

Abstract
Objective:
This paper is a proposal for an update of the Iron Hypothesis of Alzheimer’s disease (AD), based on large-scale emerging evidence.
Background:
Iron featured historically early in AD research efforts for its involvement in the amyloid and tau proteinopathies, APP processing, genetics, and one clinical trial, yet iron neurochemistry remains peripheral in mainstream AD research. Much of the effort investigating iron in AD has focused on the potential for iron to provoke the onset of disease, by promoting proteinopathy though increased protein expression, phosphorylation, and aggregation.
New/updated Hypothesis:
We provide new evidence from a large post-mortem cohort that brain iron levels within the normal range were associated with accelerated ante-mortem disease progression in cases with underlying proteinopathic neuropathology. These results corroborate recent findings that argue for an additional downstream role for iron as an effector of neurodegeneration, acting independently of tau or amyloid pathologies. We hypothesize that the level of tissue iron is a trait that dictates the probability of neurodegeneration in AD by ferroptosis, a regulated cell death pathway that is initiated by signals such as glutathione depletion and lipid peroxidation.
Major Challenges for the Hypothesis:
While clinical biomarkers of ferroptosis are still in discovery, the demonstration of additional ferroptotic correlates (genetic or biomarker derived) of disease progression is required to test this hypothesis. The genes implicated in familial AD are not known to influence ferroptosis, although recent reports on APP mutations and ApoE have shown impact on cellular iron retention. Familial AD mutations will need to be tested for their impact on ferroptotic vulnerability. Ultimately, this hypothesis will be substantiated, or otherwise, by a clinical trial of an anti-ferroptotic/iron compound in AD patients.
Linkage to other Major Theories:
Iron has historically been linked to the amyloid and tau proteinopathies of AD. Both tau, APP and ApoE have been implicated in physiological iron homeostasis in the brain. Iron is biochemically the origin of most chemical radicals generated in biochemistry and thus closely associated with the Oxidative Stress Theory of AD. Iron accumulation is also a well-established consequence of ageing and inflammation, which are major theories of disease pathogenesis.
1. Objective (brief paragraph to orient the read to the purpose).
This paper is a proposal for an update of the Iron Hypothesis of Alzheimer’s disease (AD), based on emerging evidence. Historically, the focus of iron research in AD has concentrated on the role of iron in promoting the hallmark pathology (plaque, neurofibrillary tangles; NFT) of AD, and while more recent findings do not invalidate this concept, it is becoming clear that iron effects downstream of proteinopathy. We seek input from other investigators in our effort to broaden the understanding of iron pathophysiology in AD, and how this relates to other recognized disease mechanisms. In reassessing the role of iron in AD, we seek to clarify therapeutic opportunities that target iron pathways, including when in the natural history of AD iron-based therapeutics may be beneficial.
2. Background
a. Historical evolution
Being first identified in 1953 [1], and subsequently in 1960 [2], iron deposition is one of the earliest reported chemical changes in the AD brain. Iron enrichment was observed in plaque, tangle-bearing neurons, as well as microglia. While both the importance of iron to brain health, as well as the potential for iron to cause tissue damage by oxidative chemistry (as the most abundant catalytic source of free radicals) became increasingly recognized in the ensuing decades, it was not until the 1990s that iron regained some attention in AD research. An early clinical trial of the iron chelator, desferrioxamine, reported slowing of AD progression in 1991, however this was never followed up [3]. While iron regulating proteins were shown to be altered in the AD brain [4, 5], which suggested iron dyshomeostasis, more attention was directed toward the potential for iron to be enriched in plaques and tangles [6, 7], as a source of oxidative radical damage emitting from the pathology [8–10] or as a chemical that cemented amyloid and tau protein deposition [11, 12]. Enrichment of iron in plaques and tangles has since been demonstrated by multiple groups using modern techniques [13–15]. The iron within plaque may be supplied by ferritin, which is a major iron binding protein that accumulates in the pathology [6, 16].
Iron as a priority in AD research was sidelined during the AD genetic revolution of the 1990s, which re-orientated research towards Aβ, and to a lesser extent tau, and APOE. Yet, polymorphism in transferrin, the main iron transporting protein, was one of the earliest (1993) genetic risk factors identified for AD [17]. It was later reported that there is an epistatic interaction between the hemochromatosis risk allele, H63D, and APOE ε4 in conferring AD risk [18, 19], with more recent reports showing elevation of iron biomarkers in ε4 carriers [20–22].
While the causal genetic mutations in APP and presenilin were co-opted into the Aβ Theory, early evidence implicated iron biochemistry in this pathway. In 1995 it was shown [23] (and later supported [24–28]) that iron regulates processing of APP, and an iron-responsive element in 5’-untranslated region of the APP transcript promotes the translation of APP in response to iron [29]. Iron regulation of APP expression was consistent with subsequent findings that implicated APP in iron homeostasis. In 2000 the Snyder laboratory reported that APP inhibits heme oxygenase, which liberates iron from heme [30]. It was later shown that APP acts to stabilize the iron exporting protein, ferroportin, to promote neuronal iron export [31–33], and that tau protein promotes surface trafficking of APP to ferroportin [34]. The ability of APP to support iron efflux is impacted by its processing, since it was recently shown that either genetic (Italian mutation) or pharmacological promotion of β-cleavage of APP causes iron retention in neurons, whereas the opposite response was observed with genetic (Icelandic mutation) or pharmacological promotion of APP α-cleavage [33].
The links between APP and iron biology offer an intriguing possibility for iron biochemical changes to feature in AD pathogenesis. Recent unbiased single cell transcriptomics and proteomics analyses have prominently implicated iron pathways in AD [35, 36]. While iron is reported as elevated in grey matter in AD, a meta-analysis revealed variability in iron levels between cortical areas, and across studies [37]. Within brain regions, iron has been shown to be redistributed in AD in a diffuse laminar pattern when different techniques have been used to measure iron in tissue sections [38–41]; the inhomogeneous deposition of iron may contribute to variance between regions/studies.
Accumulating data links iron burden as a trait to the rate of AD progression. Brain iron levels reflected by ferritin in CSF [20, 21, 42, 43], by Quantitative Susceptibility Mapping-MRI [44, 45], or directly measured post mortem [46], predict longitudinal cognitive deterioration and brain atrophy in people with underlying AD pathology. This correlation between iron and disease progression occurs even in the absence of bulk iron elevation; that is, relatively high biomarkers of iron, but within the normal range, were associated with risk of neurodegeneration when the underlying pathology of AD was present [20, 21, 42–44]. Taken together, the data indicate that brain iron burden is a trait associated with accelerated decline, even though iron values in cases overlap with healthy.
The association of iron with clinical progression rather than pathology deposition, are consistent with a role for iron downstream of proteinopathy accumulation in AD. Indeed, mediation analysis has revealed evidence that iron acts downstream of tangle pathology to influence neurodegeneration [47] and cognitive impairment [46]. The potential for iron, which can undergo redox cycling between Fe2+ and Fe3+, to cause tissue damage by oxidative chemistry has long been recognized as a potential neurotoxic mechanism. But the discovery of ferroptosis, an iron-dependent and non-apoptotic programmed cell-death pathway [48], provided an intriguing mechanism to consider as an explanation for the neurodegeneration in AD, which occurs after the appearance of the proteinopathy. Importantly, ferroptosis is not caused by poisonous elevation of iron to toxic levels, rather, iron becomes activated during ferroptosis to cause toxicity (e.g. liberated from ferritin). Ferroptosis may occur at all levels of iron, but cells with higher levels are more susceptible to initiating this form of regulate cell death. Iron therefore acts as moderator of susceptibility, so that cell death is more likely to occur once ferroptosis is triggered (e.g. by a decrease in glutathione). Ferroptosis would therefore be consistent with recent clinical findings linking iron with risk of decline, and offers a mechanistic explanation for neurodegeneration (synaptic and neuronal loss) in AD that has yet to be established for Aβ and tangles.
b. Rationale
While NFT and plaque are the defining pathological features of AD, the variability in the timing and extent of burden of these pathologies and their association with cognitive performance and neurodegeneration [49] imply that additional factors impact on functional deterioration. The mechanisms linking plaque to NFT pathology, and ultimately to neuronal toxicity remain inconclusive, and other neurochemical changes that occur in concert with these proteinopathies warrant interrogation. Iron has historically been implicated in AD proteinopathy, and while this remains of interest, the new understanding of ferroptosis has introduced iron as a potential downstream effector of neurodegeneration.
3. New or updated hypothesis
We propose an updated hypothesis for iron in AD – that iron-dependent ferroptosis is a relevant mechanism of neurodegeneration in AD. This updated hypothesis differs to prior hypotheses that iron acts upstream in AD to alter the processing of APP, or to accelerate the formation of plaque and NFTs. Our hypothesis does not oppose the former, rather complements this by asserting an additional toxic mechanism for iron, which may also result from pro-oxidant chemistry emanating from iron-enriched protein pathology.
This hypothesis would be supported if iron concentrations within the brain were associated with accelerated AD progression. Iron would need not be elevated in AD subjects, rather, people with relatively high iron (but within the normal range) would be expected to have poorer outcomes. Using a sub-set of cases (n=209) from the Memory and Aging Project (MAP; an ongoing clinical-neuropathological cohort study of older adults), we have previously reported that iron measured in post mortem inferior temporal cortex was elevated in people with AD pathology (that met CERAD or other neuropathological criteria) who had a clinical diagnosis of dementia; furthermore, temporal cortex iron levels were associated with more aggressive antemortem cognitive decline [46]. Here, we test these conclusions in a larger sample across more brain regions, by investigating iron in 645 post-mortem brains from the MAP cohort. We measured iron in three brain areas prominently affected in AD: anterior cingulate cortex, mid-frontal cortex, and inferior temporal cortex, as well as the comparatively spared cerebellar cortex, to test for the association between brain iron and pathological and antemortem clinical changes in AD.
a. Early experimental or observational data
Iron was measured using inductively coupled plasma mass spectrometry in post-mortem cortical tissue from inferior temporal, mid frontal, anterior cingulate, and cerebellum from 645 brains (see supplementary files for procedures). The values of iron in each region were similar to our findings in another cohort of control brains, with anterior cingulate containing approximately half the value of other cortical regions [50]. Iron levels in each region were modestly but significantly associated, within individuals, with iron levels in each other region (0.10<r<0.27).
Donors were classified clinically during life as having Alzheimer’s Dementia (using NINCDS-ADRDA criteria), and, at autopsy, classified pathologically using dichotomous stratifications of CERAD, Braak, or NIA-Reagan scales (low/high). CERAD criteria were used in the primary analysis, but the modelling was replicated using each of the criteria. The classification according to clinical and pathological criteria is important because individuals with AD pathology may not have a clinical diagnosis (e.g. prodromal disease); others with a clinical diagnosis may not have the pathology (i.e. clinical misdiagnosis or non-AD dementia).
The clinical and pathological stratifications resulted in assignment of 166 low pathology (CERAD) subjects without dementia, 40 low pathology subjects with dementia, 212 subjects with high pathology but without dementia, and 227 subjects with high pathology and dementia (demographics: Table 1). In subjects without dementia, iron levels were not associated with pathological status (neither the composite, nor each region assessed separately). In people classified low for AD pathology, the composite iron score did not differ with a clinical (non-AD) dementia diagnosis, but there was a small increase in the cerebellum (P=0.026) in this group. The composite iron score was elevated in people with pathology-confirmed clinical AD (multiple regression: P=0.002); a result mostly attributable to the inferior temporal cortex, P=2×10−4), since iron levels in other regions were not associated with clinical diagnosis (Figure 1). These findings were consistent when Braak and NIA/Reagan criteria of pathological diagnosis were applied (data not shown). Therefore, except for the inferior temporal cortex, iron was not elevated in other regions in subjects with a clinical diagnosis of AD, nor was there an association between pathological diagnosis and iron in any brain region.
Table 1.
Subject demographics
| Pathology (CERAD) | Negative | Negative | Positive | Positive |
| Clinical dementia Dx | No | Yes | No | Yes |
| N | 166 | 40 | 212 | 227 |
| Age at death (SD) | 88.1 (6.58) | 91.8 (5.97) | 90.4 (6.19) | 91.5 (5.62) |
| Female sex (%) | 110 (66.3%) | 25 (62.5%) | 151 (71.2%) | 164 (72.2%) |
| APOE ε4 +ve (%) | 16 (9.6%) | 7 (17.5%) | 50 (23.6%) | 88 (38.8%) |
| Education years (SD) | 15.0 (2.74) | 14.4 (2.86) | 14.5 (2.91) | 14.5 (2.98) |
| Presence of infarcts (%) | 46 (27.7%) | 21 (52.5%) | 79 (37.3%) | 97 (42.7%) |
| Presence of Lewy bodies (%) | 28 (16.9%) | 15 (37.5%) | 36 (17.0%) | 72 (31.7%) |
| Aβ plaques: mm2 (SD) | 0.87 (1.59) | 0.59 (1.62) | 6.62 (4.12) | 7.04 (3.92) |
| NFT: mm2 (SD) | 2.77 (2.68) | 3.71 (3.04) | 6.34 (5.87) | 13.3 (11.5) |
Figure 1. Iron levels in different brain regions of donors stratified by clinical and pathological (CERAD) diagnosis of AD.
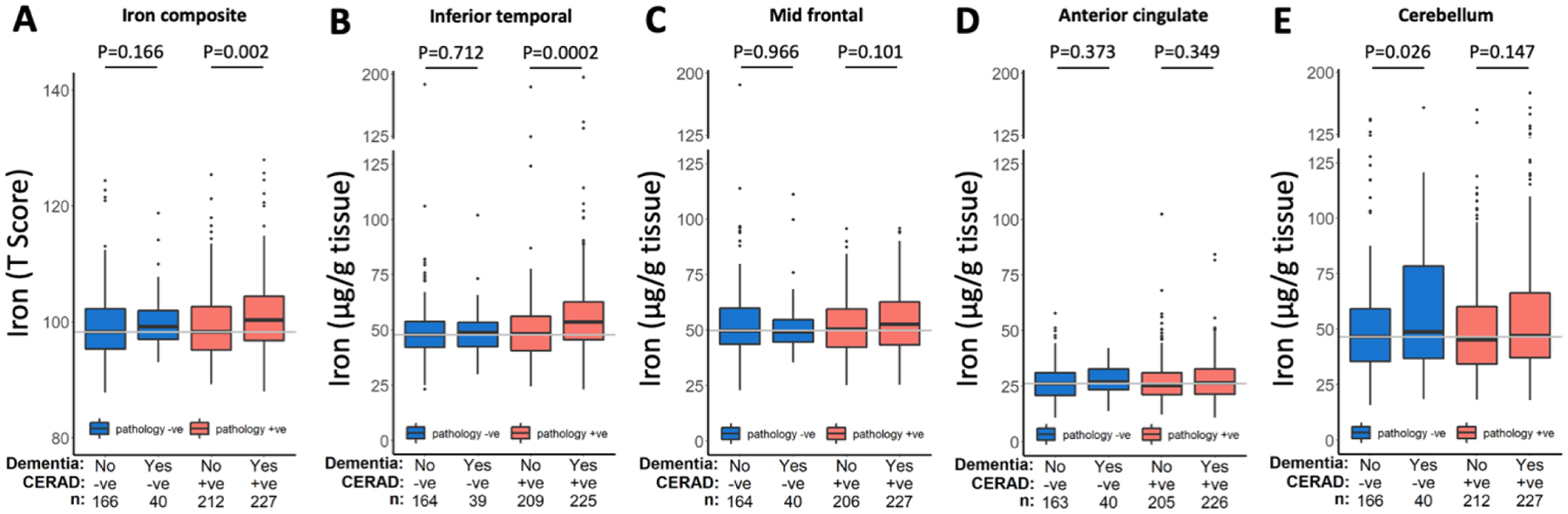
Statistics are from multiple regression models of iron in each region in strata of pathological diagnosis, and including the following covariates: age, sex, APOE ε4 and clinical diagnosis.
To explore further whether iron was associated with the plaque and NFT pathology of AD, we included the iron composite (primary analysis), and iron in each brain region (exploratory) in separate multiple regression models of NFT and plaque in people classified positive or negative for AD pathology by CERAD criteria. The iron composite was not associated with plaque or NFT burden (scores for the entire brain). Iron in the inferior temporal cortex was moderately associated with global NFT burden (p=0.008) but not global plaque burden, while iron levels in the other regions were not associated with global values for either pathology (Table 2). We replicated this analysis using regional measures of NFT and amyloid pathology that were available for mid-frontal and inferior temporal cortices. Similarly, iron in the inferior temporal cortex was associated with NFT burden in the inferior temporal cortex of donors with high CERAD (p=0.028), but not in those with low CERAD scores (Supplementary Table 1). Inferior temporal iron levels were not associated with amyloid burden of this region regardless of CERAD designation. Iron in the midfrontal was not associated with amyloid and NFT pathology in this region of either CERAD positive or negative subjects (Supplementary Table 1).
Table 2. Association of iron in different brain regions with global plaque and NFT counts in people who were positive or negative for AD pathology (CERAD).
Data are from multiple regressions of plaque and NFT included age, sex, APOE ε4 status, clinical dementia status, and in separate models, iron levels in each brain regions, and composite iron score of all regions. β-coefficients are the association between iron in each region and either plaque or NFT (as dependent variables) represented in standardized units (z-score).
| Pathology (CERAD) | Brain region | Plaque | NFT | ||||
|---|---|---|---|---|---|---|---|
| β | SE | P | β | SE | P | ||
| Positive | Composite | −0.072 | 0.059 | 0.220 | 0.092 | 0.072 | 0.208 |
| Inferior Temporal | −0.021 | 0.035 | 0.548 | 0.115 | 0.043 | 0.008 | |
| Mid Frontal | −0.051 | 0.038 | 0.177 | 0.028 | 0.047 | 0.794 | |
| Anterior Cingulate | 0.0063 | 0.853 | 0.853 | 0.019 | 0.042 | 0.656 | |
| Cerebellum | −0.049 | 0.036 | 0.180 | −0.039 | 0.045 | 0.381 | |
| Negative | Composite | 0.010 | 0.020 | 0.608 | −0.011 | 0.038 | 0.768 |
| Inferior Temporal | −4.0×10−4 | 0.013 | 0.977 | −0.022 | 0.024 | 0.376 | |
| Mid Frontal | 0.0029 | 0.011 | 0.549 | 0.026 | 0.021 | 0.211 | |
| Anterior Cingulate | 0.011 | 0.014 | 0.415 | −0.036 | 0.027 | 0.184 | |
| Cerebellum | 0.0013 | 0.012 | 0.913 | 4.7×10−4 | 0.022 | 0.984 | |
Bold values represent P<0.05.
We next investigated whether iron levels in brains with high AD pathology were associated with clinical deterioration in the decade prior to death, using mixed effects models of annual cognitive performance. A composite of memory tests for Global Cognition was used in our primary analysis, while composites for Episodic Memory, Perceptual Organization, Perceptual Speed, Semantic Memory, and Working Memory were investigated in exploratory analysis. We investigated change in cognition in donors who had positive AD pathology by CERAD criteria but did not have dementia during life, as well as donors who met both pathological and clinical criteria for a diagnosis of AD dementia before they died (all volunteers were cognitively normal when they entered the study). When inspecting the raw cognitive data, we noted that the cognitive trajectories were non-linear, so we included quadratic and linear terms for each variable, which resulted in an improved fit (determined by Akaike information criterion, Bayesian information criterion, and log likelihood [LL]) compared to when we only included linear terms.
In donors who were positive for AD pathology by CERAD criteria and suffered dementia, decline in Global Cognition was strongly and independently associated with the composite iron score (Fig 2A; Table 3) and NFT burden (Fig 2B), while plaque counts were not predictive (Fig 2C). The association between the iron composite and decline in Global Cognition was very similar when NIA/Reagan (Supplementary Fig 1B) and Braak (Supplementary Fig 1C) criteria of pathological diagnosis were applied. Iron in each brain region was predictive of decline in Global Cognition (Supplementary Fig 2; Table 3), regardless of whether iron was found to be elevated in these pathologically confirmed-AD subjects (i.e. inferior temporal). In exploring other cognitive domains, the iron composite was associated with cognitive decline in all but episodic memory and perceptual orientation (Table 3).
Figure 2. Association between brain iron and cognitive change in donors with CERAD-confirmed AD pathology, with and without clinical diagnosis of dementia.
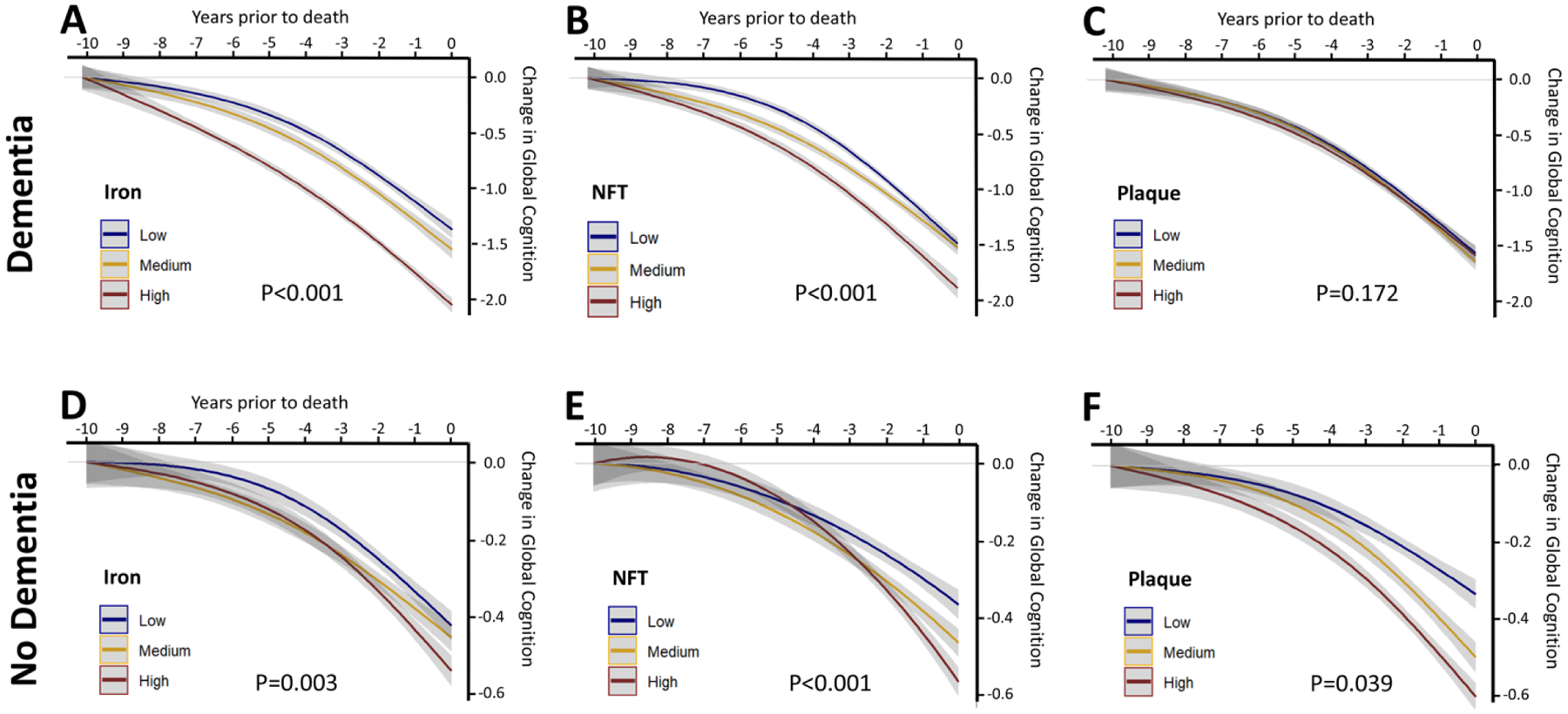
Association between linear and quadratic terms of (A,D) iron composite, (B,E) NFTs and (C,F) plaque deposition with change in the Global Cognitive composite in the 10 years prior to death of donors who had CERAD-confirmed AD pathology with (A-C) and without (D-F) a clinical diagnosis of dementia. Data was modelled using continuous variables, but these were represented in tertiles for visual display.
Table 3. Association between brain iron composite, plaque and NFTs, with different domains of cognition in subjects who had AD neuropathology with or without dementia at time of death.
Data were obtained from mixed-effects models of cognitive performance (e.g. Global Cognition) that included linear and quadratic terms for the following covariates, interacted with time: age, sex, APOE-ε4, years of education, plaque counts, tangle counts, presence of Lewy bodies, presence of gross infarcts and, in separate models, iron levels in the brain regions listed. Data are differences in model fit (change in log likelihood: ΔLL) and associated levels of significance determined by χ2 analysis between models with and without each individual variable (both linear and quadratic terms).
| Global Cognition | Working Memory | Semantic Memory | Perceptual Speed | Perceptual Organization | Episodic Memory | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dementia | Variable | ΔLL | χ2 | P | ΔLL | χ2 | P | ΔLL | χ2 | P | ΔLL | χ2 | P | ΔLL | χ2 | P | ΔLL | χ2 | P |
| Yes | Aβ plaques | −1.8 | 3.5 | 0.172 | −1.2 | 2.3 | 0.310 | −3.1 | 6.3 | 0.043 | 0.0 | 0.0 | 0.996 | −0.8 | 1.7 | 0.429 | −2.3 | 4.6 | 0.101 |
| NFT | −20.6 | 41.2 | <0.001 | −2.1 | 4.2 | 0.120 | −21.7 | 43.4 | <0.001 | −2.8 | 5.5 | 0.062 | −0.5 | 0.9 | 0.634 | −11.2 | 22.3 | <0.001 | |
| Iron composite | −9.3 | 18.6 | <0.001 | −5.1 | 10.2 | 0.006 | −3.2 | 6.5 | 0.039 | −13.1 | 26.1 | <0.001 | −2.4 | 4.9 | 0.087 | −0.7 | 1.3 | 0.518 | |
| Inferior temporal iron | −4.4 | 8.9 | 0.012 | −3.3 | 6.5 | 0.039 | −3.3 | 6.7 | 0.035 | −9.9 | 19.8 | <0.001 | −0.1 | 0.3 | 0.866 | −0.1 | 0.1 | 0.945 | |
| Mid Frontal iron | −4.9 | 9.9 | 0.007 | −2.9 | 5.8 | 0.055 | −2.2 | 4.4 | 0.111 | −4.3 | 8.5 | 0.014 | −0.9 | 1.7 | 0.417 | −0.9 | 1.9 | 0.388 | |
| Anterior cingulate iron | −5.6 | 11.2 | 0.004 | −1.7 | 3.4 | 0.181 | −4.7 | 9.4 | 0.009 | −8.7 | 17.4 | <0.001 | −1.1 | 2.1 | 0.348 | −1.9 | 3.8 | 0.149 | |
| Cerebellum iron | −7.1 | 14.1 | 0.001 | −5.0 | 10.0 | 0.007 | −2.9 | 5.8 | 0.054 | −12.2 | 24.4 | <0.001 | −2.9 | 5.8 | 0.056 | −3.3 | 6.7 | 0.036 | |
| No | Aβ plaques | −3.2 | 6.5 | 0.039 | −2.5 | 4.9 | 0.084 | −0.5 | 1.1 | 0.591 | −3.9 | 7.8 | 0.020 | −3.1 | 6.1 | 0.047 | −1.2 | 2.3 | 0.312 |
| NFT | −17.0 | 34.0 | <0.001 | −3.5 | 7.0 | 0.030 | −11.7 | 23.4 | <0.001 | −9.1 | 18.2 | <0.001 | −4.3 | 8.7 | 0.013 | −23.6 | 47.3 | <0.001 | |
| Iron composite | −5.9 | 11.9 | 0.003 | −5.2 | 10.3 | 0.006 | −2.3 | 4.7 | 0.097 | −0.8 | 1.6 | 0.448 | −2.1 | 4.2 | 0.122 | −3.6 | 7.1 | 0.029 | |
| Inferior temporal iron | −3.4 | 6.8 | 0.033 | −2.3 | 4.7 | 0.098 | −3.3 | 6.6 | 0.037 | −0.3 | 0.5 | 0.775 | −0.1 | 0.3 | 0.863 | −2.6 | 5.2 | 0.075 | |
| Mid Frontal iron | −12.9 | 25.9 | <0.001 | −5.6 | 11.2 | 0.004 | −7.7 | 15.5 | <0.001 | −1.4 | 2.8 | 0.244 | −2.5 | 5.0 | 0.081 | −7.6 | 15.3 | <0.001 | |
| Anterior cingulate iron | −0.8 | 1.6 | 0.459 | −1.5 | 3.1 | 0.213 | −0.6 | 1.1 | 0.565 | −0.4 | 0.9 | 0.648 | −0.4 | 0.9 | 0.647 | −0.1 | 0.2 | 0.899 | |
| Cerebellum iron | −0.8 | 1.7 | 0.435 | −1.5 | 3.0 | 0.223 | −0.4 | 0.8 | 0.674 | −0.5 | 0.9 | 0.636 | −0.9 | 1.7 | 0.421 | −1.3 | 2.6 | 0.274 | |
Bold values represent P<0.05.
In CERAD-positive donors without dementia, the iron composite was associated with cognitive decline in global cognition (Figure 2D; Table 3), and working memory and episodic memory. This result was mostly attributable to iron in the inferior temporal and mid frontal cortices, since exploratory analysis revealed significant associations between iron in these regions and decline on these scales (Table 3), despite these CERAD-positive non-demented cases having no change in iron levels in any region when compared to CERAD negative cases. Notably, in these CERAD-positive non-demented cases plaque and NFT burden was, like iron burden, also significantly associated with antecedent cognitive decline (Figure 2E,F; Table 3).
This large post-mortem cohort ratifies an elevation of iron in the temporal lobe in persons with dementia and AD pathology, but not other regions investigated (mid frontal, anterior cingulate, cerebellum). This pattern is similar to a recent report investigating iron by quantitative susceptibility mapping MRI [47]. Iron was not elevated merely by the presence of AD-pathology (i.e. AD-pathology positive, clinically silent cases), nor in people with dementia without AD-pathology (i.e. non-AD dementia).
This large dataset corroborates that, while brain regions outside of the inferior temporal lobe were not elevated in iron, the iron burden throughout the brain was still strongly associated with decline. Iron burden influencing disease progression even when not pathologically elevated is consistent with the mechanism of ferroptotic neurodegeneration (Fig 3). Ferroptotic cell death does not require or induce elevated total iron levels, rather, cells with greater iron levels have increased susceptibility to undergo ferroptosis [48], which is consistent with our findings. If iron were merely increased with pathological or clinical diagnosis and not associated with cognitive trajectory, then the change in iron may be an epiphenomenon reflecting other primary disease mechanisms (e.g. inflammation). The association of iron burden with rate of deterioration is unlikely to be attributable to canonical AD pathology since iron itself was not elevated in most brain regions studied. While still correlative, our results are more consistent with iron acting as a risk factor for disease progression, while also being increased in some brain regions causally or epiphenomenologically.
Figure 3. Current and Proposed Iron hypothesis of AD (simplified).
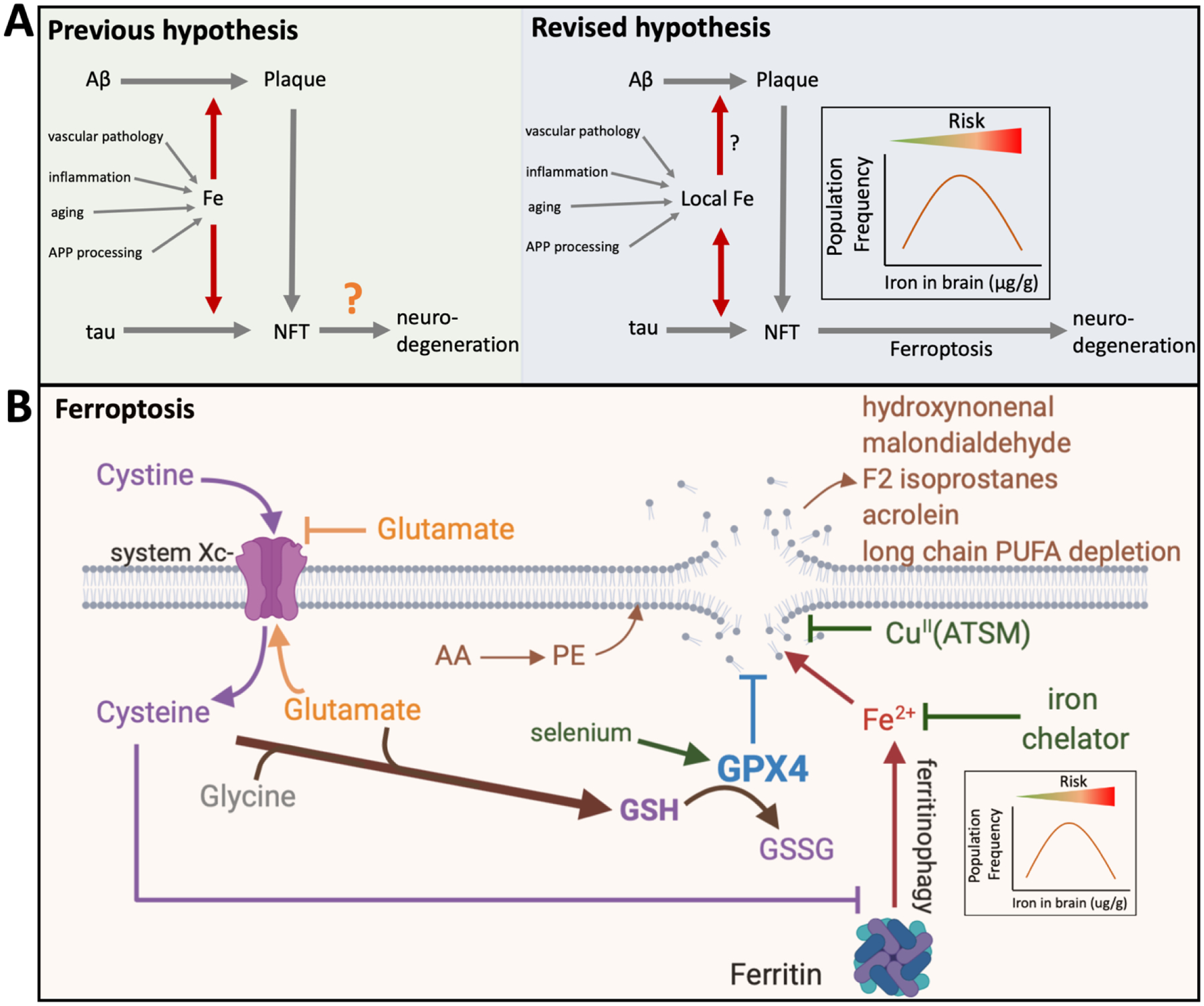
(A)The previous hypothesis was that iron promotes plaque and tangle formation by promoting APP and tau production as well as aggregation of Aβ and phospho-tau. Brain iron levels may be impacted by aging, changes in APP processing, inflammation and vascular damage. Iron trapped within these protein pathologies augmented toxicity through generating reactive oxygen species. Our current findings from a large longitudinal cohort confirmed that tissue iron burden may impact on the formation of NFTs, at least in the inferior temporal cortex. Our current investigation of bulk tissue levels of iron does not uphold a relationship between bulk tissue iron burden and amyloid formation. However, this study was unable to test the impact of iron in microvicinities, where previous findings indicate that iron is markedly enriched in plaques and tangles. Loss of tau function can, in its own right, cause neuronal iron accumulation. The new data lead us to further update the hypothesis, where iron has an additional role downstream of proteinopathy, by influencing the susceptibility of neurons to die (or cause synaptic damage) due to ferroptosis. Abnormal iron elevation does not itself cause ferroptosis, rather, the burden of iron in the tissue renders neurons more susceptible to ferroptotic stress. Therefore, people with relatively more iron, but still within the normal range, are likely to deteriorate faster during disease. (B) Ferroptosis pathways. Glutathione, which is depleted in AD brain tissue [51, 60, 68], prevents ferroptosis by providing substrate for the ferroptosis checkpoint selenoenzyme, GPX4. GPX4 detoxifies lipid hydroperoxides that are formed by cytoplasmic iron reacting with membrane PUFAs, otherwise cell rupture ensues. Cystine enters neurons via the system Xc− antiporter in exchange for glutamate that is exported. Cystine is reduced to cysteine within the cell, which is the rate limiting amino acid for glutathione synthesis. Low cysteine levels promote ferroptosis by both depleting glutathione, and by promoting ferritin degradation that releases cytoplasmic iron to fuel the peroxidation reaction [69]. The more iron in the cell, the greater the vulnerability toward ferroptosis. Lipid peroxides in ferroptosis lead to downstream products such as hydroxynonenal, malondialdehyde, F2-isoprostanes, acrolein, with subsequent depletion of long-chain PUFA depletion- all changes that are reported for AD brain tissue [70–76]. The sites of putative therapeutics are noted.
The reason for iron elevation in the inferior temporal cortex and not the other brain regions is unknown. Iron elevation can be a consequence of vascular damage [50], inflammation [51], or change in APP processing [33], as discussed below. One clue is that this region has severe NFT pathology. Low soluble tau levels as a consequence of deposition within NFT could lead to neuronal iron retention [34].
b. Future experiments and validation studies
While it is clear that substantial neuronal loss occurs in AD [49], determining the cause of this cell death is challenging. There are currently no definitive reporters of ferroptosis, but even if they did exist their assessment would be confounded by agonal changes. Certain lipid peroxidation products may be markers for ferroptosis and their assessment at a similar scale in post-mortem tissue awaits. However, like any programmed cell death pathway, it may be difficult to pick up specific signatures of the mechanism of death in such an indolent disease where most of the dead cells met their demise years earlier. Nonetheless, the association of iron levels, or other constituent risk/protective ferroptotic factors (glutathione, selenium, mono/poly unsaturated fatty acids), with disease outcomes provide evidence for ferroptosis.
An in vivo biomarker of ferroptotic stress (e.g. elevation of a specific lipid peroxidation marker) could provide more compelling evidence of ferroptosis in AD. While no such biomarker currently exists, fluid and imaging biomarkers of iron have been shown predict disease progression [20, 21, 42, 44, 45], and glutathione, a major anti-ferroptotic molecule, is reported to be decreased in AD [51].
Yet, all such biomarker studies are correlative, and even if ferroptotic biomarkers demonstrate elevation in disease, it would remain unknown if this elevation is cause or effect of another more damaging disease process. Our findings relating iron burden to cognitive decline, while correlative, are still consistent with a damaging role for iron in AD since it is unlikely that disease pathology induces a change in iron because the levels of iron were mostly unchanged. Rather, it is more likely that iron acts as a risk factor of decline. By analogy, ambient temperature does not cause bushfires, but temperature has a major bearing on the severity of the fire.
More definitive evidence of ferroptosis as a pathogenic mechanism in AD would be obtained by a clinical trial of an anti-ferroptotic drug that slowed or stopped disease progression. There are currently no specific anti-ferroptotic compounds that are clinically available, however iron chelators that are in current clinical use are anti-ferroptotic, albeit with low potency. An early clinical trial of the iron chelator, desferrioxamine, showed some promise of slowing disease progression [3], and a phase II AD clinical trial of the orally available and BBB penetrant iron chelator, deferiprone, is currently underway (NCT03234686). When used at a lower dose than for treating iron overload in thalassemia (30mg/kg/day vs ~100mg/kg/day), deferiprone has been found in clinical trials of several neurological disorders not to deplete systemic iron, but nonetheless to lower biomarkers of brain iron burden (e.g. [52]). Neutropenia is an infrequent (~3%) adverse effect, not related to iron chelation, that is managed by temporarily suspending treatment. Vitamin E, which is a weak ferroptosis inhibitor, also has reported benefits in phase 3 testing [53]. Much more specific and potent anti-ferroptotic agents are currently in development and may provide proof of concept. CuII(ATSM) is a BBB penetrant orally available anti-ferroptotic agent that has reported promising outcomes in early studies of Parkinson’s disease and amyotrophic lateral sclerosis (ALS), where ferroptosis is also implicated [54]. It is currently undergoing proof of concept phase 2 testing for ALS (NCT04082832) and may warrant testing in AD.
In the case where a drug targeting the ferroptosis pathway slowed AD progression, this would add considerable weight to the hypothesis we outline here. However, any one drug may have multiple mechanisms of action, and it is possible that the putative anti-ferroptotic compound exerts its effect by another pathway. Therefore, testing the hypothesis would require further demonstration of multiple drugs targeting different steps in the ferroptosis pathway each conferring benefit in clinical trials with concomitant improvements in ferroptosis biomarkers.
It is also of interest to know whether brain iron has a specific role in AD, or whether it risks accelerated disease progression across the dementias. We did not pursue the non-AD dementia (AD-pathology negative) group to the same extent because this group was comparatively small, consisted of subjects with varying types of dementia with variable trajectories of cognitive decline, and with different brain regions implicated. While less explored, tissue iron burden is also implicated in dementia with Lewy bodies/Parkinson’s disease dementia [55], frontotemporal dementia [56] and dementia with vascular pathology [57], and its role in these contexts also warrants further investigation.
4. Major challenges for the hypothesis
In this large study of post-mortem tissue, we did not find iron elevation at the bulk tissue level in most brain areas studied, except for inferior temporal cortex. Also, we found that the bulk iron levels in the regions were not related to plaque burden and were related to NFT burden only, again, in the inferior temporal cortex. While we find strong evidence that bulk tissue iron levels propel cognitive deterioration, how this fits in with the canonical proteinopathy is not clear. Also, the iron burden clearly does not need to be markedly elevated on the bulk level (like hemochromatosis) to cause organ failure. So, a mechanism for how iron burden might induce accelerate cognitive deterioration is speculative, but, in all fairness, the mechanism for how amyloid or NFTs might cause neurodegeneration is no more certain.
Critics of the iron hypothesis of AD have pointed to the lack of association between canonical iron-related genes in the genetics of AD. But iron-loading diseases such as β-thalassemia and hemochromatosis affect peripheral organs, and there is little communication between peripheral and brain iron levels (e.g. [20]). Patients with uncontrolled iron levels will exhibit damage to peripheral organs resulting in disease before any consequence to the brain, and patients who have their iron levels therapeutically controlled will not risk brain iron elevation. The neurodegenerative class of diseases termed Neurodegeneration with Brain Iron Accumulation (NBIA) demonstrate the potential for brain iron loading (by various genetic lesions) to be sufficient cause of dementia.
Do iron-related genes feature in the genetic architecture of AD? While the genetic links in iron-related genes such as transferrin [17] and HFE [18, 19] have been reported, it is clear that their effect is comparatively minor. Yet such a view may ignore the possibility that canonical AD genes, such as APP, could be cryptic regulators of brain iron metabolism. Indeed, APP has been shown to have important functions relating to heme metabolism [30] and neuron iron export [31–34], which may directly link iron in AD genetics. This argument is strengthened by findings that iron promotes the expression [29] and α-cleavage [23] of APP, such that iron is both regulated by, and regulates APP biology.
The hypothesis that we put forward here actually does not require genetic evidence implicating iron in AD. The hypothesis that ferroptotic stress underlies neurodegeneration in AD does not imply that iron or ferroptosis is the upstream cause of the disease, rather it is a downstream mechanism of toxicity. It is reasonable to expect true causes of the disease to feature in AD genetics, but if iron/ferroptosis exerts its influence as a consequence of other factors (e.g. Aβ, tau), then iron/ferroptotic genes need not be genetic causes of the disease. Instead, we may expect that genetic variation in iron/ferroptotic genes would influence the risk for cognitive decline, which has been much less explored.
If indeed ferroptosis is a mechanism of neurodegeneration in AD, the biochemical trigger for this in AD needs to be determined. There are now many known mechanisms for inducing ferroptosis, with the original trigger of glutathione depletion being best described [48]. Low glutathione is observed in AD brain tissue [51], and GWAS reveals that variants in glutathione homeostatic genes predict cognitive decline upon underlying AD pathology [58]. When glutathione levels fall, the iron levels of the cell dictate the susceptibility to ferroptosis [59], consistent with our current findings. But what causes glutathione depletion in AD? – could this involve plaque or tangles? The molecular events linking plaques to tangles, and tangles to neurodegeneration remain undescribed. This is a major challenge for the entire AD field. Ferroptotic pathways, such as glutathione depletion, present possible explanations. While the mechanisms linking pathology to glutathione remain to be elucidated, low glutathione has been shown to correlate with increased plaque pathology in cognitively normal subjects [60].
5. Linkage to other major theories
Protein aggregation
A wealth of prior research has demonstrated that iron accelerates the aggregation of Aβ and tau proteins, and that iron is enriched in plaque and NFT pathology [6–15]. The influence of iron on promoting AD pathology may not be surprising given that protein aggregation is a generalized response to iron stress [61]. However, in our cohort, we did not observe bulk iron enrichment in any brain region from subjects with AD pathology unless they were demented. In the inferior temporal cortex, iron was associated with NFT counts but not plaque pathology in cases that met CERAD criteria, in agreement with prior ex vivo and in vivo findings [46, 47]. While the association between pathology and iron we observed was not as prominent as with other reports, iron changes at the histopathological level are likely to be more prominent and may have been missed on the bulk tissue scale. Indeed, when investigated at the microscopic scale using low throughput/high spatial resolution techniques, iron elevation has been observed with laminar distribution, and associated with plaque pathology [38–41]. We did not intend to resolve this association with our bulk analysis techniques, so we cannot determine whether iron, acting in the local microenvironment, impacts on plaque or NFT pathology. However, if this is the case, it is unlikely that the iron burden of the tissue would explain the pathology, based on our current data, except for tangles in the inferior temporal cortex.
APP metabolism
That familial AD mutations in APP promote β-secretase cleavage (required for Aβ production) underscores the importance of APP processing in AD pathogenesis. The impact of iron on APP expression [29] may make sense given the function of APP to regulate heme metabolism [30] and iron export via ferroportin [31–34]. Recently, either genetic or pharmacologically induced β-secretase cleavage of APP prevented APP stabilization of ferroportin, causing neuronal iron retention. In contrast, inducing α-secretase cleavage promoted surface expression of ferroportin and iron release [33]. These findings introduce the possibility that FAD causative or protective mutations of APP might act by changing iron retention in neurons, with testable ramifications for ferroptosis.
Aging
Aging is the greatest risk factor for AD by an uncertain neurochemical mechanism. It has been repeatedly demonstrated that brain iron levels rise with age in rodents, primates and humans [62], which may prime the tissue for disease. Indeed, in a multivariate genomic scan reported that APOE, heme-regulation and plasma iron levels were the strongest factors associated with human healthspan and lifespan [63]. It is therefore not surprising that iron also could impact on AD as a major complication of aging.
Inflammation
Inflammatory changes, including microglial activation in AD, are associated with cellular iron retention [64]. This is part of the innate immune response so that extracellular pathogens are deprived of iron for their growth. Conversely, iron elevation may promote a pro-inflammatory state. Production of the pro-inflammatory cytokine, IL-1β, which is induced in microglia by the NALP3-inflammasome, is enhanced by elevated iron [65] or heme [66]. Microglial cells with higher iron produce more IL-1β in response to Aβ [67]. So, brain tissue with more iron might have a lower threshold for inflammation-related damage, which may explain why higher iron levels in brain tissue are associated with faster disease progression.
6. Concluding remarks
Iron accumulation was one of the earliest chemical changes identified in AD [1, 2]. Iron featured historically early in the research efforts of protein pathology characterization [1, 2, 6, 7], APP processing [23], genetics [17], and a clinical trial [3], yet iron remains of peripheral interest to AD research compared to proteinopathy. Much of the effort investigating iron in AD has focused on the potential for iron to be a casual factor in the onset of disease, by promoting pathology deposition though increased protein expression, phosphorylation, and aggregation. These pathways warrant serious consideration and investigation. However, the observation that iron levels within the normal range risk accelerated disease progression when the proteinopathy is already present argue for a downstream role for iron as an effector of neurodegeneration. This has implications for the understanding of iron in AD, understanding of neurodegenerative mechanisms in AD, and, importantly, therapeutic interventions. That iron is temporally associated with neurodegeneration and cognitive impairment may allow for the possibility to target iron/ferroptosis at this symptomatic stage to alter the disease course.
Supplementary Material
Research in context.
Systematic Review:
We reviewed the evidence that links brain iron levels to the pathologies and clinical outcomes of Alzheimer’s disease (AD), including a large new set of post-mortem values. While iron has been linked to the proteinopathy of AD for many years, we find evidence for iron acting downstream of pathology, associated with neurodegeneration.
Interpretation:
In addition to these recent findings, we provide new data from 645 post-mortem brains with antecedent cognitive data where iron was measured in 4 regions. We report that iron was modestly associated with clinical diagnosis and neuropathology, but strikingly associated with the rate of cognitive decline in the decade prior to death.
Future Directions:
Mechanisms underlying the role of iron as an effector of damage in AD, for example by ferroptosis, or enhancing neuroinflammatory processes, warrant further investigation for therapeutic discovery. The role of iron as correlative or causative may be determined by a clinical trial of an anti-ferroptotic drug candidate or blood-brain barrier permeable iron chelator.
Acknowledgments
This study was supported by grants from the National Institute of Health (R01AG017917, R21E2021290, and RF1AG054057). Analysis was supported by funds from the Australian Research Council, the Australian National Health & Medical Research Council (NHMRC), the Cooperative Research Centre for Mental Health (the Cooperative Research Centre program is an Australian Government Initiative) and the Gandioli-Fumagali Foundation. The Florey Institute of Neuroscience and Mental Health acknowledges support from the Victorian Government, in particular, funding from the Operational Infrastructure Support Grant. No funder of this study had any role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Abbreviations
- AD
Alzheimer’s disease
- CERAD
Consortium to Establish a Registry for Alzheimer Disease
- NFT
Neurofibrillary Tangles
- MAP
Memory and Aging Project
- NINCDS-ADRDA
National Institute of Neurologic and Communicative Disorders and Stroke and the AD and Related Disorders Association
Footnotes
Conflict of interest statement
AIB is a shareholder in Prana Biotechnology Ltd, Cogstate Ltd, Brighton Biotech LLC, Grunbiotics Pty Ltd, Eucalyptus Pty Ltd, and Mesoblast Ltd. He is a paid consultant for, and has a profit share interest in, Collaborative Medicinal Development Pty Ltd.
References
- [1].Goodman L. Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. The Journal of nervous and mental disease. 1953;118:97–130. [PubMed] [Google Scholar]
- [2].Hallgren B, Sourander P. The non-haemin iron in the cerebral cortex in Alzheimer’s disease. J Neurochem. 1960;5:307–10. [DOI] [PubMed] [Google Scholar]
- [3].Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337:1304–8. [DOI] [PubMed] [Google Scholar]
- [4].Connor JR, Menzies SL, St Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. Journal of neuroscience research. 1992;31:75–83. [DOI] [PubMed] [Google Scholar]
- [5].Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ. Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimer’s disease. Journal of neuroscience research. 1992;31:327–35. [DOI] [PubMed] [Google Scholar]
- [6].Grundke-Iqbal I, Fleming J, Tung YC, Lassmann H, Iqbal K, Joshi JG. Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta neuropathologica. 1990;81:105–10. [DOI] [PubMed] [Google Scholar]
- [7].Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:9866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J Neurochem. 2000;74:270–9. [DOI] [PubMed] [Google Scholar]
- [9].Schubert D, Chevion M. The role of iron in beta amyloid toxicity. Biochemical and biophysical research communications. 1995;216:702–7. [DOI] [PubMed] [Google Scholar]
- [10].Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–16. [DOI] [PubMed] [Google Scholar]
- [11].Yamamoto A, Shin R-W, Hasegawa K, Naiki H, Sato H, Yoshimasu F, et al. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J Neurochem. 2002;82:1137–47. [DOI] [PubMed] [Google Scholar]
- [12].Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, et al. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem. 1993;61:1171–4. [DOI] [PubMed] [Google Scholar]
- [13].Gong NJ, Dibb R, Bulk M, van der Weerd L, Liu C. Imaging beta amyloid aggregation and iron accumulation in Alzheimer’s disease using quantitative susceptibility mapping MRI. NeuroImage. 2019. [DOI] [PubMed] [Google Scholar]
- [14].Everett J, Collingwood JF, Tjendana-Tjhin V, Brooks J, Lermyte F, Plascencia-Villa G, et al. Nanoscale synchrotron X-ray speciation of iron and calcium compounds in amyloid plaque cores from Alzheimer’s disease subjects. Nanoscale. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Telling ND, Everett J, Collingwood JF, Dobson J, van der Laan G, Gallagher JJ, et al. Iron Biochemistry is Correlated with Amyloid Plaque Morphology in an Established Mouse Model of Alzheimer’s Disease. Cell chemical biology. 2017: 10.1016/j.chembiol.2017.07.014. [DOI] [PubMed] [Google Scholar]
- [16].Raha AA, Vaishnav RA, Friedland RP, Bomford A, Raha-Chowdhury R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta neuropathologica communications. 2013;1:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van Rensburg SJ, Carstens ME, Potocnik FC, Aucamp AK, Taljaard JJ. Increased frequency of the transferrin C2 subtype in Alzheimer’s disease. Neuroreport. 1993;4:1269–71. [DOI] [PubMed] [Google Scholar]
- [18].Combarros O, Garcia-Roman M, Fontalba A, Fernandez-Luna JL, Llorca J, Infante J, et al. Interaction of the H63D mutation in the hemochromatosis gene with the apolipoprotein E epsilon 4 allele modulates age at onset of Alzheimer’s disease. Dementia and geriatric cognitive disorders. 2003;15:151–4. [DOI] [PubMed] [Google Scholar]
- [19].Percy M, Moalem S, Garcia A, Somerville MJ, Hicks M, Andrews D, et al. Involvement of ApoE E4 and H63D in sporadic Alzheimer’s disease in a folate-supplemented Ontario population. Journal of Alzheimer’s disease : JAD. 2008;14:69–84. [DOI] [PubMed] [Google Scholar]
- [20].Ayton S, Faux NG, Bush AI, Alzheimer’s Disease Neuroimaging I. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nature communications. 2015;6:6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ayton S, Faux NG, Bush AI. Association of Cerebrospinal Fluid Ferritin Level With Preclinical Cognitive Decline in APOE-ε4 Carriers. JAMA neurology. 2017;74:122–5. [DOI] [PubMed] [Google Scholar]
- [22].van Bergen JM, Li X, Hua J, Schreiner SJ, Steininger SC, Quevenco FC, et al. Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci Rep. 2016;6:35514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bodovitz S, Falduto MT, Frail DE, Klein WL. Iron levels modulate alpha-secretase cleavage of amyloid precursor protein. J Neurochem. 1995;64:307–15. [DOI] [PubMed] [Google Scholar]
- [24].Chen YT, Chen WY, Huang XT, Xu YC, Zhang HY. Iron dysregulates APP processing accompanying with sAPPalpha cellular retention and beta-secretase inhibition in rat cortical neurons. Acta pharmacologica Sinica. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Guo LY, Alekseev O, Li Y, Song Y, Dunaief JL. Iron increases APP translation and amyloid-beta production in the retina. Experimental eye research. 2014;129:31–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Banerjee P, Sahoo A, Anand S, Ganguly A, Righi G, Bovicelli P, et al. Multiple mechanisms of iron-induced amyloid beta-peptide accumulation in SHSY5Y cells: protective action of negletein. Neuromolecular medicine. 2014;16:787–98. [DOI] [PubMed] [Google Scholar]
- [27].Frackowiak J, Potempska A, Mazur-Kolecka B. Formation of amyloid-beta oligomers in brain vascular smooth muscle cells transiently exposed to iron-induced oxidative stress. Acta neuropathologica. 2009;117:557–67. [DOI] [PubMed] [Google Scholar]
- [28].Guo C, Wang T, Zheng W, Shan ZY, Teng WP, Wang ZY. Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiology of aging. 2013;34:562–75. [DOI] [PubMed] [Google Scholar]
- [29].Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, et al. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. The Journal of biological chemistry. 2002;277:45518–28. [DOI] [PubMed] [Google Scholar]
- [30].Takahashi M, Dore S, Ferris CD, Tomita T, Sawa A, Wolosker H, et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron. 2000;28:461–73. [DOI] [PubMed] [Google Scholar]
- [31].Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142:857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dlouhy AC, Bailey DK, Steimle BL, Parker HV, Kosman DJ. Fluorescence resonance energy transfer links membrane ferroportin, hephaestin but not ferroportin, amyloid precursor protein complex with iron efflux. The Journal of biological chemistry. 2019;294:4202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tsatsanis A, Wong BX, Gunn AP, Ayton S, Bush AI, Devos D, et al. Amyloidogenic processing of Alzheimer’s disease beta-amyloid precursor protein induces cellular iron retention. Molecular psychiatry. 2020;25:1958–66. [DOI] [PubMed] [Google Scholar]
- [34].Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nature medicine. 2012;18:291–5. [DOI] [PubMed] [Google Scholar]
- [35].Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nature medicine. 2020;26:131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bai B, Wang X, Li Y, Chen PC, Yu K, Dey KK, et al. Deep Multilayer Brain Proteomics Identifies Molecular Networks in Alzheimer’s Disease Progression. Neuron. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tao Y, Wang Y, Rogers JT, Wang F. Perturbed iron distribution in Alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: a systematic review and meta-analysis. Journal of Alzheimer’s disease : JAD. 2014;42:679–90. [DOI] [PubMed] [Google Scholar]
- [38].Bulk M, Abdelmoula WM, Geut H, Wiarda W, Ronen I, Dijkstra J, et al. Quantitative MRI and laser ablation-inductively coupled plasma-mass spectrometry imaging of iron in the frontal cortex of healthy controls and Alzheimer’s disease patients. NeuroImage. 2020:116808. [DOI] [PubMed] [Google Scholar]
- [39].Bulk M, Kenkhuis B, Graaf LMV, Goeman JJ, Natte R, Weerd LV. Postmortem T2*- Weighted MRI Imaging of Cortical Iron Reflects Severity of Alzheimer’s Disease. Journal of Alzheimer’s disease : JAD. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bulk M, Abdelmoula WM, Nabuurs RJA, van der Graaf LM, Mulders CWH, Mulder AA, et al. Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early- and late-onset Alzheimer’s disease. Neurobiology of aging. 2017;62:231–42. [DOI] [PubMed] [Google Scholar]
- [41].Kenkhuis B, Jonkman LE, Bulk M, Buijs M, Boon BDC, Bouwman FH, et al. 7T MRI allows detection of disturbed cortical lamination of the medial temporal lobe in patients with Alzheimer’s disease. NeuroImage Clinical. 2019:101665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Diouf I, Fazlollahi A, Bush AI, Ayton S, Alzheimer’s disease Neuroimaging I. Cerebrospinal fluid ferritin levels predict brain hypometabolism in people with underlying beta-amyloid pathology. Neurobiology of disease. 2019;124:335–9. [DOI] [PubMed] [Google Scholar]
- [43].Ayton S, Diouf I, Bush AI, Alzheimer’s disease Neuroimaging I. Evidence that iron accelerates Alzheimer’s pathology: a CSF biomarker study. Journal of neurology, neurosurgery, and psychiatry. 2018;89:456–60. [DOI] [PubMed] [Google Scholar]
- [44].Ayton S, Fazlollahi A, Bourgeat P, Raniga P, Ng A, Lim YY, et al. Cerebral quantitative susceptibility mapping predicts amyloid-beta-related cognitive decline. Brain : a journal of neurology. 2017;140:2112–9. [DOI] [PubMed] [Google Scholar]
- [45].Damulina A, Pirpamer L, Soellradl M, Sackl M, Tinauer C, Hofer E, et al. Cross-sectional and Longitudinal Assessment of Brain Iron Level in Alzheimer Disease Using 3-T MRI. Radiology. 2020:192541. [DOI] [PubMed] [Google Scholar]
- [46].Ayton S, Wang Y, Diouf I, Schneider JA, Brockman J, Morris MC, et al. Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Molecular psychiatry. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Spotorno N, Acosta-Cabronero J, Stomrud E, Lampinen B, Strandberg OT, van Westen D, et al. Relationship between cortical iron and tau aggregation in Alzheimer’s disease. Brain : a journal of neurology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Annals of neurology. 1997;41:17–24. [DOI] [PubMed] [Google Scholar]
- [50].McAllum EJ, Hare DJ, Volitakis I, McLean CA, Bush AI, Finkelstein DI, et al. Regional iron distribution and soluble ferroprotein profiles in the healthy human brain. Prog Neurobiol. 2019:101744. [DOI] [PubMed] [Google Scholar]
- [51].Mandal PK, Saharan S, Tripathi M, Murari G. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biological psychiatry. 2015;78:702–10. [DOI] [PubMed] [Google Scholar]
- [52].Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxidants & redox signaling. 2014;21:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, et al. Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. Jama. 2014;311:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Southon A, Szostak K, Acevedo KM, Dent KA, Volitakis I, Belaidi AA, et al. Cu(II) (atsm) inhibits ferroptosis: Implications for treatment of neurodegenerative disease. British journal of pharmacology. 2020;177:656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Thomas GEC, Leyland LA, Schrag AE, Lees AJ, Acosta-Cabronero J, Weil RS. Brain iron deposition is linked with cognitive severity in Parkinson’s disease. Journal of neurology, neurosurgery, and psychiatry. 2020;91:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sheelakumari R, Kesavadas C, Varghese T, Sreedharan RM, Thomas B, Verghese J, et al. Assessment of Iron Deposition in the Brain in Frontotemporal Dementia and Its Correlation with Behavioral Traits. AJNR American journal of neuroradiology. 2017;38:1953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fazlollahi A, Raniga P, Bourgeat P, Yates P, Bush AI, Salvado O, et al. Restricted Effect of Cerebral Microbleeds on Regional Magnetic Susceptibility. Journal of Alzheimer’s disease : JAD. 2020;76:571–7. [DOI] [PubMed] [Google Scholar]
- [58].Kim HR, Lee T, Choi JK, Jeong Y, Alzheimer’s Disease Neuroimaging I. Genetic variants beyond amyloid and tau associated with cognitive decline: A cohort study. Neurology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jenkins NL, James SA, Salim A, Sumardy F, Speed TP, Conrad M, et al. Changes in ferrous iron and glutathione promote ferroptosis and frailty in aging Caenorhabditis elegans. eLife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chiang GC, Mao X, Kang G, Chang E, Pandya S, Vallabhajosula S, et al. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR American journal of neuroradiology. 2017;38:1130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Vasconcellos LR, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, et al. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E7474–E82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hare D, Ayton S, Bush A, Lei P. A delicate balance: Iron metabolism and diseases of the brain. Frontiers in aging neuroscience. 2013;5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Timmers P, Wilson JF, Joshi PK, Deelen J. Multivariate genomic scan implicates novel loci and haem metabolism in human ageing. Nature communications. 2020;11:3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet neurology. 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nakamura K, Kawakami T, Yamamoto N, Tomizawa M, Fujiwara T, Ishii T, et al. Activation of the NLRP3 inflammasome by cellular labile iron. Exp Hematol. 2016;44:116–24. [DOI] [PubMed] [Google Scholar]
- [66].Erdei J, Toth A, Balogh E, Nyakundi BB, Banyai E, Ryffel B, et al. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxidative medicine and cellular longevity. 2018;2018:4310816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nnah IC, Lee CH, Wessling-Resnick M. Iron Potentiates Microglial Interleukin-1beta Secretion Induced by Amyloid-beta. J Neurochem. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. Journal of neuropathology and experimental neurology. 2010;69:155–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell research. 2016;26:1021–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hajimohammadreza I, Brammer M. Brain membrane fluidity and lipid peroxidation in Alzheimer’s disease. Neuroscience letters. 1990;112:333–7. [DOI] [PubMed] [Google Scholar]
- [71].Bradley MA, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med. 2010;48:1570–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer’s disease. Neurobiology of aging. 2006;27:1094–9. [DOI] [PubMed] [Google Scholar]
- [73].Montine TJ, Kaye JA, Montine KS, McFarland L, Morrow JD, Quinn JF. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Archives of pathology & laboratory medicine. 2001;125:510–2. [DOI] [PubMed] [Google Scholar]
- [74].Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Annals of neurology. 2005;58:730–5. [DOI] [PubMed] [Google Scholar]
- [75].Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, et al. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiology of disease. 2008;30:107–20. [DOI] [PubMed] [Google Scholar]
- [76].Baldeiras I, Santana I, Proenca MT, Garrucho MH, Pascoal R, Rodrigues A, et al. Oxidative damage and progression to Alzheimer’s disease in patients with mild cognitive impairment. Journal of Alzheimer’s disease : JAD. 2010;21:1165–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.