

Abstract
Nonalcoholic fatty liver disease (NAFLD) has become the most common chronic liver disease in children and adolescents, increasing the risk of its progression toward nonalcoholic steatohepatitis (NASH), cirrhosis, and cancer. There is an urgent need for noninvasive early diagnostic and prognostic tools such as epigenetic marks (epimarks), which would replace liver biopsy in the future. We used plasma samples from 67 children with biopsy‐proven NAFLD, and as controls we used samples from 20 children negative for steatosis by ultrasound. All patients were genotyped for patatin‐like phospholipase domain containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), membrane bound O‐acyltransferase domain containing 7 (MBOAT7), and klotho‐β (KLB) gene variants, and data on anthropometric and biochemical parameters were collected. Furthermore, plasma cell‐free DNA (cfDNA) methylation was quantified using a commercially available kit, and ImageStream(X) was used for the detection of free circulating histone complexes and variants. We found a significant enrichment of the levels of histone macroH2A1.2 in the plasma of children with NAFLD compared to controls, and a strong correlation between cfDNA methylation levels and NASH. Receiver operating characteristic curve analysis demonstrated that combination of cfDNA methylation, PNPLA3 rs738409 variant, coupled with either high‐density lipoprotein cholesterol or alanine aminotransferase levels can strongly predict the progression of pediatric NAFLD to NASH with area under the curve >0.87. Conclusion: Our pilot study combined epimarks and genetic and metabolic markers for a robust risk assessment of NAFLD development and progression in children, offering a promising noninvasive tool for the consistent diagnosis and prognosis of pediatric NAFLD. Further studies are necessary to identify their pathogenic origin and function.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is currently considered the most common chronic liver disease worldwide in both adults and children. NAFLD prevalence has increased significantly in relation to the obesity prevalence.[ 1 , 2 ] Based on its link with obesity and other metabolic derangements, a new name has been proposed for NAFLD, which is metabolic associated fatty liver disease (MAFLD). This new definition clearly establishes this disease as a metabolic disorder, including the evidence of hepatic steatosis accompanied by (i) overweight or obesity, (ii) type 2 diabetes, and (iii) lean/normal weight associated with metabolic abnormalities, such as dyslipidaemia.[ 3 , 4 ]
In many cases, NAFLD/MAFLD further progresses to nonalcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and hepatocellular carcinoma.[ 5 ] NASH comprises different histological traits including steatosis, inflammation, ballooning and eventually fibrosis, and is a major leading cause of advanced liver disease, liver transplantation, cardiovascular morbidity, and mortality; thus, its early diagnosis and treatment are now key challenges in hepatology.[ 6 , 7 ] Currently, liver biopsy remains the gold standard for NASH diagnosis, but it is impractical as a diagnostic tool because it is invasive and expensive.[ 8 ] In this context, noninvasive approaches to liver biopsy may prove crucial for the early diagnosis, effective management, and future treatment of patients with NASH. Imaging‐based approaches and circulating biomarkers have been used in NAFLD/MAFLD or NASH, but recent studies have shown that multivariate indexes based on blood biomarkers represent alternative translatable strategies for the stratification of patients at diagnosis and during follow‐up.[ 9 ]
Liquid biopsy to detect circulating cell‐free DNA (cfDNA) is emerging as an important diagnostic tool in several diseases, such as cancer and type 2 diabetes.[ 10 , 11 ] Different amounts of cfDNA have been described in various pathophysiological states.[ 12 , 13 , 14 ] Moreover, recently it has emerged that the diagnostic utility of cfDNA as a noninvasive biomarker could be enhanced by including the parallel characterization of epigenetic traits, such as global DNA methylation, histone modifications, and histone variants.[ 15 , 16 ] Indeed, cell death may cause genome fragmentation in the form of nucleosomes that are able to preserve histone modifications and are released into the circulation.[ 17 , 18 ]
It has been reported that, in liver diseases, changes occur in DNA methylation, nucleosome histone variants (notably H2A variant macroH2A1), posttranslational histone modifications, and noncoding RNAs, which may be crucial for noninvasive early diagnosis and treatment.[ 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 ] Accumulating evidence suggests the involvement of epigenetic mechanisms, which affect gene expression without altering the DNA sequence, on the pathogenesis of NAFLD, indicating the potential use of epigenetic factors as noninvasive biomarkers of the disease.[ 29 , 30 ]
Because cfDNA generally is fragmented and released from dying cells into blood, it is conceivable that the analysis of its pattern (e.g., amount, fragmentation, methylation) and epigenetic modifications (e.g., histone complexes, variants, modifications) could unveil efficient circulating biomarkers also for NAFLD. In line with this evidence, it has been reported that circulating cfDNA fragments are associated with NAFLD severity, especially in patients with increased liver stiffness and a high risk of disease progression.[ 31 ] Moreover, Hardy et al.[ 32 ] demonstrated that plasma DNA methylation signatures at the level of peroxisome proliferator‐activated receptor gamma (PPARγ) promoter might correlate with fibrosis stage in NASH. More recently, our pilot study identified a circulating histone pattern that may be useful to diagnose the severity of steatosis in adults with lean MAFLD.[ 33 ] These findings highlight that further studies are needed to improve not only the mechanistic understanding of the role of epigenetic marks in NAFLD/MAFLD and its progressive form, but also their potential use as noninvasive tools for diagnosis and follow‐up. Here, we evaluated whether cfDNA methylation and circulating histone signature may correlate with the severity of disease in a cohort of Italian children with NAFLD/MAFLD.
METHODS
Patients
Bio‐banked samples of 67 young patients with biopsy‐proven NAFLD/MAFLD were used for the present study. Bio‐banked samples of 20 children negative to steatosis by ultrasound and without other liver diseases were also used as controls. The samples were stored at Bambino Gesù Children's Hospital from March 2018 to June 2021 and included in the EuPNAFLD protocol (1774_OPBG_2019). Written, informed consent for biobanking and future use was obtained from each child's parent/legal guardian at the time of enrollment. Patients' anthropometrical, biochemical, and histological data were withdrawn from electronic medical records.
At time of the enrollment, the patients' height, weight, and body mass index (BMI) were measured using standard procedures as already reported,[ 34 ] and as BMI z score, based on the Centers for Disease Control and Prevention growth charts (https://www.cdc.gov/growthcharts/zscore.htm). Alanine aminotransferase (ALT), aspartate aminotransferase, gamma‐glutamyltransferase, total cholesterol, high‐density lipoprotein cholesterol (HDL‐C), low‐density lipoprotein cholesterol, and triglycerides were measured by standard laboratory methods. Glucose and insulin were measured at fasting. The homeostasis model assessment index of insulin resistance (HOMA‐IR) [ 35 ] was calculated as a surrogate marker of insulin resistance.
Liver biopsy and histology
In all patients, liver biopsies were performed using an automatic core biopsy 16‐gauge or 18‐gauge needle under general anesthesia and ultrasound guidance. The histological features of steatosis (0–3), lobular inflammation (0–2), and hepatocyte ballooning (0–2) were combined in the NAFLD activity score (NAS), ranging from 0 to 8 using the criteria of the NAFLD Clinical Research Network as previously described by Kleiner et al.[ 36 ] In particular, hepatic steatosis was graded as 0 = steatosis involving fewer than 5% of hepatocytes, 1 = steatosis involving up to 33% of hepatocytes, 2 = steatosis involving 33%–66% of hepatocytes, and 3 = steatosis involving more than 66% of hepatocytes. Lobular inflammation was graded as 0 = none, 1 = mild, and 2 = moderate. Hepatocyte ballooning was graded as 0 = no balloon cells, 1 = few balloon cells, and 2 = many/prominent balloon cells. The sum of the scores for steatosis, lobular inflammation, and ballooning was performed to calculate NAS. Cases with NAS ≥ 4 were evaluated by two expert pathologists to confirm or exclude diagnosis of NASH.
The stage of hepatic fibrosis was quantified with a five‐point scale: 0 = no fibrosis; 1 = perisinusoidal or periportal fibrosis ([1a] mild, zone 3, perisinusoidal; [1b] moderate, zone 3, perisinusoidal; and [1c] portal/periportal); 2 = perisinusoidal and portal/periportal fibrosis; 3 = bridging fibrosis; and 4 = cirrhosis.
Genotyping
The patatin‐like phospholipase domain containing 3 (PNPLA3) rs738409, transmembrane 6 superfamily member 2 (TM6SF2) rs58542926, membrane bound O‐acyltransferase domain containing 7 (MBOAT7) rs641738, and klotho‐β (KLB) rs17618244 variants were genotyped by allelic discrimination using TaqMan 5′‐nuclease assays (Life Technologies). Genomic DNA was isolated from venous blood using a Blood DNA Extraction Kit (Qiagen). Real‐time polymerase chain reaction (PCR) was performed using Applied Biosystems 7900HT Fast Real‐Time PCR System. Positive and negative controls were included on each reaction plate, to verify the reproducibility of the results.
Global DNA methylation assay
DNA was extracted from 500 μl of plasma using the QIAamp Circulating Nucleic Acid Kit (QIAGEN) and a vacuum manifold according to the manufacturer's protocol. Quantitative and qualitative DNA analysis was performed using the Nanodrop spectrophotometer. Global methylation of cfDNA was performed by using 50 ng of extracted DNA that was analyzed by adapting the manufacturer's instructions for the Methylamp Global DNA Methylation Quantification Ultra kit (Epigentek). Briefly, DNA was immobilized to the well coated with DNA affinity substance; then the methylated fraction of DNA was recognized by 5‐methylcytosine antibody, thus measuring total DNA methylation level as a percentage of total DNA present in the sample through an enzyme‐linked immunosorbent assay–like reaction and quantification. The amount of methylated DNA is proportional to the optical density intensity. Samples, standard curve, and both the positive and negative controls were run in duplicate.
Detection of histone complexes in the plasma of children with NAFLD
For the detection of histones and histone complexes in the plasma, we used multispectral imaging flow cytometer ImageStream(X) (AMNIS, part of Luminex Corp.), which is designed for the acquisition of multiparametric cellular imagery in up to 6 spectral channels.
Imaging flow cytometry combines the speed and statistical power of flow cytometry with high‐throughput imaging features of fluorescence microscopy.
As previously described,[ 33 ] we used three staining sets, each consisting of four different primary antibodies and four appropriate secondary antibodies. Six channels of detection are available in ImageStream(X), but for fluorescence detection, only four are available, as one channel is dedicated for bright‐field images acquisition and for dark‐field scattering.
In the first staining set, the primary antibodies were anti‐macroH2A1.1 (39871; ActiveMotif), anti‐histone H2B (Ab134211; Abcam,), anti‐histone H4 (Ab31830; Abcam), and anti‐histone H3 (Ab12079; Abcam). Secondary antibodies were anti‐rabbit immunoglobulin G (IgG) H&L‐Alexa Fluor 488 (Ab150077; Abcam), anti‐chicken immunoglobulin Y (IgY) H&L‐DyLight 594 (Ab96953; Abcam), anti‐mouse IgG H&L‐Alexa Fluor 647 (Ab150115; Abcam), and anti‐goat IgG H&L Alexa Fluor 555 (Ab150130; Abcam).
In the second staining set, the primary antibodies were anti‐macroH2A1.2 (61427; ActiveMotif), anti‐histone H2B (Ab134211; Abcam), anti‐histone H4 (Ab31830; Abcam), and anti‐histone H3 (Ab12079; Abcam). Secondary antibodies were anti‐rabbit IgG H&L‐Alexa Fluor 488 (Ab150077; Abcam), anti‐chicken IgY H&L‐DyLight 594 (Ab96953; Abcam), anti‐mouse IgG H&L‐Alexa Fluor 647 (Ab150115; Abcam), and anti‐IgG H&L Alexa Fluor 555 (Ab150130; Abcam).
For the third staining set, the primary antibodies were anti‐macroH2A (Ab18255; Abcam), anti‐histone H2B (Ab134211; Abcam), anti‐histone H4 (Ab31830; Abcam), and anti‐histone H3 (Ab12079; Abcam). Secondary antibodies were anti‐rabbit IgG H&L‐Alexa Fluor 488 (Ab150077; Abcam), anti‐chicken IgY H&L‐DyLight 594 (Ab96953; Abcam), anti‐mouse IgG H&L‐Alexa Fluor 647 (Ab150115; Abcam), and anti‐goat IgG H&L Alexa Fluor 555 (Ab150130; Abcam).
The procedure for using each of the sets was as follows (same for each type). Plasma sample (50 μl) from each pediatric patient was incubated with four primary antibodies overnight at 4°C (1 μl of each primary antibody was added from stock solution 1 mg/ml, sequentially). Following the primary incubation, the samples were incubated with four compatible fluorescent secondary antibodies for 2 h at 24°C (1 μl of each secondary antibody was added from stock solution 0.05 mg/ml, sequentially).
The samples prepared in this way were analyzed by imaging flow cytometer ImageStream(X) with the following parameters. For each stained patient plasma sample, 10,000 objects were collected using the following acquisition setup: excitation laser 488 nm (5 mW) for Alexa Fluor 488, fluorescence imaging in channel 2 (480–560 nm); excitation laser 561 nm (20 mW) for Alexa Fluor 555 and DyLight 594, fluorescence imaging in channel 3 (560–595 nm) and channel 4 (595–642 nm), respectively; and excitation laser 642 nm (5 mW) for Alexa Fluor 647, fluorescence imaging in channel 5 (642–745 nm) (bright‐field imaging in channel 1 and the dark‐field scatter imaging in channel 6).
The histone level was quantified as relative abundances of detected histone species among all gated objects in each patient sample. Particularly, macroH2A1.1, macroH2A1.2, H2A, H2B, H3, H4, dimers, and tetramers were determined by gating of plasma objects with gates applied to discriminate (a) focused objects (using the gradient root mean square method for focused objects filtering) and (b) objects with fluorescence from secondary antibodies. Single‐color controls and fluorescence minus one controls were used to find the threshold for a secondary antibody fluorescence. The same controls were used to calculate a spectral crosstalk matrix that was applied to the image files in order to isolate probed images to single imaging channels. The resulting compensated image files were analyzed using image‐based algorithms available in the IDEAS statistical analysis software package (AMNIS, part of Luminex Corp.), and analysis of the results was done with the same software.
Statistics
Significant differences of histone expression and methylation levels between groups of patients were found using the Mann–Whitney test. Kendall's Tau test was used to measure the correlation between NASH or histological variables and both histones expression and methylation levels. Fisher's exact test was used to evaluate the difference between frequencies of genotypes. Each statistical result was considered to be significant if its associated p value was lower than 0.05. Histones profiled in more than one staining set were averaged to produce a single‐point expression per patient. Patients with missing histone data were excluded from tests in which they could influence the outcome.
To evaluate the performance of our noninvasive parameters in differentiating NASH from no NASH, we implemented a K‐nearest‐neighbors (KNN) binary classifier. The parameter K was set to be equal to the number of patients used as the training set. The weights of the training patients in the predictions were assigned as the inverse of their Euclidean distance from the query point in the features space. The model was tested by leave‐one‐out cross‐validation, and an area under the curve (AUC) score was computed on the leave‐one‐out predicted probabilities. Various combinations of features were explored, still removing all samples that exhibited at least one missing value in the feature set. For each training cycle, the features were standardized on the mean and SD of the training set. We binary‐encoded the sex feature as 1 or 0, respectively, for males and females; we also encoded the single‐nucleotide polymorphism features as allele count (0, 1 or 2, respectively, for wild‐type, heterozygous, and homozygous patients). For each receiver operating characteristic (ROC) curve, we tested the alternative hypothesis that AUC > 0.5 by bootstrapping the leave‐one‐out predictions 1000 times, each time computing the AUC on the bootstrapped data. The resulting p value was the number of bootstrapped AUC values lower than 0.5 over the total number of bootstrapped AUC values (1000).
RESULTS
Global methylation of cfDNA and histone signatures are altered in plasma of pediatric patients with NAFLD/MAFLD
We evaluated the plasma cfDNA methylation and circulating histone signatures in 20 control children without liver diseases, and in 67 children with biopsy‐proven NAFLD/MAFLD. Anthropometrics, biochemical parameters, and genetics of controls and children with NAFLD are reported in Tables S1 and S2, respectively.
Our data showed that median cfDNA levels in patients with NAFLD/MAFLD were not significantly higher than in controls (Figure S1). Moreover, our data revealed a significant increase of methylated cfDNA levels in children affected by NAFLD/MAFLD compared to controls (Mann–Whitney test, p = 5.58e‐6) (Figure 1A).
FIGURE 1.
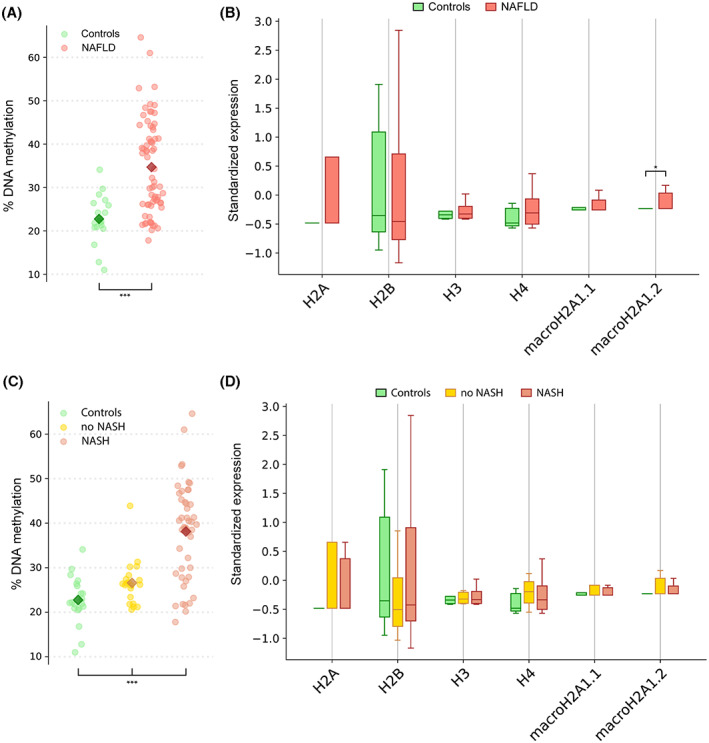
Global DNA methylation and histone signatures in plasma of pediatric patients with nonalcoholic fatty liver disease (NAFLD). (A) Percentage of cell‐free DNA (cfDNA) methylation in NAFLD (red) and control (green) patients. ***p < 0.001 vs. controls. (B) Box plots of the standardized individual histones expression in NAFLD (red) and controls (green); outliers were removed; the horizontal bars represent the medians and the whiskers. *p < 0.05 vs. controls. (C) Percentage of cfDNA methylation in patients with nonalcoholic steatohepatitis (NASH) NAFLD (red) and no NASH (yellow). ***p < 0.001 vs. controls. (D) Box plots of the standardized histones expression in patients with NASH (red) and no NASH (yellow); outliers were removed; the horizontal bars represent the medians and the whiskers.
According to the dynamics and intermediates on the process of histone assembly into nucleosomes, we assayed six individual histones (H2A, H2B, H3, H4, macroH2A1.1, and macroH2A1.2) together with the following biological dimers: H2A/H2B, macroH2A1.1/H2B, and macroH2A1.2/H2B by ImageStream(X), as previously reported.[ 33 ] We found a significant enrichment of the levels of histone variant macroH2A1.2 in the plasma of children with NAFLD/MAFLD versus controls (Mann–Whitney test, p = 0.02) (Figure 1B). None of the other individual histones or histone complexes significantly differed (Figure S2).
Next, we stratified the patients with NAFLD, differentiating children without NASH (no NASH, n = 20) from children with NASH (NASH, n = 47). As indicated in Table 1, in the comparison between the no‐NASH group and NASH group, only the BMI z score and levels of ALT significantly increased (p = 0.034 and p = 0.027, respectively). No statistical differences were found in median cfDNA levels in patients with NASH compared to no NASH (Figure S3). In addition, the plasma levels of methylated cfDNA were significantly higher in children with NASH than in children with no NASH (Mann–Whitney test, p = 7.49e‐5) (Figure 1C).
TABLE 1.
Comparison of characteristics in children without (no NASH) versus children with NASH
| Parameters | No NASH (n = 20) | NASH (n = 47) |
|---|---|---|
| Sex (M/F) | 15/5 | 24/23 |
| Age (years) | 12.5 (7.0–19.0) | 12.0 (6.0–17.0) |
| Weight (kg) | 63.3 (29.0–120.0) | 60.5 (20.1–194.0) |
| Height (cm) | 159.5 (127.6–173.7) | 151.0 (118.0–187.5) |
| BMI (kg/m2) | 24.3 (16.8–49.3) | 25.9 (14.4–57.9) |
| BMI z score a | 0.26 (0.1–0.7) | 1.7 (1.2–2.3) |
| WC (cm) | 87.0 (54.0–108.0) | 88.0 (65.0–145.0) |
| Cholesterol (mg/dl) | 163.5 (124.0–203.0) | 149.0 (108.0–214.0) |
| HDL‐C (mg/dl) | 49.0 (38.0–73.0) | 43.0 (29.0–71.0) |
| LDL‐C (mg/dl) | 101.0 (48.0–132.0) | 93.0 (36.0–163.0) |
| Triglycerides (mg/dl) | 83.5 (40.0–211.0) | 85.0 (44.0–268.0) |
| Fasting insulin (μUI/ml) | 15.2 (4.6–80.0) | 16.0 (2.6–87.0) |
| Fasting glucose (mg/dl) | 83.5 (77.0–94.0) | 81.0 (68.0–147.0) |
| HOMA‐IR | 3.0 (0.9–15.2) | 3.5 (0.5–26.1) |
| AST (IU/L) b | 27.5 (13.0–153.0) | 31.0 (13.0–152.0) |
| ALT (IU/L) | 23.5 (10.0–79.0) | 38.0 (10.0–95.0) |
| GGT (IU/L) | 15.0 (6.0–62.0) | 16.0 (0.5–53.0) |
| MAF (G) PNPLA3 rs738409 | 0.17 | 0.46 |
| PNPLA3‐CC | 8 (40%) | 15 (31.9%) |
| PNPLA3‐CG | 8 (40%) | 21 (44.7%) |
| PNPLA3‐GG | 4 (20%) | 11 (23.4%) |
| MAF (T) TM6SF2 rs5854292 | 0.15 | 0.08 |
| TM6SF2‐CC | 18 (90%) | 41 (87.2%) |
| TM6SF2‐CT | 1 (5%) | 4 (8.5%) |
| TM6SF2‐TT | 1 (5%) | 2 (4.3%) |
| MAF (T) MBOAT7 rs64173826 | 0.52 | 0.46 |
| MBOAT7‐CC | 10 (50%) | 15 (31.9%) |
| MBOAT7‐CT | 10 (50%) | 21 (44.7%) |
| MBOAT7‐TT | — | 11 (23.4%) |
| MAF (A) KLB rs17618244 | 0.25 | 0.19 |
| KLB‐AA | 2 (10%) | 2 (4.3%) |
| KLB‐GA | 5 (25%) | 14 (59.8%) c |
| KLB‐GG | 13 (65%) | 31 (66%) |
Note: Data are expressed as number, median, and range (min–max); Mann–Whitney test was applied for comparison between no‐NASH and NASH anthropometric and biochemical variables.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; F, female; GGT, gamma‐glutamyltransferase; HDL‐C, high‐density lipoprotein cholesterol; HOMA‐IR, homeostasis model assessment index of insulin resistance; KLB, klotho‐β; LDL, low‐density lipoprotein; M, male; MAF, minor allele frequency; MBOAT7, membrane bound O‐acyltransferase domain containing 7; TM6SF2, transmembrane 6 superfamily member 2; WC, waist circumference.
p = 0.034.
p = 0.027. Genotypes are reported as number and percentage, and Fisher's exact test was applied for comparison.
p < 0.05.
On the other hand, individual histones or histone complexes did not correlate with disease progression, as their levels did not differ between patients with no NASH and with NASH (Figure 1D and Figure S4).
Plasma cfDNA methylation and histone signatures are correlated with histological traits in children with NAFLD/MAFLD
Because cfDNA methylation increased with NAFLD progression, we sought to determine its correlation with the histological traits. Table S3 lists the histological traits in our study cohort. Among the patients, 11.9% had severe steatosis (grade 3); 76.1% exhibited lobular inflammation; 86.6% presented ballooning; and 16.4% had severe fibrosis (F3). As shown in Figure 2A–C, correlation analysis indicated a moderate (Kendall's coefficient of concordance 0.23–0.27) but significant positive correlation between cfDNA methylation levels and steatosis (p = 0.01), lobular inflammation (p = 0.02), and liver fibrosis (p = 0.003). Conversely, as reported in Figure 2D, no correlation was found between cfDNA methylation levels and hepatocyte ballooning (Kendall's coefficient of concordance 0.11; p = 0.26). Of note, the strongest positive correlation was found between the NAS and cfDNA methylation levels (Kendall's coefficient of concordance 0.3; p = 0.001) (Figure 2E).
FIGURE 2.
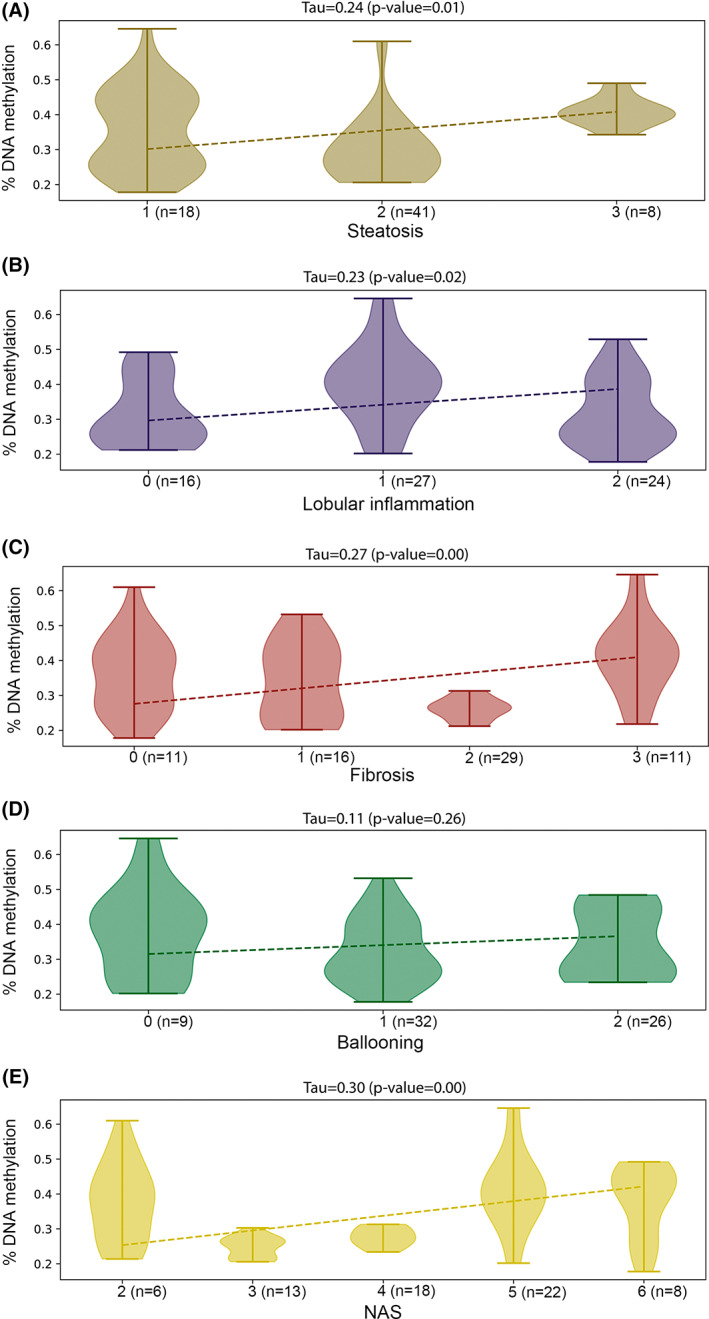
Correlation between circulating cfDNA methylation and histological traits in patients with NAFLD. Correlation between percentages of cfDNA methylation in patients with NAFLD (n = 67) stratified by steatosis (A), lobular inflammation (B), fibrosis (C), ballooning (D), and NAFLD activity score (NAS) (E).
Moreover, we assayed the correlation between circulating histones (individual: H2A, H2B, H3, H4, macroH2A1.1, and macroH2A1.2; dimers: H2A + H2B, H3 + H4, macroH2A1.1 + H2B, and macroH2A1.2 + H2B; nucleosomes: H2A + H2B + H3 + H4, macroH2A1.1 + H2B + H3 + H4, and macroH2A1.2 + H2B + H3 + H4), and the histological traits in children with NAFLD/MAFLD (Figure S5). The levels of the analyzed histones were not correlated with histological features, except the histone variant macroH2A1.2. Indeed, this histone variant was found significantly and inversely correlated (p < 0.05) with steatosis and NAS (Figure 3A,B), thus reflecting the enrichment of its levels in the plasma of children with NAFLD/MAFLD.
FIGURE 3.
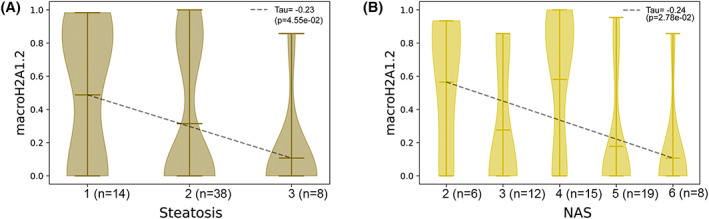
Correlation between circulating histones and histological traits in patients with NAFLD. Correlation between macroH2A1.2 histone expression and steatosis (A) or NAS (B) in patients with NAFLD (n = 60); macroH2A1.2 histone expressions were quantile‐normalized to produce these plots.
Genetic and epigenetic signatures predicting NASH phenotype in children with NAFLD/MAFLD
In children affected by NAFLD/MAFLD, genetic background has a strong impact on severity of disease.[ 1 ] Indeed, the gene variants PNPLA3 rs738409 and TM6SF2 rs58542926 are associated with NASH progression[ 37 , 38 ] and KLB rs17618244 is associated with an increased risk of ballooning and lobular inflammation,[ 39 , 40 ] while MBOAT7 rs641738 does not show any association with the disease.[ 41 ] Here, we analyzed the correlation of these variants with NASH in our study cohort. As reported in Figure S6, only PNPLA3 rs738409 significantly correlated with NASH. Therefore, we sought to test the ability of cfDNA methylation, histone macroH2A1.2, and PNPLA3 rs738409 to discriminate among pediatric patients with NAFLD and those with NASH, using ROC curve analysis. Notably, ROC curve analysis demonstrated that cfDNA methylation was the more efficient variable to discriminate children with NASH with an AUC of 0.83, with respect to PNPLA3 and macroH2A1.2 (considered separately) (Figure 4 and Figure S7). The evaluation of a model considering altogether cfDNA methylation, histone macroH2A1.2, and PNPLA3 rs738409 showed that this combination could discriminate children with NASH with an AUC of 0.81, but the removal of macroH2A1.2 from this model improved the AUC up to 0.86 (Figure 4).
FIGURE 4.
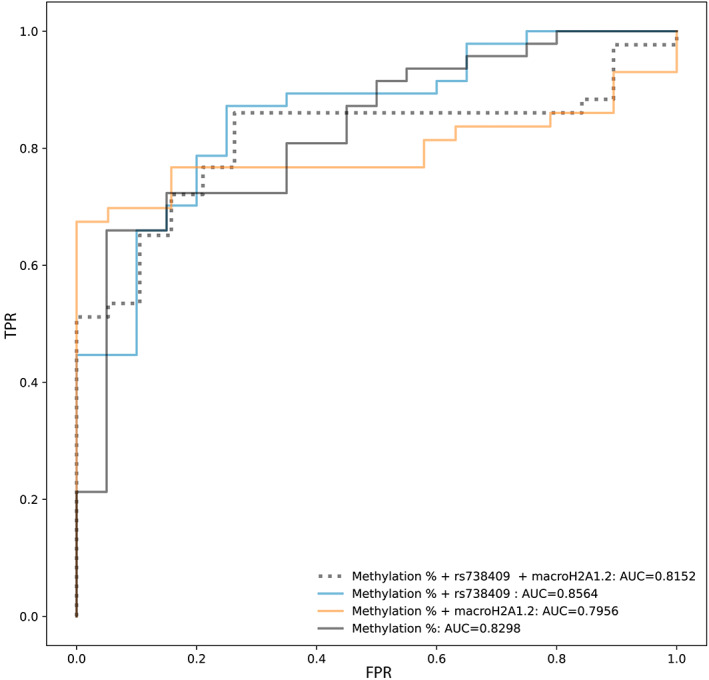
Performance of cfDNA methylation, patatin‐like phospholipase domain containing 3 (PNPLA3) rs738409, and macroH2A1.2 in predicting children with NASH. K‐nearest‐neighbors (KNN) receiver operating characteristic (ROC) curves achieved combining cfDNA methylation, PNPLA3 rs738409, and macroH2A1.2 features. True positive rate (TPR) and false positive rate (FPR) are indicated in the graph. Abbreviation: AUC, area under the curve.
Combining cfDNA methylation, PNPLA3 variant, and clinical features to identify the best‐fit model for predicting NASH
Next, we assessed which one among the anthropometrical and biochemical features could be used as best predictor to discriminate NASH in our cohort of children with NAFLD/MAFLD. As shown in Figure 5A, the four parameters most significantly correlated with NASH were ALT, HDL‐C and total cholesterol, in addition to the epigenetic parameter of cfDNA methylation. Therefore, we included all possible combinations of ALT, HDL‐C, and total cholesterol to the cfDNA methylation + PNPLA3 rs738409 model. As shown in Figure 5B, when we added HDL‐C or ALT, the AUC of the ROC was the highest (>0.87), demonstrating that the combination of cfDNA methylation and PNPLA3 rs738409, with the addition of either HDL‐C or ALT levels, strongly predicted the progression of pediatric NAFLD to NASH.
FIGURE 5.
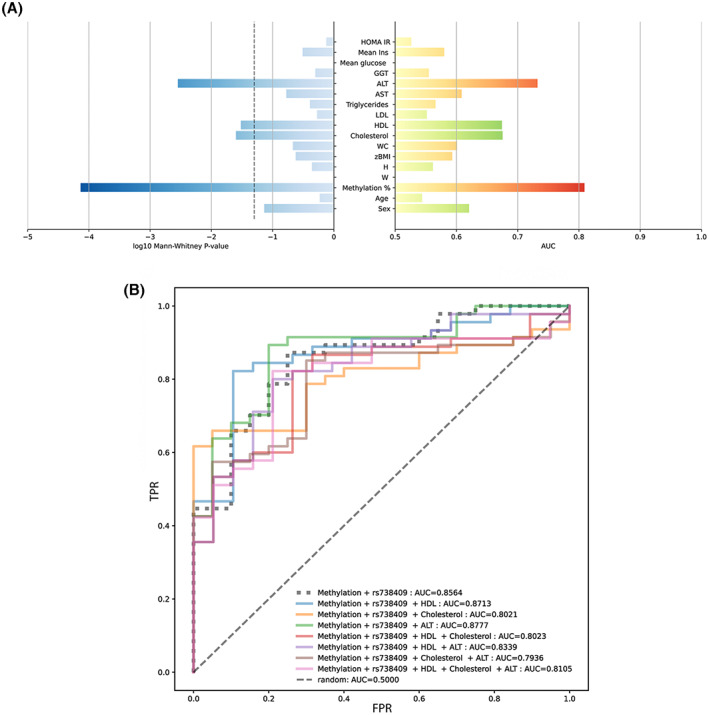
Identification of the best model for predicting NASH. (A) Right bars represent the AUC achieved using the single feature values to predict the NASH status; red and green bars indicate, respectively, the positive and negative associations between the feature and the NASH status; left bars represent the log p value from the Mann–Whitney test for the same features performed on the NASH and no‐NASH NAFLD groups. (B) KNN ROC curves achieved by combining additional features to the cfDNA methylation and PNPLA3 rs738409 features. TPR and FPR are indicated in the graph.
DISCUSSION
In this study, we performed the analyses of circulating levels of histones and cfDNA methylation and assessed whether these two parameters add value to the prediction of NAFLD/MAFLD severity on top of other well‐known risk factors, such as well‐known metabolic and genetic polymorphisms. We obtained different multivariate prediction models that combined these parameters with other disease‐related variables.
cfDNA methylation and circulating histones have now been shown to be useful biomarkers for diagnosis, prognosis, and/or response to therapy in different type of cancers, and recently also in some chronic diseases, such as obesity and type 2 diabetes.[ 11 , 42 , 43 ] These findings are of particular relevance for diseases in which biomarkers may help in understanding their natural history, such as for NAFLD/MAFLD.
The “natural history” of pediatric NAFLD/MAFLD is a complex topic, in that there is a paucity of longitudinal data in children with NAFLD/MAFLD. A better understanding of the severity range, variability, and associations of pediatric NAFLD/MAFLD is important, as children may represent different time points on a history continuum that may involve their whole life. It is of the utmost importance to stratify pediatric patients with NAFLD and discriminate NASH, primarily to offer them the best management and to reduce the risk of fibrosis exacerbation. Unfortunately, because of the paucity of data on noninvasive imaging and biomarkers, liver biopsy remains the gold standard for diagnosis of NASH in children.[ 44 , 45 ]
Practice guidelines for the initial screening of pediatric NAFLD/MAFLD include ALT and ultrasound to assess changes in steatosis over time, but these methods appear to be unable to provide an indirect measure of other liver histological features.[ 45 ] Indeed, the ALT levels were not found to be elevated in obese patients with NAFLD diagnosed by imaging procedures, suggesting that ALT elevation could occur at more advanced stages of NAFLD.[ 46 ] Draijer et al. recently compared ALT and ultrasound accuracy in a cohort of 99 children with NAFLD, demonstrating that alone or in combination these two parameters are even incapable of detecting steatosis in children with obesity.[ 47 ] Therefore, novel screening tools are needed to replace liver biopsy and to identify which children with NAFLD/MAFLD have, or will progress into, NASH and fibrosis.
Genetic and epigenetic factors play a key role in the development of pediatric NAFLD/MAFLD and its progression to NASH, and they have a strong potential as noninvasive disease biomarkers.[ 45 ] In this respect, liquid biopsies, such as cfDNA and circulating histones/nucleosomes (“epimarks”), have been proposed as promising biomarkers for adult NAFLD.[ 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 31 , 32 , 33 ] Although previous evidence in adults with NAFLD demonstrated that circulating levels of total cfDNA are different in comparing patients with different stages of disease,[ 48 ] we found that total cfDNA concentration in children was unchanged among the different groups of comparison (controls, NASH, and no NASH). Even if this finding suggests that the levels of absolute cfDNA could not be a good parameter for monitoring disease progression, further studies on a larger number of patients are required to support this assumption.
The major finding of this study is that cfDNA methylation is not only significantly associated with NASH features, but also with fibrosis. These results are in agreement with previous studies demonstrating that the evaluation of liver tissue and plasma DNA methylation at particular CpG loci in the human PPARγ gene promoter can be used to stratify fibrosis severity in NAFLD.[ 32 , 49 ] A previous study found that DNA methylation in the serum of patients with NAFLD was significantly higher in the subjects with cirrhosis than those without.[ 48 ] We found a similar trend in children with NAFLD who exhibited a more severe pattern of diseases, but the significance and the origin of this methylated DNA remains to be explored.
This study explores circulating epimarks as biomarkers of disease progression in a cohort of children with biopsy‐proven NAFLD. We found that circulating levels of histone variant macroH2A1.2 strongly correlates with the early stages of NAFLD/MAFLD, but not with the progression toward NASH. This correlation was strongest with early stages of steatosis and lower levels of NAS, and therefore decreased with disease severity, consistent with our previous pilot findings observed in adults affected by NAFLD/MAFLD—independent of body weight.[ 33 ] We have previously shown that hepatocytes accumulate high levels of macroH2A1.2 during NAFLD/MAFLD pathogenesis.[ 23 , 24 ] Although the tissue(s) of origin of circulating macroH2A1.2 remain unknown, we speculate that the differences in the circulating levels of macroH2A1.2 may reflect the amount of those histones remained “trapped” within hepatocytes or other cell types during disease progression.
An additional factor with a clinically relevant impact on pediatric NAFLD onset and progression is the genetic background.[ 1 ] Several genetic variants have been found associated with NAFLD, but only few of them, including PNPLA3 rs738409, TM6SF2 rs58542926, MBOAT7 rs641738 and KLB rs17618244, have been correlated with the risk of NASH in children.[ 1 , 39 , 40 , 41 ] In the present cohort, we found that PNPLA3 rs738409 is the gene variant mostly associated with a NASH phenotype. Although this variant may explain a large part of the total heritability of NAFLD, it is possible that its combination with epigenetic factors could provide a score that improves NASH diagnosis. However, to date, there is no evidence about the utility and the accuracy of the combination of epimarks, gene variants, and metabolic biomarkers in models that may help to discriminate children with progressive disease.[ 45 ] Hence, whether the combination of these biomarkers could improve the accuracy in distinguishing NASH and what form of combination could achieve better diagnostic accuracy are key questions.
Here, we have shown that the combination of genetic, epigenetic, and metabolic variables, using a KNN algorithm that is very fast and does not require a rigid decision of the hyperparameters, may help in predicting the NASH onset. The best combination of variables included methylation, the PNPLA3 rs738409 variant, and the ALT concentration, but also considering HDL‐C to the place of ALT led to good performance. Our findings are in agreement with previous studies reporting that diagnostic efficiency of the combined models with composite biomarkers might be higher than that observed with single biomarkers.[ 50 ] Stratification of children with epimarks could be a promising tool for the consistent diagnosis and prognosis in pediatric NAFLD/MAFLD, but may also improve patient management and refine the prediction of their response to a given drug or combination of drugs.[ 45 ]
Our study has two main limitations, including the fact that liver biopsies have been performed in NAFLD but not in control subjects (unavoidable because the liver biopsy in these subjects is unethical); and that the sample includes only Caucasian subjects, thus suggesting the need for additional studies on a larger cohort of children/adolescents with different ethnicity.
CONCLUSIONS
We propose a combinatorial panel of biomarkers to assess the risk of NAFLD and its progression in pediatric patients, suggesting that the early stage of NAFLD might be monitored by macroH2A1.2 histone variant, while the presence of NASH could be detected by a composite algorithm including cfDNA methylation and PNPLA3, together with HDL‐C or ALT levels.
FUNDING INFORMATION
Supported by the Ministry of Education and Science of Bulgaria under the National Scientific Programme “Excellent Research and People for the Development of European Science” 2021 of the Bulgarian National Science Fund (#KP‐06‐DV/4); the Bulgarian National Science Fund (#KP‐06‐N53/6); the European Regional Development Fund—Project MAGNET (CZ.02.1.01/0.0/0.0/15_003/0000492); the Italian Ministry of Health (5‰ 2022); and a fellowship granted by the Associazione Italiana per lo Studio del Fegato.
CONFLICT OF INTEREST
Nothing to report.
Supporting information
Appendix S1 Supporting information
Buzova D, Braghini MR, Bianco SD, Lo Re O, Raffaele M, Frohlich J, et al. Profiling of cell‐free DNA methylation and histone signatures in pediatric NAFLD: A pilot study. Hepatol Commun. 2022;6:3311–3323. 10.1002/hep4.2082
Diana Buzova, Maria Rita Braghini, and Salvatore Daniele Bianco contributed equally to this work.
Contributor Information
Tommaso Mazza, Email: t.mazza@css-mendel.it.
Anna Alisi, Email: anna.alisi@opbg.net.
Manlio Vinciguerra, Email: manlio.vinciguerra@fnusa.cz.
REFERENCES
- 1. Nobili V, Alisi A, Valenti L, Miele L, Feldstein AE, Alkhouri N. NAFLD in children: new genes, new diagnostic modalities and new drugs. Nat Rev Gastroenterol Hepatol. 2019;16:517–30. [DOI] [PubMed] [Google Scholar]
- 2. Lazarus JV, Mark HE, Anstee QM, Arab JP, Batterham RL, Castera L, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol. 2022;19:60–78. [DOI] [PubMed] [Google Scholar]
- 3. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero‐Gomez M, et al. A new definition for metabolic dysfunction‐associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73:202–9. [DOI] [PubMed] [Google Scholar]
- 4. Eslam M, Alkhouri N, Vajro P, Baumann U, Weiss R, Socha P, et al. Defining paediatric metabolic (dysfunction)‐associated fatty liver disease: an international expert consensus statement. Lancet Gastroenterol Hepatol. 2021;6:864–73. [DOI] [PubMed] [Google Scholar]
- 5. Kleiner DE, Makhlouf HR. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin Liver Dis. 2016;20:293–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mantovani A, Scorletti E, Mosca A, Alisi A, Byrne CD, Targher G. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism. 2020;111S:154170. [DOI] [PubMed] [Google Scholar]
- 7. Majumdar A, Tsochatzis EA. Changing trends of liver transplantation and mortality from non‐alcoholic fatty liver disease. Metabolism. 2020;111S:154291. [DOI] [PubMed] [Google Scholar]
- 8. Bedossa P. Diagnosis of non‐alcoholic fatty liver disease/non‐alcoholic steatohepatitis: why liver biopsy is essential. Liver Int. 2018;38(Suppl 1)):64–6. [DOI] [PubMed] [Google Scholar]
- 9. Harrison SA, Ratziu V, Boursier J, Francque S, Bedossa P, Majd Z, et al. A blood‐based biomarker panel (NIS4) for non‐invasive diagnosis of non‐alcoholic steatohepatitis and liver fibrosis: a prospective derivation and global validation study. Lancet Gastroenterol Hepatol. 2020;5:970–85. [DOI] [PubMed] [Google Scholar]
- 10. Luo H, Wei W, Ye Z, Zheng J, Xu RH. Liquid biopsy of methylation biomarkers in cell‐free DNA. Trends Mol Med. 2021;27:482–500. [DOI] [PubMed] [Google Scholar]
- 11. Arosemena M, Meah FA, Mather KJ, Tersey SA, Mirmira RG. Cell‐free DNA fragments as biomarkers of islet beta‐cell death in obesity and type 2 diabetes. Int J Mol Sci. 2021;22:2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tran NH, Kisiel J, Roberts LR. Using cell‐free DNA for HCC surveillance and prognosis. JHEP Rep. 2021;3:100304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jackson AM, Amato‐Menker C, Bettinotti M. Cell‐free DNA diagnostics in transplantation utilizing next generation sequencing. Hum Immunol. 2021;82:850–8. [DOI] [PubMed] [Google Scholar]
- 14. Yuwono NL, Warton K, Ford CE. The influence of biological and lifestyle factors on circulating cell‐free DNA in blood plasma. Elife. 2021;10:e69679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell‐type methylation atlas reveals origins of circulating cell‐free DNA in health and disease. Nat Commun. 2018;9:5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ungerer V, Bronkhorst AJ, Van den Ackerveken P, Herzog M, Holdenrieder S. Serial profiling of cell‐free DNA and nucleosome histone modifications in cell cultures. Sci Rep. 2021;11:9460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Miranda FS, Barauna VG, Dos Santos L, Costa G, Vassallo PF, Campos LCG. Properties and application of cell‐free DNA as a clinical biomarker. Int J Mol Sci. 2021;22:9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sadeh R, Sharkia I, Fialkoff G, Rahat A, Gutin J, Chappleboim A, et al. ChIP‐seq of plasma cell‐free nucleosomes identifies gene expression programs of the cells of origin. Nat Biotechnol. 2021;39:586–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arechederra M, Recalde M, Garate‐Rascon M, Fernandez‐Barrena MG, Avila MA, Berasain C. Epigenetic biomarkers for the diagnosis and treatment of liver disease. Cancers (Basel). 2021;13:1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Borghesan M, Fusilli C, Rappa F, Panebianco C, Rizzo G, Oben JA, et al. DNA hypomethylation and histone variant macroH2A1 synergistically attenuate chemotherapy‐induced senescence to promote hepatocellular carcinoma progression. Cancer Res. 2016;76:594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lo Re O, Douet J, Buschbeck M, Fusilli C, Pazienza V, Panebianco C, et al. Histone variant macroH2A1 rewires carbohydrate and lipid metabolism of hepatocellular carcinoma cells towards cancer stem cells. Epigenetics. 2018;13:829–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lo Re O, Mazza T, Giallongo S, Sanna P, Rappa F, Vinh Luong T, et al. Loss of histone macroH2A1 in hepatocellular carcinoma cells promotes paracrine‐mediated chemoresistance and CD4(+)CD25(+)FoxP3(+) regulatory T cells activation. Theranostics. 2020;10:910–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pazienza V, Borghesan M, Mazza T, Sheedfar F, Panebianco C, Williams R, et al. SIRT1‐metabolite binding histone macroH2A1.1 protects hepatocytes against lipid accumulation. Aging (Albany NY). 2014;6:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Podrini C, Koffas A, Chokshi S, Vinciguerra M, Lelliott CJ, White JK, et al. MacroH2A1 isoforms are associated with epigenetic markers for activation of lipogenic genes in fat‐induced steatosis. FASEB J. 2015;29:1676–87. [DOI] [PubMed] [Google Scholar]
- 25. Rappa F, Greco A, Podrini C, Cappello F, Foti M, Bourgoin L, et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis‐associated hepatocellular carcinoma. PLoS One. 2013;8:e54458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lo Re O, Fusilli C, Rappa F, Van Haele M, Douet J, Pindjakova J, et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology. 2018;67:636–50. [DOI] [PubMed] [Google Scholar]
- 27. Podrini C, Borghesan M, Greco A, Pazienza V, Mazzoccoli G, Vinciguerra M. Redox homeostasis and epigenetics in non‐alcoholic fatty liver disease (NAFLD). Curr Pharm Des. 2013;19:2737–46. [DOI] [PubMed] [Google Scholar]
- 28. Benegiamo G, Vinciguerra M, Mazzoccoli G, Piepoli A, Andriulli A, Pazienza V. DNA methyltransferases 1 and 3b expression in Huh‐7 cells expressing HCV core protein of different genotypes. Dig Dis Sci. 2012;57:1598–603. [DOI] [PubMed] [Google Scholar]
- 29. Sinton MC, Hay DC, Drake AJ. Metabolic control of gene transcription in non‐alcoholic fatty liver disease: the role of the epigenome. Clin Epigenetics. 2019;11:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sodum N, Kumar G, Bojja SL, Kumar N, Rao CM. Epigenetics in NAFLD/NASH: targets and therapy. Pharmacol Res. 2021;167:105484. [DOI] [PubMed] [Google Scholar]
- 31. Karlas T, Weise L, Kuhn S, Krenzien F, Mehdorn M, Petroff D, et al. Correlation of cell‐free DNA plasma concentration with severity of non‐alcoholic fatty liver disease. J Transl Med. 2017;15:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hardy T, Zeybel M, Day CP, Dipper C, Masson S, McPherson S, et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non‐alcoholic fatty liver disease. Gut. 2017;66:1321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buzova D, Maugeri A, Liguori A, Napodano C, Lo Re O, Oben J, et al. Circulating histone signature of human lean metabolic‐associated fatty liver disease (MAFLD). Clin Epigenetics. 2020;12:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Manco M, Panera N, Crudele A, Braghini MR, Bianchi M, Comparcola D, et al. Angiopoietin‐2 levels correlates with disease activity in children with nonalcoholic fatty liver disease. Pediatr Res. 2021;91:1781–6. [DOI] [PubMed] [Google Scholar]
- 35. Stepanek L, Horakova D, Stepanek L, Cibickova L, Karasek D, Vaverkova H, et al. Associations between homeostasis model assessment (HOMA) and routinely examined parameters in individuals with metabolic syndrome. Physiol Res. 2019;68:921–30. [DOI] [PubMed] [Google Scholar]
- 36. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. [DOI] [PubMed] [Google Scholar]
- 37. Valenti L, Alisi A, Galmozzi E, Bartuli A, Del Menico B, Alterio A, et al. I148M patatin‐like phospholipase domain‐containing 3 gene variant and severity of pediatric nonalcoholic fatty liver disease. Hepatology. 2010;52:1274–80. [DOI] [PubMed] [Google Scholar]
- 38. Li J, Hua W, Ji C, Rui J, Zhao Y, Xie C, et al. Effect of the patatin‐like phospholipase domain containing 3 gene (PNPLA3) I148M polymorphism on the risk and severity of nonalcoholic fatty liver disease and metabolic syndromes: a meta‐analysis of paediatric and adolescent individuals. Pediatr Obes. 2020;15:e12615. [DOI] [PubMed] [Google Scholar]
- 39. Dongiovanni P, Crudele A, Panera N, Romito I, Meroni M, De Stefanis C, et al. Beta‐klotho gene variation is associated with liver damage in children with NAFLD. J Hepatol. 2020;72:411–9. [DOI] [PubMed] [Google Scholar]
- 40. Panera N, Meroni M, Longo M, Crudele A, Valenti L, Bellacchio E, et al. The KLB rs17618244 gene variant is associated with fibrosing MAFLD by promoting hepatic stellate cell activation. EBioMedicine. 2021;65:103249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zusi C, Morandi A, Maguolo A, Corradi M, Costantini S, Mosca A, et al. Association between MBOAT7 rs641738 polymorphism and non‐alcoholic fatty liver in overweight or obese children. Nutr Metab Cardiovasc Dis. 2021;31:1548–55. [DOI] [PubMed] [Google Scholar]
- 42. Yan YY, Guo QR, Wang FH, Adhikari R, Zhu ZY, Zhang HY, et al. Cell‐free DNA: hope and potential application in cancer. Front Cell Dev Biol. 2021;9:639233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Duforestel M, Briand J, Bougras‐Cartron G, Heymann D, Frenel JS, et al. Cell‐free circulating epimarks in cancer monitoring. Epigenomics. 2020;12:145–55. [DOI] [PubMed] [Google Scholar]
- 44. Vos MB, Abrams SH, Barlow SE, Caprio S, Daniels SR, Kohli R, et al. NASPGHAN clinical practice guideline for the diagnosis and treatment of nonalcoholic fatty liver disease in children: recommendations from the expert committee on NAFLD (ECON) and the north American Society of Pediatric Gastroenterology, hepatology and nutrition (NASPGHAN). J Pediatr Gastroenterol Nutr. 2017;64:319–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mosca A, Panera N, Crudele A, Alisi A. Noninvasive diagnostic tools for pediatric NAFLD: where are we now? Expert Rev Gastroenterol Hepatol. 2020;14:1035–46. [DOI] [PubMed] [Google Scholar]
- 46. Ibacahe C, Correa‐Burrows P, Burrows R, Barrera G, Kim E, Hirsch S, et al. Accuracy of a semi‐quantitative ultrasound method to determine liver fat infiltration in early adulthood. Diagnostics (Basel). 2020;10:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Draijer LG, Feddouli S, Bohte AE, Vd Baan Slootweg O, Pels Rijcken TH, Benninga MA, et al. Comparison of diagnostic accuracy of screening tests ALT and ultrasound for pediatric non‐alcoholic fatty liver disease. Eur J Pediatr. 2019;178:863–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chrysavgis L, Papatheodoridi A, Cholongitas E, Koutsilieris M, Papatheodoridis G, Chatzigeorgiou A. Significance of circulating cell‐free DNA species in non‐alcoholic fatty liver disease. Int J Mol Sci. 2021;16:8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zeybel M, Hardy T, Robinson SM, Fox C, Anstee QM, Ness T, et al. Differential DNA methylation of genes involved in fibrosis progression in non‐alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics. 2015;7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Daniels SJ, Leeming DJ, Eslam M, Hashem AM, Nielsen MJ, Krag A, et al. ADAPT: an algorithm incorporating PRO‐C3 accurately identifies patients with NAFLD and advanced fibrosis. Hepatology. 2019;69:1075–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting information