

Abstract
Cysteine bioconjugation serves as a powerful tool in biological research and has been widely used for the chemical modification of proteins, constructing antibody-drug conjugates, and enabling cell imaging studies. Cysteine conjugation reactions with fast kinetics and exquisite selectivity have been under heavy pursuit as they would allow clean protein modification with just stoichiometric amounts of reagents, which minimizes side reaction, simplifies purification and broadens the functional group tolerance. In this concept, we summarize the recent advances in fast cysteine bioconjugation, and discuss the mechanism and chemical principles that underlie the high efficiencies of the newly developed cysteine reactive reagents.
Keywords: Cysteine bioconjugation, Fast kinetic, Protein modification, Reaction rate constant, N-terminal cysteine
Graphical Abstract
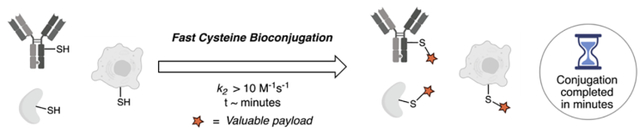
This Concept article summarizes the recently developed fast cysteine bioconjugation reactions, with a focus on the mechanisms and design principles that underlie the high efficiency of the new cysteine-modifying reagents.
Graphical Abstract
Introduction
Cysteine bioconjugation refers to the use of cysteine as a reactive linker to conjugate proteins with synthetic functional groups under physiological conditions. The synthetic functional groups could be fluorophores, pharmacophores, or bioorthogonal handles (Figure 1A). Cysteine bioconjugation has been one of the default choices for modifying proteins in biological research as well as in the development of protein-based therapeutics.[1] Among the 20 canonical amino acids, cysteine has a relatively low abundance (1.9% in protein, mainly exists as disulfide) and a strong nucleophilic side chain (pKa ~ 8.5 for cysteine sulfhydryl group).[2] These features make cysteine an ideal residue for the chemo-selective modification of proteins. Cysteine-specific bioconjugation of proteins in vivo remains quite challenging due to the high concentration of endogenous cysteine species: glutathione is present in live cells with a concentration of 1–10 mM, in addition to some other thiol species at lower concentrations (e.g., free cysteine, 30–200 μM).[3] However, much success has been documented for cysteine bioconjugation in in vitro settings. For example, 11 antibody-drug conjugates (ADCs) have been approved by the Food and Drug Administration (FDA) to date, and most of these ADCs were constructed via cysteine-specific bioconjugation to link cytotoxic drugs to a monoclonal antibody (mAb), which allows selective delivery of the drugs to tumor cells.[4] Other than ADCs, cysteine bioconjugation has also been used for installing protein post-translational modifications (PTMs) mimics[5], peptide stapling[6], phage library modification[7], and living cell imaging[1a, 8].
Figure 1.
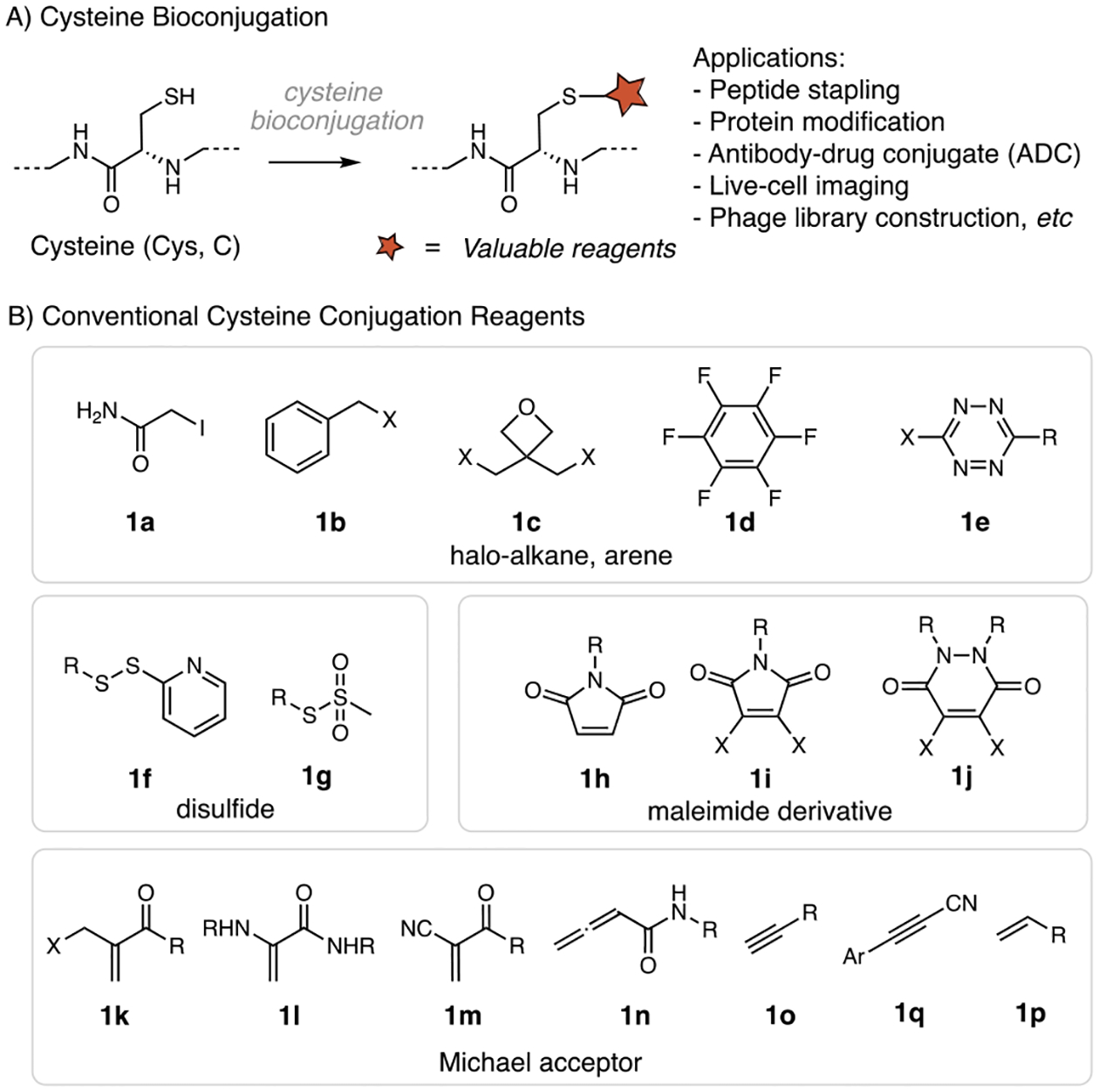
Cysteine bioconjugation and some commonly used reagents.
Numerous papers in the literature document the development and characterization of cysteine-reactive reagents.[1a, 9] Some of the most commonly used cysteine-modifying reagents include activated alkyl halides[10] (such as iodoacetamide 1a) and maleimide derivatives 1h-1j[11] (Figure 1B). Various other electrophilic reagents are also developed for cysteine labeling, such as active disulfides 1f-1g[12], perfluoroarene 1d[6], and Michael acceptors 1k-1p[13]. Iodoacetamides are frequently used for cysteine conjugation, although the reaction rate is relatively slow and typically requires a high (millimolar) concentration of reagents. Cysteine conjugation to maleimides shows high efficiency and fast kinetics. The maleimide reagents are easy to synthesize and handle, and various derivatives are commercially available that carry fluorophore labels, biotin tags, and bio-orthogonal handles. However, the thiol-succinimide adducts show poor stability towards hydrolysis in aqueous media, which can complicate biological applications.[11] New cysteine conjugation methods with fast kinetics and enhanced product stability are still highly desirable to overcome the limitations mentioned above.
Ideally, a bioconjugation reaction should be performed with just one equivalent of the coupling reagent to the protein substrate within a short time frame. As protein solutions are generally low in concertation, the conjugation reaction would have to be fast in kinetics to ensure speedy completion of the conjugation reaction. Importantly, a fast cysteine bioconjugation would only require a low concentration of reagents, which can minimize side reactions, simplify purification and broaden the functional group tolerance. For live cell applications, a low concentration of the labeling reagents would minimize their cytotoxicity as well. Finally, in comparison to iodoacetamide, a fast cysteine-reactive warhead was able to reveal a greater number of reactive cysteines in proteomic profiling studies.[14] With this Concept, we seek to summarize the recent advances in fast cysteine conjugation reactions with a special focus on the design principles of the fast thiol-reactive reagents. Biological applications are not the focus of this contribution and are only discussed briefly in relevant contexts.
General Considerations of Fast Cysteine Bioconjugation
Definition.
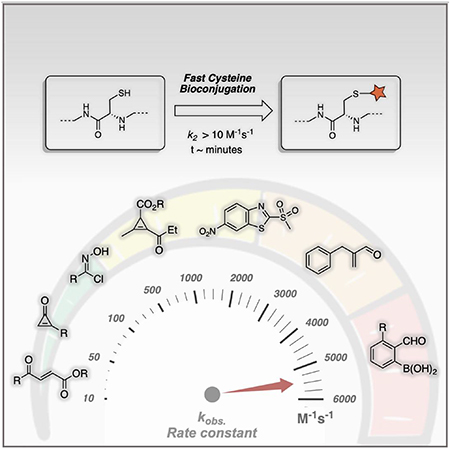
There is no clear agreement on how to define fast cysteine bioconjugation. In this concept, we define fast cysteine bioconjugation as cysteine conjugation reactions with secondary rate constants (k2) greater than 10 M−1s−1 (Figure 2A). With this kinetics, 10 μM of the substrate could be modified with 97% of conversion within 1 hour in the presence of 10 equivalents of conjugation reagent. This reaction rate is more than 15 folds faster than the most commonly used iodoacetamide chemistry for cysteine bioconjugation (k2 ~ 0.6 M−1s−1). As another comparison, we note the widely used strain-promoted alkyne–azide cycloadditions (SPAAC) exhibit secondary rate constants of 10−2 to 1 M−1s−1.[15] According to this definition, the conventional maleimide-cysteine chemistry (k2 ~ 102 M−1s−1) would be considered a fast cysteine bioconjugation,[11] although the poor conjugate stability limits its applications.
Figure 2.
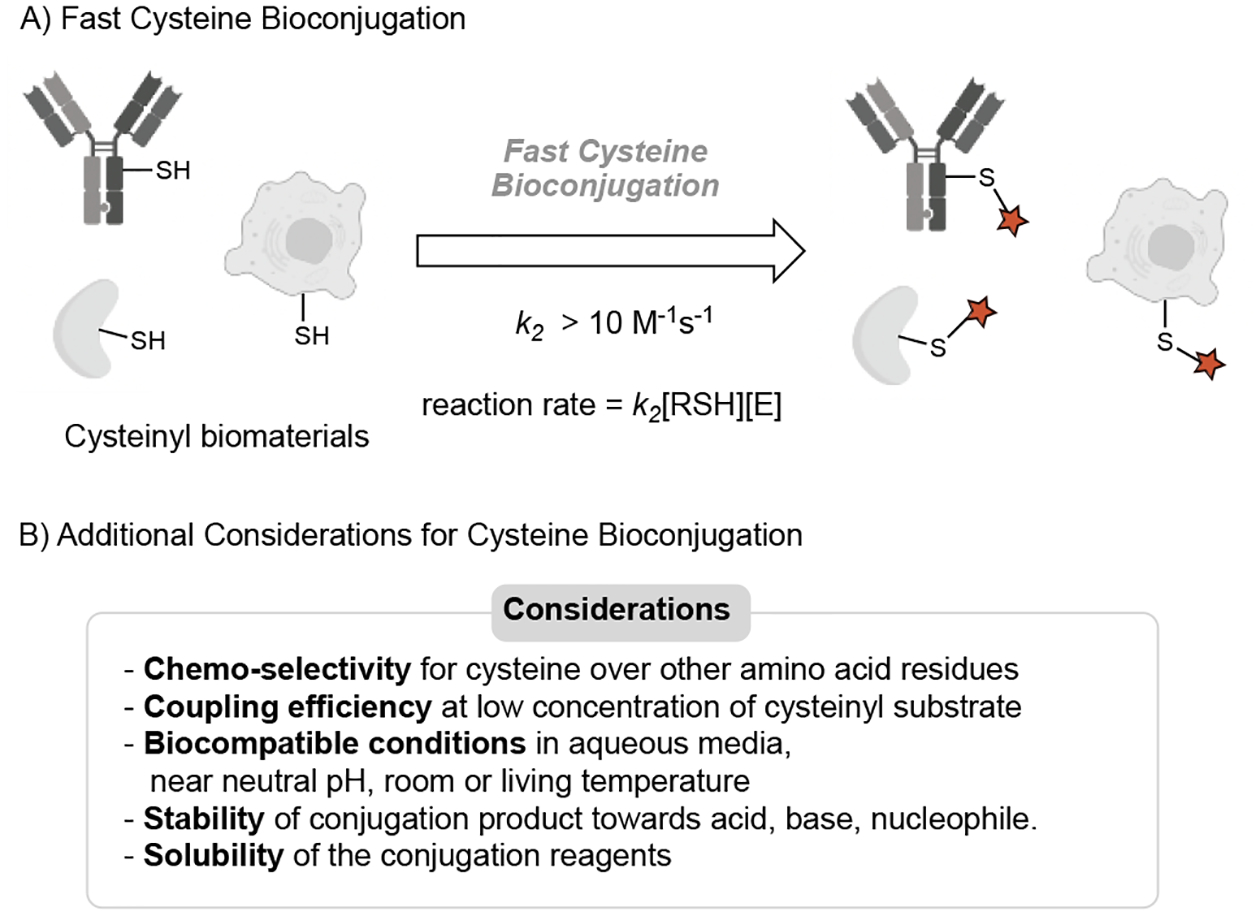
Developing fast cysteine bioconjugation chemistries. (A) Illustration of fast cysteine conjugation on proteins and cells. E: electrophile. k2: secondary rate constant. SH: thiol group. (B) General considerations in addition to reaction rate.
Generally speaking, the rate-determining step (RDS) for a cysteine bioconjugation reaction is the initial nucleophilic addition of cysteine sulfhydryl group or thiolate to an electrophile. The reaction pathways can be SN2 replacement, Michael addition, SNAr substitution, or radical-involved reaction. The reaction rate in principle can be tuned by the reactivities of both reactants as well as the pH and the temperature of the reaction. However, the nucleophilicity of a cysteine substrate is limited by its inherent reactivity. Similarly, the pH and temperature of the reactions are largely fixed to physiologic conditions as well. Thus, the electrophilicity of electrophiles is the main parameter to be modulated when designing fast cysteine bioconjugation methods. Besides the reaction kinetics, other general factors for biocompatible conjugation also need to be considered: the chemo-selectivity for cysteine residue in the presence of other potential competing nucleophiles, coupling efficiency at requisite substrate concentration, biocompatible reaction conditions (aqueous media, near-neutral pH, room or living temperature), stability of the conjugate, solubility, and availability of the reagents (Figure 2B).
Guided by these general principles, a number of reagents have been developed for fast cysteine bioconjugation. Representative reagents and the reaction rate are listed in Figure 3 with those modifying cysteine thiols (internal or in-chain cysteines) on top. The reagents show on the bottom are developed to target N-terminal cysteines (NCys), which can be engineered into a protein of interest to enable site-specific conjugations. Interestingly, few reports exist in the current literature that describe conjugation reactions targeting C-terminal cysteines. These fast reactions show wide range of reaction rate constants (10 M−1s−1 to 5500 M−1s−1). Among them, the 2-formyl phenylboronic acid (2-FPBA) chemistry developed independently by the Gao[16] and Gois[17] labs shows extremely fast kinetics (k2 = 5500 M−1s−1), approaching the rate of enzymatic processes. For cysteine thiol modifications, heteroaromatic sulfone[14] represents the fastest conjugation reactions with a rate constant of 1651 M−1s−1. Chlorooxime[18] and cyclopropenyl ketone[19] also show comparable reaction rates (k2 > 102 M−1s−1) to conventional cysteine-maleimide chemistry. In the following sections, we will discuss the fast bioconjugation reactions that target internal cysteine and NCys respectively. For selected literature reports that lack explicit kinetics data but show fast conjugation profiles, we estimated the reaction rate according to the reported experimental conditions.
Figure 3.
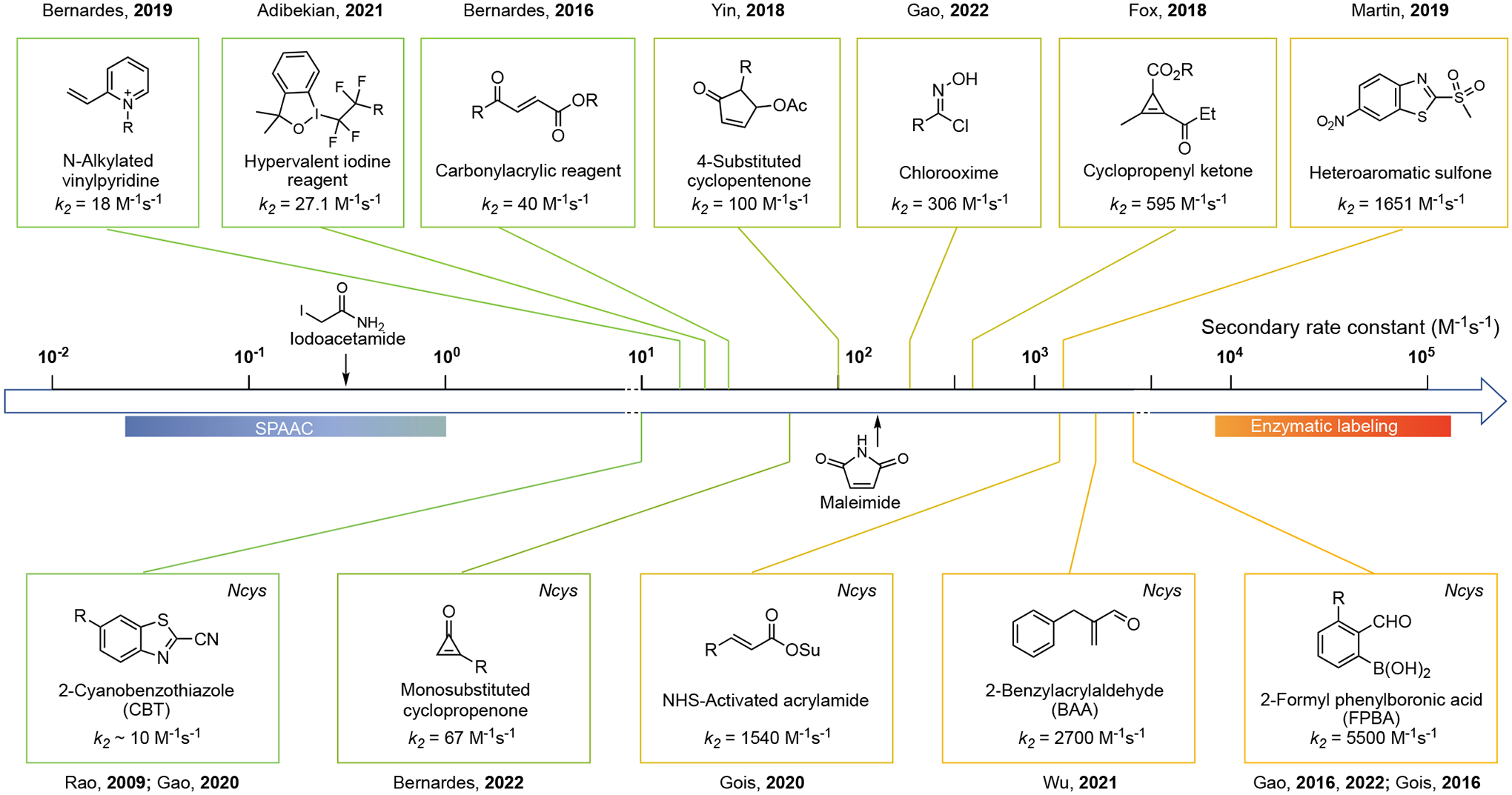
Overview of reaction rates of fast cysteine conjugation protocols. SPAAC: strain-promoted alkyne−azide cycloaddition.
Fast Bioconjugation of Internal Cysteines
For internal cysteine bioconjugation, the sulfhydryl group of the cysteine side chain is the reactive site for modification. The sulfhydryl group can easily be deprotonated to generate a more nucleophilic thiolate, which could react with an activated electrophile to form conjugate rapidly. The thiol form was used as the nucleophile to simplify the mechanistic discussion in the following sections. A summary of protocols for fast internal cysteine bioconjugation is shown in Figure 4. These cysteine-specific reagents include activated Michael acceptors, activated heteroaromatic compounds, strain-releasing reagents, cation-activated electrophiles, and hypervalent iodine reagents. The cysteinyl substrates react with the coupling partners through S-alkylation, S-arylation, S-alkynylation, and thiohydroxymate formation. Generally, these reactions are carried out in aqueous media at near-neutral pH under mild conditions (room temperature to 37 °C), and they could be completed within 1 hour at a 5–50 micromolar (μM) concentration of cysteinyl substrate. In some cases, small portions of organic solvent are used to improve the solubility of the reagents, such as N, N-dimethylformamide (DMF), and tetrahydrofuran (THF).
Figure 4.
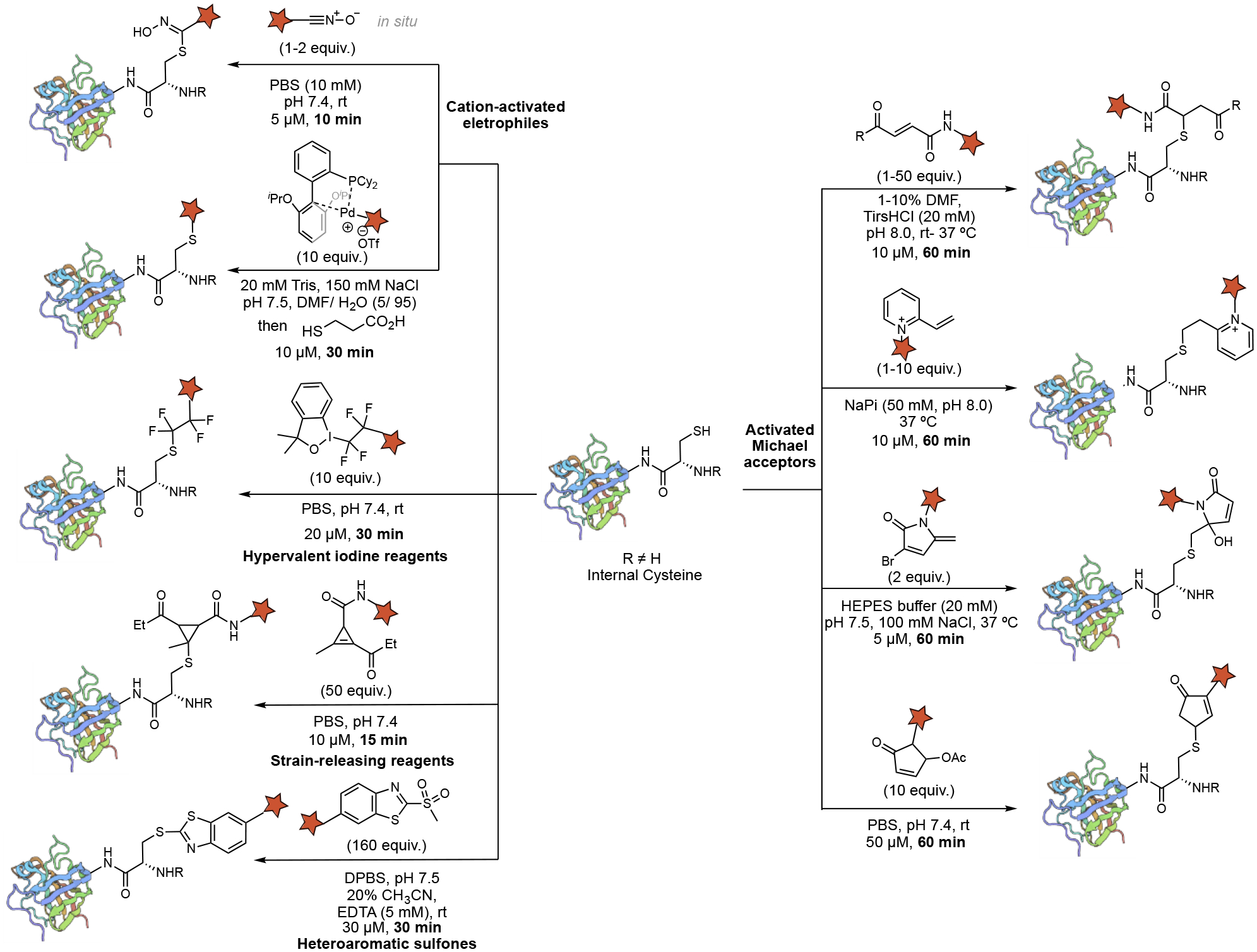
Fast bioconjugation chemistries of internal cysteines
Activated Michael acceptors.
Michael acceptors such as maleimide derivatives represent one of the most frequently used reagents for cysteine bioconjugation. For the Michael addition type of cysteine conjugation, the rate-determining step is the addition of thiol to electron-deficient alkene or alkyne of Michael acceptors. The nucleophilicity of the cysteine residue is dictated by protein structure and working pH, hence less tunable. In contrast, the electrophilicity of the Michael acceptor can be tuned to improve the conjugation efficiency. The introduction of the electron-withdrawing group (EWG) is a straightforward strategy to activate the electrophile through delocalizing the negative charge and stabilizing the anionic adduct intermediate (Figure 5A). The stabilizing effect leads to a decreased Gibbs free energy of activation ΔG≠ and an increased reaction rate, which is nicely demonstrated by the recent work of Bernardes and co-workers.[20] With the help of quantum mechanical calculations, they recognized that the introduction of an electron-withdrawing carbonyl group to acrylic reagent can significantly increase the reaction rate for thiol-Michael addition (Figure 5B, top). Compared to the trans-1, 2-disubstituted alkene containing two amide functionalities, the alkene bearing one amide group and one ketone is computationally predicted to be more reactive by more than 6 million folds. The efficiency of this activated acrylate was then confirmed experimentally by the observation of rapid thiol-Michael-addition of protected cysteine (N-Boc-Cys-OMe) to carbonylacrylate 6. The reaction (with 50 mM reagent) provides the thioether conjugate in 99% yield within just 2 min. Importantly, proteins bearing surface-exposed cysteine residues, such as Annexin V, were labeled completely using carbonylacrylic reagent 3 at 10 μM concentration after 1-hour incubation. The reaction rate constant of this fast reaction was determined to be 40.2 M−1s−1, showing a reactivity comparable to that of maleimide. Unlike thiol-maleimide conjugate, this linear conjugate is proven stable to hydrolysis and thio-exchange in plasma, showing promise for potential in vivo applications. Indeed, fast irreversible cysteine bioconjugation methods as such have been used for precise fluorescent labeling of proteins and constructing cysteine-tagged antibodies in various formats (full-length IgGs, nanobodies).[21]
Figure 5.
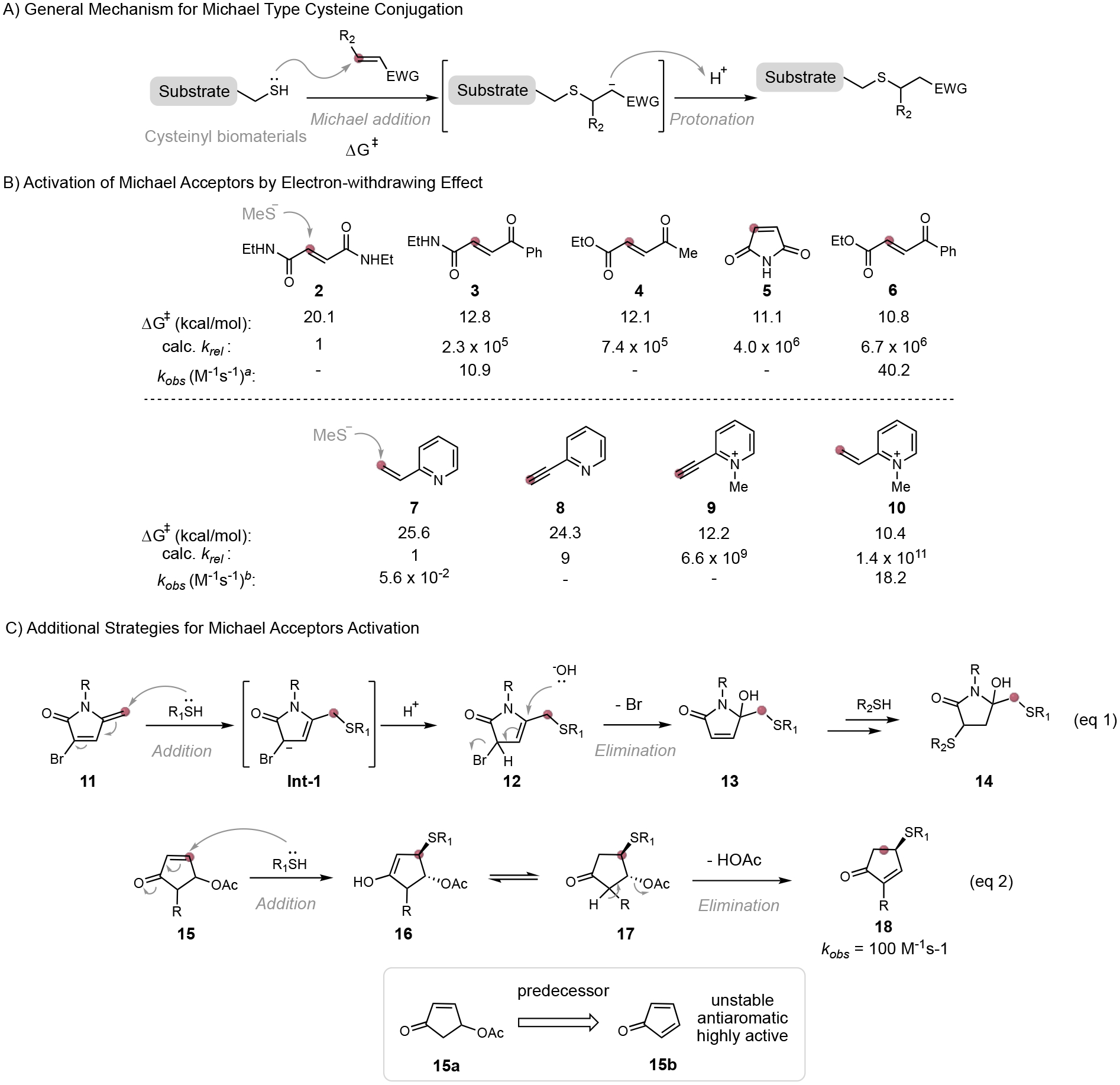
Activated Michael acceptors for fast cysteine bioconjugation. aObserved reaction rate constant for N-Boc-Cys-OMe. bObserved reaction rate constant for N-Ac-Cys-NH2. ΔG≠:Gibbs free energy of activation
Similar to the acrylamide-based reagents, a vinylheteroarene type Michael acceptor was recently shown to exhibit tunable reactivity for thiol addition as well. As expected, the alkene becomes more reactive for cysteine conjugation when the electron density of the heteroarene is reduced. The reactivity of vinylpyridine can be dramatically enhanced by introducing a positive charge to the pyridine nitrogen. Through theoretical calculations of the Gibbs free energy of activation ΔG≠ for nucleophilic addition, Bernardes’s group identified that quaternized pyridiniums 9 and 10 were ~109 times more reactive with thiolate than their non-quaternized analogs 7 and 8 (Figure 5B, bottom).[22] The model reaction of N-methylated vinylpyridine 12 with protected cysteine (N-Ac-Cys-NH2) gave 93% of conversion to conjugate within less than 4 minutes at 3 mM concentration. The observed thiol addition rate constant for N-methylated vinylpyridine 10 is 18.2 M−1s−1, which is around 325 folds more reactive than non-quaternized vinylpyridine 7 (k2 = 0.056 M−1s−1). These fast cysteine bioconjugation methods using quaternized vinyl- and alkynyl-pyridine reagents were applied to create a homogenous antibody-drug conjugate with a precise drug-to-antibody ratio at cysteine residues. These cysteine-crosslinked antibody-drug conjugates were proved to be stable in human plasma and retained their specificity towards HER2+ cells. Another interesting application of this method is the modulation of protein electrophoretic mobility through the introduction of a positive charge to the overall net charge of the protein (+1 charge per modified cysteine).
Another strategy for fast cysteine conjugation explores carefully designed maleimide analogs. For example, Zhou and co-workers developed a novel 3-bromo-5-methylene pyrrolone (3Br-5MP) reagents 11,[23] which is structurally similar to bromomaleimide,[24] but with one carbonyl group replaced by one methylene group (Figure 5C, eq 1). This designed molecule allows the allyl anion intermediate Int-1 formed through 1,6-thio-Michael addition to be stabilized by both the electron-withdrawing carbonyl group and the bromine atom. This stabilized intermediate Int-1 was then protonated followed by the addition of water molecule and elimination of the bromine to form a stable mono-functionalized conjugate 14. The 5-methylene pyrrolone (5MP) analog without bromide substituent shows a much slower reaction rate.[23b] The labeling of a model protein, histone H3 mutant (H3-V35C), using a 3Br-5MP reagent showed fast kinetics and completed the mono-functionalization within 1 hour at 100 μM concentration. Interestingly, the mono-functionalization product can further react with a second thiol nucleophile to form a dual-functionalized conjugate, though the second thiol addition is much slower. The dual-functionalized conjugate was liable to loss of the secondary modification via thiol-exchange reaction, but stable linear conjugation products can be generated by the reduction with NaBH3CN. Disulfide rebridging of goat anti-human IgG Fab fragment was achieved using this efficient 3Br-5MP chemistry. Other activated Michael acceptors are also developed for rapid cysteine-specific conjugation, such as 2-chloromethyl acrylamide or acrylate[25], and 4-substituted cyclopentenone reagent[26]. 4-Substituted cyclopentenone reagent 15a was designed as the predecessor for unstable and antiaromatic cyclopentadienone 15b, which reacts rapidly with cysteine with a rate constant of 100 M−1s−1 at the peptide level (Figure 5C, eq 2). Interestingly, the protein modified by 4-substituted cyclopentenone 11 is prone to thio-exchange, and conjugate 18 could be cleaved by an external thiol nucleophile to regenerate the initial protein, making the reagent be served as a protecting group for cysteine residue.
Activated heteroaromatic compounds.
Similar to Michael acceptors, normally nucleophilic heteroaromatic rings could also become electrophilic reagents through the electron-withdrawing effect. The EWG-activated heteroaromatic compounds reacted with cysteinyl substrates to form conjugates through the nucleophilic aromatic substitution (SNAr) pathway. Mechanistically, the cysteinyl substrate adds to the electron-deficient heteroaromatic cycle at the site of leaving group (LG) to form an anionic σ complex Int-2, followed by elimination of LG, and aromatization of the heterocycle to give the conjugation product (Figure 6A). The rate-determining step is the nucleophilic thiol addition to electron-deficient heteroaromatic compounds.
Figure 6.
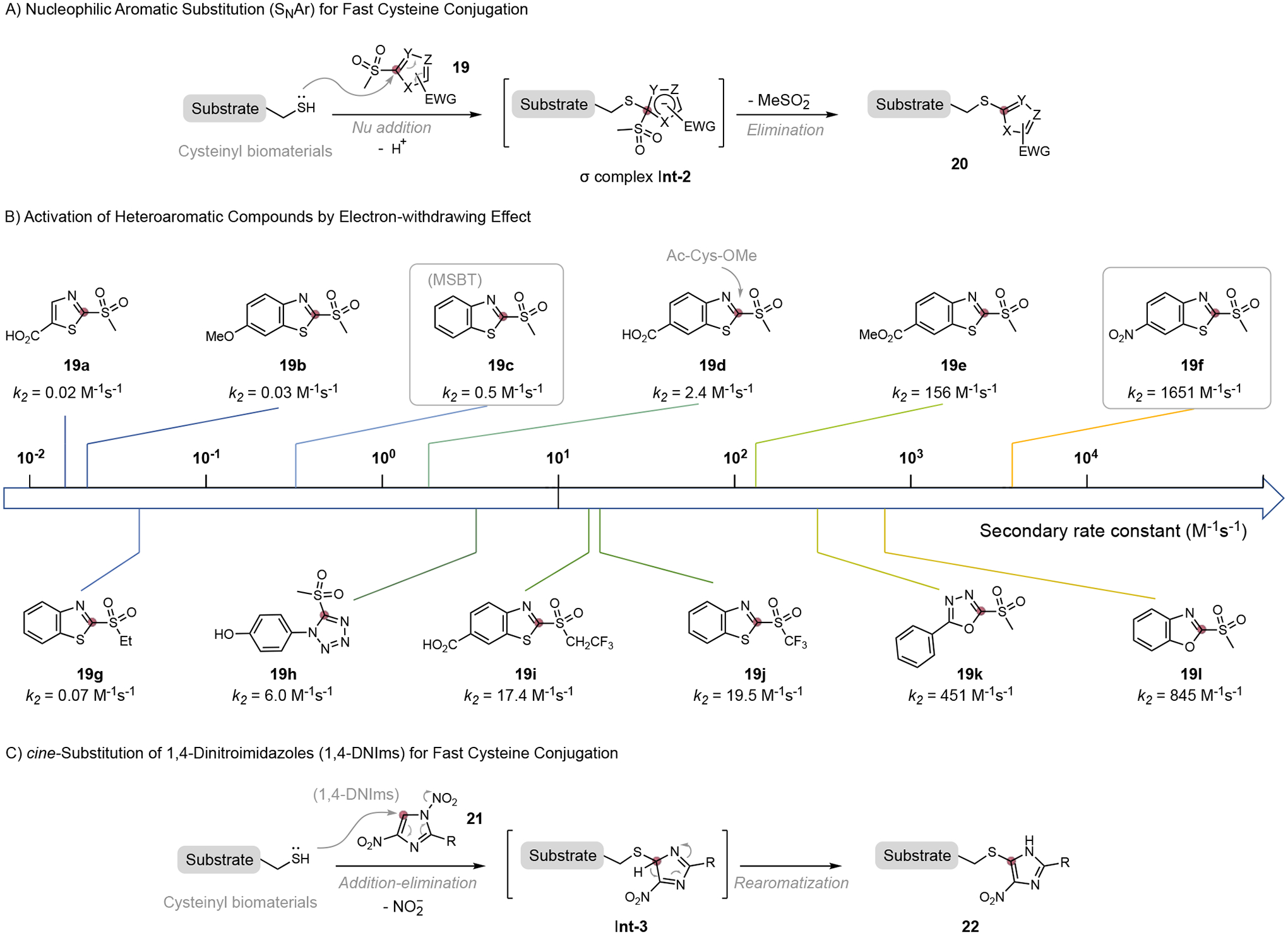
Activated heteroaromatic compounds for fast cysteine bioconjugation. Observed reaction rate constant for N-Ac-Cys-OMe.
Recently, Martin and co-workers revealed an extremely fast heteroaromatic sulfone-cysteine conjugation chemistry by systematic investigation of substitutions effect for thiol SNAr reaction.[14] Inspired by the elegant discovery of methylsulfonylbenzothiazole (MSBT) for thiol blocking,[27] they evaluated a library of heteroaromatic sulfones with different substituents and electron deficiency for cysteine labeling (Figure 6B). It was found that the electron-withdrawing groups at benzothiazole derivatives significantly accelerate the cysteine conjugation. For example, the cysteine conjugation with nitro-substituted benzothiazole sulfone 19f is 3300 times faster (k2 = 1651 M−1s−1) than the traditional non-substituted MSBT 19c (k2 = 0.5 M−1s−1). This reactivity enhancement was also observed in other electron-withdrawing groups, such as ester (19e, 300 folds) and carboxylic acid (19d, 5 folds). A linear Hammett plot across the benzothiazole substitutions gave a positive slope value (ρ=4.464), clearly indicating the activation effect of EWG in the rate-determining step of the thiol SNAr reaction.[14] Sulfone scaffolds with good leaving ability benefit the conjugation, as exemplified in the reaction rate with an order of SO2CF3 (19j) > SO2CH2CF3 (19i) > SO2CH3 (19c) > SO2Et (19g). More electronegative oxygen or nitrogen atom substituents in heterocycle cores also enhance the reactivity compared to benzothiazole 19c, such as benzoxazole 19l (k2 = 845 M−1s−1,1690 folds), and tetrazole 19h (k2 = 6.0 M−1s−1, 12 folds).[28] 10 folds of acceleration were observed when comparing sulfone MSBT with heteroaromatic azole thioether (HAT) developed by Monika and co-workers.[29] Benefiting from their rapid kinetics and high reactivity, these cysteine conjugations with activated heteroaromatic compounds can greatly enhance the detection limit for in-cell profiling, revealing greater number of active thiol species than classical iodoacetamide chemistry.[14]
Alternatively, electron-deficient heteroaromatic compounds could conjugate with cysteine through a cine-substitution mechanism, where the thiol substitution happens at the adjacent carbon of leaving group. A selected example is the 1,4-dinitroimidazoles (1,4-DNIms) reagent developed by Wang and co-workers for rapid cysteine bioconjugation.[30] In 1,4-DNIms, the imidazole becomes highly electrophilic for thiol addition due to the substitutions of two electron-withdrawing nitro groups. The addition of thiol to 1,4-DNIms 21 and elimination of adjacent nitro group lead to the formation of intermediate Int 3, which was re-aromatized to form a cine-substituted conjugate 22 (Figure 6C). The 1,4-DNIms was able to react with cysteine with rapid kinetics, affording the conjugate in full conversion in less than 5 mins at 0.1 mM substrate concentration. 1,4-DNIms derivatives carrying fluorescent, clickable alkyne handle, and biologically active RGD (Arg-Gly-Asp) peptide could be conjugated with cysteine-containing proteins with good specificity and high efficiency. Interestingly, the chemo-selectivity of 1,4-DNIms was shifted to lysine residue when the reaction was carried out in an organic solvent. This lysine conjugation occurred through a distinct ANRORC (Addition of the Nucleophile, Ring Opening, and Ring Closure) mechanism. These unique properties make 1,4DNIms to be appealing bifunctional reagents for peptide macrocyclization. [30]
Electrophilicity enhancement via α-cation activation.
In addition to Michael acceptors and SNAr reagents, searching for other reactive electrophilic centers with deficient electron density also proves suitable for developing fast cysteine conjugation. Introducing a cation species or positive charge near the electrophilic center can significantly decrease the electron density nearby and increase its electrophilicity, activating it for fast thiol addition. Examples of such cation-activated electrophiles include nitrile oxide, and organometallic reagent (Figure 8A).
Figure 8.
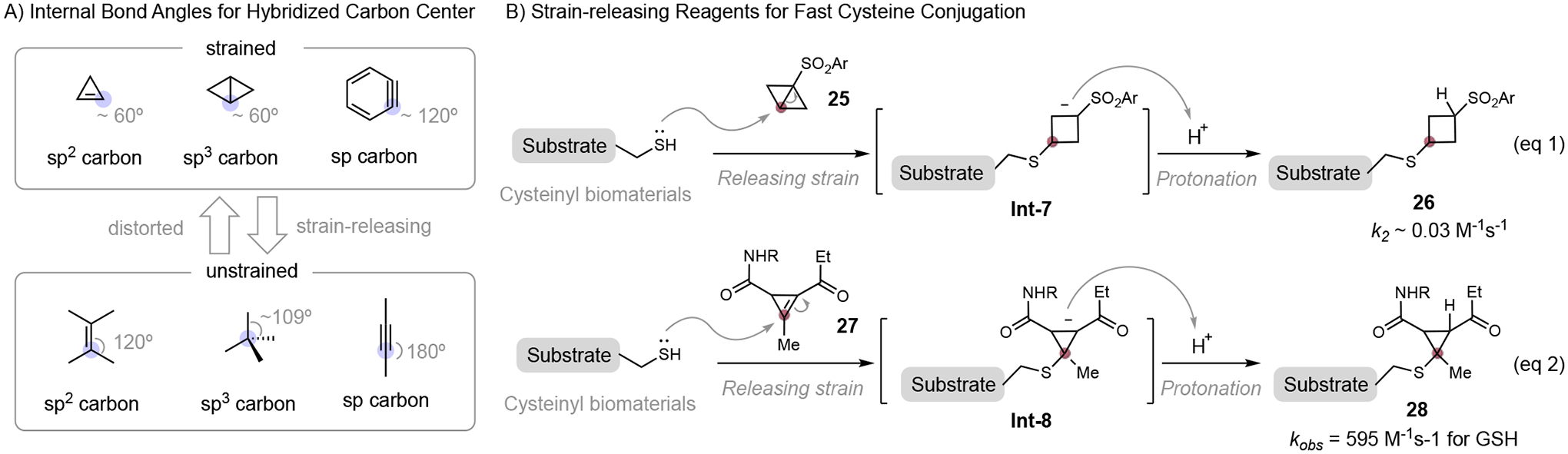
Strain-releasing reagents for fast cysteine bioconjugation
Most recently, we and Wang group designed a highly reactive nitrile oxide species for fast cysteine bioconjugation under physiological conditions (Figure 8, eq 1).[18] Readily available chlorooxime was used as the easily-handle precursor for in situ nitrile oxide generation. The oxide dramatically decreases the electron density of nitrile functionality, turning it into high electrophilic species. In the absence of an external nucleophile, the reactive nitrile oxide can dimerize through [2+3] dipolar cycloaddition. The reactive nitrile oxide species can be captured rapidly by thiol nucleophile to form a stable thiohydroximate 23. An experimental model study shows that chlorooxime-glutathione conjugation under physiological conditions was completed in less than 3 min at 50 μM concentration with an apparent rate constant up to 306 M−1s−1. This fast chlorooxime-cysteine bioconjugation was applied to the cysteine-cysteine stapling of unprotected peptide, modification of Tobacco Etch Virus (TEV) protease, and M13 bacteriophage pIII coat protein, as well as the construction of phage libraries, demonstrating the utility of this method in the biological application. Robust for in vitro biomolecule engineering experiment, however, the application of this chlorooxime-cysteine conjugation in vivo is still changeling due to the high concentration of competing cysteine species (such as glutathione) in the living cell. A masked nitrile oxide species could be a solution for this problem, as studied by Schreiber and co-workers on selective covalent targeting of glutathione peroxidase 4 (GPX4).[31]
In 2015, Buchwald reported a novel organometallic palladium reagent for fast cysteine bioconjugation (Figure 9A, bottom).[32] Thiol group with strong nucleophilicity and coordination ability allows the rapid oxidative addition of cysteine to the cationic palladium center followed by reductive elimination, forming stable thioether conjugate 24 (Figure 9B, eq 2). When competing with maleimide, the organometallic palladium reagent reacts with cysteine favorably to give the aromatic thioether conjugate as the major product, showing a conjugation with a faster kinetic than that of maleimide (k2 ~ 102 M−1s−1). Compared to cysteine conjugate formed by conventional iodoacetamide and maleimide, the thioether conjugate shows high stability towards acid, base, oxidant, and external thiol nucleophiles. This the organometallic platform was applied to peptide stapling[33], protein labeling, and antibody-drug conjugate. The drawback of this method is the use of a stoichiometric amount of palladium reagent, which might be problematic when dealing with living-cell or animal experiments due to its high cellular toxicity. For this reason, methods using a catalytic amount of palladium reagents or less toxic metal to promote cysteine coupling would be of great significance. Some progress has been made in this pursuit in recent years. For example, rapid cysteine arylations are achieved by Spokoyny[34] and Ball team[35] using less toxic organometallic gold(III) reagents, and nickel in combination with arylboronic acid, respectively. Worth to note, Ball group’s protocol doesn’t require the use of a complex ligand for the metal center, though its substrates are limited to boronic acid with an electron-withdrawing group at 2-position.
Figure 9.
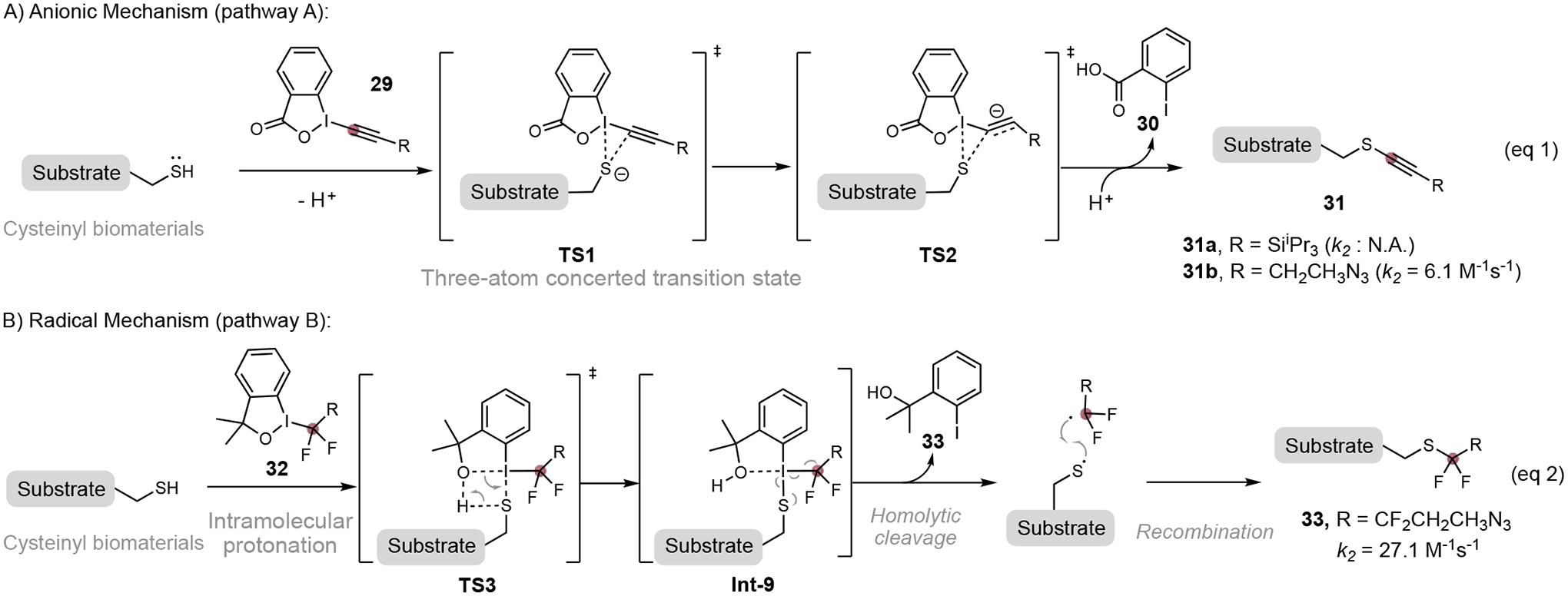
Hypervalent iodine reagents for fast cysteine bioconjugation. Observed reaction rate constant for Ellman’s reagent 2-nitro-5-thiobenzoic acid anion (TNB2-).
Strain-releasing reagents.
An ingenuous strategy to accelerate cysteine conjugation is to introduce a ring strain to the conjugation reagents. The strained compounds are unstable due to their distorted geometries and they are highly reactive to thiol nucleophiles. The chemical bonds in strained compounds are greatly distorted, for example, the internal angle of the sp2-hybridized carbon in cyclopropene is around 60°, which is almost 60° smaller than normal sp2-hybridized carbon (120°) (Figure 8A). This highly distorted structure has a great tendency for ring expansion and reacts with an external nucleophile to form a less strained product. Baran group elegantly applied this strain-releasing strategy for cysteine-selective peptide cyclobutylation using a bicyclobutylsulfone 25, while the reaction rate is still low (estimated k2 ~ 0.03 M−1s−1) (Figure 8B, eq 1).[36] Integrating the concept of strain-releasing with an activated Michael acceptor results in a highly reactive cyclopropenyl ketones reagent 27 for cysteine bioconjugation as demonstrated by Fox’s group (Figure 10, eq 2).[19] The nucleophilic addition of thiol to cyclopropenyl ketones 27 generates intermediate Int-8, which was protonated to form the conjugate 28. The thiol addition was greatly accelerated by the strain-releasing of the alkene carbon (sp2→sp3 rehybridization). The developed cyclopropenyl ketones reacted with reduced glutathione efficiently with a secondary rate constant up to 595 M−1s−1, which is around 60 folds faster than that of carbonylacrylate 3 without the strain-releasing motif (Figure 5). An additional advantage of this method is the facile installation of various useful functionalities to the reagents using a cyclopropenyl ketone precursor bearing an N-hydroxysuccinimide (NHS) motif. Modified proteins and PEG-based hydrogels were synthesized using this efficient chemistry. Perhaps not surprisingly, the highly reactive cyclopropenyl ketone reagent did show non-specific reactivity with water, as well as other nucleophilic functionalities. Compared to the well-studied Michael acceptor, strained electrophiles for fast cysteine bioconjugation methods are underdeveloped. One of the reasons might be the difficulty to synthesize these strained compounds. Recent progress on one-pot synthesis of strain-releasing reagent 1-sulfonyl bicyclo[1.1.0]butanes and housanes make this type of reagent more available, providing an opportunity for broader study on this chemistry and applications in complex biomaterials.[37]
Figure 10.
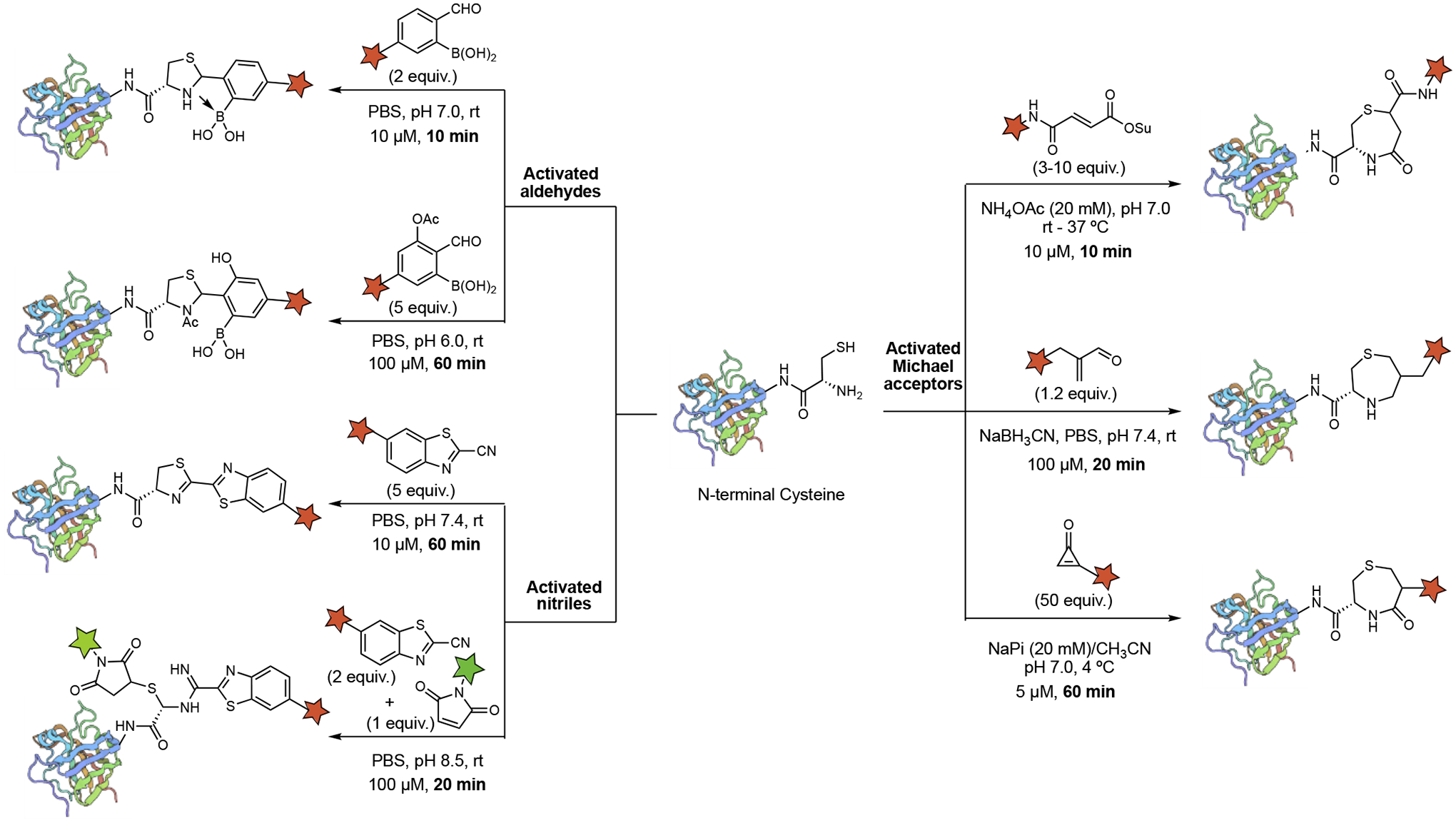
Fast bioconjugation chemistries of N-terminal cysteines
Hypervalent iodine reagents.
Hypervalent iodine reagents have become a valuable toolbox for cysteine-selective peptide and protein functionalizations upon their discovery.[38] The thiol-alkynylation using hypervalent iodine reagent TIPS-ethynyl- benziodoxolone (29) proceeded rapidly in the organic solvent to form conjugate 31a, completing the peptide alkynylation within 30 seconds (Figure 9A).[38a] In aqueous media, however, the reaction proceed much slower (k2 = 6.1 M−1s−1 for 31b). A three-atom concerted transition state TS1 and TS2 were proposed based on computational studies conducted by Waser and co-workers.[38b] As a comparison, the corresponding halogen alkyne analog was unreactive to thiol nucleophile. Most recently, the toolbox of hypervalent iodine reagent was expanded to aqueous media for fast cysteine bioconjugation.[39] Adibekian and co-workers identified a highly reactive hypervalent iodine reagent, tetrafluoroalkyl benziodoxole (TFBX) 32, for cysteine-selective chemoproteomic profiling.[39a] The cysteine fluoroalkylation using TFBX 33 show fast reaction kinetics with a secondary rate constant of 27.1 M−1s−1 (Figure 9, eq 2). The fluoroalkylation is proposed to proceed through a radical pathway.[40] The intramolecular protonation of hypervalent iodine reagent 32 via rate-determining transition state TS3 gives intermediate Int-9, which is converted to the conjugate 33 through homolytic cleavage and subsequential radical recombination (Figure 10, pathway B). This fast cysteine labeling reagent shows great improvements in the aspect of target occupancy, reaction kinetics, and proteomic coverage compared to classical cysteine-reactive probes (such as iodoacetamide derivative). The TFBX-cysteine conjugation enables fast cystine profiling in live cells at submillimolar concentration. For example, the chemoproteomic profiling of cysteine with 33 in HeLa cells allowed the identification of the target protein (X-ray repair cross-complementing protein 5, XRCC5) for antitumor antibiotics (−)-myrocin G,[41] and reveal its role in inhibiting the nonhomologous end joining (NHEJ) DNA repair pathway. [39a]
Fast Bioconjugation of N-Terminal Cysteines
N-Terminal cysteine (NCys) is also a popular target for site-specific bioconjugation as they are typically solvent-exposed and provide distinct 1,2-aminothiol functionality for chemical modification.[42] Native chemical ligation (NCL), which represents a widely used method for protein ligation and assembly, relied on the reaction of a peptide having an NCys with thioesters.[43] Generally, bioconjugation protocols targeting internal cysteine also work for NCys. Unlike an internal cysteine, an NCys has an adjacent nucleophilic amino group, which could participate in the conjugation process along with the side chain thiol. To achieve fast NCys-selective conjugation, the initial thiol (or N-terminal amine) addition to the labeling reagent needs to be a reversible and fast process. Irreversible initiation would lead to permanent “off-target” labeling at internal cysteine or lysine residue. Following the first nucleophilic addition, the second addition of 1,2-aminothiol functionality occurs intramolecularly, and the reaction rate could be greatly enhanced by the proximity effect. Indeed, several NCys conjugations with a reaction rate over 103 M−1s−1 have been reported, while such a rapid reaction targeting internal cysteine is rare except in enzymatic processes.[15a, 44] Although NCys is extremely rare in native proteins, it can readily be constructed after protein expression through proteolytic cleavage with Factor Xa, Tobacco Etch Virus (TEV), and thrombin proteases.[45] For example, Tobacco Etch Virus (TEV) protease can be used to generate the N-terminal cysteinyl protein through precise recognition of the amino acid sequence ENLYFQ/C and specifically cleavage of the amide bond between the glutamine (Q) and cysteine (C) residue.[46] This fact provides access for site-specific modification of protein and biomolecules engineering using fast N-terminal cysteine conjugation.
An overview of recently developed protocols for fast NCys bioconjugation was listed in Figure 10. These bioconjugation reagents are classified into three main categories: activated aldehydes (such as 2-formylphenyl boronic acid, 2-FPBA), activated nitriles (such as 2-cyanobenzothiazole, CBT), and activated Michael acceptors. These methods show good selectivity for N-terminal cysteine over internal cysteine and other nucleophilic amino acid residues. Generally, high conversions of conjugation could be obtained in less than 1 hour with low micromolar (μM) concentrations of reagents. In most cases, the 1,2-aminothiol motif of NCys was converted into a 5-member or 7-member cyclic structure through tandem nucleophilic additions.
Activated aldehydes.
NCys is known to react with aldehydes selectively to form thiazolidines in the presence of other endogenous nucleophiles.[47] The reaction initiates with nucleophilic addition of α-amino group of NCys to aldehyde carbonyl group to form a hemiaminal intermediate 34 (Figure 11A). The dehydration of 34 leads to the formation of imine species 35. Intramolecular imine thiolation generates the thiazoline product 36. Thiol could also react with the aldehyde to form a thiohemiacetal intermediate, however, the thiohemiacetal is not stable and could easily disassociate back to starting materials. The rate-determining step is likely to be the imine formation step.
Figure 11.
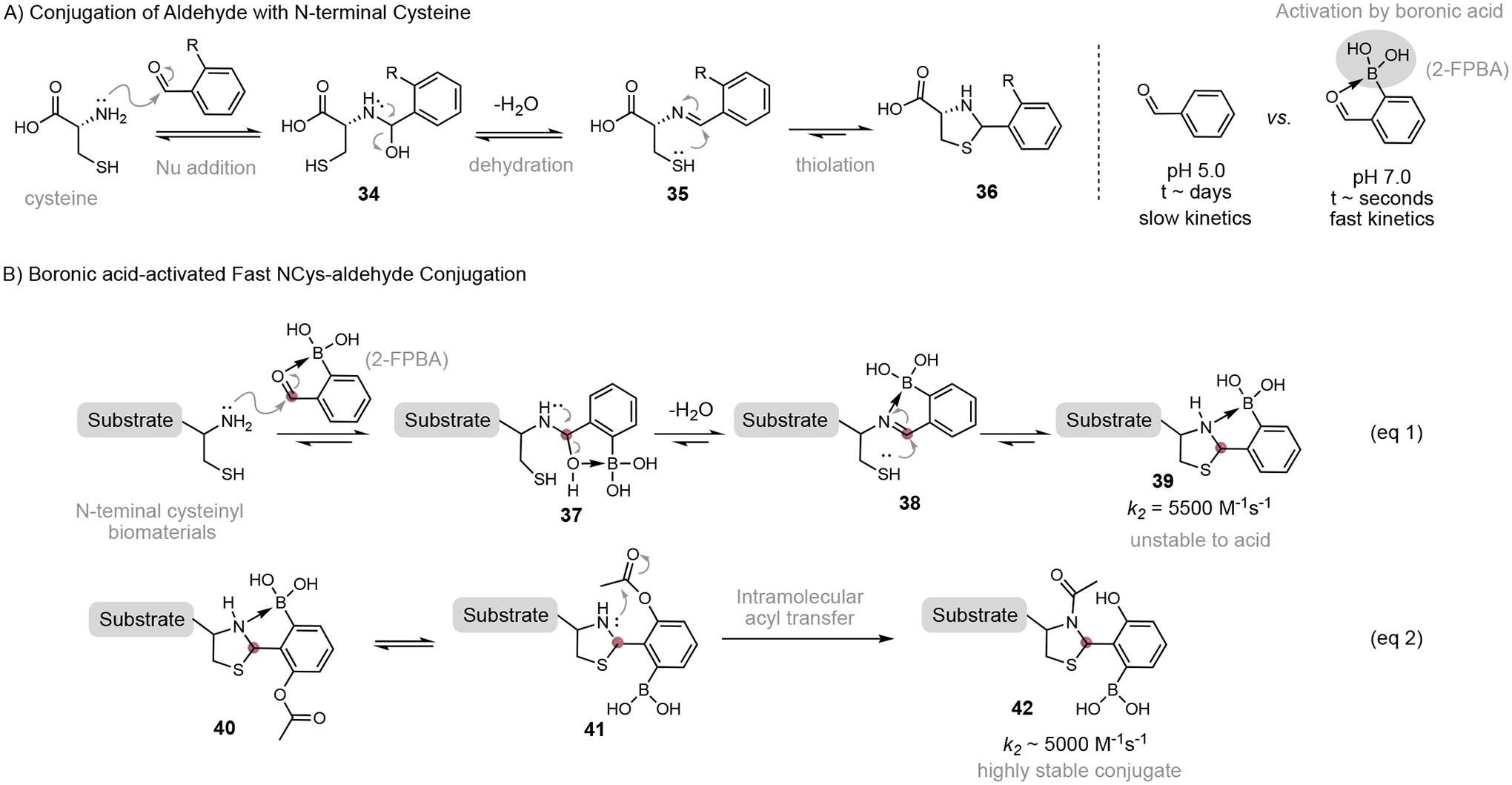
Activated aldehyde for fast N-terminal cysteine bioconjugation
Hence, increasing the electrophilicity of aldehyde could be a solution for fast NCys bioconjugation. The NCys conjugation with unsubstituted benzaldehyde suffers from slow kinetics, which generally takes days to complete, and requires acidic reaction conditions (pH 4–5) (Figure 11A). Recently, we and the Gois group developed a novel iminoboronate chemistry for aldehyde activation.[16–17] By introducing a boronic acid substituent into the ortho-position of benzaldehyde, the aldehyde group was activated by the coordination of the oxygen lone pair electron to a nearby boronic acid moiety, enhancing the electrophilicity of the carbonyl group. For example, 2-formyl phenylboronic acid (2-FPBA) was able to react with NCys rapidly to form thiazolidine conjugate at near-neutral pH within seconds (Figure 11A). As shown in Figure 11B (eq 1), the 2-formyl phenylboronic acid (2-FPBA) 35 reacts instantaneously with the amino group to form an imine intermediate 38 through boronic acid-promoted nucleophilic addition and dehydration of 37. The imine was then activated by boronic acid through N-B coordination, facilitating the intramolecular thiol addition to form a thiazolidine product 39. The NCys conjugation of 2-FPBA proceeds extremely rapidly and gives a reaction rate constant up to 5500 M−1s−1, affording one of the fastest bioconjugation reactions for protein labeling. The stability of conjugate was also improved significantly by the N-B coordination compared to the aldehyde analogous without the boronic acid motif, favoring the equilibrium for conjugate formation. The thiazolidinoboronate complex, however, was observed to dissociate upon mild acidification, and 2-FPBA labeled proteins occur slowly thiol-exchange with external free cysteine species. To overcome these limitations, our group optimized the protocol and developed a thiazolidino boronate-mediated acyl transfer strategy to get a more stable N-acyl thiazolidines conjugate 42 without significant loss of efficiency (k2 ~ 5000 M−1s−1) (Figure 11B, eq 2).[48] It is worth noting that 2-FPBA reagents display excellent solubility in aqueous media due to the hydrogen bonding of boronic acid to water molecules. The fast NCys conjugation elicited by iminoboronate chemistry has been successfully applied to site-specific protein labeling as well as chemical modification of bacteriophage libraries.[48]
Activated nitriles.
Nitriles are commonly seen in small molecule drugs and bioactive compounds, and they have been used as aldehyde analogs for cysteine protease inhibition.[49] A nitrile is a weaker electrophile than aldehydes, while its reactivity to thiol addition can be enhanced when the cyano group is linked to an electron-withdrawing group (Figure 12A).[50] 2-cyanobenzothiazole (CBT), a heteroarene activated nitrile, has been one of the most popular reagents for fast NCys labeling since its discovery.[51] The nucleophilic addition of the thiol group to CBT at the cyanocarbon leads to the rapid and reversible formation of thioimidate intermediate 44, which is then cyclized by the intramolecular nucleophilic addition of amino group to give 2-aminothiazolidine intermediate 45. The irreversible elimination of ammonia from 45 generates the stable luciferin conjugate 46 (Figure 12, eq 1, pathway A). α-Amino group, on the other hand, is unreactive for nucleophilic addition to CBT nitrile. This conjugation could be carried out under physiological conditions with a fast reaction rate (k2 ~ 10 M−1s−1).[51a] A side chain thiol does show conjugation with CBT, however, the thioimidate conjugate is prone to hydrolysis. Consequently, CBT exhibits excellent selectivity to NCys over internal cysteines. A wide range of applications using CBT chemistry has been demonstrated, including site-specific protein ligation, and protein labeling in vitro and on live HeLa cell surfaces.[52] Recently, our group expanded the rapid CBT-NCys toolbox by investigating an alternative condensation pathway (Figure 12B, pathway B).[53] Interestingly, we discovered that the CBT-NCys conjugation is peptide sequence-dependent and pH-dependent. Distinct conjugates could be generated from the same intermediate 45. Under relatively basic conditions (pH 8.5), intermediate 45 undergoes elimination of thiol and deprotonation to form an amidine intermediate 47 bearing thiolate anion. The tripeptide tag Cys-Ile-Ser (CIS) in the substrate is able to stabilize the thiolate intermediate Int-10 through an intramolecular hydrogen bonding between the serine hydroxyl group and thiolate, favoring the formation of non-luciferin products 49. With the tripeptide, N, S-double labeling of NCys was achieved by capturing the free-thiol intermediate using a maleimide reagent 48 through Michael addition. The rate constant for amidine formation was estimated to be 20 M−1s−1. Structurally similar to luciferin, thiazoline conjugation products of NCys can also be constructed using a 2-((alkylthio)(aryl)methylene)malononitrile) (TAMM) reagent recently developed by Wu group[54] and chlorooxime chemistry by our group[18a], although the reaction is relatively slow. Cysteine-specific protein labeling in vitro, labeling of a cell-surface protein on mammalian cells, and phage modification were studied using these NCys-specific conjugation chemistries.[54]
Figure 12.
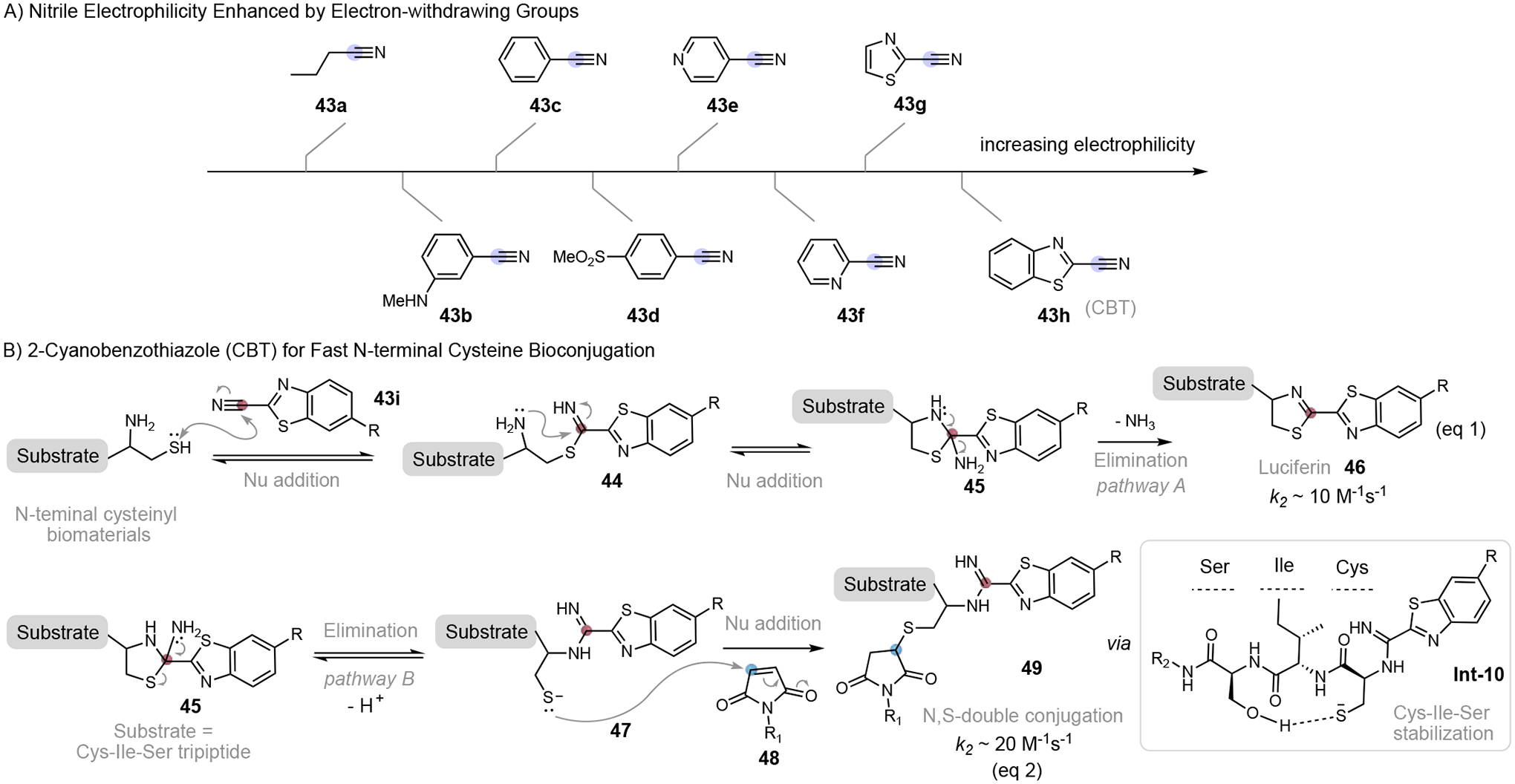
Activated nitriles for fast N-terminal cysteine conjugation.
Activated Michael acceptors.
Activated bifunctional Michael acceptors react with 1,2-aminothiol were developed for fast NCys conjugation. Examples of these newly developed reagents include the N-hydroxysuccinimide (NHS)-activated acrylamides by Gois[55] and 2‐benzylacrylaldehyde (BAA) by Wu[56]. Both the NHS-activated acrylamides and 2‐benzylacrylaldehyde showed exceptional fast kinetics (k2 = 1540 M−1s−1 and 2700 M−1s−1, respectively) and form a 7-member ring conjugation product (Figure 13). The NHS-activated acrylamide 50 initiates the reaction with a nucleophilic attack of the thiol group to the Michael acceptor, followed by macrocyclization of the amino group with an activated ester to form the cyclized conjugate 53 (Figure 13, eq 1). With different reaction mechanisms, the 2‐benzylacrylaldehyde chemistry initials with fast reversible 1,2-addition of the amino group towards the aldehyde functionality of 54 (Figure 13, eq 2). Rapid proximity-driven thiol addition to the alkene of 55 followed by dehydration gives 7-member cyclic imine 58, which is then reduced by NaBH3CN to afford the stable product 59. 2‐Benzylacrylaldehyde can give NCys-selective modifications without interference from the internal cysteine and lysine residues. Interestingly, NHS-activated acrylamides can also be used for the cyclization of peptides or proteins bearing an internal cysteine with a nearby lysine. These fast bioconjugation methods have wide applicability, such as the synthesis of peptide drug conjugate, lysosome imaging in microglia cells, and labeling of proteins.
Figure 13.
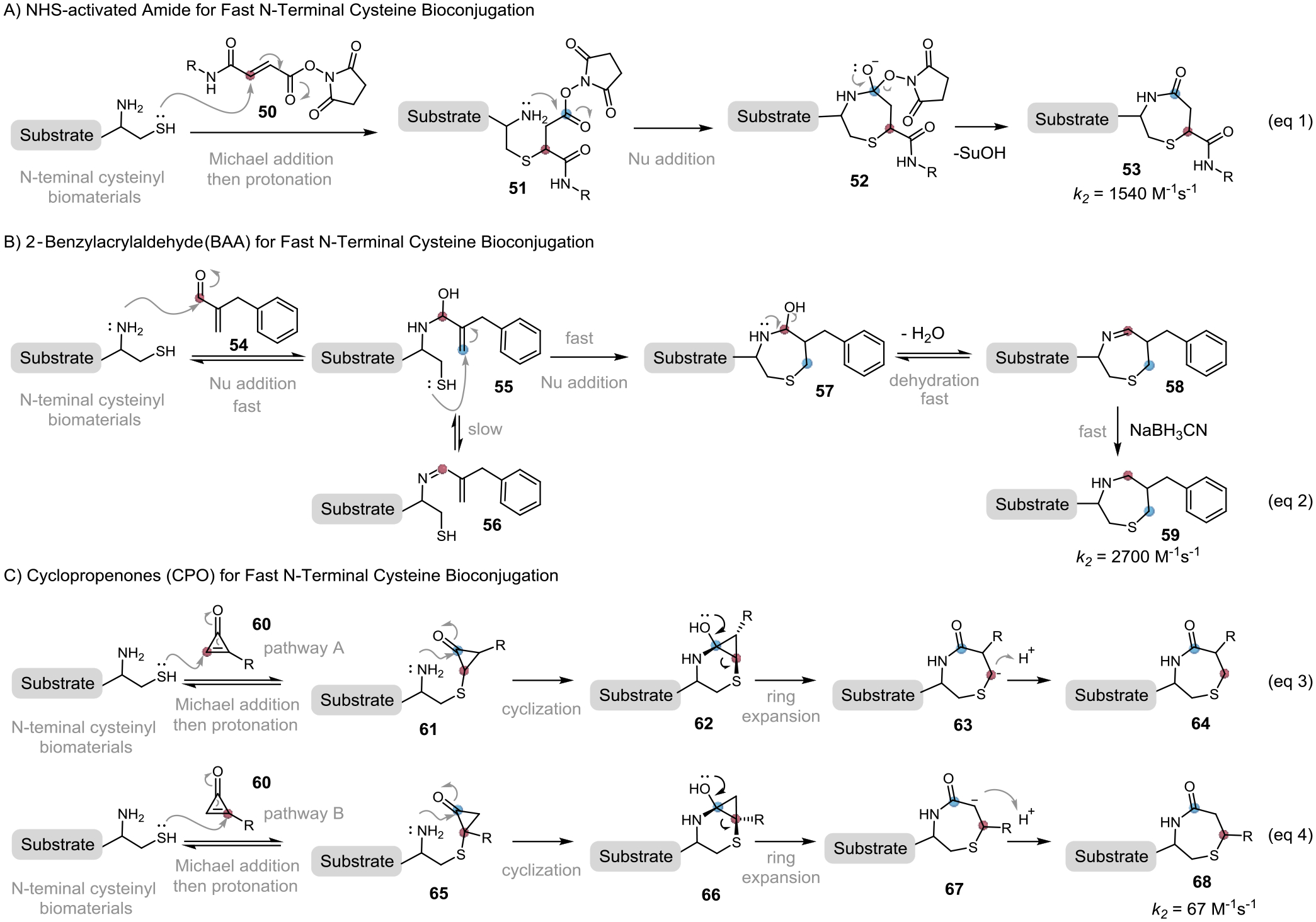
Activated Michael acceptors for fast N-terminal cysteine bioconjugation
Most recently, Bernardes and co-workers developed an NCys-selective modification using a strain-releasing bifunctional Michael acceptor, monosubstituted cyclopropenones (CPO) 60.[57] This reagent allows efficient NCys modification in the presence of solvent-exposed cysteines with fast kinetics (k2 = 67 M−1s−1), forming a 7-member amide 64 or 68 with excellent stability (Figure 13C). Mechanistically speaking, the thiolate initiates with nucleophilic Michael addition to the CPO 60 at the relatively less sterically hindered carbon to form cyclopropanone 61 (pathway A). The intramolecular amine addition to the cyclopropanone carbonyl group generates a hemiaminal intermediate 62 driven by the strain-releasing of the carbonyl carbon (sp2→sp3 rehybridization). Irreversible ring expansion of 62 gives a 7-member species 63, which is protonated to afford the conjugate 64. Amino group, as a competitive nucleophile, is not reactive for conjugation addition of CPO compound. The reversibility of initial thio addition enables the selective labeling of NCys over internal cysteine. It should be noted that the initial thiol Michael addition could also occur at tri-substituted alkene carbon of 60 (pathway B) with activation barriers only 1 kcal/mol higher than that of pathway A, resulting in the formation of a mixture of regio-isomers. By carrying out the reaction under mild conditions (aqueous buffer, pH 7, 4–25 °C), they demonstrated the versatilities of this chemistry in peptides and proteins modifications using cyclopropenone reagents that carry various valuable payloads, including fluorescent dyes, biotin, and biorthogonal handles. This efficient CPO-NCys bioconjugation was also applied to the construction of protein-protein conjugate.
Summary and Outlook
In this concept, we discussed recent developments in fast cysteine conjugation chemistries. The field has seen a number of highly efficient conjugation reactions that display reaction rates ranging from 10 M−1s−1 to 5500 M−1s−1 under physiological conditions. These elegant and powerful new reactions show clear advantages over the venerable thiol-reactive reagents, such as iodoacetamide and maleimide, thereby expanding our capacity for protein engineering tremendously. The fast cysteine conjugation chemistries have been successfully implemented in a wide range of biological applications, including protein labeling, antibody-drug conjugates construction, and phage libraries modification. This contribution provides a summary of the most notable advances in this field, with an intentional focus on the reaction mechanisms and the design principles of the newly developed reagents for fast cysteine conjugation. Our mechanistic analysis shows that the fast reaction kinetics can be understood by considering the electrophilicity of the reagents using physical organic chemistry terms. Discrete strategies to achieve fast cysteine bioconjugation include the introduction of electron-withdrawing groups, strain-releasing reagents, cation activation, and coordination functionalities activation. We note many other attractive cysteine bioconjugation methods have also been developed, while their fast versions remained to be explored, including umpolung strategy[58], π-clamp mediated conjugation[59], reactive peptide interface conjugation,[60] and phosphorus (V) electrophiles[61]. Enzymatic, organo-, or metal-catalytic coupling reactions might also provide new opportunities for fast cysteine bioconjugation. With the rapid progress in this field in recent years, we imagine that there will be even more efficient cysteine bioconjugation protocols developed in the future to broaden the scope of protein engineering and enable novel biological applications.
Figure 7.
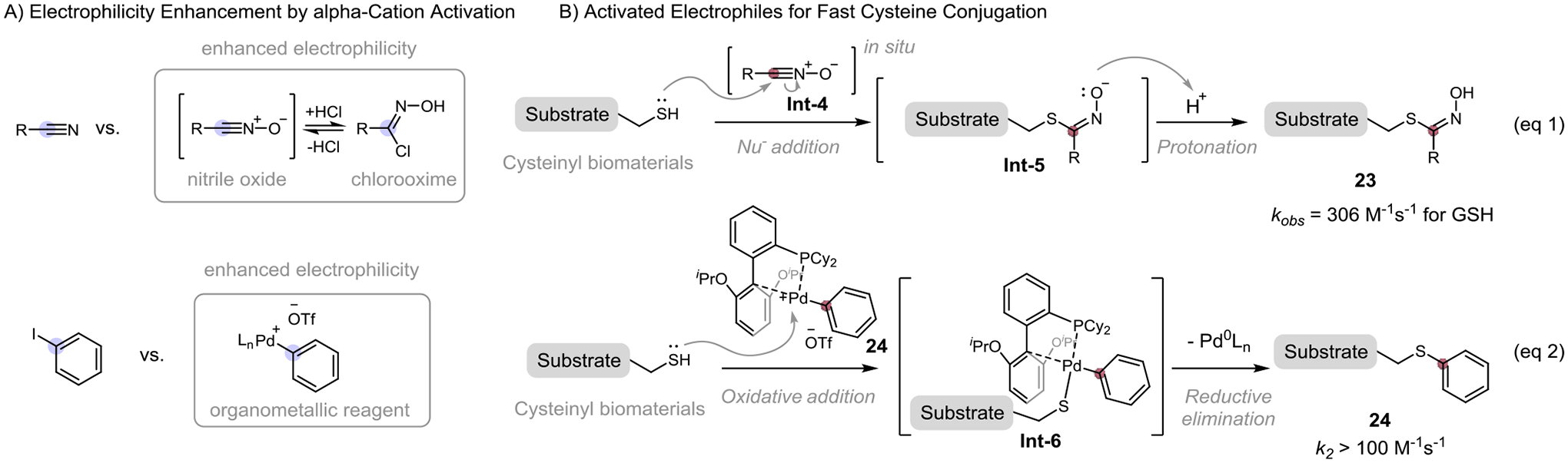
Cation-activated electrophiles for fast cysteine bioconjugation
Acknowledgements
We acknowledge the generous financial support of our research from NSF (CHE-1904874/2117246), NIH (GM102735/124231), and the Ono Pharma Foundation.
Biographies

Jianmin Gao.
Jianmin Gao received his B.S. degree in 1999 from University of Science and Technology of China. He completed his Ph.D. in 2004 with Prof. Eric T. Kool from Stanford University. In 2004, he joined the group of Prof. Jeffery W. Kelly at the Scripps Research Institute as a postdoctoral fellow. In 2007, he started his independent career at Boston College, where he remains as a Full Professor of Chemistry.

Fa-Jie Chen
Fa-Jie Chen received his B.S. degree in 2011 from Fuzhou University (China.). He completed his Ph.D. in 2016 in chemistry with Prof. Bing-Feng Shi and Prof. Jun Wu from Zhejiang University (China). His research focused on transition metal-catalyzed carbon-hydrogen bond functionalization using a removable directing group. After postdoctoral training at Fujian Institute of Research on the Structure of Matter (with Prof. Weiping Su) and University of Illinois at Chicago (with Prof. Daesung Lee), he joined the group of Prof. Jianmin Gao in 2020 as a postdoctoral fellow and currently works on cysteine-specific peptide and protein modifications.
References
- [1].a) Ochtrop P, Hackenberger CPR, Curr. Opin. Chem. Biol 2020, 58, 28–36; [DOI] [PubMed] [Google Scholar]; b) Spicer CD, Davis BG, Nat. Commun 2014, 5, 4740; [DOI] [PubMed] [Google Scholar]; c) Boutureira O, Bernardes GJL, Chem. Rev 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- [2].Poole LB, Free Radical Biol. Med 2015, 80, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND, J. Nutr 2004, 134, 489–492. [DOI] [PubMed] [Google Scholar]
- [4].Tong JTW, Harris PWR, Brimble MA, Kavianinia I, Molecules 2021, 26, 5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Laserna V, Abegg D, Afonso CF, Martin EM, Adibekian A, Ravn P, Corzana F, Bernardes GJL, Angew. Chem. Int. Ed 2021, 60, 23750–23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Spokoyny AM, Zou Y, Ling JJ, Yu H, Lin Y-S, Pentelute BL, J. Am. Chem. Soc 2013, 135, 5946–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Zheng X, Liu W, Liu Z, Zhao Y, Wu C, Bioconjugate Chem 2020, 31, 2085–2091; [DOI] [PubMed] [Google Scholar]; b) Chen S, Rentero Rebollo I, Buth SA, Morales-Sanfrutos J, Touati J, Leiman PG, Heinis C, J. Am. Chem. Soc 2013, 135, 6562–6569; [DOI] [PubMed] [Google Scholar]; c) Oppewal TR, Jansen ID, Hekelaar J, Mayer C, J. Am. Chem. Soc 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Gunnoo SB, Madder A, ChemBioChem 2016, 17, 529–553; [DOI] [PubMed] [Google Scholar]; b) Chalker JM, Bernardes GJL, Lin YA, Davis BG, Chem. Asian J 2009, 4, 630–640. [DOI] [PubMed] [Google Scholar]
- [9].Gehringer M, Laufer SA, J. Med. Chem 2019, 62, 5673–5724. [DOI] [PubMed] [Google Scholar]
- [10].Lindley H, Biochem. J 1962, 82, 418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ravasco JMJM, Faustino H, Trindade A, Gois PMP, Chem. Eur. J 2019, 25, 43–59. [DOI] [PubMed] [Google Scholar]
- [12].a) Roberts DD, Lewis SD, Ballou DP, Olson ST, Shafer JA, Biochemistry 1986, 25, 5595–5601; [DOI] [PubMed] [Google Scholar]; b) King TP, Li Y, Kochoumian L, Biochemistry 1978, 17, 1499–1506. [DOI] [PubMed] [Google Scholar]
- [13].Costa AM, Bosch L, Petit E, Vilarrasa J, J. Org. Chem 2021, 86, 7107–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Motiwala HF, Kuo Y-H, Stinger BL, Palfey BA, Martin BR, J. Am. Chem. Soc 2020, 142, 1801–1810. [DOI] [PubMed] [Google Scholar]
- [15].a) Lang K, Chin JW, ACS Chem. Biol 2014, 9, 16–20; [DOI] [PubMed] [Google Scholar]; b) Saito F, Noda H, Bode JW, ACS Chem. Biol 2015, 10, 1026–1033. [DOI] [PubMed] [Google Scholar]
- [16].a) Cambray S, Gao J, Acc. Chem. Res 2018, 51, 2198–2206; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bandyopadhyay A, Cambray S, Gao J, Chem. Sci 2016, 7, 4589–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Faustino H, Silva MJSA, Veiros LF, Bernardes GJL, Gois PMP, Chem. Sci 2016, 7, 5052–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Chen F-J, Zheng M, Nobile V, Gao J, Chem. Eur. J, n/a, e202200058; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Q, Long T, Zheng J, Sheng W, Sun S, Wei W, Zhao J, Wang H, CCS Chemistry 2022, Just Published. DOI: 10.31635/ccschem.31021.202101386. [DOI] [Google Scholar]
- [19].Smith NJ, Rohlfing K, Sawicki LA, Kharkar PM, Boyd SJ, Kloxin AM, Fox JM, Org. Biomol. Chem 2018, 16, 2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bernardim B, Cal PMSD, Matos MJ, Oliveira BL, Martínez-Sáez N, Albuquerque IS, Perkins E, Corzana F, Burtoloso ACB, Jiménez-Osés G, Bernardes GJL, Nat. Commun 2016, 7, 13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bernardim B, Matos MJ, Ferhati X, Compañón I, Guerreiro A, Akkapeddi P, Burtoloso ACB, Jiménez-Osés G, Corzana F, Bernardes GJL, Nat. Protoc 2019, 14, 86–99. [DOI] [PubMed] [Google Scholar]
- [22].Matos MJ, Navo CD, Hakala T, Ferhati X, Guerreiro A, Hartmann D, Bernardim B, Saar KL, Compañón I, Corzana F, Angew. Chem. Int. Ed 2019, 58, 6640–6644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].a) Zhang Y, Zang C, An G, Shang M, Cui Z, Chen G, Xi Z, Zhou C, Nat Commun 2020, 11, 1015; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Y, Zhou X, Xie Y, Greenberg MM, Xi Z, Zhou C, J. Am. Chem. Soc 2017, 139, 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Smith MEB, Schumacher FF, Ryan CP, Tedaldi LM, Papaioannou D, Waksman G, Caddick S, Baker JR, J. Am. Chem. Soc 2010, 132, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu L, Silva MJSA, Gois PMP, Kuan SL, Weil T, Chem. Sci 2021, 12, 13321–13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yu J, Yang X, Sun Y, Yin Z, Angew. Chem. Int. Ed 2018, 57, 11598–11602. [DOI] [PubMed] [Google Scholar]
- [27].Zhang D, Devarie-Baez NO, Li Q, Lancaster JR, Xian M, Org. Lett 2012, 14, 3396–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen X, Wu H, Park C-M, Poole TH, Keceli G, Devarie-Baez NO, Tsang AW, Lowther WT, Poole LB, King SB, Xian M, Furdui CM, ACS Chem. Biol 2017, 12, 2201–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tang KC, Maddox SM, Backus KM, Raj M, Chem. Sci 2022, 13, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Luo Q, Tao Y, Sheng W, Lu J, Wang H, Nat. Commun 2019, 10, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, Kramm A, Chen S, Hillig RC, Clemons PA, Gradl S, Montagnon C, Lazarski KE, Christian S, Bajrami B, Neuhaus R, Eheim AL, Viswanathan VS, Schreiber SL, Nat. Chem. Biol 2020, 16, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vinogradova EV, Zhang C, Spokoyny AM, Pentelute BL, Buchwald SL, Nature 2015, 526, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rojas AJ, Zhang C, Vinogradova EV, Buchwald NH, Reilly J, Pentelute BL, Buchwald SL, Chem. Sci 2017, 8, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Messina MS, Stauber JM, Waddington MA, Rheingold AL, Maynard HD, Spokoyny AM, J. Am. Chem. Soc 2018, 140, 7065–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hanaya K, Ohata J, Miller MK, Mangubat-Medina AE, Swierczynski MJ, Yang DC, Rosenthal RM, Popp BV, Ball ZT, Chem. Commun 2019, 55, 2841–2844. [DOI] [PubMed] [Google Scholar]
- [36].Lopchuk JM, Fjelbye K, Kawamata Y, Malins LR, Pan C-M, Gianatassio R, Wang J, Prieto L, Bradow J, Brandt TA, Collins MR, Elleraas J, Ewanicki J, Farrell W, Fadeyi OO, Gallego GM, Mousseau JJ, Oliver R, Sach NW, Smith JK, Spangler JE, Zhu H, Zhu J, Baran PS, J. Am. Chem. Soc 2017, 139, 3209–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jung M, Lindsay VNG, J. Am. Chem. Soc 2022, 144, 4764–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].a) Frei R, Waser J, J. Am. Chem. Soc 2013, 135, 9620–9623; [DOI] [PubMed] [Google Scholar]; b) Frei R, Wodrich MD, Hari DP, Borin P-A, Chauvier C, Waser J, J. Am. Chem. Soc 2014, 136, 16563–16573; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Allouche EMD, Grinhagena E, Waser J, Angew. Chem. Int. Ed 2022, 61, e202112287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].a) Abegg D, Tomanik M, Qiu N, Pechalrieu D, Shuster A, Commare B, Togni A, Herzon SB, Adibekian A, J. Am. Chem. Soc 2021, 143, 20332–20342; [DOI] [PubMed] [Google Scholar]; b) Tessier R, Nandi RK, Dwyer BG, Abegg D, Sornay C, Ceballos J, Erb S, Cianférani S, Wagner A, Chaubet G, Adibekian A, Waser J, Angew. Chem. Int. Ed 2020, 59, 10961–10970. [DOI] [PubMed] [Google Scholar]
- [40].Sala O, Santschi N, Jungen S, Lüthi HP, Iannuzzi M, Hauser N, Togni A, Chem. Eur. J 2016, 22, 1704–1713. [DOI] [PubMed] [Google Scholar]
- [41].Economou C, Tomanik M, Herzon SB, J. Am. Chem. Soc 2018, 140, 16058–16061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].a) Rosen CB, Francis MB, Nat. Chem. Biol 2017, 13, 697–705; [DOI] [PubMed] [Google Scholar]; b) Asiimwe N, Al Mazid MF, Murale DP, Kim YK, Lee J-S, Peptide Science 2022, 114, e24235. [Google Scholar]
- [43].Kulkarni SS, Sayers J, Premdjee B, Payne RJ, Nat. Rev. Chem 2018, 2, 0122. [Google Scholar]
- [44].Zhang Y, Park K-Y, Suazo KF, Distefano MD, Chem. Soc. Rev 2018, 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a) De Rosa L, Di Stasi R, Romanelli A, D’Andrea LD, Molecules 2021, 26, 3521; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Romanelli A, Shekhtman A, Cowburn D, Muir TW, Proc. Natl. Acad. Sci. USA 2004, 101, 6397–6402; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu D, Xu R, Dutta K, Cowburn D, FEBS Lett 2008, 582, 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Carrington JC, Dougherty WG, Proc. Natl. Acad. Sci. USA 1988, 85, 3391–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].a) Casi G, Huguenin-Dezot N, Zuberbühler K, Scheuermann J, Neri D, J. Am. Chem. Soc 2012, 134, 5887–5892; [DOI] [PubMed] [Google Scholar]; b) Bernardes GJL, Steiner M, Hartmann I, Neri D, Casi G, Nat. Protoc 2013, 8, 2079–2089. [DOI] [PubMed] [Google Scholar]
- [48].Li K, Wang W, Gao J, Angew. Chem., Int. Ed 2020, 59, 14246–14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].a) Brogi S, Ibba R, Rossi S, Butini S, Calderone V, Gemma S, Campiani G, Molecules 2022, 27, 2561; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dufour E, Storer AC, Menard R, Biochemistry 1995, 34, 9136–9143. [DOI] [PubMed] [Google Scholar]
- [50].a) Berteotti A, Vacondio F, Lodola A, Bassi M, Silva C, Mor M, Cavalli A, ACS Medicinal Chemistry Letters 2014, 5, 501–505; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oballa RM, Truchon J-F, Bayly CI, Chauret N, Day S, Crane S, Berthelette C, Bioorganic & Medicinal Chemistry Letters 2007, 17, 998–1002. [DOI] [PubMed] [Google Scholar]
- [51].a) Ren H, Xiao F, Zhan K, Kim Y-P, Xie H, Xia Z, Rao J, Angew. Chem. Int. Ed 2009, 48, 9658–9662; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) White EH, McCapra F, Field GF, J. Am. Chem. Soc 1963, 85, 337–343. [Google Scholar]
- [52].Zhang M, Liang G, Sci. China Chem 2018, 61, 1088–1098. [Google Scholar]
- [53].Wang W, Gao J, J. Org. Chem 2020, 85, 1756–1763. [DOI] [PubMed] [Google Scholar]
- [54].Zheng X, Li Z, Gao W, Meng X, Li X, Luk LYP, Zhao Y, Tsai Y-H, Wu C, J. Am. Chem. Soc 2020, 142, 5097–5103. [DOI] [PubMed] [Google Scholar]
- [55].Silva M, Faustino H, Coelho JAS, Pinto MV, Fernandes A, Companon I, Corzana F, Gasser G, Gois PMP, Angew. Chem. Int. Ed 2021, 60, 10850–10857. [DOI] [PubMed] [Google Scholar]
- [56].Wu Y, Li C, Fan S, Zhao Y, Wu C, Bioconjugate Chem 2021, 32, 2065–2072. [DOI] [PubMed] [Google Scholar]
- [57].Istrate A, Geeson MB, Navo CD, Sousa BB, Marques MC, Taylor RJ, Journeaux T, Oehler SR, Mortensen MR, Deery MJ, Bond AD, Corzana F, Jiménez-Osés G, Bernardes GJL, J. Am. Chem. Soc 2022, DOI: 10.1021/jacs.1022c02185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].White AM, Palombi IR, Malins LR, Chem. Sci 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang C, Welborn M, Zhu T, Yang NJ, Santos MS, Van Voorhis T, Pentelute BL, Nat. Chem 2016, 8, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Tuang S, Dieppa-Matos D, Zhang C, Shugrue CR, Dai P, Loas A, Pentelute BL, Chem. Commun 2021, 57, 3227–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].a) Baumann AL, Schwagerus S, Broi K, Kemnitz-Hassanin K, Stieger CE, Trieloff N, Schmieder P, Hackenberger CPR, J. Am. Chem. Soc 2020, 142, 9544–9552; [DOI] [PubMed] [Google Scholar]; b) Stieger CE, Park Y, de Geus MAR, Kim D, Huhn C, Slenczka JS, Ochtrop P, Müchler JM, Süssmuth R, Broichhagen J, Baik M-H, Hackenberger C, Angew. Chem. Int. Ed 2022, DOI: 10.1002/anie.202205348; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kasper M-A, Glanz M, Oder A, Schmieder P, von Kries JP, Hackenberger CPR, Chem. Sci 2019, 10, 6322–6329; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Park Y, Baumann AL, Moon H, Byrne S, Kasper M-A, Hwang S, Sun H, Baik M-H, Hackenberger CPR, Chem. Sci 2021, 12, 8141–8148; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kasper M-A, Glanz M, Stengl A, Penkert M, Klenk S, Sauer T, Schumacher D, Helma J, Krause E, Cardoso MC, Leonhardt H, Hackenberger CPR, Angew. Chem. Int. Ed 2019, 58, 11625–11630. [DOI] [PubMed] [Google Scholar]