

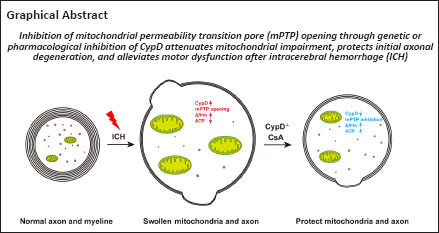
Keywords: axonal injury, corticospinal tract, cyclophilin D, cyclosporin A, intracerebral hemorrhage, mitochondrial impairment, mitochondrial permeability transition pore, motor dysfunction, retraction bulb, white matter
Abstract
The mitochondrial permeability transition pore is a nonspecific transmembrane channel. Inhibition of mitochondrial permeability transition pore opening has been shown to alleviate mitochondrial swelling, calcium overload, and axonal degeneration. Cyclophilin D is an important component of the mitochondrial permeability transition pore. Whether cyclophilin D participates in mitochondrial impairment and axonal injury after intracerebral hemorrhage is not clear. In this study, we established mouse models of intracerebral hemorrhage in vivo by injection of autologous blood and oxyhemoglobin into the striatum in Thy1-YFP mice, in which pyramidal neurons and axons express yellow fluorescent protein. We also simulated intracerebral hemorrhage in vitro in PC12 cells using oxyhemoglobin. We found that axonal degeneration in the early stage of intracerebral hemorrhage depended on mitochondrial swelling induced by cyclophilin D activation and mitochondrial permeability transition pore opening. We further investigated the mechanism underlying the role of cyclophilin D in mouse models and PC12 cell models of intracerebral hemorrhage. We found that both cyclosporin A inhibition and short hairpin RNA interference of cyclophilin D reduced mitochondrial permeability transition pore opening and mitochondrial injury. In addition, inhibition of cyclophilin D and mitochondrial permeability transition pore opening protected corticospinal tract integrity and alleviated motor dysfunction caused by intracerebral hemorrhage. Our findings suggest that cyclophilin D is used as a key mediator of axonal degeneration after intracerebral hemorrhage; inhibition of cyclophilin D expression can protect mitochondrial structure and function and further alleviate corticospinal tract injury and motor dysfunction after intracerebral hemorrhage. Our findings provide a therapeutic target for preventing axonal degeneration of white matter injury and subsequent functional impairment in central nervous diseases.
Introduction
Intracerebral hemorrhage (ICH) is a non-traumatic hemorrhage in the brain parenchyma (Samarasekera et al., 2015). Among all ICH events, ICH in the basal ganglia easily leads to disability or even death because of injury to the corticospinal tracts (CSTs) and myelin of white matter (WM) (Koyama et al., 2012; Venkatasubramanian et al., 2013; Cheng et al., 2015; Tao et al., 2017; Jiang et al., 2019; Xu et al., 2022). Despite numerous approaches to develop strategies that promote axon regeneration and remyelination after WM injury (WMI), the behavioral improvements after these treatments are suboptimal (He and Jin, 2016; Hilton and Bradke, 2017). Our previous study revealed severe axonal degeneration around the hematoma at 24 hours after ICH (Yang et al., 2022), but the molecular mechanisms remain unclear. Therefore, more promising strategies are exploring axonal injury mechanisms and potential therapeutic targets to prevent axonal degeneration within 24 hours after ICH.
Adenosine triphosphate (ATP) produced by mitochondria is critical for the development, maintenance, and regeneration of axons (Chamberlain and Sheng, 2019). However, reactive oxygen species (ROS) are a direct byproduct of ATP production and can cause direct damage to neurons (van Hameren et al., 2019). Previous studies demonstrated that ROS released from injured mitochondria increased within 2 hours after peripheral nerve injury (Hervera et al., 2018) and 6 hours after ICH (Kim-Han et al., 2006; Qu et al., 2019) near the injury site. Moreover, treatment with oxidant scavengers at the lesion site immediately before injury blocked the increase in ROS signaling and axonal degeneration (Hervera et al., 2018). Additionally, an elevation in the concentration of free intracellular Ca2+ released from injured mitochondria and the endoplasmic reticulum triggers axonal degeneration after axotomy (Villegas et al., 2014). The above evidence suggests that mitochondrial injury is a critical initial event for axonal degeneration in WM.
The mitochondrial permeability transition pore (mPTP) is a nonspecific transmembrane channel, and cyclophilin D (CypD) is an important component of mPTP (Kalani et al., 2018; Bonora et al., 2020). The mPTP is normally closed but opens under mitochondrial stress conditions, which causes the release of Ca2+, ROS, and proapoptotic proteins from mitochondria, ultimately leading to cell death (Kalani et al., 2018). A previous study found that inhibiting the opening of the mPTP alleviated mitochondrial swelling, calcium overload, and axonal degeneration after spinal cord injury (Barrientos et al., 2011). However, the involvement of mPTP activation-associated axonal degeneration after ICH is largely unknown.
Here, we investigated the function of the mPTP and CypD in mitochondrial swelling and axonal injury after ICH in vitro and in vivo.
Methods
Animals
Thy1-YFP mice (Stock No. 003782, RRID: IMSR_JAX:003782) were provided from The Jackson Laboratory (Bar Harbor, ME, USA), CypD–/– (Ppif–/–) mice were gifts from Peking University, and C57/BL6 wild-type (WT) mice were obtained from the Experimental Animal Center of the Third Military Medical University (license No. SCXK (Yu) 20170002). Thy1-YFP::CypD–/– mice were generated by crossing Thy1-YFP mice with CypD–/– mice, and Thy1-YFP::WT mice were generated by crossing Thy1-YFP mice with WT mice.
The mice were fed ad libitum and housed in a room with specific-pathogen-free conditions and a standard 12-hour light/dark cycle at 22°C. All experiments are reported in compliance with the Animal Research: Reporting in vivo Experiments (ARRIVE) guidelines (Percie du Sert et al., 2020). The experimental protocols were approved by the Laboratory Animal Welfare and Ethics Committee of Third Military Medical University (approval No. AMUWEC2020761; on April 9, 2020) and performed according to the Guide for the Care and Use of Laboratory Animals. The mice were anesthetized using 2% isoflurane (RWD Life Science, Shenzhen, China)/air (1–2 L/min) mixture during modeling or execution.
ICH model and CsA treatment
This study initially included 199 adult male mice (20–25 g, 8–10 weeks old); 21 mice were excluded (3 mice died from an anesthetic accident, 14 mice died after surgery, and 4 mice were excluded because of unsuccessful ICH modeling). Finally, 178 adult male mice were used in this study (Figure 1A and B). We did not use female mice for three reasons. First, estrogen fluctuates with the menstrual cycle and affects behavior. Second, estrogen has a neuroprotective effect (Petrovska et al., 2012). Third, to keep the homogeneity of mice and avoid the behavioral bias caused by different gonadal hormones, only male mice were used in this study.
Figure 1.
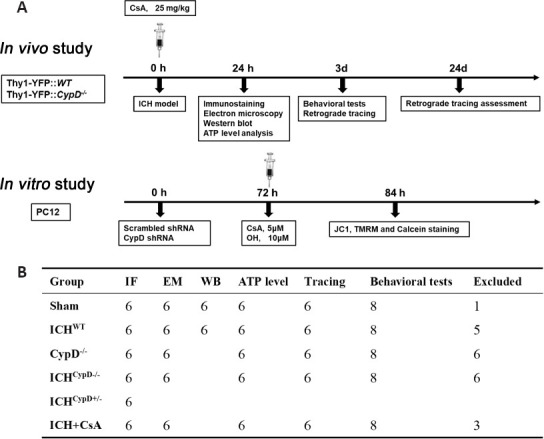
The experimental groups and sample sizes.
(A) The timelines for in vivo and in vitro experiments. (B) The sample size in each group. ATP: Adenosine triphosphate; CypD: cyclophilin D; CsA: cyclosporin A; EM: electron microscope; ICH: intracerebral hemorrhage; IF: immunofluorescence; JC1: 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; OH: oxyhemoglobin; shRNA: short hairpin RNA; TMRM: tetramethylrhodamine methyl ester; WB: western blot; WT: wild type.
The Thy1-YFP::CypD–/– and Thy1-YFP::WT mice were divided into six groups: ShamWT, ShamCypD–/–, ICHWT, ICHCypD–/–, ICHCyp+/–, and ICHWT + cyclosporin A (CsA) groups. The details of animal groupings are shown in Figure 1. Randomization was carried out using odd/even numbers.
To establish the ICH model, the mice were anesthetized. Under stereotactic guidance (RWD Life Science), a small cranial burr hole was made above the striatum (Bregma coordinates: anteroposterior = +0.8 mm, mediolateral = 2 mm). The autologous arterial blood was obtained by puncturing the central tail artery with a sterile needle (26 gauge) and collecting blood into an unheparinized capillary tube. The blood sample (25 μL) was transferred quickly into the glass barrel of a sterile syringe (33 Gauge; Hamilton, Bonaduz, Switzerland). The blood was injected into the right striatum (Bregma coordinate: dorsoventral = –3 mm) at a rate of 2 μL/min with a microinfusion pump (Harvard Apparatus, Holliston, MA, USA) (Krafft et al., 2012). In other experiments, to mimic the ICH model, 25 μL oxyhemoglobin (OH, 10 μM, Sangon Biotech Co., Ltd., Shanghai, China) was also injected into the same location. Sham-operated mice underwent the same surgery without blood or saline injection. For the ICH + CSA group, ICHWT mice intraperitoneally received CsA (one dose, 15 mg/kg, dissolved in corn oil and ethanol at a ratio of 9:1; Cat# HY-B0579, MCE, Shanghai, China) or an equal volume (5 mL/kg) of the vehicle immediately after the surgery. The brain tissue around the hematoma was harvested at different time points after ICH for morphological and biochemical experiments (Figure 1A).
Cell lines and culture
PC12 cells (American Type Culture Collection (ATCC), Rockville, MD, USA, Cat# CRL-1721.1, RRID: CVCL_F659) are an extensively used cell model in neurobiology as they exhibit some features of mature neurons (Greene and Tischler, 1976). PC12 cells were cultured in Dulbecco’s modified Eagle media/nutrient mixture F-12 (DMEM/F12) supplemented with 10% fetal bovine serum medium.
Short hairpin RNA interference
CypD (ppif) short hairpin RNA (shRNA) and scrambled shRNA lentiviral particles (Origene, Rockville, MD, USA, Cat# TL713307 V) were used in PC12 cells following the manufacturer’s instructions. CypD knockdown was confirmed in PC12 cells at 72 hours after lentiviral transfection using western blotting (Yang et al., 2018b; Figure 1A).
Measurement of mitochondrial function
To mimic ICH in PC12 cells, OH was added to the PC12 cell culture medium at a concentration of 10 μM for 12 hours. CsA was added (5 μM) at the same time. After 12 hours, the function of mitochondria in the PC12 cells in each group (Figure 1A) was examined as described in a previous study (Sarkar et al., 2020). Mitochondrial membrane potential (ΔΨm) were quantified in live cells using confocal microscopy. Mitochondria were labeled with MitoTracker®Green (Cat# 9074, Cell Signaling Technology, Boston, MA, USA), and nuclei were stained using Hoechst 33342 (Cat# 14533, Sigma, St. Louis, MO, USA). We used 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, 0.5 μg/mL, Beyotime, Shanghai, China, Cat# C2006) and tetramethylrhodamine methyl ester (TMRM; 30 nM; Cat# T668, Invitrogen, Carlsbad, CA, USA) to measure ΔΨm. JC-1poly emits red fluorescence and labels mitochondria with high ΔΨm, and JC-1mono emits green fluorescence and labels mitochondria with low ΔΨm. The changes in ΔΨm are expressed as changes in the JC-1poly (red)/JC-1mono (green) fluorescence ratio. A decreased TMRM signal indicates a low ΔΨm.
Electron microscopy imaging
To examine the microstructure of axons and mitochondria, transmission electron microscopy was performed at 24 hours after surgery as previously described (Yang et al., 2020). Briefly, mice were perfused with 2.5% glutaraldehyde/2% paraformaldehyde buffer and small pieces (1 mm3 cube) around the hematoma were collected. The pieces were washed in 0.1 M cacodylate buffer, postfixed with 1% osmium tetroxide (OsO4)/1.5% potassium ferrocyanide (KFeCN6) for 1 hour, and subsequently dehydrated in serial grades of alcohol. The samples were embedded in TAAB Epon and polymerized at 60°C for 48 hours. The samples were sectioned (60 nm thick) using an ultramicrotome (LKB-V, LKB Produkter AB, Bromma, Sweden) and examined using a transmission electron microscope (TECNAI10, Philips, Eindhoven, The Netherlands). Low magnification views were used to find the perihematomal areas, and the regions of interest were recorded with an AMT 2k CCD camera. The WM was evaluated by analyzing axonal diameters, g-ratios (axon diameter/axon-plus-myelin diameter), and mitochondrial diameter using ImageJ (V1.8.0.112; National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012). Six mice in each group were examined.
Immunofluorescence staining
We used immunofluorescence staining to examine the morphology of axons and mitochondria. The mice were anesthetized and then perfused. Cryosections of brain with hematoma were made and blocked with 3% bovine serum albumin (Cat# ST025, Beyotime Biotechnology, Shanghai, China) for 1 hour. The sections were then incubated with the following primary antibodies overnight at 4°C: goat anti-myelin basic protein (1:1000, Abcam, Cambridge, UK, Cat# ab106583, RRID: AB_10859573) and rabbit anti-CypD (1:200, Cat# ab155979; Abcam). After washing in phosphate buffer saline for three times, the sections were incubated with donkey anti-goat Alexa Fluor® 555 (1:500, Abcam, Cat# ab150134, RRID: AB_2715537) or goat anti-rabbit Alexa Fluor® 555 (1:500, Abcam, Cat# ab150078, RRID: AB_2722519) at 25 ± 1°C for 2 hours. Finally, the sections were mounted with an anti-fluorescence quencher (Santa Cruz Biotechnology, Dallas, Texas, USA, Cat# sc-24941, RRID: AB_10189288). Images were acquired by confocal microscopy (Carl Zeiss, Weimar, Germany) and examined using Zen 2011 software. There were six mice in each group, and five randomly selected microscopic fields were examined at 10× and 40× magnification (ImageJ) in a double-blind manner.
Western blot assay
The brain tissues around the hematoma were collected using an inverted microscope (RWD Life Science) after mice were anesthetized. Brain tissues and treated PC12 cells were lysed in precooled radioimmunoprecipitation assay buffer (Sigma, Cat# R0278) supplemented with a protease inhibitor cocktail as described in a previous study (Yang et al., 2018a). After quantifying protein concentration, 20 μg of protein was loaded with loading buffer was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA, USA, Cat# M1660023) and transferred onto polyvinylidene fluoride membranes (Bio-Rad, Cat# 1620184). The membranes were blocked in 3% bovine serum albumin for 2 hours. The membranes were incubated with rabbit anti-CypD (1:1000, Abcam, Cat# ab155979) and mouse anti-voltage-dependent anion channel (VDAC, 1:1000, Abcam, Cat# ab14734, RRID: AB_443084) antibodies overnight at 4°C. After washing in TBST buffer (Tris-buffered + saline + Tween) several times, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody (1:1000, Abcam, Cat# ab6721, RRID: AB_955447) or rabbit anti-mouse antibody (1:1000, Abcam, Cat# ab6728, RRID: AB_955440) at room temperature for 1 hour. Finally, the membranes were processed using WesternBright ECL Kits (Advansta, Menlo Park, CA, USA) and imaged by a ChemiDoc™ XRS C imaging system (Bio-Rad). Optical density of bands was evaluated using Image Lab™ software (Bio-Rad).
Assessment of mPTP opening
Calcein acetoxymethyl ester (calcein-AM) and CoCl2 (Hausenloy et al., 2004) were used to test the opening of mPTP in PC12 cells. Cells were incubated with 1 μM calcein-AM for 30 minutes at 37°C to label calcein. Next, 1 mM CoCl2 was added to the culture medium to quench cytosolic calcein and selectively label the mitochondria. Finally, the PC12 cells were treated with OH and CsA. After 30 minutes, calcein fluorescence was detected using confocal microscopy (Carl Zeiss, Weimar, Germany) (emission at 488 nm and detection at 505 nm). Reduction in the mitochondrial calcein signal indicates mPTP opening. Calcein fluorescence measurement was repeated in six separate experiments per treatment group.
ATP level analysis
ATP in the tissue around the hematoma was measured using an ATP assay kit (Abcam, Cat# ab83355) following a previously described protocol (Tennessen et al., 2014). Briefly, brain tissues around the hematoma were washed in cold phosphate buffer saline and homogenized with a mortar. After centrifugation, the collected supernatants were mixed with assay buffer and ATP reaction mix. The solution was loaded into a dark 96-well plate and incubated for 30 minutes. The plate was read using a microplate reader (Shenzhen Reagen Technology, Shenzhen, China). The intracellular ATP concentration was estimated by the mean optical density using a standard curve.
Retrograde tracking of the CST
Briefly, mice were anesthetized, dehaired, and placed in the prone position on a stereotaxic frame. A midline incision was made over the lumbar vertebrae and the lumbar vertebrae were fixed with a spinal cord fixator (RWD Life Science, Shenzhen, China, Cat# 68097). The lumbar spinal cord (L2–L4) was exposed by removing the spinal process. For lumbar spinal cord retrograde tracking of the CST, the retro-CAG-GFP plasmid (BrainVTA Technology, Wuhan, China) was packaged using adeno-associated virus (AAV) (titer was adjusted to 1 × 1012 copies/mL for injection; BrainVTA Technology) and injected into the spinal cord (Chen et al., 2018a; Yang et al., 2022) using a pulled glass pipette and a Micro4 injection pump controller (World Precision Instruments, Sarasota, FL, USA). The injection sites were 0.2 mm lateral to the midline and separated by 1 mm. At each injection site, one injection was performed with a depth of 0.6 mm following the spinal cord atlas (Anderson et al., 2009) (three injections on the contralateral side, 100 nL per injection, 1 nL/s). The labeled cortical neurons were counted and analyzed 3 weeks after injection, and the groups were compared.
Behavioral analysis
Beam walking and ladder rung walking tasks were used in this study following the procedure described previously (Yang et al., 2019, 2022). These tasks are used to assess the capacity of precise paw placement associated with the CST. Beam walking was performed on a narrow beam (0.6 cm wide, 120 cm long, and 60 cm high). The horizontal ladder rung walking test was performed using an apparatus (100 cm long, 19 cm high, and 10 cm wide) with 100 metal rungs (3 mm diameter). Metal rungs were randomly inserted on bilateral clear Plexiglas walls to create a floor with a minimum distance of 1 cm and a maximum distance of 3 cm between the rungs. During training and assessments, the mice were recorded by a video camera, and paws slipping down the horizontal surface of the beam or ladder were identified as foot faults. The number of contralateral forelimb and hindlimb foot faults within 50 steps were counted and analyzed after ICH.
The following two exclusion criteria were used to exclude mice from the analysis of behavioral data. In the evaluation before ICH surgery, mice that slipped more than 10 times per 50 steps in beam walking and ladder rung walking were excluded because they were unable to learn the behavioral tasks. In the evaluation after ICH, mice that walked fewer than 50 steps were excluded because they failed to complete these behavioral tests.
Statistical analysis
No statistical methods were used to predetermine sample sizes; however, our sample sizes were similar to those reported in a previous publication (Yang et al., 2022). The evaluator was blind to the grouping. All data were analyzed using SPSS 19.0 (IBM, Armonk, NY, USA) and GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, USA, www.graphpad.com) statistical software. Two-tailed Student’s t-test was used for comparisons between two groups. One-way analysis of variance followed by Tukey’s post hoc test was used for comparisons of more than two groups. Error bars in all figures represent the mean ± standard error of the mean (SEM). A P value < 0.05 was considered statistically significant.
Results
Morphological changes in the WM around the hematoma after ICH
To better visualize morphological changes in the axons after ICH, we used Thy1-YFP mice, in which layer 5 pyramidal cells in the cerebral cortex and their descending axons were labeled with bright green fluorescence (Porrero et al., 2010; Guo et al., 2015). Consistent with our previous results (Yang et al., 2022), we found a large number of degenerated axons around the hematoma, and some axons degenerated into retraction bulbs (RBs) that had round or oval tips at 24 hours after ICH (Figure 2A). In addition, the diameters of myelin were increased and myelin sheaths (detected by staining for myelin basic protein) were drastically compressed (Figure 2A). A total of 153.6 ± 10.1 degenerated axons per mm2 were observed around the hematoma in the ICH group (Figure 2B). The diameters of normal axons in the Sham group were less than 2 μm, while the distribution of axonal diameters in the ICH group was 0–2 μm (9.7%), 2–4 μm (42.7%), 4–6 μm (36.4%), and > 6 μm (11.2%) (Figure 2C). Notably, RBs are the typical pathomorphological feature of degenerated axons after ICH. Moreover, the bright YFP fluorescence made the diameters of normal and degenerated axons (including RBs) appear much larger than the real diameters in electron microscopy images (Figure 3A).
Figure 2.
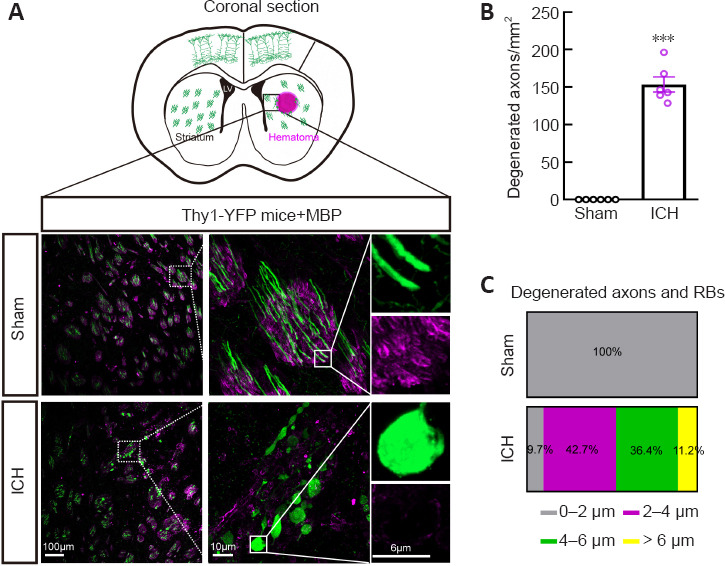
Severe axonal injury around the hematoma at 24 hours after ICH.
(A) Schematic diagram and representative images of the white matter of mice in each group. No degenerated axons were observed in the Sham group, but there were a large number of degenerated axons around the hematoma in the ICH group. MBP (purple) indicates myelin sheaths. Scale bars: 100 μm (left), 10 μm (middle), 6 μm (right). (B) Number of degenerated axons per mm2 around the hematoma in each group. Data are shown as the mean ± SEM (n = 6 animals for each group). ***P < 0.001, vs. Sham group (two-tailed Student’s t-test). (C) Distribution of diameter of degenerated axons in each group. ICH: Intracerebral hemorrhage; MBP: myelin basic protein.
Figure 3.
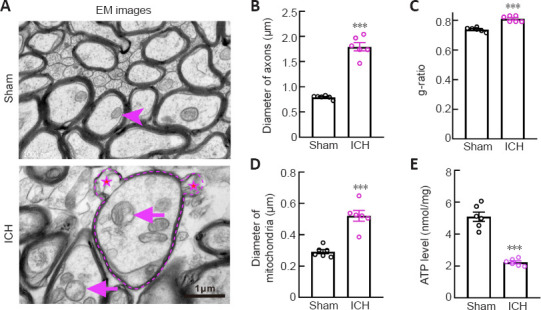
Changes in mitochondria in axons after ICH.
(A) Representative EM images of normal and degenerated axons and mitochondria morphology in the Sham group and at 24 hours after ICH. The diameters of axons and mitochondria were markly increased in the ICH group. Arrowhead indicates normal mitochondria; star indicates broken myelin sheath; arrows indicate swollen mitochondria. Scale bar: 1 μm. (B, C) Diameters of axons (B) and g-ratios of the white matter (C) around the hematoma in each group. (D) Quantification of the diameters of mitochondria in each group. (E) ATP levels in tissue around the hematoma in each group. For data collection, three sections of each mouse and three visual fields around the hematoma of each section were observed. At least 50 degenerated axons and swollen mitochondria were measured for each mouse. Data are shown as the mean ± SEM (n = 6 animals for each group). ***P < 0.001, vs. Sham group (two-tailed Student’s t-test). ATP: Adenosine triphosphate; EM: electron microscopy; ICH: intracerebral hemorrhage.
Impaired mitochondria in degenerated axons after ICH in vivo and in vitro
Electron microscopy images showed that degenerated axons were marked by significantly enlarged diameters (P < 0.001; Figure 3A and B). Compared with axons in the Sham group, axons with hypomyelination showed an increased g-ratio in the ICH group (P < 0.001; Figure 3C). Notably, myelin sheaths around swollen axons remained relatively intact (Figure 3A). However, similar to the myelin basic protein staining in Figure 2A, we observed thin, or partially broken, myelin sheaths around the swollen axons (Figure 3A). These results were quite different from the unconsolidated status of myelin sheaths observed several days after ICH (Xia et al., 2019; Yang et al., 2020). These phenomena suggested that axons were more vulnerable than their myelin sheath at the acute phase after ICH.
To examine the morphology and function of mitochondria in the Sham and ICH groups, we first measured the mitochondrial diameter using coronal electron microscopy sections and found that the diameter was significantly increased in the ICH group (P < 0.001; Figure 3A and D). We also collected tissues around the hematoma to assess the ATP level and found that the ATP level in the ICH group was significantly lower than that in the Sham group (P < 0.001; Figure 3E).
Our previous study showed that OH, a neurotoxic hemolysate after ICH, caused axonal degeneration of primary cultured cortical neurons (Yang et al., 2018b). Therefore, to mimic the ICH model, we injected OH into the striatum. Axonal injury was found around the injection area (Additional Figure 1A (525.3KB, tif) and B (525.3KB, tif) ). To investigate the underlying mechanisms of how mitochondria might be involved in the acute phase of ICH, we used OH to treat PC12 cells. ΔΨm is a key indicator of mitochondrial activity because it reflects the processes of electron transport and oxidative phosphorylation, the driving forces behind ATP production (Zorova et al., 2018). We performed live-imaging of ΔΨm using TMRM and JC-1. We found that JC-1mono was increased and the JC-1poly/JC-1mono fluorescence ratio was significantly decreased in PC12 cell axons in the OH group (P < 0.001; Figure 4A and B). Moreover, the fluorescence density and absorbance of TMRM in the OH group were significantly lower than those in the Control group (P < 0.001; Figure 4C and D). The JC-1 and TMRM results indicated a marked decrease in ΔΨm in PC12 cells after OH treatment.
Figure 4.
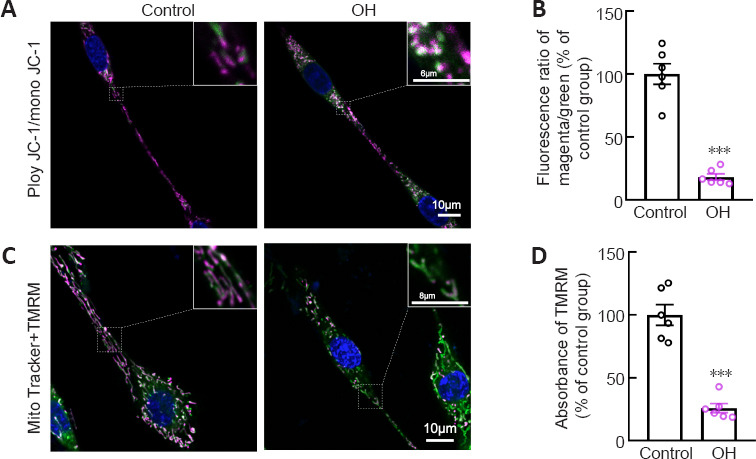
Mitochondria impairment in PC12 cells after OH treatment.
(A) Representative images showing JC-1poly (magenta), JC-1mono (green,) and Hoechst (blue) staining for mitochondrial membrane potential measurement and nuclei in each group. The green fluorescence of mono JC-1 in mitochondria was increased in PC12 cell axons in the OH group compared with that in the Control group. (B) Quantification of the fluorescence ratio of JC-1poly/JC-1mono in each group. (C) Representative pictures showing TMRM (magenta), MitoTracker green (green) and Hoechst (blue) staining for mitochondrial membrane potential measurement, mitochondrial structure, and nuclei in each group. TMRM was significantly decreased in mitochondria in the OH group compared with that in the Control group. Scale bar: 10 μm, 6 μm (enlarged part in A), 8 μm (enlarged part in C). (D) Quantification of the fluorescence absorbance of TMRM in each group. The fluorescence intensity was normalized to that in the Control group. Data are shown as the mean ± SEM (n = 6 independent cell cultures). ***P < 0.001, vs. Control group (two-tailed Student’s t-test). JC1: 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; OH: oxyhemoglobin; TMRM: tetramethylrhodamine methyl ester.
CypD upregulation in degenerated axons after ICH
CypD is the mitochondrial cyclophilin isoform in the peptidylprolyl cis-trans isomerase cyclophilin chaperone family. Recent studies have demonstrated that CypD is the essential component of mPTP and regulates the mPTP opening (Kroemer et al., 2007; Bock and Tait, 2020). Overexpression of CypD mediates mitochondrial dysfunction and promotes the opening of the mPTP in various neurological diseases (Schinzel et al., 2005; Sullivan et al., 2005; Du et al., 2008; Warne et al., 2016; Chen et al., 2018b). Our immunofluorescent staining and western blot results showed that the expression of CypD was increased around the hematoma at 12 and 24 hours after ICH (12 hours: P < 0.05, 24 hours: P < 0.01; F(2, 12) = 24.37; Figure 5A–C). Using Thy1-YFP mice, we found that CypD was upregulated especially in degenerated axons (Figure 5B).
Figure 5.
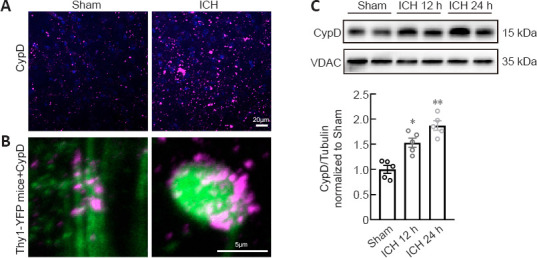
CypD upregulation in degenerated axons.
(A) Representative immunofluorescence micrograph of CypD (magenta) around the hematoma in each group. The expression of CypD was much higher in the ICH group compared with that in the Sham group. (B) Representative immunofluorescence micrograph of CypD (magenta) in normal and degenerated axons (green) around the hematomas in Thy1-YFP mice in each group. The expression of CypD was much higher in the degenerated axons in the ICH group compared with that in the normal axons in the Sham group. Scale bar: 20 μm in A, 5 μm in B. (C) Representative western blot and quantification of CypD in tissues around the hematoma 12 and 24 hours after ICH. VDAC is the internal reference. Data are shown as the mean ± SEM (n = 5 animals for each group). *P < 0.05, **P < 0.01, vs. Sham group (one-way analysis of variance followed by Tukey’s post hoc test). CypD: Cyclophilin D; ICH: intracerebral hemorrhage; VDAC: voltage-dependent anion channel.
CypD deficiency and CsA treatment protect against OH-induced mPTP opening and mitochondrial injury in PC12 cells
To further confirm the CypD activation-induced mPTP opening-mediated mitochondrial injury, pharmacological and genetic inhibition of CypD was performed using CsA (Crompton et al., 1998) and CypD shRNA, respectively, in PC12 cells.
We next used calcein-AM to assess mPTP opening in PC12 cells. Calcein fluorescence was significantly decreased in the OH + scrambled shRNA group compared with that in the scrambled shRNA group (P < 0.001, F(4, 25) = 61.82; Figure 6A and E). Notably, the calcein fluorescence in the OH + CypD shRNA and OH + scrambled shRNA + CsA groups was increased compared with that in the OH + scrambled shRNA group (P < 0.001; Figure 6A and E). These results proved the opening of mPTP after OH treatment and further showed that the mPTP opening could be reversed by CypD knockdown or CsA treatment.
Figure 6.
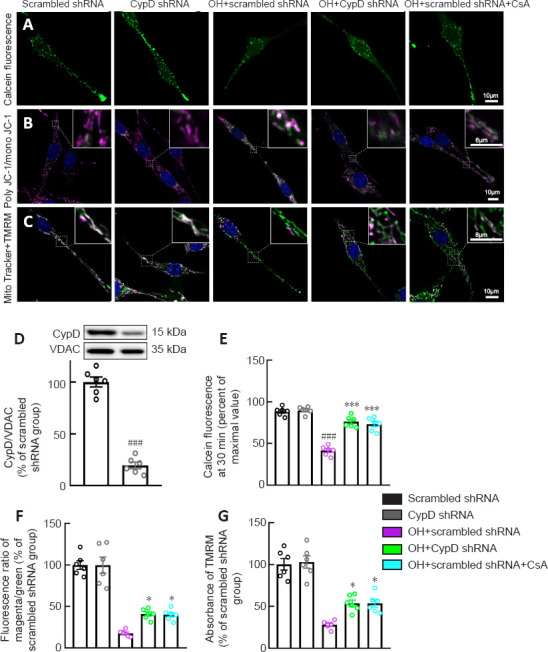
CypD deficiency and CsA treatment protect against OH-induced mitochondrial injury in vitro.
(A) Immunofluorescence images showing mitochondrial calcein fluorescence (green in PC12 cells loaded with calcein-AM and CoCl2 in each group). The calcein fluorescence in the OH + CypD shRNA and OH + scrambled shRNA + CsA groups was increased compared with that in the OH + scrambled shRNA group. (B) Representative immunofluorescence images showing JC-1poly (magenta), JC-1mono (green) and Hoechst (blue) staining for mitochondrial membrane potential measurement and nuclei in each group. The JC-1poly/JC-1mono fluorescence ratio in the OH + CypD shRNA and OH + scrambled shRNA + CsA groups were significantly increased compared with that in the OH + scrambled shRNA group. (C) Representative immunofluorescence images showing TMRM (magenta), MitoTracker green (green) and Hoechst (blue) staining for mitochondrial membrane potential measurement, mitochondrial structure, and nuclei in each group. The fluorescent absorbance of TMRM in the OH + CypD shRNA and OH + scrambled shRNA + CsA groups were significantly increased compared with those in the OH + scrambled shRNA group. Scale bar: 10 μm, 6 μm (enlarged part in B), 8 μm (enlarged part in C). (D) Representative western blot and quantification of CypD and VDAC in PC12 cells in the Scrambled shRNA and CypD shRNA groups. (E) Quantification of calcein fluorescence at 30 minutes in each group. (F) Quantification of the fluorescence ratio of JC-1poly/JC-1mono in each group. (G) Quantification of the fluorescence absorbance of TMRM in each group. Data are shown as the mean ± SEM (n = 6 independent cell cultures). ###P < 0.001, vs. scrambled shRNA group; *P < 0.05, ***P < 0.001, vs. OH + scrambled shRNA group (one-way analysis of variance followed by Tukey’s post hoc test). CsA: Cyclosporin A; CypD: cyclophilin D; JC1: 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide; OH: oxyhemoglobin; TMRM: tetramethylrhodamine methyl ester; VDAC: voltage-dependent anion channel.
The collapse of the ΔΨm is an early event of mitochondrial dysfunction and leads to the opening of the mPTP (Springer et al., 2018). TMRM and JC-1 were used to measure ΔΨm. We found that the JC-1poly/JC-1mono fluorescence ratio (P < 0.05, F(4, 25) = 51.59) and the fluorescent absorbance of TMRM (P < 0.05, F(4, 25) = 37.79) in the OH + CypD shRNA and OH + scrambled shRNA + CsA groups were significantly increased compared with those in the OH + scrambled shRNA group (Figure 6B, C, F and G). Indeed, the expression of CypD was significantly decreased at 72 hours after lentiviral transfection of CypD shRNA (P < 0.001; Figure 6D). These results suggest that mPTP inhibition caused by CypD knockdown or CsA treatment could protect against decreased mitochondrial ΔΨm in PC12 cells after OH intervention.
CypD deficiency and CsA treatment alleviate axonal degeneration after ICH
We crossed Thy1-YFP with CypD–/– lines to generate Thy1-YFP::CypD–/– mice. Thy1-YFP::CypD–/– mice, Thy1-YFP::CypD+/– mice, and Thy1-YFP::WT mice treated with the CypD inhibitor CsA were used to assess the involvement of CypD and mPTP opening in axonal degeneration after ICH. Notably, the number of degenerated axons (F(4, 25) = 64.73; Figure 7A–C) around the hematoma (F(4, 25) = 89.58; Additional Figure 1A (525.3KB, tif) and B (525.3KB, tif) ) was significantly decreased in the ICHCypD–/–, ICHCypD+/–, and ICHWT + CsA groups compared with that in the ICHWT group. We next used electron microscopy to investigate the pathomorphological changes in axons and mitochondria. We found that the membrane structure of mitochondria was more integrated in the ICHCypD–/– and ICHWT + CsA groups (Figure 7B). The mitochondrial (P < 0.001, F(4, 25) = 58.50; Figure 7D) and axonal diameters (P < 0.01, F(4, 25) = 42.89; Figure 7E) in the ICHCypD–/– and ICHWT + CsA groups were smaller than those in the ICHWT group. The WM in the ICHCypD–/– and ICHWT + CsA groups showed a decreased g-ratio compared with that in the ICHWT group (P < 0.01, F(4, 25) = 21.26; Figure 7F). In addition, the ATP level around the hematoma was significantly increased in the ICHCypD–/– (P < 0.001, F(4, 25) = 22.12) and ICHWT + CsA (P < 0.01; Figure 7G) groups compared with that in the ICHWT group, suggesting that inhibiting the opening of the mPTP led to significant alleviation of axonal swelling and degeneration after ICH.
Figure 7.
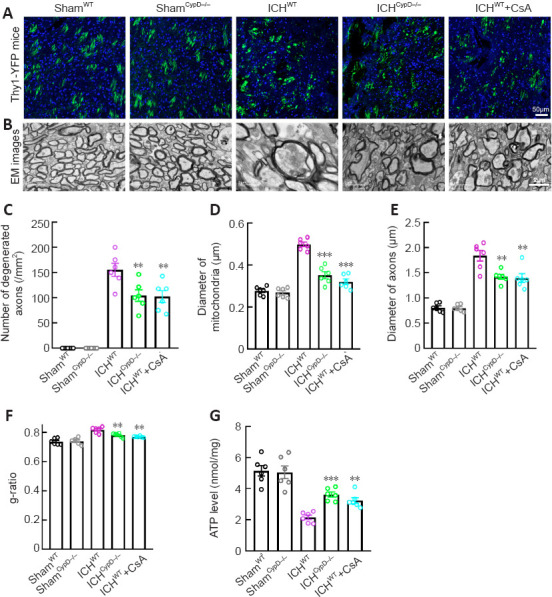
Effects of CypD deficiency and CsA treatment on axonal injury after ICH.
(A) Representative immunofluorescence micrographs of normal and degenerated axons in each group. (B) EM images of normal and degenerated axons and normal and swollen mitochondria morphology in each group. The mitochondrial and axonal diameters in the ICHCypD–/– and ICHWT + CsA groups were smaller than those in the ICHWT group. Scale bars: 50 μm in A, 2 μm in B. (C) Number of degenerated axons per mm2 around the hematoma in each group. The number of degenerated axons around the hematoma was significantly decreased in the ICHCypD–/– and ICHWT + CsA groups compared with that in the ICHWT group. (D) Quantification of the diameter of mitochondria in each group. (E, F) The axonal diameters (E) and mean g-ratio of the WM (F) around the hematoma in each group. (G) Quantification of ATP levels in tissue around the hematoma in each group. For data collection, three sections of each mouse and three visual fields around the hematoma of each section were observed. At least 50 degenerated axons and swollen mitochondria were measured for each mouse. Data are shown as the mean ± SEM (n = 6 animals for each group). **P < 0.01, ***P < 0.001, vs. ICHWT group (one-way analysis of variance followed by Tukey’s post hoc test). CsA: Cyclosporin A; CypD: cyclophilin D; EM: electron microscopy; ICH: intracerebral hemorrhage; WT: wild type.
CypD deficiency and CsA treatment protect CST integrity and motor function after ICH
Motor outcome after stroke is dependent on the integrity of the CST (Jang, 2010). To assess the integrity of the CST and CST-associated fine motor function after ICH, viral retrograde tracking (AAV2/Retro-CAG-GFP) from the lumbar spinal cord and fine motor behavioral tests were performed. The number of retrogradely labeled GFP-positive corticospinal neurons (Bregma: –0.7 mm) in the ICHCypD–/– (P < 0.01, F(4, 25) = 32.73) and ICHWT + CsA (P < 0.05; Figure 8A and B) groups was greater than that in the ICHWT group. The slip rates in the beam walking (F(4, 35) = 121.6) and irregular ladder walking tests (F(4, 35) = 48.97) were decreased in the ICHCypD–/– and ICHWT + CsA groups at 3 days after ICH compared with those in the ICHWT group (Figure 8C and D). These results indicate that inhibition of CypD facilitated the integrity of the CST and promoted functional recovery after ICH.
Figure 8.
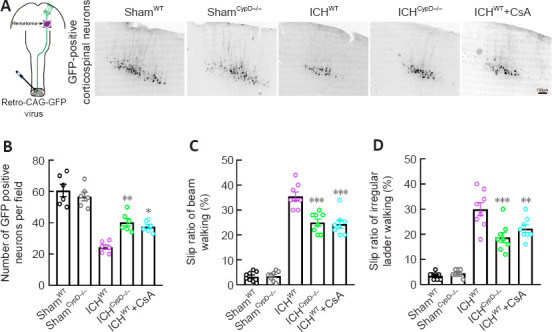
CypD deficiency and CsA treatment protect the CST and alleviate motor dysfunction after ICH.
(A) Diagram of retrograde tracking of the CST (left) and GFP-positive corticospinal neurons (right, Bregma: –0.7 mm) in each group. The number of retrograde labeled GFP-positive corticospinal neurons in the ICHCypD–/– and ICHWT + CsA groups was greater than that in the ICHWT group. Scale bar: 100 μm. (B) Number of labeled corticospinal neurons in each group. (C, D) The slip ratio of the contralateral limbs in beam walking (C) and ladder rung walking (D) at 3 days post-ICH in each group. Data are shown as the mean ± SEM (n = 6–8 animals for each group). *P < 0.05, **P < 0.01, ***P < 0.001, vs. ICHWT group (one-way analysis of variance followed by Tukey’s post hoc test). CsA: Cyclosporin A; CST: corticospinal tract; CypD: cyclophilin D; GFP: green fluorescent protein; ICH: intracerebral hemorrhage; WT: wild type.
Discussion
WMI contributes to neurological deficits after ICH (Zuo et al., 2017; Jiang et al., 2019). The CST is the most important top-down motor control tract in humans. Recent studies showed that the CST, as detected by diffusion tensor imaging, could be used to accurately predict the motor outcomes at the acute stage after ICH (DeVetten et al., 2010; Koyama et al., 2012; Puig et al., 2019). This is beneficial for family counseling and patient selection for rehabilitation or neuroprotective trials. In our previous studies, we discovered that microtubule disassembly led to axonal injury after ICH. Moreover, mitochondria are preferentially bundled with acetylated α-tubulin within axons (Yang et al., 2022). Overexpression of acetylated α-tubulin alleviates microtubule disassembly, axonal injury, and mitochondrial swelling in degenerated axons by enhancing the axonal transport of mitochondria after ICH, indicating that mitochondrial swelling might also be a potential therapeutic target for axonal degeneration after ICH (Yang et al., 2022). Mechanistically, this work showed that CypD activation- and mPTP opening-induced mitochondrial impairment triggered axonal degeneration in the WM. Furthermore, genetic or pharmacological inhibition of CypD attenuated mitochondrial impairment and alleviated axonal degeneration after ICH.
Direct compression by hematoma is proposed as the physical mechanism for early brain injury after ICH (Jiang et al., 2019). Recent studies also found that early hemolysis occurs in the hematoma within 24 hours after ICH in rodents and humans, which might be the triggering factor that contributes to the development of early perihematomal edema, oxidative stress, neuronal loss, and inflammation. Furthermore, early compression by hematoma also leads to red blood cell breakdown (Liu et al., 2019; Wang et al., 2021). Thus, to verify the damage effects of early hemolysis on axons and mitochondria, we used OH, a neurotoxic hemolysate, to treat primary cultured cortical neurons and for injections into the striatum. Although the OH-treated model could not mimic the compression effect of hematoma, it will help further our understanding of the possible mechanism of brain injury after ICH.
A better understanding of the characteristics of WMI at the acute stage following ICH will contribute to the discovery of early therapeutic targets to avoid a worse prognosis (Cui et al., 2021). WMI is characterized by axonal degeneration, demyelination, and oligodendrocyte loss, which frequently occur within 3 days of the onset of ICH (Tao et al., 2016; Zhuo et al., 2016; Xia et al., 2019). Interestingly, instead of the unconsolidated demyelination that was found several days after ICH, we found that the myelin sheaths remained relatively intact 24 hours after ICH (Xia et al., 2019; Yang et al., 2020). This phenomenon suggested that axons were more vulnerable than myelin sheaths at the acute phase after ICH, and therefore inhibiting axonal swelling at an early stage might alleviate demyelination and subsequent severe motor dysfunction.
A previous study revealed that intra-axonal Ca2+ overload triggers the initial disintegration of the axoplasm and axonal degeneration (George et al., 1995). Furthermore, one study reported that a very early Ca2+ increase in axonal mitochondria after injury precedes an axoplasmic increase in Ca2+ (Stirling and Stys, 2010). While mitochondria are important regulators of intracellular Ca2+ homeostasis, mitochondrial Ca2+ overloading leads to mitochondrial swelling and ROS generation, which acts as a positive feedback loop for further mitochondrial injury (Halestrap et al., 1997; Brookes et al., 2004). In our study, electron microscopy results revealed the presence of swollen mitochondria along with swollen axons 24 hours after ICH. mPTP opening is traditionally linked to mitochondrial impairment because it leads to the depolarization of ΔΨm, cessation of ATP synthesis, release of Ca2+, generation of ROS, and swelling of the matrix (Kalani et al., 2018). The above evidence strongly suggests that early mitochondrial impairment in degenerated axons is associated with the opening of the mPTP. Several studies have demonstrated that NAD and pyruvate treatment protect against axonal injury, and each has been shown to inhibit the activation of the mPTP (Wang et al., 2005). Thus, the mPTP is a potential therapeutic target for alleviating initial mitochondrial impairment and axonal degeneration.
The mPTP is a nonspecific channel in the inner membrane of mitochondria. CypD, a mitochondrial matrix protein with cis-trans peptidyl-prolyl isomerase activity, is an important component of the mPTP (Kalani et al., 2018). Mechanistically, CypD translocates from the matrix to the inner mitochondrial membrane to promote mPTP opening under oxidative stress conditions, which causes the positive feedback release of large amounts of Ca2+, ROS, and pro-apoptotic proteins from mitochondria (Brookes et al., 2004; Kalani et al., 2018). Previous studies have revealed that the inhibition of mPTP opening by inactivating CypD has therapeutic potential in Alzheimer’s disease (Du et al., 2008), multiple sclerosis (Warne et al., 2016), sciatic nerve injury (Barrientos et al., 2011), traumatic brain injury (Halestrap et al., 1997), and stroke (Schinzel et al., 2005; Yang et al., 2022). Our results showed that CypD was significantly upregulated around the hematoma, especially in degenerated axons and RBs, indicating that the activation of CypD and opening of the mPTP mediated mitochondrial impairment. Inhibition of mPTP opening through genetic knockout of CypD attenuated mitochondrial impairment and axonal degeneration. CsA is a specific inhibitor of CypD (Karch et al., 2019). Although several cases have reported that CsA treatment–induced renal failure and acute hypertension lead to ICH and posterior reversible encephalopathy syndrome in transplant patients (Mori et al., 2000; Loar et al., 2013), some clinical trials and basic studies have shown that CsA is safe and has promising prospects for clinical transformation (Manno et al., 1997). Our study, together with published data, demonstrated that CsA treatment could ameliorate mitochondrial impairment and axonal degeneration. The fact that CsA and CypD knockout protects CST integrity after ICH suggests that mPTP activation is a central mediator of axonal degeneration.
The mPTP is involved in cell death processes such as apoptosis and necrosis (Fayaz et al., 2015), and inhibiting neuronal apoptosis and necrosis alleviates neurological disorders after spinal cord injury, traumatic brain injury, and stroke (Schinzel et al., 2005; Kilbaugh et al., 2011; Chen et al., 2018b; Springer et al., 2018). Therefore, our strategy of targeting CypD-regulated mPTP opening might also play a protective role by inhibiting apoptosis and necrosis after ICH. This possibility will be investigated in our future work.
This study has several limitations. First, we silenced CypD expression using only global genetic and pharmacological methods. Although pharmacological methods are meaningful for clinical transformation, conditional CypD activation and silencing experiments would be helpful to consolidate our findings and conclusions. Second, to exclude the neuroprotective effect of estrogen (Petrovska et al., 2012), we used only adult male mice in this study. Female and older ICH mice also need to be investigated in future studies. Finally, the present work mainly focused on axonal degeneration after ICH. Myelin is also an important structure of WM, and the function of mPTP inhibition in remyelination after ICH should also be explored.
Our results revealed a key mechanism by which CypD- and mPTP activation-induced mitochondrial injury triggers the initial axonal degeneration of WM after ICH. The inhibition of mPTP opening through CsA treatment and CypD knockout attenuated mitochondrial impairment and alleviated WMI and motor dysfunction after ICH. As WMI is increasingly recognized as the most critical aspect of motor dysfunction, our work provides an important therapeutic target for preventing WMI and subsequent functional impairment in the central nervous system.
Additional files:
Additional Figure 1 (525.3KB, tif) : The effect of oxyhemoglobin and expression level of CypD on the extent of axonal injury after ICH.
The effect of oxyhemoglobin and expression level of CypD on the extent of axonal injury after ICH.
(A) Representative immunofluorescence micrographs of axons and retraction bulbs in each group. The number of degenerated axons around the hematoma was increased in the OH treatment and ICHCypD+/+ groups, but significantly decreased in the ICHCypD+/- and ICHCypD-/- groups. Scale bar: 20 μm. (B) Number of degenerated axons per mm2 around the hematoma in each group. Data are shown as the mean ± SEM (n = 6 animals for each group). *P < 0.05, **P < 0.01, vs. ICHCypD+/+ group; #P < 0.05, vs. ICHCypD+/- group (one-way analysis of variance followed by Tukey’s post hoc test). CypD: cyclophilin D; ICH: intracerebral hemorrhage; OH: oxyhemoglobin; WT: wild type.
Additional file 1: Open peer review report 1 (92.4KB, pdf) .
Acknowledgments:
We thank Prof. He-Ping Cheng (Peking University, China) for providing CypD–/–(Ppif–/–) mice and Prof. Tong-Hui Xu (Huazhong University of Science and Technology, China) for providing Thy1-YFP mice.
Footnotes
Funding: This work was supported by the National Natural Science Foundation of China, Nos. 81901267 (to YY), 82001263 (to WXC), 81901193 (to HLZ); a grant from State Key Laboratory of Trauma, Burn and Combined Injury, No. SKLYQ202002 (to YJC); a grant from Wuxi Municipal Health Commission No. 2020ZHYB19 (to YY); and a grant from Wuxi Science and Technology Bureau, No. Y20212045 (to LKY).
Conflicts of interest: The authors declare that they have no competing interests
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open peer reviewer: Wen Hu, New York State Institute for Basic Research in Developmental Disabilities, USA.
P-Reviewer: Hu W; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Yu J, Song LP; T-Editor: Jia Y
References
- 1.Anderson CR, Ashwell KWS, Collewijn H, Conta A, Harvey A, Heise C, Hodgetts S, Holstege G, Kayalioglu G, Keast JR, McHanwell S, McLachlan EM, Paxinos G, Plant G, Scremin O, Sidhu A, Stelzner D, Watson C. The spinal cord:a christopher and dana reeve foundation text and atlas. In: Watson C, Paxinos G, Kayalioglu G, editors. In:The spinal cord. p v. San Diego: Academic Press; 2009. [Google Scholar]
- 2.Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100. doi: 10.1038/s41580-019-0173-8. [DOI] [PubMed] [Google Scholar]
- 4.Bonora M, Patergnani S, Ramaccini D, Morciano G, Pedriali G, Kahsay AE, Bouhamida E, Giorgi C, Wieckowski MR, Pinton P. Physiopathology of the permeability transition pore:molecular mechanisms in human pathology. Biomolecules. 2020;10:998. doi: 10.3390/biom10070998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium ATP and ROS:a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 6.Chamberlain KA, Sheng ZH. Mechanisms for the maintenance and regulation of axonal energy supply. J Neurosci Res. 2019;97:897–913. doi: 10.1002/jnr.24411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen B, Li Y, Yu B, Zhang Z, Brommer B, Williams PR, Liu Y, Hegarty SV, Zhou S, Zhu J, Guo H, Lu Y, Zhang Y, Gu X, He Z. Reactivation of dormant relay pathways in injured spinal cord by KCC2 manipulations. Cell. 2018a;174:521–535. doi: 10.1016/j.cell.2018.06.005. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen ZR, Ma Y, Guo HH, Lu ZD, Jin QH. Therapeutic efficacy of cyclosporin A against spinal cord injury in rats with hyperglycemia. Mol Med Rep. 2018b;17:4369–4375. doi: 10.3892/mmr.2018.8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng CY, Hsu CY, Huang YC, Tsai YH, Hsu HT, Yang WH, Lin HC, Wang TC, Cheng WC, Yang JT, Lee TC, Lee MH. Motor outcome of deep intracerebral haemorrhage in diffusion tensor imaging:comparison of data from different locations along the corticospinal tract. Neurol Res. 2015;37:774–781. doi: 10.1179/1743132815Y.0000000050. [DOI] [PubMed] [Google Scholar]
- 10.Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur J Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- 11.Cui Y, Jin X, Choi JY, Kim BG. Modeling subcortical ischemic white matter injury in rodents:unmet need for a breakthrough in translational research. Neural Regen Res. 2021;16:638–642. doi: 10.4103/1673-5374.295313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeVetten G, Coutts SB, Hill MD, Goyal M, Eesa M, O'Brien B, Demchuk AM, Kirton A MONITOR and VISION study groups. Acute corticospinal tract Wallerian degeneration is associated with stroke outcome. Stroke. 2010;41:751–756. doi: 10.1161/STROKEAHA.109.573287. [DOI] [PubMed] [Google Scholar]
- 13.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fayaz SM, Raj YV, Krishnamurthy RG. CypD:the key to the death door. CNS Neurol Disord Drug Targets. 2015;14:654–663. doi: 10.2174/1871527314666150429113239. [DOI] [PubMed] [Google Scholar]
- 15.George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo L, Xiong H, Kim JI, Wu YW, Lalchandani RR, Cui Y, Shu Y, Xu T, Ding JB. Dynamic rewiring of neural circuits in the motor cortex in mouse models of Parkinson's disease. Nat Neurosci. 2015;18:1299–1309. doi: 10.1038/nn.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halestrap AP, Woodfield KY, Connern CP. Oxidative stress thiol reagents and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 19.Hausenloy D, Wynne A, Duchen M, Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 20.He Z, Jin Y. Intrinsic control of axon regeneration. Neuron. 2016;90:437–451. doi: 10.1016/j.neuron.2016.04.022. [DOI] [PubMed] [Google Scholar]
- 21.Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, Hutson T, Danzi MC, Perry RB, Santos CXC, Kapustin AN, Fleck RA, Del Río JA, Carroll T, Lemmon V, Bixby JL, Shah AM, Fainzilber M, Di Giovanni S. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol. 2018;20:307–319. doi: 10.1038/s41556-018-0039-x. [DOI] [PubMed] [Google Scholar]
- 22.Hilton BJ, Bradke F. Can injured adult CNS axons regenerate by recapitulating development? Development. 2017;144:3417–3429. doi: 10.1242/dev.148312. [DOI] [PubMed] [Google Scholar]
- 23.Jang SH. Prediction of motor outcome for hemiparetic stroke patients using diffusion tensor imaging:A review. NeuroRehabilitation. 2010;27:367–372. doi: 10.3233/NRE-2010-0621. [DOI] [PubMed] [Google Scholar]
- 24.Jiang YB, Wei KY, Zhang XY, Feng H, Hu R. White matter repair and treatment strategy after intracerebral hemorrhage. CNS Neurosci Ther. 2019;25:1113–1125. doi: 10.1111/cns.13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalani K, Yan SF, Yan SS. Mitochondrial permeability transition pore:a potential drug target for neurodegeneration. Drug Discov Today. 2018;23:1983–1989. doi: 10.1016/j.drudis.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, Peixoto PM, Molkentin JD. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv. 2019;5:eaaw4597. doi: 10.1126/sciadv.aaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kilbaugh TJ, Bhandare S, Lorom DH, Saraswati M, Robertson CL, Margulies SS. Cyclosporin A preserves mitochondrial function after traumatic brain injury in the immature rat and piglet. J Neurotrauma. 2011;28:763–774. doi: 10.1089/neu.2010.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim-Han JS, Kopp SJ, Dugan LL, Diringer MN. Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke. 2006;37:2457–2462. doi: 10.1161/01.STR.0000240674.99945.4e. [DOI] [PubMed] [Google Scholar]
- 29.Koyama T, Tsuji M, Miyake H, Ohmura T, Domen K. Motor outcome for patients with acute intracerebral hemorrhage predicted using diffusion tensor imaging:an application of ordinal logistic modeling. J Stroke Cerebrovasc Dis. 2012;21:704–711. doi: 10.1016/j.jstrokecerebrovasdis.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Krafft PR, Rolland WB, Duris K, Lekic T, Campbell A, Tang J, Zhang JH. Modeling intracerebral hemorrhage in mice:injection of autologous blood or bacterial collagenase. J Vis Exp. 2012:e4289. doi: 10.3791/4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 32.Liu R, Li H, Hua Y, Keep RF, Xiao J, Xi G, Huang Y. Early hemolysis within human intracerebral hematomas:an MRI study. Transl Stroke Res. 2019;10:52–56. doi: 10.1007/s12975-018-0630-2. [DOI] [PubMed] [Google Scholar]
- 33.Loar RW, Patterson MC, O'Leary PW, Driscoll DJ, Johnson JN. Posterior reversible encephalopathy syndrome and hemorrhage associated with tacrolimus in a pediatric heart transplantation recipient. Pediatr Transplant. 2013;17:E67–70. doi: 10.1111/petr.12039. [DOI] [PubMed] [Google Scholar]
- 34.Manno EM, Gress DR, Ogilvy CS, Stone CM, Zervas NT. The safety and efficacy of cyclosporine A in the prevention of vasospasm in patients with Fisher grade 3 subarachnoid hemorrhages:a pilot study. Neurosurgery. 1997;40:289–293. doi: 10.1097/00006123-199702000-00010. [DOI] [PubMed] [Google Scholar]
- 35.Mori A, Tanaka J, Kobayashi S, Hashino S, Yamamoto Y, Ota S, Asaka M, Imamura M. Fatal cerebral hemorrhage associated with cyclosporin-A/FK506-related encephalopathy after allogeneic bone marrow transplantation. Ann Hematol. 2000;79:588–592. doi: 10.1007/s002770000192. [DOI] [PubMed] [Google Scholar]
- 36.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, Pearl EJ, et al. The ARRIVE guidelines 2.0:Updated guidelines for reporting animal research. PLoS Biol. 2020;18:e3000410. doi: 10.1371/journal.pbio.3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petrovska S, Dejanova B, Jurisic V. Estrogens:mechanisms of neuroprotective effects. J Physiol Biochem. 2012;68:455–460. doi: 10.1007/s13105-012-0159-x. [DOI] [PubMed] [Google Scholar]
- 38.Porrero C, Rubio-Garrido P, Avendaño C, Clascá F. Mapping of fluorescent protein-expressing neurons and axon pathways in adult and developing Thy1-eYFP-H transgenic mice. Brain Res. 2010;1345:59–72. doi: 10.1016/j.brainres.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 39.Puig J, Blasco G, Terceño M, Daunis IEP, Schlaug G, Hernandez-Perez M, Cuba V, CarbóG , Serena J, Essig M, Figley CR, Nael K, Leiva-Salinas C, Pedraza S, Silva Y. Predicting motor outcome in acute intracerebral hemorrhage. AJNR Am J Neuroradiol. 2019;40:769–775. doi: 10.3174/ajnr.A6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu X, Wang N, Chen W, Qi M, Xue Y, Cheng W. RNF34 overexpression exacerbates neurological deficits and brain injury in a mouse model of intracerebral hemorrhage by potentiating mitochondrial dysfunction-mediated oxidative stress. Sci Rep. 2019;9:16296. doi: 10.1038/s41598-019-52494-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samarasekera N, Fonville A, Lerpiniere C, Farrall AJ, Wardlaw JM, White PM, Smith C, Al-Shahi Salman R. Influence of intracerebral hemorrhage location on incidence characteristics and outcome:population-based study. Stroke. 2015;46:361–368. doi: 10.1161/STROKEAHA.114.007953. [DOI] [PubMed] [Google Scholar]
- 42.Sarkar TJ, Quarta M, Mukherjee S, Colville A, Paine P, Doan L, Tran CM, Chu CR, Horvath S, Qi LS, Bhutani N, Rando TA, Sebastiano V. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat Commun. 2020;11:1545. doi: 10.1038/s41467-020-15174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ:25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Springer JE, Prajapati P, Sullivan PG. Targeting the mitochondrial permeability transition pore in traumatic central nervous system injury. Neural Regen Res. 2018;13:1338–1341. doi: 10.4103/1673-5374.235218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stirling DP, Stys PK. Mechanisms of axonal injury:internodal nanocomplexes and calcium deregulation. Trends Mol Med. 2010;16:160–170. doi: 10.1016/j.molmed.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma:cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- 48.Tao C, Hu X, Li H, You C. White matter injury after intracerebral hemorrhage:pathophysiology and therapeutic strategies. Front Hum Neurosci. 2017;11:422. doi: 10.3389/fnhum.2017.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tao C, Zhang R, Hu X, Song L, Wang C, Gao F, You C. A novel brainstem hemorrhage model by autologous blood infusion in rat:white matter injury magnetic resonance imaging and neurobehavioral features. J Stroke Cerebrovasc Dis. 2016;25:1102–1109. doi: 10.1016/j.jstrokecerebrovasdis.2016.01.025. [DOI] [PubMed] [Google Scholar]
- 50.Tennessen JM, Barry WE, Cox J, Thummel CS. Methods for studying metabolism in Drosophila. Methods. 2014;68:105–115. doi: 10.1016/j.ymeth.2014.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Hameren G, Campbell G, Deck M, Berthelot J, Gautier B, Quintana P, Chrast R, Tricaud N. In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies. Acta Neuropathol Commun. 2019;7:86. doi: 10.1186/s40478-019-0740-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Venkatasubramanian C, Kleinman JT, Fischbein NJ, Olivot JM, Gean AD, Eyngorn I, Snider RW, Mlynash M, Wijman CA. Natural history and prognostic value of corticospinal tract Wallerian degeneration in intracerebral hemorrhage. J Am Heart Assoc. 2013;2:e000090. doi: 10.1161/JAHA.113.000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Villegas R, Martinez NW, Lillo J, Pihan P, Hernandez D, Twiss JL, Court FA. Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J Neurosci. 2014;34:7179–7189. doi: 10.1523/JNEUROSCI.4784-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang M, Xia F, Wan S, Hua Y, Keep RF, Xi G. Role of complement component 3 in early erythrolysis in the hematoma after experimental intracerebral hemorrhage. Stroke. 2021;52:2649–2660. doi: 10.1161/STROKEAHA.121.034372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warne J, Pryce G, Hill JM, Shi X, Lennerås F, Puentes F, Kip M, Hilditch L, Walker P, Simone MI, Chan AW, Towers GJ, Coker AR, Duchen MR, Szabadkai G, Baker D, Selwood DL. Selective inhibition of the mitochondrial permeability transition pore protects against neurodegeneration in experimental multiple sclerosis. J Biol Chem. 2016;291:4356–4373. doi: 10.1074/jbc.M115.700385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia M, Chen W, Wang J, Yin Y, Guo C, Li C, Li M, Tang X, Jia Z, Hu R, Liu X, Feng H. TRPA1 activation-induced myelin degradation plays a key role in motor dysfunction after intracerebral hemorrhage. Front Mol Neurosci. 2019;12:98. doi: 10.3389/fnmol.2019.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu TQ, Lin WZ, Feng YL, Shen FX, Chen J, Wu WW, Zhu XD, Gu L, Fu Y. Leukoaraiosis is associated with clinical symptom severity poor neurological function prognosis and stroke recurrence in mild intracerebral hemorrhage:a prospective multi-center cohort study. Neural Regen Res. 2022;17:819–823. doi: 10.4103/1673-5374.322469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang Y, Zhang K, Zhong J, Wang J, Yu Z, Lei X, Chen X, Quan Y, Xian J, Chen Y, Liu X, Feng H, Tan L. Stably maintained microtubules protect dopamine neurons and alleviate depression-like behavior after intracerebral hemorrhage. Sci Rep. 2018a;8:12647. doi: 10.1038/s41598-018-31056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Y, Zhang X, Ge H, Liu W, Sun E, Ma Y, Zhao H, Li R, Chen W, Yuan J, Chen Q, Chen Y, Liu X, Zhang JH, Hu R, Fan X, Feng H. Epothilone b benefits nigrostriatal pathway recovery by promoting microtubule stabilization after intracerebral hemorrhage. J Am Heart Assoc. 2018b;7:e007626. doi: 10.1161/JAHA.117.007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang Y, Zhang K, Chen X, Wang J, Lei X, Zhong J, Xian J, Quan Y, Lu Y, Huang Q, Chen J, Ge H, Feng H. SVCT2 promotes neural stem/progenitor cells migration through activating CDC42 after ischemic stroke. Front Cell Neurosci. 2019;13:429. doi: 10.3389/fncel.2019.00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, Zhang K, Yin X, Lei X, Chen X, Wang J, Quan Y, Yang L, Jia Z, Chen Q, Xian J, Lu Y, Huang Q, Zhang X, Feng H, Chen T. Quantitative iron neuroimaging can be used to assess the effects of minocycline in an intracerebral hemorrhage minipig model. Transl Stroke Res. 2020;11:503–516. doi: 10.1007/s12975-019-00739-2. [DOI] [PubMed] [Google Scholar]
- 63.Yang Y, Chen X, Feng Z, Cai X, Zhu X, Cao M, Yang L, Chen Y, Wang Y, Feng H. MEC17-induced α-tubulin acetylation restores mitochondrial transport function and alleviates axonal injury after intracerebral hemorrhage in mice. J Neurochem. 2022;160:51–63. doi: 10.1111/jnc.15493. [DOI] [PubMed] [Google Scholar]
- 64.Zhuo F, Qiu G, Xu J, Yang M, Wang K, Liu H, Huang J, Lu W, Liu Q, Xu S, Huang S, Sun S. Both endoplasmic reticulum and mitochondrial pathways are involved in oligodendrocyte apoptosis induced by capsular hemorrhage. Mol Cell Neurosci. 2016;72:64–71. doi: 10.1016/j.mcn.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 65.Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–59. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zuo S, Pan P, Li Q, Chen Y, Feng H. White matter injury and recovery after hypertensive intracerebral hemorrhage. Biomed Res Int. 2017;2017:6138424. doi: 10.1155/2017/6138424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The effect of oxyhemoglobin and expression level of CypD on the extent of axonal injury after ICH.
(A) Representative immunofluorescence micrographs of axons and retraction bulbs in each group. The number of degenerated axons around the hematoma was increased in the OH treatment and ICHCypD+/+ groups, but significantly decreased in the ICHCypD+/- and ICHCypD-/- groups. Scale bar: 20 μm. (B) Number of degenerated axons per mm2 around the hematoma in each group. Data are shown as the mean ± SEM (n = 6 animals for each group). *P < 0.05, **P < 0.01, vs. ICHCypD+/+ group; #P < 0.05, vs. ICHCypD+/- group (one-way analysis of variance followed by Tukey’s post hoc test). CypD: cyclophilin D; ICH: intracerebral hemorrhage; OH: oxyhemoglobin; WT: wild type.