

Abstract
Background:
Several studies have suggested genetic variants associated with acetaminophen induced liver injury (DILI) following overdose. Genetic variation associated with acetaminophen induced alanine aminotransferase elevation during therapeutic dosing has not been examined.
Methods:
We performed genetic analyses on patients that ingested therapeutic doses of 4 grams of acetaminophen for up to 16 days. We examined 20 genes previously implicated in the metabolism of acetaminophen or the development of immune mediated DILI using the Illumina Multi-Ethnic Global Array 2. Autosomes were aligned and imputed using TOPMed. A candidate gene region analysis was performed by testing each gene individually using linkage disequilibrium (LD) pruned variants with the adaptive sum of powered scores (aSPU) test from the aSPU R package. The highest measured ALT during therapy, the maximum ALT, was used as the outcome.
Results:
192 subjects taking therapeutic APAP were included in the genetic analysis. 136 (70.8%) were female, 133 (69.2%) were Caucasian race, and the median age was 34 years (IQR: 26, 46). Age > than 50 years was the only clinical factor associated with maximum ALT increase. Variants in SULT1E1, the gene responsible for Sulfotransferase Family 1E Member 1 enzyme production, was associated with maximum ALT. No single variant drove this association, but rather the association was due to the additive effects of numerous variants within the gene. No other genes were associated with maximum ALT increase in this cohort.
Conclusion:
Acetaminophen induced ALT elevation at therapeutic doses was not associated with variation in most genes associated with acetaminophen metabolism or immune induced DILI in this cohort. The role of SULT1E1 polymorphism in acetaminophen induced elevated ALT needs further examination.
Keywords: Acetaminophen, drug induced liver injury, genetic, hepatotoxicity, pharmacogenetics, therapeutic dose
Introduction
Drug induced liver injury (DILI) is the most common reason for pharmaceutical removal from development or the market once approved.[1] DILI can range from asymptomatic mild elevation in hepatic transaminases to fulminant liver failure. The prognosis of the condition is dependent upon several important factors such as the drug, the pathophysiologic mechanism, patient factors, and treatment. The predominant pathophysiology of DILI is felt to be immune mediated, though many DILI causes remain idiosyncratic. 4% of these idiosyncratic DILI cases progress to acute liver failure,[1] therefore understanding the pathophysiology of the condition, as well as hepatic adaptation may allow for improved recognition and treatment.
Demographic risk factors have not adequately explained the risk of DILI; female sex was associated with the condition in a US based cohort[2], while male sex predominated in a Spanish cohort[3]. A UK cohort suggested that sex was a modifying effect dependent upon the drug that caused the injury[2]. DILI is characterized by the microscopic patterns it imparts; hepatocellular, cholestatic, or mixed. While most DILI is categorized as predominantly one microscopic subtype, some drugs may yield different patterns in different patients. Thus, the pathophysiology of the injury may vary among patients. In addition to these clinical and demographic factors, genetic differences between patients may predispose some patients to elevated transaminases when challenged with some medications. In fact, genetic variants have been associated with the development of hepatotoxicity for such drugs as fluvoxamine[4], isoniazid[5], and may explain some of these differences between patients that develop DILI. Genetic variation may also explain different hepatic injury outcomes following acetaminophen overdose.
Acetaminophen is the leading cause of acute liver failure in the United States, and while the majority of this liver failure is due to overdose, it is known that some patients developed transient elevated transaminases even at therapeutic doses. Acetaminophen is the prototypical hepatocellular toxin resulting from through production of N-acetyl-p-benzoquinone imine (NAPQI) predominantly through CYP2E1 metabolism of the parent compound. When glutathione detoxification is overwhelmed, NAPQI leads to hepatocyte damage and death. However, there is evidence that immune factors may also contribute to elevation of transaminases in the setting of therapeutic dosing[6]. This reaction is likely polygenic, given the complexity of acetaminophen metabolism (Figure 1) [7] and the range of clinical response observed in patients taking the drug. Expanded cohorts are necessary to capture the relatively uncommon clinical outcome of transaminase elevation from therapeutic dosing, given that it is frequently asymptomatic and transient. The primary objective of this study was to examine genetic variants in metabolic enzymes and immune genes associated with acetaminophen induced liver injury in a large, well-controlled cohort of patients taking therapeutic acetaminophen.
Figure 1:
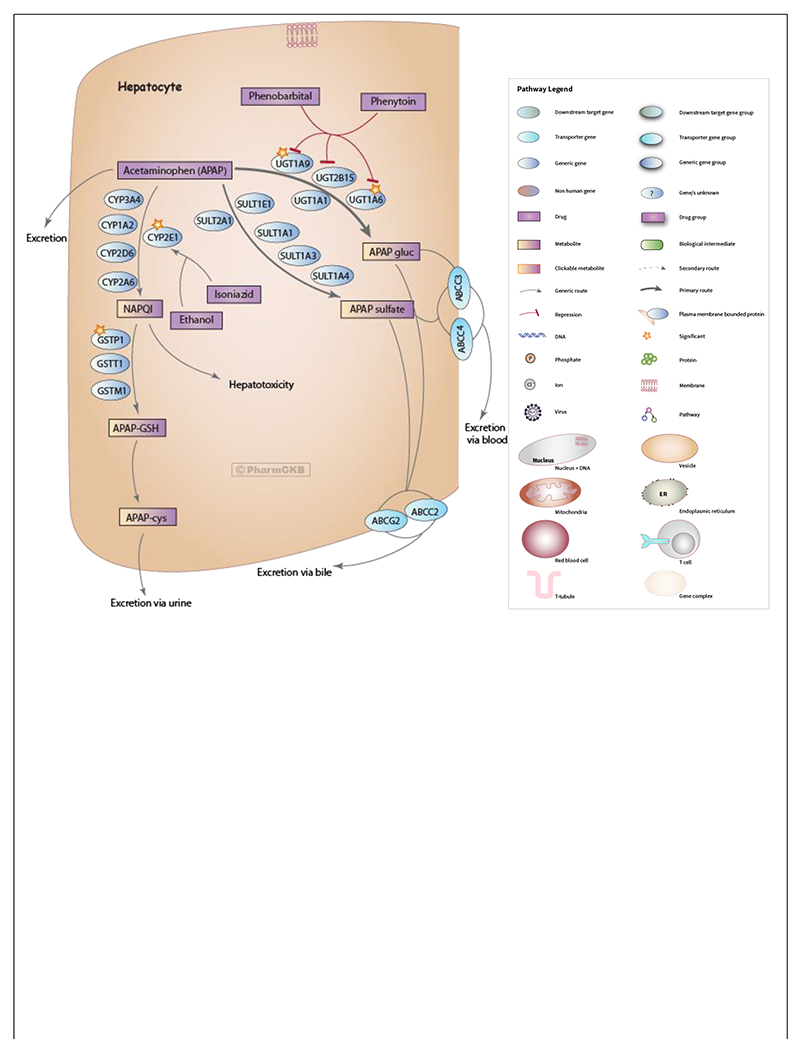
Acetaminophen Pathway (therapeutic doses), Pharmacokinetics. Reproduced with permission from PharmGKB[7].
Methods:
We performed genetic analyses on patients that ingested a therapeutic dose of 4 grams of acetaminophen for up to 16 days as part of a previously completed randomized controlled clinical trial (NCT00743093).[8] Genes examined were advised by 1) PharmGKB metabolic pathway of acetaminophen[7], prior whole genome sequence[6] and metabolomic[9] analyses from subsets of this cohort. Both the parent study and this genomic analysis were approved by the Colorado Multiple Institutional Review Board.
Parent study:
The parent study was an outpatient placebo controlled trial in which subjects were recruited from research list-serves, posters, a classified advertisement website (Craigslist), and media advertising. All subjects provided informed consent. We included male and female subjects who were age 18 years or older and who did not have any of the following exclusion criteria: history of acetaminophen ingestion on any of the four days preceding study enrollment or a measurable serum acetaminophen concentration at time of enrollment; laboratory testing suggesting active viral hepatitis A, B, or C infection; any of the following tests greater than the upper limit of normal at screening: serum aminotransferase or total bilirubin, International Normalized Ratio (INR) or alkaline phosphatase activity; platelet count less than 125,000/mL; positive pregnancy test; history of cholelithiasis (without cholecystectomy); history of heavy ethanol use defined as consuming more than an average of 3 alcohol containing drinks daily or 3 or more alcohol containing drinks on any given day over the preceding 2 weeks prior to study enrollment; new prescription medication started within the previous 30 days; taking isoniazid or warfarin; currently has anorexia nervosa or reports a fasting type diet; clinically intoxicated, psychiatrically impaired or unable to give informed consent for any reason; known hypersensitivity or allergy to acetaminophen.
Intervention:
Subjects were administered a therapeutic dose of acetaminophen, 4 g/day. Subjects were instructed to take 2 x 500 mg tablets every 4 hours for 4 doses each day (the exact timing varied but subjects were asked to take the doses every 4 hours after the first dose each day). The subjects noted the time of ingestion for each dose in a study diary. They also recorded other medications and alcohol consumption. Compliance was verified by study diary, pill counts at each study visit, and confirmation of acetaminophen in the plasma. ALT was drawn on study days 0 (baseline), 4, 7, 10, 13 and 16 and, when elevated, until it returned to baseline. Patients did not change their diet throughout the study. The parent study also included a placebo arm, whose patients were not investigated as part of this study.
Genetic analyses:
Genomic DNA was extracted from peripheral blood collected in 4mL EDTA tubes on an Autogen FlexSTAR+ instrument utilizing Qiagen Flexigene chemistry. Nucleic acid yield was determined by fluorescence (QuantiFluor ds DNA system, Promega) and UV absorbance. Microarray genotyping was performed on the custom Multi-Ethnic Global Array 2 (MEGA-2, Illumina, Inc.). Cluster files for each version (.egt) were developed with the same 708-sample training set comprising 79 samples purchased from Coriell and 629 deidentified discarded clinical blood samples. The resulting calls were evaluated and underperforming probes, generally defined as those with call frequencies <0.97 or cluster separation ≤0.3 or intensity means ≤0.2, were removed (n=12,577 removed from MEGAv2). Sample-level and 96-well plate-level QC of microarray data was performed. Post-genotypic quality control (QC) procedures where implemented: checking sample and variant call rates, sex discordance, restriction to bi-allelic SNPs, departure from Hardy Weinberg Equilibrium, plate batch effects, removal of related/duplicate samples (Figures S1 – S6), and control sample concordance with 1000 genomes (TGP)[10] and Genome in a Bottle (GIAB)[11] reference samples. All QC steps were completed in PLINK 1.9 [12], with the exception of sample kinship estimation, which was completed in the GENESIS R Package[13].
Autosomes were aligned and imputed using the TOPMed imputation server, which lifted over all information to Genome Reference Consortium human genome version 38 (GRCh38). In total, 49,708,367 sites passed the post-imputation quality filters, where variants needed to be genotyped or have an Imputed Rsq ≥ 0.7 with minor allele frequency ≥ 0.01 to be retained for further analyses. Copy number variants (CNVs) in CYP2E1 were captured using the PennCNV v1.0.5 software[14].
Statistical analyses:
Our primary outcome was highest ALT measured throughout the study period, treated as a continuous variable. For clinical covariates with a non-Gaussian distribution, median values were reported with their associated IQRs. We performed linear multivariate logistic regression including age in years, ancestry principle component analyses, and target genes to determine association of clinical variables with max ALT elevation. Age was assessed as a categorical variable to determine the age associated with elevated ALT.
Ancestry representative principal components (PCs) and empirical kinship adjusted for population structure were estimated using the PC-Relate/PC-AiR pipeline within the GENESIS R package[15], as relatedness of the first, second, and third degree was present between samples. Clinically relevant variants (Appendix 1) in the pathways for APAP induced liver injury were examined and genes with preliminary data suggesting a role in APAP induced liver injury were used to create a candidate gene list[6]. A candidate gene region analysis was performed by testing each gene individually using linkage disequilibrium (LD) pruned variants (Rsq > 0.1) with the adaptive sum of powered scores (aSPU) test from the aSPU R package[16].
The aSPU test leverages a Monte Carlo framework to flexibly test for the association between the SNPs in a gene region and a trait of interest. This test facilitates high power for identifying SNP associations via an adaptive framework. This adaptive approach maintains power under various genetic association architectures, ranging from one SNP being associated to all SNPs being associated. The test provides a properly calibrated p-value within the adaptive framework. A significant association in this context should be interpreted as a rejection of the ‘global null’ hypothesis, which is that no SNP in the region is associated with the trait. Given a significant association, we can investigate which component test (or tests) are driving the association, thus allowing us to draw post-hoc inference about the likely genetic architecture. We conducted aSPU tests for our genes of interest using LD pruned variants from imputed genotypes and correcting for age (treated as a categorical variable with three levels), gender, and population structure (accounted for with the first five principal components). Gene regions were identified using the NIH NCBI GRCh38 database. Statistical significance for the candidate gene analysis is established using a Bonferroni correction (0.05/20=0.0025). Individual variants within significant genes were tested using linear regression, also accounting for age in years, gender and population stratification.
Results:
Subject demographic and clinical profiles:
192 subjects of the entire study population (n=204) taking therapeutic APAP dosing passed quality control analysis for genotyping. Demographics are provided in Table 1. The mean maximum ALT during the study period was 38 IU/L (St Dev: 26, range 11-191 IU/L) (Figure 2). The only clinical variable associated with maximum ALT was age older than 50 years (17 IU/L increase in ALT [7 to 27 IU/ml, 95% CI] compared to age 30-49 years p=0.0013, and 13 IU/ml increase in ALT [3 to 24 IU/L, 95% CI] compared to age 18-29 years p=0.0114). The majority of this cohort were women (n=136, 71%). Frequencies of gene variants examined in this study do not change based upon gender.
Table 1. Subject demographic and clinical profiles:
There were no significant differences in demographics between ALT elevation and No ALT elevation groups. ALT was significantly elevated at baseline for the ALT elevation group. IQR = Interquartile range, st.dev. = standard deviation
| Demographic/Clinical Parameter | |
|---|---|
| N (%) | 192 |
| Female, n (%) | 136 (70.8) |
| Race, Caucasian, n (%) | 133 (69.2) |
| Race, Other, n (%) | 59 (30.8) |
| Ethnicity, Non-Hispanic, n (%) | 192 (84.4) |
| Age, median (IQR) | 34 (26,46) |
| BMI (kg/m2), mean (st.dev.) | 26.7 (6.0) |
| Baseline ALT, mean (st.dev.) | 21 (7.6) |
| Max ALT during study, mean (st dev, range) | 38 (26, 11-191) |
Figure 2.
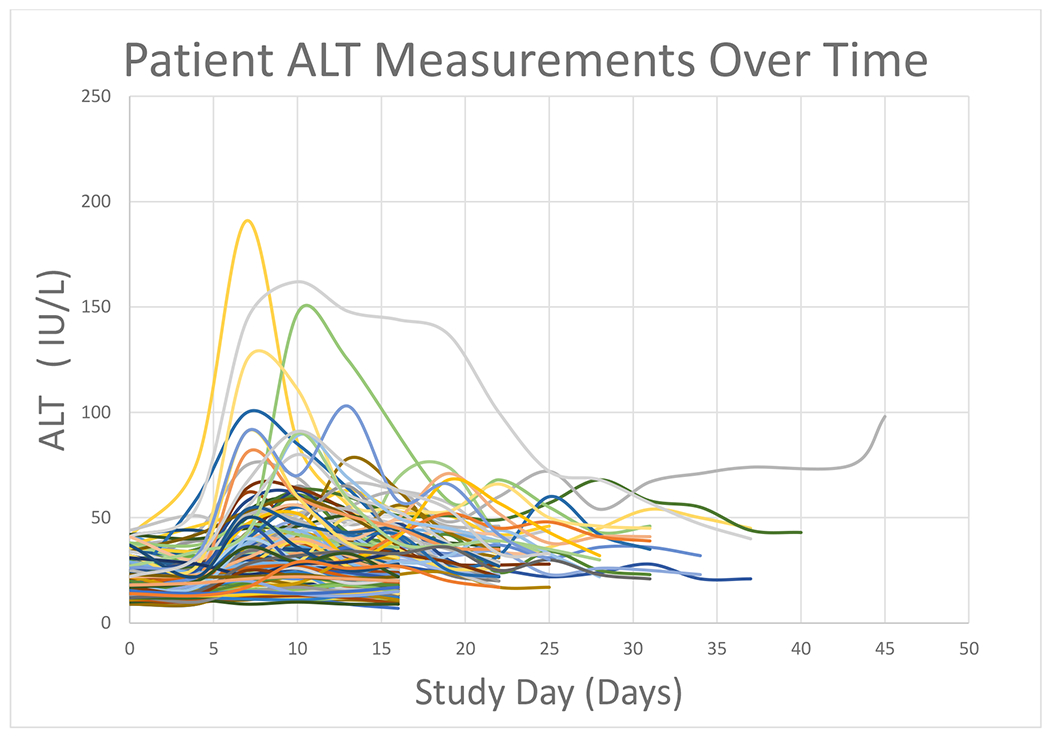
ALT progression over time during the study period. Each line represents an individual patient’s ALT measurements during the study period. Patients remained on protocol for a minimum of 16 days, subsets of the cohort were followed for 22 and 31 days, or until their transaminases returned to normal.
Targeted Genetic Analyses:
19 out of the 20 targeted genes associated with acetaminophen metabolism or previously associated with drug induced liver injury were not associated with maximum ALT elevation in this cohort. CYP2E1 polymorphisms did not predict ALT elevation at therapeutic doses. There were no full gene CYP2E1 copy number variants identified in this cohort.
SULT1E1, the gene responsible for Sulfotransferase Family 1E Member 1 enzyme production, was associated with maximum ALT (Table 2). Table 3 describes the p-values for each of the component tests of the aSPU; it is apparent that the significance of the adaptive test is being driven by the SPU(1) test. The SPU(1) test is equivalent to a burden test[17], which has the comparatively strongest power under a genetic architecture of multiple causal SNPs. The single variant regression p-values (Table 4) show that no single SNP within the SULT1E1 gene is significantly associated with maximum ALT. Taken in conjunction and given the significant adaptive test, these two results – that is, the best performance of the SPU(1) component test and the lack of an individually associated SNP – suggest a genetic architecture where multiple SNPs in the SULT1E1 gene region are associated with the outcome.
Table 2.
Genes involved with acetaminophen metabolism or drug induced liver injury examined for association with maximum ALT elevation over 16 days of daily therapy.
| Gene | CHR | LD Pruning at 0.1, Variant coverage | aSPU P-Value |
|---|---|---|---|
| CYP2E1 | chr10 | 3 | 0.1838 |
| CYP2E1 Promoter | chr10 | 1 | 0.3076 |
| UGT1A | chr2 | 37 | 0.3796 |
| HLA-DRB1 | chr6 | 1 | 0.1318 |
| HLA-DRB5 | chr6 | 1 | 0.8841 |
| FZD5 | chr2 | 3 | 0.0969 |
| SULT1A1 | chr16 | 2 | 0.6893 |
| SULT1E1 | chr4 | 8 | 0.0019* |
| SULT2A1 | chr19 | 4 | 0.1218 |
| CYP2A6 | chr19 | 1 | 0.2997 |
| CYP2D6 | chr22 | 2 | 0.5744 |
| CYP3A4 | chr7 | 6 | 0.2547 |
| UGT2B15 | chr4 | 7 | 0.0949 |
| GSTP1 | chr11 | 2 | 0.6413 |
| CYP1A2 | chr15 | 3 | 0.4655 |
| CD44 | chr11 | 25 | 0.0539 |
| XBP1 | chr22 | 3 | 0.6663 |
| BTNL2 | chr6 | 20 | 0.4775 |
| GRHL3 | chr1 | 12 | 0.8441 |
| ANXA11 | chr10 | 19 | 0.6093 |
Following Bonferroni correction with genes with variants included after LD pruning, level of significance was 0.0025. GSTT1, GSTM1, SULT1A4, and SULT1A3 were also tested though after LD pruning, no variants remained and thus, were not included in these analyses.
Table 3.
Gamma p-values for the SULT1E1 aSPU Test. These values demonstrate that no individual component drives the ASPU test, but rather it is the summation of each of the components.
| GAMMA | P-VALUE |
|---|---|
| 1 | 0.001 |
| 2 | 0.034 |
| 3 | 0.015 |
| 4 | 0.022 |
| 5 | 0.017 |
| 6 | 0.02 |
| 7 | 0.018 |
| 8 | 0.019 |
| INFINITY | 0.018 |
| ASPU | 0.001998 |
Table 4.
Single variant linear regression p-values from SULT1E1
| SNP | ESTIMATE | STANDARD ERROR | P-VALUE |
|---|---|---|---|
| CHR4:69843078:C:T | 0.69 | 6.93 | 0.920 |
| CHR4:69843639:C:T | 2.64 | 4.00 | 0.510 |
| CHR4:69846443:A:T | 5.59 | 2.51 | 0.026 |
| CHR4:69850788:T:C | 7.67 | 15.96 | 0.631 |
| CHR4:69852851:G:A | 4.66 | 5.22 | 0.373 |
| CHR4:69854148:T:A | −10.06 | 11.67 | 0.389 |
| CHR4:69859231:G:A | 8.30 | 10.63 | 0.436 |
| CHR4:69859383:C:T | −2.44 | 11.99 | 0.838 |
In patients with an ALT of > 80 IU/L, there were 5 patients with variants in SULT1E1 and 7 patients without variants in the gene. In patients with ALT peak less than 80 IU/L, there were 16 patients with variants in SULT1E1 and 164 without variants in the gene. The odds of having an ALT>80 IU/L in this cohort among those with variants in SULT1E1 was 7.32 (95% CI: 2.08-25.74).
Limitations:
There are several limitations to this study. Elevated ALT at therapeutic doses is an uncommon outcome, thus we may have missed rare genetic variants associated with this outcome in our relatively small sample. Due to the limited power in only 192 subjects we may not have captured all genetic underpinnings of ALT elevation in this cohort. Only approximately 25% of the cohort had dynamic changes in ALT during the study period (Figure 2). There was only one subject that developed ALT elevation that did not return to baseline during the study timeline. This study was unable to control for diet or lifestyle choices which often significantly alter liver function. However, these factors are present in the general population taking APAP and are thus representative of real-world drug dosing. We were able to examine 20 genes associated with APAP metabolism and drug induced liver injury. These genes were chosen based upon prior studies and the known metabolic pathways of APAP. The aSPU analysis considered the gene as the unit of analysis, rather than individual variants. This maximizes our ability to detect a gene specific effect, though it still may be underpowered by low frequency of an important variant. Thus, thousands of patients with whole genome sequencing may be necessary to generate a predictive polygenic risk score for this clinical outcome.
Discussion
In this study, we have demonstrated that elevated transaminases associated with therapeutic acetaminophen are not associated with the most common genes associated with acetaminophen metabolism or genes previously associated with drug induced liver injury. This is likely because metabolism of this drug is polygenic with significant redundancy in the metabolic pathways. Variants in only one gene, SULT1E1, were associated with maximum ALT elevation.
The SULT1E1 sulfotransferase enzyme catalyzes the sulfate conjugation of hormones, neurotransmitters, and some pharmaceuticals. This sulfotransferase utilizes 3′-phospho-5′-adenylyl sulfate as a sulfonate donor to catalyze the sulfate conjugation of a wide variety of acceptor molecules bearing a hydroxyl or an amine group, including acetaminophen. Sulfonation increases the water solubility, and therefore their renal excretion. Acetaminophen sulfation is saturated at high doses in rats at high doses[18], and differences in sulfation have been associated with hepatotoxicity in overdose[19]. There is some concern that this finding is a false positive association, given the redundancy in the acetaminophen sulfation system in humans. In fact, SULT1A1, SULT1A3 and SULT1A4, SULT1C4[20], SULT2A are all involved in acetaminophen sulfation[21]. This would suggest that deleterious variants in the gene for SULT1E1 could be mitigated by other sulfation pathways as well as other detoxification mechanisms. Genes associated with glutathione production and glucuronidation were not altered in individuals with deleterious variants in SULT1E1 thus alternative detoxification and conjugation pathways remained intact supporting our hypothesis that this is not a clinically relevant finding. However, if this variant leads to significantly reduced sulfation capacity, this finding may, in fact, lead to shunting to toxic metabolites, even at therapeutic doses. There are 13 SULT isoforms in humans[22], though the % contribution of each SULT enzyme in acetaminophen metabolism in humans is unknown. Individual SULT enzyme contribution likely varies between patients due to genetic variation, diet, and comorbidity[23]. Therefore, we will seek to examine this variant in other cohorts and capture urine metabolite levels to assess this finding further.
We also examined genes associated with immune mediated drug induced liver injury, HLA-DRB1[24], HLA-DRB5[24], CD44[25, 26], ANXA11[27], and FZD5[6]. These genes were also not associated with elevated ALT in this targeted genetic analysis. Immune mediated hepatitis injury due to acetaminophen is also likely a polygenic condition. Genes associated with hepatic adaption to xenobiotic insult were not examined as part of this study. Examination of genes associated with hepatic adaption in other physiologic stressful conditions, such as pregnancy[28], should be examined in future studies. Genes associated with hepatic regeneration and cell proliferation will be the focus of our future studies.
Past work using the acute liver failure study group that examined genetic variants associated with acetaminophen induced liver injury did not adequately confirm acetaminophen induced liver failure[29, 30] nor did they control for other pertinent clinical parameters. However, that work provided initial hypothesis generating data suggesting that genetic variants may contribute to discordant clinical presentation. Observations from that group suggested that patients with UGT2B15 *2/*2 genotype had higher acetaminophen protein adduct formation and those with CYP2E1*1D/*1D genotype had lower oxidative clearance; those findings were not confirmed in this study. However, our cohort had fewer African American patients, and the CYP2E1*1D variant may be more common in those patients. A larger and ancestrally diverse patient population will be necessary to generate a polygenic risk score that predicts acetaminophen mediated drug induced liver injury.
In summary, this work suggests that acetaminophen induced ALT elevation at therapeutic doses is not associated with variants in most genes associated with acetaminophen metabolism. We will examine SULT1E1 and sulfation metabolites to assess that variant association. Future studies examining drug-induced liver injury at therapeutic doses should examine genes in hepatic adaption pathways.
Appendix 1: Genes and genome location examined in the targeted genetic analysis.
| Gene | Location: GRCh38.p13 Assembly |
|---|---|
| CYP2E1 | NC_000010.11 (133527363..133539123) |
| CYP2E1 Promoter | NC_000010.11 (133523651..133527458) |
| UGT1A | NC_000002.12 (233585439..233773299) |
| HLA-DRB1 | NC_000006.12 (32578775..32589848, complement) |
| HLA-DRB5 | NC_000006.12 (32517343..32530316, complement) |
| FZD5 | NC_000002.12 (207753889..207769906, complement) |
| SULT1A1 | NC_000016.10 (28605258..28623395, complement) |
| SULT1E1 | NC_000004.12 (69841212..69860151, complement) |
| SULT2A1 | NC_000019.10 (47870467..47886315, complement) |
| SULT1A3 | NC_000016.10 (30199255..30204310) |
| SULT1A4 | NC_000016.10 (29459913..29464966) |
| CYP2A6 | NC_000019.10 (40843541..40850447, complement) |
| CYP2D6 | NC_000022.11 (42126499..42130810, complement) |
| CYP3A4 | NC_000007.14 (99756967..99784184, complement) |
| UGT2B15 | NC_000004.12 (68646597..68670652, complement) |
| GSTP1 | NC_000011.10 (67583812..67586653) |
| GSTT1 | NT_187633.1 (270308..278486, complement) |
| GSTM1 | NC_000001.11 (109687817..109693745) |
| CYP1A2 | NC_000015.10 (74748845..74756607) |
| CD44 | NC_000011.10 (35139168..35232402) |
| XBP1 | NC_000022.11 (28794560..28800569, complement) |
| BTNL2 | NC_000006.12 (32393339..32408879, complement) |
| GRHL3 | NC_000001.11 (24319333..24364482) |
| ANXA11 | NC_000010.11 (80150889..80205808, complement) |
Data Availability Statement:
Genetic data from individual subjects are not available due to their identifying nature. However, deidentified summary data from this study are available upon request of the authors.
References:
- 1.Katarey D and Verma S, Drug-induced liver injury. Clin Med (Lond), 2016. 16(Suppl 6): p. s104–s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chalasani N, et al. Features and Outcomes of 899 Patients With Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology, 2015. 148(7): p. 1340–52 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robles-Diaz M, et al. Use of Hy’s law and a new composite algorithm to predict acute liver failure in patients with drug-induced liver injury. Gastroenterology, 2014. 147(1): p. 109–118 e5. [DOI] [PubMed] [Google Scholar]
- 4.Daly AK, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet, 2009. 41(7): p. 816–9. [DOI] [PubMed] [Google Scholar]
- 5.Levano KS, et al. Allelic and genotypic frequencies of NAT2, CYP2E1, and AADAC genes in a cohort of Peruvian tuberculosis patients. Mol Genet Genomic Med, 2021: p. e1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monte AA, et al. The Genomics of Elevated ALT and Adducts in Therapeutic Acetaminophen Treatment: a Pilot Study. J Med Toxicol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazaleuskaya LL, et al. PharmGKB summary: pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet Genomics, 2015. 25(8): p. 416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heard K, et al. A randomized, placebo-controlled trial to determine the course of aminotransferase elevation during prolonged acetaminophen administration. BMC Pharmacol Toxicol, 2014. 15: p. 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sonn BJ, et al. Metabolomic markers predictive of hepatic adaptation to therapeutic dosing of acetaminophen. Clin Toxicol (Phila), 2021: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.1000 Genomes reference dataset. 2021. [cited 2021.
- 11.Genome in a Bottle reference dataset. 2021.
- 12.Chang C PLINK 1.90 beta. 2020. [cited 2021.
- 13.Conomos MP, et al. Model-free Estimation of Recent Genetic Relatedness. Am J Hum Genet, 2016. 98(1): p. 127–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res, 2007. 17(11): p. 1665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gogarten SM, et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics, 2019. 35(24): p. 5346–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan W, Kwak IY, and Wei P, A Powerful Pathway-Based Adaptive Test for Genetic Association with Common or Rare Variants. Am J Hum Genet, 2015. 97(1): p. 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang T and Elston RC, Improved power by use of a weighted score test for linkage disequilibrium mapping. Am J Hum Genet, 2007. 80(2): p. 353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L and Klaassen CD, Different mechanism of saturation of acetaminophen sulfate conjugation in mice and rats. Toxicol Appl Pharmacol, 1996. 139(1): p. 128–34. [DOI] [PubMed] [Google Scholar]
- 19.Li J, et al. Sulfate conjugation may be the key to hepatotoxicity in paracetamol overdose. Br J Clin Pharmacol, 2021. 87(5): p. 2392–2396. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto A, et al. Sulphation of acetaminophen by the human cytosolic sulfotransferases: a systematic analysis. J Biochem, 2015. 158(6): p. 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adjei AA, et al. Interindividual variability in acetaminophen sulfation by human fetal liver: implications for pharmacogenetic investigations of drug-induced birth defects. Birth Defects Res A Clin Mol Teratol, 2008. 82(3): p. 155–65. [DOI] [PubMed] [Google Scholar]
- 22.Lindsay J, et al. Structure, function and polymorphism of human cytosolic sulfotransferases. Curr Drug Metab, 2008. 9(2): p. 99–105. [DOI] [PubMed] [Google Scholar]
- 23.McGill MR and Jaeschke H, Metabolism and disposition of acetaminophen: recent advances in relation to hepatotoxicity and diagnosis. Pharm Res, 2013. 30(9): p. 2174–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hautekeete ML, et al. HLA association of amoxicillin-clavulanate--induced hepatitis. Gastroenterology, 1999. 117(5): p. 1181–6. [DOI] [PubMed] [Google Scholar]
- 25.Johnson P and Ruffell B, CD44 and its role in inflammation and inflammatory diseases. Inflamm Allergy Drug Targets, 2009. 8(3): p. 208–20. [DOI] [PubMed] [Google Scholar]
- 26.Liang JB, et al. CD8(+) T cells actively penetrate hepatocytes via the CD44/p-ERM/F-actin pathway in autoimmune hepatitis. J Dig Dis, 2021. 22(6): p. 351–362. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, et al. Annexin A11 in disease. Clin Chim Acta, 2014. 431: p. 164–8. [DOI] [PubMed] [Google Scholar]
- 28.Bustamante JJ, et al. Gene profiling of maternal hepatic adaptations to pregnancy. Liver Int, 2010. 30(3): p. 406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Court MH, et al. The UDP-glucuronosyltransferase (UGT) 1A polymorphism c.2042C>G (rs8330) is associated with increased human liver acetaminophen glucuronidation, increased UGT1A exon 5a/5b splice variant mRNA ratio, and decreased risk of unintentional acetaminophen-induced acute liver failure. J Pharmacol Exp Ther, 2013. 345(2): p. 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Court MH, et al. Candidate gene polymorphisms in patients with acetaminophen-induced acute liver failure. Drug Metab Dispos, 2014. 42(1): p. 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Genetic data from individual subjects are not available due to their identifying nature. However, deidentified summary data from this study are available upon request of the authors.