.69 ± 12.24 pA, n = 6 cells from 5 rats, ∗∗∗p < 0.001; day 28: 47.50 ± 10.12 pA, n = 7 cells from 5 rats, ∗∗∗p < 0.001.
(G) Summarized amplitude data of mIPSCs. Sham: 66.17 ± 11.90 pA, n = 8 cells from 5 rats; CCI-E: 48.20 ± 15.47 pA, n = 6 cells from 4 rats, p = 0.4808; CCI-L: 14.54 ± 3.10 Hz, n = 5 cells from 4 rats, ∗p = 0.0187. Significant differences were assessed using one-way ANOVA with Dunnett’s multiple comparisons test.
(H and I) Decay time of mIPSCs in CCI-E and CCI-L rats. Histograms of cumulative miniature IPSCs from a representative neuron showing the shift to the right after PNI. Kolmogorov-Smirnov test. n.s., not significant.
Post-PNI temporal structural changes in the SDH: Excitatory and inhibitory neuronal loss associated with astrocyte proliferation
We had demonstrated the detailed temporal changes in the functional plasticity of SDH neurons after PNI. However, the dynamics of structural plasticity and the causal relationship between functional and structural plasticity remain unresolved. Astrocyte-neuron interactions have been implicated in the development and maintenance of chronic pain.4,5,29 Reactive astrocytes are strongly induced by CNS injury and diseases; in the brain, activated microglia induce astrocyte activation by releasing IL-1α and TNF, and astrocytes have been suggested to strongly contribute to CNS neuronal death.30 Previous studies showed that astrocyte proliferation after PNI is accompanied by increased extracellular glutamate concentration, resulting in neuron loss and/or degeneration.8,26 To clarify the relationship between astrocyte proliferation and functional and structural plasticity in the SDH after PNI, we first evaluated astrocyte proliferation immunohistochemically over time from PNI in CCI rats. Astrocyte was detected based on GFAP immunoreactivity.5 As previously reported,4 marked astrocyte proliferation was observed from the CCI-E phase and continued for at least 4 weeks (Figures 5 and S3).
Figure 5.

PNI induces persistent upregulation of GFAP in SDH astrocytes
(A) Low and high magnification images of GFAP-positive cells in the SDH of Sham, CCI-E, and CCI-L rats. Scale bars are 100 μm (left panels) and 50 μm (right panels).
(B) GFAP quantification. The percentage area occupied by GFAP positive cells in the SDH was analyzed. Histograms indicate the relative mean area occupied by GFAP-positive cells in Sham, CCI-E, and CCI-L rats (Sham: 11.48 ± 1.99%; CCI-E: 27.37 ± 1.30%, ∗∗∗p < 0.001; CCI-L: 18.83 ± 1.13%, ∗∗p = 0.0064; n = 5 rats in Sham group, and n = 6 rats in CCI-E and CCI-L group respectively). One-way ANOVA followed by Dunnett’s post hoc test.
(C) Representative images of the SDH L4 or L5 of Sham, E-CCI, and L-CCI rats. Neurons were immunostained for NeuN, Pax2, or both NeuN and Pax2. White arrowheads indicate typical excitatory (Pax2-/NeuN+) neurons, and light blue arrowheads indicate typical inhibitory (Pax2+/NeuN+) neurons. Scale bar: 100 or 50 μm for lower and higher magnification images, respectively.
(D) Comparison of the number of neurons (NeuN+) in SDH between CCI-E, CCI-L, and Sham rats (n = 5 rats for Sham group, and n = 6 rats for CCI-E and CCI-L group. Sham: 192.4 ± 22.07; CCI-E: 135.5 ± 12.65, ∗p = 0.0222; CCI-L: 78.83 ± 3.74, ∗∗∗p = 0.001.
(E) Comparison of the number of excitatory neurons (Pax2-/NeuN+) in SDH between CCI-E, CCI-L, and Sham rats. Sham: 123.0 ± 15.13; CCI-E: 76.33 ± 5.99, ∗∗p = 0.0037; CCI-L: 46.67 ± 2.08, ∗∗∗p < 0.001.
(F) Comparison of the number of inhibitory interneurons (Pax2+/NeuN+) in SDH between CCI-E, CCI-L and Sham rats. Sham: 68.40 ± 11.27; CCI-E: 59.17 ± 7.37, p = 0.5337; CCI-L: 32.17 ± 2.01, ∗∗p = 0.0064. Significant differences were assessed using one-way ANOVA followed by Dunnett’s post hoc test. n.s., not significant.
Next, we examined the effect of astrocyte proliferation on the structural plasticity of SDH neurons. The degeneration or loss of neurons in SDH after PNI has been focused mainly on inhibitory interneurons.7,26 We have shown that glutamate release from primary afferents is transiently but dramatically increased only in the early stages of PNI. Thus, we hypothesized that excitatory neurons must also be affected by glutamate toxicity and that not only inhibitory neurons but also excitatory neurons may be eliminated. Based on previous reports, we identified excitatory neurons indirectly based on the absence of Pax2 staining and positive labeling for the pan-neuronal marker NeuN31; in other words, excitatory neurons were identified as Pax2-/NeuN+ cells whereas inhibitory neurons were identified as Pax2+/NeuN+ cells. Pax2 is a marker exclusively expressed in virtually all GABAergic and glycinergic SDH interneurons in both embryonic and adult rodents.32 Subsequent analyses were performed in the superficial dorsal horn, at the same level as the neurons analyzed in the electrophysiological experiments.
The number of NeuN-positive neurons was significantly reduced in both CCI-E and CCI-L. However, the reduction was greater in CCI-L than in CCI-E (Figures 6A and 6B). The number of excitatory neurons (Pax2-/NeuN+ cells) was also significantly reduced in CCI rats and was particularly remarkable in CCI-L (Figures 5A and 5C), whereas inhibitory neuron (Pax2+/NeuN+) was reduced only in CCI-L, and the extent of their reduction was less than that of excitatory neurons (Figures 6A and 6D). Thus, we found that with astrocyte proliferation, not only inhibitory neuronal loss but rather excitatory neuronal loss in a greater proportion occurred over a long time after PNI. This histological change possibly plays a crucial role in the mechanism of chronic pain development and maintenance.
Figure 6.

PNI increases the number of RIP3-expressing neurons
(A) Immunohistological staining of RIP3-expressing neurons in SDH. Light blue arrowheads indicate typical NeuN-positive and RIP3-positive neurons. Scale bar: 50 μm for both lower and higher magnification images. The lower right panel shows a magnified view of representative NeuN-positive and RIP3-positive neurons in the ipsilateral side of a CCI-L rat. RIP3 is expressed around the plasma membrane. Scale bar: 20 μm.
(B) Comparison of the number of RIP3 expressing neurons in SDH of CCI-L rats between CBX- and PBS-treated groups. Ipsilateral PBS-treated CCI-L: 8.40 ± 1.86; Ipsilateral CBX-treated CCI-L rats: 3.00 ± 0.71, ∗p = 0.0265. The number of RIP3-expressing neurons was significantly increased ipsilateral compared to the contralateral side in the PBS group, but no difference was observed in the CBX group. Contralateral PBS-treated CCI-L: 2.20 ± 0.73, ∗p = 0.0147; Contralateral CBX-treated CCI-L: 1.83 ± 0.79, p = 0.2268. Both are compared to the ipsilateral side of each. Significant differences were assessed using the two-sided unpaired Student’s t test. n.s., not significant.
PNI-induced neuronal loss is accompanied by necroptosis
We have shown directly for the first time in vivo that PNI induces activation of NMDA receptors, which are essential for induction of LTP, and enhancement of excitatory synaptic transmission, as represented by LTP. LTP is elicited by TNFα and IL-1 from glial cells.33 TNFα also induces necroptosis, a regulated necrotic cell death.17 Therefore, we investigated immunohistochemically whether the degeneration and loss of neurons after PNI is accompanied by necroptosis. Necroptosis is regulated by receptor-interacting protein 3 (RIP3) and mixed linear kinase domain-like protein (MLKL). In particular, RIP3 is upregulated in the spinal cord after PNI, thus potentially providing a new therapeutic target for chronic pain.9 Meanwhile, although increased expression of RIP3 after PNI in microglia has been reported,9 its expression level in neurons and its relationship with astrocyte proliferation is unknown. Therefore, we first compared the number of RIP3-expressing neurons between Sham, CCI-E, and CCI-L rats. Because it has been reported that the expression level of RIP3 increases within 3 days after PNI, we also checked the number of RIP3-expressing neurons in rats 3 days after CCI.
The results showed that 3 days after CCI, CCI-E and CCI-L rats all had increased numbers of RIP3-positive neurons in the ipsilateral SDH compared to Sham rats (Figures 6A and 6B). Comparisons between ipsilateral and contralateral rats showed a significantly ipsilateral increase in the number of RIP3 neurons (Figures 6A and 6B). Of interest, RIP3 expression was found around the plasma membrane in most ipsilateral neurons (Figure 6A, bottom). We show for the first time that PNI increases the number of RIP3-expressing neurons.
Astrocyte regulation in the early post-PNI phase suppresses neuronal loss and necroptosis
If astrocyte proliferation and interactions between astrocytes and neurons contribute to the development and maintenance of chronic pain, preventing astrocyte proliferation should suppress the transition to chronic pain. CBX, an astrocyte gap junction blocker, effectively suppresses astrocyte proliferation after PNI and temporarily elevates pain thresholds when administered immediately before behavioral assessment.4,34 However, astrocytes are characterized by a marked proliferation from approximately one week after PNI, which is maintained for several weeks.5,33 Therefore, we hypothesized that inhibition of astrocyte proliferation only immediately after PNI might prevent the transition to chronic pain in the long term by suppressing functional and structural plasticity.
CBX (10 mg/kg) or the same amount of PBS was intraperitoneally administered to Sham rats and CCI rats every day from the first to seventh day after surgery. Behavioral analysis was performed on postoperative days 5, 7, 14, 21, and 28, with the preoperative paw withdrawal threshold defined as the control value, and immunohistological and electrophysiological analyses on CCI-E and CCI-L (Figure 7A). No difference in the mechanical threshold was observed between the CBX and PBS group in Sham rats, whereas in CCI rats, the mechanical thresholds on the 14th, 21st, and 28th postoperative days were significantly higher in the CBX group than in the PBS group (Figure 7B). As previously reported,4 CBX treatment significantly inhibited post-PNI astrocyte proliferation. Of interest, when CBX was administered for 1 week immediately after PNI, the GFAP-positive area percentage after 4 weeks was significantly decreased as compared to that in control rats (Figures 7C and 7D). Furthermore, we found that CBX treatments reliably inhibited neuron loss associated with PNI both in excitatory (Pax2-/NeuN+ cells) and inhibitory neurons (Pax2+/NeuN+ cells) in CCI-L rats (Figures 7E and 7F).
Figure 7.

CBX administration only in the early post-PNI stages leads to an increase in persistent pain threshold and inhibition of astrocyte proliferation and neuronal loss
(A) Schematic timeline for intraperitoneal injection and subsequent von Frey test, electrophysiological experiments, and fixation after PNI.
(B) Paw withdrawal threshold to mechanical stimulation by von Frey filaments measured before (day 1) and on days 5, 7, 14, 21, and 28 after PNI. Early inhibition of astrocyte activation significantly increased the withdrawal threshold in CCI rats as compared to that in CCI rats injected with PBS (p = 0.9808 at day 5, p = 0.9994 at day 7, ∗∗p = 0.0055 at day 14, ∗p = 0.0300 at day 21, and ∗p = 0.0373 at day 28 after PNI; n = 5 rats each in the CCI-PBS and Sham-PBS groups; n = 6 rats each in the CCI-CBX and Sham-CBX groups.).Significant differences were assessed using two-way ANOVA with Tukey’s post hoc test.
(C) Representative images of GFAP-positive cells in the SDH of CCI-L rats after intraperitoneal administration of CBX or PBS immediately after CCI. Scale bars are 100 μm (left panels) and 50 μm (right panels).
(D) GFAP quantification. The percentage area occupied by GFAP-positive cells in the SDH. The relative mean area occupied by GFAP-positive cells in CCI-L rats was 22.22 ± 1.06% (n = 6) in the PBS group and 13.71 ± 1.23% (n = 6) in the CBX group. ∗∗p = 0.0022. Significant differences were assessed using the Mann-Whitney U test.
(E) Representative images of the SDH L4 or L5 in L-CCI rats treated with PBS or CBX. Neurons were immunostained for NeuN, Pax2, or both NeuN and Pax2. White arrowheads indicate typical excitatory (Pax2-/NeuN+) neurons, and light blue arrowheads indicate typical inhibitory (Pax2+/NeuN+) neurons. Scale bars are 100 and 50 μm for lower and higher magnification images, respectively.
(F) Analyses of number of cells (NeuN+, Pax2-/NeuN+, and Pax2+/NeuN+) in the SDH (n = 6 rats for each group; 3–5 sections/rat). The number of NeuN + cells was 77.17 ± 3.71 in the PBS group and 107.51 ± 3.99 in the CBX group (∗∗∗p = 0.0002). The number of excitatory neurons indirectly identified by Pax2-and NeuN+ immunostaining was 46.67 ± 2.11 in the PBS group and 63.00 ± 5.63 in the CBX group (∗p = 0.0216). The number of inhibitory interneurons identified by Pax2 positive and NeuN positive immunostaining was 32.50 ± 2.42 in the PBS group and 44.50 ± 3.32 in the CBX group (∗p = 0.0153). Significant differences were assessed using the two-sided unpaired Student’s t test. n.s., not significant.
We have shown an increase in the number of RIP3-expressing neurons after PNI, that is, necroptosis, but the relationship between astrocytes and necroptosis is unclear. Necroptosis is a kind of necrosis that triggers an innate immune response by rupturing dead cells to release intracellular components, which can be triggered by Toll-like receptor agonists or TNF.17 Because astrocytes contribute to pain gating in SDH after PNI35 and TNF receptor 1, a TNFα receptor that induces necroptosis, is expressed more in astrocytes than in microglia,15 we hypothesized that astrocyte regulation may suppress necroptosis. We compared the number of RIP3-expressing neurons in the CBX-treated and PBS-treated groups immediately after CCI with the protocol shown in Figure 7A. In ipsilateral versus contralateral comparisons, there was a significant ipsilateral increase in RIP3- and NeuN-positive neurons in the PBS-treated group, but no difference between the ipsilateral and contralateral groups in the CBX-treated group (Figure 8B).
Figure 8.

Expression of RIP3-positive neurons after PNI is suppressed by CBX administration
(A) Neurons were immunostained for NeuN, RIP3, or both NeuN and RIP3. Light blue arrowheads indicate typical NeuN-positive and RIP3-positive neurons. Scale bar: 50 μm for both lower and higher magnification images.
(B) Comparison of the number of RIP3 expressing neurons in SDH between PBS CCI-L and CBX-CCI-L rats. PBS CCI-L: 8.4 ± 1.86; CBX-CCI-L: 3.0 ± 0.71, ∗p = 0.0265. The number of RIP3-expressing neurons was significantly increased ipsilateral compared to contralateral in the PBS group, but not in the CBX group. Contralateral CCI-L of the PBS group: 2.2 ± 0.73, †p = 0.0147; Contralateral CCI-L of CBX group: 1.8 ± 0.58, p = 0.2268. Compare with each ipsilateral side. Significant differences were assessed using the two-sided unpaired Student’s t test. n.s., not significant.
Astrocyte regulation in the early phase after PNI is key to controlling chronic pain
Finally, to determine whether these structural plasticities are associated with functional changes, we analyzed excitatory synaptic transmission and RF size in response to innoxious stimuli in CCI-L rats treated with CBX or PBS using the in vivo patch-clamp recording. In CBX-treated CCI-L rats, sEPSC amplitude was significantly reduced to the level of Sham rats compared to the PBS group. On the other hand, there was no difference in frequency between the PBS and CBX groups (Figures 9A and 9B). RF expansion was also significantly suppressed by CBX administration (Figures 9C and 9D). Taken together, we showed that early post-PNI suppression of astrocyte proliferation inhibits the transition to chronic pain by suppressing SDH neurodegeneration.
Figure 9.

Early post-PNI inhibition of astrocyte proliferation in the SDH prevents the development of chronic pain
(A) Representative electrophysiological recordings of CCI-L rats treated with PBS or CBX (holding potential = −70 mV).
(B) Comparative analysis data of the frequency (left) and amplitude (right) of sEPSCs between the PBS-treated and CBX-treated CCI-L rat groups (Frequency - CCI-L PBS: 22.7 6 ± 5.55 Hz; CCI-L CBX: 20.66 ± 3.77 Hz, p = 0.7598. Amplitude - CCI-L PBS: 284.0 ± 34.02 pA versus CCI-L CBX: 187.5 ± 25.89 Hz, ∗p = 0.0434. n = 7 cells from 5 rats in each group).
(C) Significant differences between CCI-L PBS and CCI-L CBX groups were assessed using the two-sided unpaired Student’s t test.
(D) Comparison of evoked EPSC-based RF maps before and after CBX administration. The mean RF area was 1.15 ± 0.14 cm2 (n = 18 cells from 7 rats) in the CCI-L-PBS group and 0.68 ± 0.19 cm2 (n = 11 cells from 6 rats, ∗p = 0.0344) in the CCI-L-CBX group. In the Sham group, the mean RF area was 0.36 ± 0.05 cm2 (n = 22 cells from 10 rats). Significant differences between the CCI-L PBS and CCI-L CBX groups were assessed using the Mann–Whitney test. n.s., not significant.
Discussion
Our study demonstrated that the time course of functional and structural plasticity in the SDH from PNI to the establishment of chronic pain and the causal relationship between them, and showed that these plasticities are astrocyte-dependent. Using in vivo patch-clamp recording, we found that RF expansion, allodynia, and changes in neuronal properties, which are characteristic of chronic pain, were observed from early post-PNI, whereas the synaptic transmission was significantly different between early and late post-PNI. Presynaptic glutamate release and GABA and/or glycine release increased transiently in the early post-PNI period but returned to control levels in the late period. On the other hand, in the postsynaptic receptor response, the excitatory response increased and the inhibitory response dramatically decreased toward the late post-PNI period. Thus, the form of symptom expression after PNI is the same in the early and late stages, but the synaptic transmission changes in a time-dependently during the process to chronic pain. These functional plasticities were accompanied by structural plasticity consisting of the loss of neurons with necroptosis. Furthermore, inhibition of astrocyte proliferation only initially after PNI prevents long-term pain threshold reduction, neuronal loss and altered synaptic transmission. These findings indicated that early therapeutic interventions post-PNI can suppress the transition to chronic pain, suggesting that manipulation of early astrocyte activity could be an effective treatment strategy for chronic pain.
The balance between excitatory and inhibitory synaptic transmission changes over time post-PNI
In the early post-PNI period, the release of glutamate and inhibitory transmitters (GABA and/or Glycine) increases temporarily, but both return to control levels in the late period. On the other hand, changes in receptor sensitivity, increasing sensitivity of glutamate receptors and decreasing sensitivity of GABA and/or glycine receptors, were found in the late post-PNI period. Increased SDH neuron excitability is suggested as a probable pathogenic mechanism of chronic pain, because the excitatory neurotransmitter administration to the spinal cord induces chronic pain-like pathology36; however, whether this excitability persists for a long time after PNI remains unclear. We have shown that spontaneous glutamate release in SDH neurons is transient in the early post-PNI period. Conversely, the increased sensitivity of postsynaptic receptors was marked toward the late post-PNI period. Thus, the transient increase in presynaptic glutamate in the early post-PNI period may have triggered the functional changes of postsynaptic receptors over time.
Disinhibition of inhibitory neurons in SDH is also considered to be one of the pathogenesis of chronic pain. In vitro electrophysiological analysis has not given a consensus on changes in IPSC after PNI.7,26 In this study, IPSC frequency did not decrease, but rather temporarily increased in the early post-PNI period. This may be a reasonable result, considering that it reflects the physiological role of the inhibitory system in the onset of pain. On the other hand, IPSC amplitude was dramatically suppressed, and the suppression was greater in the late post-PNI period. The decay time of IPSC has been shown to be highly dependent on GABA receptor isoforms,37 and chronic pain is accompanied by altered expression of some GABA receptor subunits,38 and our results support these reports. One of the main reasons why our results differ from previous electrophysiological reports of inhibitory synaptic transmission after PNI is the difference between in vivo and in vitro. In vivo animals maintain a strong inhibitory input to the SDH from the superior CNS, such as the locus coeruleus, which is involved in pain modulation. In fact, in vivo IPSC frequency and amplitude are more than ten times higher than those in vitro. Further in vivo studies will be necessary to comprehensively understand the changes in the inhibitory system after PNI.
Allodynia and changes in response properties as functional plasticity of SDH neurons
We used whole-cell voltage-clamp recording to classify neurons by recording EPSCs and showed that the proportion of neurons that respond only to innoxious stimuli decreases and the proportion of neurons that respond to both innoxious and noxious stimuli increases (Figure 1C). Under extracellular recording or whole-cell current clamp recording, it has been reported that SG neurons can be classified into several types based on their response profiles to both innoxious and noxious stimuli.12,24 Medrano et al. used extracellular recording to classify SDH neurons into (1) low-threshold (LT) neurons which respond equally or more to innoxious touch than to noxious pinch, (2) wide-dynamic-range (WDR) neurons which respond more to pinch than to touch, and (3) high-threshold (HT) neurons responded only to pinch, and reported that the proportion of LT neurons increased in PNI animals. The difference in response profiles between our results and theirs may be because of the recording of subthreshold EPSCs in this study. In fact, under in vivo patch-clamp recording, many SDH layer II neurons have been shown to respond to both innoxious and noxious stimuli.23 In addition, it is difficult to record from superficial layers using extracellular recording methods, which may be another reason why the profiles differ from our results of recording from the superficial layer.
We have shown that PNI elicits a stronger glutamatergic response to innoxious stimuli, that is, allodynia. Because excitatory interneurons receive excitatory input via Aδ/C fibers and inhibitory input via Aβ fibers and inhibitory interneurons,39 the balance between these two inputs may change the form of pain expression in response to stimuli. Aδ and C fibers, which transmit noxious stimuli, input the superficial layers (layers I and II) of the SDH, whereas Aβ fibers, which transmit innoxious stimuli, input the deep layers (layers II to V).40 Our results of postsynaptic inhibitory attenuation of inhibitory interneurons after PNI may explain the mechanism of allodynia, which results from enhanced relative Aβ fiber-mediated responses.
PNI-induced astrocyte proliferation leads to neuronal loss accompanied by necroptosis
The molecular biological mechanisms of chronic pain are complex, and current research indicates that inflammatory factors, neuronal loss, altered receptors and ion channels, peripheral sensitization, and central sensitization are involved in the induction and maintenance of chronic pain.41,42 Among these factors, loss of spinal cord neurons has been found to play an important role in the induction and maintenance of chronic pain.8 Loss of neurons associated with PNI has been reported to occur in all SDH neurons, including excitatory neurons, but to date, the focus has been solely on loss of inhibitory neurons.8,26 Because it is clear that excitatory neurons account for a large proportion of the SDH lamina II,31 their loss cannot be ignored. One reason for the absence of clarity on the role of excitatory neuron loss is the lack of reliable immunocytochemical markers for the glutamatergic neuron cell bodies. Therefore, based on previous reports,31 we counted non-inhibitory neurons as excitatory neurons. Our finding that the number of excitatory neurons is reduced while their excitability to stimuli is increased, suggests that receptor sensitivity may be compensatorily altered in surviving neurons. In view of the magnitude of the excitatory neuron loss ratio, it is important to clarify the significance of this change in the future. Manipulation of spinal cord excitatory neurons alone by chemogenetic techniques and other methods will further clarify their functional significance.
In recent years, necroptosis, which is distinct from apoptosis among cell deaths, has attracted much attention. However, there are still few reports on the relationship between necroptosis and chronic pain. Although elevated RIP3 protein levels and increased co-expression with microglia have been shown in SDH after PNI,9 we are the first to show that PNI causes an increase in the number of RIP3-expressing neurons, in other words, necroptosis of neurons. Necroptosis can be initiated by several stimuli, in particular, TNFR1-mediated activation has been the most widely studied; TNF-α is increased in PNI rats.9 Furthermore, because TNF-α contributes to LTP as well as to increased glutamate release,15 our electrophysiological results may also be viewed as the result of a series of TNF-α-induced responses. Further studies are needed to understand the physiological significance of neuronal necroptosis.
Astrocyte activation, which is strongly induced by CNS injury or disease, has been reported as a cause of neuronal death and neurodegeneration.30 Our results, as in previous reports,12,43 of an increased proportion of neurons without identifiable RF after PNI, also indicate neuronal death or degeneration in SDH.
Early post-PNI astrocyte regulation is a key to structural and functional plasticity changes in the SDH in chronic pain
In this study, we showed that astrocyte proliferation after PNI in SDH leads to neuronal loss with necroptosis and that inhibition of astrocyte proliferation prevents neuronal loss and the transition to chronic pain. Astrocytes are the most abundant type of glial cell type in the CNS, and accumulating evidence suggests that they highly contribute to neuropathic pain development and maintenance through the release of astrocytic mediators that increase the activity of spinal cord nociceptive neurons (Chen et al., 2014; Kohro et al., 2020; Takano et al., 2020). Astrocyte activation, strongly induced by CNS injury or disease, has been reported as a cause of neuronal degeneration and neuronal death.30 In this study, we showed that the inhibition of astrocyte activation suppresses neuronal necroptosis, but it is unclear whether astrocyte activation alone induces necroptosis. On the other hand, optogenetic activation of astrocytes is sufficient to elicit microglial activation, and it is known that there is a bidirectional interaction between microglia and astrocytes in the induction of pain.44 Thus, inhibition of astrocyte activation may have suppressed the production of TNFα, which could be a potential therapeutic target for chronic pain. Of course, microglial regulation contributes to the prevention of chronic pain,14,45 but long-term regulation of activated astrocytes may be more consistent with clinical therapeutic targets.
Notably, we have shown that simply inhibiting astrocyte proliferation early after PNI can reduce decreased neuronal loss in the long term. This is consistent with the fact that clinically early treatment reduces the transition to chronic pain.46 Nevertheless, astrocytes responding to nerve injury are also known to play a neuroprotective role,5 therefore further data are needed to determine the duration and timing of proliferation inhibition to provide therapeutic benefit.
In conclusion, this study provides the first direct evidence that astrocyte proliferation after PNI induces loss of SDH neurons with necroptosis and is involved in the development of chronic pain. Specifically, in vivo techniques were applied to a chronic pain model to establish a causal relationship between SDH structural plasticity and pain levels during the temporal progression process after PNI. Importantly, structural plasticity explains the long-term persistence of changes that occur in pathological pain states. We offer several reasons why chronic pain is so difficult to treat. Because suppression of astrocyte activity reduces neuronal decreases and the transition to chronic pain, its control early after PNI could lead to a new treatment strategy for chronic pain with fewer adverse events.
Limitations of the study
In this study, we revealed that inhibition of astrocyte proliferation suppresses neuronal necroptosis, but it is unclear from this study whether astrocyte proliferation alone induces necroptosis. Suppression of microglial activation may also regulate necroptosis induction, because microglia are activated before astrocyte activation after PNI. In addition, because microglia and astrocytes interact in both directions, both may be involved, but this remains to be elucidated. Another limitation is that CBX used in this study is a non-selective gap junction blocker. Therefore, studies with more selective inhibitors of astrocyte proliferation will be needed.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Anti-GFAP | Abcam | Cat# ab7260; RRID:AB_10793236 |
| Rabbit monoclonal Anti-Pax2 | Abcam | Cat# ab79389; RRID:AB_1603338 |
| Mouse monoclonal, clone A60 Anti-NeuN | Millipore | Cat# MAB377; RRID:AB_2298772 |
| Goat anti-rabbit IgG Cy3-conjugated | Jackson Immuno Research | Cat# 111-167-003; RRID:AB_2313593 |
| Donkey anti-mouse FITC-conjugated | Millipore | Cat# AP192F; RRID:AB_92643 |
| Rabbit polyclonal Anti-RIP3 | Sigma-Aldrich | Cat# PRS2283; RRID:AB_1856303 |
| Chemicals, peptides, and recombinant proteins | ||
| AMPA | FUJIFILM Wako Pure Chemical Corporation | Cat# 012-18491; CAS: 74341-63-2 |
| CNQX | FUJIFILM Wako Pure Chemical Corporation | Cat# 032-23121; CAS: 115066-14-3 |
| Bicuculline | FUJIFILM Wako Pure Chemical Corporation | Cat# B382500; CAS: 485-49-4 |
| NMDA | Sigma-Aldrich | Cat# M3262; CAS: 6384-92-5 |
| Carbenoxolone | FUJIFILM Wako Pure Chemical Corporation | Cat# C0167; CAS: 5697-56-3 |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Axon™ pClamp®10.6.2 | Molecular Devices | https://www.moleculardevices.com/ |
| Mini Analysis6.0.3 | Synaptosoft | Synaptosoft Inc., Decatur, GA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Miyuki Kurabe (miyuki915@med.niigata-u.ac.jp).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
All animal experiments were conducted in accordance with the international guidelines on the ethical use of animals, and all efforts were made to minimise animal pain and discomfort. Animal housing protocols and surgical procedures were approved by the Institutional Animal Care and Use Committee of Niigata University Graduate School of Medical and Dental Science. Male Wistar rats (8–12 weeks old, Charles River Laboratories Japan) were housed in plastic cages at 22 ± 2°C on a standard 12-h light/dark cycle and with free access to food and water. All viable efforts were made to minimize the use of animals and reduce their suffering from experimental procedures.
Method details
Surgery for chronic pain models
Chronic pain was induced using the sciatic nerve CCI model.4,16 Animals were anaesthetised using isoflurane (2–3%), the right sciatic nerve was exposed under a microscope, and four loose 4/0 silk ligatures were placed around the nerve proximal to the trifurcation with 1 mm between each ligature. The ligatures were loosely tied until a short flick of the ipsilateral hind limb was observed. The sham-operated animals used as controls underwent only sciatic nerve exposure without ligation.
Behavioural test
Mechanical sensitivity was assessed based on the von Frey method using an automatic dynamic plantar aesthesiometer (37450; Ugo Basile, Comerio, Italy). Briefly, animals were placed in a ventilated Plexiglass cage on an elevated wire mesh grid and allowed to acclimatise. Once exploratory behaviour had ceased, an actuator filament (diameter, 0.5 mm) under computer control delivered a 50 s ramp of increasing force (0–50 g) to the plantar surface of the hind paw until the animal withdrew the paw. Paw withdrawal thresholds were averaged over at least five measurements at 5-min intervals.
Electrophysiological recordings (in vivo patch-clamp recording from SG neurons)
The method used for in vivo patch clamp recordings from SG neurons was the same as we have performed previously.22 Adult Wistar rats were anaesthetised using urethane (1.2–1.5 g/kg, intraperitoneally). When withdrawal reflexes were observed, a supplemental dose of urethane was given during surgery or the data collection period. A heating pad was placed beneath the rat to maintain its body temperature. Thoracolumbar laminectomy was performed at the T10 to L2 level to expose the lumbar enlargement of the spinal cord. Briefly, the rat was placed in a stereotaxic apparatus (Model STS-B; Narishige, Tokyo, Japan), and after the dura mater was opened, the pia-arachnoid membrane was removed to make a window large enough to allow the patch electrode to enter the spinal cord. The surface of the spinal cord was continuously perfused with Krebs solution (117 mM NaCl, 3.6 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 1.2 mM NaH2PO4, 25 mM NaHCO3, and 11 mM D-glucose) equilibrated with a 95% O2 and 5% CO2 gas mixture at a flow rate of 10–15 mL/min maintained at 36°C using a temperature controller (TC-324B; Warner Instruments, Hamden, CT, USA). The patch electrodes were pulled from borosilicate glass capillaries (OD, 1.5 mm) using a puller (p-97; Sutter Instruments, Novato, CA, USA). The resistance of a typical patch pipette was 6–10 MΩ when filled with internal solution (110 mM Cs2SO4, 5 mM tetraethylammonium, 0.5 mM CaCl2, 2 mM MgCl2, 5 mM EGTA, 5 mM HEPES, and 5 mM ATP-Mg; pH 7.2). The electrode was advanced into the SDH through the window in the pia-arachnoid membrane using a micromanipulator (Model MWS-32S; Narishige, Tokyo, Japan). A giga-ohm seal (resistance >5 GΩ) was then formed with neurons at a depth of 50–150 μm from the spinal cord surface; thereafter, the patch membrane was ruptured by a brief period of negative pressure, resulting in a whole-cell configuration. Signals were obtained using an Axopatch 200B amplifier in conjunction with a Digidata 1440A A/D converter (Molecular Devices, Sunnyvale, CA, USA) and stored on a personal computer using the pCLAMP 10 data acquisition program (Molecular Devices). The data were analyzed using Mini Analysis 6.0 (Synaptosoft, Fort Lee, NJ, USA) or pCLAMP 10.6.2 All experiments were performed in voltage-clamp mode. For excitatory post-synaptic current (EPSC) and inhibitory post-synaptic current (IPSC) recordings, the holding potential was held at −70 mV (calculated from Cl– reversal potential) and 0 mV (reversal potential for EPSC), respectively.
Cutaneous sensory stimulation
A noxious mechanical stimulus was applied to the ipsilateral hindlimb by pinching the skin manually using toothed forceps as previously reported.22,23 Innocuous touch stimuli were applied to the ipsilateral hind paw by brushing the skin. Both stimuli were given for 5 s each. To clearly distinguish between noxious and innoxious stimuli to hair, we shaved the fur from the rat hindquarters (the lumbar, gluteal, and hindlimb regions). In voltage-clamp mode, we first confirmed the response to innoxious stimuli by brushing, and marked the response area with a marker. Then, we examined the response to the toothed forceps-induced noxious stimulus, and marked the corresponding response area. For the subsequent area under the curve (AUC) analysis, the stimulus was applied to the part of the response area that responded more. To maintain a fixed strength of noxious stimulation, the toothed forceps were clamped during skin pinching for a constant duration of 5 s to induce pinching pain. Drug application for electrophysiological recordings, drugs were dissolved in Krebs solution and applied to the dorsal surface via the perfusate. For the experiments in Figure 2, the selective α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor antagonist 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX; 20 μM), the selective GABAA receptor antagonist bicuculline (20 μM), and the selective glycine receptor antagonist strychnine (2 μM) were used to analyse only N-methyl-D-aspartate (NMDA) receptor-induced evoked EPSCs. To record both miniature EPSCs (mEPSCs) and miniature IPSCs (mIPSCs) in the experiments in Figures 2 and 3, TTX (0.5 μM) was used. All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) or FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan).
Carbenoxolone (CBX) administration
CBX inhibits astrocyte gap junctions and suppresses reactive astrocytes. Briefly, the baseline paw-withdrawal threshold was measured 1 day before CCI surgery, and CBX (10 mg/kg) or the same amount of PBS was intraperitoneally administered once daily 1–7 days after surgery as shown in Figure 4A. Paw-withdrawal thresholds were measured 5, 7, 14, 21, and 28 days after CCI surgery.
Immunohistochemistry
Immunohistochemistry was performed on transverse sections of the rat lumbar spinal cord. As a general neurochemical marker that reliably labels all dorsal horn glutamatergic neurons is not available, based on previous reports, we identified excitatory neurons indirectly based on the absence of staining for Pax2 and positive labelling with the pan-neuronal marker NeuN (Mullen et al., 1992). Pax2 is expressed in the majority of spinal inhibitory interneurons not only during rodent development,47 but also in adults, and has proved to be a suitable marker for dorsal horn inhibitory interneurons.31,48
Rats were euthanised by isoflurane inhalation overdose. Immediately, 100 mL of saline followed by an equal amount of 4% paraformaldehyde (Mildform® 10N, Fujifilm Wako Pure Chemical Company) were injected for transcardial perfusion. The L4–L5 segments of the spinal cord were removed and post-fixed in 4% paraformaldehyde overnight at 4°C. Then, the samples were cryoprotected using 20% sucrose in 0.1 M phosphate buffer overnight at 4°C. The spinal tissues were covered entirely with FSC22 frozen section media (Leica biosystems), and then stored at −70°C until ready for sectioning. Spinal sections (16 μm) were serially cut using a cryostat (Leica CM1520, Leica microsystems), mounted on APS-coated glass slides (Matsunami Glass), and stored at −70°C until ready for staining. Sections were washed twice with TNT buffer (0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.03% Tween 20), blocked with Blocking One Histo (Nacalai Tesque) at room temperature (23–25°C) for 1 h, and, then, incubated with anti-glial acidic fibrillary protein (GFAP) antibody (rabbit polyclonal; 1:1,000; Abcam, ab7260), anti-Pax2 antibody [EP3251] (rabbit monoclonal; 1:100; Abcam, ab79389), Anti-RIP3 antibody (rabbit polyclonal; 1:200; Sigma-Aldrich, PRS2283), and anti-NeuN antibody (mouse monoclonal, clone A60; 1:800; Millipore, MAB377), as appropriate, in TNB buffer (0.1 M Tris-HCl, pH 7.5, 0.15 M NaCl, 0.1% Tween 20, 0.5% blocking reagent; Roche, 11096176001) at 4°C for 48 h. Anti-Pax2 and anti-NeuN antibodies and-anti RIP3 and anti-NeuN antibodies were also used for double immunostaining. Thereafter, the sections were washed twice with TNT buffer and incubated with Cy3-conjugated goat anti-rabbit IgG (1:400 or 1:1,000; Jackson Immuno Research, 111-167-003) or FITC-conjugated donkey anti-mouse (1:400; Millipore, AP192F) in TNB buffer overnight at 4°C. Then, the sections were washed twice with TNT buffer, embedded using VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA), and visualised using a fluorescence microscope (BX 53, Olympus, Tokyo, Japan) equipped with a digital camera system (DP73, Olympus) or an all-in-one fluorescence microscope (BZ-X810, Keyence, Osaka, Japan).The density of GFAP-labelled astroglial cells in the SDH was measured using ImageJ (National Institutes of Health, version 1.52). Measurements were obtained from 100 × 100 μm2 regions of the SDH layer II (Okada-Ogawa et al., 2009).
Cell numbers were also counted using ImageJ. To quantify the immunoreactivity profile of the spinal cord, three to four L4–L5 spinal cord sections per rat were randomly selected for each group. All cells in the SDH layer II (50–150 μm from the surface of the spinal cord) were counted in a randomly selected spinal cord section. For quantification of colocalization, we counted cells using the corresponding colour channel, and then among those, the number of cells co-expressing the marker of interest. A cell was considered positive for a given marker if the corresponding signal was significantly higher than the background fluorescence.
Quantification and statistical analysis
Data are presented as means ± standard errors of the mean (s.e.m.). Statistical analysis was performed using GraphPad Prism 8 (version 8.0.2; GraphPad Software, Inc., USA) and specific tests are identified in the figure legends. Statistical significance was determined with an overall significance level of p < 0.05 (n.s. for p > 0.05, ∗p < 0.05, †p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ‡p< 0.001). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications.6,18 Data collection was not randomised, and data analysis and collection were not performed blind to the experimental conditions, except for quantification of cellular expression.
Acknowledgments
We thank Y. Sato for starting the initial experiment for in vivo patch clamp recording. This work was supported by a Grant-in-Aid for Exploratory Research 18K08811 (to M.K.), 18H02897 (to Y.K.), and 19K22652 (to Y.K.) from the Japan Society for the Promotion of Science, Tokyo, Japan. We would like to thank Editage [http://www.editage.com] for editing and reviewing this manuscript for English language.
Author contributions
M.K. developed the project. M.K. and M.S. conducted experiments; M.S., K.F., H.F., Y.K., and H.B. advised on the project. M.K. wrote the manuscript. All authors discussed the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: December 22, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105555.
Supplemental information
Data and code availability
This study did not generate any unique datasets or code.
References
- 1.Basbaum A.I., Bautista D.M., Scherrer G., Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolf C.J., Salter M.W. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 3.Kuner R., Flor H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2016;18:20–30. doi: 10.1038/nrn.2016.162. [DOI] [PubMed] [Google Scholar]
- 4.Chen G., Park C.K., Xie R.G., Berta T., Nedergaard M., Ji R.R. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain. 2014;137:2193–2209. doi: 10.1093/brain/awu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji R.R., Donnelly C.R., Nedergaard M. Astrocytes in chronic pain and itch. Nat. Rev. Neurosci. 2019;20:667–685. doi: 10.1038/s41583-019-0218-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuda M., Kohro Y., Yano T., Tsujikawa T., Kitano J., Tozaki-Saitoh H., Koyanagi S., Ohdo S., Ji R.R., Salter M.W., Inoue K. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain. 2011;134:1127–1139. doi: 10.1093/brain/awr025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore K.A., Kohno T., Karchewski L.A., Scholz J., Baba H., Woolf C.J. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J. Neurosci. 2002;22:6724–6731. doi: 10.1523/jneurosci.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholz J., Broom D.C., Youn D.H., Mills C.D., Kohno T., Suter M.R., Moore K.A., Decosterd I., Coggeshall R.E., Woolf C.J. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J. Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang P., Sun G., Wang J. RIP3-mediated necroptosis increases neuropathic pain via microglia activation: necrostatin-1 has therapeutic potential. FEBS Open Bio. 2021;11:2858–2865. doi: 10.1002/2211-5463.13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuner R. Central mechanisms of pathological pain. Nat. Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- 11.Ma C., Shu Y., Zheng Z., Chen Y., Yao H., Greenquist K.W., White F.A., LaMotte R.H. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J. Neurophysiol. 2003;89:1588–1602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- 12.Medrano M.C., Dhanasobhon D., Yalcin I., Schlichter R., Cordero-Erausquin M. Loss of inhibitory tone on spinal cord dorsal horn spontaneously and nonspontaneously active neurons in a mouse model of neuropathic pain. Pain. 2016;157:1432–1442. doi: 10.1097/j.pain.0000000000000538. [DOI] [PubMed] [Google Scholar]
- 13.Yan X., Jiang E., Gao M., Weng H.R. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J. Physiol. 2019;591:2001–2019. doi: 10.1113/jphysiol.2012.250522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoue K., Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018;19:138–152. doi: 10.1038/nrn.2018.2. [DOI] [PubMed] [Google Scholar]
- 15.Gruber-schoffnegger D., Drdla-schutting R., Ho C., Wunderbaldinger G., Gassner M. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J. Neurosci. 2013;33:6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett G.J., Xie Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 17.Yu Z., Jiang N., Su W., Zhuo Y., Walker C.S. Necroptosis : a novel pathway in neuroin fl ammation. Front. Pharmacol. 2021;12:1–13. doi: 10.3389/fphar.2021.701564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohno T., Moore K.A., Baba H., Woolf C.J. Peripheral nerve injury alters excitatory synaptic transmission in laminal II of the rat dorsal horn. J. Physiol. 2003;548:131–138. doi: 10.1113/jphysiol.2002.036186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook A.J., Woolf C.J., Wall P.D., Mcmahon S.B. Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature. 1987;325:151–153. doi: 10.1038/325151a0. [DOI] [PubMed] [Google Scholar]
- 20.Saito K., Hitomi S., Suzuki I., Masuda Y., Kitagawa J., Tsuboi Y., Kondo M., Sessle B.J., Iwata K. Modulation of trigeminal spinal subnucleus caudalis neuronal activity following regeneration of transected inferior alveolar nerve in rats. J. Neurophysiol. 2008;99:2251–2263. doi: 10.1152/jn.00794.2007. [DOI] [PubMed] [Google Scholar]
- 21.Baba H., Doubell T.P., Moore K.A., Woolf C.J. Silent NMDA receptor-mediated synapses are developmentally regulated in the dorsal horn of the rat spinal cord. J. Neurophysiol. 2000;83:955–962. doi: 10.1152/jn.2000.83.2.955. [DOI] [PubMed] [Google Scholar]
- 22.Kurabe M., Furue H., Kohno T. Intravenous administration of lidocaine directly acts on spinal dorsal horn and produces analgesic effect: an in vivo patch-clamp analysis. Sci. Rep. 2016;6:26253–26312. doi: 10.1038/srep26253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Furue H., Narikawa K., Kumamoto E., Yoshimura M. Responsiveness of rat substantia gelatinosa neurones to mechanical but not thermal stimuli revealed by in vivo patch-clamp recording. J. Physiol. 1999;521(Pt 2):529–535. doi: 10.1111/j.1469-7793.1999.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graham B.A., Brichta A.M., Callister R.J. In vivo responses of mouse superficial dorsal horn neurones to both current injection and peripheral cutaneous stimulation. J. Physiol. 2004;561:749–763. doi: 10.1113/jphysiol.2004.072645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikeda H., Heinke B., Ruscheweyh R., Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- 26.Inquimbert P., Moll M., Latremoliere A., Tong C.K., Whang J., Sheehan G.F., Smith B.M., Korb E., Athié M.C.P., Babaniyi O., et al. NMDA receptor activation underlies the loss of spinal dorsal horn neurons and the transition to persistent pain after peripheral nerve injury. Cell Rep. 2018;23:2678–2689. doi: 10.1016/j.celrep.2018.04.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawasaki Y., Xu Z.Z., Wang X., Park J.Y., Zhuang Z.Y., Tan P.H., Gao Y.J., Roy K., Corfas G., Lo E.H., Ji R.R. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 2008;14:331–336. doi: 10.1038/nm1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polgár E., Hughes D.I., Arham A.Z., Todd A.J. Loss of neurons from laminas I-III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. J. Neurosci. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milligan E.D., Watkins L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liddelow S.A., Guttenplan K.A., Clarke L.E., Bennett F.C., Bohlen C.J., Schirmer L., Bennett M.L., Münch A.E., Chung W.S., Peterson T.C., et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. doi: 10.1038/nature21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Punnakkal P., von Schoultz C., Haenraets K., Wildner H., Zeilhofer H.U. Morphological, biophysical and synaptic properties of glutamatergic neurons of the mouse spinal dorsal horn. J. Physiol. 2014;592:759–776. doi: 10.1113/jphysiol.2013.264937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dimidschstein J., Chen Q., Tremblay R., Rogers S.L., Saldi G.A., Guo L., Xu Q., Liu R., Lu C., Chu J., et al. A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat. Neurosci. 2016;19:1743–1749. doi: 10.1038/nn.4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji R.R., Berta T., Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain. 2013;154:S10–S28. doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H., Cao Y., Chiang C.Y., Dostrovsky J.O., Sessle B.J. The gap junction blocker carbenoxolone attenuates nociceptive behavior and medullary dorsal horn central sensitization induced by partial infraorbital nerve transection in rats. Pain. 2014;155:429–435. doi: 10.1016/j.pain.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu Q., Ford N.C., He S., Huang Q., Anderson M., Chen Z., Yang F., Crawford L.K., Caterina M.J., Guan Y., et al. Astrocytes contribute to pain gating in the spinal cord. Sci. Adv. 2021;7:eabi6287. doi: 10.1126/sciadv.abi6287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nichols M.L., Allen B.J., Rogers S.D., Ghilardi J.R., Honore P., Luger N.M., Finke M.P., Li J., Lappi D.A., Simone D.A., Mantyh P.W. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- 37.Syed P., Durisic N., Harvey R.J., Sah P., Lynch J.W. Effects of GABAA receptor α3 subunit epilepsy mutations on inhibitory synaptic signaling. Front. Mol. Neurosci. 2020;13:602559–602612. doi: 10.3389/fnmol.2020.602559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iura A., Takahashi A., Hakata S., Mashimo T., Fujino Y. Reductions in tonic GABAergic current in substantia gelatinosa neurons and GABAA receptor δ subunit expression after chronic constriction injury of the sciatic nerve in mice. Eur. J. Pain. 2016;20:1678–1688. doi: 10.1002/ejp.891. [DOI] [PubMed] [Google Scholar]
- 39.Baba H., Ji R.R., Kohno T., Moore K.A., Ataka T., Wakai A., Okamoto M., Woolf C.J. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol. Cell. Neurosci. 2003;24:818–830. doi: 10.1016/S1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- 40.Todd A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010;11:823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu D., Zhao H., Gao H., Zhao H. Participation of pro-inflammatory cytokines in neuropathic pain evoked by chemotherapeutic oxaliplatin via central GABAergic pathway. Mol. Pain. 2018;14 doi: 10.1177/1744806918783535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou D., Zhang S., Hu L., Gu Y., Cai Y., Wu D., Liu W., Jiang C. Inhibition of apoptosis signal-regulating kinase by paeoniflorin attenuates neuroinflammation and ameliorates neuropathic pain. J. Neuroinflamm. 2019;6:1–11. doi: 10.1186/s12974-019-1476-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hao J.X., Kupers R.C., Xu X.J. Response characteristics of spinal cord dorsal horn neurons in chronic allodynic rats after spinal cord injury. J. Neurophysiol. 2004;92:1391–1399. doi: 10.1152/jn.00121.2004. [DOI] [PubMed] [Google Scholar]
- 44.Nam Y., Kim J.H., Kim J.H., Jha M.K., Jung J.Y., Lee M.G., Choi I.S., Jang I.S., Lim D.G., Hwang S.H., et al. Reversible induction of pain hypersensitivity following optogenetic stimulation of spinal astrocytes. Cell Rep. 2016;17:3049–3061. doi: 10.1016/j.celrep.2016.11.043. [DOI] [PubMed] [Google Scholar]
- 45.Tsuda M., Shigemoto-mogami Y., Koizumi S., Mizokoshi A., Kohsaka S., Salter M.W., Inoue K. P2X 4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01859.1. [DOI] [PubMed] [Google Scholar]
- 46.Kehlet H., Jensen T.S., Woolf C.J., Centre M. Persistent postsurgical pain: risk factors and prevention. Lancet. 2006;367:1618–1625. doi: 10.1016/S0140-6736(06)68700-X. [DOI] [PubMed] [Google Scholar]
- 47.Maricich S.M., Herrup K. Pax-2 expression defines a subset of GABAergic interneurons and their precursors in the developing murine cerebellum. J. Neurobiol. 1999;41:281–294. doi: 10.1002/(SICI)1097-4695(19991105)41:2<281::AID-NEU10>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Balázs A., Mészár Z., Hegedűs K., Kenyeres A., Hegyi Z., Dócs K., Antal M. Development of putative inhibitory neurons in the embryonic and postnatal mouse superficial spinal dorsal horn. Brain Struct. Funct. 2017;222:2157–2171. doi: 10.1007/s00429-016-1331-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.