

Short abstract
Content available: Audio Recording
Answer questions and earn CME
Listen to an audio presentation of this article.
INTRODUCTION
Biliary atresia (BA) is a congenital disease characterized by obliteration of the extrahepatic bile duct and periductal inflammation and progressive destruction of the intrahepatic bile ducts in infants. BA is believed to occur in approximately 1 in 13,000 live births. 1 Currently, the gold standard treatment for BA is a Kasai hepatoportoenterostomy (HPE), a surgical procedure performed ideally within the first 3 months of life in an attempt to restore mechanical bile flow and clearance. Approximately half of infants who undergo HPE do not achieve clearance of their jaundice and require early liver transplantation (“early failure”), while another subset of infants with BA continue to develop progressive fibrosis after HPE despite restoration of bile flow and ultimately require liver transplantation (“late failure”). Given the aggressive progression of liver fibrosis in this disease and limited therapeutic options, ultimately approximately 70%–80% of children with BA require liver transplantation, making it the most common indication for liver transplantation among children. Although traditionally thought to be idiopathic, increasing evidence has demonstrated an important immune dysregulation component to the pathogenesis of BA, with dysregulated macrophages, neutrophils, dendritic cells, natural killer cells, B cells, and T cells all having been implicated as playing a role in the destruction of cholangiocytes. 1 , 2
In this regard, it is likely that galectin‐3 plays an important role in the immunopathology and fibrotic progression of BA. Galectin‐3 is a member of a family of β‐galactoside–binding lectins and is unique among this family in its ability to form pentamers and lattice structures through its N‐terminal domain. 3 Galectin‐3 is found both intracellularly and extracellularly, with the latter the result of nonclassical secretion pathways that bypass the endoplasmic reticulum and Golgi apparatus. Intracellular galectin‐3 is known to regulate the cell cycle, growth, differentiation, and apoptosis, while extracellular galectin‐3 is implicated in cell adhesion, chemoattraction, and activation. 3 Galectin‐3 is well known to be involved in inflammation, as well as in organ fibrosis, including the kidney, heart, and liver. Honsawek et al. 4 demonstrated that galectin‐3 is elevated in the serum of children with BA in comparison with healthy control subjects, and that it corresponds to markers of disease severity, such as bilirubin levels, transaminases, and presence of portal hypertension. This review will serve to draw attention to the links between what is known about the pathogenesis of BA and the known inflammatory and profibrotic functions of galectin‐3 in response to cholestatic liver injury, as well as highlight the therapeutic potential of galectin‐3 inhibitors in improving long‐term outcomes for children with BA.
GALECTIN‐3 AND THE IMMUNE RESPONSE
In BA, it is thought that an initial injury to the bile duct epithelium triggers a response by local antigen‐presenting cells, such as dendritic cells and Kupffer resident macrophages, which then trigger an intensive immune cell periductal infiltration and inflammatory cascade involving natural killer cells, neutrophils, helper and cytotoxic T cells, and autoantibody‐producing B cells, which further propagate bile duct injury and ultimately mediate its destruction. 1 , 2 This has been well demonstrated both in human tissue samples and in the widely used Rhesus rotavirus A mouse model of BA. Galectin‐3 is in general considered to be proinflammatory due to its ability to activate immune cells by binding and cross‐linking surface markers, in particular in neutrophils and macrophages (Figure 1). 3 It is also known to serve as a chemoattractant for neutrophils and macrophages and to promote their adhesion and phagocytic capabilities. Specifically in response to liver injury, galectin‐3 knockout mice and mice treated with galectin‐3 inhibitors demonstrated fewer liver infiltrating mononuclear cells, including B cells, dendritic cells, T cells, natural killer cells, and proinflammatory macrophages. 5 , 6 In addition, galectin‐3 knockout or inhibitor‐treated mice had a significant increase in M2 macrophages, which are traditionally thought to be an anti‐inflammatory subset of this cell type. In an autoimmune cholangitis cholestatic liver injury model, galectin‐3 was critical to macrophage inflammasome activation and IL‐17 expression, important mediators of biliary epithelial injury in BA. 2 , 6 , 7 Conversely, galectin‐3 is known to be expressed in cholangiocytes, and it has been demonstrated that knockout mice suffer more severe apoptosis of biliary epithelial cells in response to cholestatic liver injury, emphasizing the contrasting roles and effects galectin‐3 has in different cell types and when expressed intracellularly versus extracellularly. 8
FIGURE 1.
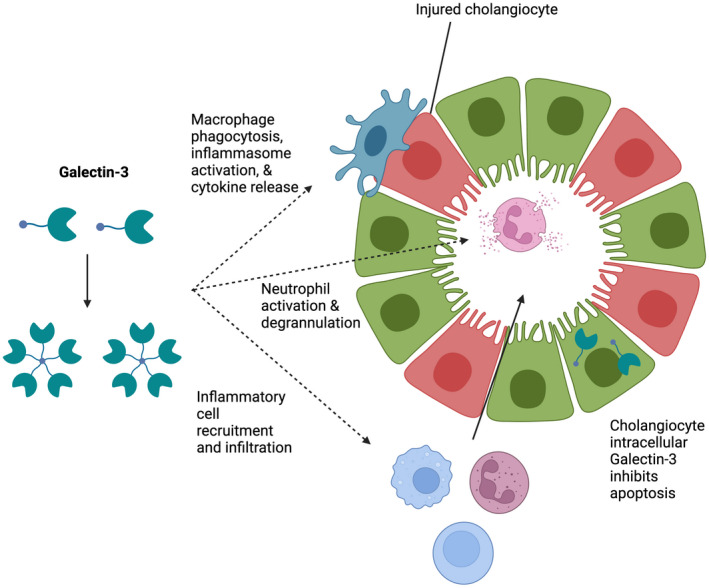
Effects of galectin‐3 on immune cell response to biliary disease. Extracellular galectin‐3 is critical to the activation and function of macrophages and neutrophils, as well as to the recruitment of various inflammatory cells in response to liver and bile duct injury. Intracellular galectin‐3, known to be expressed in cholangiocytes, has antiapoptotic effects. Figure was created with BioRender.com. All rights and ownership of BioRender content are reserved by BioRender. BioRender content included in the completed graphic is not licensed for any commercial uses beyond publication in a journal.
GALECTIN‐3 AND LIVER FIBROSIS
BA is characterized by aggressive progression of liver fibrosis to cirrhosis and portal hypertension, with approximately 50% of patients requiring a liver transplant by 2 years of age. 1 While galectin‐3 expression in healthy livers is minimal and limited to cholangiocytes and Kupffer cells, galectin‐3 is upregulated in cirrhotic livers, especially in the regenerative nodules. 9 This has been shown to be consistent across various etiologies of liver disease in adults, including primary biliary cholangitis, although this has not yet been investigated in tissue samples of patients with BA. Hepatic stellate cells (HSCs) are the key regulators of liver fibrosis. In response to liver injury, HSCs phagocytose apoptotic bodies, thereby differentiating into an active, myofibroblast phenotype that produces transforming growth factor‐β1 (TGF‐β1) and releases collagen into the extracellular matrix within the liver, leading to the formation of fibrotic ridges and bridging fibrosis characteristic of advanced liver fibrosis and cirrhosis. Galectin‐3 is critical to this profibrotic HSC activation, with HSCs from galectin‐3 knockout mice demonstrating decreased phagocytosis, TGF‐β1 production, and procollagen production in response to bile duct ligation or carbon tetrachloride administration (Figure 2). 9 , 10 This effect can be reversed with the addition of recombinant galectin‐3 in an in vitro assay, indicating that it is extracellular galectin‐3 that is critical for HSC activation. Extracellular blockage of galectin‐3 via competitive binding of the carbohydrate recognition domain using oral modified citrus pectin achieved decreased HSC activation and liver fibrosis. 11
FIGURE 2.
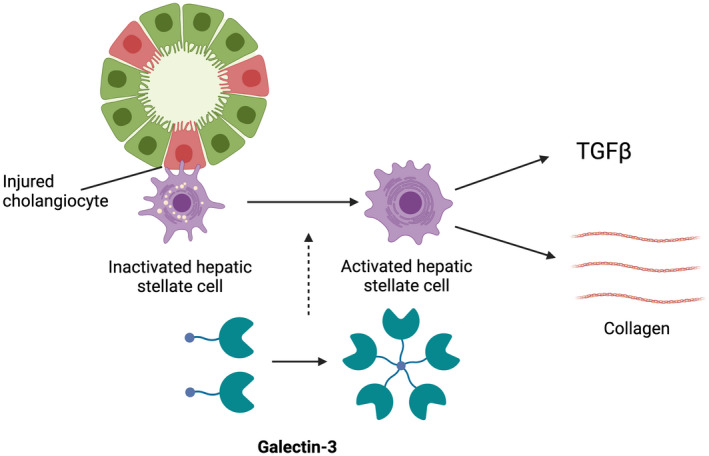
Role of galectin‐3 in liver fibrosis in cholestatic disease. Galectin‐3 is crucial to the activation of HSCs into a myofibroblast phenotype in response to hepatocyte or cholangiocyte injury. Activated HSCs release collagen and TGF‐β leading to liver fibrosis. Figure was created with BioRender.com. All rights and ownership of BioRender content are reserved by BioRender. BioRender content included in the completed graphic is not licensed for any commercial uses beyond publication in a journal.
THERAPEUTIC APPLICATIONS OF GALECTIN‐3 INHIBITORS
Inhibition of galectin‐3 may be beneficial to the treatment of progressive liver diseases by mitigating immune‐mediated injury and slowing liver fibrosis, in particular BA, given the major role of the inflammatory response in its pathogenesis and the disease's aggressive fibrotic profile. Galectin‐3 inhibitors predominantly act on extracellular, rather than intracellular, galectin‐3, as can be elucidated by comparing differences in the effect of galectin‐3 inhibitors and galectin‐3 gene knockout models, the latter of which removes both the extracellular and intracellular effects of galectin‐3. In animal models, galectin‐3 inhibitors have largely been demonstrated to block macrophage and HSC activation and chemoattraction of infiltrating immune cells, which are functions associated with extracellular galectin‐3 (Table 1). The preservation of intracellular galectin‐3 function could be beneficial, especially in BA, because intracellular galectin‐3 in cholangiocytes has been demonstrated to decrease apoptosis in the biliary epithelial cells in response to injury. 8 Anti‐galectin‐3–directed therapies, in theory, would hamper the proinflammatory effects of extracellular galectin‐3, while preserving the beneficial antiapoptotic attributes of intracellular galectin‐3 in cholangiocytes, although research on the latter has been limited to knockout models, and thus further research on the effect of galectin‐3 inhibitors on cholangiocyte survival is required.
TABLE 1.
Known functions of galectin‐3 in cell populations relevant to BA
| Cell population | Galectin‐3 function in response to liver injury | Impact of knocking out galectin‐3 | Impact of blocking galectin‐3 |
|---|---|---|---|
| Cholangiocytes |
Expressed at baseline in healthy livers 3 Antiapoptotic/protective 8 |
Greater apoptosis | Unknown |
| Kupffer cells | Expressed at baseline 3 | Fewer proinflammatory macrophages and cytokine release, larger anti‐inflammatory M2 macrophage population 5 | Fewer proinflammatory macrophages and cytokine release, larger anti‐inflammatory M2 macrophage population 5 |
| Critical for activation, phagocytosis, adhesion, and inflammasome activation 6 , 7 | No inflammasome activation 6 , 7 | ||
| Infiltrating immune cells | Critical for immune cell recruitment and infiltration of injured liver 3 , 5 | Decreased | Decreased |
| HSCs | Critical for phagocytosis, activation, TGF‐β1 release, and collagen deposition 9 , 10 | Less fibrosis 9 , 10 | Less fibrosis 11 |
While the benefits of galectin‐3 inhibition have been consistently demonstrated in animal models of liver disease, studies on applications of anti‐galectin‐3–directed therapies in humans are limited. 5 , 11 A phase 2b randomized trial of GR‐MD‐02, a galectin‐3 inhibitor, in patients with nonalcoholic steatohepatitis cirrhosis and portal hypertension failed to achieve a significant benefit based on the primary endpoint of reversing portal hypertension. 12 It is yet to be determined, however, if earlier administration of anti‐galectin‐3 therapy could slow the progression of fibrosis and portal hypertension, even if it could not reverse it when administered in an already highly advanced disease state. The notion that earlier galectin‐3 inhibitor therapy may be more beneficial was bolstered by the subanalysis finding from this trial that GR‐MD‐02 significantly reduced the development of new varices in patients without esophageal varices at baseline. Anti‐galectin‐3–directed therapeutics may be an especially useful adjunct for the subset of patients with BA who successfully achieve restored bile flow following HPE but still experience progressive fibrosis as a result of intrahepatic involvement. If started at time of HPE, galectin‐3 inhibition may be effective in slowing down the disease progression, thereby delaying, if not preventing, the need for liver transplantation in these children (Figure 3). Even if galectin‐3 inhibition could help delay the need for transplant past the first year of life, this would be an important achievement, because there is a significant waiting list and posttransplant survival benefit has been documented if the transplant can be delayed past this time point. 13
FIGURE 3.
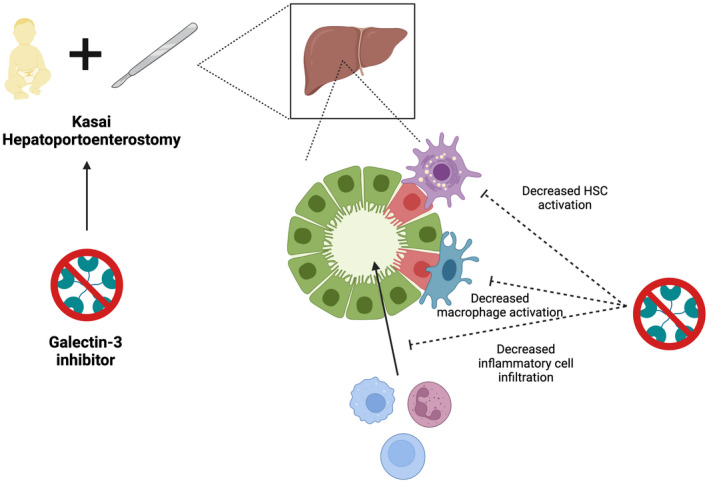
Therapeutic potential of galectin‐3 inhibition in BA. Use of a galectin‐3 inhibitor at time of Kasai HPE could slow the progression of liver fibrosis by decreasing macrophage and HSC activation, as well as overall immune cell infiltration of the liver. Figure was created with BioRender.com. All rights and ownership of BioRender content are reserved by BioRender. BioRender content included in the completed graphic is not licensed for any commercial uses beyond publication in a journal.
CONCLUSION
BA is characterized by immune‐mediated destruction of intrahepatic and extrahepatic bile ducts, resulting in progressive liver fibrosis that ultimately requires liver transplantation in most children with this disease. Galectin‐3 is heavily involved in the activation of immune cells and HSCs in response to liver injury and may be an important therapeutic target to improve both pretransplant and posttransplant survival in this patient population. Although parallels can be drawn from other animal models of cholestatic liver injury, such as bile duct ligation and autoimmune cholangitis, dedicated studies are required to explore the role of galectin‐3 in BA using patient tissue samples and the Rhesus rotavirus A mouse model of BA, which more closely mimics the specific disease process in BA.
FUNDING INFORMATION
This work was supported by National Institutes of Health/National Center for Advancing Translational Sciences Colorado CTSA Grant TL1 TR002533 (to D.Y.). The contents of this review are the authors' sole responsibility and do not necessarily represent official National Institutes of Health views.
CONFLICT OF INTEREST
C.L.M. consults for Albireo.
Yoeli D, Mack CL, Navarro‐Alvarez N. Role of galectin‐3 in the pathogenesis and progression of biliary atresia. Clinical Liver Disease. 2022;20:170–174. 10.1002/cld.1245
REFERENCES
- 1. Bezerra JA, Wells RG, Mack CL, Karpen SJ, Hoofnagle JH, Doo E, et al. Biliary atresia: clinical and research challenges for the twenty‐first century. Hepatology. 2018;68:1163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ortiz‐Perez A, Donnelly B, Temple H, Tiao G, Bansal R, Mohanty SK. Innate immunity and pathogenesis of biliary atresia. Front Immunol. 2020;11:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dumic J, Dabelic S, Flogel M. Galectin‐3: an open‐ended story. Biochim Biophys Acta. 2006;1760:616–35. [DOI] [PubMed] [Google Scholar]
- 4. Honsawek S, Chongsrisawat V, Praianantathavorn K, Theamboonlers A, Poovorawan Y. Elevation of serum galectin‐3 and liver stiffness measured by transient elastography in biliary atresia. Eur J Pediatr Surg. 2011;21:250–4. [DOI] [PubMed] [Google Scholar]
- 5. Volarevic V, Milovanovic M, Ljujic B, Pejnovic N, Arsenijevic N, Nilsson U, et al. Galectin‐3 deficiency prevents concanavalin A‐induced hepatitis in mice. Hepatology. 2012;55:1954–64. [DOI] [PubMed] [Google Scholar]
- 6. Arsenijevic A, Milovanovic J, Stojanovic B, Djordjevic D, Stanojevic I, Jankovic N, et al. Gal‐3 deficiency suppresses Novosphyngobium aromaticivorans inflammasome activation and IL‐17 driven autoimmune cholangitis in mice. Front Immunol. 2019;10:1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tian J, Yang G, Chen HY, Hsu DK, Tomilov A, Olson KA, et al. Galectin‐3 regulates inflammasome activation in cholestatic liver injury. FASEB J. 2016;30:4202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arsenijevic A, Milovanovic M, Milovanovic J, Stojanovic B, Zdravkovic N, Leung PS, et al. Deletion of galectin‐3 enhances xenobiotic induced murine primary biliary cholangitis by facilitating apoptosis of BECs and release of autoantigens. Sci Rep. 2016;6:23348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Henderson NC, Mackinnon AC, Farnworth SL, Poirier F, Russo FP, Iredale JP, et al. Galectin‐3 regulates myofibroblast activation and hepatic fibrosis. Proc Natl Acad Sci U S A. 2006;103:5060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang JX, Chen X, Hsu DK, Baghy K, Serizawa N, Scott F, et al. Galectin‐3 modulates phagocytosis‐induced stellate cell activation and liver fibrosis in vivo. Am J Physiol Gastrointest Liver Physiol. 2012;302:G439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abu‐Elsaad NM, Elkashef WF. Modified citrus pectin stops progression of liver fibrosis by inhibiting galectin‐3 and inducing apoptosis of stellate cells. Can J Physiol Pharmacol. 2016;94:554–62. [DOI] [PubMed] [Google Scholar]
- 12. Chalasani N, Abdelmalek MF, Garcia‐Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin‐3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. 2020;158:1334–45.e5. [DOI] [PubMed] [Google Scholar]
- 13. Yoeli D, Choudhury RA, Sundaram SS, Mack CL, Roach JP, Karrer FM, et al. Primary vs. salvage liver transplantation for biliary atresia: a retrospective cohort study. J Pediatr Surg. 2022. 10.1016/j.jpedsurg.2021.12.027 [DOI] [PubMed] [Google Scholar]