

Abstract
SARS-CoV-2 is the infectious agent responsible for the coronavirus disease since 2019, which is the viral pneumonia pandemic worldwide. The structural knowledge on SARS-CoV-2 is rather limited. These limitations are also applicable to one of the most attractive drug targets of SARS-CoV-2 proteins – namely, main protease Mpro, also known as 3C-like protease (3CLpro). This protein is crucial for the processing of the viral polyproteins and plays crucial roles in interfering viral replication and transcription. In fact, although the crystal structure of this protein with an inhibitor was solved, Mpro conformational dynamics in aqueous solution is usually studied by molecular dynamics simulations without special sampling techniques. We conducted replica exchange molecular dynamics simulations on Mpro in water and report the dynamic structures of Mpro in an aqueous environment including root mean square fluctuations, secondary structure properties, radius of gyration, and end-to-end distances, chemical shift values, intrinsic disorder characteristics of Mpro and its active sites with a set of computational tools. The active sites we found coincide with the currently known sites and include a new interface for interaction with a protein partner.
Keywords: SARS-CoV-2 main protease, Dynamics, Replica exchange MD simulations
Graphical abstract
Highlights
-
•
We conducted extensive replica exchange molecular dynamics (REMD) simulations on Mpro in water, and report the dynamic structures of Mpro in an aqueous environment including its secondary structure properties, radius of gyration, and end-to-end distances.
-
•
We also studied intrinsic disorder characteristics of Mpro and its active sites with a set of computational tools.
-
•
The active sites we found coincide with the currently known sites and also include a new interface for interaction with a protein partner.
Humans and animals are regularly infected by coronaviruses (CoVs). While typically this type of infection has rather mild respiratory symptoms, and most coronaviruses are not dangerous, there are also instances when a CoV infection can cause severe respiratory illness. Prior to the end of 2019, illustrative examples of such severe forms of CoV infection were given by the outbreaks of Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) caused by the SARS-CoV and MERS-CoV infections. Although at the time of their appearance (2003 for SARS and 2012 for MERS) these CoVs were considered as a major threat, their danger was not even close to that posed by the newest representative of the Coronaviridae family, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) causing the coronavirus disease 2019 (COVID-19). This is reflected in the values of the corresponding morbidity and mortality rates. In fact, by the time of its containment in 2003, SARS-CoV spread to 26 countries, where a total of 8,098 people became sick and 774 people died (Chan-Yeung and Xu, 2003). Similarly, MERS-CoV, which appeared initially in Jordan, spread over 24 countries mostly in or near the Arabian Peninsula, but also in Asia, Europe, and America (Subbaram et al., 2017). From June 2012 to January 2020, there were 2,519 confirmed MERS cases, with 866 people dying from the disease (data from WHO). Therefore, MERS-CoV infection is characterized by a case mortality rate of 34.4%, which is 4-fold higher than that of SARS (Ford et al., 2020). Recently, we presented the structural characteristics of MERS-CoV macro domain in water (Akbayrak et al., 2021). However, the culprit of the current COVID-19 pandemic, SARS-CoV-2, is killing a larger number of people each day than SARS and MERS did. Currently, COVID-19 is spreading and affecting every country in the world, and infecting millions of people. Specifically, according to the Worldometer, https://www.worldometers.info/coronavirus/, as of October 4th, 2022, there were 624,083,852 COVID-19 cases in 230 countries.
SARS-CoV-2's RNA genome embraces 29,903 nucleotides and encodes three proteins (spike glycoprotein (S), membrane protein (M), and nucleocapsid protein (N)) and several non-structural proteins (NSPs) (Khailany et al., 2020). A single large replicase gene is responsible for the encoding of the proteins that are at the center of viral replication. The replicase gene encodes the overlapping polyproteins called pp1a and pp1ab that are necessary for viral transcription and replication. The larger replicase polyprotein 1 ab has 7,073 amino acid residues, which contains fifteen non-structural proteins. Nsp1, Nsp2, and Nsp3 are released from polyprotein through proteolytic processing using a viral papain-like proteinase (Nsp3/PL-Pro), while the rest are cleaved by viral 3C-like proteinase, Nsp5/3CLPro or main protease Mpro, uses eleven or more conserved sites for digesting the protein. The digestion is initiated by an autocatalytic cleavage of this enzyme from pp1a and pp1ab. The functional importance of Mpro in the viral life cycle is the main reason for it being an attractive target for antiviral treatment design.
Yang and co-workers used computer-aided drug design (CADD) to create a series of Michael acceptor inhibitors including an inhibitor named N3 that efficiently inhibits various CoV Mpro species from SARS-CoV to SARS-CoV-2. This inhibitor forms a reversible complex with the protease, which in turn is prone to a chemical step that yields the formation of a stable covalent bond (Matthews et al., 1999). Recently, researchers solved a crystal structure of the Mpro from SARS-CoV-2 in a complex with its inhibitor N3, reporting an active peptide fragment (Ala-Val-Leu) that binds specifically to the substrate-binding pocket of SARS-CoV-2 (Jin et al., 2020a). Immediately after its publication, this study raised significant interest from the scientific community, and since then guides some of the efforts towards the design of treatment for COVID-19 infection.
A crystal structure represents only static snapshots of a dynamic system, and therefore does not embrace the impacts of structural dynamics and the bulk water effects on Mpro structure in water. This is a serious problem since protein dynamics can hold some important keys to the protein's structure and function. A solution to this problem can be provided by computational studies employing molecular dynamics (MD) simulations in bulk water environment. However, MD simulation scenarios require ergodic sampling of trajectories characterized by complex energy landscapes that possess minima and energy barriers between varying minima which can be challenging for crossing at ambient temperatures over currently available simulation time-scales. Despite this, replica-exchange molecular dynamics (REMD) simulations aim to enhance the conformational sampling via running independent temperature-dependent replicas and periodically exchanging the coordinates of the temperature-dependent replicas (Coskuner and Uversky, 2017; Coskuner and Wise-Scira, 2013; Coskuner-Weber and Uversky, 2019). Even though various MD simulations without special sampling techniques have been conducted on the SARS-CoV-2 main protease, these simulations did not utilize enhanced sampling algorithms (Suares and Diaz, 2020; Kneller et al., 2020a; Diaz and Suarez, 2021; Komatsu et al., 2020). For instance, the structural flexibility of SARS-CoV-2 main protease was investigated by means of the classical MD simulations without special sampling technique applications using ff14SB version of AMBER parameters for the protein and the TIP3P model for water (Suares and Diaz, 2020). Based on these simulations, the native state configurations of the enzyme and those of its noncovalent complex with a model peptide for mimicking the polyprotein sequence were recognized at the active site. For each configuration, the authors also investigated the monomeric and dimeric states and showed that the domain III is not stable in the monomeric state, whereas in the presence of the peptide substrate, the monomeric protease exhibits a stable interdomain arrangement (Suares and Diaz, 2020). Furthermore, they looked at the catalytic impact of the enzyme dimerization and concluded that the active site flexibility was induced by substrate binding (Suares and Diaz, 2020). Furthermore, in a different study, the structural plasticity of SARS-CoV-2 3CL Mpro active site cavity was investigated at room temperature via X-ray crystallography and MD simulations (Kneller et al., 2020a). In this study, the conformational flexibility of the enzyme active site was detected by the comparisons between the room temperature ligand-free structure and the low temperature ligand-free and inhibitor-bound structures. This analysis indicated that the room-temperature structure of the 3CL Mpro ligand-free form may be a more physiologically relevant structure for performing docking studies (Kneller et al., 2020a). In addition, there have been various docking and drug binding studies on the SARS-CoV-2 main protease (Diaz and Suarez, 2021; Komatsu et al., 2020). For instance, Komatsu et al. performed MD simulations on dimeric SARS-CoV-2 main protease to examin the binding dynamics of small ligands (Komatsu et al., 2020). They used seven HIV inhibitors, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, and tipranavir were utilized as potential lead drugs for studying access to the drug binding sites in the structures of Mpro (Komatsu et al., 2020). The active sites were identified based on the probability calculations and contacts. Results showed differences in the shapes of the binding sites and binding poses of the ligands (Komatsu et al., 2020).
Different from the existing studies, we conduct extensive and computationally expensive REMD simulations and link these to several state-of-the-art structural bioinformatics tools for investigating the structural characteristics of the SARS-CoV-2 Mpro. Specifically, we report herein the root mean square fluctuations, secondary structure element abundances per residue, K-means clustering results, chemical shift values as well as possible binding sites for peptides/proteins in general. Knowledge obtained from these structural analyses may help in designing more efficient COVID treatments including novel small molecule drugs.
1. Methods
All-atom REMD simulations of SARS-CoV-2 main protease were conducted in water with temperatures distributed between 280 K and 320 K using 40 replicas, which were exponentially distributed between these temperatures. For the SARS-CoV-2 main protease, we used the CHARMM36 parameters (Akbayrak et al., 2021). We selected the TIP3P parameters to model the solvent (Akbayrak et al., 2021). We used a water layer of 10 Å for solvating the main protease utilizing a cubic box along with periodic boundary conditions and minimum image convention. We utilized the GROMACS 5.1.4 package to simulate the system. We used the crystal structure determined by Yang and co-workers (PDB ID: 6LU7) due to the higher resolution in their experiments to isolate the initial Mpro structure in our REMD simulations (Jin et al., 2020a). After solvating the structure in water, we initially conducted equilibration simulations using the NVT ensemble for 20 ns and next using the NPT ensemble for additional 20 ns per replica. Simulations were conducted for a total simulation time of 4.0 μs. Exchanges between replicas are attempted every 5 ps with a time step of 2 fs. Following our recent studies (Akbayrak et al., 2021), we use the Langevin dynamics for maintaining the temperature of each replica with a collision frequency of 2 ps−1. Also, we applied the particle mesh Ewald (PME) method for treating the long-range interactions (Akbayrak et al., 2021). We utilized the SHAKE algorithm for constraining the bonds to hydrogen atoms and counterions were added for neutralizing the system (Akbayrak et al., 2021).
We calculated the structural properties of SARS-CoV-2 Mpro from the conformations - obtained after convergence - from the replica closest to 310 K. We computed the contents of the secondary structure elements with the DSSP program linked to our own script (Akbayrak et al., 2021). Moreover, we calculated the end-to-end distance and radius of gyration of SARS-CoV-2 main protease in water. We applied the k-means clustering method to partition the structural observations into clusters, where each analysis belongs to the cluster with the nearest centroid. Therefore, we partitioned the data space onto Voronoi cells such that k-means clustering minimizes using squared Euclidean distances within cluster variances. To relate the ensemble generated in this study to available experimental data, experimental and calculated Cα and Hα chemical shifts were compared by the shifts 4.3 program that uses a database of density functional shifts for more than 2000 peptides and empirical formulas to predict the chemical shift values in Mpro (Xu. and Case, 2001).
We combined and aligned the above atomic-level analysis with residue-level studies that rely on a number of modern bioinformatics tools. We calculated and compared the REMD-derived root mean square fluctuations (RMSF) for each residue of the homodimer main protease in the vicinity of the inhibitor's peptide fragment in water with findings from several popular residue-level intrinsic disorder predictors: PONDR® VLXT (Romero et al., 2001), PONDR® VSL2 (Obradovic et al., 2005; Peng et al., 2006), PONDR® FIT (Xue et al., 2010), and IUPred capable of predicting long and short disordered regions (Dosztányi et al., 2005a, 2005b; Mészáros et al., 2018). We evaluated predisposition of the SARS-CoV-2 Mpro for interaction with proteins and peptides with two complementary methods: HybridPBRpred (Zhang et al., 2020) and PepBCL (Wang et al., 2022). HybridPBRpred predicts protein-binding residues by combining outputs of the two tools: SCRIBER that targets predictions for structured proteins (Zhang and Kurgan, 2019) and DisoRDPbind that focuses on the intrinsically disordered proteins (Peng and J'Kurgan, 2015; Peng et al., 2017). PepBCL is one of the most recent tools that predicts peptide binding residues (Wang et al., 2022). We also evaluate the nucleic acid binding potential of the SARS-CoV-2 Mpro with the DRNApred predictor (Yan and Kurgan, 2017). Combining HybridPBRpred, PepBCL and DRNApred allows us to annotate putative interactions with proteins, peptides, DNA and RNA along the SARS-CoV-2 Mpro sequence. Importantly, these methods generate predictions directly from the protein sequence, without the use of the homology modelling, and were shown to produce accurate results even in the absence of similarity to training proteins (Zhang et al., 2020; Wang et al., 2022; Yan and Kurgan, 2017). This means that they do not merely identify binding residues based on a similar protein complex, but are capable of finding new binding regions.
2. Results and discussion
Fig. 1 presents the selected structures for SARS-CoV-2 Mpro in water that were retrieved from our REMD simulations. The figure shows that the protein possesses noticeable structural flexibility, as evidenced by noticeable difference in spatial organization of these selected structures.
Fig. 1.

Selected conformations of the SARS-CoV-2 Mpro in water from REMD simulations.
Fig. 2 represents the calculated root mean square fluctuation (RMSF) values that represent a range of fluctuations of a dynamical system about an average position for each residue of SARS-CoV-2 Mpro in water measured at several temperatures (280, 290, 300, 310, and 320 K). The figure shows that the degree of this protein per-residue flexibility was minimally affected by changes in temperature within the studied temperature interval. The mean RMSF value for the overall dynamics of Mpro in water at 310 K is 1.52 ± 1.31 Å. The maximum RMSF value of Mpro is 10.86 Å, while the minimum value is 0.55 Å.
Fig. 2.

REMD-obtained structural flexibility of the SARS-CoV-2 Mpro in water with its intrinsic disorder pre-disposition. The figure shows root mean square fluctuations (RMSF) of SARS-CoV-2 Mpro in water by REMD simulations at 280, 290, 300, 310, and 320 K and intrinsic disorder profile generated by PONDR® VLXT, PONDR® VSL2, PONDR® FIT, IUPred_short, and IUPres_long.
Fig. 2, Fig. 3 summarize the residue-level analyses. Fig. 2 shows that the SARS-CoV-2 Mpro is predicted mostly as an ordered protein by the majority of disorder predictors, since only short fragments of the N- and C-terminal regions and a short region in the vicinity of residue 95 have disorder scores exceeding the threshold of 0.5. However, we find several flexible regions in this protein (i.e., regions with disorder scores from 0.2 to 0.25). Fig. 2 shows that the majority of the dynamics features observed in our REMD simulations are correlated with the intrinsic disorder predisposition of the SARS-CoV-2 Mpro. The relation between these two independent results indicates that several peaks in disorder profile serve as envelopes which enclose the local RMSF peaks. Despite, in several regions, the heights of the RMSF peaks noticeably exceed the heights of the corresponding disorder peaks (e.g. residues 60–80 and 210–240). There are also several regions (e.g., residues 25–40 and 150–160), which are classified as ordered, however, still present significant structural variations/fluctuations. This may mean that the structural fluctuations of some regions of SARS-CoV-2 Mpro in water may be related to their intrinsic disorder predispositions, whereas other structural fluctuations of other regions are independent of their intrinsic disorder predisposition. Furthermore, the levels of local intrinsic disorder predisposition do not always scale up with the corresponding levels of structural flexibility.
Fig. 3.

Identification of potential protein/peptide and nucleic acid binding residues in the SARS-CoV-2 Mpro. Panel A. Predisposition of Mpro to interact with proteins and peptides predicted by HybridPBRpred (dark green lines) and PepBCL (light green lines), and with RNA (red lines) and DNA (orange lines) predicted by DRNApred. Putative propensity for binding is shown at the top of the figure while the horizontal bars directly underneath denote the location of the predicted protein binding residues (dark and light green bars). No DNA and RNA binding residues were predicted. The native protein-binding residues associated with the interface of the SARS-CoV-2 Mpro dimer are shown using the dark blue horizontal bar (PDB id: 6lu7). The residues that interact with peptide ligands are color-coded to denote the source complex structures and shown at the bottom of the panel. These annotations were extracted using the BioLip resource from 28 structures of Mpro in complex with peptides (PDB ids: 3atw, 3avz, 3aw0, 6lu7, 7bqy, 6xa4, 6xbg, 6xbh, 6xbi, 6xch, 6xfn, 7c8b, 7kvg, 7cut, 7mgr, 7mgs, 7n6n, 7n89, 7m2p, 7ein, 7s82, 7rnw, 7dvp, 7dvw, 7dvx, 7dvy, 7dw0, and 7dw6). They are grouped by the data of deposition. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
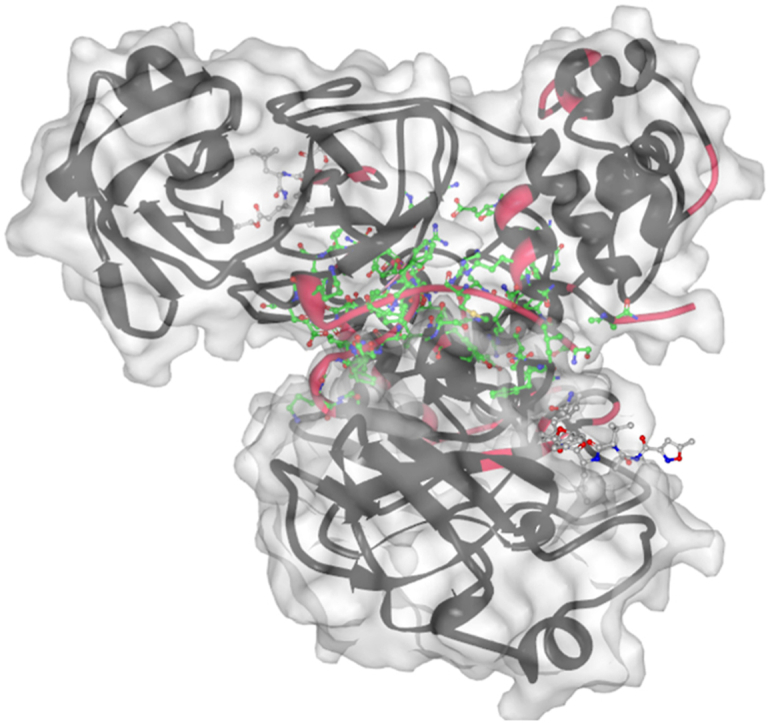
Next, we looked at the predicted protein/peptide and nucleic acid binding sites in the SARS-CoV-2 Mpro. Results of this analysis are summarized in Fig. 3A, showing that the SARS-CoV-2 Mpro contains over 60 protein/peptide binding residues (residues Ser1, Gly2, Arg4-Gly11, Glu14, Gln19, Thr21, Thr24-Leu27, His41, Thr45, Leu46, Met 49, Leu50, Ty54, Leu67, Gln 69, Tyr118, Asn119, Ser123-Tyr126, Lys137-Cys145, His163-Pro168, His172, Asp187-Gln192, Ala285, Leu286, Glu290, Arg298, Gln299, Ser301, Val303, and Thr304). We extract these data from a comprehensive collection of SARS-CoV-2 Mpro structures in complex with various ligands that we collect from PDB and annotate using the BioLip tool (Yang et al., 2013). These PDB structures include 3atw (Akaji et al., 2011), 3avz (Akaji et al., 2011), 3aw0,30 6lu7,31 7bqy (Jin et al., 2020b), 6xa4,32 6xbg (Sacco et al., 2020), 6xbh (Sacco et al., 2020), 6xbi (Sacco et al., 2020), 6xch (Kneller et al., 2020b), 6xfn (Sacco et al., 2020), 7c8b, 7kvg (Noske et al., 2021), 7cut (Wang et al., 2021), 7mgr (MacDonald et al., 2021), 7 mgs (MacDonald et al., 2021), 7n6n (Noske et al., 2021), 7n89,37 7m2p (Li et al., 2021), 7ein (Fu et al., 2021), 7s82, 7rnw (Johansen-Leete et al., 2022), 7dvp (Zhao. et al., 2022), 7dvw (Zhao. et al., 2022), 7dvx (Zhao. et al., 2022), 7dvy (Zhao. et al., 2022), 7dw0,41 and 7dw6 (Zhao. et al., 2022). Using HybridPBRpred and PepBCL we identify 55 putative protein and peptide binding residues (Ser1, Phe2-Lys12, Gln19, Thr24, Asp48, Arg60, His80, Cys85, Pro122, Tyr126, Gln127, Arg131, Phe140-Cys145, Cys160-Met165, Pro168, Thr169, His172, Asp197, Thr198, Arg217, Arg222-Thr224, Tyr239, Pro241-Gln244, His246, Pro252, Phe294, Arg298, Set301, and Thr304-Gln306). Moreover, our analysis with the DRNApred tool suggests that there are no DNA- or RNA-binding residues/regions in this protein. The predictions are in a good agreement with the experimental data, with sensitivity (rate of correct prediction among native binding residues) of 43%, specificity (rate of correct predictions among the non-binding residues) of 88%, precision (rate of correct predictions among predicted binding residues) of 47%, and F1 score (harmonic mean of precision and sensitivity) of 0.45. We note that the current annotations of native binding residues are likely incomplete, which is why we consider the above metrics as relatively good. This is supported by an observation that the native annotations have grown from 45 binding residues (using data from 2011), to 51 residues (data from 2020 and earlier), and finally to 61 residues (using current data); Fig. 3A shows progression of these annotations using the color-coded horizontal lines at the bottom. Fig. 3B annotates these results onto the structure of the dimer complexed with the N3 inhibitor (PDB id: 6lu7), which is the structure used in this analysis. The predictions, which we show in green and yellow, accurately identify binding pocket for the N3 inhibitor and the interface of the dimer. We also identify a novel putative binding region, shown in yellow in Fig. 3B (Arg131, Asp197, Thr198, Tyr239, Pro241-Gln244, His246, and Phe294).
We expect that this newly discovered putative peptide/protein binding site can be used by SARS-CoV-2 Mpro for interaction with yet to be discovered partners. The importance of the discovery of this putative site is difficult to overestimate, as it provides a novel target for small molecules that can affect functionality of SARS-CoV-2 Mpro site. This hypothesis is in line with the aforementioned notion that the current annotations of native binding residues of the SARS-CoV-2 Mpro are likely incomplete, as evidenced by the steady increase in the number of the experimentally validated binding residues of this protein.
Panel B. Visualization of the putative protein/peptide-binding residues predicted by HybridPBRpred and PepBCL (shown in green and yellow) in the tertiary structure of SARS-CoV-2 Mpro dimer complexed with N3 inhibitor (shown in blue). One of the Mpro chains is shown in black while the other is shown using a semi-transparent gray. The putative binding residues in green identify binding pocket for the N3 inhibitor and the interface of the dimer. The putative binding residues in yellow correspond to the new putative binding pocket. The right side of panel B zooms in on the predicted pocket. The images were produced with Mol∗ (Sehnal et al., 2021) using the 6lu7 PDB structure.
Next, we computed the per residue secondary structure propensities of SARS-CoV-2 Mpro in water with dynamics. Fig. 4 illustrates the results of this analysis and shows the calculated α-helix, 310-helix, β-sheet, and turn structure abundances per residue with dynamics. Based on these calculations, we find that seven regions of Mpro adopt an α-helix conformation. These regions are: Ser10-Gly15, Tyr54-Arg60, Thr201-Asn214, Leu227-Tyr237, Gln244-Thr257, Val261-Asn274, and Pro293-Cys300. On the other hand, two regions (Ser46-Asp48 and Asn63-Asn65) in the N-terminal region of Mpro adopt a 310-helix formation. Also, 13 regions located in the N-terminal and mid-domain regions of Mpro form β-sheet structures. These regions are: Met17-Cys22, Thr25-Leu32, Val35-Pro39, Phe66-Ala70, Val73-Leu75, Val77-Gln83, Val86-Val91, Tyr101-Phe103, Gln110-Tyr118, Ser121-Met130, Ser147-Ile152, Cys156-Leu167, and Val171-Thr175. There are 18 regions with turn structure formed in Mpro: Gly23-Thr24, Asp33-Asp34, Arg40-Pro52, Gly71, Asn72, Asn84, Cys85, Asn95-Thr98, Gln107-Gly109, Asn119, Gly120, Pro132-Phe134, Leu141-Gly146, Asp153-Asp155, Pro168-Gly170, Asp176-Gly179, Tyr182-Pro184, Arg217-Leu220, Met276-Thr280, Leu282-Gly283, and Ser301-Val303. Note that there are currently no experimental data (which include nuclear magnetic resonance spectroscopy measurements) on structural dynamics of the SARS-CoV-2 Mpro. Therefore, we cannot compare our findings with the experiments.
Fig. 4.

Calculated per residue propensities for α-helix, 310-helix, β-sheet, and turn secondary structure of the SARS-CoV-2 Mpro in water with dynamics.
We show the k-means clustering algorithm results of the structures of the SARS-CoV-2 Mpro in water in Fig. 5.
Fig. 5.

K-means clustering along with Rg and REE values of the SARS-CoV-2 Mpro in water. five k values were utilized and centroids were found to be located at Rg = 22.54 Å, REE = 20.83 Å (Centroid1), Rg = 22.47 Å, REE = 30.49 Å (Centroid 2), Rg = 22.52 Å, REE = 11.29 Å (Centroid 3), Rg = 22.54 Å, REE = 15.89 Å (Centroid 4), Rg = 22.51 Å, REE = 5.89 Å (Centroid 5).
The average REE value for the SARS-CoV-2 Mpro is 16.01 ± 7.07 Å, with a maximum value of 35.46 Å and a minimum value of 3.93 Å, meaning that, based on this property, SARS-CoV-2 Mpro is extremely flexible. Furthermore, the SARS-CoV-2 Mpro average Rg value is 22.54 Å ± 0.18 Å and varies only between 22 and 24 Å, indicating that the fluctuations in the compactness of Mpro in water are of rather low scale.
To gain further insights into the performance of REMD simulations, we compared NMR chemical shift values for Cα and Hα atoms of Mpro with the experimental data (Cantrelle et al., 2021) (Fig. 6) in water since co-solvents impact the predicted secondary structure and chemical shift values. This analysis revealed that, in general, there is a strong correlation between the predicted and experimental data, indicating that REMD simulations generate relatively realistic picture.
Fig. 6.

The simulated Cα and Hα chemical shift values (red circles) by REMD simulations and their comparison to experiments (black circles). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Conclusions
We conducted REMD simulations of SARS-CoV-2 Mpro in aqueous solution and present the results for body temperature replica. We also analyzed several structural characteristics, including RMSF values with varying temperature, secondary structure propensities, and k-means clustering, Cα and Hα chemical shift values and we performed residue-level analysis of several key structural and functional characteristics that include propensity for intrinsic disorder and for protein-protein and protein-nucleic acids binding.
Our findings show that some of the residues of the SARS-CoV-2 Mpro are flexible based on RMSF values, and these results are supported by REE values and deviations. We also find that the flexibility of Mpro is related to the given temperature. However, the overall degree of structural compactness does not change much and varies only by 2.0 Å in water. We detect seven α-helix, two 310-helix, thirteen β-sheet, and eighteen turn structure regions in the structures of the SARS-CoV-2 Mpro homodimer in water with dynamics in the presence of the inhibitor's active peptide fragment. We also detect intra-molecular residue interactions between the mid-domain and N- or C-terminal regions, as well as between the N- and C-terminal regions. The calculated Cα and Hα chemical shift values are in excellent agreement with available experiments. To the best of our knowledge, we present herein the first REMD simulations results for SARS-CoV-2 Mpro in water. The reported results can be useful in the long run for developing COVID-19 treatments including small drug molecules.
4. Availability of data
The data that supports the findings of this study are available within this article. Further data that supports the findings are available from the corresponding author upon request.
CRediT authorship contribution statement
Ibrahim Yagiz Akbayrak: Software, Data curation. Sule Irem Caglayan: Data curation, Visualization. Lukasz Kurgan: Data curation, Visualization. Vladimir N. Uversky: Data curation, writing. Orkid Coskuner-Weber: Supervision, Data curation, Visualization, writing, Conceptualization.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
The numerical calculations reported in this paper were performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources).
Contributor Information
Lukasz Kurgan, Email: lkurgan@vcu.edu.
Vladimir N. Uversky, Email: vuversky@health.usf.edu.
Orkid Coskuner-Weber, Email: weber@tau.edu.tr.
Data availability
Data will be made available on request.
References
- Akaji K., et al. Structure-based design, synthesis, and evaluation of peptide-mimetic SARS 3CL protease inhibitors. J. Med. Chem. 2011;54:7962–7973. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- Akbayrak I.Y., Caglayan S.I., Durdagi S., Kurgan L., Uversky V.N., Ulver B., Dervisoglu H., Haklidir M., Hasekioglu O., Coskuner-Weber O. Structures of MERS-CoV macro domain in aqueous solution with dynamics: impacts of parallel tempering simulation techniques and CHARMM36m and AMBER99SB force field parameters. Proteins: Struct., Funct., Bioinf. 2021;89(10):1289–1299. doi: 10.1002/prot.26150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrelle F.-X., et al. NMR spectroscopy of the main protease of SARS-CoV-2 and fragment-based screening identify three protein hotspots and an antiviral fragment. Angew. Chem., Int. Ed. 2021;60:25428–25435. doi: 10.1002/anie.202109965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Yeung M., Xu R.-H. SARS: Epidemiology. Respirol. Carlton Vic. 2003;8(Suppl. l):S9–S14. doi: 10.1046/j.1440-1843.2003.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner O., Uversky V.N. Tyrosine regulates β-sheet structure formation in amyloid-Β42: a new clustering algorithm for disordered proteins. J. Chem. Inf. Model. 2017;57(6):1342–1358. doi: 10.1021/acs.jcim.6b00761. [DOI] [PubMed] [Google Scholar]
- Coskuner O., Wise-Scira O. Arginine and disordered amyloid-β peptide structures: molecular level insights into the toxicity in alzheimer's disease. ACS Chem. Neurosci. 2013;4(12):1549–1558. doi: 10.1021/cn4001389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskuner-Weber O., Uversky V.N. Alanine scanning effects on the biochemical and biophysical properties of intrinsically disordered proteins: a case study of the histidine to alanine mutations in amyloid-Β42. J. Chem. Inf. Model. 2019;59(2):871–884. doi: 10.1021/acs.jcim.8b00926. [DOI] [PubMed] [Google Scholar]
- Diaz N., Suarez D. Influence of charge configuration on substrate binding to SARS-CoV-2 main protease. Chem. 2021;57:5314–5317. doi: 10.1039/d1cc01449h. [DOI] [PubMed] [Google Scholar]
- Dosztányi Z., Csizmok V., Tompa P., Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinforma. Oxf. Engl. 2005;21(16):3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Dosztányi Z., Csizmók V., Tompa P., Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005;347(4):827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- Ford N., Vitoria M., Rangaraj A., Norris S.L., Calmy A., Doherty M. Systematic review of the efficacy and safety of antiretroviral drugs against SARS, MERS or COVID-19: initial assessment. J. Int. AIDS Soc. 2020;23(4) doi: 10.1002/jia2.25489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L., et al. Mechanism of microbial metabolite leupeptin in the treatment of COVID-19 by traditional Chinese medicine herbs. mBio. 2021;12 doi: 10.1128/mBio.02220-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Jin Z., et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Johansen-Leete J., et al. Antiviral cyclic peptides targeting the main protease of SARS-CoV-2. Chem. Sci. 2022;13:3826–3836. doi: 10.1039/d1sc06750h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khailany R.A., Safdar M., Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020 doi: 10.1016/j.genrep.2020.100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D.W., Phillips G., et al. Structural plasticity of SARS-CoV-2 3CL Mpro active site reveraled by room temperature X-ray crystallography. Nat. Commun. 2020;11:3202. doi: 10.1038/s41467-020-16954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D.W., et al. Malleability of the SARS-CoV-2 3CL M(pro) active-site cavity facilitates binding of clinical antivirals. Structure. 2020;28:1313–1320. doi: 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T.S., Okimoto N., et al. Drug binding dynamics of the dimeric SARS-CoV-2 main protease, determined by molecular dynamics simulation. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-74099-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., et al. Self-masked aldehyde inhibitors: a novel strategy for inhibiting cysteine proteases. J. Med. Chem. 2021;64:11267–11287. doi: 10.1021/acs.jmedchem.1c00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald E.A., et al. Recognition of divergent viral substrates by the SARS-CoV-2 main protease. ACS Infect. Dis. 2021;7:2591–2595. doi: 10.1021/acsinfecdis.1c00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews D.A., Dragovich P.S., Webber S.E., Fuhrman S.A., Patick A.K., Zalman L.S., Hendrickson T.F., Love R.A., Prins T.J., Marakovits J.T., Zhou R., Tikhe J., Ford C.E., Meador J.W., Ferre R.A., Brown E.L., Binford S.L., Brothers M.A., DeLisle D.M., Worland S.T. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U.S.A. 1999;96(20):11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mészáros B., Erdos G., Dosztányi Z. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018;46(W1):W329–W337. doi: 10.1093/nar/gky384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noske G.D., et al. A crystallographic snapshot of SARS-CoV-2 main protease maturation process. J. Mol. Biol. 2021;433 doi: 10.1016/j.jmb.2021.167118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradovic Z., Peng K., Vucetic S., Radivojac P., Dunker A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl. 7):176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- Peng Z., J’Kurgan L. High-thorughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015;43:e121. doi: 10.1093/nar/gkv585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng K., Radivojac P., Vucetic S., Dunker A.K., Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinf. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z., Wang C., Uversky V.N., Kurgan L. Prediction of disordered RNA, DNA and protein binding regions using DisoRDPbind. Methods Mol. Biol. 2017;1484:187–203. doi: 10.1007/978-1-4939-6406-2_14. [DOI] [PubMed] [Google Scholar]
- Romero P., Obradovic Z., Li X., Garner E.C., Brown C.J., Dunker A.K. Sequence complexity of disordered protein. Proteins. 2001;42(1):38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co. ;2-3. [DOI] [PubMed] [Google Scholar]
- Sacco M.D., et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020:6. doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnal D., et al. Mol∗ Viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021;49:W431–W437. doi: 10.1093/nar/gkab314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suares D., Diaz N. SARS-CoV-2 main protease: a molecular dynamics study. J. Chem. Inf. Model. 2020;60:5815–5831. doi: 10.1021/acs.jcim.0c00575. [DOI] [PubMed] [Google Scholar]
- Subbaram K., Kannan H., Khalil Gatasheh M. Emerging developments on pathogenicity, molecular virulence, epidemiology and clinical symptoms of current Middle East respiratory syndrome coronavirus (MERS-CoV) Hayati J. Biosci. 2017;24(2):53–56. doi: 10.1016/j.hjb.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., et al. Identification of proteasome and caspase inhibitors targeting SARS-CoV-2 M(pro) Signal Transduct. Targeted Ther. 2021;6:214. doi: 10.1038/s41392-021-00639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R., Jin L., Zou Q., Nakai K., Wei L. Predicting protein-peptide binding residues via interpretable deep learning. Bioinformatics. 2022;38:3351–3360. doi: 10.1093/bioinformatics/btac352. [DOI] [PubMed] [Google Scholar]
- Xu X.P., Case D.A. Automated prediction of 15N, 13Ca, 13Cb and 13C' chemical shifts in proteins using a density functional database. J. Biomol. NMR. 2001;21:321–333. doi: 10.1023/a:1013324104681. [DOI] [PubMed] [Google Scholar]
- Xue B., Dunbrack R.L., Williams R.W., Dunker A.K., Uversky V.N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta. 2010;1804(4):996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J., Kurgan L. DRNApred, Fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res. 2017;45:e84. doi: 10.1093/nar/gkx059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Roy A., Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res. 2013;41:D1096–D1103. doi: 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Kurgan L. SCRIBER: Accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinforma. Oxf. Engl. 2019;35(14):i343–i353. doi: 10.1093/bioinformatics/btz324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Ghadermazi S., Kurgan L. Prediction of protein-BInding residues: dichotomy of sequence-based methods developed using structured complexes versus disordered proteins. Bioinformatics. 2020;36:4729–4738. doi: 10.1093/bioinformatics/btaa573. [DOI] [PubMed] [Google Scholar]
- Zhao Y., et al. Structural basis for replicase polyprotein cleavage and substrate specificity of main protease from SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2022;119 doi: 10.1073/pnas.2117142119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that supports the findings of this study are available within this article. Further data that supports the findings are available from the corresponding author upon request.
Data will be made available on request.