

Abstract
Targeted covalent inhibitors (TCIs) are considered to be an important component in the toolbox of drug discovery and about 30% of currently marketed drugs are TCIs. Although these drugs raise concerns about toxicity, their high potencies and prolonged effects result in less-frequent drug dosing and wide therapeutic margins for patients. This leads to increased interests in developing new computational methods to identify novel covalent inhibitors. The implementation of successful in silico docking algorithms have the potential to provide significant savings of time and money in the discovery of lead compounds. In this paper, we describe the implementation and testing of a covalent docking methodology in Rigid CDOCKER and the optimization of the corresponding physics-based scoring function with an additional customizable covalent bond grid potential which represents the free energy change of bond formation between the ligand and the receptor. We optimize the covalent bond grid potential for different common covalent bond formation reaction in TCIs. The average runtime for docking one covalent compound is 15 minutes which is comparable or faster than other well-established covalent docking methods. We demonstrate comparable top rank accuracy compared with other covalent docking algorithms using the pose prediction benchmark dataset for covalent docking algorithms developed by the Keserű group. Finally, we construct a retrospective virtual screening benchmark dataset containing 8 different receptor targets with different covalent bond formation reactions. To our knowledge, this is the largest dataset for benchmarking covalent docking methods. We show that our new covalent docking algorithm has the ability to identify lead compounds among a large chemical space. The largest AUC value is 0.909 for the target receptor CATK and the warhead chemistry of the covalent inhibitors is addition to the aldehyde functionality.
Keywords: Rigid CDOCKER, Covalent Docking, Virtual Screening
1. Introduction
Targeted covalent inhibitors (TCIs) have gained increased interest in drug discovery in the last two decades, with nearly 30% of currently marketed drugs known to be covalently bound to the therapeutic target.[1–3] TCIs are designed such that the initial, reversible association is followed by the formation of a covalent bond between the ligand and receptor, which strengthens the interactions and increases the potency.[2, 3] Tethered docking methods have become an efficient means of structure based TCI design, and has been widely used in identifying lead compounds.[4–6] Generally speaking, docking involves two main components: searching and scoring.[7–9] In one element, the searching generates multiple docking poses of a ligand within the constraints of the receptor binding site. The application of a scoring function then ranks these poses and is expected to identify the correct binding pose through the assumption the correct binding pose is at the top rank. Today, multiple off-the-shelf protein-ligand covalent docking programs, either commercial or free, are available for use, such as DOCKovalent[10], GOLD[11–13], AutoDock4[14, 15], CovDock[16, 17], FITTED[18], ICM-Pro[19] and MOE.[20] In a recent pose prediction challenge to reproduce the binding mode of 207 cysteine-bound covalent complexes, ICM-Pro showed the best performance with the top-ranking accuracy of 62%, followed by CovDock(59%), FITTED(56%), AutoDock4(55%), and GOLD(53%).[21]
The covalent docking methods DOCKovalent, GOLD, AutoDock4, ICM-Pro and MOE handle the ligand in its bound state (i.e. covalent form).[10–13, 15, 19, 20] These methods form a physical bond between the ligand reactive atom to the receptor reactive atom and before searching for the binding pose, which have two potential issues: (1) the sampling space is reduced and does not include the initial, reversible association for ligands from an unbound form, which is essential for TCIs, (2) and the ligand preparation or reaction type generally requires manual definition which can cause difficulties in high throughput screening.
CovDock and FITTED use the ligand in its non-covalent form.[16–18] CovDock has two different versions which are Lead Optimization mode (CovDock-LO)[16] and Virtual Screening mode (CovDock-VS).[17] The first has a better accuracy with higher computational cost and was used in a previous pose prediction challenge.[21] Typically, CovDock-LO requires 1 ~ 3 hours to dock one compound.[17] The CovDock-VS mode is designed to address high throughput needs with a lower docking accuracy. Both methods will automatically identify the ligand warhead atoms and form a covalent bond during the docking simulations if the warhead is predicted to be in close proximity to the targeted residues.[16–18] On the other hand, FITTED does not allow customization of the warhead, which makes it unable to recognize certain covalent bond formation reactions (i.e., nucleophilic substitution, ring opening and disulfide bridge formation).[18, 21] Thus, both docking methods have limitations in real applications for identification of TCIs.
Rigid CDOCKER is a grid based MD docking algorithm where the ligand-receptor interaction energy is precomputed and stored on a grid.[22] This grid-based representation of interactions has been applied widely in many docking protocols which provides computational efficiency while maintaining much of the accuracy of the full force field method. Rigid CDOCKER uses a physics-based scoring function (eq 1) and was originally designed for docking reversible inhibitors.[22] In the current work, we implement a covalent docking module with Rigid CDOCKER by introducing an customizable covalent bond grid potential in the scoring function to mimic the free energy change of bond formation between the ligand and the receptor as described below.
| (1) |
2. Methods
Benchmark Dataset
Two sets of protein-ligand complexes and the ZINC12 compound library[23, 24] are used for optimizing and benchmarking the protein-ligand docking method described below.
Dataset used for optimizing the covalent CDOCKER scoring function and evaluating pose prediction accuracy.
We employed the same dataset as the one used in the previous pose prediction challenge, which contains 207 complexes representing 54 protein targets.[21] This dataset contains 7 different chemical reaction types: addition to aldehyde, disulfide bond formation, addition to ketone, Michael addition, addition to nitrile, nucleophilic substitution and ring opening, that are common for TCIs.[2, 21] We select this dataset to optimize the scoring function and compare the pose prediction accuracy of the proposed covalent docking algorithm with other covalent docking programs.
Retrospective virtual screening dataset.
One major application of docking is to identify lead compounds for a given target. A successful in silico docking protocol can save a large amount of time and money in the drug discovery process. Thus, it is important to evaluate the virtual screening performance for a newly proposed docking algorithm. Many research groups constructed different retrospective virtual screening datasets in developing their covalent docking methods.[10, 16, 17, 25] However, there are different issues with these datasets: (1) they do not include different covalent bond formation reactions of TCIs, (2) there is no additional process for filtering out decoys that are physico-chemically dissimilar with the actives for some of the datasets, and (3) the size of both the active sets and decoy sets are very small for some of the datasets. Therefore, we decided to construct our own retrospective virtual screening dataset.
We did not select the standard ZINC15 library because it has been filtered by ZINC12 clean filters.[24, 26] This removes aldehydes and thiols in the ZINC15 standard library, which is contradictory to the purpose of constructing virtual screening dataset of the reaction addition to aldehyde. Thus, the entire standard ZINC12 library containing 16 million compounds is used to construct subsets with different electrophiles that correspond to different covalent bond formation reactions. This classification is performed based on SMARTS regular expression (Table 1). If the same functional group appears more than once in a compound, then this compound is excluded in the corresponding electrophile subset. Nucleophilic substitution reactions could involve different functional groups and leaving groups, thus it is hard to maintain both specificity and generalization by using only one SMARTS regular expressions. Therefore, we decided to not include a benchmark set for the nucleophilic substitution reaction. Overall, this provides large compound libraries for each of the remaining reaction types and is used for our curation of the decoy sets.
Table 1.
Summary of the ZINC12 Subsets of Different Electrophiles.
| Electrophile | Reaction type | SMARTS expression | Number of compounds |
|---|---|---|---|
| Thiol | Disulfide Bond Formation | [#6][#16X2H] | 10196 |
| Epoxide | Ring opening | C1OC1 | 9975 |
| Aldehydea | Addition to aldehyde | [CX3H1](=O)[#6] | 48841 |
| Ketoneb | Addition to ketone | [#6][CX3](=O)[#6] | 595283 |
| Nitrile | Addition to nitrile | [NX1]#[CX2] | 745350 |
| α, β-unsaturated carbonyl | Michael addition | [CX3](=O)[CX3]=[CX3] | 986697 |
Compounds with both the functional group aldehyde and the functional group α,β-unsaturated carbonyl are excluded for the ZINC12 aldehyde subset.
Compounds with both the functional group ketone and the functional group α,β-unsaturated carbonyl are excluded for the ZINC12 ketone subset.
For each of the remaining reaction types, we intended to construct 1 ~ 2 benchmark sets with different target receptors. All experimentally validated binders for each target receptor are collected from BindingDB.[27] For the curation of active sets with a desired electrophile, the collected ligand libraries are filtered using the same SMARTS regular expressions (Table 1). We assume these filtered actives with the intended electrophile are TCIs (i.e., these inhibitors form a covalent bond upon binding.) We also perform an additional filtering to only include lead-like molecules (250 ≤ molecular weight ≤ 350; LogP ≤ 3.5; number of rotatable bonds ≤ 7) for the chemical reaction type addition to ketone, addition to nitrile and Michael addition. This allows us to limit the docking library size and avoid potential docking issues from large and flexible compounds.[10]
To construct the decoy set for each target receptor that we choose, we selected physico-chemically similar compounds from the corresponding electrophile subsets. It is understood that some of the compounds in the decoy set might actually be true binders, and our choice of using high physico-chemcially similar compounds make the retrospective virtual screening test even more challenging. A more detailed explanation of dataset construction is documented in the Supplementary Information and the similarity cutoff is also listed in the Supplementary Table S1. A summary of the retrospective virtual screening dataset is listed in Table 2.
Table 2.
Summary of the Retrospective Virtual Screening Dataset.
| Receptor name | Number of actives | Number of decoys | PDB | Reaction type |
|---|---|---|---|---|
| CATK | 56 | 3050 | 2AUX | Addition to aldehyde |
| PAPAIN | 21 | 2525 | 1CVZ | Ring opening |
| ALDH3A1 | 36 | 11678 | 4L1O | Addition to ketone |
| CASP3 | 79 | 3392 | 1RHJ | Addition to ketone |
| EGFR | 151 | 10755 | 5UG8 | Michael addition |
| JAK3 | 356 | 11773 | 5TTS | Michael addition |
| CATS | 150 | 18312 | 1MS6 | Addition to nitrile |
| CATK | 215 | 23596 | 2F7D | Addition to nitrile |
Unfortunately, we did not identify receptor targets with adequate actives (i.e., more than 10 experimentally validated inhibitors) for the chemical reaction disulfide bond formation mainly because only about 2% of TCIs undergo disulfide bond formation with the target receptor. Overall, we construct a retrospective virtual screening benchmark dataset modeling 5 common covalent bond formation reactions of TCIs. To our knowledge, this is the largest benchmark dataset for covalent docking methods.
Rigid CDOCKER Algorithm Overview
There are three main elements to the Rigid CDOCKER algorithm: the receptor and ligand representation, a searching algorithm, and the newly developed scoring function, which includes an additional energy term that approximates the free energy change of covalent bond formation.
Receptor and Ligand Representation
All protein structure files were acquired from the Protein Data Bank (PDB). Both protein structures and ligand structures are manually examined and reverted to pre-reaction form. The receptor structure is represented implicitly by grids with a grid space of 0.5 Å. Ligand structure files are manually examined and reverted to pre-reaction form. MOE (Molecular Operating Environment)[28] was used to predict the protonation state of the ligands at pH 7.4. The dominant protonation state of the compound is selected for the following docking experiments. RDKit[29] was used to generate random ligand conformations using the EDKTG method[30, 31], ParamChem[32, 33] was used to prepare the ligand topology and parameter files and the MMTSB tool set[34] was used to cluster the binding poses. A more detailed explanation of the conformer generation is documented in the Supplementary section Ligand Conformer Generation. The corresponding scripts are provided in the Supplementary Infromation and can be acquired through GitHub. Clustering used the tool cluster.pl with K-means clustering. The CHARMM C36 force fields[35] were used and docking was performed in CHARMM[36] with the CHARMM/OpenMM parallel simulated annealing feature.[37] The RMSD cutoff to identify native-like poses is set to be 2 Å for rigid docking, to be consistent with the evaluation criteria in the previous studies.[22, 37]
Docking Searching Algorithm
One general problem in docking is how to place the ligand in the vicinity of the binding site and what initial ligand internal conformation to chosen. In the original CDOCKER docking protocol, we typically use 500 docking trials for each ligand.[37] This might be redundant for a rigid compound, while at the same time insufficient for a highly flexible compound. It is endemic to docking methodology that a non-exhaustive searching of ligand conformational space and initial placement will result in a decrease in top-ranking accuracy. Hence, this will also affect the performance of docking methods in virtual screening as observed by the Shoichet group in their retrospective virtual screening test when the ligand has many rotatable bonds.[10]
Therefore, after we generate N conformers for a given ligand, we perform 100 initial placements for each conformer (i.e., 100N docking trials in total). These initial starting poses are then optimized by the molecular dynamics (MD) based simulated annealing algorithm[7, 8, 22, 37, 38] and scored with the scoring function described below. In the current study, the parameters for the van der Waals and electrostatic interactions are the same as reported in our previous studies and are included in Supplementary Table S2.[37, 38] The CHARMM scripts used for docking covalent inhibitors with Rigid CDOCKER can be acquired through GitHub and a more detailed explanation of the covalent docking searching algorithm is provided in the Supplementary Information.
Optimizing the Covalent Docking Scoring Function
The binding free energy can be written as eq 2. In cases of pose prediction, the same ligand is docked to the receptor multiple times and generates a distribution of docking poses. Since the initial state (Ginitial) is the same for all docking trials (i.e., both ligand and receptor are presented separately in the solution). Thus, only Gfinal needs to be calculated for pose prediction. The enthalpic contribution (Hfinal) can be separated into ligand internal energy (Eligand), van der Waals interaction (Evdw), electrostatic interactions (Eelec) and free energy for the chemical reaction that forms the covalent bond (Ecovalent). The energy terms Eligand, Evdw and Eelec have been well-established in the CDOCKER scoring function.[22, 38] The entropic contribution (Sfinal) can be separated into contributions from solvation and conformational entropy. Since we consistently dock the same ligand to the same binding pocket in one measurement, the change in conformational entropy for the same ligand is a constant, and we assume the solvation contribution is approximately the same for different docking poses. Thus, we suggest that the entropic contribution and the solvation contribution can be neglected. Therefore, the scoring function for Rigid CDOCKER in covalent docking can be written as eq 3.
| (2) |
| (3) |
Rigid CDOCKER is a grid based MD docking algorithm.[22] Here, we introduce a customizable grid potential (eq 4) to mimic the free energy change for the chemical reaction that forms the covalent bond. We adopt the idea of the two-point attractor method[15], where the ligand is modeled as a free ligand, and this covalent bond grid potential is used to bring together the ligand reactive atom and the targeted receptor reactive atom. As shown in Figure 1, the parameters r1, r2 and Emax determine the width and well depth of the covalent bond grid potential. The variable r is the distance between the grid point and receptor reactive atom. When the ligand reactive atom is close to the receptor reactive atom (i.e., between r1 and r2), this potential (Emax) will favors the interaction between the ligand and receptor.
| (4) |
Fig. 1.
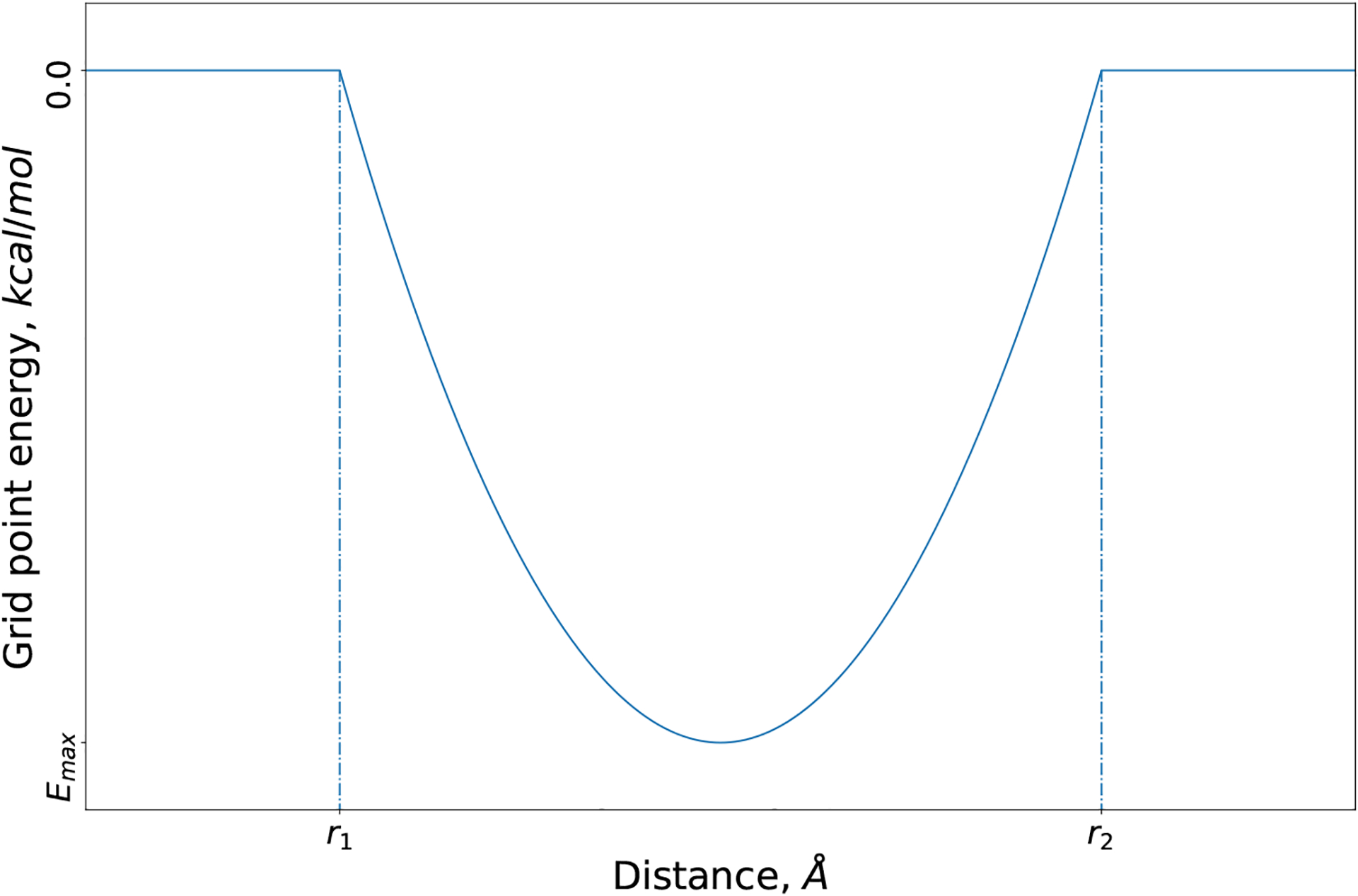
Covalent bond grid potential as a function of grid point distance.
The two-point attractor method in AutoDock treats different bond formation reaction types with the same grid potential.[15] However, for different covalent bond formation reactions, one would expect the covalent bond grid potential parameters should adopt different values. To identify the best parameters for different reaction types, we perform the docking experiments on the pose prediction dataset using different covalent bond grid potentials. This dataset contains 207 different protein-ligand complexes and is used in previous comparative docking analysis of the covalent docking methods. The different parameter values used in this experiment are listed in Table 3. Thus, we have a total of 100 different covalent bond grid potentials (i.e., 100 different combinations of the parameters). Therefore, we perform 100 docking experiments with different covalent bond grid potential against each protein-ligand complex and record the corresponding docking result. This docking experiment is repeated for 3 times.
Table 3.
Values of Parameter for the Covalent Bond Grid Potential.
| Parameter | Value |
|---|---|
| r 1 | 0, 0.5, 1, 1.5 and 2 |
| r 2 | 3, 3.5, 4, 4.5 and 5 |
| E max | −2.5, −5, −7.5 and − 10 |
Because the number of receptor structures in each reaction type is relative small (i.e., only 5 receptor structures in the chemical reaction disulfide bond formation) and the randomness of dividing a dataset into for training and testing components does not exist[39], we adopt the idea of leave-one-out cross validation and analyze the docking results with the following procedure:
For a reaction type with N receptor structures, we separate the dataset into training sets with N − 1 receptors and testing sets with one receptor (i.e., leave-one-out approach).
Calculate the cumulative ranking accuracy averaged across the 3 independent docking experiments for the training set.
Record the corresponding top-ranking accuracy and area under curve (AUC) value of the cumulative ranking accuracy plot.
Select the covalent bond grid potential parameters with the best top-ranking accuracy. If multiple sets of the grid potentials give the same highest top-ranking accuracy result, we then select the one with the largest AUC value and record the corresponding average ranking result in the testing set.
Repeat step 2 ~ 4 for all training and testing sets.
Thus, for each reaction type, we will have a set of grid parameters. We consider the most frequent set of grid parameter as the optimized grid parameters for this reaction type and compute the corresponding top-ranking accuracy (Table 4). The parameters r1 and r2 reflect the average bulkiness of the reactive leaving group and the targeted connection point within the benchmark dataset. The potential Emax is the empirical representation of the average difference in bond formation free energy of a given warhead chemistry in solution versus in the protein environment and is probably dominated by the entropic localization bias. For completeness, we also record and plot the heat-maps of the top-ranking accuracy and AUC results in the Supplementary Figure S2 and S3. A complete docking result is listed in the supplementary files rank-result.tsv and can be acquired through GitHub.
Table 4.
Covalent Bond Grid Potential for Different Reaction Types
| Reaction Type | r1, Å | r2, Å | E max , kcal/mol |
|---|---|---|---|
| Addition to aldehyde | 1.5 | 4.5 | −10 |
| Disulfide bond formation | 0 | 3.5 | −10 |
| Addition to ketone | 0 | 5 | −10 |
| Michael addition | 0 | 4.5 | −10 |
| Addition to nitrile | 1.5 | 4 | −10 |
| Nucleophilic substitution | 1 | 5 | −7.5 |
| Ring opening | 1 | 5 | −10 |
As shown in Table 4, for each of the chemical reactions, we identified the best covalent bond grid potential, and the top-ranking accuracy for each of the chemical reactions is listed in Table 5. The standard Rigid CDOCKER non-covalent docking methodology is used for direct comparison. We demonstrate that the additional covalent energy term (Ecovalent) significantly improves the docking performance. In the initial random placement of the ligand, we only include the poses with Ecovalent smaller than zero (i.e., Ecovlanet acts as an additional filter). This might also explain why we observe more improvement in pose prediction accuracy compared with other comparison work (i.e., Glide vs. CovDock and Autodock vs. Autodock covalent docking method).
Table 5.
Top Ranking Accuracy for Covalent and Non-covalent Docking in Rigid CDOCKER
| Reaction Type | Non-covalent docking | Covalent docking |
|---|---|---|
| Addition to aldehyde | 18.06% | 58.33% |
| Disulfide bond formation | 40.00% | 53.33% |
| Addition to ketone | 8.33% | 55.56% |
| Michael addition | 23.05% | 53.90% |
| Addition to nitrile | 51.06% | 66.67% |
| Nucleophilic substitution | 23.53% | 56.86% |
| Ring opening | 20.83% | 45.83% |
The top-ranking accuracy against the pose prediction dataset is comparable with other covalent docking methods used in the same benchmark dataset.[21] We recorded the top-ranking accuracy for each of the reaction types and covalent docking methods in the Supplementary Table S3. The average docking runtime is about 15 minutes, which is comparable or faster than other covalent docking methods.[14–19] We notice that we have a relative lower pose prediction accuracy for the reaction type ring opening. On the other hand, in the pose prediction challenge for the reaction type ring opening, all of the other five covalent docking methods have a top rank accuracy below 25% (Table 6). This indicates there might be potential issues in dataset construction or modeling ring opening in general for tethered docking methods. We also compared the seaching and scoring accuracy for our covalent docking method. As shown in Table 7, the relatively higher searching accuracy indicates future work should focus on further optimization of the scoring function.
Table 6.
Top Ranking Accuracy for the Chemical Reaction Ring Opening with Differenct Covalent Docking Method
| Covalent Docking Methods | Top ranking accuracy |
|---|---|
| Covalent docking in CDOCKER | 45.83% |
| AutoDock4a | 25.00% |
| CovDocka | 12.50% |
| GOLDa | 25.00% |
| ICM-Proa | 12.50% |
| MOEa | 25.00% |
Top ranking accuracy reported in the previous pose prediction challenge.[21]
Table 7.
Searching vs. Scoring for Covalent Docking in Rigid CDOCKER
| Reaction Type | Searching accuracya | Scoring accuracyb |
|---|---|---|
| Addition to aldehyde | 86.11% | 58.33% |
| Disulfide bond formation | 80.00% | 53.33% |
| Addition to ketone | 77.78% | 55.56% |
| Michael addition | 84.04% | 53.90% |
| Addition to nitrile | 89.36% | 66.67% |
| Nucleophilic substitution | 82.35% | 56.86% |
| Ring opening | 58.33% | 45.83% |
Searching accuracy is defined as number of docking experiments that we successfully identified native-like poses divided by total number of docking experiments.
Scoring accuracy is top-ranking accuracy.
Augmented Scoring Function for Virtual Screening
One of the most common applications of docking is to identify novel inhibitors for a given target. Covalent docking methods have been used to rank compounds with the same warhead chemistry and succeed in identifying covalent inhibitors.[10, 40–42] The proposed scoring function estimates the total energy of the protein-ligand complex upon binding. Therefore, in order to compare different small molecules, we need to augment our scoring function to consider the system in the unbound state. Because these compounds are docked to the same protein target, the protein energy in the unbound state is a constant and cancels out in comparative studies. Thus, to complete the scoring function for this situation, we only need to include contributions to the ligand internal energy and conformational entropy of the unbound state (eq 5).
| (5) |
The top rank docked pose is used to calculate the total energy of the protein-ligand complex using the scoring function just proposed in (eq 5). The conformers for each of the ligands used for docking are minimized in vacuum. The ligand internal energy in the unbound state is then calculated by computing the ensemble average of the internal energy of these conformers. The ligand conformational entropy (Sligand) is calculated based on the number of the rotatable bonds of the ligand using the microscopic definition of entropy (eq 6). We assume that the rotatable bonds of the ligand are independent of each other and all three states (i.e., trans, gauche- and gauche+) can be equally sampled. The temperature (T) is set to be room temperature (298 K).
| (6) |
The solvation free energy difference is computed using two different approaches: (1) implicitly represented in the proposed scoring function by the distance dielectric constant of 3r.[22, 37, 38] and (2) rescoring the system using the FACTS implicit solvent model.[43] In the rescoring approach with the FACTS implicit solvent model, the ligand internal energy in the unbound state is calculated by the same approach. The rescoring of the protein-ligand complex at the bound state is performed by minimizing the top rank docked pose with the FACTS implicit model. The coordinates of the protein atoms and the ligand reactive atoms are fixed for two reasons: (1) reduce the computational cost of the rescoring, and (2) the distance between the protein and ligand reactive atoms remains unchanged. Therefore, we do not need to re-estimate the covalent bond formation energy (Ecovalent).
As shown in Figure 2, the dominant effects in the ranking orders is the van der Waals and electrostatic energy differences, which reflects the structure complimentary between the receptor and ligand. The difference of the covalent bond formation energy (Ecovalent) is relatively small for different compounds. This reflects the likelihood of forming a covalent bond between the corresponding ligands and receptor and shifts the ranking orders for compounds with similar structural and chemical properties.
Fig. 2.
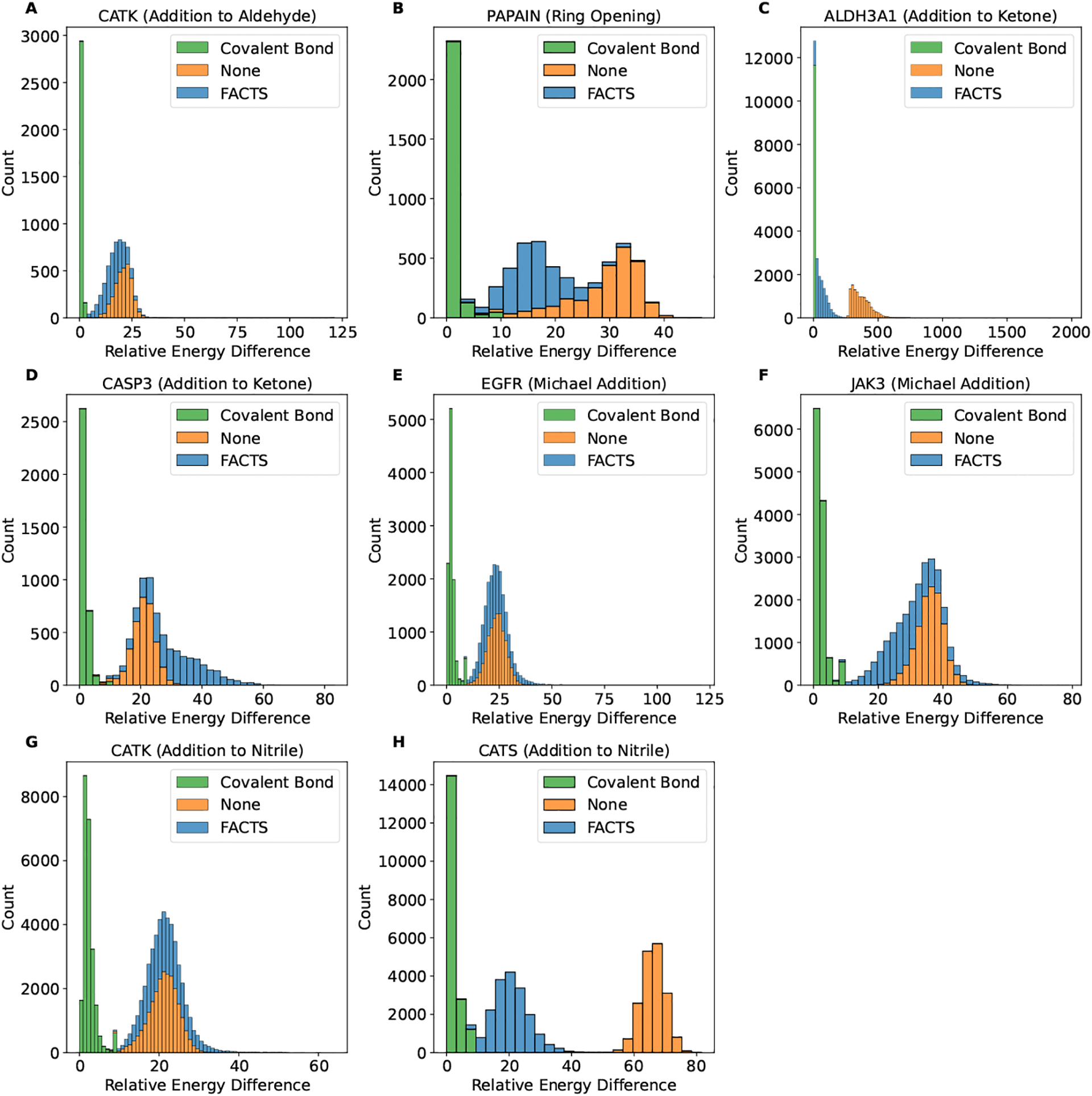
The distribution of the relative energy difference observed in the retrospective virtual screening for the receptor targets and the corresponding reaction type (A) CATK (addition to aldehyde), (B) PAPAIN (ring opening), (C) ALDH3A1 (addition to ketone), (D) CASP3 (addition to ketone), (E) EGFR (Michael addition), (F) JAK3 (Michael addition), (G) CATK (addition to nitrile) and (H) CATS (addition to nitrile). The energy differences plotted here are covalent bond formation energy Ecovalent (green), change of the system energy upon binding after subtracting the covalent bond formation energy (i.e., ΔGbinding−Ecovalent) in vacuum (orange) or using FACTS implicit solvent model (blue). The minimum energy for all comparison is set to be zero.
3. Result
Virtual Screening Performance with Generic Parameters
As mentioned above, we constructed a retrospective virtual screening dataset containing 7 receptor targets modeling 5 common TCI warheads. We perform covalent docking experiments with the docking methods just described against both ligands and non-binding decoys. We use the generic covalent bond grid potential parameters, which are the ones optimized in the pose prediction just described (Table 4). The area under the curve (AUC) value of the receiver operating characteristic (ROC) curve (Figure 3) and two enrichment factors (EF1 and EF20) are used to evaluate its performance in distinguishing the non-binders from binders (Table 8).
Fig. 3.
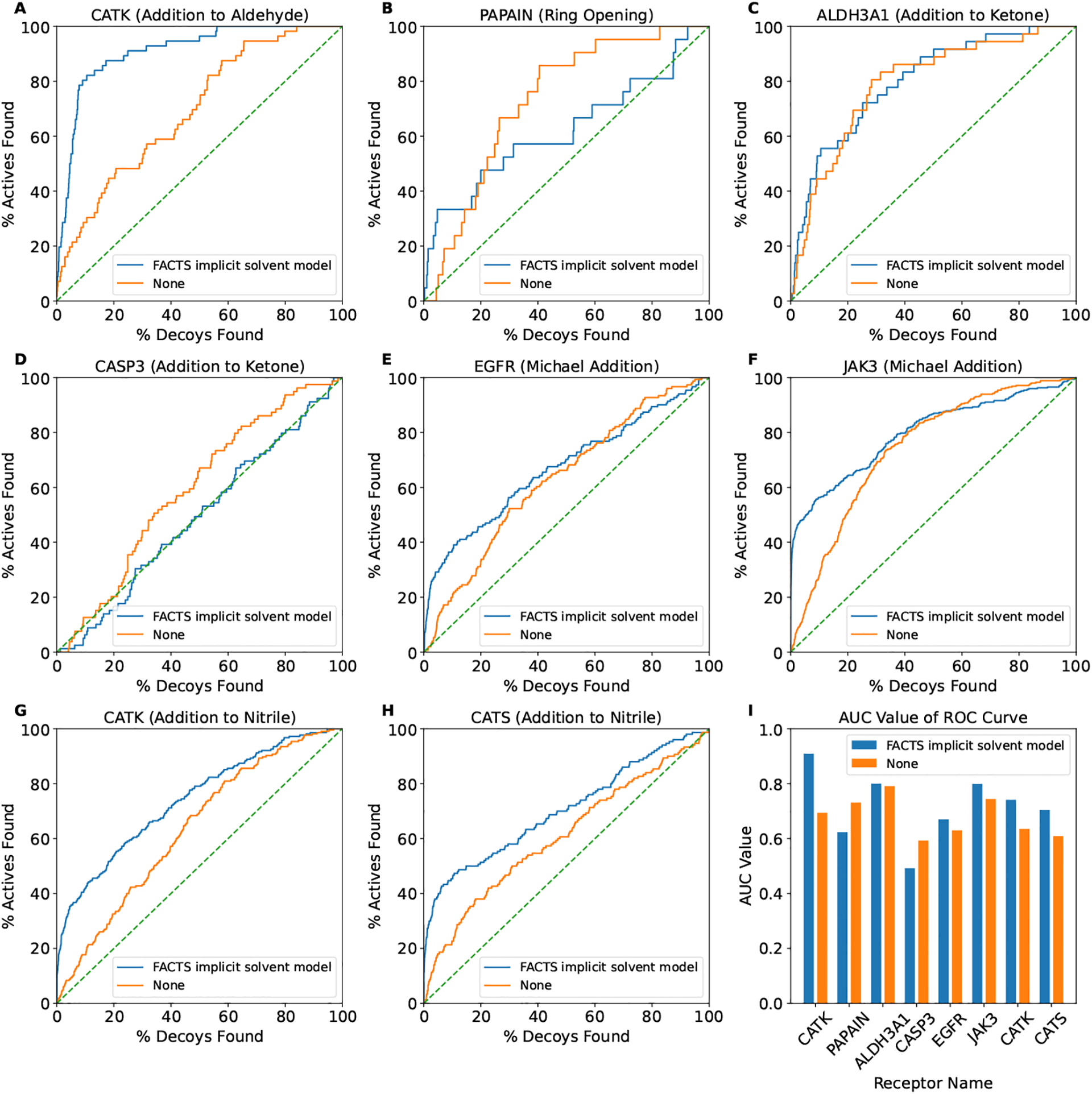
The receiver operating characteristic (ROC) curves of the receptor targets and the corresponding reaction type (A) CATK (addition to aldehyde), (B) PAPAIN (ring opening), (C) ALDH3A1 (addition to ketone), (D) CASP3 (addition to ketone), (E) EGFR (Michael addition), (F) JAK3 (Michael addition), (G) CATK (addition to nitrile) and (H) CATS (addition to nitrile). (I) Summary of the AUC value for each of the ROC curve.
Table 8.
Summary of the Retrospective Virtual Screening Performance.
| Receptor | AUCa | AUCb | EF 1 a | EF 1 b | EF 20 a | EF 20 b | Reaction type |
|---|---|---|---|---|---|---|---|
| CATK | 0.909 | 0.694 | 16.071 | 7.143 | 4.375 | 2.232 | Addition to aldehyde |
| PAPAIN | 0.623 | 0.731 | 4.762 | 0.0 | 2.143 | 2.143 | Ring opening |
| ALDH3A1 | 0.8 | 0.791 | 2.778 | 2.778 | 2.917 | 3.056 | Addition to ketone |
| CASP3 | 0.492 | 0.593 | 0.0 | 0.0 | 0.759 | 1.013 | Addition to ketone |
| EGFR | 0.67 | 0.63 | 11.258 | 0.662 | 2.285 | 1.556 | Michael addition |
| JAK3 | 0.799 | 0.744 | 28.324 | 2.601 | 3.165 | 2.471 | Michael addition |
| CATK | 0.741 | 0.635 | 18.605 | 1.86 | 2.674 | 1.628 | Addition to nitrile |
| CATS | 0.704 | 0.609 | 22.667 | 3.333 | 2.533 | 1.9 | Addition to nitrile |
Binding free energy is estimated after rescoring with the FACTS implicit solvent model.
Binding free energy is estimated using the proposed scoring function with the distance dielectric constant of 3r.
As demonstrated in Figure 3 and Table 8, the proposed covalent docking method with both solvation models has the ability to distinguish binders from non-binders in general. The best performance is for the target receptor CATK with an AUC value of 0.909 and the warhead chemistry of the covalent inhibitors is addition to the aldehyde functionality. As we noted earlier, some of the compounds in the decoy set might be true binders and this could skew our success measures. Thus, the reported values (AUC, EF1 and EF20) are actually a lower bound. Using the FACTS implicit solvent model shows improved performance, especially in the early enrichment of binders (i.e., EF1 value). The largest improvement of the EF1 value is against the receptor target JAK3 (28.324 vs. 2.601). The computational cost of rescoring with the FACTS implicit solvent model is about 5 ~ 10% of the average runtime of the proposed docking algorithm.
We noted that the FACTS implicit solvent model has a disadvantage in the case of PAPAIN (i.e., lower AUC value). We calculated the average solvent accessible surface area (SASA) of the binding pocket using CHARMM. The binding pocket is defined as any protein atom within a 4 Å distance cutoff of any of the crystal ligand atoms. When the binding pocket has a larger average SASA (i.e., ligand is more exposed to solvent upon binding), the solvation free energy change upon binding is smaller. As shown in Table 9, the receptor target PAPAIN has the largest average SASA. Therefore, this might explain why using FACTS implicit solvent model does not improve the virtual screening result.
Table 9.
Average Solvent Accessible Surface Area (SASA) of the Receptor Target Binding Pocket.
| Receptor | PDB | Average SASA | Reaction type |
|---|---|---|---|
| CATK | 2AUX | 14.07 | Addition to aldehyde |
| PAPAIN | 1CVZ | 16.23 | Ring opening |
| ALDH3A1 | 4L1O | 13.42 | Addition to ketone |
| CASP3 | 1RHJ | 14.05 | Addition to ketone |
| EGFR | 5UG8 | 13.74 | Michael addition |
| JAK3 | 5TTS | 15.71 | Michael addition |
| CATK | 2F7D | 14.26 | Addition to nitrile |
| CATS | 1MS6 | 13.35 | Addition to nitrile |
For the warhead chemistry of addition to ketone, as shown in Figures 4A and 4B, the properties of the compounds (i.e., number of rotatable bonds and molecular weight) are similar to each other. However, the binding pocket for the receptor target ALDH3A1 is small and buried (Figure 4C) and the binding pocket for the receptor target CASP3 is large and open (Figure 4D). Thus, the searching space in the case of CASP3 is relatively larger and the surface binding pocket requires less structural complimentary. This may be why we observe difference virtual screening performance for the warhead chemistry of addition to ketone and consistent with the observation that structural complimentary is important in docking.
Fig. 4.
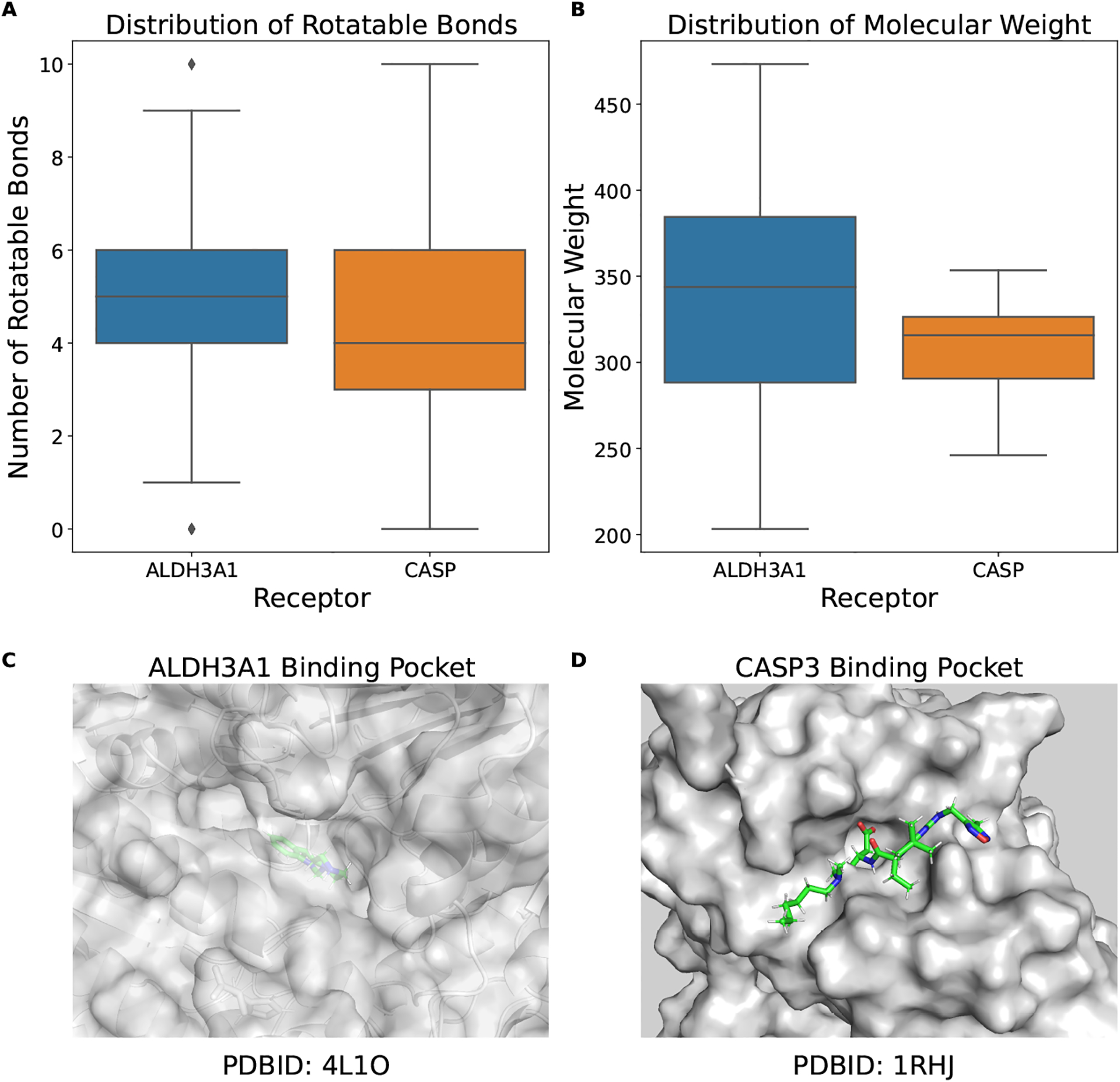
(A) ALDH3A1 binding pocket (PDBID: 4L1O), (B) CASP3 binding pocket (PDBID: 1RHJ), Distribution of the compound property: (C) rotatable bonds, (D) molecular weight.
4. Conclusions and Discussions
Targeted covalent inhibitors (TCIs) are designed such that the covalent warheads can target rare, non-conserved residues of a particular target protein and lead to the development of highly selective inhibitors with high potency and extended duration of action. In this work, we introduced a customizable covalent bond grid potential in the original Rigid CDOCKER scoring function.
In the covalent docking in Rigid CDOCKER, the covalent bond grid potential acts as an attractor if the ligand reactive atom and the protein reactive atom are close to each other and provides an estimate of the free energy change upon covalent bond formation. This is clearly evident by our comparison of the rigid docking protocol with and without the covalent bond grid potential shown in Table 5. Different bond formation reactions (warhead chemistries) should be modeled differently. We optimized and provided a set of generic parameters for the covalent bond grid potential for different reaction types. One could use QM/MM approaches to parameterize the covalent terms, but that is beyond the scope of the current work and may end up being too system specific and costly to be useful in a high-throughput scheme as we have implemented. Our covalent docking algorithm shows comparable pose prediction accuracy with other popular docking algorithms. The average docking runtime is about 15 minutes, which is comparable or faster than other covalent docking methods.
We also constructed a benchmark dataset for evaluating the ability to identify novel TCIs for tethered docking methods. To our knowledge, this is the largest retrospective virtual screening benchmark set for evaluating tethered docking methods. We demonstrate that the proposed covalent docking algorithm has the ability to discriminate binders from non-binders with both solvent models. The covalent bond grid potential in this test uses the generic parameters. This suggests that the proposed covalent docking algorithm can be widely used for different targets for lead compound identification. Using the FACTS implicit solvent model with a small additional computational cost shows better performance, especially in the early enrichment of the active compounds.
In real applications, one could further optimize or adjust this covalent bond grid potential to achieve further improvements. Recently, we developed python package of the CDOCKER family as a workflow functionality in pyCHARMM (i.e., CHARMM through a python interface), which reduces the complexity in using CDOCKER for potential users unfamiliar with CHARMM or CDOCKER. This allows one to easily modify the grid parameters and integrate CDOCKER based docking workflows with other commonly used python packages. Finally, we suggest that the proposed docking algorithm with the FACTS implicit solvent model can effectively be applied in the real-world applications of identifying novel TCIs.
Supplementary Material
Declarations
This work is supported by grants from the NIH(GM130587, GM037554, and GM107233).
Data availability
The Supporting Information (Virtual screening dataset construction; Ligand preparation workflow; Covalent docking searching algorithm; Pose prediction results.) is available free of charge via the Internet. CHARMM license is free for academic users. The full source code and license information for CHARMM are available at http://charmm.chemistry.harvard.edu/ The benchmark dataset and code examples are provided on Github https://github.com/wyujin/Covalent-Docking-in-CDOCKER and are listed below
Example of covalent docking in CDOCKER with CHARMM scripting language
Example of covalent docking in pyCHARMM CDOCKER;
SMILES strings of the retrospective virtual screening datasets used in this study;
Pose prediction result (rank-result.tsv);
Scripts for general ligand preparation.
References
- [1].Kumalo HM, Bhakat S, Soliman ME: Theory and applications of covalent docking in drug discovery: merits and pitfalls. Molecules 20(2), 1984–2000 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baillie TA: Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed 55(43), 13408–13421 (2016) [DOI] [PubMed] [Google Scholar]
- [3].Scarpino A, Ferenczy GG, Keserű GM: Covalent docking in drug discovery: Scope and limitations. Curr. Pharm. Des (2020) [DOI] [PubMed] [Google Scholar]
- [4].Wan X, Yang T, Cuesta A, Pang X, Balius TE, Irwin JJ, Shoichet BK, Taunton J: Discovery of lysine-targeted eif4e inhibitors through covalent docking. JACS 142(11), 4960–4964 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chowdhury SR, Kennedy S, Zhu K, Mishra R, Chuong P, Nguyen A. u., Kathman SG, Statsyuk AV: Discovery of covalent enzyme inhibitors using virtual docking of covalent fragments. Bioorg. Med. Chem 29(1), 36–39 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shraga A, Olshvang E, Davidzohn N, Khoshkenar P, Germain N, Shurrush K, Carvalho S, Avram L, Albeck S, Unger T, et al. : Covalent docking identifies a potent and selective mkk7 inhibitor. Cell Chem. Biol 26(1), 98–108 (2019) [DOI] [PubMed] [Google Scholar]
- [7].Kitchen DB, Decornez H, Furr JR, Bajorath J: Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug 3(11), 935–949 (2004) [DOI] [PubMed] [Google Scholar]
- [8].Yuriev E, Agostino M, Ramsland PA: Challenges and advances in computational docking: 2009 in review. J. Mol. Regonit 24(2), 149–164 (2011) [DOI] [PubMed] [Google Scholar]
- [9].Taylor RD, Jewsbury PJ, Essex JW: A review of protein-small molecule docking methods. J. Comput. Aided Mol. Des 16(3), 151–166 (2002) [DOI] [PubMed] [Google Scholar]
- [10].London N, Miller RM, Krishnan S, Uchida K, Irwin JJ, Eidam O, Gibold L, Cimermančič P, Bonnet R, Shoichet BK, et al. : Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol 10(12), 1066–1072 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jones G, Willett P, Glen RC, Leach AR, Taylor R: Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 267(3), 727–748 (1997) [DOI] [PubMed] [Google Scholar]
- [12].Jones G, Willett P, Glen RC: Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol 245(1), 43–53 (1995) [DOI] [PubMed] [Google Scholar]
- [13].Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD: Improved protein–ligand docking using gold. Proteins 52(4), 609–623 (2003) [DOI] [PubMed] [Google Scholar]
- [14].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ: Autodock4 and autodocktools4: Automated docking with selective receptor flexibility. J. Comput. Chem 30(16), 2785–2791 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bianco G, Forli S, Goodsell DS, Olson AJ: Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Science 25(1), 295–301 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, Harder E: Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J. Chem. Inf. Model 54(7), 1932–1940 (2014) [DOI] [PubMed] [Google Scholar]
- [17].Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, Kalid O: Structure-based virtual screening approach for discovery of covalently bound ligands. J. Chem. Inf. Model 54(7), 1941–1950 (2014) [DOI] [PubMed] [Google Scholar]
- [18].Corbeil CR, Englebienne P, Moitessier N: Docking ligands into flexible and solvated macromolecules. 1. development and validation of fitted 1.0. J. Chem. Inf. Model 47(2), 435–449 (2007) [DOI] [PubMed] [Google Scholar]
- [19].Abagyan R, Totrov M, Kuznetsov D: Icm—a new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem 15(5), 488–506 (1994) [Google Scholar]
- [20].Vilar S, Cozza G, Moro S: Medicinal chemistry and the molecular operating environment (moe): application of qsar and molecular docking to drug discovery. Current topics in medicinal chemistry 8(18), 1555–1572 (2008) [DOI] [PubMed] [Google Scholar]
- [21].Scarpino A, Ferenczy GG, Keserű GM: Comparative evaluation of covalent docking tools. J. Chem. Inf. Model 58(7), 1441–1458 (2018) [DOI] [PubMed] [Google Scholar]
- [22].Wu G, Robertson DH, Brooks Charles L. III., Vieth M: Detailed analysis of grid-based molecular docking: A case study of cdocker – a charmm-based md docking algorithm. J. Comput. Chem 24(13), 1549–1562 (2003) [DOI] [PubMed] [Google Scholar]
- [23].Irwin JJ, Shoichet BK: Zinc- a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 45(1), 177–182 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG: Zinc: a free tool to discover chemistry for biology. J. Chem. Inf. Model 52(7), 1757–1768 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ouyang X, Zhou S, Su CTT, Ge Z, Li R, Kwoh CK: Covalent-dock: automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J. Comput. Chem 34(4), 326–336 (2013) [DOI] [PubMed] [Google Scholar]
- [26].Sterling T, Irwin JJ: Zinc 15–ligand discovery for everyone. J. Chem. Inf. Model 55(11), 2324–2337 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK: Bindingdb: a web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 35(suppl 1), 198–201 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Inc, C.C.G.: Molecular operating environment (MOE). Chemical Computing Group Inc. 1010 Sherbooke St. West, Suite# 910, Montreal: … (2016) [Google Scholar]
- [29].Landrum G: RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Academic Press; (2013) [Google Scholar]
- [30].Riniker S, Landrum GA: Better informed distance geometry: using what we know to improve conformation generation. J. Chem. Inf. Model 55(12), 2562–2574 (2015) [DOI] [PubMed] [Google Scholar]
- [31].Wang S, Witek J, Landrum GA, Riniker S: Improving conformer generation for small rings and macrocycles based on distance geometry and experimental torsional-angle preferences. J. Chem. Inf. Model 60(4), 2044–2058 (2020) [DOI] [PubMed] [Google Scholar]
- [32].Vanommeslaeghe K, MacKerell AD Jr: Automation of the charmm general force field (cgenff) i: bond perception and atom typing. J. Chem. Inf. Model 52(12), 3144–3154 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Vanommeslaeghe K, Raman EP, MacKerell AD Jr: Automation of the charmm general force field (cgenff) ii: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model 52(12), 3155–3168 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Feig M, Karanicolas J, Brooks Charles L. III.: Mmtsb tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph 22(5), 377–395 (2004) [DOI] [PubMed] [Google Scholar]
- [35].Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. : Charmm general force field: A force field for drug-like molecules compatible with the charmm all-atom additive biological force fields. J. Comput. Chem 31(4), 671–690 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brooks BR, Brooks Charles L. III., Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. : Charmm: the biomolecular simulation program. J. Comput. Chem 30(10), 1545–1614 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ding X, Wu Y, Wang Y, Vilseck JZ, Brooks Charles L. III.: Accelerated cdocker with gpus, parallel simulated annealing, and fast fourier transforms. J. Chem. Theory Comput 16(6), 3910–3919 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gagnon JK, Law SM, Brooks Charles L. III.: Flexible cdocker: Development and application of a pseudo-explicit structure-based docking method within charmm. J. Comput. Chem 37(8), 753–762 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Wong T-T: Performance evaluation of classification algorithms by k-fold and leave-one-out cross validation. Pattern Recognit. 48(9), 2839–2846 (2015) [Google Scholar]
- [40].Luo YL: Mechanism-based and computational-driven covalent drug design. J. Chem. Inf. Model 61(11), 5307–5311 (2021) [DOI] [PubMed] [Google Scholar]
- [41].Li A, Sun H, Du L, Wu X, Cao J, You Q, Li Y: Discovery of novel covalent proteasome inhibitors through a combination of pharmacophore screening, covalent docking, and molecular dynamics simulations. J. Mol. Model 20(11), 1–13 (2014) [DOI] [PubMed] [Google Scholar]
- [42].London N, Farelli JD, Brown SD, Liu C, Huang H, Korczynska M, Al-Obaidi NF, Babbitt PC, Almo SC, Allen KN, et al. : Covalent docking predicts substrates for haloalkanoate dehalogenase superfamily phosphatases. Biochem. 54(2), 528–537 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Haberthür U, Caflisch A: Facts: Fast analytical continuum treatment of solvation. J. Comput. Chem 29(5), 701–715 (2008) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Supporting Information (Virtual screening dataset construction; Ligand preparation workflow; Covalent docking searching algorithm; Pose prediction results.) is available free of charge via the Internet. CHARMM license is free for academic users. The full source code and license information for CHARMM are available at http://charmm.chemistry.harvard.edu/ The benchmark dataset and code examples are provided on Github https://github.com/wyujin/Covalent-Docking-in-CDOCKER and are listed below
Example of covalent docking in CDOCKER with CHARMM scripting language
Example of covalent docking in pyCHARMM CDOCKER;
SMILES strings of the retrospective virtual screening datasets used in this study;
Pose prediction result (rank-result.tsv);
Scripts for general ligand preparation.