

Abstract
Background & Aim:
While certain nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) are efficacious in treating HBV infection, their effects are yet to be optimized and the emergence of NRTI-resistant HBVs remains to be a challenging issue since life-long medication is needed, making it crucial to develop agents that more profoundly suppress wild-type and drug-resistant HBVs and have a long-acting feature for patients’ greater quality of life.
Methods:
(1S,3S,5S,E)-3-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxymethyl)cyclopentane-1-carbonitrile (E-CFCP) was newly synthesized. E-CFCP’s in vitro anti-HBV activity was evaluated. In vivo anti-HBV activity was examined using HBV-infected human-liver-chimeric mice (PXB-mice). E-CFCP’s long-acting features and E-CFCP-triphosphate’s interactions with HBV-reverse transcriptase (HBV-RT) were examined.
Results:
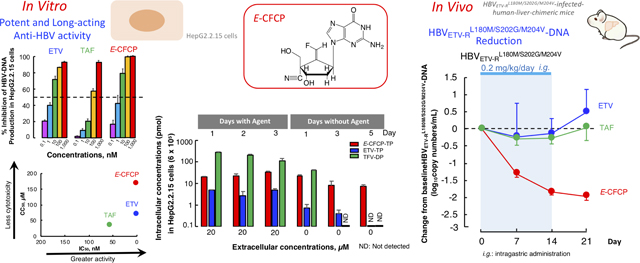
E-CFCP potently blocked HBVWTD1 production (IC50qPCR_cell=1.8 nM) in HepG2.2.15 cells and HBVWTC2 (IC50SB_cell=0.7 nM), ETV-resistant HBVETV-RL180M/S202G/M204V (IC50SB_cell=77.5 nM), and ADV-resistant HBVADV-RA181T/N236T production (IC50SB_cell=14.1 nM) in Huh7 cells. E-CFCP profoundly inhibited intracellular HBV-DNA production down to below detection limit, but ETV and TAF failed to do so. E-CFCP also showed less toxicity than ETV and TAF. E-CFCP better penetrated hepatocytes and was better tri-phosphorylated and E-CFCP-triphosphate intracellularly persisted longer compared to ETV-triphosphate. Once-daily peroral E-CFCP administration over 2 weeks (0.02∼0.2 mg/kg/day) reduced HBVWTC2-viremia by 2–3 logs in PXB-mice without significant toxicities and the reduction persisted over 1–3 weeks following treatment cessation, suggesting once-weekly dosing capabilities. E-CFCP also reduced HBVETV-RL180M/S202G/M204V-viremia by 2 logs over 2 weeks, while ETV completely failed HBVETV-RL180M/S202G/M204V-viremia reduction. None of E-CFCP-treated mice had significant adverse changes. Van der Waals interactions of E-CFCP’s 4’-cyano and polar interactions of the fluorine with HBV-RT are important for E-CFCP’s anti-HBV potency and that both 4’-cyano and fluorine help retain E-CFCP-triphosphate’s interactions of HBVETV-RL180M/S202G/M204V-RT.
Conclusion:
E-CFCP represents the first reported potential long-acting NRTI potent against HBVs.
Keywords: HBV, long-acting anti-HBV therapeutic, NRTI, drug resistant HBVs, human liver chimeric mice
Lay summary
A novel long-acting anti-HBV agent, E-CFCP, potently reduces wild-type and drug-resistant HBV viremia without toxicities in HBV-infected humanized mice with possibly once-weekly oral dosing capabilities, which may give better QOL to patients living with HBV and improve the otherwise poor adherence to current QD regimens in the low-income countries and regions, where no city water or electricity services are available. To the best of our knowledge, E-CFCP represents the first reported potential long-acting and highly potent nucleoside analog for treating wild-type and drug-resistant HBV infection.
Introduction
Hepatitis B virus (HBV) is a double-stranded DNA virus, which contains reverse transcriptase (RT) and causes various liver diseases including chronic hepatitis B (CHB) and hepatocellular carcinoma in humans.1,2 Despite the availability of effective vaccine and therapeutics, CHB continues to be a substantial public health burden, accounting for 30% of deaths from liver cirrhosis and 40% of hepatocellular-carcinoma-related deaths globally.3 The present common chemo-therapy for HBV infection is the use of nucleos(t)ide reverse transcriptase inhibitors (NRTIs), 4 which profoundly inhibit HBV-DNA synthesis and reduce serum HBV-DNA to undetectable levels. In fact, the development of resistance against ETV, TDF, and TAF has not been a major concern when used as an initial therapy although ETV is not indicated in certain patients with HBV resistant to 3TC. However, their efficacy is yet to be optimized and the emergence of NRTI-resistant HBVs may become eventually problematic due to life-long medication period, making it crucial to develop agents capable of more profoundly suppressing the replication of wild-type HBV (HBVWT) and existing drug-resistant variants. Moreover, the cure of HBV infection or the elimination of HBV is as yet elusive and CHB treatment is thought to be life-long and as in the case of treatment of HIV-1 infection, development of long-acting therapeutics for treating CHB should be considered.
We have previously reported islatravir (ISL; 4’-ethynyl-2-fluoro-2’-deoxyadenosine or EFdA/MK-8591), which has a unique 4’-ethynyl moiety and exerts highly potent anti-HIV-1 activity in vitro and in vivo5,6 and is presently under phase 3 clinical trials. An implant containing ISL has recently been shown to block HIV-1 effectively for at least 3 months, possibly a full year,7 implying that ISL is highly non-toxic and has very low bioaccumulation potential. In this regard, we have previously demonstrated that three EFdA congeners, 4’-C-cyano-2-amino-2’-deoxyadenosine (CAdA), 4’-C-cyano-2’-deoxyguanosine (CdG), and 4’-cyano-methylenecarbocyclic-2’-deoxyguanosine (CMCdG), exert potent activity against HBVWT and drug-resistant variants in cell culture and in cDNA-uPA/SCID chimeric mice with humanized liver (PXB-mice).8,9
Here, we describe a novel 4’-modified NRTI, (1S,3S,5S,E)-3-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxy-methyl)cyclopentane-1-carbonitrile (E-CFCP), which potently blocks the production of various wild-type and drug-resistant HBV strains. E-CFCP’s anti-HBV activity, like ISL’s anti-HIV-1 activity,5 persists over 1–3 weeks following treatment cessation, suggesting at least once-weekly dosing capabilities, which may give better QOL to patients living with HBV and improve the otherwise poor adherence to current QD regimens particularly in the low-income countries and regions, where no city water or electricity services are presently available. __
Materials and Methods
Cells, viruses, and antiviral agents
Six different cell lines, 3 different human primary cells [fresh human hepatocytes (PXB-cells), primary human hepatocytes, and human peripheral blood mononuclear cells (hPBMCs)], 4 different HBV clones [wild type HBV genotype C2 (HBVWTC2), wild type HBV genotype D1 (HBVWTD1), ETV-resistant HBV (HBVETV-RL180M/S202G/M204V), and ADV-resistant HBV (HBVADV-RA181T/N236T)], one HIV clone, and 5 different antiviral agents were used. More details are described in Supplementary information (SI) and the CTAT table. The synthetic methods of Z-CFCP and E-CFCP will be published elsewhere.
Anti-HBV and -HIV assays, cytotoxicity assays, cell growth assay, and mitochondrial DNA assay
Antiviral activity evaluated was expressed as IC50 values depending on the methods as follows: . when real-time-qPCR was employed, as IC50qPCR_cell or IC50qPCR_sup; Southern blot assay was employed, as IC50SB_cell; while MTT assay was employed, as IC50MTT. Drug-associated 50% cytotoxicity concentrations (CC50) were determined with a water-soluble MTT assay (CC50MTT), LDH release assay (CC50LDH), and Neutral Red staining assay (CC50NR); while drug-associated mitochondrial toxicity (Mitotox50) was determined using real time qPCR. Details are described in SI.
Anti-HBV activity of E-CFCP combined with GLS4 and E-CFCP induction of interferon production
Combination effect of E-CFCP with a capsid assembly modulator, GLS4, was examined using the reduction of HBV-DNA production by HepG2.2.15 cells as an endpoint. In order to examine the induction of Type III interferon (IFN) by E-CFCP, IFNλ2/3 induction ability of E-CFCP was assessed. Details are described in SI.
Evaluation of the persistence of anti-HBV activity of E-CFCP in HepG2.2.15 cells
The persistence of anti-HBV activity of various compounds was evaluated in HepG2.2.15 cells over various incubation periods. Details are described in SI.
Determination of intracellular concentrations of E-CFCP
The levels of intracellular concentrations of ETV, Z-CFCP and E-CFCP, which represent the balance between the penetration through the membrane and intracellular degradation of the compounds, were determined in HepG2.2.15 cells. More details are described in SI.
Determination of intracellular concentrations of ETV-TP, TFV-DP, Z-CFCP-TP, and E-CFCP-TP
HepG2.2.15 cells and/or PXB-cells were incubated with ETV, TAF, Z-CFCP, or E-CFCP for various periods of time, washed with drug-free medium, and further cultured without compounds to determine intracellular concentrations of ETV-triphosphate (TP), TFV-DP, Z-CFCP-TP, and E-CFCP-TP. Details of the method are described in SI.
Determination of the acid resistance of E-CFCP
E-CFCP was exposed to 0.01 N (0.01mol/L) hydrochloric acid (pH2) to test E-CFCP’s chemical decomposition resistance. Details of the method are described in SI.
Pharmacokinetic studies of E-CFCP in PXB-mice and its antiviral effects in HBV-infected PXB-mice
E-CFCP was administered to PXB-mice (PhoenixBio Co., Ltd., Hiroshima, Japan). All animal protocols described in this study were performed in accord with Guide for the Care and Use of Laboratory Animals (National Research Council 2011) and approved by the Animal Welfare Committee of PhoenixBio Co., Ltd. (approval no. 1148, 1725, 1726, 1727, 2341, 2505). Details are described in SI.
Statistical analyses of the changes in the viral loads and selected biomarkers in treated mice
Statistical analyses of the changes in the viral loads and selected biomarkers in treated mice were compared using repeated measures analysis of variance with two-tailed P values reported. Details are described in SI.
Molecular modeling
Structure models of the interactions of E-CFCP and Z-CFCP with HBV-RTWT and HBVETV-RL180M/S202G/M204V-RT were built using Maestro version 10.7.015 (Schrödinger, LLC, New York) as previously described.8,9 Details of the method are described in SI.
Results
E-CFCP is potent against HBVWTD1 and HBVWTC2 and has favorable cytotoxicity profiles
Since 4’-modified CAdA, CdG, and CMCdG potently blocked the replication of wild-type and various drug resistant variants,8,9 we designed and synthesized approximately 100 NRTIs containing 4’-modification and ultimately identified E-CFCP (Fig. 1A). E-CFCP structurally bears resemblance to CAdA, CdG, and CMCdG in that all the four analogs have a 4’-cyano moiety and E-CFCP also resembles ETV and CMCdG in that ETV, CMCdG, and E-CFCP have a methylidene in the 4’-position of their cyclopentyl moiety (Fig. 1A). As shown in Fig. 1B, ETV and E-CFCP were virtually equipotent. However, the inhibition of HBV-DNA production by high E-CFCP concentrations (100 and 1,000 nM) was virtually complete to detection limit (by 100%), while that by ETV never reached 100% even at its highest concentrations (100 and 1,000 nM). The inhibition of HBV-DNA production by high concentrations of TAF never reached 100%, either (Fig. 1B, left panel). We also examined the activity of ETV, TAF, and E-CFCP against HBVWTC2 using Southern blot assay, employing HBVWTC2-plasmid-transfected Huh7 cells. As shown in Figures 1C-a, -d, and -g, the IC50SB_cell values of ETV, TAF, and E-CFCP were 7.0, 25.0, and 0.2 nM, respectively. The average IC50SB_cell values of ETV and E-CFCP against HBVWTCe were 16 and 0.7 nM (Fig. 1C, right panel). We also synthesized Z-CFCP that has a fluorine atom at the Z-position of the methylidene moiety (Fig. 1A). Z-CFCP showed much less activity against HBVWTD1 (IC50qPCR_cell=414 nM) and HBVWTC2 (IC50SB_cell =938 nM) compared to E-CFCP (Tables S1 and S2). Furthermore, we examined anti-HBV activity of E-CFCP using non-cancerous hepatocytes, PXB-cells and HBVWTC2 (Fig. S1). E-CFCP exerted the most potent anti-HBV activity in HBV-exposed PXB-cells among three anti-HBV agents. The concentrations of ETV, TAF, and E-CFCP that blocked the production of intracellular HBV-DNA by IC50qPCR_cell were 0.13, 0.59, and 0.01 nM and the concentrations that blocked HBV-DNA production by 95% (IC95qPCR_cell) were 859, 513, and 339 nM, respectively. The IC95qPCR_sup of ETV, TAF, and E-CFCP, that block HBV-DNA in culture medium by 95% were 79, 90, and 3.5 nM. Of note, anti-HBV activity of the three agents as tested in PXB-cells was approximately 10 to 100 times more potent than as tested in HepG2.2.15 cells, because the HBV replication level in HBV-infected PXB cells are lower than that in HepG2.2.15 cells.
Fig. 1. Structure and anti-HBV potency of E-CFCP.

(A) Structures of entecavir (ETV), 4’-ethynyl-2-flouro-2’-deoxyadenosine (EFdA/MK-8591/ISL), 4’-cyano-methylenecarbocyclic-2’-deoxyguanosine (CMCdG), (1S,3S,5S,Z)-3-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxymethyl)cyclopentane-1-carbonitrile (Z-CFCP), and (1S,3S,5S,E)-3-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxymethyl)cyclopentane-1-carbonitrile (E-CFCP). (B) E-CFCP inhibits DNA synthesis of HBVWTD1 and shows favorable safety feature in HepG2.2.15 cells. Intracellular HBV-DNA levels were determined using real-time qPCR and expressed as % inhibition (no-drug control=100%), while cytotoxicity determined using MTT assay. Each quantitative data set is shown. (C) E-CFCP exhibits potent activity against drug-resistant variants. HBV-DNA synthesis reduction in Huh7 cells was examined using Southern blotting. Representative image of single-stranded replicative intermediate DNA (ssDNA)(left) and the quantitative mean values are shown (right).
The CC50MTT values of E-CFCP were mostly much greater in all cell lines and PXB-cells examined compared to those of ETV (Fig. 1B and Table S1). When we conducted extended cytotoxicity analyses with Neutral Red staining and LDH release assays using primary hepatocytes, E-CFCP (CC50NR>100 and CC50LDH>100) was less toxic compared to TAF (CC50NR=26 µM and CC50LDH=97 µM) (Table S3). We also performed cell proliferation assays using MOT-4 cells and human primary peripheral blood mononuclear cells in the presence of ETV, TAF, and E-CFCP and E-CFCP turned out to have highly favorable safety profiles (Fig. S2). Thus, the specificity index (S.I.) of E-CFCP was much greater (S.I.=93,889) than those of ETV and TAF (S.I.=40,556 and 638, respectively) (Fig. 1B).
To examine other properties of E-CFCP, we performed anti-HBV assays in combination of E-CFCP with GLS4, one of HBV capsid assembly modulators (CAMs), using HepG2.2.15 cells, and also examined whether E-CFCP activates type III IFN production using immune-competent hepatocytes, PXB-cells, and colon cancer (WiDr) cells. When 1 and 10 nM E-CFCP was combined with GLS4 (at the same concentration), synergism was observed as assessed with the Chou-Talalay Method10, while when 100 and 1,000 nM E-CFCP was combined with GLS4 (at the same concentration), no synergism was detected (Fig. S3). The type III interferon group consists of four IFN-λ (lambda) molecules called IFN-λ1, IFN-λ2, IFN-λ3, and IFN-λ4. Among them, we assessed the induction of IFN-λ2 and λ3. All nucleic acid analogs tested induced no detectable production within the assay range of IFNλ 2/3 in either of PXB-cells or WiDr cells (Fig. S4).
E-CFCP exerts potent activity against ETV-resistant and ADV-resistant HBV variants
We examined whether E-CFCP exerted activity against HBVETV-RL180M/S202G/M204V and HBVADV-RA181T/N236T, employing HBVETV-RL180M/S202G/M204V- and HBVADV-RA181T/N236T-containing-plasmid-transfected Huh7 cells. In a representative image data set (Fig 1C, left panel), E-CFCP exerted potent activity against HBVETV-RL180M/S202G/M204V (IC50SB_cell=57.2 nM, Fig. 1C-h). The activity of TAF against HBVETV-RL180M/S202G/M204V appeared to be comparable (IC50SB_cell =53.7 nM, Fig. 1C-e) to that of E-CFCP. As expected, ETV virtually failed to block HBVETV-RL180M/S202G/M204V replication (IC50SB_cell=53,090 nM, Fig. 1C-b). E-CFCP was also potent against HBVADV-RA181T/N236T (IC50SB_cell=6.2 nM, Fig. 1C-i), while IC50SB_cell values of ETV and TAF were 184 and 86.5 nM, respectively (Fig. 1C-c and -f). The greater potency of E-CFCP against HBVETV-RL180M/S202G/M204V and HBVADV-RA181T/N236T compared to the activity of ETV and the comparable activity of E-CFCP against HBVADV-RA181T/N236T compared to the activity of TAF were well corroborated by Southern blot assays repeated on different occasions (Fig. 1C and Table S2). However, the CC50MTT values of E-CFCP in MT-2 cells, HepG2 cells, Huh7 cells, and PXB-cells were 306, 115, >500, and >500 µM, although those of TAF were 21, 22, 34, and 8.2 µM, respectively (Table S1).
Neither ETV nor E-CFCP shows significant mtDNA synthesis inhibition in PBMCs and PXB-cells
Various NRTI-triphosphates inhibit the function of human gamma-polymerase, which is responsible for mitochondrial DNA (mtDNA) production in human cells. If the cellular mtDNA content declines or is depleted, various serious and potentially fatal adverse effects occur11. The inhibition by E-CFCP of mtDNA production in human PBMCs and PXB-cells was comparable (Mitotox50=38.2 and 29.6 µM) with that by ETV (Mitotox50=40.4 and 35.2 µM), respectively, while zalcitabine/ddC known to cause serious mitochondrial toxicity strongly blocked mtDNA production. CMCdG proved to be non-toxic as previously reported.9 Considering that ETV does not reportedly cause significant mitochondrial damages in clinical settings12, E-CFCP is expected not to interfere with mitochondrial functions (Table S4).
Anti-HBV activity of E-CFCP against HBVWTC2 persists longer than that of ETV and TAF
Currently available anti-HBV agents are orally administered once daily (QD); however, if anti-HBV therapeutics are orally administered less often, such therapeutics would improve the adherence to the treatment and quality of life (QOL) of CHB patients. Thus, we examined whether E-CFCP’s activity persisted longer than ETV and TAF, by exposing HepG2.2.15 cells to 0.01–10 µM of each NRTI over various periods of time (3–25 days), removing the compounds from the culture, continuing the culture in compound-free medium for 22 to 0 days, harvesting the cells on day 25, and determining HBVWTD1-DNA (Fig. 2A). Culture medium was replaced with compound-containing or non-compound-containing fresh medium on days 3, 9 and 17 (Fig. 2A). For example, when we exposed HepG2.2.15 cells to E-CFCP (10 µM) for 11 days, removed E-CFCP from the culture, and further cultured the cells without E-CFCP, HBVWTD1-DNA production was virtually completely blocked (by 95–100%) for up to 14 days (Fig. 2B, right panel), while 10 µM ETV-induced inhibition under the same conditions deteriorated down to ∼40% by day 14 (Fig. 2B, left panel). Since TAF was cytotoxic at 10 µM, 1 µM TAF was examined. The % inhibition by 1 µM TAF decreased to ∼55% by day 14 (middle panel, Fig. 2B), while the inhibition by 1 µM E-CFCP was substantial (∼85%) and persisted significantly longer compared to that by 1 µM ETV and TAF. These data suggest that ETV and TAF at 1 µM do not exert potent anti-HBV activity and do not continuously block HBVWTD1-DNA production. It was also noted that, the inhibition of HBV-DNA production by the highest E-CFCP was virtually complete (by 100%) but that by ETV never reached 100% even the cells were cultured with its highest concentrations. The inhibition of HBV-DNA production by highest possible concentrations of TAF never reached 100%, either (Fig. 1B).
Fig. 2. E-CFCP well penetrates HepG2.2.15 cells and is efficiently tri-phosphorylated.

(A) Protocol for evaluation of anti-HBV activity persistence. (B) E-CFCP’s anti-HBV activity (>80% Inhibition) persisted ∼16-days after E-CFCP removal, while that of ETV/TAF was lost early. Intracellular HBV-DNA levels were quantified using qPCR and shown as % controls (no drug control value=100%). (C) Intracellular concentrations of E-CFCP in HepG2.2.15 cells are greater than those in ETV. (D) E-CFCP is more efficiently tri-phosphorylated in HepG2.2.15 cells (a) and PXB-cells (b) than ETV. Each concentration in (C) and (D) was determined using TOF-LC/MS system.
Intracellular concentration of E-CFCP is greater as compared to that of ETV
We then examined the intracellular concentration of ETV and E-CFCP, which represent the balance between the penetration through the membrane and intracellular degradation of the compounds, by incubating HepG2.2.15 cells for 1–6 hours and determining intracellular levels of ETV and E-CFCP using the TOF-LC/MS system. After 1-hour incubation at 100 µM, the intracellular concentrations of ETV and E-CFCP were comparable (Fig. 2C). However, in 2 hours, intracellular E-CFCP levels more than doubled. In 6 hours, E-CFCP concentrations further increased to reach approximately 200 pmol/2×106 cells. By contrast, the intracellular concentrations of ETV did not significantly increase even when the cells were incubated for 6 hours as compared to its 1-hour incubation concentrations, suggesting that E-CFCP’s cellular penetration might involve non-transporter-mediated entry such as rapid passive diffusion, although ETV’s penetration might involve transporter-mediated entry so that ETV penetration occurs slowly compared to E-CFCP. Since Z-CFCP was significantly less potent against HBV (Fig. 1B and Tables S1 and S2), we asked if Z-CFCP’s intracellular concentration was compromised compared to E-CFCP. As shown in Supplementary Figure 5A, the amounts of intracellular E-CFCP increased in a dose-response manner; however, the increase of Z-CFCP’s intracellular concentrations was much slower compared to that of E-CFCP.
E-CFCP is efficiently converted to its triphosphate form and persists longer intracellularly
The acquisition of inhibitory activity of NRTIs against HIV-1 and HBV requires tri-phosphorylation mediated by cellular kinases.13,14 Therefore, we examined how efficiently E-CFCP is intracellularly converted to E-CFCP-TP compared to ETV by quantifying the intracellular E-CFCP-TP and ETV-TP levels. HepG2.2.15 cells were incubated with 20 µM E-CFCP or ETV for 1–3 days and at the conclusion of each incubation, intracellular concentrations of E-CFCP-TP and ETV-TP were quantified. Additionally, after 3 day’s pre-incubation of the cells with E-CFCP or ETV, the cells were washed, and the culture was further continued in the absence of compounds for 1, 3, and 5 days, and intracellular E-CFCP-TP and ETV-TP concentrations were determined. Intracellular E-CFCP-TP concentrations were much higher than those of ETV-TP and progressively increased when the culture was continued over 3 days, while the ETV-TP concentrations were low and remained substantially the same over three days’ culture (Fig. 2D-a). When E-CFCP was removed from the culture and the cells were further cultured over 1, 3, and 5 days, E-CFCP-TP concentrations gradually decreased, although ETV-TP concentrations became very low after one day and became insignificant or undetected on day 3 and beyond (Fig. 2D-a). When we conducted similar assays using PXB-cells, intracellular E-CFCP-TP on day 3 after incubation with 100 µM E-CFCP was readily detectable and persisted by day 7 of culture in the absence of E-CFCP (Fig. 2D-b). However, intracellular ETV-TP concentrations were very low even on day 3 and became virtually insignificant by day 7 when cultured without ETV (Fig. 2D-b). These data should explain at least in part the reason the anti-HBV activity of E-CFCP persisted for extended periods of time compared to that of ETV (Fig. 2B). Z-CFCP did not well penetrate HepG2.2.15 cells and intracellular Z-CFCP-TP was not visibly identified (Fig. S5B), in line with its much less anti-HBV activity observed (Fig. 1B). We also examined the phosphorylation efficiency of TAF and its intracellular persistence in Hep2.2.15 cells and compared TFV-DP’s profiles with those of E-CFCP-TP and ETV-TP as seen in the Figure S6. Intracellular TFV-DP concentration was the highest among the three agents tested when the cells were exposed to TAF, ETV, or E-CFCP. The reason why TFV-DP levels were the highest among the three agents is likely that TAF is already monophosphorylated and is readily further phosphorylated to give TFV-DP. However, when TAF was removed from the culture and the cells were further cultured, TFV-DP concentrations rapidly downed and TFV-DP was not detected by day 3 and beyond, while ETV-TP was detected on day 3 and good levels of E-CFCP-TP were detected on day 5 in the absence of E-CFCP in culture. These data should explain the relatively rapid loss of anti-HBV activity upon removal of TAF shown in Figure 2B compared to E-CFCP.
E-CFCP potently blocks HBVWTC2 and HBVETV-RL180M/S202G/M204V replication in PXB-mice without significant adverse effects
Unlike pyrimidine nucleosides, purine nucleosides are in general susceptible to low pH and tend to get decomposed when orally administered.15,16 Thus, we examined whether E-CFCP decomposed when exposed to low pH using hydrochloric acid. E-CFCP was incubated in a pH 2 culture medium for 5 and 60 minutes at 37 °C and the E-CFCP-containing culture medium was gradually neutralized with NaOH to pH 7. Subsequently, anti-HBV activity of the once pH 2-exposed E-CFCP was determined in HepG2 2.15 cells. The inhibition of HBVWTD1-DNA production by the pH 2-exposed E-CFCP was comparable to pH 2-unexposed E-CFCP (Fig. 3A). When we performed the PK study of E-CFCP in PXB-mice, the oral bioavailability value was determined quite favorable as 70% (Fig. 3B).
Fig. 3. E-CFCP is acid-resistant and shows favorable PK profiles.

(A) Anti-HBV activity of once 0.01 N HCl (pH2)-exposed and acid-unexposed E-CFCP was evaluated in HepG2.2.15 cells (no-drug control=100%). E-CFCP does not lose its anti-HBV activity after pH2 for 5 min (a) and 60 min (b) exposure at 37°C. (B) Time course of the plasma concentrations of E-CFCP and its pharmacokinetic parameters after intravenous (i.v.) and intragastric (i.g.) administration in PXB-mice. t1/2, half-life; MRT, mean residence time; AUC, area under the concentration-time curve; CL, clearance; Vdss, distribution volume.
With these data, we administered ETV and E-CFCP (0.02 mg/kg/day) to chronically-HBVWTC2-infected PXB-mice over two weeks. ETV reduced serum HBVWTC2-DNA copy numbers by 2.0±0.07 log by the conclusion of the two-week administration. When ETV administration was terminated on day 14, viral breakthrough occurred by the end of two-week follow-up (upper left, Fig. 4A). E-CFCP administration also reduced serum HBVWTC2-DNA copies; however, the potency of E-CFCP was significantly greater than that of ETV between ETV and E-CFCP (2.2±0.06 log reduction) on day 21 (Fig. 4A, P=0.0005).
Fig. 4. E-CFCP reduces serum HBVWTC2- and HBVETV-RL180M/S202G/M204V-DNA in PXB-mice.

(A) HBVWTC2- and HBVETV-RL180M/S202G/M204V-DNA viremia reduction by E-CFCP over 14 days was greater than in ETV-receiving mice (P=0.0005 and P=0.012, respectively). (B) Serum human albumin levels of E-CFCP-treated, HBVWTC2- or HBVETV-RL180M/S202G/M204V-infected mice were comparable to those in ETV-receiving mice (P=0.3 and P=0.34, respectively). Body weights of E-CFCP-treated, HBVWTC2-infected mice were comparable to those of ETV-receiving mice (P=0.77), while those of HBVETV-RL180M/S202G/M204V-infected mice were greater than in ETV-receiving mice (P=0.0031), although such an apparent difference came from the difference when assigned and was thought insignificant. Thick bars denote 14-day administration period.
We also administered ETV and E-CFCP to chronically HBVETV-RL180M/S202G/M204V-infected PXB-mice at a dose of 0.2 mg/kg/day over two weeks. ETV totally failed to reduce serum HBVETV-RL180M/S202G/M204V-DNA throughout the 2-week administration (lower left, Fig. 4A,). However, E-CFCP significantly reduced DNA copy numbers by 1.84±0.09 log by the end of two-week administration (lower right, Fig. 4A, P=0.012). Following administration cessation, HBVETV-RL180M/S202G/M204V-DNA copy numbers kept going down for another week as seen in the HBVWTC2-infected PXB-mice receiving 0.02 mg/kg/day E-CFCP (upper right, Fig. 4A). Since TAF proved to be active against both HBVWTC2 and HBVETV-RL180M/S202G/M204V in vitro (IC50SB_cell=37 and 56 nM, respectively, Fig. 1C), we administered 0.2 mg/kg/day of TAF to chronically HBVETV-RL180M/S202G/M204V-infected PXB-mice for 2 weeks. However, no detectable viremia reduction was seen over the 2 weeks (Fig. S7). Serum human albumin levels in E-CFCP-treated HBVWTC2- or HBVETV-RL180M/S202G/M204V-infected mice were comparable to those in ETV-treated mice (P=0.3 and P=0.34, respectively). Body weights of E-CFCP-treated HBVWTC2-infected mice were comparable to those of ETV-treated mice (P=0.77). Body weights of ETV -treated HBVETV-RL180M/S202G/M204V-infected mice were lesser than those of E-CFCP-treated mice (P=0.0031); however, such apparent difference came from the weight difference when assigned and was thought insignificant.
When we administered E-CFCP at a higher dose (0.2 mg/kg/day) to PXB-mice, we observed greater reduction (2.6±0.1 log reduction on day 21) in serum HBVWTC2-DNA copies (Fig. 5A). Most notably, the reduction in the mice receiving 0.2 mg/kg/day E-CFCP persisted over 3 weeks after administration cessation and no significant viral breakthrough was seen in any of the three mice (Fig. 5A). It should also be noted that the greatest reduction of serum HBVWTC2-DNA was observed a week later following the treatment cessation with both E-CFCP dosing regimens. No significant changes in body weights or serum human albumin levels were seen in any of those mice (Fig. 5B).
Fig. 5. E-CFCP (0.2 mg/kg/day) exerts further potent and persistent reduction of serum HBVWTC2-DNA.

(A) Serum HBV-DNA levels were determined in additional mouse group experiments conducted different occasion from Figure 4. The reduction level (2.6±0.1 log reduction, day 21) of serum HBVWTC2-DNA was greater in 0.2mg/kg/day-receiving mice than that in the 0.02 mg/kg/day-receiving group (2.2±0.06 log reduction, Figure 4). Notably, the reduction persisted over 3 weeks after E-CFCP administration cessation. (B) No significant changes in body weights or serum human albumin levels were seen in any of the 3 PXB-mice treated with E-CFCP (0.2 ng/kg/day) for 2 weeks.
When we also monitored the changes of body weights and human serum albumin data over 8 weeks in PXB-mice treated with E-CFCP or ETV at a once daily dose of 0.2 mg/kg/day, no significant changes in both indicators between E-CFCP-receiving mice and ETV-receiving mice were observed (Fig. S8).
Structural analyses of E-CFCP-TP interactions with HBVWT-RT and HBVETV-R L180M/S202G/M204V-RT
We generated molecular models starting from previous homology-based structural models that used the sequence similarity of the active site region between HBV-RT and HIV-RT.8,9 The intramolecular hydrogen (H)-bond interactions stabilizing the β-sheet conformation around the active site of HBV-RT is shown in Figure 6A. There are multiple H-bond interactions responsible for the binding of ETV-TP and E-CFCP-TP with HBVWT-RT. Both ETV-TP and E-CFCP-TP have an amino-substituted purine group, which forms H-bond interactions with the backbone carbonyl oxygen of Met-171, and the phosphate has H-bond interactions with Ser-85 of HBVWT-RT. Moreover, a Mg2+ ion forms polar interactions with the phosphate group and a cluster of residues around the active site (Fig. 6B-a). Both E-CFCP-TP and Z-CFCP-TP have an additional 4′-cyano substituent, which ETV-TP does not have (Fig. 1A). The 4′-cyano moiety forms a H-bond interaction with the backbone NH of Phe-88 (Fig. 6B-b and -c). The E and Z isomers of CFCP-TP differ in the orientation of the fluorine on the ethylene substituent (Fig. 6B-c). This has important implications on the interactions of these NRTIs with HBVWT-RT. E-CFCP-TP forms a halogen bond interaction with the backbone NH of Asp-205, whereas no such interaction with either Asp-205 or any other HBVWT-RT residues is possible for Z-CFCP (Figs. 6B-b and -c). The van der Waals interactions are shown in Figures. S9-a through -d. Not only do the cyano and fluorine groups of E-CFCP-TP contribute to additional polar interactions, but they contribute to better van der Waals interactions with HBVWT-RT over ETV-TP. Overall, these additional polar and better van der Waals interactions of E-CFCP-TP appear to be responsible for its potency against HBVWT over ETV. The polar interactions of ETV-TP and E-CFCP-TP with HBVETV-RL180M/S202G/M204V-RT are also shown in Figs S9-e and -f. Apparently, HBVETV-RL180M/S202G/M204V-RT has subtle but important changes in van der Waals interactions. Notably, Leu-180 and Met-204 of HBVWT-RT are in close proximity and Leu-180 also has van der Waals interactions with Phe-88 (Figs. S9-g and -i). All these residues are important for the shape of the binding site. In HBVETV-RL180M/S202G/M204V-RT, the nature of these interactions is likely different (Fig. S9-j). Also, the S202G substitution may introduce the so-called “glycine kink” and change the conformation around the active site (Fig. S9-j). These changes in the active site conformation might be partially responsible for the loss of binding of ETV-TP to HBVETV-RL180M/S202G/M204V-RT. While these changes also affect E-CFCP-TP binding to HBVETV-RL180M/S202G/M204V-RT, the additional interactions derived from the 4′-cyano and fluorine should be able to mitigate the effect of such changes. We also carried out a modeling analysis about the possible interactions of a second magnesium in the active site of HBVWT-RT in complex with E-CFCP. Our modeling suggests that there is enough room in the active site to accommodate the second magnesium ion, and it can form polar interactions with the 3′-OH of 2′-dG-MP, Asp-83 and Asp-205 (Fig. S10). This magnesium might be responsible for the nucleotidyl transfer reaction.
Fig. 6. Structural modeling analyses of E- and Z-CFCP-TP interactions with HBVWT-RT.

(A) ETV-TP (a) and E-CFCP-TP (b) bound in the active site of HBVWT-RT. The active site is defined in part by YMDD residues that form two beta-sheets joined by a loop. The hydrogen-bond interactions between residues of the beta sheets are shown by yellow-dotted lines. (B) The polar interactions of ETV-TP, E-CFCP-TP, and Z-CFCP-TP with HBVWT-RT are shown in a, b, and c, respectively. The carbon atoms of ETV and E/Z-CFCP are shown in green, while the carbon atoms of HBVWT-RT in gray.
Discussion
E-CFCP potently blocked the infectivity and replication of various wild-type and drug-resistant HBV isolates in PXB-mice and its activity persisted over 1–3 weeks following treatment cessation, suggesting a long-acting feature with potential QW dosing schedule capabilities. A long post-withdrawal efficacy has been seen in clinical trials with nucleic acid polymers and therapies with IFN-based regimens. Nevertheless, nucleic acid polymers have not been of clinical utility and the use of IFN-based regimens have not been used often due to their inherent adverse effects. Lately, tenofovir has been used because of its favorable efficacy and clinically negligible resistance development; however, the efficacy of the currently available anti-HBV therapeutics including tenofovir is yet to be optimized and the emergence of resistant HBVs may become eventually problematic due to life-long medication of any anti-HBV drugs. In this context, E-CFCP is the first reported potentially long-acting and profoundly potent nucleoside analog for treating HBV infection as compared to the currently available therapeutics.
The IC50qPCR_cell values of E-CFCP and ETV against HBVWTD1 were comparable (Fig. 1B); however, it was noted that the inhibition of HBVWTD1 production by ETV failed to reach 100% even at its high concentrations used, although E-CFCP virtually completely blocked the production to below-detection-limit at 100 and 1,000 nM (Fig. 1B). Regarding this potency difference between E-CFCP and ETV, the intracellular concentration of ETV in HepG2.2.15 cells apparently reached its maximal levels after 1-hr-incubation of the cells with 20 and 100 µM and the intracellular concentrations stayed almost the same even with further exposure for 2–6 hours. By contrast, the intracellular concentrations of E-CFCP continued to increase when the cells were incubated longer (Fig. 2C). Moreover, the intracellular concentrations of ETV-TP, the active form of ETV, stayed almost the same when HepG2.2.15 cells were incubated with ETV for 1–3 days, while E-CFCP-TP levels progressively increased over the 3-day incubation period (Fig. 2D-a and Fig. S6). Intracellular E-CFCP-TP concentrations were also much higher and persisted longer than ETV-TP in PXB-cells (Fig. 2D-b). The two factors: (i) the low intracellular concentrations of ETV compared to E-CFCP and (ii) the poor build-up of ETV-TP should contribute to the difference of potency between ETV and E-CFCP. It was also noted that the inhibition of HBV-DNA production by high concentrations of TAF never reached 100% (Figs. 1B and 2B). In addition, the anti-HBVWTD1 and anti-HBVWTC2 potency of TAF was significantly less by 32–53-fold as compared to that of E-CFCP (Fig. 1B and 1C). These data suggest that HBV production inhibition by ETV and TAF is not potent enough even with its higher dosing regimens, while E-CFCP could completely block HBV’s infectivity and replication, although such potential advantage of E-CFCP should be confirmed in well-controlled clinical studies with more sensitive quantitative HBV-DNA tests built-in.
When HepG2.2.15 cells were exposed to 10 µM E-CFCP for 3–23 days, then E-CFCP was removed, and the culture was further continued without E-CFCP for 2–22 days (Fig. 2A), HBV-DNA synthesis was completely (100%) blocked by E-CFCP over 6-day culture without E-CFCP and >90% blockade was seen after 14 days (Figs. 2A and 2B). Moreover, >80% blockade was observed even after 16 days’ culture without E-CFCP (Fig. 2B, right panel). By contrast, ETV, at the same concentration (10 µM), never blocked HBV-DNA production completely throughout the culture period (Figs. 2A and 2B). In fact, when the cells were cultured for 2 days without ETV, its HBV-DNA production inhibition was by 90% (Fig. 2B). When cultured without ETV over 6 days, the inhibition downed to <80% (Fig. 2B, left panel). TAF at 10 µM was overly cytotoxic to HepG2.2.15 cells and 1 µM-TAF’s anti-HBV activity was examined. Notably, when HepG2.2.15 cells were cultured with 1 µM TAF, only 90% inhibition was gained (very left in Fig. 2B’s middle panel). Furthermore, when HepG2.2.15 cells were exposed to 1 µM TAF and cultured without TAF for 6 days, the inhibition by TAF was ∼65%. These data strongly suggest that anti-HBV activity of E-CFCP is more potent and could be more complete than ETV and TAF.
In the present study, TAF was as effective as E-CFCP against HBVETV-RL180M/S202G/M204V as examined in Huh7 cells (Fig. 1C). However, TAF at 0.2 mg/kg/day failed to block HBVETV-RL180M/S202G/M204V-DNA production in chronically-HBVETV-RL180M/S202G/M204V-infected PXB-mice (Fig. S7), while E-CFCP at the same dose significantly blocked HBVETV-RL180M/S202G/M204V viremia in PXB-mice (lower right, Fig. 4A). In this regard, Murakami et al. have reported that 3-week administration of as much as 90 mg/kg/day TDF (about 18-fold greater dose than the current clinical dose) brought about the viral load reduction by only approximately 1.5 logs in HBV-infected PXB-mice.17 Thus, we assume that while TDF and TAF are active against both wild-type and existing resistant HBV variants and do not allow the development of TFV-resistant variants in HBV-infected individuals, both agents are moderately effective against HBV as far as examined in PXB-mice. The mechanism, by which TDF does not exert good activity in PXB-mouse system remains to be elucidated and this should be an interesting topic for future research.
E-CFCP was acid-resistant and exhibited favorable oral bioavailability at around 70% (Fig. 3), comparable to that in ETV18, explaining the potent activity against HBVs in PXB-mice (Figs. 4A and 5). While it remains to determine how the fluorine atom of E-CFCP contributed to its favorable intracellular concentrations and efficient intracellular tri-phosphorylation, considering the poor anti-HBV activity of Z-CFCP, the E-positioned fluorine in the cyclopentyl moiety likely contributed to E-CFCP’s favorable biochemical features. The halogen bond interaction of the E-positioned fluorine with Asp-205 (Figs. 6B-b and -c) should also contribute to E-CFCP’s potent anti-HBV activity (Figs. 6B-b and -c).
One can say that E-CFCP is an analog of ISL.5 The mechanisms of the highly potent activity of ISL against wild-type HIV-1 and multi-drug-resistant variants5 is likely that ISL is efficiently tri-phosphorylated and ISL-TP serves as the first-in-class the first-in-class translocation-defective delayed type HIV-1-RT inhibitor.19 ISL is now under Phase III clinical trials in multiple countries including US and Japan. Moreover, an implant containing ISL has been shown to block HIV-1 effectively for at least 3 months, possibly for a full year,20 implying that ISL is highly non-toxic and has very low bioaccumulation potential. E-CFCP also serves as a nucleoside HIV-1-RT inhibitor and exerts activity against HIV-1 (IC50MTT of E-CFCP against HIV-1LAI=205±23.8 nM) as shown in Table S1. Thus, it is likely that E-CFCP also serves as the first-in-class translocation-defective HBV-RT inhibiter as well as a delayed-type HBV proviral DNA chain terminator.
In conclusion, the present data warrant that further study be conducted toward clinical development of E-CFCP as a potential therapeutic for infection with wild-type HBV and drug-resistant variants with potential long-acting dosing regimen capabilities. If E-CFCP’s clinical utility is proven, E-CFCP should improve the otherwise poor adherence to current QD regimens, particularly, in the low-income countries and regions, where no city water or electricity services are presently available. Moreover, E-CFCP’s high anti-HBV potency could completely and more durably shut-off HBV-DNA synthesis compared to the presently existing anti-HBV therapeutics.
Supplementary Material
Highlights.
E-CFCP potently reduces HBV viremia without toxicities in human liver chimeric mice.
E-CFCP efficiently penetrates into and is tri-phosphorylated in human hepatocytes.
E-CFCP represents the first reported long-acting NRTI for treating HBV infection.
Acknowledgements:
We are grateful to Asuka Fujiwara for her technical help. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH, Bethesda, MD (https://hpc.nih.gov).
Financial Support
The present work was supported in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (HM, DV); a grant for Development of Novel Drugs for Treating HBV Infection from Japan Agency for Medical Research and Development (YT; JP20fk0310101, HM:JP16fk0310501 and JP20fk0310113); grants for the Research Program on HIV/AIDS from the Japan Agency for Medical Research and Development under grant number JP18fk0410001 (HM); and grants from Japan Society for the Promotion of Sciences (HM:17H04222 and 17K19577).
Data availability statement:
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.
List of Abbreviations:
- ADV
adefovir pivoxil
- CAdA
4’-C-cyano-2-amino-2’-deoxyadenosine
- CC50
50% cytotoxicity concentration
- CdG
4’-C-cyano-2’-deoxyguanosine
- CHB
chronic hepatitis B
- CMCdG
4’-cyano- methylenecarbocyclic-2’-deoxyguanosine
- E-CFCP
(1S,3S,5S,E)-3-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy1-(hydroxymethyl)cyclopentane-1-carbonitrile
- ETV
entecavir
- HBVADV-RA181T/N236T
ADV-resistant HBV
- HBVETV-RL180M/S202G/M204V
ETV-resistant HBV
- HBV
Hepatitis B virus
- HIV-1
human immunodeficiency virus type 1
- HBVWTC2
wild type HBV genotype C2
- HBVWTD1
wild type HBV genotype D1
- HBVWT
wild-type HBV
- hPBMCs
human peripheral blood mononuclear cells
- IC50
50% inhibitory concentration
- ISL
islatravir or 4’-ethynyl-2-fluoro-2’-deoxyadenosine or EFdA/MK-8591
- Mitotox50
50% mitochondria toxicity concentration
- mtDNA
mitochondrial DNA
- qPCR
quantitative polymerase chain reaction
- PXB-cells
fresh human hepatocytes
- PXB-mice
cDNA-uPA/SCID chimeric mice with humanized liver or human-liver-chimeric mice/ humanized mice
- RT
reverse transcriptase
- S.I.
specificity index
- SI
supplementary information
- TAF
tenofovir alafenamide
- TDF
tenofovir disoproxil fumarate
- TP
triphosphate
- Z-CFCP
(1S,3S,5S,Z)-3-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-2-(fluoromethylene)-5-hydroxy-1-(hydroxymethyl)cyclopentane-1-carbonitrile
Footnotes
Disclosures:
Yasuhito Tanaka has received personal fees from Fujirebio Inc., personal fees and grants from Gilead Sciences Inc., and grants from the Board of Trustees of the Leland Stanford Junior University. Except for Yasuhito Tanaka, none of the co-authors has issues to disclose. Please refer to the accompanying ICMJE disclosure forms for further details.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yuen MF, Chen DS, Dusheiko GM, Janssen HLA, Lau DTY, Locarnini SA,et al. Hepatitis B virus infection. Nat Rev Dis Primers 2018; 4:18035. [DOI] [PubMed] [Google Scholar]
- 2.Likhitsup A, Razumilava N, Parikh ND. Treatment for Advanced Hepatocellular Carcinoma: Current Standard and the Future. Clin Liver Dis (Hoboken) 2019; 13:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vittal A, Ghany MG . WHO Guidelines for Prevention, Care and Treatment of Individuals Infected with HBV: A US Perspective. Clin Liver Dis 2019; 23:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopatin U Drugs in the Pipeline for HBV. Clin Liver Dis 2019; 23:535–555. [DOI] [PubMed] [Google Scholar]
- 5.Nakata H, Amano M, Koh Y, Kodama E, Yang G, Bailey CM, et al. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4’-ethynyl-2-fluoro-2’-deoxyadenosine. Antimicrob Agents Chemother 2007; 51(8):2701–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molina JM, Yazdanpanah Y, Saud AA, Bettacchi C, Anania CC, DeJesus E, et al. MK-8591 at doses of 0.25 to 2.25 mg QD, in combination with doravirine establishes and maintains viral suppression through 48 weeks in treatment-naïve adults with HIV-1 infection. IAS 2019 WEAB0402LB.
- 7.Game-changer for treatment and prophylaxis of HIV infection: http://www.independent.co.uk/news/health/hiv-cure-implant-prevent-aids-drugs-pills-lgbt-safe-sex-a9020551.html. [Google Scholar]
- 8.Takamatsu Y, Tanaka Y, Kohgo S, Murakami S, Singh K, Das D, et al. 4’-modified nucleoside analogs: potent inhibitors active against entecavir-resistant hepatitis B virus. Hepatology 2015; 62:1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higashi-Kuwata N, Hayashi S, Das D, Kohgo S, Murakami S, Hattori SI, et al. CMCdG, a Novel Nucleoside Analog with Favorable Safety Features, Exerts Potent Activity against Wild-Type and Entecavir-Resistant Hepatitis B Virus. Antimicrob Agents Chemother 2019; 63: e02143-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006. 58(3):621–81. [DOI] [PubMed] [Google Scholar]
- 11.Mazzucco CE, Hamatake RK, Colonno RJ, Tenney DJ. Entecavir for treatment of hepatitis B virus displays no in vitro mitochondrial toxicity or DNA polymerase gamma inhibition. Antimicrob Agents Chemother 2008; 52:598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furihata T, Morio H, Zhu M, Suzuki Y, Ide H, Tsubota A, et al. Human organic anion transporter 2 is an entecavir, but not tenofovir, transporter. Drug Metab Pharmacokinet 2017; 32:116–119. [DOI] [PubMed] [Google Scholar]
- 13.Mitsuya H, Jarrett RF, Matsukura M, Di Marzo Veronese F, DeVico AL, Sarngadharan MG, et al. Long-term inhibition of human T-lymphotropic virus type III/lymphadenopathy-associated virus (human immunodeficiency virus) DNA synthesis and RNA expression in T cells protected by 2’,3’-dideoxynucleosides in vitro. Proc Natl Acad Sci U S A 1987; 84:2033–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kassianides C, Hoofnagle JH, Miller RH, Doo E, Ford H, Broder S, et al. Inhibition of duck hepatitis B virus replication by 2’,3’-dideoxycytidine. A potent inhibitor of reverse transcriptase. Gastroenterology 1989; 97:1275–80. [DOI] [PubMed] [Google Scholar]
- 15.Marquez VE, Tseng CK, Mitsuya H, Aoki S, Kelley JA, Ford H Jr, et al. Acid-stable 2’-fluoro purine dideoxynucleosides as active agents against HIV. J Med Chem 1990; 33:978–985. [DOI] [PubMed] [Google Scholar]
- 16.Abdel-Hamid M, Novotny L, Hamza H. Stability study of selected adenosine nucleosides using LC and LC/MS analyses. J Pharm Biomed Anal 2000; 22:745–755. [DOI] [PubMed] [Google Scholar]
- 17.Murakami E, Tsuge M, Hiraga N, Kan H, Uchida T, Masaki K, et al. Effect of tenofovir disoproxil fumarate on drug-resistant HBV clones. J Infect. 2016;72:91–102. [DOI] [PubMed] [Google Scholar]
- 18.Yan JH, Bifano M, Olsen S, Smith RA, Zhang D, Grasela DM, et al. Entecavir pharmacokinetics, safety, and tolerability after multiple ascending doses in healthy subjects. J Clin Pharmacol 2006; 46:1250–1258. [DOI] [PubMed] [Google Scholar]
- 19.Salie ZL, Kirby KA, Michailidis E, Marchand B, Singh K, Rohan LC, et al. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4’-ethynyl-2-fluoro-2’-deoxyadenosine (EFdA). Proc Natl Acad Sci U S A. 2016; 113:9274–9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grobler JA, Fillgrove KL, Hazuda DJ, Huang Q, Lai MT, Matthews RP, et al. MK-8591 Potency and PK Provide High Inhibitory Quotients at Low Doses QD and QW. Conference on Retroviruses and Opportunistic Infections, Seattle, Washington, March 4-7, 2019. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article [and/or] its supplementary materials.