

Abstract
Sepsis remains the single most common cause of mortality and morbidity in hospitalized patients requiring intensive care. Although earlier detection and improved treatment bundles have reduced in-hospital mortality, long-term recovery remains dismal. Sepsis survivors who experience chronic critical illness often demonstrate persistent inflammation, immune suppression, lean tissue wasting, and physical and functional cognitive declines which often last in excess of one year. Older patients and those with pre-existing comorbidities may never fully recover, and have increased mortality compared to individuals who restore their immunological homeostasis. Many of these responses are shared with individuals with advanced cancer, active autoimmune diseases, chronic obstructive pulmonary disease and chronic renal disease. Here we propose that this resulting immunological endotype is secondary to a persistent maladaptive reprioritization of myelopoiesis and pathological activation of myeloid cells. Driven in part by the continuing release of endogenous alarmins from chronic organ injury and muscle wasting, as well as by secondary opportunistic infections, ongoing myelopoiesis at the expense of lymphopoiesis and erythropoiesis leads to anemia, recurring infections and lean tissue wasting. Early recognition and intervention are required to interrupt this pathological activation of myeloid populations.
Keywords: emergency myelopoiesis, CCI, sepsis, sepsis survivors, bone marrow
Summary Sentence:
Critically ill sepsis survivor patients show specific immunological endotypes, secondary to a persistent maladaptive reprioritization of myelopoiesis and pathological activation of myeloid cells.
Graphical Abstract
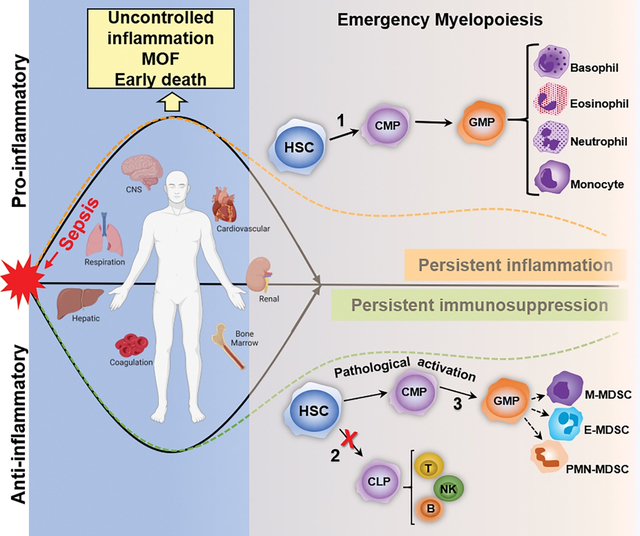
Sepsis occurs when an overwhelming pro-inflammatory response is generated and a compensatory anti-inflammatory response fail to reach homeostasis, which results in organ dysfunction and risk of death. Aberrant emergency myelopoiesis may have a critical role in the development of chronic critical illness in sepsis survivors. During sepsis, hematopoietic stem cells (HSC) respond to PAMPs/DAMPs via PRR activation and undergo expansion and self-renewal. Cytokine and chemokine production promote the rapid release of myeloid cells from bone marrow (emergency myelopoiesis [EM]), that is critical for controlling the infection. Myelopoiesis (1) predominates at the expense of common lymphoid progenitor production and lymphopoiesis (2). Critically ill patients have persistent PAMP and DAMP release, in which EM and myeloid cells are released with an immature phenotype, including MDSCs (3). Persistent MDSC (M- , PMN- , E -MDSC) expansion dampens the adaptive immune response by inhibiting T-cell proliferation and promoting Treg expansion. Persistently elevated MDSCs in sepsis survivors with CCI is a strong, independent predictor of secondary infections and worse outcomes. MOF: Multi-organ failure, CMP: Common myeloid progenitor cell, GMP: Granulocyte-monocyte progenitor cell, MDSC: CLP: Common lymphoid progenitor cell, Myeloid-derived suppressor cell, M-MDSC: Monocytic MDSC; E-MDSC: Early MDSC, PMN-MDSC: Granulocytic MDSC, T: T-cell, B: B-cell, NK: Natural killer cell. Created with Biorender.com.
1. Introduction
Sepsis is a leading cause of death from infection worldwide. In the United States, about 1.7 million sepsis cases and 270,000 sepsis-related deaths per year are reported, with estimated costs around $20 billion dollars (1–3). Sepsis, defined as a life-threatening organ dysfunction to infection, is caused by a complex host response to bacterial, viral, parasitic or fungal infections, and is significantly influenced by host factors, such as comorbidities, age, sex, and ethnicity/race, as well as their exposome and microbiome. As a highly heterogeneous syndrome, the definition of sepsis has been modified and updated multiple times since its first clinical definition in the early 1990s when the first consensus definition of sepsis was published (4). The current iteration, Sepsis-3 (Box 1), has de-emphasized the specific contributions of systemic inflammation per se, but has more broadly defined sepsis as organ injury and immune dyscrasia produced by microbial infection (5).
Box 1. Sepsis- 3: New terms and Definitions 5.
Sepsis is a life threatening organ dysfunction caused by a dysregulated host response to infection.
Organ dysfunction can be identified as an acute change in total SOFA score ≥2 as a consequence of the infection.
During sepsis, there are recognized alterations in the host immune response that results in simultaneously, both an inflammatory and immunosuppressive state. The assumption that inflammation precedes immunosuppression has been discarded only to be replaced with the concept that inflammation and immunosuppression occur simultaneously, and possibly independently (6,7). With that said, however, there are several recognized clinical trajectories that the septic patient can traverse during hospitalization. When a septic patient is admitted into the critical care unit today, approximately 20% will manifest an irreversible ‘cytokine storm’ and progress into MOF and early death (8). Fortunately, with earlier diagnoses and evidence-based treatment bundles, the majority of septic patients will recover and be discharged from the critical care unit having achieved some degree of immune homeostasis (9). Unfortunately, approximately 20–40% of sepsis survivors will not fully recover, but will progress to a phenotype of chronic critical illness (CCI) and often exhibit the persistent inflammation, immunosuppression, protein catabolism syndrome (PICS) endotype (10,11). This syndrome presents with persistent inflammation, suppression of both adaptive and innate immunity, lean tissue wasting and cachexia, and an increased frequency of secondary infections (Figure 1) (9,11–13).
Figure 1.
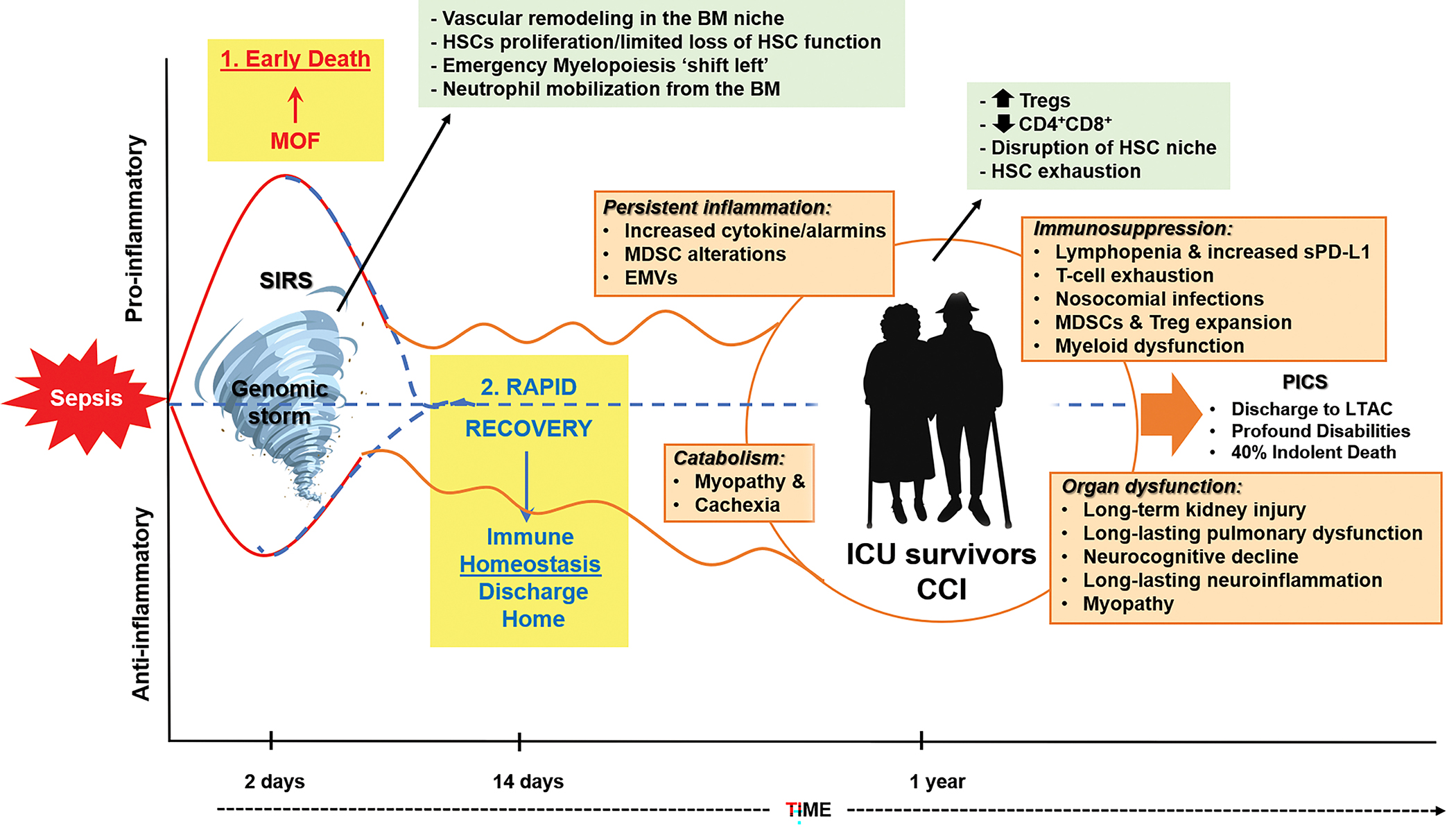
Bone Marrow changes associated with the clinical trajectories of sepsis. BM: Bone marrow; EMVs: Exosomes and microvesicles; PICS: Persistent inflammation-immunosuppression and catabolism syndrome, LTAC: Long-term acute care, MOF: Multi-organ failure, SIRS: Systemic inflammatory response syndrome, HSCs: Hematopoietic stem cells, Tregs: Regulatory T-cells, sPD-L1: Soluble programmed death-ligand 1, MDSC: Myeloid-derived suppressor cells (10–13).
The endogenous signals that drive CCI and the PICS endotype are currently unknown, but are likely multiple. Microbial products resulting from secondary infections are clearly one source, but CCI and PICS can progress in the absence of any documented infection. This has led to speculation that the initiating signals are host-derived, most likely by the release of endogenous alarmins from injured organs and atrophied skeletal muscle (9,14,15). There are several categories of these alarmins (Table 1), and in many cases are organ nonspecific, but rather reflect damaged cells and organelles, especially mitochondria. Since most of these alarmins signal through the same receptors and pathways that recognize microbial products (16), it is not surprising that many of the clinical manifestations between secondary microbial infection and chronic organ injury, secondary to trauma, burns and even severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections are frequently indistinguishable.
Table 1.
Major alarmins in sepsis.
| Alarmin | Receptors | Biological effects in sepsis | Ref. |
|---|---|---|---|
|
| |||
| HMGB1 | RAGE TLR2,4,9 | Activates innate immune effector cells. Induce a proinflammatory response | (17) |
| eCIRP | TLR4/MD2 | Induces macrophage production of proinflammatory cytokines and HMGB1 | (18) |
| Histones | TLR4 | Induce proinflammatory response | (19) |
| ATP | Purinergic receptors (P2XR, P2YR, P1R) | Disrupt neutrophil function. T-cell suppression | (20) |
| cfDNA | TLR9, AIM2, cGAS, IFI16 | Promote the release of proinflammatory cytokines. Prolong neutrophil lifespan | (21,22) |
| IL-33 | ST2 | Induces immunosuppression via induction of Tregs | (23) |
| S100 proteins | RAGE, TLRs | Induce the production of proinflammatory cytokines and neutrophils trafficking | (24,25) |
HMGB1: high mobility group box 1; RAGE: receptor for advanced glycation end-products; Toll-like receptor (TLR); eCIRP: cold-inducible RNA-binding protein; ATP: adenosine triphosphate; cfDNA: cell-free DNA; AIM2: absent in melanoma 2; cGAS cyclic GMP-AMP synthase; IFI16: interferon gamma inducible protein 16; ST-2: suppression of tumorigenicity 2
2. The immunopathology of sepsis: An overview.
2.1. Host inflammatory response to sepsis.
Microbial infection initiates a rapid release and expansion of myeloid cells owing to the release of inflammatory mediators and adrenergic stimulation (26,27). The integrated host response to sepsis is complex, shaped not only by exogenous pathogenic factors but also by a non-homeostatic host response to infections, which depends on several factors, including genetics, age, sex, pre-existing comorbidities, environment, virulence and other factors. The “dysregulated host response to an infection” included in the Sepsis-3 definition no longer relates exclusively to the initial hyperinflammatory response characterized by hyper-metabolism, shock and fever, but can now include the marked immunosuppressive phase and a failure to return to homeostasis, due to a marked acute cellular and metabolic reprogramming (5). More recently, the novel SARS-CoV-2 (cause of a serious life-threatening coronavirus disease of 2019 [COVID-19] pandemic), has emerged as a complex disease that created a major discussion about the definition of sepsis, as this new etiologic viral agent causes clinical features and hematopathological changes strikingly similar to those seen in sepsis (e.g. MOF, coagulopathy, cytokine storm, leukocytosis/leukopenia, respiratory failure, hypotension) (28). A highly dysregulated myeloid cell response, characterized by early activation of inflammatory monocytes and a later immunosuppressive myeloid cell phenotype, combined with dysregulated myelopoiesis, cause persistent inflammation, dysfunctional host defense and the development of severe COVID-19 (29).
2.2. The cytokine storm and hyperinflammation.
After infection, host innate immune cells recognize the invading pathogens via interaction between pattern recognition receptors (PRRs) and pathogen-associated molecular patterns (PAMPs), to initiate the inflammatory response. Multiple innate immune effector cells (neutrophils, monocytes/macrophages, dendritic cells [DCs], natural killer cells [NK], innate lymphoid cells [ILCs]) are involved in this initial phase of the inflammatory response, in order to reduce pathogen replication, and its dissemination from the site of infection. While these cells have a central role in eliminating the invading pathogens via phagocytosis, the cross-talk between these cells induces the production of pro-inflammatory cytokines (IL-6, IL-12, TNF-α), whose integrated goal is to delay microbial colonization and expansion (30). This sepsis-induced cytokine storm is the consequence of an uncontrolled auto-amplifying phenomenon which results in systemic effects, including, vasodilatation and hypotension, fever, neutrophil activation/degranulation, increased gluconeogenesis and lipolysis, and the generation of acute phase proteins (31).
Similarly, excessive complement activation and the uncontrolled production of anaphylatoxins have been associated with sepsis severity and enhanced long-term mortality (32). Although inhibition of the complement activation has been considered as a treatment for sepsis, it is not clear which component might be the best target to achieve protection against pathogens without compromising tissue integrity.
2.3. Dysregulated erythropoiesis and thrombocytopenia.
Bone marrow steady-state erythropoiesis maintains red blood cell homeostasis throughout life; however, this process is significantly affected in pathological conditions. During erythropoiesis, the erythroid lineage-committed cells move through different compartments (erythroid progenitors, precursors) to generate mature red blood cells (RBCs) (33). Any acute physiologic insult (e.g. inflammation) accompanied by increased oxygen demand, increases RBC production (stress erythropoiesis). This process is inhibited during infection, due to production of proinflammatory cytokines that alter bone marrow hematopoiesis (via regulating proliferation and differentiation of HSPCs) to increase the production of innate immune effector cells, at the expense of RBC production. In this regard, IL-1β and IFN-γ production, increases the expression of Pu.1 in multipotent progenitors (MPPs) leading to myeloid skewing of hematopoiesis and inhibits erythropoiesis (34). Even more, long-term epigenetic demethylation of Pu.1 has been showed after sepsis in humanized mice, which reflects alterations in the immunological milieu that leads to the emergence of comorbidities in sepsis survivors (35).
Thrombocytopenia is a common and multifactorial phenomenon occurring in critical ill patients and is particularly high in septic patients, which has been associate to poor prognosis. The mechanism behind thrombocytopenia in sepsis remains unknown. While inflammatory signaling during acute infection drives rapid maturation of megakaryocyte progenitors (MkPs) and increased platelet production (via STAT1/mTOR and TLR4/MyD88/TRIF downstream pathways), repeated cycles of proinflammatory cytokines accentuate HSC bias towards the megakaryocytic lineage, and trigger a constant maturation and a partial exhaustion of MkPs (36). Thrombocytopenia in sepsis is a consequence of a combination of several causes, including: decreased platelet production, increased platelet consumption (thrombin-mediated platelet activation, disseminated intravascular coagulation [DIC]) and platelet immune-mediated destruction (37).
2.4. Immunosuppression.
Sepsis-induced immunosuppression is characterized by T-cell exhaustion and innate immune effector cell reprogramming, increased effector T-cell apoptosis and expansion of regulatory T cells (Tregs) (38). Innate and adaptive immune effector cell apoptosis is a well-described phenomenon that contributes to immunosuppression. Early in sepsis, apoptosis causes a profound depletion of CD4+, CD8+ T cells, B cells and DCs via PD1 death receptor and/or mitochondrial-mediated pathways, leading the host more vulnerable to secondary infections and death (6). In addition, phagocytosis of apoptotic cells lead to immunotolerance via induction of anergy and epigenetic reprogramming of immune cells and the production of anti-inflammatory cytokines (IL-10 and TGFβ) (39). Sepsis-induced immunosuppression is the result of epigenetic and metabolic modifications that contribute to the reprogramming of immune cells, and that persist in subsequent cell generations; thus, it may have long-term consequences in host morbidity and mortality, even years later after the initial disease has resolved.
3. Sepsis induces dysregulated myelopoiesis.
3.1. Sepsis-mediated changes in the bone marrow niche.
The main function of the bone marrow (BM) microenvironment is to provide the necessary signals to regulate the production of blood cells required to maintain host homeostasis. This anatomical space provides the supportive environment containing mesenchymal stromal cells (leptin receptor positive [LepR+] and vascular endothelial cells) required not only for maintaining the hematopoietic cell progenitors (HPCs) and its precursors (hematopoietic stem cells, [HSCs]), but it also facilitates its differentiation into mature blood cells (through endocrine signals and cell-to-cell interactions). These life-long processes of continuous formation and turnover of blood cells generally meet host demands. In a steady-state BM niche, the HSCs have self-renewal ability and also produce common myeloid progenitors (CMPs) that give rise to megakaryocyte/erythroid progenitors (MEPs) and granulocyte-macrophage progenitors (GMPs) (Figure 2). GMPs are able to generate neutrophils, macrophages, mast cells and eosinophils. Similarly, common lymphoid progenitors (CLPs) are the source of the precursors of B, NK and T lymphocytes. The intrinsic characteristics at each stage will determine the properties of the HSCs to support their expansion and differentiation into one type of cell or another.
Figure 2.
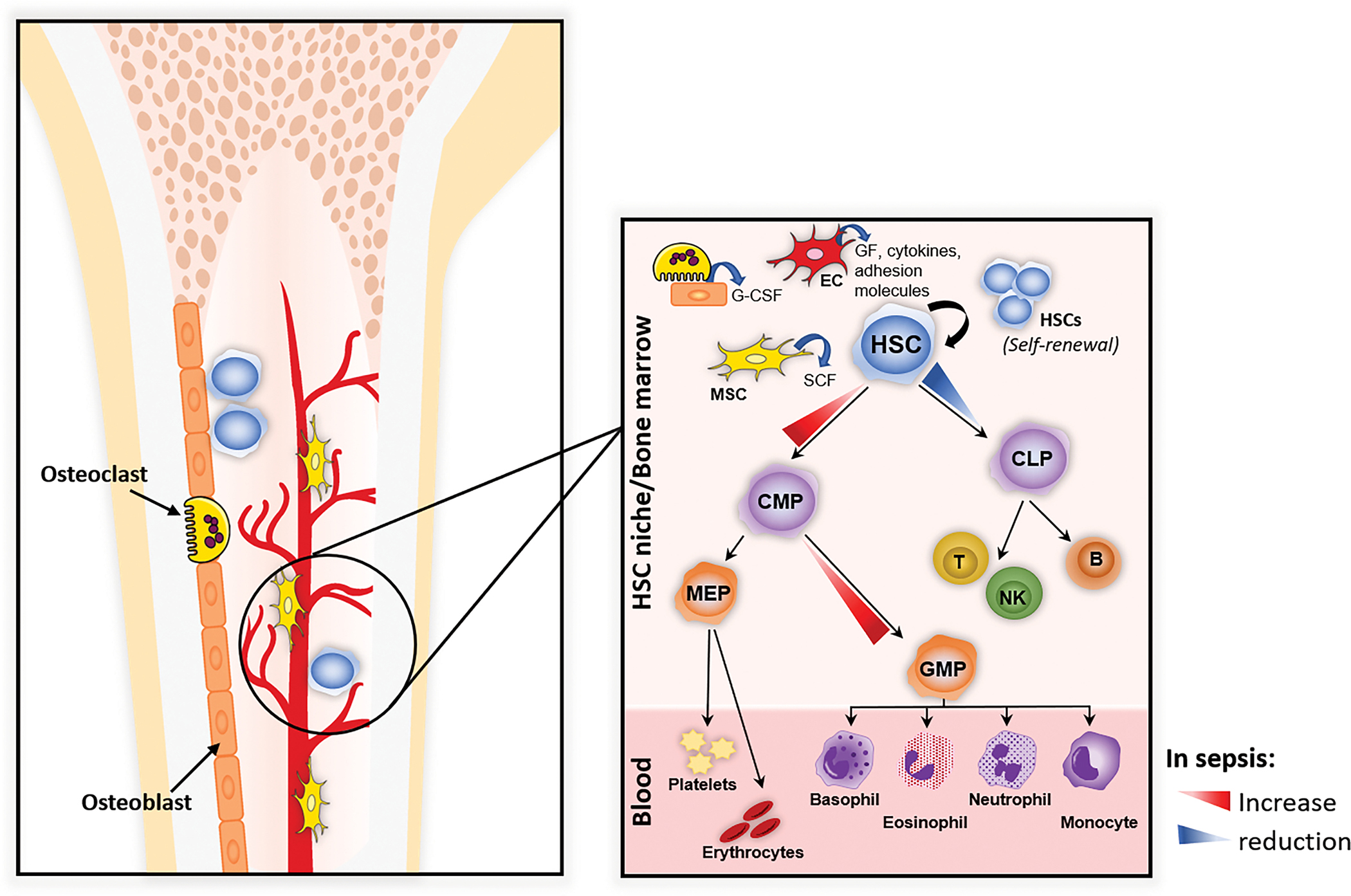
In a steady-state bone marrow (BM), the HSC niche is dynamic and provides a specialized architecture (mesenchymal stromal LepR+ cells [MSC], vascular endothelial cells [EC]) that regulates self-renewal of stem cells and the quiescent/proliferative balance of HSCs. Sepsis significantly affects the BM niche leading to quick HSC activation, which is regulated by BM stromal cells (MSC, EC, osteolineage cells), to induce multiple HSC responses (emergency granulopoiesis) and maintain its self-renewal. HSC: Hematopoietic stem cell, GF: Growth factors, SCF: Stem cell factor, G-CSF: Granulocyte colony stimulating factor, CLP: Common lymphoid progenitor cell, T: T-cell, NK: Natural killer cell, B: B-cell, CMP: Common myeloid progenitor cell, GMP: Granulocyte-monocyte progenitor cell, MEP: Megakaryocyte-erythrocyte progenitor.
However, the BM niche is highly dynamic and after acute or chronic stress (e.g. irradiation, inflammation, infection, cancer) can be significantly remodeled by the production of chemokine gradients, which organize the different cell populations into “microniches” to induce multiple responses in the HSCs to regulate hematopoiesis and maintain its self-renewal. Furthermore, PRR expression and engagement on HSCs promote their exit from quiescence and proliferation that results in their differentiation (40). The release of proinflammatory cytokines after infection rapidly induces remodeling of the vascular niche and recruitment of quiescent HSCs into cell cycle (41,42). Different stressors induce HSC activation and emergency myelopoiesis. Microbial endotoxins, for example, induce significant transcriptomic changes toward to a proinflammatory signature, pathological changes of the BM sinusoidal endothelium, HSC proliferation/renewal and neutrophil mobilization from the BM to the periphery (43). Similarly, sepsis-induced changes in BM niche have a major impact on hematopoiesis. Inflammation leads to a massive induction of granulopoiesis to fight infection but also induces anemia owing to inhibition of erythropoiesis production and a significant reduction of their life span (44). Anemia in the context of sepsis is associated with poor outcomes.
As previously mentioned, immunosuppression is one of the most important causes of late mortality in sepsis, which has been related to the expansion and enhanced function of CD4+CD25+FoxP3+ Tregs. In healthy conditions, a high frequency of Tregs in BM (compared with other T cell subpopulations), may provide the HSC niche with immune privilege mechanisms to protect HSCs from excessive inflammation (45). During sepsis, a significant reduction of Tregs and an increased number of CD4+CD8+ effector cells in the BM has been described, which is correlated with an increased number of Tregs in the periphery due to alterations of the CXCL12/CXCR4 (SDF1) axis (45). These sepsis-induced BM changes on Tregs numbers may be related to disruptions of HSC niche immune privilege status, which result in BM hematopoietic dysfunction (46).
3.2. Emergency myelopoiesis.
The hematopoietic system rapidly adapts to stress due to injury or infection by increasing the differentiation of HSCs into the cell lineages that are required in order to meet the higher cell demands. Sepsis significantly affects HSC BM niche and cell-to-cell interactions leading to a quick activation, known as ‘emergency or demand-adapted myelopoiesis’, to give rise to mature myeloid cells (neutrophils, monocytes, macrophages, DCs) as a part of an optimal response to eliminate pathogens. However, there is speculation that promotion of the acute expansion of myeloid progenitors during sepsis causes HSC dysregulation that results in failure to generate mature myeloid cells in septic mice (47). On the other hand, human data have suggested that changes on HSCs early after sepsis have a negative impact with the later production of mature myeloid cells. Furthermore, low counts of circulating hematopoietic cells have been correlated with greater probability of survival to sepsis (48,49).
During severe infection, recruitment and extravasation of neutrophils into infected tissues lead to neutrophil depletion that needs to be counterbalanced. Thus, steady-state granulopoiesis switches to emergency granulopoiesis (‘left shift’) to secure the de novo generation of neutrophils, which results from the increased MCP’s proliferation in the BM as a consequence of the systemic infection (44). Replenishment of neutrophil pools after systemic inflammation requires increased levels of growth factors to promote myelopoiesis. G-CSF, for example, is critical for neutrophil differentiation and proliferation of its precursors, as it has been shown that G-CSF deficient mice show reduced numbers of neutrophils and neutrophil precursors (granulocyte-monocyte progenitor cells [GMPs]), and have impaired neutrophil migration (50,51). GMPs (Lin−cKit+CD34hiCD16/32hi) derive from common myeloid progenitor cells (CMPs) and generate monocytes and neutrophils. In murine models of infection, emergency myelopoiesis is regulated by G-, GM- and M- CSF expression, mainly produced by endothelial cells, which not only drives myelopoiesis but also promotes HSC proliferation (50).
Similarly, proinflammatory cytokines regulate the activation of hematopoietic progenitor cells in emergency myelopoiesis. IL-1β is a major proinflammatory cytokine that induces a rapid host innate immune response against pathogens, but also has been involved in autoimmunity. IL-1β induces proliferation of HSCs via induction of the PU.1 transcription factor, which regulates the expression of GM-CSF and M-CSF receptors, and myeloid-biased hematopoiesis. On the other hand, chronic IL-1 exposure may also significantly impair HSC self-renewal and lineage output (52). Similarly, IL-27, a member of the IL-6/IL-12 family, promotes the early expansion and differentiation of human CD34+ HSCs into myeloid progenitor cells, which have high potential to differentiate into neutrophils, myeloid DCs (mDC) and mast cells (53). IL-27 is a unique cytokine in emergency myelopoiesis as it directly acts on the most primitive HSCs (LT-HSCs) (53). Together with IL-27, IL-6 plays a critical role in hematopoiesis under inflammatory conditions (54). Human CD34+ cells isolated from peripheral blood, produce IL-6 after endotoxin stimulation and further promote their own differentiation in vitro; while in vivo studies using IL-6 null mice show dysregulated HSC proliferation and reduced capacity of self-renewal (54). In addition, IL-6, IL-3, M-CSF and GM-CSF, synergistically promote proliferation of HSCs in vitro and these cytokines show pleiotropic effects on the maturation and survival of myeloid cells in vivo (55).
3.3. Hematopoietic stem cell (HSC) exhaustion.
In healthy conditions, HSCs maintain a quiescent state that rapidly turn into a highly proliferative phase in response to acute infection (56). During sepsis, we speculate that there is often an inadequate host BM response to infection that initiates with a dramatic neutrophilia and hyper-inflammatory response, followed by neutropenia and leukocyte dysfunction with the subsequent inability to control the infection. The exit of HSC homeostasis is a highly regulated process by inflammatory/anti-inflammatory mediators and cell-to-cell interactions in the BM niche, and it is proposed that aberrant activation of these cells may induce hematopoietic failure that results in chronic inflammatory blood diseases and malignancies (57,58). During localized infection, innate immune effector cells are quickly activated and consumed but efficiently replenished by HSC activation. In contrast, it is proposed that severe systemic infection or sepsis induces rapid ablation of osteoblasts, which correlates with a reduced numbers of common lymphoid progenitors (CLPs), BM aplasia, dysfunctional HSCs and inefficient hematopoiesis (44).
Though inflammatory cytokines induce HSC division, self-renewal and mobilization, we have speculated that if this process persists it can lead to the exhaustion of HSCs over time. Chronic infection and persistent inflammation can lead to pancytopenia, depletion of BM HSCs and impaired self-renewal of these cells (59), thus HSCs quiescence-proliferation may represent a balance during infection to sustain hematopoiesis and host long-term survival. The prolonged exposure to IL-1, TNF and type-I IFNs induces chronic activation of HSCs that results in impaired function and exhaustion in vivo (41,52,60). In addition, modifications of the epigenome (e.g. DNA methylation, histone acetylation, non-coding RNAs) have been shown to have a major role influencing immune cell differentiation (61,62). After sepsis, murine peripheral monocyte/macrophages show an impaired inflammatory phenotype that results in delayed wound repair and these changes are related to epigenetic modifications of HSCs in the BM (63). Human data have shown similar findings, as patients who survive sepsis show decreased MLL1 expression (histone methyltransferase that regulates cytokine expression in wound healing) (63). Even more, septic patients have increased numbers of circulating primitive CD34+CD38− and Lin−CD133+CD45+ (49), which has been correlated with an increased risk of death (63,64); however, the mechanistic link between dysfunctional hematopoiesis and sepsis course is still unknown.
3.4. Myeloid-derived suppressor cell expansion.
Even after full clinical recovery, one (of many) important long-term complication in patients with sepsis is their impaired immune response that leads to secondary infections, greater risk of rehospitalization and death. Many of the myeloid cells produced in the BM, as a consequence of sepsis, never reach a mature phenotype and, therefore, these cells are released into the circulation as immature myeloid cells (IMCs).
During systemic infection, soluble mediators (CSF, IL-6, IL-1, IFNs, ROS) trigger the mobilization of IMCs from bone marrow to activate the emergency myelopoiesis response to replenish mature leukocytes with IMCs, which are morphologically similar to fully mature myeloid cells but they are immunosuppressive (65). Dysregulated inflammation, epigenetic modifications, metabolic imbalance and BM niche remodeling occurring in sepsis, result in the need of HSCs to adapt their proliferation/differentiation rate to meet the increased demand of the host immune response to the infection.
Myeloid-derived suppressor cells (MDSCs) in adults are phenotypically and functionally non-fully matured myeloid cells that are immunosuppressive and expand during chronic and acute inflammation from long-term HSCs. The expansion of MDSCs starts with a significant production of IMCs in the BM directed by the host immune response to eliminate the pathogen. Later, these myeloid cells become ‘pathologically activated’ via transcriptional regulators of MDSC biology, including NF-kB, STAT1, STAT3, STAT6, PGE2, COX2 (66).
MDSCs disrupt T-cell function via multiple pathways, including arginase-mediated arginine depletion with consecutive inhibition of the T-cell and NK cell metabolism (67). In addition, though MDSCs show impaired phagocytosis they can produce high amounts of reactive oxygen/nitrogen species (ROS, RNS), NO, anti-inflammatory cytokines (IL-10, TGF-β) and activate Tregs (Figure 3) (67). Within sepsis, MDSC expansion during the early stage may be beneficial by protecting the host from hyper-inflammation and organ dysfunction. On the other hand, MDSCs can be detrimental as their persistent expansion inhibits the adaptive immune response via T-cell suppression that leads to long-term complications (10,68).
Figure 3.
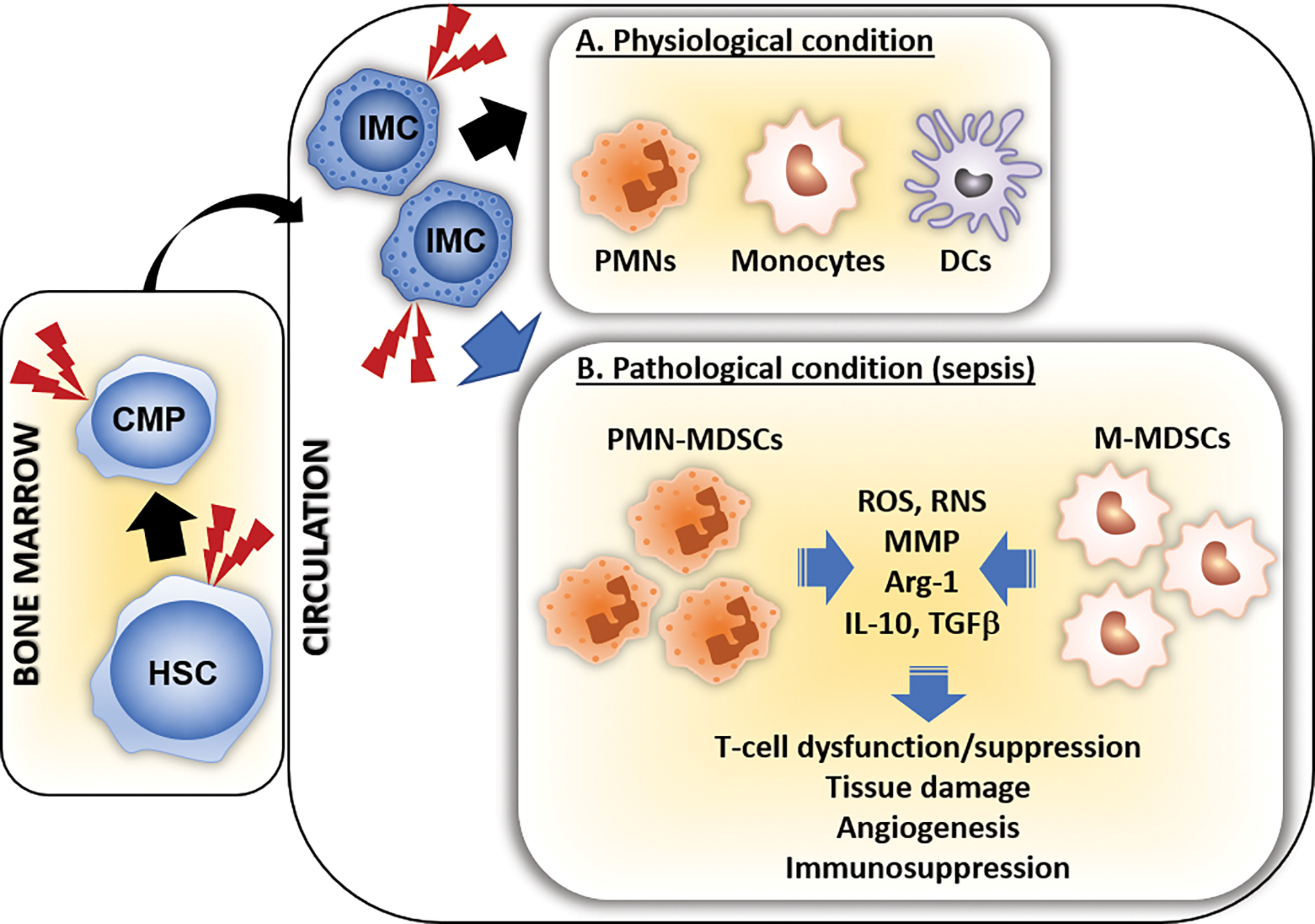
MDSC expansion in sepsis. A. During physiological conditions of myelopoiesis, hematopoietic stem cells (HSCs) differentiate into common myeloid progenitor cells (CMPs) and then into mature innate immune effector cells (PMNs, monocytes, DCs). B. The expansion of MDSCs starts with the increased production of immature myeloid cells (IMCs), initially directed by the host immune response to eliminate the pathogen. The prolonged inflammatory response (red lightning bolts) in sepsis induces the release of soluble mediators (growth factors, cytokines, PAMPs, DAMPs) that induce pathologically activation/differentiation of these IMCs into MDSCs. Activated MDSCs have strong immunoregulatory effects via production of reactive oxygen/nitrogen species (ROS, RNS), matrix metalloproteinases (MMP), Arginase-1 (Arg-1) and proinflammatory mediators (59). PMN-MDSCs: Granulocytic myeloid-derived suppressor cells, M-MDSCs: Monocytic myeloid-derived suppressor cells.
The balance between the pro-inflammatory and immunosuppressive roles of MDSCs is modulated by multiple environmental signals that also regulate their expansion. MDSCs are highly plastic as they can maintain their phenotype and show less or more immunosuppressive function, and also, they can differentiate into more mature myeloid cells with varying activities. Thus, different conditions or pathologies may induce these cells to adapt to a particular milieu, which would result in a unique polarization that will determine their function (69).
3.5. Chronic critical illness and long-term morbidity and mortality after sepsis.
Chronic critical illness (CCI) is an emerging phenotype of approximately 30% of sepsis survivors, characterized by prolonged intensive care unit (ICU) stays, low-grade persistent inflammation and chronic immunosuppression. It has been reported that 5–8% of all ICU admissions develop CCI, accounting for more than 300,000 annual cases and over USD $26 billion in health care costs (70). CCI patients have shown poor long-term outcomes, including post-discharge health care resource utilization, low health-related quality of life and higher long-term mortality rates (71). The underlying pathobiology of CCI is a maladaptive self-perpetuating cycle of persistent inflammation, where an acquired BM failure is no longer able to support blood homeostasis.
Following sepsis, the rapid mobilization of neutrophils from the BM to the site of infection and the activation of BM microenvironment cells, lead to self-renewal and differentiation of HSCs to supply the required myeloid cells to fight the infection. CCI is the clinical manifestation of the disrupted ability of the host immune system to return to homeostasis. The immunological endotype that best defines this response has been termed “Persistent Inflammatory, immunosuppressive and protein Catabolic Syndrome (PICS)”, characterized not only by a persistent inflammation, but also the massive release of alarmins (as result of organ injury) which induces the expansion of MDSCs and Tregs as a compensatory anti-inflammatory mechanism, with a subsequent increase of secondary infections (10,11,72). This persistent inflammation is further associated with a significant lean muscle mass wasting associated with: decreased muscle autophagia, aberrant up-regulation of genes involved in structural and functional muscle development, decreased numbers of satellite cells, impaired muscle regeneration, mitochondrial dysfunction (satellite cells) and abnormalities in calcium homeostasis (channelopathies). Thus, catabolism, malnutrition and persistent inflammation could be a possible explanation for the observed accelerated aging of limb muscles and sarcopenia in ICU survivors (inflamm-aging) (Figure 4) (73,74).
Figure 4.
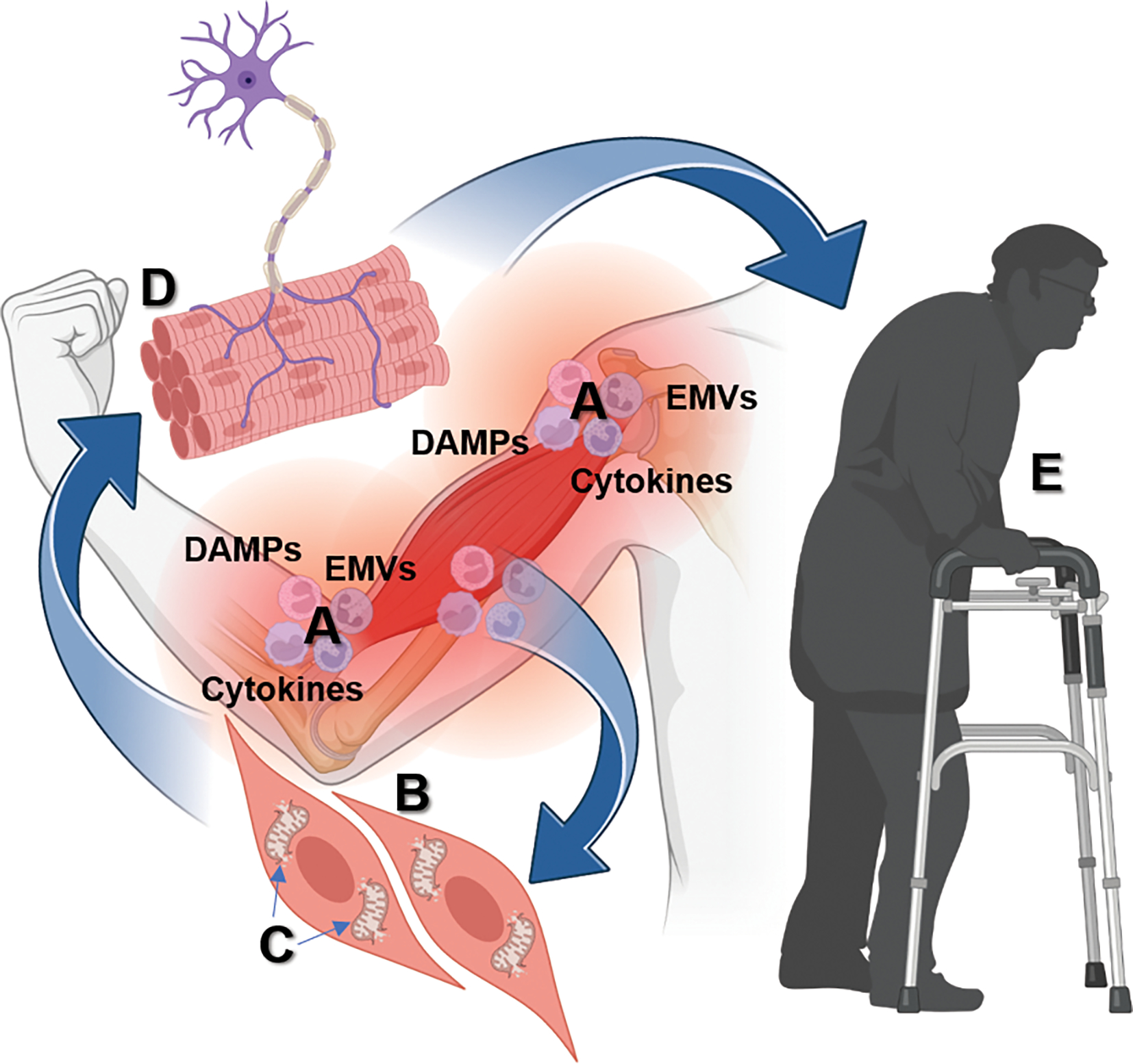
Sepsis survivors exhibit an imbalance between anabolism and catabolism that leads to muscle weakness and wasting. Persistent inflammation (A) after sepsis is associated with an impaired regenerative capacity of muscle cells (B), damaged mitochondria that result in the release of alarmins, which promotes more inflammation and MDSC expansion (B), altered neuromuscular junction (D), which trigger persistent myopathy in sepsis survivors characterized by skeletal muscle weakness (E). DAMPs: Damage-associated molecular patters, EMVs: Exosomes and microvesicles. Created with Biorender.com.
The maladaptive immunological dyscrasia shown in CCI/PICS is caused by multiple cellular and redundant molecular pathways that promote the expansion and persistence of MDSCs. In addition, these patients show suppression of protective immunity via lymphopenia, T-cell exhaustion, increased appearance of Tregs, increased production of soluble PD-L1, and reduced HLA-DR expression, which have been also associated to an increased susceptibility to secondary infections and all-cause mortality (13). The simultaneous activation of these cellular and molecular mechanisms in the CCI patient, creates the ideal environment to promote aberrant myelopoiesis which in turn prolongs immunosuppression and the subsequent susceptibility to secondary infections. More recently, gene expression and histone modification changes of exhausted murine monocytes have shown expression profiles of pathogenic inflammation and immunosuppression, characterized by CD38 expression, NAD+ depletion, ROS production and impaired mitochondria respiration via TRAM-dependent pathway of TLR4 (75). Furthermore, TRAM may diverge with TRIF under exhaustive conditions (e.g. sepsis) to facilitate monocyte exhaustion through elevated STAT1/IRF7 (75,76).
Sepsis causes multiple alterations in cellular function that lead to the development of tissue and organ dysfunction. Despite more than 30 years of research and multiple clinical trials, and significant advances in the understanding of the immunopathology of sepsis, long-term sepsis mortality remains significantly high and there is no single treatment that persistently save lives in septic patients (77); instead, sepsis treatment includes early administration of antimicrobials and fluids, ventilation, and management of blood flow to protect vital organ function (78). As not all individuals respond to the same antigen challenge similarly, individualized genomics and/or immune effector cell profiling may provide a clear analysis of the patient health status to design the best treatment for them.
Concluding remarks.
Post-sepsis immunopathology is complex and often associated with long-term dismal outcomes in sepsis survivors. A persistent pro-inflammatory/anti-inflammatory interplay characterized by highly dynamic changes in the BM (caused by inflammatory mediators), induce multiple responses in HSCs and a maladaptive immunological dyscrasia seen in post-septic critically ill patients.
Currently, there are no specific therapeutic interventions for CCI/PICS; nevertheless, precision medicine has emerged as an attractive approach to incorporate different factors (genomic, environmental, biologic) into a better therapeutic strategy to improve outcomes of patients with sepsis. With the increasing availability of epigenomic, metabolomic, and proteomic-based tools to elucidate the molecular basis of host variability, we are getting closer to providing septic patients a personalized treatment. Nevertheless, it is critically important to determine the functional immune status of each septic patient, which is a major unmet medical need to succeed in the development of new sepsis therapeutics.
Acknowledgments
Supported in part by grants RM1 GM139690, awarded to LLM, and R35 GM140806, awarded to PAE, from the National Institute of General Medical Sciences, NIH HHS.
Abbreviations
- MOF
multi-organ failure
- CCI
chronic critical illness
- PICS
persistent inflammation, immunosuppression, protein catabolism syndrome
- TLRs
toll-like receptors
- NLRs
nod-like receptors
- CLRs
C-type lectins receptors
- RLRs
RIG-I-like receptors
- cGas/STING
cyclic GMP-AMP synthase – stimulator of interferon genes
- PRRs
patter recognition receptors
- PAMPs
pathogen-associated molecular patterns
- DAMPs
damage-associated molecular patterns
- NETs
neutrophil extracellular traps
- ROS
reactive oxygen species
- RBCs
red blood cells
- HSPCs
hematopoietic stem and progenitor cells
- Pu.1
transcription factor with multiple roles in hematopoiesis
- MPPs
multipotent progenitors
- PD1
programmed cell death protein 1
- PD-L1
programmed cell death protein 1 ligand
- HPCs
hematopoietic progenitor cells
- HSCs
hematopoietic stem cells
- CMPs
common myeloid progenitor cells
- GMPs
granulocyte-monocyte progenitor cells
- CLPs
common lymphoid progenitor cells
- MEPs
megakaryocyte-erythroid progenitor cells
- IMCs
immature myeloid cells
- MLL1
mixed lineage leukemia 1/histone-H3 lysine-4 (H3K4) methyltransferase
- MDSCs
myeloid-derived suppressor cells
- EMVs
exosomes and microvesicles
Footnotes
Disclosures
The authors declare no conflict of interest
References
- 1.Paoli CJ, Reynolds MA, Sinha M, Gitlin M, Crouser E. Epidemiology and Costs of Sepsis in the United States-An Analysis Based on Timing of Diagnosis and Severity Level. Crit Care Med. 46(12):1889–1897, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Judd WR, Stephens DM, Kennedy CA. Clinical and economic impact of a quality improvement initiative to enhance early recognition and treatment of sepsis. Ann Pharmacother. 48(10):1269–1275, 2014. [DOI] [PubMed] [Google Scholar]
- 3.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. Jama. 318(13):1241–1249, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 20(6):864–874, 1992. [PubMed] [Google Scholar]
- 5.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama. 315(8):801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 13(12):862–874, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, et al. A genomic storm in critically injured humans. J Exp Med. 208(13):2581–2590, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchman TG, Simpson SQ, Sciarretta KL, Finne KP, Sowers N, Collier M, Chavan S, Oke I, Pennini ME, Santhosh A, et al. Sepsis Among Medicare Beneficiaries: 3. The Methods, Models, and Forecasts of Sepsis, 2012–2018. Crit Care Med. 48(3):302–318, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, Efron PA, Bihorac A, Segal M, Moore FA, et al. Chronic Critical Illness and the Persistent Inflammation, Immunosuppression, and Catabolism Syndrome. Front Immunol. 9:1511, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, Moore FA, Moldawer LL. Sepsis Pathophysiology, Chronic Critical Illness, and Persistent Inflammation-Immunosuppression and Catabolism Syndrome. Crit Care Med. 45(2):253–262, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA. Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg. 72(6):1491–1501, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stortz JA, Mira JC, Raymond SL, Loftus TJ, Ozrazgat-Baslanti T, Wang Z, Ghita GL, Leeuwenburgh C, Segal MS, Bihorac A, et al. Benchmarking clinical outcomes and the immunocatabolic phenotype of chronic critical illness after sepsis in surgical intensive care unit patients. J Trauma Acute Care Surg. 84(2):342–349, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stortz JA, Murphy TJ, Raymond SL, Mira JC, Ungaro R, Dirain ML, Nacionales DC, Loftus TJ, Wang Z, Ozrazgat-Baslanti T, et al. Evidence for Persistent Immune Suppression in Patients Who Develop Chronic Critical Illness After Sepsis. Shock. 49(3):249–258, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brudecki L, Ferguson DA, McCall CE, El Gazzar M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect Immun. 80(6):2026–2034, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 19(2):108–119, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang D, Han Z, Oppenheim JJ. Alarmins and immunity. Immunol Rev. 280(1):41–56, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Ward MF, Sama AE. Targeting HMGB1 in the treatment of sepsis. Expert Opin Ther Targets. 18(3):257–268, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiang X, Yang WL, Wu R, Zhou M, Jacob A, Dong W, Kuncewitch M, Ji Y, Yang H, Wang H, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 19(11):1489–1495, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ekaney ML, Otto GP, Sossdorf M, Sponholz C, Boehringer M, Loesche W, Rittirsch D, Wilharm A, Kurzai O, Bauer M, et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit Care. 18(5):543, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ledderose C, Bao Y, Kondo Y, Fakhari M, Slubowski C, Zhang J, Junger WG. Purinergic Signaling and the Immune Response in Sepsis: A Review. Clin Ther. 38(5):1054–1065, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harrington JS, Choi AMK, Nakahira K. Mitochondrial DNA in Sepsis. Curr Opin Crit Care. 23(4):284–290, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng Z, Abrams ST, Toh J, Wang SS, Wang Z, Yu Q, Yu W, Toh CH, Wang G. The Critical Roles and Mechanisms of Immune Cell Death in Sepsis. Front Immunol. 11:1918, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrow KN, Coopersmith CM, Ford ML. IL-17, IL-27, and IL-33: A Novel Axis Linked to Immunological Dysfunction During Sepsis. Front Immunol. 10:1982, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding Z, Du F, Averitt VR, Jakobsson G, Rönnow CF, Rahman M, Schiopu A, Thorlacius H. Targeting S100A9 Reduces Neutrophil Recruitment, Inflammation and Lung Damage in Abdominal Sepsis. Int J Mol Sci. 22(23), 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Timmermans K, Kox M, Scheffer GJ, Pickkers P. DANGER IN THE INTENSIVE CARE UNIT: DAMPS IN CRITICALLY ILL PATIENTS. Shock. 45(2):108–116, 2016. [DOI] [PubMed] [Google Scholar]
- 26.Cosentino M, Marino F, Maestroni GJ. Sympathoadrenergic modulation of hematopoiesis: a review of available evidence and of therapeutic perspectives. Front Cell Neurosci. 9:302, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, Moore FA, Brakenridge SC, Brumback BA, Moldawer LL, et al. Human Myeloid-derived Suppressor Cells are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann Surg. 265(4):827–834, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olwal CO, Nganyewo NN, Tapela K, Djomkam Zune AL, Owoicho O, Bediako Y, Duodu S. Parallels in Sepsis and COVID-19 Conditions: Implications for Managing Severe COVID-19. Front Immunol. 12:602848, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulte-Schrepping J, Reusch N, Paclik D, Baßler K, Schlickeiser S, Zhang B, Krämer B, Krammer T, Brumhard S, Bonaguro L, et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell. 182(6):1419–1440.e1423, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivera A, Siracusa MC, Yap GS, Gause WC. Innate cell communication kick-starts pathogen-specific immunity. Nat Immunol. 17(4):356–363, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. 17(7):407–420, 2017. [DOI] [PubMed] [Google Scholar]
- 32.Mollnes TE, Huber-Lang M. Complement in sepsis-when science meets clinics. FEBS Lett. 594(16):2621–2632, 2020. [DOI] [PubMed] [Google Scholar]
- 33.An X, Mohandas N. Erythroblastic islands, terminal erythroid differentiation and reticulocyte maturation. Int J Hematol. 93(2):139–143, 2011. [DOI] [PubMed] [Google Scholar]
- 34.Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 130(15):1693–1698, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lapko N, Zawadka M, Polosak J, Worthen GS, Danet-Desnoyers G, Puzianowska-Kuźnicka M, Laudanski K. Long-term Monocyte Dysfunction after Sepsis in Humanized Mice Is Related to Persisted Activation of Macrophage-Colony Stimulation Factor (M-CSF) and Demethylation of PU.1, and It Can Be Reversed by Blocking M-CSF In Vitro or by Transplanting Naïve Autologous Stem Cells In Vivo. Front Immunol. 8:401, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haas S, Hansson J, Klimmeck D, Loeffler D, Velten L, Uckelmann H, Wurzer S, Prendergast ÁM, Schnell A, Hexel K, et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell. 17(4):422–434, 2015. [DOI] [PubMed] [Google Scholar]
- 37.Claushuis TA, van Vught LA, Scicluna BP, Wiewel MA, Klein Klouwenberg PM, Hoogendijk AJ, Ong DS, Cremer OL, Horn J, Franitza M, et al. Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients. Blood. 127(24):3062–3072, 2016. [DOI] [PubMed] [Google Scholar]
- 38.Cao C, Yu M, Chai Y. Pathological alteration and therapeutic implications of sepsis-induced immune cell apoptosis. Cell Death Dis. 10(10):782, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green DR, Beere HM. Apoptosis. Gone but not forgotten. Nature. 405(6782):28–29, 2000. [DOI] [PubMed] [Google Scholar]
- 40.Capitano ML. Toll-like receptor signaling in hematopoietic stem and progenitor cells. Curr Opin Hematol. 26(4):207–213, 2019. [DOI] [PubMed] [Google Scholar]
- 41.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 458(7240):904–908, 2009. [DOI] [PubMed] [Google Scholar]
- 42.Prendergast ÁM, Kuck A, van Essen M, Haas S, Blaszkiewicz S, Essers MA. IFNα-mediated remodeling of endothelial cells in the bone marrow niche. Haematologica. 102(3):445–453, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandoorne K, Rohde D, Kim HY, Courties G, Wojtkiewicz G, Honold L, Hoyer FF, Frodermann V, Nayar R, Herisson F, et al. Imaging the Vascular Bone Marrow Niche During Inflammatory Stress. Circ Res. 123(4):415–427, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glatman Zaretsky A, Engiles JB, Hunter CA. Infection-induced changes in hematopoiesis. J Immunol. 192(1):27–33, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Day RB, Bhattacharya D, Nagasawa T, Link DC. Granulocyte colony-stimulating factor reprograms bone marrow stromal cells to actively suppress B lymphopoiesis in mice. Blood. 125(20):3114–3117, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 474(7350):216–219, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang J, Saijo K, Skola D, Jin C, Ma Q, Merkurjev D, Glass CK, Rosenfeld MG. Histone demethylase LSD1 regulates hematopoietic stem cells homeostasis and protects from death by endotoxic shock. Proc Natl Acad Sci U S A. 115(2):E244–e252, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsaganos T, Giamarellos-Bourboulis EJ, Kollias S, Zervakis D, Karagianni V, Pelekanou A, Tampaki EC, Kontogiorgi M, Koroneos A, Drakoulis N, et al. Kinetics of progenitor hemopoetic stem cells in sepsis: correlation with patients survival? BMC Infect Dis. 6:142, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skirecki T, Mikaszewska-Sokolewicz M, Godlewska M, Dołęgowska B, Czubak J, Hoser G, Kawiak J, Zielińska-Borkowska U. Mobilization of Stem and Progenitor Cells in Septic Shock Patients. Sci Rep. 9(1):3289, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 84(6):1737–1746, 1994. [PubMed] [Google Scholar]
- 51.Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. 5(5):491–501, 1996. [DOI] [PubMed] [Google Scholar]
- 52.Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 18(6):607–618, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seita J, Asakawa M, Ooehara J, Takayanagi S, Morita Y, Watanabe N, Fujita K, Kudo M, Mizuguchi J, Ema H, et al. Interleukin-27 directly induces differentiation in hematopoietic stem cells. Blood. 111(4):1903–1912, 2008. [DOI] [PubMed] [Google Scholar]
- 54.Bernad A, Kopf M, Kulbacki R, Weich N, Koehler G, Gutierrez-Ramos JC. Interleukin-6 is required in vivo for the regulation of stem cells and committed progenitors of the hematopoietic system. Immunity. 1(9):725–731, 1994. [DOI] [PubMed] [Google Scholar]
- 55.Mitroulis I, Kalafati L, Hajishengallis G, Chavakis T. Myelopoiesis in the Context of Innate Immunity. J Innate Immun. 10(5–6):365–372, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol. 10(3):201–209, 2010. [DOI] [PubMed] [Google Scholar]
- 57.Mirantes C, Passegué E, Pietras EM. Pro-inflammatory cytokines: emerging players regulating HSC function in normal and diseased hematopoiesis. Exp Cell Res. 329(2):248–254, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clapes T, Lefkopoulos S, Trompouki E. Stress and Non-Stress Roles of Inflammatory Signals during HSC Emergence and Maintenance. Front Immunol. 7:487, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matatall KA, Jeong M, Chen S, Sun D, Chen F, Mo Q, Kimmel M, King KY. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep. 17(10):2584–2595, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pronk CJ, Veiby OP, Bryder D, Jacobsen SE. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J Exp Med. 208(8):1563–1570, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 15(2):163–176, 2005. [DOI] [PubMed] [Google Scholar]
- 62.Stability Reik W. and flexibility of epigenetic gene regulation in mammalian development. Nature. 447(7143):425–432, 2007. [DOI] [PubMed] [Google Scholar]
- 63.Davis FM, Schaller MA, Dendekker A, Joshi AD, Kimball AS, Evanoff H, Wilke C, Obi AT, Melvin WJ, Cavassani K, et al. Sepsis Induces Prolonged Epigenetic Modifications in Bone Marrow and Peripheral Macrophages Impairing Inflammation and Wound Healing. Arterioscler Thromb Vasc Biol. 39(11):2353–2366, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nacionales DC, Szpila B, Ungaro R, Lopez MC, Zhang J, Gentile LF, Cuenca AL, Vanzant E, Mathias B, Jyot J, et al. A Detailed Characterization of the Dysfunctional Immunity and Abnormal Myelopoiesis Induced by Severe Shock and Trauma in the Aged. J Immunol. 195(5):2396–2407, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schrijver IT, Théroude C, Roger T. Myeloid-Derived Suppressor Cells in Sepsis. Front Immunol. 10:327, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Condamine T, Mastio J, Gabrilovich DI. Transcriptional regulation of myeloid-derived suppressor cells. J Leukoc Biol. 98(6):913–922, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 5(1):3–8, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. 14(2):121–137, 2018. [DOI] [PubMed] [Google Scholar]
- 69.Sanchez-Pino MD, Dean MJ, Ochoa AC. Myeloid-derived suppressor cells (MDSC): When good intentions go awry. Cell Immunol. 362:104302, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kahn JM, Le T, Angus DC, Cox CE, Hough CL, White DB, Yende S, Carson SS. The epidemiology of chronic critical illness in the United States*. Crit Care Med. 43(2):282–287, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gardner AK, Ghita GL, Wang Z, Ozrazgat-Baslanti T, Raymond SL, Mankowski RT, Brumback BA, Efron PA, Bihorac A, Moore FA, et al. The Development of Chronic Critical Illness Determines Physical Function, Quality of Life, and Long-Term Survival Among Early Survivors of Sepsis in Surgical ICUs. Crit Care Med. 47(4):566–573, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, Pachot A, Brooks TL, Deych E, Shannon WD, et al. Reactivation of multiple viruses in patients with sepsis. PLoS One. 9(2):e98819, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voiriot G, Oualha M, Pierre A, Salmon-Gandonnière C, Gaudet A, Jouan Y, Kallel H, Radermacher P, Vodovar D, Sarton B, et al. Chronic critical illness and post-intensive care syndrome: from pathophysiology to clinical challenges. Ann Intensive Care. 12(1):58, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E, De Benedictis G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 908:244–254, 2000. [DOI] [PubMed] [Google Scholar]
- 75.Pradhan K, Yi Z, Geng S, Li L. Development of Exhausted Memory Monocytes and Underlying Mechanisms. Front Immunol. 12:778830, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naler LB, Hsieh YP, Geng S, Zhou Z, Li L, Lu C. Epigenomic and transcriptomic analyses reveal differences between low-grade inflammation and severe exhaustion in LPS-challenged murine monocytes. Commun Biol. 5(1):102, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rello J, Valenzuela-Sánchez F, Ruiz-Rodriguez M, Moyano S. Sepsis: A Review of Advances in Management. Adv Ther. 34(11):2393–2411, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Evans L, Rhodes A, Alhazzani W, Antonelli M, Coopersmith CM, French C, Machado FR, McIntyre L, Ostermann M, Prescott HC, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 47(11):1181–1247, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]