

Abstract
Gas chromatography–mass spectrometry (GC-MS) platforms are typically run in electron ionization (EI) mode for mass spectral matching and metabolite annotation. With the advent of high resolution mass spectrometry (HRMS), soft ionization techniques such as chemical ionization (CI) may provide additional coverage for compound identification. We evaluated NIST SRM 1950 pooled plasma reference sample using a HRGC-MS instrument [GC-Orbitrap-MS with electron ionization (EI), positive chemical ionization (PCI), and negative CI (NCI) capabilities] for metabolite annotation and quantification to assess the suitability of the platform for routine discovery metabolomics. Using both open source and vendor workflows, we validated the spectral matches with an in-house spectral library (Wake Forest CPM GC-MS spectral and retention time libraries) of EI-MS and CI-MS/MS spectra obtained from chemical standards. We confidently [metabolomics standards initiative (MSI) confidence level 2] identified 263, 93, and 65 metabolites using EI, PCI, and NCI modes, respectively, of which 270 metabolites (64%) were validated using our Wake Forest CPM GC-MS spectral libraries. When compared to published LC-MS-based efforts using the same NIST SRM 1950 plasma sample, there was only 17% overlap between the two platforms. In addition, the metabolomics analysis of community approved standard human plasma demonstrated the ability of EI- and CI-MS modes of analysis using a HRGC-MS platform to enable reproducible and interoperable spectral matching.
Keywords: metabolomics, chemical ionization, Orbitrap, GC-MS, plasma, high resolution
INTRODUCTION
Metabolomics refers to the comprehensive study of small molecules in the molecular weight range of 50–2000 Da in biological systems (cells, tissues, organs, biofluids, or whole organisms).1 Platforms for metabolomics typically include chromatography coupled with mass spectrometry (GC-MS, LC-MS, and CE-MS), and spectroscopy technologies (NMR).2 GC in particular is very amenable to polar primary metabolites and fatty acids,3 in addition to its excellent chromatographic resolution, thereby lending itself to routine quantitative metabolomic applications. High resolution mass spectrometry (HRMS) refers to mass analyzers which have a mass resolving power (Rp determined at full-width half-height maximum, fwhm) higher than 10 000.4 Fourier transformation mass spectrometry (FTMS) is the most powerful technology not only thanks to its highest resolving power (i.e., Rp values greater than 100 000 up to several million), but also due to its extended dynamic mass range. Newer high-resolution (HR) technologies such as Orbitrap mass spectrometers are capable of sub-ppm mass accuracy at high mass resolutions (>50 000), and hence allow calculation of predicted molecular formulas based on the mass defect of a detected ion5 from spectral data at high resolving power with mass accuracies <1 ppm,6–8 but are yet to be explored for human metabolomics and biomedical research applications, especially in combination with GC.9 In the past, mass spectral and retention time index (MSRI) libraries for the GOLM Database were generated using identical types of capillary GC columns, and using two independent GC-MS detection technologies, namely, quadrupole-GC-MS and time-of-flight mass spectrometry (GC-ToF-MS).10 Spectral libraries (e.g., FiehnLib) were generated using both high resolution GC-ToF-MS and GC quadrupole mass spectrometry (GC-Quad) spectral comparisons.11 Moreover, recently we compared high resolution MS detectors such as Orbitrap MS and unit resolution ToF MS for characterization of the baboon serum metabolome,1 in our GC-MS based metabolomics research efforts.
Over the past decade, advances in liquid chromatography–mass spectrometry (LC-MS) have revolutionized untargeted metabolomics analyses for biomarker discovery. Indeed, HPLC-MS-based untargeted approaches have been shown to be useful for preliminary diagnostic screening when canonical processes do not reveal disease etiology nor establish a clear diagnosis.12 However, major challenges limit the broad application of untargeted GC-MS based metabolomics for several reasons discussed in detail elsewhere.13 While GC potentially has an advantage over HPLC due to superior chromatographic resolution and peak capacity, especially for polar primary metabolites and fatty acids,3 its application for discovery metabolomics has been limited, and high resolution GC-MS instruments have only recently become available.
Numerous analytical tools have been developed for the analysis of GC-MS and LC-MS metabolomics data. Both commercial and open source tools offer a wide range of approaches for compound annotation, including SpectConnect, Metabolite Detector 2.01a, MetAlign, MZmine 2.0, TagFinder 04, and XCMS Online. In a recent comparison of GC-MS data set analysis, multiple software packages such as SpectConnect, MetAlign, and Metabolite Detector correctly identified ≥90% of the true positives independent of the tool used.14 For untargeted LC-MS analysis, commercial software tools, such as MarkerLynx (Waters), MarkerView (Sciex), SIEVE (Thermo Scientific), Progenesis QI (Waters Corporation), MetaboScape (Bruker), Compound Discoverer (Thermo Scientific), Mass Profiler Pro (Agilent Technologies Inc.), and Metabolic Profiler (Bruker), allow work only on their own proprietary data formats and commercial proprietary spectral libraries, and are incapable of using raw MS spectra files generated from mass spectrometers of other brands. However, multiple free data analysis tools use open data formats are available and provide any user much more flexibility during each step of the analysis, such as XCMS, MZmine, OpenMS, XAlign, MZedDB, MetAlign, MAVEN, and MetaboAnalyst.15 Akin to the CI-MS data, as they are both soft ionization techniques, resources amenable to the analysis of ESI-MS spectral data are adoptable to the analysis of GC-EI-MS data, just that the majority of the metabolites/features are silylated. Moreover, both positive and negative modes of ionization have been shown to be useful in LC-MS based metabolomics research efforts, using positive/negative ion polarity switching in a targeted fashion,16 using Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometers in an untargeted manner in cerebrospinal fluid (CSF),17 and in the extensive analysis of methanolic extracts of human serum where using ESI for both positive and negative ions resulted in >90% additional unique ions being detected in the negative ESI mode.18
To date, most published studies have relied on a (GC)-high resolution accurate mass (HRAM) mass spectrometer (MS) only in EI-MS mode of operation. Chemical ionization (CI) is an alternative soft-ionization technique that can lead to enhanced annotation and robust quantification of metabolites. NCI is an electron capture process from electrons transferred from the reagent gas, whereas PCI is based on proton transfer from ion–molecule collisions. The availability of CI capabilities on HRMS platforms allows the expansion of the chemical space of biomolecules that can be identified and quantified in clinical metabolomics research. Historically, chemical ionization (CI) has been used extensively in targeted analysis of chemical classes from a wide range of biological matrices, i.e., on a single chemical class of compounds from blood, such as serum testosterone,19 quinolic acid in rat blood, plasma, and brain,20 estrogens,21 glycolipids,22 and prostanoids in urine,23 but not for broad-based discovery metabolomics applications.
There is an intrinsic need to increase the known chemical space of biofluids and tissues, such as blood serum and plasma, in search of the blood-based biomarker metabolites for disease characterization, toward confident and sensitive quantification of metabolites in the clinical research arena. Therefore, we examined whether using soft ionization techniques for gasphase analyses (volatile compounds) using CI, and usage of an in-house spectral library with spectra and retention time (RT) information (Wake Forest CPM GC-MS spectral libraries) can aid in the annotation and quantification of additional metabolites compared to EI-MS analysis. To our knowledge, this study is the first attempt to annotate and quantify plasma metabolites in all three modes, EI, PCI, and NCI, using a high mass resolution mass spectrometer (GC-Orbitrap-MS) on a comparative basis. The principal aims were to assess the capabilities of the CI using GC-Orbitrap-MS for annotation of metabolites in a standard reference material as a test sample, and to assess the EI-MS capabilities with a chemical standards generated library in order to exploit the full capabilities of chemical derivatization alongside HRMS for untargeted metabolomics. Finally, we compared our GC-Orbitrap-MS findings to published results from LC-HRMS metabolomics analyses of the same plasma samples to define overlap and platform-specific features.
MATERIALS AND METHODS
Chemicals
Acetonitrile, isopropanol, water, and pyridine were of HPLC grade, and methoxyamine hydrochloride (MeOX), 1% TMCS in N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA), and adonitol, were obtained from Sigma-Aldrich, St. Louis, MO, USA.
Chemical Standards for Generation of EI-MS, PCI-MS/MS, and NCI-MS Libraries
A collection of 635 small molecule metabolite standards on seven 96-well plates (Mass Spectrometry Metabolite Library of Standards, MSMLS from IROA Technologies) was purchased from Sigma-Aldrich, supplied as dried standards distributed across seven 96-well plates (Sigma-Aldrich; St. Louis, MO), where each well contained 5 μg aliquot of dried analytical standards. For reconstitution, samples in plates one to four were dissolved in 400 μL water, those in plate five were dissolved in 400 μL 40% methanol in water (v/v), while for plate six, some standards were dissolved in 400 μL 40% methanol in water and some in 400 μL water (mostly simple carbohydrates/sugars), and for plate seven the standards were dissolved in 400 μL of a 1:1 v/v water:ethanol mixture (mostly lipids), as recommended by the manufacturer. All compounds were reconstituted in the solvents as mentioned above, diluted into aliquot stocks of 25 ng/μL working concentrations, and were further vacuum-dried to moisture-free conditions prior to trimethylsilylation (TMS) derivatization reactions. The oncolumn injection amount was 60 ng for each compound across EI, PCI-MS/MS, and NCI-MS modes of analysis.
Reference NIST SRM 1950 Human Plasma Sample
The National Institute of Standards and Technology (NIST) Standard Reference Material (SRM) 1950 – Metabolites in Frozen Human Plasma24 was procured through Sigma-Aldrich (St. Louis, MO, USA) and used as a complex biological matrix for validation of the prepared library as well as for annotation of additional unknown metabolites. Briefly, the sample was developed by NIST in collaboration with the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) from pooled plasma of 100 fasted individuals representing average composition of the US population by race sex and health. The SRM 1950 human plasma was handled at a Biosafety Level (BSL) 2 laboratory (defined as laboratories that work with agents associated with human diseases that pose a moderate health hazard, or pathogenic or infectious organisms. Due to the potential risk, all procedures are performed biological safety cabinets, using personal protective equipment, available decontamination methods, and available means of biohazard disposal) for processing. Plasma samples were thawed on ice just once prior to all analyses.
Plasma Sample Extraction and Derivatization for GC-EI-MS, PCI-MS/MS, and NCI-MS Analysis in Three Modes
NIST SRM 1950 human plasma samples (30 μL) were subjected to sequential solvent extraction once each with 1 mL of acetonitrile:isopropanol:water (3:3:2, v/v ratio) and 500 μL of acetonitrile:water (1:1, v/v ratio) mixtures at 4 °C as described.25 Adonitol (5 μL from 10 mg/mL stock) was added to each aliquots as an internal standard prior to the extraction. The pooled extracts (∼1500 μL) from the two steps were combined and dried under vacuum at 4 °C prior to processing for GC-MS analysis. Detailed protocols are provided elsewhere.26 Dummy extractions performed on blank tubes served as extraction blanks to account for background (sample tubes, extraction solvent, derivatization reagents) noise and other sources of contamination (septa, liner, column, vials, handling etc.). Blanks were only used to see that no carryover occurred during randomized run orders and to manually filter out contaminating chemicals from the combined list of features. Samples were prepared such that EI-MS and CI-MS analysis (in both positive and negative modes) could be run from derivatization to data acquisition within a 24 h period due to the limited stability of silylated products. For GC-MS analysis, the samples were then sequentially derivatized with methoxyamine hydrochloride (MeOX) and 1% TMCS in N-methyl-N-trimethylsilyl-trifluoroacetamide (MSTFA) as described elsewhere.1,27,28 Steps involved addition of 10 μL of MeOX (20 mg/mL) in pyridine incubated under shaking at 55 °C for 60 min followed by trimethylsilylation at 60 °C for 60 min after adding 90 μL MSTFA.
GC-Orbitrap-MS Instrument Parameters
The acquisition sequence started with blank solvent (pyridine) injections, followed by randomized lists of extraction blanks (B), reagent blanks (R) and plasma samples (S). Pooled human plasma samples were injected at scheduled intervals for tentative annotation and monitoring shifts in retention indices (RI) as well as serving as quality control (QC) checks.
A robotic arm TriPlus RSH autosampler (Thermo Scientific, Bremen, Germany) was used to inject 1 μL of derivatized sample into a split/splitless (SSL) injector at 220 °C in a splitless mode into the TRACE 1310 gas chromatograph (Thermo Scientific, Austin, TX). For EI-MS, helium carrier gas at a flow rate of 1 mL/min was used for separation on a Thermo Scientific TraceGOLD TG-5SIL-MS 30 m length × 0.25 mm i.d. × 0.25 μm film thickness column. For both EI- and CI-MS runs, the initial oven temperature was held at 50 °C for 0.5 min, followed by an initial gradient of 20 °C/min ramp rate. The final temperature was 300 °C and held for 10 min. Eluting peaks were transferred through an auxiliary transfer line temperature of 230 °C into a Q Exactive-GC mass spectrometer (Thermo Scientific, Bremen, Germany). GC conditions were identical for all three modes, EI-MS, PCI-MS/MS, and NCI-MS, for authentic chemical standards and human plasma samples.
The mass spectrometer has an ExtractaBrite Ion Source technology, patented RF lens, with resolving power (RP) 120 000 fwhm at m/z 200 with EI, CI, full-scan and MS/MS capabilities. From ion source, an AQT quadrupole is used for precursor ion isolation for the CI analysis, which leads to Orbitrap Mass analyzer or higher-energy collisional dissociation (HCD) cell for MS/MS fragmentation. Electron ionization (EI) at 70 eV energy, emission current of 50 μA with an ion source temperature of 250 °C was used in all experiments. A filament delay of 5.7 min was selected to prevent excess reagents from being ionized for all 3 modes of acquisition. High resolution EI spectra were acquired using 60 000 resolution (fwhm at m/z 200) with a mass range of m/z 50–650. Data acquisition and instrument control were carried out using Xcalibur 4.3 and TraceFinder 4.1 softwares (Thermo Scientific). High resolution CI-MS/MS spectra were acquired with the following conditions: CI gas flow of 1 mL/min (methane, CH4), emission current: 50.0 μA, MS1 resolution: 120 000, automatic gain control (AGC) target: 106, scan range of 60–900 m/z in profile mode, followed by data dependent (dd)-MS/MS using 60 000 resolution, AGC target of 105, isolation window 2 m/z, scan range of 50–500 m/z, and a stepped normalized (N) collision (CE) energy of 15, 25, 35; in both positive or negative modes for PCI-MS, and NCI-MS data acquisition, respectively.
We obtained MS/MS fragmentation spectra for compounds run in PCI-MS, while NCI-MS did not result in compound fragmentation (hence only MS1 data). EI-MS spectra were acquired in typical full scan mode, and the same strategy was followed for compound identification in NIST SRM 1950 plasma as well.
For metabolites identified through all three modes of data acquisition, reported annotations were considered level 2 (putative annotation based on spectral library similarity) or 3 (putatively characterized compound class based on spectral similarity to known compounds of a chemical class) by the 2007 metabolomics standard initiative (MSI).29 Compounds identified in NCI spectra were assigned level 2 for all compounds for which authentic standards were available, based on their molecular ion (m/z) [M–H]− and RT annotations. We did not include and report any annotations solely based on MS1 obtained accurate masses from PCI-MS and NCI-MS acquisition from the NIST SRM 1950 plasma, where authentic standards, RT, and MS/MS fragmentation spectra were not available.
Data Analysis Using Various Open Source and Commercial Software Tools
TraceFinder 4.1.
The acquired EI-MS data were processed using TraceFinder 4.1 software (Thermo Scientific, Bremen, Germany). Initially, background-corrected full-scan data were deconvoluted and RT aligned using the incorporated deconvolution plugin. Initial analysis of collected spectra included baseline correction, peak filtering, quantification, assignment of a unique mass and retention index, signal-to-noise calculation and compound annotation based on the mass spectral pattern as compared to EI spectral libraries such as the NIST Mass Spectral Reference Library (NIST17/2017; National Institute of Standards and Technology, USA; with EI-MS data of 242 466 compounds), the Wiley Registry of Mass Spectra 11th Edition, the MSRI spectral libraries from the GOLM Metabolome Database,30 available from Max-Planck-Institute for Plant Physiology, Golm, Germany (http://csbdb.mpimp-golm.mpg.de/csbdb/gmd/gmd.html; 2594 spectra), MassBank (60 114 spectra),31 MoNA (Mass Bank of North America, (http://mona.fiehnlab.ucdavis.edu/; total 18 608 spectra and 1118 spectra from RTX Fiehn library: FiehnLib), and a supplied high resolution (HR)-MS spectral library for the GC-MS data set using proprietary TraceFinder 4.1 software, which contains high resolution electron impact MS data of > 800 primary and secondary metabolites; Thermo Scientific, Bremen, Germany). The returned list with tentative matches was manually evaluated for positive annotations. The decision on whether or not a match was a true analytical positive result was based on the search index score and the high resolution filtering value as well as on visual comparison of the acquired spectrum with the suggested library spectrum. Within this process, up to five top library hits were also considered where relevant.
Compound Discoverer 2.1.
For the data obtained in PCI and NCI modes for HR-GC-CI-MS, we used Compound Discoverer 2.1 software (Thermo Fisher Scientific, Bremen, Germany). An example workflow for this proprietary software is provided as Figure S1. For replication of the study, some of the important parameters used are listed below. For “select spectra” we used 100–5000 Da for min and max. precursor mass, with MS resolution of 60 000 and MSn resolution of 30 000. For “align retention times” adaptive curve alignment model was followed with maximum allowed shift of 0.5 min, and mass tolerance of 5 ppm. To “detect unknown compounds”, mass tolerance, 5 ppm, intensity tolerance, 30%, S/N threshold of 5, min peak intensity 10 000, ions searched were [M + H]+1, [M + C2H5]+1, [M + CH3]+1 (with the latter two specific for CH4 (methane) as CI gas) allowing elemental searches for C, H, N, O, P, S only. To group unknown compounds, the RT tolerance was set to 0.1 min, and mass tolerance of 5 ppm. Additionally, two local mass database lists were searched consisting of 4400 endogenous metabolites, extractables and leachables with a mass tolerance as above. mzCloud32 was consulted with ion activation energy tolerance of 20, similarity forward search and a match factor threshold cut off of 50. To predict compositions, a 5 ppm mass tolerance was allowed, for C, H, N, O, P, S, based compounds, 30% intensity tolerance, 5 ppm mass tolerance S/N = 5, for pattern and fragment matching. For searching ChemSpider33 for annotations, 21 databases were manually selected for their biological relevance with a 5 ppm mass tolerance, and #5 top hits being reported. Further, to filter out noise and less confident compounds, we discarded all compounds with a CV > 30%. Finally, all siloxane, halogen-derivatives, phthalate, acrylate, and silyloxy, borane, dioxolan, and silan, silox-derivative compounds were manually removed from the list.
MS-DIAL 3.9.
Raw data (.RAW) for EI-MS and PCI-MS, and NCI-MS from the Orbitrap instrument from a combined three different modes were converted to .mzML formats using ProteoWizard’s msConvert34 tool. The MS-DIAL with open source publicly available EI spectra library was used for raw peaks extraction and the data baseline filtering and calibration of the baseline, peak alignment, deconvolution analysis, peak annotation and integration of the peak height essentially followed as described.35 An average peak width of 20 scan and minimum peak height of 10 000 amplitudes was applied for peak detection, and sigma window value of 0.5, EI spectra cutoff of 50 000 amplitudes was implemented for deconvolution. For retention time and retention index library creation purposes, we recorded the (a) retention times, and also obtained the (b) Kovat’s retention indices (KIs) using 37 fatty acid methyl ester (FAME) mixture (Cat. No. # 47885-U, Supelco, Inc.), 10 mg/mL in methylene chloride; and the (c) linear retention time indices (RIs) using an even chain n-hydrocarbon mixture, C8–C40, 17 components (Cat. No. # 95394, Absolute Standards), 200 ng on column] for all the quantified features. For annotation setting, the retention time tolerance was 0.2 min, the m/z tolerance was 0.5 Da, the EI similarity cutoff was 80%, and the annotation score cutoff was 80%. In the alignment parameters setting process, the retention time tolerance was 0.5 min, and retention time factor was 0.5. For MS-DIAL 3.9 data annotations, we used publicly available libraries (both positive and negative ion mode) for MassBank, GNPS, RIKEN, MoNA. Detailed method to aggregate a local version of public EI spectral database is available.36 Further, a compound in negative mode is defined as a certain m/z ([M–H]−) detected at a specific retention time, as previously proposed.18
MS-FINDER 3.22.
For additional annotation of unknown features (compounds which did not match with existent public libraries and did not match any of the standards from the MSMLS library compounds) we used MS-FINDER 3.22 to determine their elemental formulas and in silico mass spectral fragmentation with MS-FINDER 3.22 (http://prime.psc.riken.jp/). Spectra were interrogated by sending queries from MS-DIAL 3.9 directly into the MS-FINDER 3.22 interface. The mass tolerance was set at 0.01 Da, relative abundance cut off for MS/MS was set at 1%, where all other parameters were kept as default. Other settings were as follows: LEWIS and SENIOR check (yes), isotopic ratio tolerance: 20%, element probability check (yes), element selection (O, N, P, S). Structure Finder setting: tree depth: 2, maximum reported number: 10, data sources (all except MINEs DBs).
Pathway Enrichment Analysis.
Pathway enrichment was performed using MetaboAnalyst 4.0 (www.metaboanalyst.ca).37 For ID conversions, the Chemical Translation Service (CTS: http://cts.fiehnlab.ucdavis.edu/conversion/batch) was used to convert the common chemical names into their KEGG, HMDB, Metlin, PubChem CID, and ChEBI identifiers.
Data Sharing.
The raw data sets and the metadata obtained from the GC-MS platforms for NIST SRM 1950 human plasma (10 replicates, S1–S10; blanks, B1, B2; solvent blanks, P1, P2, and reagents blanks, R1, R2 for all three modes EI-MS, PCI-MS, and NCI-MS acquired) are deposited at the Global Natural Product Social Molecular Networking (GNPS) Massive Server (study accession ID: MSV000084784) and is available at this link: ftp://massive.ucsd.edu/MSV000084784/ to the readers. Furthermore, the MS-FINDER 3.22 annotated structures from the HRGC-EI-MS runs, i.e., 41 unique mass spectra are submitted at the MoNA DB that are freely accessible here for the community: https://mona.fiehnlab.ucdavis.edu/ (accession IDs from MoNA011445 through to MoNA011494). All spectral libraries and data are available from the authors upon request.
RESULTS
A freshly prepared commercially available NIST SRM 1950 plasma was aliquoted, extracted, and derivatized for GC-EI-MS, GC-PCI-MS, and GC-NCI-MS analysis using a QE-GC-Orbitrap-MS instrument. Four different software tools for analysis of GC-MS data sets were used to extract the features, annotate and quantify the features in these 5 data sets, for a total of 15 sample files. Software included two commercial tools (TraceFinder 4.1 and Compound Discoverer 2.1) and two widely used open source tools (MS-DIAL 3.9 and MS-FINDER 3.22). The overall spectral data acquisition workflow using a QEGC-MS is provided in a pictorial form (Figure 1).
Figure 1.
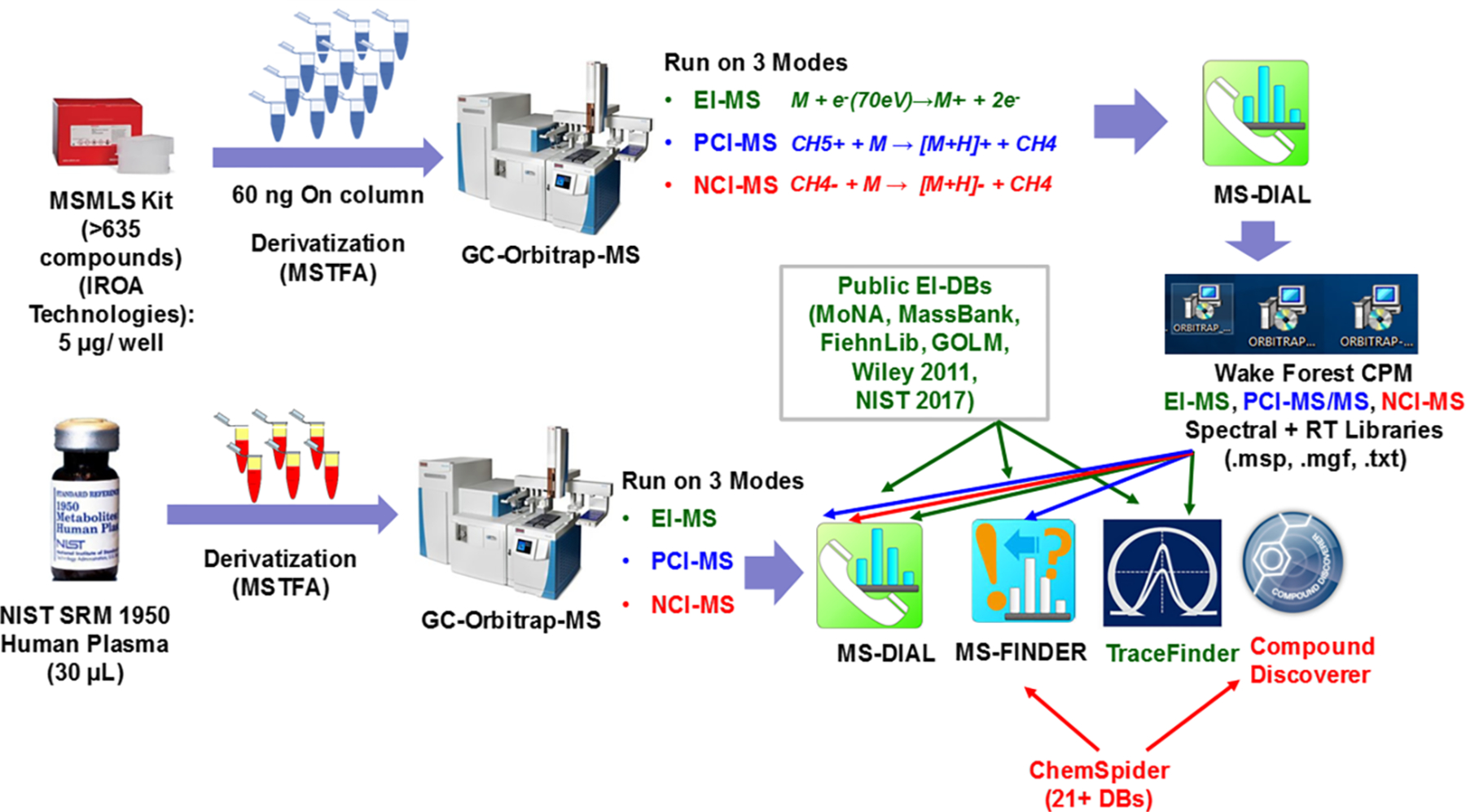
Summary workflow for the analysis of human NIST SRM 1950 human plasma using HR-GC-MS (EI, PCI, and NCI) for annotation of metabolites. The Wake Forest CPM library was first prepared using the authentic chemical standards from the MSMLS Kit (IROA Technologies) that helped annotate spectra for 330 unique compounds using EI, PCI, and NCI modes of data acquisition. Second, NIST SRM 1950 human plasma was run three times on EI, PCI, and NCI modes in a similar manner, and compounds were identified using the built Wake Forest CPM Library and public EI-MS databases (MoNA, MassBank, FiehnLib, GOLM DB, Wiley 2011, NIST 2017), and commercial workflows that used ChemSpider databases for PCI-MS/MS and NCI-MS for compound annotation. Green, blue, and red fonts and lines correspond to EI-MS, PCI-MS, and NCI-MS, respectively.
Our purpose was not to evaluate the feature calling capabilities of the tools, rather, to use their default settings for our data sets to find “consensus” and “unique” features between the platforms for HR-MS data sets. Representative total ion chromatograms (TICs) obtained for a single NIST SRM 1950 human plasma sample in EI, PCI, and NCI modes are provided in Figure 2A–C.
Figure 2.
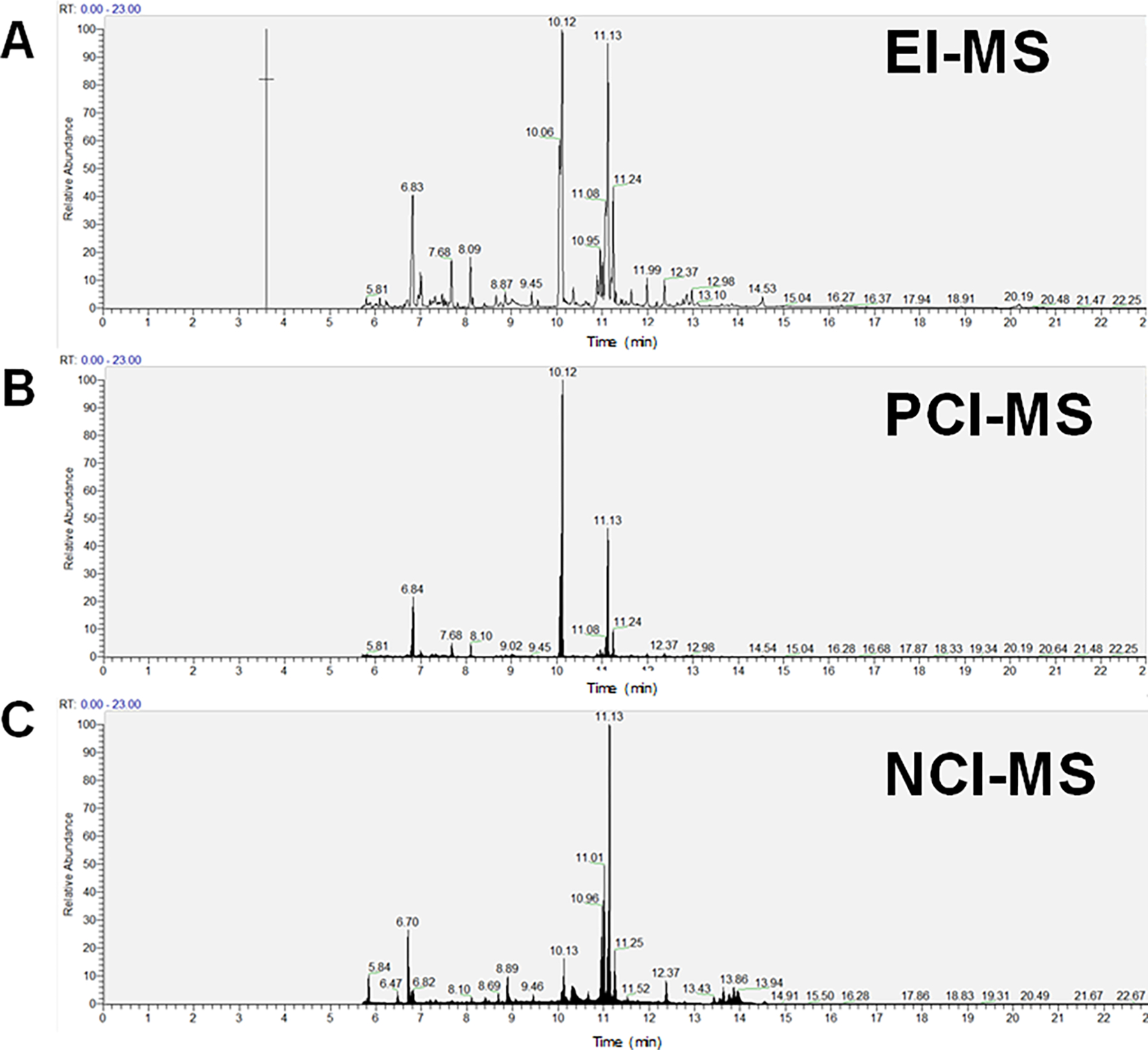
Total ion chromatograms (TICs) obtained for NIST SRM 1950 human plasma samples run on (A) electron ionization (EI), (B) positive chemical ionization (PCI), and (C) negative chemical ionization (NCI) modes.
Metabolite Library Preparation and Individual Metabolite Features Obtained from HR-GC-MS Platforms Using NIST SRM 1950 Human Plasma
Using the 635 compounds from MSMLS standards obtained from IROA Technologies (chemical list and metadata: Table S1), with our standard MSTFA derivatization procedure, we obtained EI, PCI and NCI spectra for the compounds after reconstitution. The spectra were recorded in a matrix with spectra and RT/RI using FAME and n-hydrocarbon mixtures. Of the 635 compounds, we successfully obtained EI-MS spectra for 258 compounds (40%) (EI-GC-MS Wake Forest CPM spectral library), whereas CI-MS (in both positive and negative modes) yielded spectra for 176 compounds (28%) (CI-GC-MS Wake Forest CPM spectral library). In total, these combined efforts resulted in spectral and retention time data acquisition for 330 unique metabolites. Further, 98 and 82 unique metabolites were represented in the PCI-MS and NCI-MS libraries, respectively, whereas another 69 were shared between the two CI modes (Figure 3A).
Figure 3.
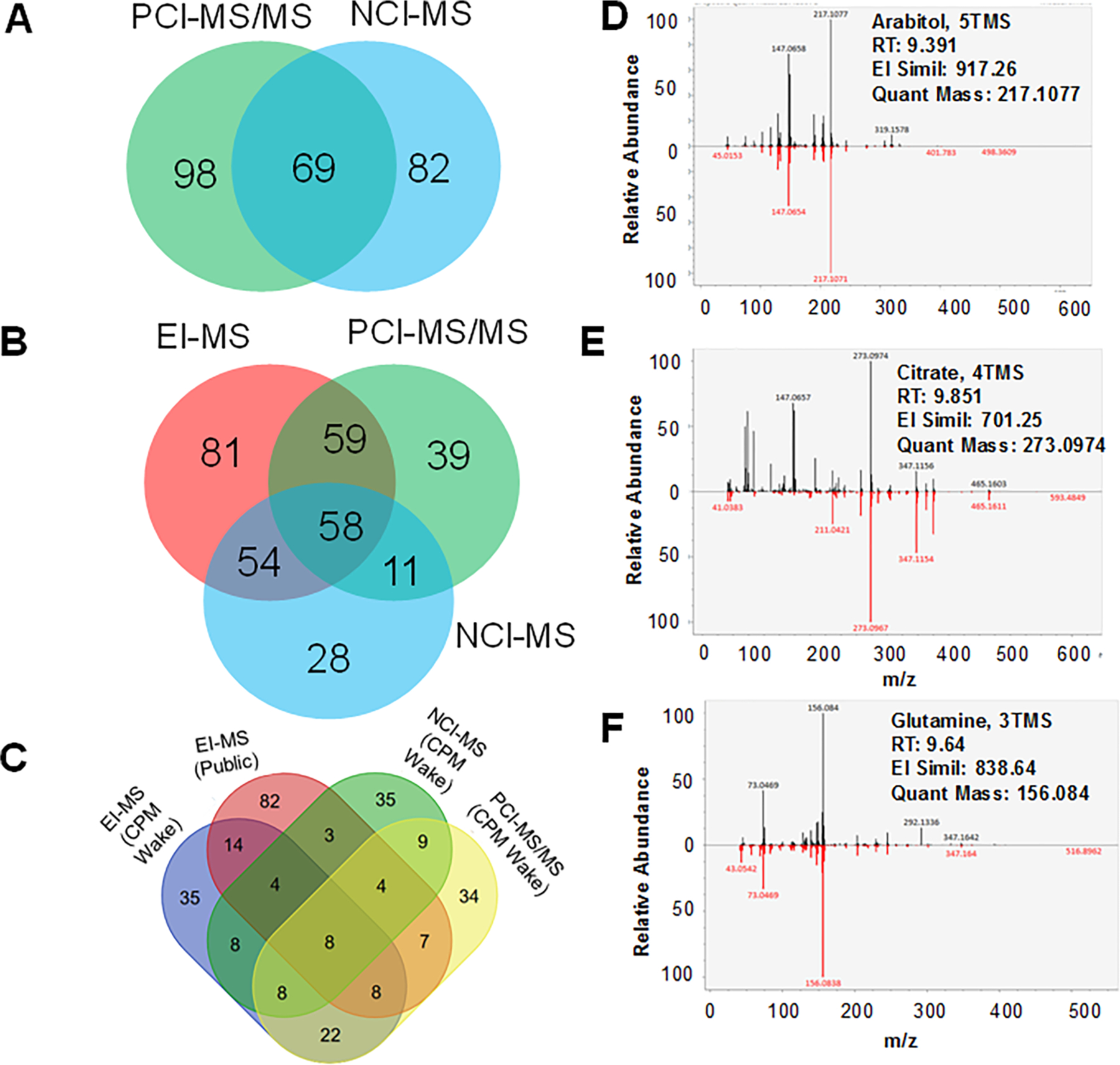
EI-MS, PCI-MS, NCI-MS spectral library preparation and spectral validation using NIST SRM 1950 human plasma. (A) Shared and unique metabolites for PCI-MS and NCI-MS spectral and retention time libraries. (B) Shared and unique metabolites for EI-MS, PCI-MS, and NCI-MS spectral and retention time libraries. (C) A four-way Venn diagram displaying the shared and unique metabolites between (C) GC-EI-MS, GC-PCI-MS, and GC-NCI-MS from in Wake Forest CPM spectral libraries and GC-EI-MS spectral hits from public libraries. HR EI-MS spectral matches for plasma sample derived spectra and library hit for (D) arabitol, 5TMS, (E) citrate, 4TMS, and (F) glutamine, 3TMS.
Overall, a total of 330 of the 635 metabolites were amenable to GC-MS analysis for spectral library generation with a combined ionization modes of EI-MS, PCI-MS, and NCI-MS, with 81, 39, and 28 unique metabolites, respectively, captured using each ionization modes (Figure 3B). Only 58 metabolites were common and amenable to all three modes of ionization for GC-MS analysis.
In the analysis of the NIST SRM 1950 plasma samples, the total number of metabolites captured using EI-MS with the EI-GC-MS Wake Forest CPM spectral library, public EI-MS library, PCI-GC-MS Wake Forest CPM spectral library, and NCI-GC-MS Wake Forest CPM spectral library are all provided with identifiers (Table S2). Using the proprietary TraceFinder 4.1 software that used the Thermo Fisher GC-Orbitrap-MS HRMS Library v 2.0, NIST 2017 and Wiley 2011 libraries, we detected 1670 EI-MS features which resulted in confident annotation (MSI confidence level 2 and with RT) of 201 metabolites, and of these, 84 were silylated following trimethylsilylation and methoxyamination reactions (Table S3). Using MS-DIAL 3.9 we further reanalyzed the data using the EI-GC-MS Wake Forest CPM spectral library and public libraries (MoNA, MassBank Japan, Fiehn library, GOLM DB, GNPS, HMDB etc.) which resulted in annotation of 112 and 151 metabolites, respectively (Figure 3C). As examples, we provide the HR EI-MS spectra for common metabolites such as citrate, 4TMS, glutamine, 3TMS, and arabitol, 5TMS (Figures 3D–F).
For PCI-MS/MS spectral data acquisition, using PCI-GC-MS Wake Forest CPM spectral library (mass spectra + retention time (RT)), we identified 93 metabolites using MS-DIAL 3.9 (Table S2), and another 21 using MS-FINDER 3.22. Representative PCI-MS/MS spectra for caffeic acid, glutarate, and 10-hydroxydecanoate are shown in Figure 4A–C.
Figure 4.
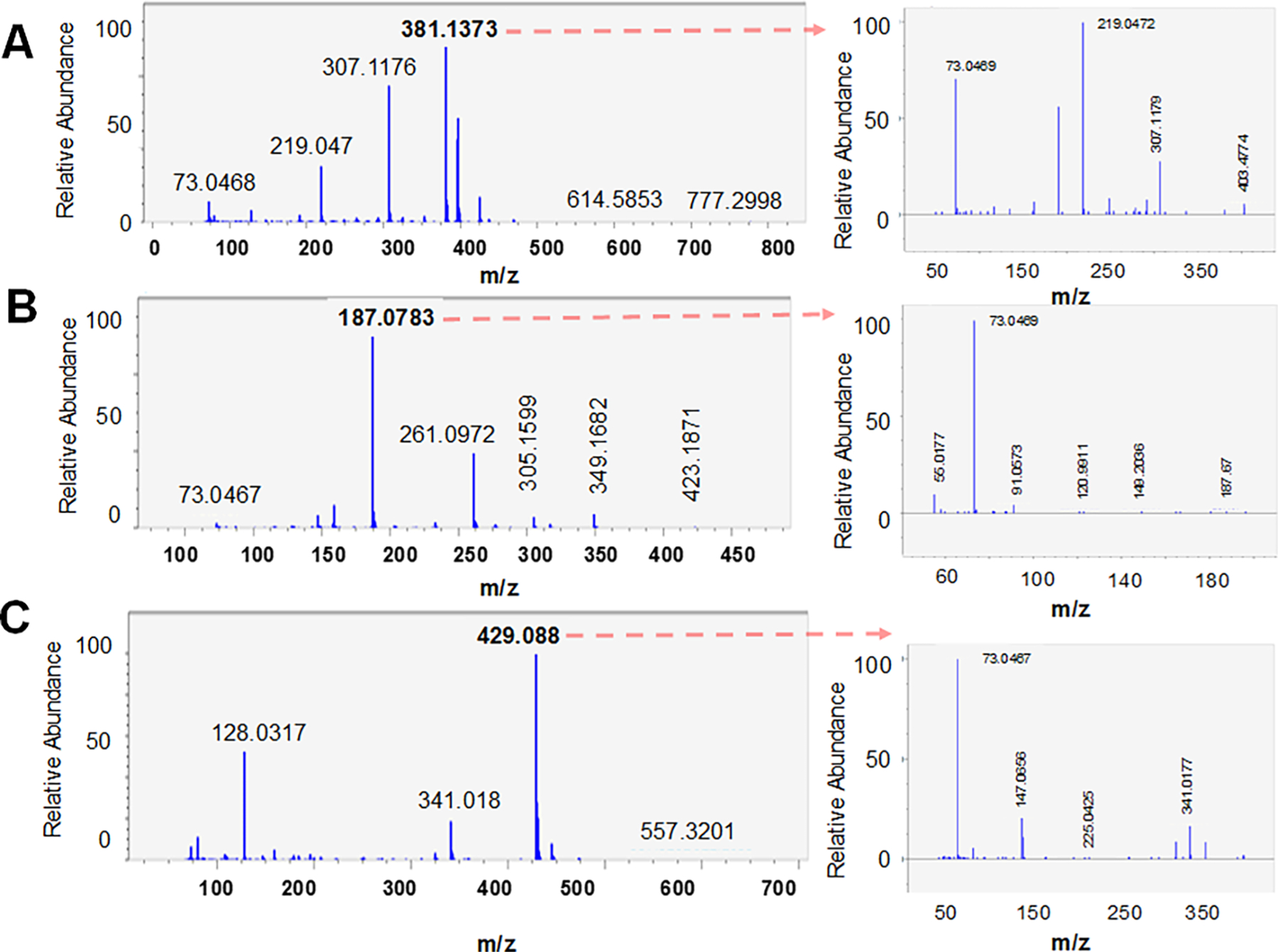
Example PCI-MS and PCI-MS/MS spectra for (A) caffeic acid, (B) glutarate, and (C) 10-hydroxydecanoate.
For NCI-MS spectral data acquisition, using the NCI-GC-MS Wake Forest CPM spectral library (high resolution MS1 and RT), we identified 65 metabolites (Table S3), and another 21 using MS-FINDER 3.22. However, it is important to note that for the NCI-mode, no fragments were obtained for MS/MS, a technological limitation, though the MS1 accurate mass profiles along with the RT represented matched authentic standards. NCI is a softer ionization process than EI so there is a tendency for less fragmentation, and in a number of cases the quantitative or qualifier ion is the molecular ion or a higher mass product ion as compared to EI; and NCI in either SIM or SRM mode has added selectivity with no response for the other matrix components that affect EI.38 Further, in NCI modes, fragmentation processes are limited to elimination reactions, and/or show dominant quasi-molecular ions, with little fragmentation, at times involving loss of combinations of the even electron neutral species H. This also renders the scan function in NCI-MS data useless due to the resulting lack of MS/MS spectra. When we consulted the Compound Discoverer 2.1 workflow, it resulted in annotation of 81 and 116 metabolites respectively, based on the NCI-MS and PCI-MS data sets (Table S3). However, the confidence on these annotations was low due to lack of retention time information for these matches and lack of fragmentation (MS/MS spectra). Moreover, the Compound Discoverer workflow did not capture any silylated molecules, as it failed to annotate Si-carrying compounds and the consulted databases did not contain silylated compounds.
Finally, using the in silico spectral data prediction tool MS-FINDER 3.22, we used the EI-MS spectral data from NIST SRM 1950 plasma to tentatively annotate 41 unique metabolites. Using the commercial workflow provided with the instrument by the mass-spec vendor, Compound Discoverer, which by virtue of the licensed databases consists of only nonsilylated chemicals, helped annotate 123 metabolites in total, 7 unique to PCI-MS and 42 unique to NCI-MS, with 74 compounds common in both modes. Comparing metabolite coverage for EI-MS, NCI-MS, and PCI-MS using only commercial workflows that used TraceFinder and Compound Discoverer, only 13 were found to be shared between all three modes (Figure S2A), with significant overlap between the PCI and NCI-modes and highest numbers identified with TraceFinder that used all possible commercial EI-MS spectral libraries available. However, when all annotated compounds IDed using commercial workflows were compared against open source workflows with our Wake Forest CPM spectral libraries, we found out that the Wake Forest CPM spectral libraries helped obtain 47% new IDs as compared to 33% new IDs using commercial workflows and databases alone (Figure S2B). However, both workflows (commercial and open source that used Wake Forest CPM spectral libraries) identified very different sets of compounds, and only shared 19.7% of metabolite IDs.
Unique and Shared Metabolic Pathways for Metabolites Identified in NIST SRM 1950 Human Plasma Using 3 HRGC-MS Runs and Public LC-MS Datasets
We asked if the metabolites identified using the spectral library would map differentially to diverse biological pathways by virtue of their chemical properties, i.e., amenability to an analysis mode, because the application of a spectral library can only be fully assessed in the context of its biological relevance. Hence we preformed a pathway analysis separately on metabolites captured using each of the three modes of analysis. When the metabolites were combined for all three GC-MS runs, the identified metabolites represented 63 different KEGG-derived metabolic pathways, with the criteria of at least 2 metabolites/pathway (Figure 5A, Table S4).
Figure 5.
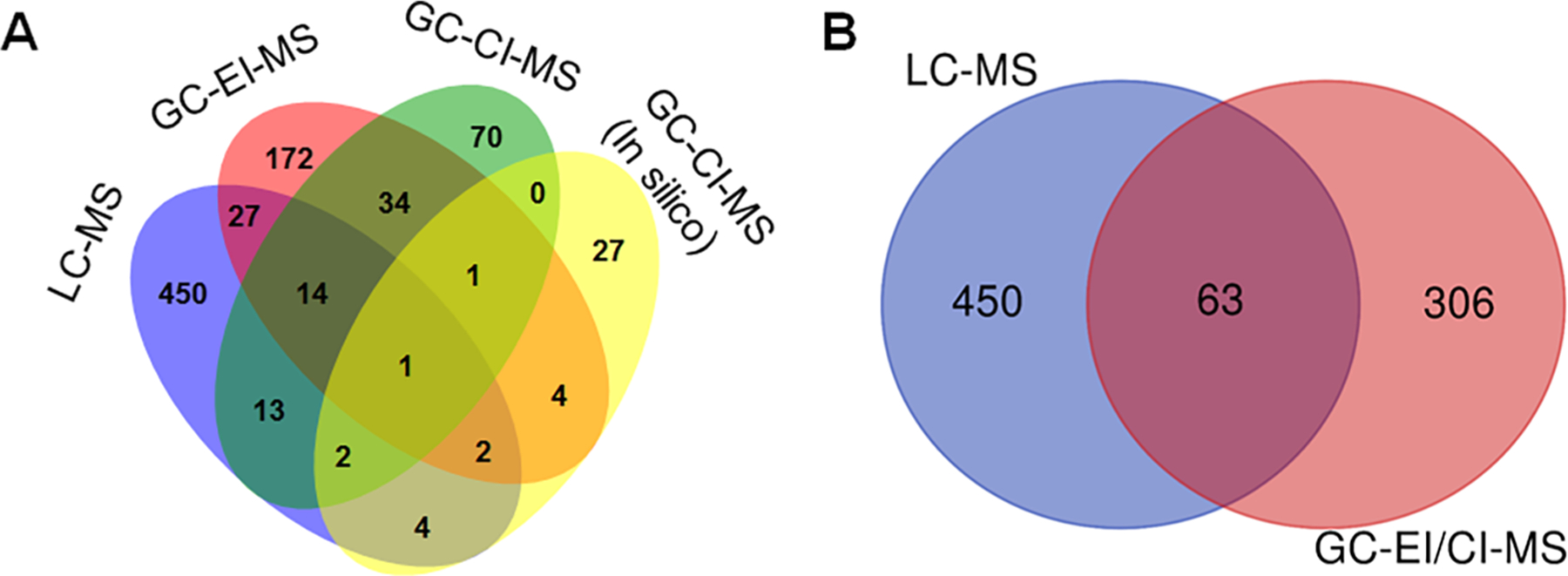
Shared and unique metabolites quantified based on ionization modes and platforms. (A) A four-way Venn–diagram displaying the coverage of metabolites for LC-MS, GC-EI-MS, GC-CI-MS, and CI-MS (MS-FINDER 3.22 based discovery). (B) A two-way Venn diagram displaying overlap of metabolites for LC-MS and GC-MS approaches in NIST SRM 1950 plasma.
The top 10 over-represented pathways (P-values <0.05, hypergeometric test) were as follows: beta-alanine metabolism, alanine, aspartate and glutamate metabolism, aminoacyl-tRNA biosynthesis, citrate cycle (TCA cycle), arginine and proline metabolism, pyrimidine metabolism, galactose metabolism, glycine, serine and threonine metabolism, pantothenate and CoA biosynthesis, and nitrogen metabolism. For metabolites with valid HMDBI IDs, 107, 130, 79, and 100 metabolites were captured using the EI-GC-MS Wake Forest CPM spectral library, the EI-MS public open-source spectral library, NCI-MS (combined NCI-GC-MS Wake Forest CPM spectral library and in silico prediction), and PCI-MS (combined PCI-GC-MS Wake Forest CPM spectral library and in silico prediction), respectively; for a total of 281 unique metabolites with HMDB identifiers identified by GC-MS (Figure 5A). The complementarity of EI and CI modes of analysis in the analysis postderivatization is evident (Figure 5B).
Together, the combined 566 metabolites from both the GC-MS analysis in this study, and LC-MS analyses obtained from published literature corresponded to 562 HMDB IDs, and 454 KEGG IDs, respectively, from NIST SRM 1950 plasma. A summary Venn diagram shows the overlap of GC-MS-derived metabolites in comparison to those for LC-MS based identified metabolites in literature24,39–41 (Figure 5A). Essentially, only 63 metabolites were identified in both approaches, while 450 and 306 unique metabolites were captured using LC-MS and GC-MS efforts, respectively (Table S2; Figure 5B). Clearly, the metabolites are very different for the LC and GC platforms, and of the total 281 metabolites captured using HR GC-MS alone, the EI-MS and CI-MS derived metabolite-based pathways present very different pictures (Figure S3).
Comparison of NIST SRM 1950 Human Plasma Identified Metabolites Using HRGC-MS Results to Public HRLC-MS Datasets
Next, we wanted to compare the metabolites identified using 3 modes of HRGC-MS analysis with publicly available LC-MS data sets. Toward this, we included available open data from 4 publications that performed NIST SRM 1950 plasma metabolomics analysis using LC-MS platforms.24,39–41 The combined LC-MS analyses identified a total of 514 individual metabolites. To examine the putative basis of the differences that are specific to modes of ionization and analytical platforms used, we investigated if the chemical properties of the metabolites such as logP values, monoisotopic masses, boiling points, or number of C atoms/metabolite showed any trends for the CI, EI, and ESI modes of analysis, as well as across the two distinct chromatography platforms (GC-MS vs LC-MS) (Figure S4A–D). The metabolites covered using the various hard (EI) and soft ionization (CI, ESI) modes are very diverse and share minimum overlap. However, the coverage of high MW compounds is higher in ESI than both EI and CI modes. Boiling points of the compounds showed no discernible patterns explaining the differences. For the number of C atoms, both EI and ESI covered a wider range than CI. Further, EI-MS did show a better linear relationship for RT (min) against log2 (S/N) of the compounds (Table S4E). Given the enormity of the human plasma metabolome (as evident from the HMDB), and the limited number of chemicals captured per analysis using GC-MS or LC-MS, it is possible that the limited representation of metabolites is biased, and could influence the outcome of this analysis.
The 513 LC-MS-identified metabolites led to representation of 64 KEGG-derived metabolic pathways, with the criteria of at least 2 metabolites per pathway (Figures 6A,B; Table S5).
Figure 6.
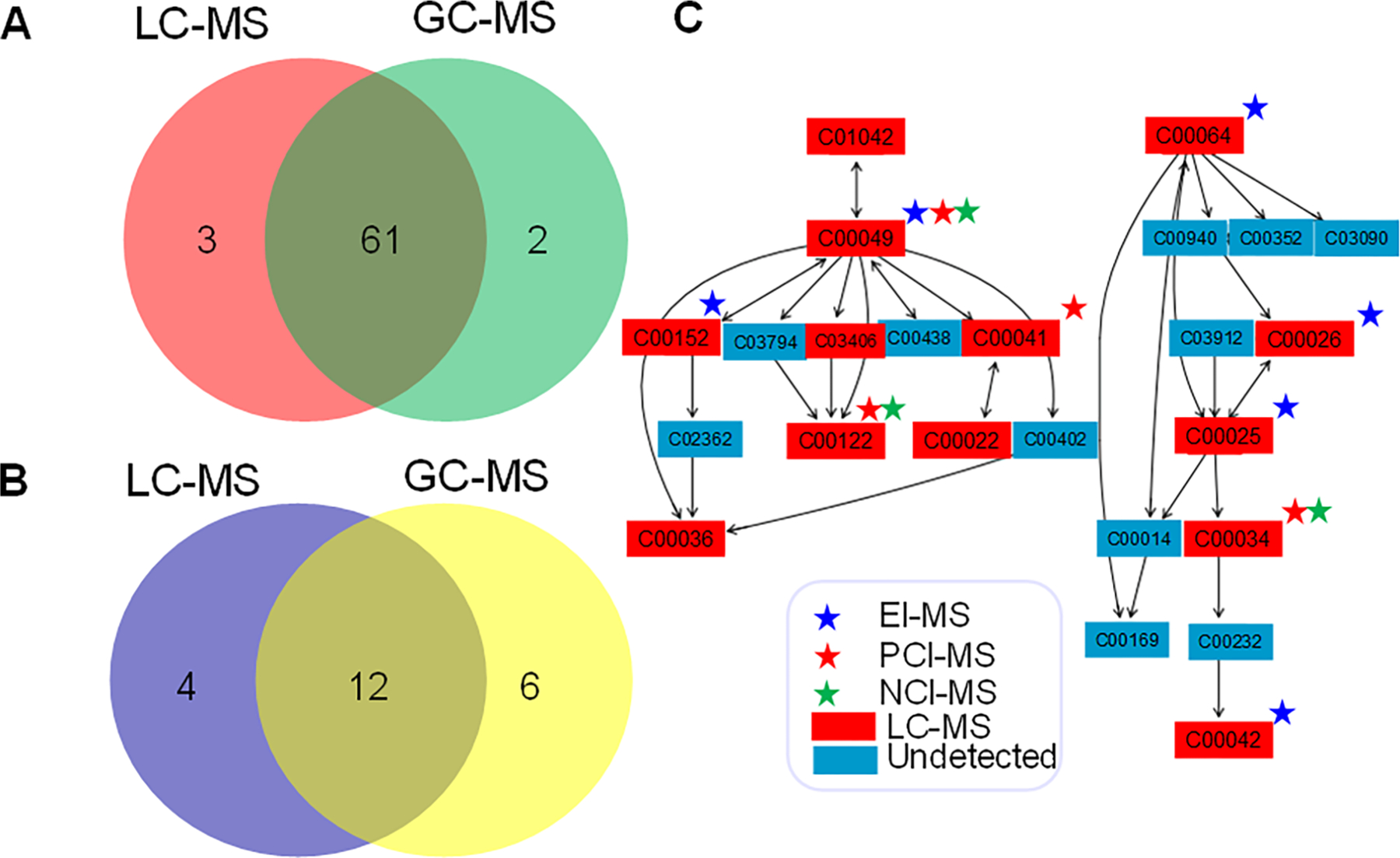
Pathway analysis of the metabolite coverage from LC-MS and GC-MS platforms. (A) KEGG-pathways shared between LC-MS and GC-MS platforms with at least 2 metabolites per pathway as a cutoff. (B) Statistically significantly enriched (P-values < 0.05) KEGG-pathways shared between LC-MS and GC-MS platforms. (C) Complementarity of the GC-MS modes of analysis (EI-MS, PCI-MS, NCI-MS) and LC-MS platforms shown as an example with mapped metabolites onto alanine, aspartate, and glutamate metabolism.
In a pathway enrichment analysis, the top 10 pathways (P < 0.05, hypergeometric test) were as follows: pyrimidine metabolism, alanine, aspartate and glutamate metabolism, glycine, serine and threonine metabolism, arginine and proline metabolism, citrate cycle (TCA cycle), aminoacyl-tRNA biosynthesis, nitrogen metabolism, purine metabolism, glutathione metabolism, and cysteine and methionine metabolism. Seven of these pathways match with the top 10 pathways identified in the analysis of the GC-MS data. Four pathways were unique to the LC-MS data sets: nicotinate and nicotinamide metabolism, caffeine metabolism, cysteine and methionine metabolism, purine metabolism. Six pathways were unique to the GC-MS derived metabolites: vitamin B6 metabolism, lysine biosynthesis, ascorbate and aldarate metabolism, pentose phosphate pathway, tyrosine metabolism, galactose metabolism.
Using an example of the KEGG pathway of alanine, aspartate, and glutamate metabolism which consists of 24 metabolites, and had highest representation of LC-MS and GC-MS data sets, we mapped all identified metabolites to examine the complementarity of the platforms and various modes of analysis (Figure 6C). Indeed, six metabolites were captured by EI-MS, four by PCI-MS, and three by NCI-MS, and 13 were captured using LC-MS. It should also be noted that 11 metabolites remain undetected with any of the platforms used. Altogether, GC-MS methods identified 9 metabolites, and 13 metabolites in this pathway were detected by LC-MS, with only 4 unique metabolites exclusively covered by LC-MS. The metabolite names, retention times, and qualifier ions for EI-MS, PCI-MS, and NCI-MS annotated metabolites are available (Tables S6–S8). In EI-MS, 20 compounds showed multiple derivatized forms, amounting to 47 entries in the library, which are specified in Table S6. In PCI-MS, 12 compounds showed multiple derivatized forms, amounting to 24 entries in the library, which are specified in Table S7. In NCI-MS, 12 compounds showed multiple derivatized forms, amounting to 24 entries in the library, which are specified in Table S8.
DISCUSSION
Complementarity of High Resolution GC-EI-MS and GC-CI-MS for NIST SRM 1950 Human Plasma Analysis
In this study, we first generated three spectral libraries using different MS modes, EI-MS, PCI-MS/MS, and NCI-MS, using a commercial kit with authentic chemical standards, followed by an application of the libraries to the analysis of a community standard reference material, NIST SRM 1950 human plasma (Figure 1). In a recent publication, we demonstrated the capabilities of a HRGC-MS platform run on EI-MS mode only for metabolomic characterization of baboon serum using publicly available open source and commercial spectral libraries,1,42 where we used Orbitrap technology for increased annotation and robust quantification of metabolites using EI mode alone. Moreover, it has also been stressed that mass accuracy and RT alone are not sufficient for metabolite annotation in untargeted metabolomics approaches.43 One widely used annotation approach is to search spectral libraries in reference databases for matching metabolites; however, this approach is limited by the coverage of these libraries.44 The entirety of chemical space of small molecules currently covered in databases such as PubChem, ChemSpider, or the Chemical Abstracts Database is larger than 120 million compounds, and those with biological relevance are estimated at 1–2 million, but up to 5–20% of metabolites discovered during untargeted metabolic profiling remain unmatched.45,46 The mass spectral databases that are available to users (i.e., either open source or from vendors) are different in coverage and their sizes, thus resulting in uncertainties of metabolite annotation and differential outputs from a given data set. Additional technical variability in MS analysis further complicates the reliable and reproducible metabolite annotation. Finally, in a recent study, the authors concluded that electronic factors related to the functioning of the ESI source, namely, the capillary and sample cone voltages, significantly influenced the recorded MS signals with regard to not only the number and abundance of features, but also the overall structure of the collected data and resulting metabolite annotation.47 Clearly, numerous factors influence the depth and accuracy of metabolite profiling in untargeted metabolomics analyses.
In the analysis presented here, we evaluated the capabilities of CI and EI for GC-MS-based metabolomics analysis using human plasma (Figure 2). For our HRMS GC-MS instrument, switching between EI and CI was easy, and a single prepared sample could be subjected to all three runs sequentially. We established novel databases of biologically relevant compounds as references for GC-MS-based metabolomics analysis using HRMS to improve the confidence in metabolite annotation, and then compared the GC-MS results to metabolomics profiles obtained by LC-HRMS in previously published studies using the same reference plasma sample. In this study, we initially performed CI-MS with ammonia (NH3) as the chemical ionization gas, but the extensive and intense fragmentation did not improve structure prediction, resulted in loss of signal in the molecular mass, and represented a computational challenge. Hence, we developed and optimized the conditions for methane (CH4) as the source for CI, and tandem mass spectrometry in PCI-MS/MS mode (Figure 4A–C), and all data presented here are based on this approach. Given that a recent publication reports on the development of an isobutene-argon CI mixture for more robust CI-MS,48 this may provide additional spectral annotation in the future in analysis of clinical samples.
A previous study suggested that NCI was 150-fold more sensitive in pg levels of melatonin quantification in human plasma.49 Neurosteroids were quantified in rat plasma and brain following stress and drug administration using NCI GC-MS, though on a selective ion monitoring (SIM) mode of analysis.50 Further, electron capture–negative chemical ionization–gas chromatography/mass spectrometry (EC-NCI GC-MS) was shown to be 100-fold more sensitive than LC-MS/MS for analyzing authentic halogenated tyrosines in human plasma in 5 min runs.51 However, most of the applications in CI have focused on analysis of pesticides and environmental/organic pollutants,52 pharmaceuticals and endocrine disruptors,53 fluorinated organics,54 drugs, i.e., cocaine in hair,55 ketamine and its derivatives in urine,56 and statins in plasma,57 and not widely untargeted metabolomics applications. As can be seen, most of these efforts used only NCI modes, not PCI mode of analysis. Moreover, earlier studies have not attempted to characterize the complex biological matrixes themselves (in a discovery analysis), but have only targeted specific chemical compounds of interest such as drugs, pesticides, and organic exogenous chemicals. Thus, our analysis represents the first combined NCI and PCI analysis of human plasma, and a detailed comparison with EI-based GC-MS analysis (Figure 3).
Usefulness of Adopted Software Tools and Workflows in HRGC-MS Analaysis
A comparative overview of some of the metabolomics tools used in our analysis (i.e., MS-DIAL, MS-FINDER) is provided elsewhere.58 As our results demonstrate, different tools lead to differences in metabolite annotations. This matches with results reported in a recent study, using plant extracts and mixtures of standard compounds, where researchers demonstrated the differences in numbers of features captured using different open source and commercial software tools for the same samples.59
We observed PCI-MS spectra displaying losses of CH4 [M + 1–16]+, 1–3 TMSOH [M + 1–n × 90]+, and combinations of CH4 and TMSOH losses [M + 1–n × 90–16]+, as reported previously,60 that were incorporated to feature finding in both MS-DIAL and Compound Discoverer. The vendor software, Compound Discoverer 2.1, relies only on large public chemical databases, e.g., ChemSpider (http://www.chemspider.com/), and limited spectral matching functionalities from mzCloud (https://www.mzcloud.org/) and helped in annotation of limited number of compounds (Figure 5A–D). For instance, we demonstrate the annotation of 2-hydroxyglutarate (C5H8O5; monoisotopic mass: 148.03717) and N-acetyl-l-glutamine (C7H12N2O4; monoisotopic mass: 188.0797) from PCI-MS analysis that used Compound Discoverer workflows (Figure S5A–D). However, these resources do not include silylated compounds, and the software does not allow for searching TMS or Si derivatives. Compound Discoverer allows a set of up to 300 databases for annotations, of which only 21 are biologically relevant (given that our matrix is known, and is NIST SRM 1950 human plasma) and were chosen (Table S9). Also, Compound Discoverer only provides access to mzCloud and ChemSpider,33 and the limited spectral databases are not downloadable, thus, we could not run a comparable analysis in MS-DIAL 3.9 workflow. Similarly, for soft ionization spectra such as HR-LC-ESI-MS analysis, the XCMS Online suite of tools allows consultation of METLIN DB for annotation, but the database does not contain silylated compounds, making METLIN not useful for GC-MS spectral annotation. Since the application programming interfaces (APIs) for METLIN online have been disabled since 2011,61 this prevented us from using METLIN for Compound Discoverer 2.1 or MS-DIAL/MS-FINDER annotations. In contrast, MS-DIAL 3.9 allows all open source libraries to be consulted for annotation. However, as METLIN, mzCloud and ChemSpider are not available for MS-DIAL 3.9, the comparisons are not informative. Also, the number of naturally occurring metabolites is still low in mzCloud compared to other popular databases.61 Thus, we relied on a workflow that used the functionalities of the MS-FINDER for spectral annotation which allowed searching of diverse chemical databases, and built custom spectral libraries (for both EI and CI modes), with formula searchability for C, H, N, O, P, S, Si compounds and their TMS and MeOX derivatives. To gain confidence in the annotations for 41 compounds identified using MS-FINDER, we selected 15 of the tentatively MS-FINDER identified compounds for validation using authentic chemical standards. Nine compounds were indeed found to match the spectrum and RT, while the remaining six showed >70% matched spectra but differences in RT, indicating they could be related compounds (Table S10). Moreover, the ability to convert RTs to Fiehn-Retention Index (RI) that can be performed using the 37 component FAME mixture enables higher confidence matches for all 3 modes of analysis. Both the RI and KI retention time (min) and carbon marker information are also provided (Table S11).
For HR-GC-MS analysis, the proprietary tool that we used was TraceFinder 4.1, which allowed consultation of NIST 2017, Wiley 2011, Thermo Fisher Scientific’s proprietary HR-MS spectral libraries as well as open source and custom ones such as MoNA (North America), MassBank (Japan), FiehnLib, and the GC-MS Wake Forest CPM spectral libraries for EI- and CI-MS data analysis. Munson and Geiger established that CH5 + and C2H5 + formed in the presence of methane do not further react with methane,62 which allowed reactions of theseions with additives, giving rise to the development of CI-MS. Using MS-DIAL, we could add these adducts while searching for annotations in soft-ionization DBs (i.e., mostly ESI-MS) such as those derived from Mass Bank, GNPS, RIKEN, HMDB, KEGG among others. In addition, for reproducible analysis of the workflow, we have also provided the detailed MS-DIAL parameters used for data processing for EI-MS, PCI-MS/MS, and NCI-MS data analysis (Table S13–S15). We used MS-FINDER 3.22 to annotate the metabolites on the basis of structure elucidation using the MS and MS/MS spectra of unknown peaks. For unknown HR-MS spectral structure elucidation, MS-FINDER (3.22 and higher) integrates structures and formulas for 224 622 known metabolites and now also includes 643 307 hypothetical compounds from the enzyme promiscuity database MINE-DB (http://minedatabase.mcs.anl.gov/).63 This in silico fragmentation tool considers multiple parameters such as bond dissociation energies, mass accuracies, fragment linkages, and various hydrogen rearrangement rules at the candidate ranking phase into a resulting scoring system range from 1 (worst candidate) to 10 (best candidate).64 We demonstrate that CI-MS generated MS1 and MS/MS spectra with retention time can provide additional orthogonal validation of metabolite annotation and quantification, using the same platform, without having to depend on LC-MS technology. Recently, CSI:FingerID was used for molecular structure search analysis of GC-MS/MS dopant-assisted atmospheric pressure chemical ionization (dAPCI) data;65 however, CSI:FingerID was not useful in predicting the formulas for silylated compounds for NCI-MS and PCI-MS. Similarly, another tool for in silico structure prediction, SIRIUS 4,66 was unable to interpret the PCI-MS/MS and NCI-MS spectral data for the silylated “known” compounds from the chemical standards. Another novel approach, the NIST hybrid search, to further annotate unknown compounds (at MSI level 3) in diverse matrices such as human urine46 and human milk67 is currently feasible for LC-ESI-MS data sets. However, for our silylated HRGC-MS data sets, the NIST hybrid search did not capture the silylated fragments from CI-MS. This clearly limits further in silico annotation of unknown CI-MS spectra using HRGC-MS efforts alone, beyond in-house mass spectral and retention time libraries.
Interestingly, we observed variabilities in peak areas (abundances) between metabolites quantified using different software packages. This could be caused by the intrinsic algorithms used for peak detection and integration and could be influenced by chromatographic alignment methods.68 Indeed, in another study raw HR LC-MS data from two types of biological samples (bile and urine), as well as a standard mixture of 84 metabolites, were processed with four peak-picking softwares: Peakview, Markerview, MetabolitePilot, and XCMS Online. Only a small proportion of all peaks (less than 10%) were common to all four software programs.69
Both ESI (coupled to LC) and CI (coupled to GC) are soft ionization techniques which enable quantification of a clear and characteristic molecular ion signal. There have been recommendations to use ESI-MS on negative mode based on an analysis of selected 33 standard compounds.70 In our analysis PCI is far more informative (based on number of identified metabolites and total signal) than the NCI for our complex matrix of NIST SRM 1950 human plasma. Moreover, a key challenge of metabolomics is high peak area (abundance) dynamic range (4–6 orders of magnitude) of metabolite concentrations present in biological samples where different chemical classes of metabolites often have distinct LC and GC affinities and ionization (mode) efficiencies on a given platform.
Overall, it is likely that untargeted metabolomics analyses in the future will be critically dependent in silico fragmentation tools such as MS-FINDER 3.22, CFM-ID, MetFrag, ChemDistiller, and CSI:FingerID that can annotate compounds from existing structure databases,45 as well as predicted retention time models from LC and the utility of collision cross-section (CCS) modeling from ion mobility experiments to improve HRMS annotations.46 For these analyses, more inclusive and synchronized workflows are needed that would only depend on freely available spectral and chemical databases to all users.
As we show in our study, a single sample (prepared only once) is amenable to three modes of runs/analysis, i.e., EI-MS, PCI-MS/MS, and NCI-MS, thereby providing additional IDs and quantification other than traditional GC-MS and standard LC-MS procedures (Figure 6). The high resolution spectra as well as molecular ions can help in annotation of novel compounds as well as aiding in annotation of reaction mechanisms (i.e., loss/addition of methyl group, oxidation, (de)protonation etc.) in the future applications as well.
Limitations of the Study
Despite the exciting findings reported above, our study suffers from several limitations. For comparison of NIST SRM 1950 human plasma GC-MS and LC-MS data sets, LC-MS data sets were publicly available from publications, for only four studies where data could be traced and data extracted. Further it needs to be appreciated that the LC-MS efforts in those studies adopted sample preparation protocols that are different from a common one adopted in our method for GC-MS analysis using three ionization modes. While there are several other publications using NIST SRM 1950 human plasma samples, they are all lipidomics studies, and given that GC-MS as a platform is not useful for detailed in depth lipidomics analysis, we refrained from including lipidomics data in the LC-MS data sets prior to the comparisons. Moreover, in our GC-MS data, the EI-MS signals were higher than the PCI and NCI signals for a comparable injection volume of sample. Future studies would have to explore how an increase in injection volume may ameliorate this reduced signal (sensitivity) in CI approaches. Finally, we understand these analyses need to be performed with much more complex tissue samples and larger numbers of clinically relevant samples to assess the robustness of our findings, and these are our ongoing efforts in the laboratory that will help determine the appropriate complementary MS-based approaches for complex matrix metabolomics
CONCLUSIONS
Clearly, each of the three MS platforms (GC-EI-MS, or GC-CI-MS, or LC-MS) captures a very different subset of metabolites from the same NIST SRM 1950 human plasma. Also, the different modes (positive and negative) of data acquisition identified different complementary subsets of metabolites. Our data demonstrate clearly that HRLC-MS and HRGC-MS are highly complementary technologies for annotation of metabolites from this reference sample. It remains a challenge, though, to consolidate “unknown” features detected in positive and negative modes, and now between CI and EI modes, to capture only true features. Clearly, improved mass spectral databases for the different ionization methodologies are essential to capture the full potential of HRMS approaches in metabolomics, and the generation of the custom Wake Forest CPM spectral libraries for EI- and CI-MS represent a first step toward this goal.
Supplementary Material
Funding
We acknowledge the National Institute on Aging, National Institutes of Health (NIH) (Award Number: U19AG057758).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Misra BB; Bassey E; Bishop AC; Kusel DT; Cox LA; Olivier M High Resolution GC-MS Metabolomics of Non-Human Primate Serum. Rapid Commun. Mass Spectrom 2018, 32, 1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fiehn O Metabolomics–the link between genotypes and phenotypes. Plant Mol. Biol 2002, 48 (1–2), 155–71. [PubMed] [Google Scholar]
- (3).Fiehn O Metabolomics by gas chromatography–mass spectrometry: Combined targeted and untargeted profiling. Curr. Protoc. Mol. Biol 2016, 114 (1), 30.4. 1–30.4. 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).de Hoffmann E; Stroobant V Mass Analyzers. In Mass Spectrometry: Principles Applications; Wiley, 2002; pp 85–174. [Google Scholar]
- (5).Kind T; Tsugawa H; Cajka T; Ma Y; Lai ZJ; Mehta SS; Wohlgemuth G; Barupal DK; Showalter MR; Arita M; Fiehn O Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev 2018, 37 (4), 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Peterson AC; Balloon AJ; Westphall MS; Coon JJ Development of a GC/Quadrupole-Orbitrap mass spectrometer, part II: new approaches for discovery metabolomics. Anal. Chem 2014, 86 (20), 10044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Peterson AC; Hauschild JP; Quarmby ST; Krumwiede D; Lange O; Lemke RAS; Grosse-Coosmann F; Horning S; Donohue TJ; Westphall MS; Coon JJ; Griep-Raming J Development of a GC/Quadrupole-Orbitrap Mass Spectrometer, Part I: Design and Characterization. Anal. Chem 2014, 86 (20), 10036–10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Peterson AC; McAlister GC; Quarmby ST; Griep-Raming J; Coon JJ Development and characterization of a GC-enabled QLT-Orbitrap for high-resolution and high-mass accuracy GC-MS. Anal. Chem 2010, 82 (20), 8618–28. [DOI] [PubMed] [Google Scholar]
- (9).Weidt S; Haggarty J; Kean R; Cojocariu CI; Silcock PJ; Rajendran R; Ramage G; Burgess KEV A novel targeted/untargeted GC-Orbitrap metabolomics methodology applied to Candida albicans and Staphylococcus aureus biofilms. Metabolomics 2016, DOI: 10.1007/s11306-016-1134-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schauer N; Steinhauser D; Strelkov S; Schomburg D; Allison G; Moritz T; Lundgren K; Roessner-Tunali U; Forbes MG; Willmitzer L; Fernie AR; Kopka J GC-MS libraries for the rapid identification of metabolites in complex biological samples. FEBS Lett 2005, 579 (6), 1332–7. [DOI] [PubMed] [Google Scholar]
- (11).Kind T; Wohlgemuth G; Lee DY; Lu Y; Palazoglu M; Shahbaz S; Fiehn O FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem 2009, 81 (24), 10038–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rochat B; Mohamed R; Sottas PE LC-HRMS Metabolomics for Untargeted Diagnostic Screening in Clinical Laboratories: A Feasibility Study. Metabolites 2018, 8 (2), 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cui L; Lu H; Lee YH Challenges and emergent solutions for LC-MS/MS based untargeted metabolomics in diseases. Mass Spectrom. Rev 2018, 37 (6), 772–792. [DOI] [PubMed] [Google Scholar]
- (14).Niu WH; Knight E; Xia QY; McGarvey BD Comparative evaluation of eight software programs for alignment of gas chromatography-mass spectrometry chromatograms in metabolomics experiments. Journal of Chromatography A 2014, 1374, 199–206. [DOI] [PubMed] [Google Scholar]
- (15).Tian H; Li BW; Shui GH Untargeted LC-MS Data Preprocessing in Metabolomics. Journal of Analysis and Testing 2017, 1 (3), 187–192. [Google Scholar]
- (16).Yuan M; Breitkopf SB; Yang XM; Asara JM A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc 2012, 7 (5), 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wishart DS; Lewis MJ; Morrissey JA; Flegel MD; Jeroncic K; Xiong YP; Cheng D; Eisner R; Gautam B; Tzur D; Sawhney S; Bamforth F; Greiner R; Li L The human cerebrospinal fluid metabolome. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2008, 871 (2), 164–173. [DOI] [PubMed] [Google Scholar]
- (18).Nordstrom A; Want E; Northen T; Lehtio J; Siuzdak G Multiple ionization mass spectrometry strategy used to reveal the complexity of metabolomics. Anal. Chem 2008, 80 (2), 421–429. [DOI] [PubMed] [Google Scholar]
- (19).Fitzgerald RL; Herold DA Serum total testosterone: Immunoassay compared with negative chemical ionization gas chromatography mass spectrometry. Clin. Chem 1996, 42 (5), 749–755. [PubMed] [Google Scholar]
- (20).Heyes MP; Markey SP Quantification of Quinolinic Acid in Rat-Brain, Whole-Blood, and Plasma by Gas-Chromatography and Negative Chemical Ionization Mass-Spectrometry - Effects of Systemic L-Tryptophan Administration on Brain and Blood Quinolinic Acid Concentrations. Anal. Biochem 1988, 174 (1), 349–359. [DOI] [PubMed] [Google Scholar]
- (21).Nakamura S; Sian TH; Daishima S Determination of estrogens in river water by gas chromatography-negative-ion chemical-ionization mass spectrometry. J. Chromatogr. A 2001, 919 (2), 275–282. [DOI] [PubMed] [Google Scholar]
- (22).Levery SB; Hakomori S Microscale Methylation Analysis of Glycolipids Using Capillary Gas-Chromatography Chemical Ionization Mass Fragmentography with Selected Ion Monitoring. Methods Enzymol 1987, 138, 13–25. [DOI] [PubMed] [Google Scholar]
- (23).Schweer H; Watzer B; Seyberth HW Determination of seven prostanoids in 1 mL of urine by gas chromatography-negative ion chemical ionization triple stage quadrupole mass spectrometry. J. Chromatogr., Biomed. Appl 1994, 652 (2), 221–7. [DOI] [PubMed] [Google Scholar]
- (24).Bowden JA; Heckert A; Ulmer CZ; Jones CM; Koelmel JP; Abdullah L; Ahonen L; Alnouti Y; Armando AM; Asara JM; Bamba T; Barr JR; Bergquist J; Borchers CH; Brandsma J; Breitkopf SB; Cajka T; Cazenave-Gassiot A; Checa A; Cinel MA; Colas RA; Cremers S; Dennis EA; Evans JE; Fauland A; Fiehn O; Gardner MS; Garrett TJ; Gotlinger KH; Han J; Huang YY; Neo AHP; Hyotylainen T; Izumi Y; Jiang HF; Jiang HL; Jiang J; Kachman M; Kiyonami R; Klavins K; Klose C; Kofeler HC; Kolmert J; Koal T; Koster G; Kuklenyik Z; Kurland IJ; Leadley M; Lin K; Maddipati KR; McDougall D; Meikle PJ; Mellett NA; Monnin C; Moseley MA; Nandakumar R; Oresic M; Patterson R; Peake D; Pierce JS; Post M; Postle AD; Pugh R; Qiu YP; Quehenberger O; Ramrup P; Rees J; Rembiesa B; Reynaud D; Roth MR; Sales S; Schuhmann K; Schwartzman ML; Serhan CN; Shevchenko A; Somerville SE; John-Williams LS; Surma MA; Takeda H; Thakare R; Thompson JW; Torta F; Triebl A; Trotzmuller M; Ubhayasekera SJK; Vuckovic D; Weir JM; Welti R; Wenk MR; Wheelock CE; Yao LB; Yuan M; Zhao XQH; Zhou SL Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res 2017, 58 (12), 2275–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fiehn O; Wohlgemuth G; Scholz M; Kind T; Lee DY; Lu Y; Moon S; Nikolau B Quality control for plant metabolomics: reporting MSI-compliant studies. Plant J 2008, 53 (4), 691–704. [DOI] [PubMed] [Google Scholar]
- (26).Misra BB; Olivier M Metabolite Extraction and Derivatization of Plasma/ Serum Samples for High Resolution GC-MS-based Metabolomics Protocols.io 2019, DOI: 10.17504/protocols.io.723hqgn. [DOI] [Google Scholar]
- (27).Lisec J; Schauer N; Kopka J; Willmitzer L; Fernie AR Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc 2006, 1 (1), 387–96. [DOI] [PubMed] [Google Scholar]
- (28).Misra BB; Upadhayay RP; Cox LA; Olivier M Optimized GC-MS metabolomics for the analysis of kidney tissue metabolites. Metabolomics 2018, DOI: 10.1007/s11306-018-1373-5. [DOI] [PubMed] [Google Scholar]
- (29).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3 (3), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kopka J; Schauer N; Krueger S; Birkemeyer C; Usadel B; Bergmuller E; Dormann P; Weckwerth W; Gibon Y; Stitt M; Willmitzer L; Fernie AR; Steinhauser D GMD@CSB.DB: the Golm Metabolome Database. Bioinformatics 2005, 21 (8), 1635–1638. [DOI] [PubMed] [Google Scholar]
- (31).Horai H; Arita M; Kanaya S; Nihei Y; Ikeda T; Suwa K; Ojima Y; Tanaka K; Tanaka S; Aoshima K; Oda Y; Kakazu Y; Kusano M; Tohge T; Matsuda F; Sawada Y; Hirai MY; Nakanishi H; Ikeda K; Akimoto N; Maoka T; Takahashi H; Ara T; Sakurai N; Suzuki H; Shibata D; Neumann S; Iida T; Tanaka K; Funatsu K; Matsuura F; Soga T; Taguchi R; Saito K; Nishioka T MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom 2010, 45 (7), 703–14. [DOI] [PubMed] [Google Scholar]
- (32).Mistrik R; Lutisan J; Huang Y; Suchy M; Wang J; Raab M In mzCloud: A Key Conceptual Shift to Understand ‘Who’s Who’ in Untargeted Metabolomics; Metabolomics Society: Glasgow, 2013; July 2013 Conference, pp 1–4. [Google Scholar]
- (33).Pence HE; Williams A ChemSpider: An Online Chemical Information Resource. J. Chem. Educ 2010, 87 (11), 1123–1124. [Google Scholar]
- (34).Adusumilli R; Mallick P Data Conversion with ProteoWizard msConvert. Methods Mol. Biol 2017, 1550, 339–368. [DOI] [PubMed] [Google Scholar]
- (35).Tsugawa H; Cajka T; Kind T; Ma Y; Higgins B; Ikeda K; Kanazawa M; VanderGheynst J; Fiehn O; Arita M MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12 (6), 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Misra BB Steps for Building an Open Source EI-MS Mass Spectral Library for GC-MS -based Metabolomics Protocols.io 2019, DOI: 10.17504/protocols.io.8txhwpn. [DOI] [Google Scholar]
- (37).Xia J; Sinelnikov IV; Han B; Wishart DS MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res 2015, 43 (W1), W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Raina R; Hall PJA c. i. Comparison of gas chromatography-mass spectrometry and gas chromatography-tandem mass spectrometry with electron ionization and negative-ion chemical ionization for analyses of pesticides at trace levels in atmospheric samples. Anal. Chem. Insights 2008, 3, ACI.S1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Benner BA; Schantz MM; Powers CD; Schleicher RL; Camara JE; Sharpless KE; Yen JH; Sniegoski LT Standard Reference Material (SRM) 2378 fatty acids in frozen human serum. Certification of a clinical SRM based on endogenous supplementation of polyunsaturated fatty acids. Anal. Bioanal. Chem 2018, 410 (9), 2321–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Ulmer CZ; Jones CM; Yost RA; Garrett TJ; Bowden JA Optimization of Folch, Bligh-Dyer, and Matyash sample-to-extraction solvent ratios for human plasma-based lipidomics studies. Anal. Chim. Acta 2018, 1037, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ulmer CZ; Ragland JM; Koelmel JP; Heckert A; Jones CM; Garrett TJ; Yost RA; Bowden JA LipidQC: Method Validation Tool for Visual Comparison to SRM 1950 Using NIST Interlaboratory Comparison Exercise Lipid Consensus Mean Estimate Values. Anal. Chem 2017, 89 (24), 13069–13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Brockbals L; Habicht M; Hajdas I; Galassi FM; Rühli FJ; Kraemer T Untargeted metabolomics-like screening approach for chemical characterization and differentiation of canopic jar and mummy samples from Ancient Egypt using GC-high resolution MS. Analyst 2018, 143 (18), 4503–4512. [DOI] [PubMed] [Google Scholar]
- (43).Chaleckis R; Meister I; Zhang P; Wheelock CE Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol 2019, 55, 44–50. [DOI] [PubMed] [Google Scholar]
- (44).Alden N; Krishnan S; Porokhin V; Raju R; McElearney K; Gilbert A; Lee K Biologically consistent annotation of metabolomics data. Anal. Chem 2017, 89 (24), 13097–13104. [DOI] [PubMed] [Google Scholar]
- (45).Blazenovic I; Kind T; Ji J; Fiehn O Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8 (2), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Blaženović I; Kind T; Sa MR; Ji J; Vaniya A; Wancewicz B; Roberts BS; Torbasinovic H; Lee T; Mehta SS; et al. Structure annotation of all mass spectra in untargeted metabolomics. Anal. Chem 2019, 91, 2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Tugizimana F; Steenkamp PA; Piater LA; Dubery IA Mass spectrometry in untargeted liquid chromatography/mass spectrometry metabolomics: Electrospray ionisation parameters and global coverage of the metabolome. Rapid Commun. Mass Spectrom 2018, 32 (2), 121–132. [DOI] [PubMed] [Google Scholar]
- (48).Newsome GA; Steinkamp FL; Giordano BC Isobutane made practical as a reagent gas for chemical ionization mass spectrometry. J. Am. Soc. Mass Spectrom 2016, 27 (11), 1789–1795. [DOI] [PubMed] [Google Scholar]
- (49).Lewy AJ; Markey SP Analysis of Melatonin in Human-Plasma by Gas-Chromatography Negative Chemical Ionization Mass-Spectrometry. Science 1978, 201 (4357), 741–743. [DOI] [PubMed] [Google Scholar]
- (50).Vallee M; Rivera JD; Koob GF; Purdy RH; Fitzgerald RL Quantification of neurosteroids in rat plasma and brain following swim stress and allopregnanolone administration using negative chemical ionization gas chromatography/mass spectrometry. Anal. Biochem 2000, 287 (1), 153–166. [DOI] [PubMed] [Google Scholar]
- (51).Gaut JP; Byun J; Tran HD; Heinecke JW Artifact-free quantification of free 3-chlorotyrosine, 3-bromotyrosine, and 3-nitrotyrosine in human plasma by electron capture-negative chemical ionization gas chromatography mass spectrometry and liquid chromatography-electrospray ionization tandem mass spectrometry (vol 300, pg 252, 2002). Anal. Biochem 2002, 304 (2), 275–275. [DOI] [PubMed] [Google Scholar]
- (52).de Almeida Azevedo D; Lacorte S; Vinhas T; Viana P; Barcelo D Monitoring of priority pesticides and other organic pollutants in river water from Portugal by gas chromatography-mass spectrometry and liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 2000, 879 (1), 13–26. [DOI] [PubMed] [Google Scholar]
- (53).Zhao JL; Ying GG; Wang L; Yang JF; Yang XB; Yang LH; Li X Determination of phenolic endocrine disrupting chemicals and acidic pharmaceuticals in surface water of the Pearl Rivers in South China by gas chromatography-negative chemical ionization-mass spectrometry. Sci. Total Environ 2009, 407 (2), 962–974. [DOI] [PubMed] [Google Scholar]
- (54).Martin JW; Muir DCG; Moody CA; Ellis DA; Kwan WC; Solomon KR; Mabury SA Collection of airborne fluorinated organics and analysis by gas chromatography/chemical ionization mass spectrometry. Anal. Chem 2002, 74 (3), 584–590. [DOI] [PubMed] [Google Scholar]
- (55).Harkey MR; Henderson GL; Zhou CH Simultaneous Quantitation of Cocaine and Its Major Metabolites in Human Hair by Gas-Chromatography Chemical Ionization Mass-Spectrometry. J. Anal. Toxicol 1991, 15 (5), 260–265. [DOI] [PubMed] [Google Scholar]
- (56).Kim EM; Lee JS; Choi SK; Lim MA; Chung HS Analysis of ketamine and norketamine in urine by automatic solid-phase extraction (SPE) and positive ion chemical ionization-gas chromatography-mass spectrometry (PCI-GC-MS). Forensic Sci. Int 2008, 174 (2–3), 197–202. [DOI] [PubMed] [Google Scholar]
- (57).Morris MJ; Gilbert JD; Hsieh JYK; Matuszewski BK; Ramjit HG; Bayne WF Determination of the Hmg-Coa Reductase Inhibitors Simvastatin, Lovastatin, and Pravastatin in Plasma by Gas-Chromatography Chemical Ionization Mass-Spectrometry. Biol. Mass Spectrom 1993, 22 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- (58).Liu XJ; Locasale JW Metabolomics: A Primer. Trends Biochem. Sci 2017, 42 (4), 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Li Z; Lu Y; Guo Y; Cao H; Wang Q; Shui W Comprehensive evaluation of untargeted metabolomics data processing software in feature detection, quantification and discriminating marker selection. Anal. Chim. Acta 2018, 1029, 50. [DOI] [PubMed] [Google Scholar]
- (60).Warren CR Use of chemical ionization for GC-MS metabolite profiling. Metabolomics 2013, 9 (1), S110–S120. [Google Scholar]
- (61).Vinaixa M; Schymanski EL; Neumann S; Navarro M; Salek RM; Yanes O Mass spectral databases for LC/MS-and GC-MS-based metabolomics: state of the field and future prospects. TrAC, Trends Anal. Chem 2016, 78, 23–35. [Google Scholar]
- (62).Munson MS; Field F-H Chemical ionization mass spectrometry. I. General introduction. J. Am. Chem. Soc 1966, 88 (12), 2621–2630. [Google Scholar]
- (63).Jeffryes JG; Colastani RL; Elbadawi-Sidhu M; Kind T; Niehaus TD; Broadbelt LJ; Hanson AD; Fiehn O; Tyo KE; Henry CS MINEs: open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminf 2015, 7 (1), 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Tsugawa H; Kind T; Nakabayashi R; Yukihira D; Tanaka W; Cajka T; Saito K; Fiehn O; Arita M Hydrogen rearrangement rules: computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem 2016, 88 (16), 7946–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Larson EA; Hutchinson CP; Lee YJ Gas chromatography-tandem mass spectrometry of lignin Pyrolyzates with dopant-assisted atmospheric pressure chemical ionization and molecular structure search with CSI: FingerID. J. Am. Soc. Mass Spectrom 2018, 29 (9), 1908–1918. [DOI] [PubMed] [Google Scholar]
- (66).hrkop K; Fleischauer M; Ludwig M; Aksenov AA; Melnik AV; Meusel M; Dorrestein PC; Rousu J; Böcker S SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16 (4), 299. [DOI] [PubMed] [Google Scholar]
- (67).Remoroza CA; Mak TD; De Leoz MLA; Mirokhin YA; Stein SE Creating a mass spectral reference library for oligosaccharides in human milk. Anal. Chem 2018, 90 (15), 8977–8988. [DOI] [PubMed] [Google Scholar]
- (68).Castillo S; Gopalacharyulu P; Yetukuri L; Oresic M Algorithms and tools for the preprocessing of LC-MS metabolomics data. Chemom. Intell. Lab. Syst 2011, 108 (1), 23–32. [Google Scholar]
- (69).Rafiei A; Sleno L Comparison of peak-picking workflows for untargeted liquid chromatography/high-resolution mass spectrometry metabolomics data analysis. Rapid Commun. Mass Spectrom 2015, 29 (1), 119–127. [DOI] [PubMed] [Google Scholar]
- (70).Liigand P; Kaupmees K; Haav K; Liigand J; Leito I; Girod M; Antoine R; Kruve A Think Negative: Finding the Best Electrospray Ionization/MS Mode for Your Analyte. Anal. Chem 2017, 89 (11), 5665–5669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.