

Abstract
Targeted protein degraders are heterobifunctional small molecules that link a target ligand or bait to an E3-ligase binder via a chemical spacer. Upon entering the cell, these ligands trigger the formation of a ternary complex between the target protein, degrader and E3-ligase, which leads to target polyubiquitination and proteasomal degradation. In recent years, TPD has expanded rapidly as a field, becoming the modality of choice in drug discovery and chemical probe development. This has been driven by the unique pharmacology of these molecules, which allows for fast and reversible knockdown of the target protein. Recent studies have demonstrated that degraders with specificity for a defined subpopulation of a protein-of-interest can be developed, giving rise to the emerging concept of protein state-specific targeting. In this article, we review advances towards developing degraders that differentiate between target protein subpopulations based on their; activation state, oligomerization state, cellular localization state, and cell type.
Keywords: Targeted protein degradation, PROTACs, Degraders, Molecular glues
Introduction
Heterobifunctional targeted protein degraders (or proteolysis-targeting chimera (PROTAC)s) are small molecules that link a target ligand or bait to an E3-ligase binder via a chemical spacer. On entering the cell, these ligands trigger the formation of a ternary complex between the target protein, degrader, and E3-ligase, which leads to target polyubiquitination and proteasomal degradation [1]. In recent years, targeted protein degradation (TPD) has expanded rapidly as a field, becoming the modality of choice in drug discovery and chemical probe development [2]. This has been driven by the unique pharmacology of these molecules, which allows for fast and reversible knockdown of the target protein. Beyond this, recent studies have demonstrated that degraders with specificity for a defined subpopulation of a protein of interest can be developed, giving rise to the emerging concept of protein state–specific targeting. In this article, we review advances toward developing degraders that differentiate between target protein subpopulations based on the activation state, oligomerization state, cellular localization state, and cell type.
Activation state–specific targeting
In order for a successful TPD event to occur via PROTACs or E3-recruiting molecular glues, the protein of interest must be able to form a ternary complex with the E3-ligase in which free lysine side chains are suitably oriented to allow for efficient polyubiquitination. This presents an opportunity for protein conformation state-specific targeting that has been exploited to create degraders of oncogenic kinase variants that spare their wild-type counterparts.
Conformation specific–targeting of BRAF
Selective degradation of activated BRAF conformations has been reported by two independent groups [3,4]. The first used BI 882370 [5], a preclinical type II BRAFV600E inhibitor that also potently binds to BRAFWT, as the BRAF-binding ligand in a CRBN-recruiting degrader. Lead degrader, P4B, was able to form ternary complexes with, and degrade BRAFV600E, but not BRAFWT [4]. The authors hypothesized this was owing to differences in the Mitogen-activated protein kinase kinase(MEK) binding status of the two proteins. The second article incorporated United States Food and Drug Administration-approved type 1.5 inhibitor vemurafenib, that binds BRAFWT as well as class 1, 2, and 3 BRAF mutants, into VHL-recruiting degrader SJF-0628 [3]. Although studies performed on SJF-0628 show evidence for ternary complex formation with both WT and V600E BRAF variants in vitro, NanoBRET experiments in cells demonstrated that BRAFWT weakly associates with the von-hippel lindau (VHL) E3-ligase complex in the cellular matrix leading to minimal ubiquitination and degradation, as opposed to BRAFV600E which has much higher ternary complex formation [3]. Activation of BRAFWT via receptor tyrosine kinase (RTK) upregulation or activation, RAS mutation, and relief of negative feedback (MEK inhibition) promoted BRAFWT degradation, whereas upstream BRAF inactivation via epidermal growth factor receptor (EGFR) or Kirsten rat sarcoma virus (K-RasG12C) inhibition abrogated SJF’s effects. Together, these results suggest targeted BRAF degradation is dependent on its activation state, independent of the E3-ligase or BRAF-binding ligand incorporated.
Selective degradation of BCR-ABL over c-ABL
The selectivity of ABL-targeted degraders for c-ABL or BCR-ABL has been reported to vary dependent on the E3-ligase recruited. Molecules that hijack CRBN or VHL induced preferential degradation of c-ABL across a series incorporating various linkers and ABL-binding bait ligands, including dasatinib [6]. However, when Ring Finger Protein 114 (RNF114)-recruiting nimbolide was incorporated, this selectivity was reversed, indicating that differences in the E3-ligase compatibility of ABL variants drive these trends [7].
Oligomerization state–specific targeting
Selective degradation of misfolded proteins in neurodegenerative disease
The accumulation of misfolded proteins concurrent with disease progression is a hallmark of neurodegenerative disorders known as proteinopathies [8]. However, the role of these aggregated species in driving disease progression is poorly understood. Genetic approaches, that knockdown all protein conformers, fail to differentiate between effects mediated by loss of endogenous function versus gain of proteotoxicity. Therefore, several groups have applied a TPD approach to achieve aggregate-specific degradation of proteinopathy-related targets in Huntington’s’ disease and frontotemporal dementia.
Huntington’s disease is caused by a CAG expansion repeat in the Huntingtin gene, resulting in a polyglutamate stretch in the mutant huntingtin (mHTT) protein which promotes misfolding and accumulation [9]. Tomoshige et al. [10] used benzothiazole derivatives, which were previously developed as candidate diagnostic agents that bind the characteristic cross beta-sheet structure of aggregated proteins, linked to a ligand for the E3 ligase Cellular Inhibitor of Apoptosis Protein 1 (cIAP1) to induce degradation of mHTT via the ubiquitin-proteasome machinery [11]. The small molecules they developed were able to degrade mHTT in both primary cells from patients with Huntington’s disease and in HeLa cells transfected with mHTT exon 1. However, their small molecules also caused degradation of wild-type HTT that had formed oligomers. Furthermore, they found that the molecules had poor selectivity for aggregated mHTT as they also degraded other misfolded proteins such as mutant ataxin-1, mutant ataxin-7, and mutant atrophin-1, mimicking the pan-aggregate selectivity of the parental ligand. To generate more selective degraders, Li et al. [12] pursued a ‘molecular glue’ strategy, to identify molecules that interact with mHTT and the autophagosome protein microtubule–associated protein 1A/1B light chain 3. The group used a small molecule microarray-based screen using wild-type huntingtin (wtHTT) as a counter screen. The compounds were not only found to degrade mHTT without affecting wild-type HTT but they also rescued disease-relevant phenotypes in cells as well as in fly and mouse models of Huntington’s disease. In addition, these compounds were found to be active against mutant ataxin-3, another disease-relevant protein that contains an expanded polyQ stretch. However, these glue compounds also contain pan-assay interference compounds (‘PAINS’) substructures that may be desirable to remove in future optimized compounds [13].
Frontotemporal dementia is characterized by the accumulation of misfolded tau, which in its soluble form binds to and stabilizes microtubules [14]. Silva et al. converted clinical positron emission tomography (PET) tracer Tauvid [15] that binds selectively to pathological tau into a heterobifunctional molecule that recruits the E3 ligase CRBN [16]. The group found that their molecule induced aberrant-tau ubiquitination and proteasomal degradation in frontotemporal dementia patient-derived neurons but had little effect on soluble tau in healthy neurons [16].
Monovalent protein degraders
An emerging degradation strategy has been to develop small molecules that take advantage of the endogenous state-specific recognition of misregulated proteins by E3-ligases in healthy cells and create degraders that act by pushing the target protein toward those states. Survivin is a homodimeric protein part of the inhibitor of apoptosis protein family and is overexpressed in cancers. The protein has been considered ‘undruggable’ because of the lack of known enzymatic activity [17]. Peery et al. [18] reported that the small molecule LQZ-7I can degrade survivin by binding to the dimerization interface of the protein and exposing the hydrophobic dimerization core giving rise to proteasomal degradation. Similarly, Slabicki et al. [19] recently discovered a new degradation mechanism of a small molecule that induces the polymerization of B-cell lymphoma 6 protein (BCL6) and its subsequent degradation [19]. BCL6 is also overexpressed in cancer acting as a master transcriptional repressor. Traditional inhibition of BCL6 has been stunted clinically owing to the high concentrations of drug necessary for inhibition [21]. Slabicki et al. found that the compound, BI-3802, binds to the domain of BCL6 that mediates the homodimerization of BCL6 and initiates the formation of filaments, which are then recognized and degraded by the E3 ligase SIAH1. The small molecule is able to activate the previously repressed BCL6 targets and induce antiproliferative effects in Diffuse large B cell lymphoma (DLBCL) cells. Both articles provide a new mechanism of action for TPD via alteration of protein dimerization or oligomerization state. These studies suggest molecular glue degraders also hold promise for targeting specific protein subpopulations.
Cellular localization state–specific targeting
Several disease-relevant proteins are postulated to play different regulatory roles depending on their intracellular localization, where they interact with different binding partners, which can be leveraged for spatial control of targeted protein degradation.
Selective degradation of microtubule-bound Aurora kinase A
Aurora A kinase (AURKA) regulates important mitotic and nonmitotic processes, and separate populations localize on centrosomes and microtubules. Want et al. [22] presented a degrader-dubbed ‘PROTAC-D’ derived from the selective AURKA inhibitor, alisertib (MLN8237). Unlike the parental inhibitor, whose action against AURKA is irrespective of subcellular localization, PROTAC-D degrades microtubule-localized protein while sparing centrosome-localized protein in both interphase and mitotic cells. PROTAC-D was also capable of degrading mutant AURKA that eluded degradation by its endogenous E3 ligase [22].
Cell cycle–dependent degradation of cyclin-dependent kinase 2 (CDK2)
Riching et al. [23] investigated and performed a detailed investigation into degradation across cyclin-dependent kinases (CDKs), which are a family of cell cycle regulatory proteins. They determined that while many CDKs were degraded by pan-kinase degrader TL12-186, degradation of CDK2 was accomplished only in G1 phase of the cell cycle, where CDK2/cyclin E complexes accumulate in the nucleus. Although TL12-186 binding to CDK2 was observed in all cell cycle stages, ternary complex formation essential to protein clearance was observed only in G1 cells. Authors surmise that because CDK2 and Cip/Kip cyclin-dependent kinase inhibitors are known to interact in S, G2, and M stages, this interaction frustrates ternary complex formation in non-G1 stages. Other cell cyclee–associated CDK proteins (1, 4, and 6) were slowly and incompletely degraded in 24-h dose–response experiments, indicating that their degradation mechanisms may also be cell cycle-dependent.
Nuclear-specific degradation
A subset of E3-ligases are restricted to defined cellular compartments and can be hijacked to deplete the local pool of a target protein. For example, Zhang et al. [24] used chemoproteomic electrophilic fragment screening to identify covalent binders of DDB1- and CUL4-associated factor 16 (DCAF16), a nuclear E3-adapter protein. Incorporation of this fragment into a bifunctional degrader enabled selective degradation of nuclear FK506-binding protein 12 (FKBP12), but not cytosolic FKBP12.
Cell type–specific targeting
Degraders that depend on ubiquitously expressed E3 ligases may be limited in their therapeutic potential by the problem of on-target toxicity. To overcome this, multiple strategies for cell type–specific targeting are in development.
Liver-specific degradation
Lysosome-targeting chimeras are a type of degraders that promote lysosomal clearance of extracellular targets by hijacking lysosome-trafficking receptors [25]. To develop tissue-specific degraders, LYTACs that bind to the liver-specific asialoglycoprotein receptor (ASGPR) have been developed [26–28]. To do so, researchers synthesized bifunctional molecules that link triantennary N-acetylgalactosamine (tri-GalNAc), a known ligand of ASGPR, to either an antibody, peptide, or small-molecule binder of the extracellular or cell surface target of interest. Ahn et al. [26] demonstrated selective clearance of EGFR in ASGPR+ Hep3B cells but not in ASGPR− HeLa cells in a coculture treated with a cetuximab- N-acetylgalactosamine LYTAC, not possible with cetuximab conjugates targeting the ubiquitously expressed cation-independent mannose-6-phosphonate receptor.
Prodrug strategies for cancer cell–specific targeted degradation
Recently, groups have developed degrader prodrugs that are targeted to specific cell surface receptors, for increased targeting to cells with high expression. Maneiro et al. [29] described an antibody-PROTAC conjugate (Ab-PROTAC) consisting of trastuzumab, a human epidermal growth factor receptor 2 (HER2) receptor binding antibody, linked to the previously described bromodomain and extraterminal (BET) bromodomain degrader MZ1 via an ester prodrug. The Ab-PROTAC effected rapid and selective Bromodomain-containing protein 4 (BRD4) degradation in HER2-positive breast cancer cells. By switching the antibody, similar Ab-PROTACs could be developed to engage other receptors. One limitation, however, is the size of the Ab-PROTAC conjugate, which limits bioavailability. Using a similar concept, Liu et al. [30] described a folate-caged PROTAC that degrades BRD4 in a folate receptor alpha (FOLR1)dependent mechanism. FOLR1 is a folate transporter highly expressed in ovarian, lung, and breast cancers. The authors inactivated BET bromodomain degrader ARV-771 by masking a hydroxyl group critical to VHL recognition with an ester-linked folate molecule. The caged PROTAC cannot form a ternary complex with VHL until taken into cells through FOLR1 where the folate moiety is liberated by intracellular hydrolases and the activated PROTAC degrades BRD4. Authors further used this approach to degrade mitogen-activated protein kinases (MEK1/2) and anaplastic lymphoma kinase fusion proteins. In a follow-up article, Chen et al. [31] used folate-caged cereblon-recruiting molecular glues and PROTACs to degrade Ikaros Zinc Finger (IKZF) neosubstrates and anaplastic lymphoma kinase fusion proteins in cancer cell lines expressing FOLR1, demonstrating the generalizability of this strategy. For cereblon-recruiting degraders, this was achieved by caging the glutarimide group on pomalidomide with a disulfide-linked folate molecule that could be cleaved by intracellular glutathione.
Conclusion
Endogenous protein homeostasis is tightly regulated, and mechanisms for controlling protein levels in response to their fold, post translational modification (PTM)s, localization, and complexation state have evolved. By tapping into these endogenous pathways, targeted protein degraders that enable precision control over protein subpopulations have been developed, setting TPD apart from both genetic and inhibitor-based approaches. This may lead to tools that enable detailed biological studies into specific protein signaling functions and provide therapeutic advantages through enhanced selectivity (see Figure 1).
Figure 1.
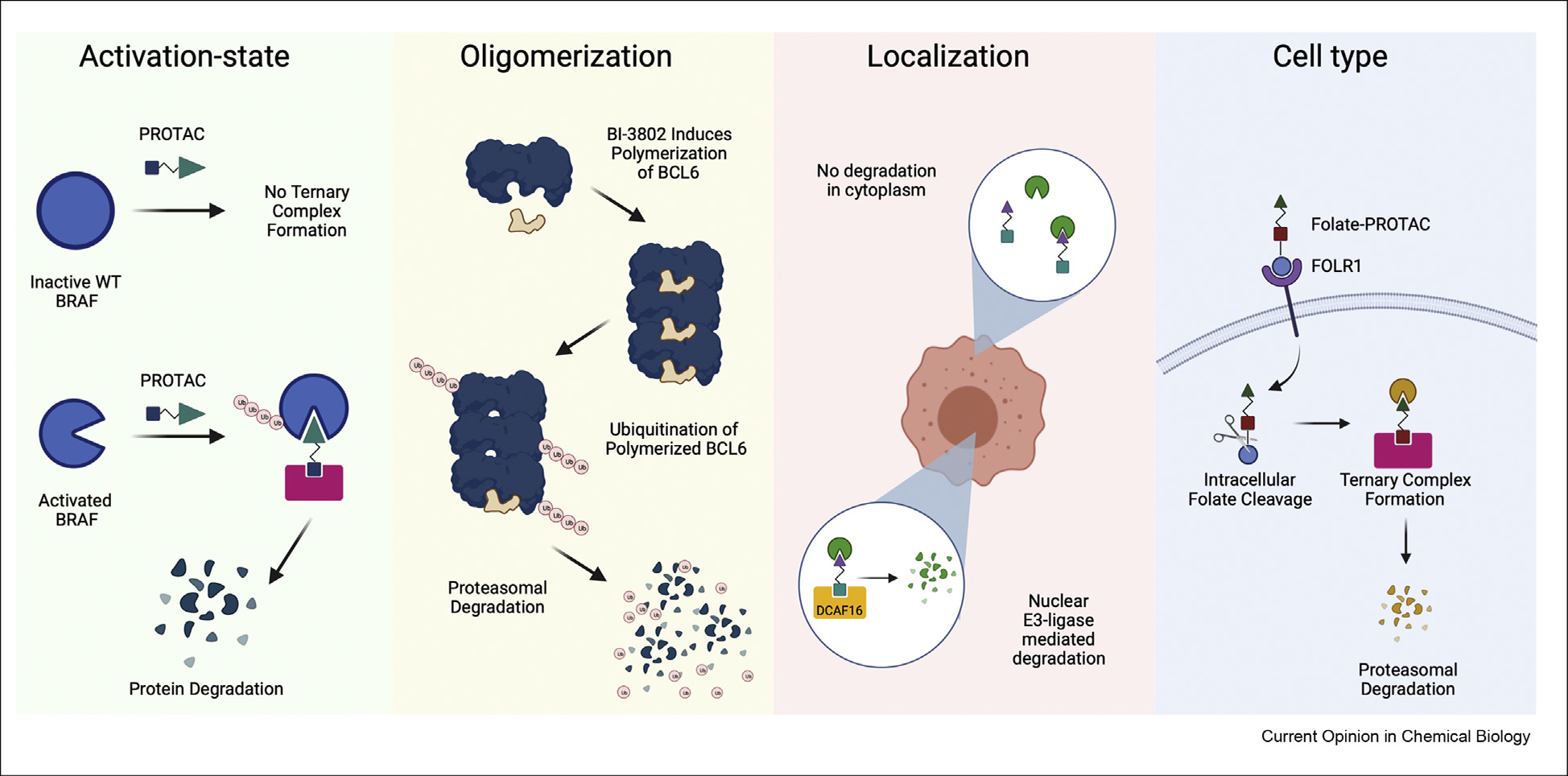
Overview of approaches.
Acknowledgements
G.E.G. acknowledges support from a matching fellowship associated with the Molecular Biophysics Training Grant.
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: F.M.F is a scientific co-founder and equity holder in Proximity Therapeutics, and a scientific advisory board member and equity holder in Santi Therapeutics. F.M.F is or was recently a consultant to or received speaking honoraria from Tocris BioTechne and RA Capital. All other authors have no known interested to declare.
References
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
- 1.*. Burslem GM, Crews CM: Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181:102–114. A review of the recent breakthroughs, applications, and current challenges in the field of targeted protein degradation.
- 2.Dale B, Cheng M, Park KS, et al. : Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer 2021, 21:638–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alabi S, Jaime-Figueroa S, Yao Z, et al. : Mutant-selective degradation by BRAF-targeting PROTACs. Nat Commun 2021, 12:920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Posternak G, Tang X, Maisonneuve P, et al. : Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat Chem Biol 2020, 16:1170–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waizenegger IC, Baum A, Steurer S, et al. : A novel RAF kinase inhibitor with DFG-out-binding mode: high efficacy in BRAF-mutant tumor xenograft models in the absence of normal tissue hyperproliferation. Mol Cancer Therapeut 2016, 15:354–365. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y, Gao H, Sun X, et al. : Global PROTAC toolbox for degrading BCR-ABL overcomes drug-resistant mutants and adverse effects. J Med Chem 2020, 63:8567–8583. [DOI] [PubMed] [Google Scholar]
- 7.*. Tong B, Spradlin JN, Novaes LFT, et al. : A nimbolide-based kinase degrader preferentially degrades oncogenic BCR-ABL. ACS Chem Biol 2020, 15:1788–1794. A paper that describes the discovery of a natural-product (nimbolide) that binds to the E3-ligase RNF114 and, when incorporated in to a dasatinib-based targeted protein degrader, enables selective degradation of oncogenic BCR-ABL over c-ABL.
- 8.Kovacs GG: Molecular pathology of neurodegenerative diseases: principles and practice. J Clin Pathol 2019, 72:725–735. [DOI] [PubMed] [Google Scholar]
- 9.Bates GP, Dorsey R, Gusella JF, et al. : Huntington disease. Nat Rev Dis Prim 2015, 1:15005. [DOI] [PubMed] [Google Scholar]
- 10.Okazawa H, Ikawa M, Jung M, et al. : Multimodal analysis using [(11)C]PiB-PET/MRI for functional evaluation of patients with Alzheimer’s disease. EJNMMI Res 2020, 10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomoshige S, Nomura S, Ohgane K, et al. : Discovery of small molecules that induce the degradation of Huntingtin. Angew Chem Int Ed Engl 2017, 56:11530–11533. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Wang C, Wang Z, et al. : Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature 2019, 575:203–209. [DOI] [PubMed] [Google Scholar]
- 13.Baell JB, Nissink JWM: Seven year itch: pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem Biol 2018, 13:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silva MC, Haggarty SJ: Tauopathies: deciphering disease mechanisms to develop effective therapies. Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li CH, Chen TF, Chiu MJ, et al. : Integrated (18)F-T807 tau PET, structural MRI, and plasma tau in tauopathy neurodegenerative disorders. Front Aging Neurosci 2021, 13:646440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silva MC, Ferguson FM, Cai Q, et al. : Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altieri DC: Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer 2008, 8:61–70. [DOI] [PubMed] [Google Scholar]
- 18.Peery R, Kyei-Baffour K, Dong Z, et al. : Synthesis and identification of a novel lead targeting survivin dimerization for proteasome-dependent degradation. J Med Chem 2020, 63:7243–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.*. Slabicki M, Yoon H, Koeppel J, et al. : Small-molecule-induced polymerization triggers degradation of BCL6. Nature 2020, 588:164–168. This study defined a new mechamism of action for small-molecule mediated protein degradation, in which the ligand BI-3802 induces highly specific polymerization of its target BCL6. The authors demonstrate BCL6 filaments are then specifically recognized by SIAH1 E3 ligase, which leads to ubiquitination and proteasomal processing.
- 20.Kerres N, Steurer S, Schlager S, et al. : Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Rep 2017, 20:2860–2875. [DOI] [PubMed] [Google Scholar]
- 21.Pearce AC, Bamford MJ, Barber R, et al. : GSK137, a potent small-molecule BCL6 inhibitor with in vivo activity, suppresses antibody responses in mice. J Biol Chem 2021, 297:100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, Ascanelli C, Abdelbaki A, et al. : Selective targeting of non-centrosomal AURKA functions through use of a targeted protein degradation tool. Commun Biol 2021, 4:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.*. Riching KM, Schwinn MK, Vasta JD, et al. : CDK family PROTAC profiling reveals distinct kinetic responses and cell cycle-dependent degradation of CDK2. SLAS Discov 2021, 26:560–569. A detailed study investigating the kinetics of cellular binding, ubiquitination and degradation of CDKs by multi-kinase degrader TL12-186. Here, CDK2 was shown to be degraded only in the G1 phase of the cell cycle.
- 24.Zhang X, Crowley VM, Wucherpfennig TG, et al. : Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol 2019, 15:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banik SM, Pedram K, Wisnovsky S, et al. : Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahn G, Banik SM, Miller CL, et al. : LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat Chem Biol 2021, 17:937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caianiello DF, Zhang M, Ray JD, et al. : Bifunctional small molecules that mediate the degradation of extracellular proteins. Nat Chem Biol 2021, 17:947–953. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Y, Teng P, Montgomery NT, et al. : Development of triantennary N-acetylgalactosamine conjugates as degraders for extracellular proteins. ACS Cent Sci 2021, 7:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maneiro MA, Forte N, Shchepinova MM, et al. : Antibody-PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem Biol 2020, 15:1306–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Chen H, Liu Y, et al. : Cancer selective target degradation by folate-caged PROTACs. J Am Chem Soc 2021, 143:7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Liu J, Kaniskan HU, et al. : Folate-guided protein degradation by immunomodulatory imide drug-based molecular glues and proteolysis targeting chimeras. J Med Chem 2021, 64:12273–12285. [DOI] [PMC free article] [PubMed] [Google Scholar]