

Abstract
Since its discovery nearly a century ago, over 100,000 studies of growth hormone (GH) have investigated its structure, how it interacts with the GH receptor and its multiple actions. These include effects on growth, substrate metabolism, body composition, bone mineral density, the cardiovascular system and brain function, among many others. Recombinant human GH is approved for use to promote growth in children with GH deficiency (GHD), along with several additional clinical indications. Studies of humans and animals with altered levels of GH, from complete or partial GHD to GH excess, have revealed several covert or hidden actions of GH, such as effects on fibrosis, cardiovascular function and cancer. In this Review, we do not concentrate on the classic and controversial indications for GH therapy, nor do we cover all covert actions of GH. Instead, we stress the importance of the relationship between GH and fibrosis, and how fibrosis (or lack thereof) might be an emerging factor in both cardiovascular and cancer pathologies. We highlight clinical data from patients with acromegaly or GHD, alongside data from cellular and animal studies, to reveal novel phenotypes and molecular pathways responsible for these actions of GH in fibrosis, cardiovascular function and cancer.
Recombinant human growth hormone (hGH) is used primarily to treat children with short stature due to GH deficiency (GHD). In addition, hGH is approved for use in children born small for gestational age, as well as children with idiopathic short stature, Turner syndrome, Noonan syndrome, Prader–Willi syndrome, short stature homeobox-containing gene deficiency and chronic renal insufficiency, and in adults with GH deficiency. Despite this success, controversial issues remain, and not all indications are approved in all countries1,2.
The Argentine clinician Bernardo Houssay discovered an adverse effect of GH nearly 90 years ago; namely its ability to inhibit insulin action, which is now known as its diabetogenic effect3. Since that time and despite the astonishing success of hGH, several puzzling actions of GH have been and continue to be described. Here, we review three interrelated covert actions reported in adult mice and humans: fibrosis, cardiovascular disease and cancer. Many have joked that ‘too much of a good thing is bad’, which in this context is too much GH. Interestingly, the absence of GH action in animals and humans, despite short stature, results in resistance to type 2 diabetes mellitus (T2DM) and cancer, and improvements in other indicators of healthspan4. With regard to longevity, mice with a reduction or absence in GH action have a robust and reproducible increase in lifespan5. Although data from humans is insufficient to draw firm conclusions, some cohorts of individuals with isolated GHD (for example, in the Brazilian Itabaianinha cohort) have attained extreme longevity despite representing a fairly small proportion of the population. Such observations suggest that the findings in rodents are relevant to humans5. The effects of the GH–insulin-like growth factor 1 (IGF1) axis on human ageing, how GH levels change during ageing and the relationship to age-related diseases are discussed in detail elsewhere6.
In this Review, we explore the possibility that too much, too little or inappropriately expressed GH might provoke covert physiological actions related to fibrosis, cardiovascular function and cancer development.
GH and fibrosis
Fibrosis is characterized by excessive accumulation of the extracellular matrix (ECM) within tissues. Although fibrosis is initially a protective and adaptive response to wound healing and tissue repair, it can become uncontrolled and lead to tissue dysfunction and pathology. Almost every tissue type can be affected by pathological fibrosis; however, heart, adipose tissue, liver, kidney, intestine and skin are among the best studied. Tissue fibrosis is thought to be a major underlying cause of death in humans, with some studies estimating that it is associated with as much as 45% of all-cause mortality in industrialized nations7. As GH has an important role in growth and collagen turnover, it is a likely factor in ECM remodelling and fibrosis formation.
Influence of GH on collagens
A compelling reason for a GH–fibrosis link is the close correlation between GH exposure and collagen production. For example, in vitro studies of GH treatment of cells indicate increases in Colla1, Col3a1 and Col6a1 expression in differentiated adipocyte-like 3T3-L1 cells (a mouse cell line)8 and type I collagen mRNA and protein in rodent intestinal primary myofibroblasts9. Animal studies have shown a similar positive correlation between collagen and GH. For example, GH treatment increases collagen IV protein in mouse neural tissue damaged by stroke10, and type I collagen RNA and protein in the jejunum of GH-treated rats9. Chronic exposure to excess GH in rodent models also increases collagen expression in other tissues such as kidneys and tendons11,12.
The data in adult humans are similar. Both acute and chronic GH treatment in older men increases collagen expression in tendon and muscle13. Furthermore, the same study failed to detect an effect of GH on muscle fibre synthesis13. Acromegaly is a rare disease usually caused by a benign GH-producing pituitary tumour and elevated serum levels of IGF1 (REF.14). Adult patients with acromegaly have increased collagen turnover and serum levels of type I collagen and procollagen III amino-terminal propeptide (PIIINP)15, which decline with remission of the disease16,17. The close association among GH, collagen synthesis and turnover, and fibrosis in clinical populations is further illustrated by several examples, including the use of validated tests to detect GH doping in sport that are based on elevated serum levels of IGF1 and PIIINP levels18. Moreover, a marker of growth plate activity and overall rate of linear bone growth (the intact trimeric non-collagenous domain of type X collagen) shows promise for reflecting GH action in children and adults with GHD19. Finally, hGH is used in a combination therapy to treat osteogenesis imperfecta, an inherited connective tissue disorder characterized by a quantitative or qualitative defect in collagen synthesis20.
GH and other fibrosis-promoting factors
GH influences many factors besides collagens that contribute to fibrosis, including ECM-modifying proteins, several proteins and pathways implicated in fibrosis (for example, the transforming growth factor-β (TGFβ) pathway and mitogen-activated protein kinase (MAPK) pathways), senescence, immune cell function, and fibroblast activation or plasticity. In general, fibrosis is also often preceded by and closely associated with inflammation7. These fibrotic effects of GH are frequently context-specific and tissue-specific and not always in a direction that favours fibrosis. For example, an ECM-degrading endopeptidase, matrix metalloproteinase 2 (MMP2), is decreased after GH treatment of individuals with GHD21, but increased in patients with active acromegaly22. Likewise, TGFβ, a master regulator and promoter of fibrosis in multiple tissues, is regulated distinctly by GH in a tissue-specific manner. For example, TGFβ expression is decreased by GH in primary cardiac fibroblasts23. Furthermore, TGFβ expression is increased in the glomeruli of bovine GH (bGH) transgenic mice, a model of GH overexpression11. Interestingly, GH action itself might also be influenced by ECM-modifying proteins. Tissue inhibitor of metalloproteinase 3 (TIMP3) is unique among the TIMP family because it alone has a high affinity for ECM proteoglycans and possesses the broadest range of substrates. TIMP3 modulates GH receptor (GHR) abundance on the cell surface in human cell lines that stably express GHR and JAK2, and dampens the GH-induced intracellular signalling cascade, revealing an interesting interplay between GH and the ECM24.
Senescence.
Cellular senescence is characterized by irreversible cell cycle arrest and a senescence-associated secretory phenotype (SASP). The SASP is the main feature of senescent cells and includes pro-inflammatory cytokines and chemokines, growth factors and proteases, which contribute to a harmful tissue microenvironment. However, whether senescence is beneficial or detrimental for the development of fibrosis remains debated. For example, senescence might be beneficial by preventing fibroblast differentiation and the ability of these fibroblasts to contribute to liver fibrosis, based on studies using senolytic chimeric antigen receptor T cells that efficiently target and ablate senescent cells in mice25. However, senescent cells could be detrimental, as removal of senescent cells in rodents either pharmacologically or genetically can reverse fibrosis at least in some tissues such as lung26, and can extend median lifespan27. Of note, GH and/or IGF1 levels are positively correlated with senescence and SASP in white adipose tissue in mice28, as well as in cardiomyocytes and skin fibroblasts from patients with acromegaly29, and in human primary fibroblasts and mouse embryonic fibroblasts30. In fact, GH can also be secreted by senescent cells, thus constituting a component of the SASP31. By contrast, however, GH treatment attenuated senescence of primary human endothelial progenitor cells32, and IGF1 overexpression in rats improved stress-induced senescence in the liver33.
Fibroblasts.
Dysregulation of fibroblasts has long been considered a root cause of fibrosis. Data from the past 5 years show marked heterogeneity in fibroblast subtypes, which localize to unique anatomical sites and have distinct physiological functions34. Many studies have shown that GH and/or IGF1 influence the differentiation and proliferation of fibroblasts35,36. This association has been best studied in wound healing. For example, GH was identified as an inhibitor of TGFβ-induced myofibroblast differentiation in the skin of bGH transgenic mice37. A 2020 study showed that fibroblast activation protein (FAP) has been linked to GH action in humans16. FAP is a cell surface protease that has an important role in the degradation of ECM and with known substrates that include type I and III collagens38. FAP expression is limited after birth except in activated fibroblasts and in conditions associated with notable ECM remodelling, such as liver fibrosis39. Interestingly, serum levels of FAP are elevated in patients with untreated acromegaly and become considerably reduced after disease management, which closely correlates with reductions in markers of collagen turnover16. Additional studies are needed to determine if GH directly influences FAP abundance and action to influence ECM remodelling, as well as the ability of FAP to serve as a biomarker for GH-induced fibrosis.
Fibrosis and GH: the yin and the yang
As outlined above, GH has a complex relationship with many factors that contribute to fibrosis and ECM remodelling. Whether GH promotes or reduces pathological tissue fibrosis depends on the experimental conditions, the tissues analysed and the degree of GH signalling.
The yin: acromegaly and GH-transgenic mice.
Acromegaly is accompanied by soft-tissue enlargement and increases in ECM constituents; for example, glycosaminoglycan deposits that contribute to generalized oedema are apparent in the skin of patients with acromegaly40. Both early autopsy studies done in the 1980s41 as well as in vivo studies from 2015 using cardiac MRI have revealed myocardial fibrosis in adult patients with acromegaly42. Furthermore, increased hepatic fibrosis has also been shown in subpopulations of adult patients with acromegaly, particularly those that are already at genetic risk of developing hepatic steatosis43. Finally, hard thyroid nodules are common in patients with acromegaly and are also thought to be caused by increased nodular fibrosis44. However, overdiagnosis due to surveillance bias is inherent with regard to thyroid nodules, and further studies are needed45.
Data derived from mice are more definitive of the actions of excess GH and IGF1 in inducing fibrosis and organ dysfunction. In fact, most tissues assessed for fibrosis in bGH mice show some increase in fibrosis at older ages (FIG. 1) compared with wild-type mice, which might reflect an unexplored benefit of somatopause (the gradual decline in GH secretion that occurs with ageing). The older bGH mice have an enlarged heart with impaired function and marked distinct perivascular and interstitial fibrosis46,47. These older transgenic mice also have renal damage with increased fibrosis and glomerular lesions47. Marked fibrosis is also apparent in their adipose tissue, more prominently in subcutaneous depots8, as well as in the intestines48. Severe skin fibrosis has also been found in bGH mice, which is more prominent in males than females37. Overall, increased fibrosis has been observed in many tissues assessed in bGH mice relative to controls, with males often exhibiting a more robust phenotype. By contrast, several mouse lines with decreased GH action (such as GHR antagonist mice8, mice with GHR disruption in adipocytes49 or mice deficient in GH50) have decreased fibrosis, at least in adipose tissue. Collectively, these data in humans with acromegaly and GH transgenic mice provide strong support of fibrosis being an underlying component of the organ dysfunction that is common with pathological increases in GH and/or IGF1 action.
Fig. 1 |. GH overexpression in mice induces tissue fibrosis.
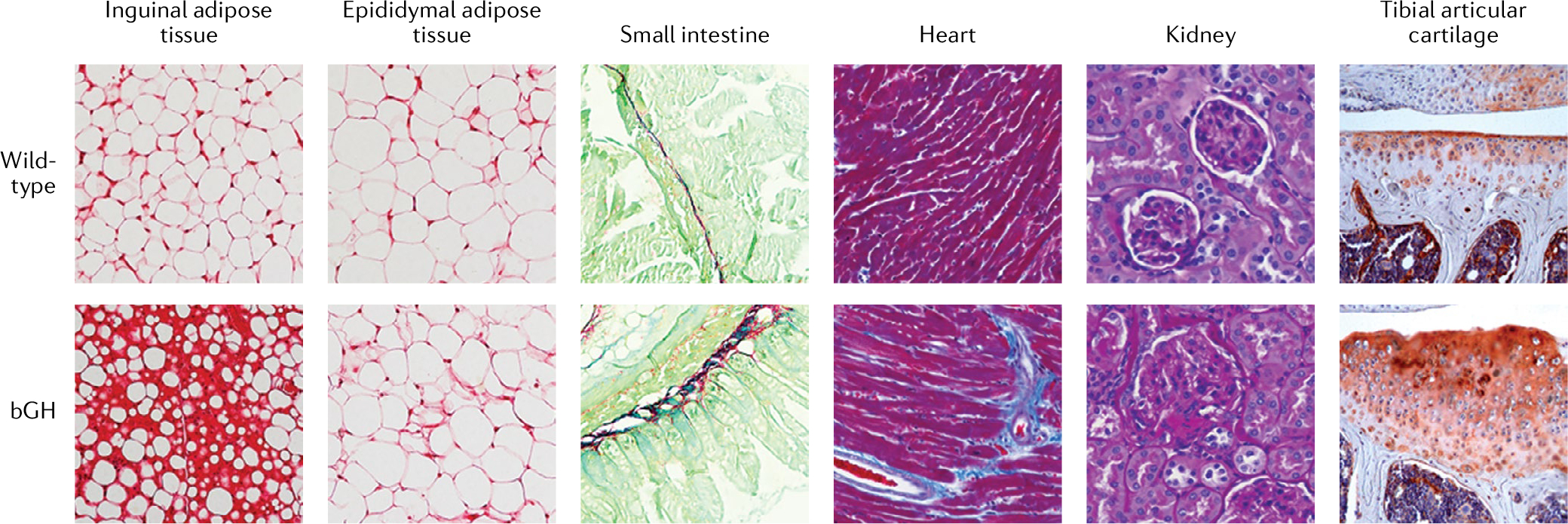
Histology of assorted tissues in aged male mice (10–13 months of age) overexpressing bovine growth hormone (bGH) and wild-type control mice. White adipose tissue from inguinal or epididymal adipose depots was stained with Sirius red (a non-specific red collagen stain), Swiss rolls of the small intestine were stained using Sirius Red and Fast Green (which stains non-collagenous protein), heart was stained with Masson’s trichrome (which stains connective tissue blue) and kidney sections were stained with periodic acid Schiff stain (which stains connective tissue a purple–magenta colour). The tibial articular cartilage images were generated via immunostaining with a type X collagen (a bone-specific collagen)-specific antibody (orange–red colour). In bGH mice, the increased GH activity results in increased fibrosis in the tissues shown. The histological images of tibial articular cartilage are courtesy of S. Zhu, Ohio University.
The yang: therapeutic potential of GH.
The benefits of GH in promoting collagen deposition are best illustrated by its effect on promoting longitudinal growth and bone acquisition51. However, GH might also have therapeutic potential for selected fibrotic diseases. These benefits might relate to restored GH levels in deficient states or to indirect influences on other disease pathologies or severe conditions. For example, studies have suggested that the dysregulation of the GH–IGF1 axis in patients with morbid obesity or in adult patients with hypopituitarism and GHD might contribute to the severity of hepatic fibrosis52,53. Furthermore, in at least one small study of adult patients with GHD, GH replacement therapy improved hepatic fibrosis54. Another example of GH action to attenuate fibrosis was provided by a rat model of intestinal inflammation, in which GH improved rather than exacerbated intestinal fibrosis55, Moreover, in a randomized controlled trial, GH therapy was beneficial in the treatment of severely burned children, in whom it promoted healing without the development of hypertrophic scarring caused by tissue fibrosis56. Initial small, randomized studies in patients with large burns treated with GH have shown promising clinical benefit. However, treatment with GH also induces hyperglycaemia in adults with severe burns, which raises questions for its utility in clinical practice in the treatment of large burns57.
Importantly, several limitations exist for the use of GH therapy for fibrotic disease. First, in most studies that showed some benefit of GH therapy on fibrosis, GH was restored to normal levels in patients with GHD. Second, many studies assessed acute or short-term GH therapy without evaluating the long-term cardiometabolic effects. Third, attenuation of fibrosis, treatment of burns and improvement in obesity and its complications are non-approved uses of GH.
Summary
Overall, the available data suggest an intricate balance between having sufficient GH action to promote favourable ECM remodelling and avoiding excess GH action that promotes ECM deposition and scarring, thereby resembling a ‘Goldilocks effect’. Although the focus of this Review is GH, disentangling the direct effects of GH versus indirect effects exerted via IGF1 is difficult in vivo. Endocrine and paracrine IGF1 also has an important role in fibrosis development. For example, local IGF1 administration stimulates in vivo tendon collagen synthesis in healthy, sedentary humans13. In addition, overexpression of IGF1 receptor (IGF1R) in human primary dermal fibroblasts suggests a role for IGF1 signalling in the development of keloid and hypertrophic scarring58. However, excess levels of GH, but not IGF1, have been shown to promote kidney glomerulosclerosis59. Together, these findings illustrate the need for additional studies or experimental systems that can help delineate the contributions of each individual hormone to fibrosis in specific tissues. In addition, further studies are needed to evaluate whether GH and IGF1 are primary drivers of fibrosis or if fibrosis occurs secondary to organ dysfunction.
GH and cardiovascular disease
GH hypersecretion: acromegaly
The onset of acromegaly is gradual, with a diagnostic delay of 5–10 years in most patients, which has negative therapeutic and prognostic implications. Patients with acromegaly are exposed to long-standing GH and IGF1 excess and have increased cardiovascular mortality60 that is associated with hypertension and heart failure14,61. Interestingly, atherosclerosis is not always present in acromegaly despite the presence of classic risk factors such as hypertension, insulin resistance and T2DM62. By contrast, typical cardiomyopathy present in active acromegaly is cardiomegaly and, in particular, hypertrophy of the left ventricle14,62. Histopathology of cardiac tissue from patients with acromegaly reveals pronounced interstitial fibrosis (FIGS. 1,2), myocardial hypertrophy and evidence of myocarditis with infiltration of inflammatory cells41. Importantly, 85% of patients with acromegaly have been observed to have interstitial fibrosis41. In addition, regurgitation of the aortic and mitral valves, as well as diastolic dysfunction, are described in acromegaly, whereas systolic dysfunction is a rare and late-onset occurrence14,62.
Fig. 2 |. The pleiotropic actions of GH–IGF1 on the cardiovascular system, and electrolyte and water balance in humans.
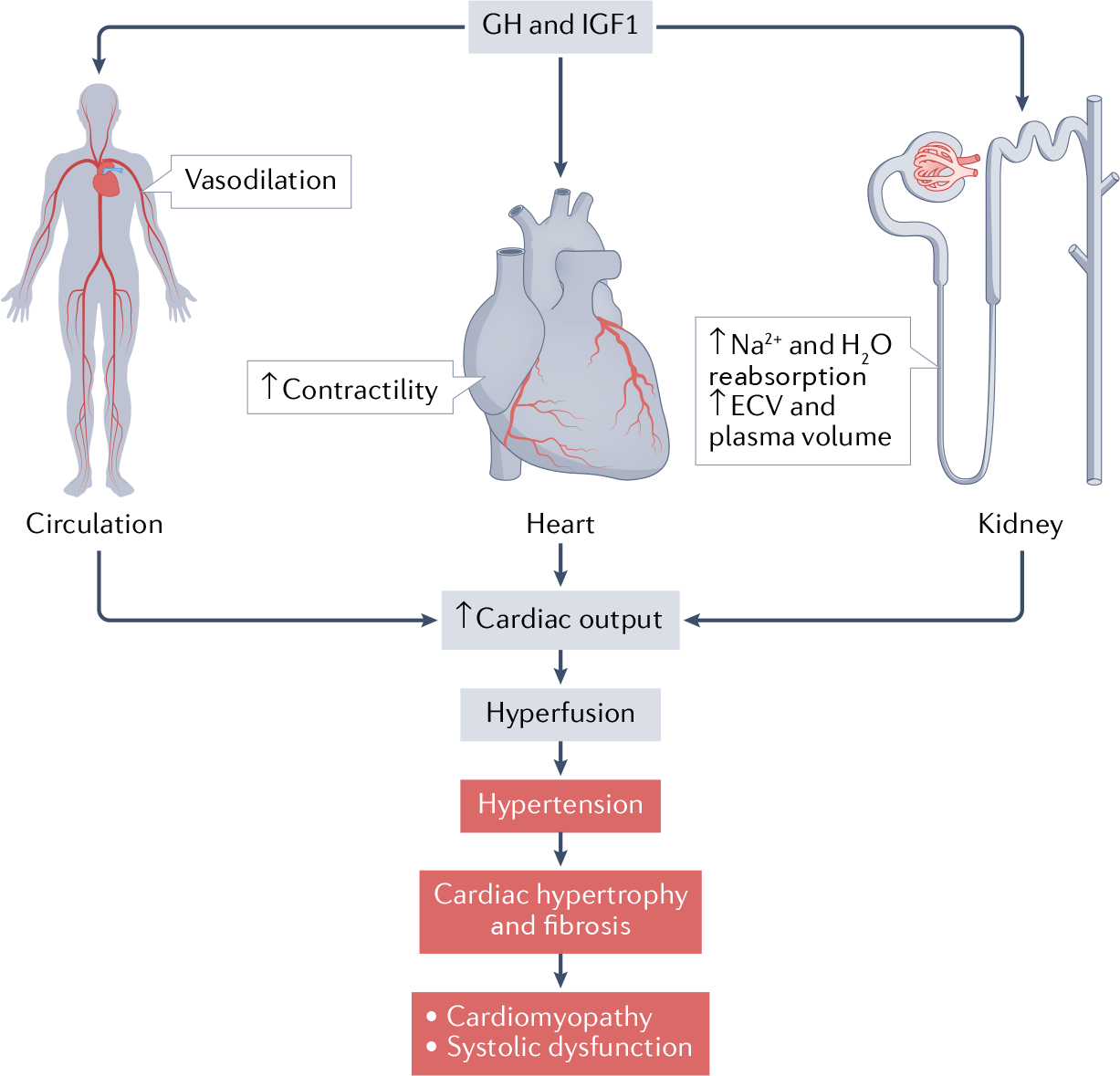
Growth hormone (GH) and insulin-like growth factor 1 (IGF1) promote renal reabsorption of sodium and water and thereby increase the extracellular volume (ECV) and plasma volume. This effect is accompanied by nitric oxide-mediated vasodilation and increased myocardial contractility, which translate into increased cardiac output. This increase results in hyperperfusion and reduced total peripheral resistance. Pathological GH–IGF1 excess (such as in uncontrolled acromegaly) results in hypertension and cardiac hypertrophy. We hypothesize that GH–IGF1-induced fibrosis eventually contributes to the development of cardiomyopathy and heart failure (systolic dysfunction). Pathological processes are highlighted in the figure in red.
Due to the gradual onset of the disease and the paucity of long-term prospective studies, the pathogenesis of the cardiovascular complications of overt acromegaly remains elusive. Individual and combined effects can also be exerted by GH and IGF1, further complicating the picture. However, short-term experimental studies in humans could provide clues. For example, 1 week of supra-physiological GH administration in healthy men aged ~30 years substantially increased fractional shortening of the left ventricle, which was mainly attributed to a reduced end-systolic diameter63. In concomitance with a significant 15% increase in resting heart rate, cardiac output increased by ~13% without a change in mean arterial blood pressure63,64, which suggests a simultaneous reduction in peripheral vascular resistance. Increased blood flow and reduced vascular resistance via a nitric oxide-dependent mechanism are also observed after short-term, high-dose GH infusion into the brachial artery of healthy individuals65. In addition, the GH–IGF1 axis is well recognized to acutely induce fluid and sodium retention66 and increases heart rate without a change in blood pressure67. Similar findings of increased myocardial contractility and cardiac output are also encountered in patients with active acromegaly68 together with sodium and fluid retention69. Taken together, sustained fluid and sodium retention, in combination with direct inotropic effects of GH–IGF1, are hypothesized to promote hyperperfusion, cardiac hypertrophy and ultimately systemic hypertension in acromegaly66. In combination with other risk factors such as insulin resistance70, T2DM and obstructive sleep apnoea, these effects might eventually cause cardiomyopathy and heart failure in the absence of coronary atherosclerosis (FIG. 2).
Treatment of acromegaly that achieves biochemical remission71, translates into a life expectancy that is close to that in the general population61. Regarding cardiovascular function, successful surgical resection of the GH-producing adenoma improves diastolic function and reduces left ventricular mass, together with reductions in heart rate and blood pressure72. Similar beneficial effects have been reported after disease control obtained by pharmacological treatment with a somatostatin analogue alone73 as well as with a GHR antagonist (pegvisomant)14. In addition, disease control in acromegaly improves certain risk factors for cardiovascular disease, such as insulin resistance and obstructive sleep apnoea; however, body composition changes towards increased adipose mass and reduced lean body mass14.
GH deficiency
GHD in adults is caused by diseases affecting the hypothalamic–pituitary region, the most common being benign tumours. The diagnosis of GHD in adults must be made in the correct clinical context (that is, in patients with a well-defined hypothalamic–pituitary disorder) using a validated GH stimulation test, unless four or more anterior pituitary hormone deficiencies exist together with a low serum concentration of IGF1 (REF.74). GHD in adults can occur in isolation but is commonly associated with other pituitary hormone deficiencies. These deficiencies can also affect patient outcomes; for example, inadequate or untreated sex steroid deficiencies in women with hypopituitarism can affect their GHD outcome, inadequate treatment of central hypothyroidism might have negative effects on many metabolic processes and inadequate treatment of adrenal insufficiency has negative effects on metabolism as well as on risk of premature mortality75–77. Cardiovascular disease is a complication in adults with GHD; adults with GHD have increased blood pressure, a phenotype resembling the metabolic syndrome78 and increased cardiovascular mortality79. Several cardiovascular risk factors are increased in adults with GHD, which can be explained by many of the effects of GH and IGF1 on the cardiovascular system (which expresses both GHR and IGF1R80), as well as effects on lipid and lipoprotein metabolism and systemic inflammation81.
Adults with GHD often have overweight or obesity, with accumulation of visceral adipose tissue82,83. Studies on their body composition show that their total body adipose mass is increased (predominantly abdominal), muscle mass is reduced and extracellular fluid is reduced. GH antagonizes the effects of insulin in several important tissues such as liver, muscle and adipose tissue83,84. Studies in children and young adults with GHD have clearly shown that these individuals have increased insulin sensitivity, and young children in particular might even experience hypoglycaemia85. Surprisingly, adults with long-standing untreated GHD deficiency showed markedly reduced insulin sensitivity, measured using the hyperinsulinaemic glucose clamp technique86,87. Their abdominal adiposity together with reduced muscle mass, reduced serum concentration of IGF1 and physical inactivity due to reduced exercise capacity might help explain this finding83,86,88. Abdominal adiposity and associated insulin resistance are therefore important mediators of premature atherosclerosis and increased risk of cardiovascular disease seen in adults with GHD.
Adults with GHD have reduced nitric oxide urinary excretion, increased peripheral vascular resistance89, increased sympathetic nervous system activity90 and increased systemic inflammation81 (FIG. 2). A consistent finding is that the extracellular fluid volume and plasma volume are decreased in GHD91. Untreated acromegaly is associated with hypertension, therefore blood pressure would be expected to increase with GH replacement in adults with GHD. However, the initial placebo-controlled trials with adult GH replacement showed that diastolic blood pressure decreases92 despite a sustained increase in extracellular fluid and plasma volume91. The mechanisms for reduced diastolic blood pressure seem to be nitric oxide-mediated vasodilation and reduced peripheral resistance89,93, reduced sympathetic nervous system activity93 and improved endothelial function94. Also, MMP and vascular endothelial growth factors, which are markers of endothelial function, are increased in adults with GHD and decline with GH replacement21. Of note, individuals with isolated GHD that is caused by an inactivating mutation of the GH-releasing hormone RH receptor (GHRHR) have obesity, elevated levels of LDL cholesterol, C-reactive protein (CRP) and mild systolic hypertension, but an absence of atherosclerosis or heart failure95.
Long-term GH replacement therapy in patients with GHD induces sustained sodium and water retention96,97. This effect is due to increased sodium and water reabsorption from the distal renal tubuli, mediated by the direct actions of GH and IGF1, but also indirectly through stimulation of the renin–angiotensin–aldosterone system96–98. This sustained increase in extracellular water is in contrast to the effects of mineralocorticoids, which after administration increase the extracellular volume for days, where after renal sodium escape occurs, extracellular volume returns to baseline levels99. A probable explanation for the sustained sodium and water retention induced by GH is the reduction of natriuretic peptides induced by GH treatment, which mitigates renal sodium escape96,100, together with increased glycosaminoglycan deposition in peripheral tissues40.
Reduced exercise capacity has consistently been shown in adults with GHD, which improves with GH replacement101,102. The mechanism responsible could be related to effects on the heart and cardiovascular system, but also due to effects on muscle size and muscle function103. GHD is associated with reduced left ventricular mass index, reduced cardiac output with both impaired diastolic and systolic left ventricular function and reduced fractional shortening104. With GH replacement, an increase occurs in the left ventricular mass index as well as improvements in systolic and diastolic function that might contribute to improving exercise capacity104.
GH has important regulatory effects on various aspects of lipid and lipoprotein metabolism. Lipid metabolism is strongly linked to atherosclerosis and local endothelial inflammation105. GH upregulates LDL receptors in the liver106, stimulates lipolysis107 and increases the synthesis and secretion of VLDL from the liver108. In addition, GH increases the ratio of plasma lecithin to cholesterol acyltransferase concentration and lipid transfer protein activities, which might explain the increase in HDL cholesterol levels seen in some GH replacement studies in adults with GHD109. Adults with GHD have increased concentrations of total cholesterol and LDL cholesterol, and in some studies also reduced HDL cholesterol110,111. Likewise, controlled studies of adult GH replacement have revealed decreased circulating levels of total cholesterol and LDL cholesterol92, whereas effects on HDL cholesterol and triglycerides are smaller and less robust. Lipoprotein(a) is a lipoprotein that is similar to LDL in terms of its lipid and apolipoprotein B-100 content, but also contains apolipoprotein(a) covalently linked to apolipoprotein B-100. Increased plasma concentration of lipoprotein(a) is associated with increased risk of cardiovascular disease and its plasma level is mainly genetically determined, with an inverse relationship between circulating levels of lipoprotein(a) and the size polymorphism of apolipoprotein(a). Adults with GH deficiency have similar lipoprotein(a) concentrations to healthy matched control individuals, but GH replacement increases the concentration of lipoprotein(a)112. However, the net outcome of these changes in lipid and lipoprotein metabolism during GH replacement cannot be determined from available data.
Systemic inflammation markers such as CRP are well recognized cardiovascular risk factors113. Adults with GHD have increased circulating markers of inflammation such as CRP and IL-6 (REF.114). In one controlled study, GH replacement reduced the circulating levels of both CRP and IL-6 (REF.115). Also, in studies in women with abdominal obesity, GH treatment in comparison with placebo reduced biomarkers of systemic inflammation116. Thus, GHD is associated with a pro-inflammatory state but not with tissue fibrosis81. Taken together, studies of GH replacement in adults with GHD have shown beneficial effects on cardiovascular risk factors, cardiovascular function and surrogate variables for cardiovascular morbidity and mortality. However, no prospective controlled studies are available demonstrating reduced vascular morbidity and mortality. Such a study is unlikely to be performed due to the low prevalence of the disorder. However, in a retrospective meta-analysis of mortality in patients with hypopituitarism (including patients receiving GH replacement), GH replacement was associated with reduced mortality, particularly in men117.
GH and endothelial cellular function
The vascular endothelium, which is the largest organ in the body, responds to various circulatory growth factors including GH. Angiogenesis is essential for organogenesis and successful embryonic and fetal development118. Disruption of the mechanisms controlling physiological angiogenesis underlies the pathophysiology and pathogenesis of various diseases including cancer, psoriasis, arthritis, retinopathies, obesity, asthma and cardiovascular disease. During angiogenesis, endothelial cells are regulated by an interplay between cells, multiple soluble factors and the ECM. For example, vascular endothelial growth factor A (VEGFA) binds to its receptor, VEGFR2, to stimulate downstream signalling to activate endothelial nitric oxide synthase (eNOS) and nitric oxide release119 (FIG. 2). Nitric oxide has a crucial role in angiogenesis and vasodilation, and also acts as an inhibitor of platelet adhesion and aggregation, monocyte adhesion and vascular smooth muscle cell growth. Endothelial cells express GHR, and GH, in part, regulates endothelial cell function and angiogenesis through VEGFA120.
As discussed earlier in the article, both in vivo and in vitro studies have shown that treatment with IGF1 stimulates eNOS expression and nitric oxide release121 (FIG. 2). Interestingly, age-dependent impairment of endothelial progenitor cells in middle-aged and older humans is corrected by treatment with hGH-mediated increases of IGF1 (REF.32), which supports the role of declining GH levels in ageing-associated cardiovascular dysfunction. Similarly, studies in adults with GHD have shown improved endothelial function and reduced vascular risk after GH replacement therapy122. As patients with GHD have reduced nitric oxide production89, a feasible explanation of increased blood flow after GH replacement could be the improvement in endothelial function123. In addition, an improvement in the arterial response to induced vasodilation is observed in adolescents with GHD after GH replacement therapy124.
Although atherosclerosis is not prevalent in acromegaly62, GH has been found in experimental studies to directly stimulate the development of atherosclerosis in endothelial cells. For example, GH stimulates VCAM1 and SELE transcripts in human umbilical vein endothelial cells (HUVECs) via the MAPK pathway, which results in augmented adhesion of a human leukaemia monocyte cell line (THP-1) and primary monocytes to HUVECs125. As the endothelium has a key role in the pathogenesis of atherosclerotic plaques, excess GH or IGF1 could play an active or passive part in atherosclerosis and cardiovascular dysfunction via effects on endothelial pathophysiology. These effects include endothelial proliferation, endothelial progenitor cell dysfunction or endothelial oxidative stress126.
The effect of GH–IGF1 on endothelial cells has also been noted in other tissues and organs. In mouse endothelioma cells, GH has mitogenic effects127. GH also affects endothelial cell morphology and augments the deposition of the ECM molecules, laminin and fibronectin, on the cell surface127. In addition, human GH at physiological concentrations stimulates human retinal microvascular endothelial cells in vitro, thereby enhancing their proliferation128. In retinopathy, the proliferative form of retinal endothelial cells is observed during a more advanced stage of the disease and is characterized by retinal neovascularization. Although now abandoned, pituitary ablation was a method to suppress GH secretion as a potential treatment for proliferative diabetic retinopathy129. Interestingly, GH has an essential role in ischaemia-induced retinal neovascularization in mice130 and a GHR antagonist prevents this effect130. Finally, topical application of GH accelerates the closure of skin wounds by accelerating re-epithelialization and collagen deposition, and stimulating angiogenesis127, which occurs primarily via local production of IGF1 in the tissue131. Overall, these studies highlight the importance of the GH–IGF system in vascularization and angiogenesis.
Regulation of cardiac function
Studies in vitro and in rats have shown that GH and IGF1 increase cardiomyocyte gene expression and protein synthesis, which translates into cardiac hypertrophy and remodelling, but they also inhibit apoptosis132. GH transgenic mice that have elevated levels of GH and IGF1 show increased organ sizes including the heart, while heart-specific IGF1 expression markedly promotes myocyte proliferation133. Furthermore, the removal of GH action in the heart of adult mice affects neither the local levels of IGF1 nor cardiac function, even though endocrine IGF1 levels are altered134. As for IGF1, loss-of-function studies using cardiac-specific Igf1r-knockout mice have shown that autocrine and paracrine IGF1 promotes heart repair in response to injury and conservation of cardiac function135.
Endothelial-specific human IGF1R-overexpressing transgenic mice show reduced basal and insulin-stimulated eNOS activity, with no change in size or weight or whole-body glucose homeostasis136. These mice show normal blood pressure, an enhanced aortic response and increased endothelial cell migration and regeneration. By contrast, the Igf1r-knockout mice show normal glucose homeostasis with enhanced basal and insulin-stimulated eNOS phosphorylation137 and vascular permeability in the endothelial lining138. These studies support an important role for IGF1R in regulating nitric oxide bioavailability and vascular repair, which are hallmarks of several human diseases involving tissue growth and vascularization. In addition, macrophage IGF1 signalling exerts anti-atherogenic effects through reducing macrophage activities, decreased atherosclerotic lesion formation and reduced plaque vulnerability139,140.
Summary
Overall, evidence indicates that GH–IGF1 pathways have an important role in endothelial cell metabolism. Both elevated (acromegaly) and low (GHD) levels of GH are associated with cardiovascular disease. GH is a critical regulator of inflammation and immune activation. Considering that inflammation and fibrosis regulate progressive cardiac dysfunction during ischaemic heart failure141, targeting mediators of GH action could provide an exciting therapeutic avenue for this disease. GH could also have a ‘Goldilocks effect’, where too little or too much can lead to insufficient or dysregulated immune activation and fibrotic mechanisms that lead to the exacerbation of cardiovascular disease. Comprehensive studies are warranted to fully investigate the involvement of the GH–IGF1 axis in the pathogenesis and pathophysiology of cardiovascular disease and its complications.
GH and cancer
Numerous studies in multiple cancer types since 1950 have shown that the intrinsic growth-promoting action of GH drives the growth and proliferation of cancer cells both in vitro and in vivo142. However, in light of our rapidly evolving understanding of different aspects of cancer as well as of GH action, a more ‘covert’ role of GH in cancer, beyond just promoting tumour growth, has also emerged. Pituitary secreted endocrine GH, critical for normal growth and development, is well known to decrease steadily in adults with age. By contrast, local or non-pituitary GH production from several non-pituitary sites (including peripheral tissues and multiple tumour types) seems to stay fairly steady or even increase with age143,144 and exerts a profound autocrine and/or paracrine action. Fibrosis seems to be a consistent underlying theme in this action, wherein GH induces extensive ECM remodelling by inducing expression of proteases, collagen and various cytokines145. Studies in cultured cells and animal models have revealed an extensive array of molecular mechanisms by which non-pituitary GH is now known to be a critical driver of a tumour supportive microenvironment and cancer therapy resistance (FIG. 3).
Fig. 3 |. Covert actions of GH in cancer.
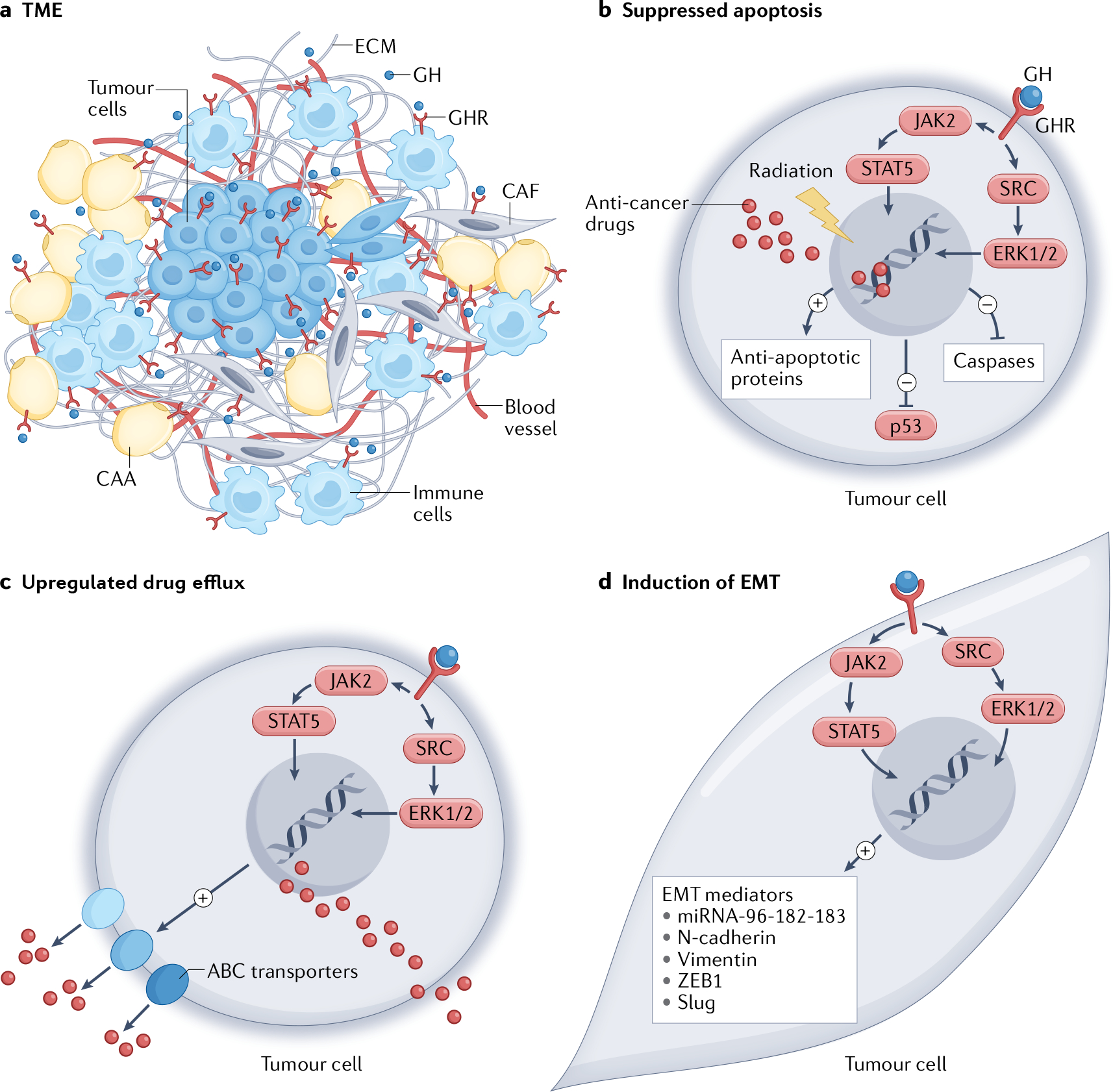
The emerging covert actions of growth hormone (GH) in cancer as evidenced from reports in the literature to date. a | GH promotes a tumour-supportive microenvironment via the crosstalk of tumour cells and multiple types of cells in the tumour microenvironment (TME), such as immune cells, cancer-associated adipocytes (CAA) and cancer-associated fibroblasts (CAF). This crosstalk occurs via autocrine and/or paracrine GH actions, which promote fibrosis and extracellular matrix (ECM) remodelling via matrix metalloproteases and collagen turnover, production of pro-inflammatory cytokines and immune-suppressive molecules such as transforming growth factor-β (TGFβ). Parts b–d illustrate the process of tumour therapy resistance through various mechanisms involving GH. b | Suppression of apoptosis due to radiotherapy-induced or chemotherapy-induced DNA damage, via downregulation of p53 and apoptotic mediators including caspases and upregulation of anti-apoptotic factors including BCL-2. c | Increased ATP-binding cassette (ABC) transporter multidrug efflux pump expression (ABCB1, ABCC1, ABCC2 and ABCG2), which actively removes multiple anti-cancer drugs out of the tumour cells. d | Induction of the metastatic process of epithelial-to-mesenchymal transition (EMT) by GH acting to upregulate EMT mediators, including vimentin, N-cadherin, ZEB1, SLUG and the microRNA (miRNA) cluster comprising miRNA-96, miRNA-182 and miRNA-183 (miRNA-96–182-183), which altogether enable a phenotype switch of the tumour cells. The above processes are induced by autocrine and/or paracrine GH binding to GH receptor (GHR)-expressing cells in the tumour and TME, activating downstream signalling mediators JAK2, STAT5, SRC and ERK1/2. Therefore, a combination of a GHR antagonist with anticancer drugs is a transformative approach for highly effective tumour clearance, which has already been validated in preclinical models.
GH in the tumour microenvironment
Local GH production in specific tissues exerts an autocrine and/or paracrine effect, distinct from its endocrine role. In tissues such as the colon and breast, local GH production increase with age143, forming a cellular niche in which oncogenic transformations could occur143,146. Furthermore, local GH production in these tissues could support the growth and survival of a tumour by affecting the tumour microenvironment (TME)147 (TABLE 1). Melmed and colleagues have elegantly elucidated the role of autocrine and/or paracrine GH in modulating the TME in support of tumour growth and survival through the ‘field cancerization’ paradigm147. This model is a much overlooked but increasingly appreciated aspect of local GH action in oncology via the TME that is intuitive and critical, and supported by several observations. First, extra-pituitary sites of GH production are found in multiple tissues144. Second, GHR is expressed in several cell types in the TME, including cancer-associated fibroblasts, adipocytes, immune cells, stromal cells147 and endothelial cells148, wherein GH exerts differential effects that are supportive of tumour growth148–151. Third, endocrine GH-induced hepatic IGF1 has well-studied mitogenic effects that act on the tumour and TME152,153.
Table 1 |.
Molecular actions of GH in human cancers
| Molecular action | Effect | Cancer type | Refs. |
| Oncogenesis and tumour microenvironment | |||
| GH is a p53 target and in turn blocks p53 by a negative feedback loop | Tumour initiation | Colon cancer | 158 |
| GH induces DNA damage and suppresses DNA damage repair in normal colon tissue | Tumour initiation | Colon cancer | 156,157,198 |
| GH resistance reduces number and size of neoplasms in C3(1)/Tag-GHRKO mice | Tumour initiation | Breast cancer, prostate cancer | 202,203 |
| GH deficiency suppress DMBA-induced tumour development | Tumour initiation | Breast cancer | 204 |
| bGH mice have a higher DEN-induced hepatoma incidence while lit/lit (Ghrhr-knockout) mice have a lower one | Tumour initiation | Liver cancer | 205,206 |
| GH supplementation enables MNU-induced tumours in GH-deficient rats; tumours regress on GH withdrawal | Tumour initiation | Breast cancer | 175 |
| GH stabilizes hTERT through αCP1 and αCP2 | Tumour initiation | Breast cancer | 207 |
| GH supports spontaneous neoplastic growth with age | Tumour initiation | Liver cancer | 208 |
| GH expression in mammary epithelia promotes oncogenic transformation via HOXA1 | Tumour initiation | Breast cancer | 209,210 |
| GH produced in ageing colon accumulates DNA damage | Tumour initiation | Colon cancer | 143 |
| Invasive tumour growth | |||
| GH increases miRNA-96, miRNA-182 and miRNA-183 cluster targeting BRMS1L | EMT induction | Breast cancer | 186,211 |
| GH induces CHOP expression via p38 MAPK activation | EMT induction | Breast cancer | 212 |
| GH supports tumour angiogenesis | Invasive growth | Breast cancer | 120 |
| GH drives migration, invasion and metastasis in tumour xenografts | Invasive growth and EMT induction | Breast cancer | 213–215 |
| GH deficiency reduces tumour growth rate in lit/lit mice | Tumour progression | Prostate cancer | 216 |
| GH increases migration, invasion, proliferation and MMP levels | Invasive growth | Prostate cancer | 217 |
| GH blockade reduces xenograft growth rate | Invasive growth | Colon cancer, meningioma | 218,219 |
| GH blockade reduces migration, invasion and markers of EMT | EMT induction | Melanoma, pancreatic cancer | 180,181 |
| GH production in lungs guides melanoma metastases in mice | Metastasis | Melanoma | 187 |
| GH inhibits tumour apoptosis via PI3K–AKT signalling | Invasive growth | Gastric cancer | 220 |
| Therapy resistance | |||
| GH increases ABC transporter expressions via JAK2-STAT5 and SRC signalling | Multidrug resistance | Melanoma | 172 |
| GH increases MITF-dependent melanogenesis and drug sequestration via JAK2–STAT5 and SRC | Multidrug resistance | Melanoma | 154 |
| GH increases ABCG2 levels via JAK2–STAT5 | Docetaxel resistance | Breast cancer | 174 |
| GH increases PI3K–AKT–MAPK and JAK2 signalling | Ruxolitinib resistance | Breast cancer | 145 |
| GH reduces tumour apoptosis and increase proliferation | Doxorubicin resistance | Breast cancer | 175 |
| GH increases post-irradiation clonogenicity and reduces radiation-induced DNA damage | Radiation resistance | Colorectal cancer, breast cancer, endometrial cancer | 171,221–223 |
| GH induces FOS expression | Doxorubicin resistance | Breast cancer | 224,225 |
| GH suppress DNA damage and apoptosis | Mitomycin-C resistance | Breast cancer, endometrial cancer | 226 |
| GH activates ERK1/2 and PKC and suppresses caspase activation | Multidrug resistance | Endometrial cancer | 227 |
| GH reduces pro-apoptotic BAX and PPARγ via STAT5B activation | PPARγ ligand resistance | Colon cancer | 228 |
| GH reduces BAX, BAD, caspase 3, caspase 8 and caspase 9 | Methylmethanosulfonate resistance | Lymphoma | 229 |
| GH increases ABCG2 and markers of cancer stem cells (NANOG, ALDH1, CD24 and CD44) | Increased stemness | Liver cancer, breast cancer, colon cancer | 163,177,178,184,213 |
| GH is a part of senescence-associated secretory phenotype | Therapy evasion and relapse | Colon cancer | 31 |
ABC, ATP-binding cassette; bGH, bovine growth hormone; C3(1)/Tag-GHRKO mice, mice from a cross between Ghr-knockout (GHRKO) mice and C3(1)/Tag mice, where females develop spontaneous mammary tumours; DEN, diethylnitrosamine; DMBA, dimethylbenz[a]anthracene; EMT, epithelial-to-mesenchymal transition; GH, growth hormone; hTERT, human telomerase reverse transcriptase; miRNA, microRNA; MMP, matrix metalloproteinases; MNU, N-methyl-N-nitrosourea.
Ageing or chemically inflicted DNA damage leads to activation of the tumour suppressor p53, which results in either apoptosis or induction of p53–p21 senescent pathway or a DNA damage repair (DDR) pathway143. Chromatin immunoprecipitation assays reveal GH as a target for p53 binding, whereas transcriptomic and proteomic analyses confirm DNA damage induces local GH production in normal colon and tumour cells154–156. In turn, locally produced GH exerts a feedback inhibition on p53 expression and diverts cellular commitment from senescence or apoptosis to proliferative survival155. Additionally, autocrine and/or paracrine GH signalling abrogates the DDR pathway, which increases the risk of oncogenic mutations, as is observed in human colonic epithelial transformation156–158. Increased colonic p53 expression was observed in Ghr-knockout (GHRKO) mice and Ames mice (which lack GH, prolactin and thyroid-stimulating hormone) compared with corresponding age-matched controls158. Furthermore, elevated expression of DDR genes were observed in γ-irradiated primary fibroblasts of GH-deficient Lewis dwarf rats compared with those from wild-type rats159. Elevated DDR gene expression was also observed in Snell mice (deficient in GH, prolactin and thyroid-stimulating hormone), GHRKO mice and Pappa-knockout mice (a model with alterations to the GH–IGF1 axis) compared with their respective wild-type controls160. Together, these findings in GH-deficient animal models support the ‘onco-promoting’ role of GH in peripheral tissues.
An analysis of the National Cancer Institute genome-wide association study identified that out of 421 pathways containing 3,962 genes, GH signalling is the third most associated pathway with breast cancer susceptibility161. Seminal work highlighted a concerted role of GH and IGF1 in facilitating the functions of oestrogen and progesterone in normal mammary development162. The neoplastic effects of non-pituitary GH in breast cancer are particularly important, given that human GH binds to both GHR and prolactin receptor (PRLR), which are highly expressed together in ductal endothelium. These binding events result in a hyperplastic signalling cascade, which normally governs mammary development but can become an onco-driver under appropriate conditions163. This signalling cascade is of particular importance in the TME in the context of fibrosis, one of the hallmarks of cancer, which enables invasive and metastatic growth and compromises antitumour immunity164,165. As discussed above, GH is a potent inducer of fibrosis in multiple tissues, whereas the activation of the PRLR is also known to promote fibrosis in cancer via STAT3-dependent pathways166,167. Downstream effectors of GH action, IGF1 and TGFβ, are also strongly implicated as autocrine and/or paracrine drivers of fibrosis168, and can therefore have profound effects in the TME. Additionally, autocrine and/or paracrine GH has been established as a prominent component of SASP, wherein it promotes DNA damage accumulation in bystander cells and predisposes senescent cells to cell cycle re-entry and neoplastic transformation31. Of note, congenital GHRKO mice and adult-onset GHRKO (at age 6 months) mice have markedly reduced fatal neoplasms in both sexes, compared with wild-type littermates169,170.
GH in cancer therapy resistance
A set of key pathways observed across all types of cancer that are unresponsive to therapy are inhibition of apoptosis, active drug efflux via ATP-binding cassette (ABC) transporters and a phenotype switch via epithelial-to-mesenchymal transition (EMT). GH expression induces resistance against apoptosis in mammary and endometrial tumour cells following irradiation171 and induces resistance against several chemotherapy treatments (mitomycin-C, doxorubicin, cisplatin, arsenic trioxide and ruxolitinib) in the same model145. Similar effects of GH in driving refractoriness against chemotherapy (doxorubicin, cisplatin and paclitaxel) and targeted (vemurafenib) therapies are also observed in melanoma172. The mechanistic validation comes from the identification that tumoural GHR activation induces a STAT5–SRC–ERK1/2-mediated upregulation of multidrug ABC transporters in human melanoma172,173. The ABC transporters impart resistance to a wide range of anticancer therapeutics by limiting their cytosolic retention through active efflux. Studies in human melanoma cells have shown autocrine GH signalling-induced upregulation of multidrug efflux transporters of the ABCB, ABCC and ABCG subtypes, which could be effectively blocked by GHR suppression172. Mouse xenograft models of human oestrogen receptor-negative breast cancer further confirm that GHR silencing reverses docetaxel resistance via downregulation of ABCG2 transporter expression174. Furthermore, chemically induced mammary tumour establishment was possible in spontaneous dwarf rats (GH-deficient) only when they were supplemented with exogenous GH. Moreover, when GH supplementation was stopped and the rats were treated with doxorubicin, tumours regressed in the dwarf animals but not in GH-sufficient wild-type animals, which confirms a GH-dependent chemoresistance175. Importantly, ABC transporters (such as ABCB1, ABCB5 and ABCG2) are known biomarkers for cancer stem cells (CSCs)176, which are highly drug-resistant and are responsible for cancer relapse. Forced GH expression in liver cancer cells increased ABCG2 expression and conferred CSC properties by a JAK2–STAT3-mediated suppression of the tight junction protein claudin 1 (REF.177). In addition forced GH expression stably increased CSC markers such as NANOG and SALL4 in both liver and colorectal cancer cells177,178.
In cancers of the breast179, colon158, liver177 and pancreas180 and in melanoma181, GH promotes successful metastasis from a primary tumour by the process of EMT. This process enables a switch from a well-defined, polarized, basement membrane-adherent cell phenotype to a depolarized, migrating invasive tumour cell phenotype182. In addition to its angiogenic and lymphangiogenic effects, GH is known to induce EMT in the overlapping pathways of tissue fibrosis and cancer metastases183. Suppression of the epithelial marker E-cadherin and upregulation of the mesenchymal markers N-cadherin and vimentin, and transcription factors ZEB1 and SLUG by recombinant hGH treatment occur in melanoma173,181 and pancreatic180 cancer cells and by autocrine GH in colorectal178 and breast cancer cells184. Attenuating the GHR reverses these effects. The most extensive evidence of GH in promoting EMT is from breast cancer, where autocrine human GH expression in mammary tumour cells leads to robust EMT induction and massive ECM remodelling. This effect occurs by GH downregulating adherence factors such as plakoglobin via hypermethylation mediated by DNA methyltransferases 3A and 3B185, by GH increasing the production of ECM-degrading MMP2 and MMP9 (REF.184) and by GH upregulating the microRNA (miRNA) cluster comprising miRNA-96, miRNA-182 and miRNA-183 leading to elevated ZEB1 and suppressed BRMS1L186. In fact, in Dj1-knockout mice, which have incidental high production of GH in the lungs, this local GH enhances the metastasis of disseminated melanoma cells187. The pronounced effect of GH in inducing fibrosis, as a part of ECM remodelling, further emphasizes a unique and critical role in promoting cancer therapy resistance188 (TABLE 1).
Inhibition of GH action in cancer
Laron syndrome is congenital GH insensitivity due to inactivating mutations of GHR189. Independent long-term follow-up studies on two of the largest cohorts of individuals with Laron syndrome in Israel and in Ecuador have shown no cases of malignancy, while the incidence rate in first-degree relatives is >20%4,190 in both cohorts. Similarly, among patients with secondary GHD due to a GHRHR defect and patients with primary congenital isolated GH deficiency, cancer incidence is considerably suppressed compared with their relatives95. The cohort of patients with isolated GH deficiency type 1B (arising due to GHRHR insufficiency), in Itabaianinha, Brazil, also show an absence of colorectal, prostate and breast cancers, unlike their relatives95. Altogether, these epidemiological findings indicate that a congenitally absent or reduced GH action seems to be onco-protective. By contrast, large-scale, long-term follow-up studies in children with GHD treated with recombinant GH do not indicate an elevated risk of neoplastic developments.
Numerous studies estimating the risks of cancer and benign neoplasia in patients with acromegaly have shown elevated risks for specific cancer types and establish cancer as a major cause of mortality in these patients191. However, confounding factors exist, including surveillance bias, normalization of circulating levels of IGF1 due to various treatment options and difficulty in adequately comparing the cause of death in individuals with well-controlled acromegaly versus healthy control individuals.
Overall, the above discussion does provoke the question: can pharmacological suppression of GH action in the ageing population help tackle oncogenicity or improve cancer prognoses? Several GHR inhibitors are currently in development, which reflects a heightened pharmaceutical interest in targeting GH action in human disease143,192,193. So far, targeting GH action in cancer using somatostatin analogues has shown no objective response in humans, whereas GHRH antagonists so far show promising effects in several preclinical models194. Of note, somatostatin–GHRH control of GH production in the pituitary is seldom maintained in the context of the tumour. Pegvisomant, the first and only FDA-approved GHR antagonist, is highly successful in efficiently reducing serum concentrations of IGF1 in 70–90% of patients with acromegaly195. Moreover, pegvisomant has shown efficacy in attenuating growth of multiple GH-expressing and GHR-expressing human cancers in preclinical models196.
Although clinical validation of the efficacy of GHR antagonists in monotherapy or in combination with other anticancer therapies is awaited, more clarity is needed in understanding the mutually exclusive roles of GH and IGF1, as well as the ratio of GH-induced IGF1 and insulin-like growth factor-binding protein 3 (IGFBP3)197 in a cancer-specific manner. Importantly, several studies have revealed that endocrine GH-induced hepatic production of IGF1 and IGFBP3 is not always observed in cultured tumour cells and mouse tumour xenografts and TME, wherein autocrine and/or paracrine GH has IGF1-independent oncogenic effects143,198. A clinical trial in patients with cancer published in 2013 for a combination of pegvisomant and figitumumab (an anti-IGF1R monoclonal antibody) against solid tumours (breast, lung, prostate and colorectal tumours, and sarcoma) was initiated. Unfortunately, the study was terminated prematurely “due to lack of operational feasibility and halt of figitumumab development”199. Therefore, whether targeting GHR in cancer is a clinically relevant option remains an open question, given the large volume of provocative studies mentioned above alongside the failure of IGF1R inhibitors and monoclonal antibodies in the treatment of cancer. Patient pre-screening for tumoural GHR overexpression and serum levels of IGF1–IGFBP3 might offer an important precision factor in this approach.
Conclusions
In this Review we emphasize the importance of GH in promoting fibrosis and its association with cardiovascular and cancer pathologies. GH induces the expression of a variety of genes encoding collagen. This is not surprising, as one of the most important actions of the hormone is stimulation of longitudinal growth in children, which involves the concerted actions of systemic GH and local IGF1 on the growth plate, a specialized connective tissue51. Moreover, GH administration also stimulates collagen synthesis and turnover in adult humans to such an extent that quantification of IGF1 and PIIINP in serum is now an approved assay for the determination of GH doping in sport18. Whether GH doping causes excessive fibrosis formation in, for example, muscle is unknown. Of note, however, GH administration in healthy adults does not increase either muscle strength or aerobic exercise capacity200. Indeed, after completion of longitudinal growth and somatic maturation, the major anabolic effect of GH administration seems to relate to collagen rather than muscle fibres13. When present in excess, GH results in fibrosis in a tissue-specific manner (FIG. 1) and the subsequent alterations to normal tissue function represent an added caution to be considered when utilizing GH supplementation in adults.
Data derived primarily from humans with elevated levels of GH (acromegaly) have shown a correlation with cardiovascular pathology, wherein fibrosis was identified as a contributor. In addition, patients with GHD often show a pro-inflammatory state without tissue fibrosis81. Together, the results suggest that a ‘normal’ amount of GH action is needed to ensure proper heart structure and cardiovascular function and either abnormally high or low levels of GH lead to cardiac pathology. An effect of the GH–IGF1 axis on vascular endothelial cells might be a common denominator of these effects that is reflected in adults with GHD with premature atherosclerosis.
In cancer, the effect of GH on fibrosis and endothelial cells makes it an important factor towards development of a detrimental TME. Clinically, cancer risks associated with GH replacement so far do not suggest any elevated risk of de novo neoplasm. However, since 1950 (REF.142), experimental and animal data clearly demonstrate the mechanistic links of how GH, and its partner IGF1, can influence the development, progression, therapy resistance and metastases of multiple human and animal cancers that express the GHR and, thus, depict a definite ‘oncodriver’ role. Therefore, the rational approach of targeting GH action in patients with cancer who have high tumoural GHR expression should be complemented with appropriate screening for the status of tumoural GH action, including testing tumour biopsy samples for GH or GHR expression.
Future studies that consider the surrounding tissue milieu and inherent subcellular differences associated with the covert actions of GH will ultimately define the molecular processes involved. Additionally, the question of local versus endocrine GH and IGF1 participation in these phenotypes must be determined. In terms of growth, GH and IGF1 have both independent and overlapping functions201. In this Review, we have updated the hidden or covert pathophysiological effects of GH and attempted to describe new molecular and cellular mechanisms responsible for them. Fibrosis is a common molecular ‘theme’ that seems to link these adverse phenotypes. Other fields of endocrinology clearly show that too much or too little exposure, or an abnormal time of exposure to a hormone, causes different adverse phenotypes. In the context of GH–IGF1, we conclude that either too much or too little of a good thing (GH) is ‘bad’ — a typical ‘Goldilocks effect’.
Key points.
Growth hormone (GH) is important for growth and tissue remodelling, extracellular matrix formation and fibrosis.
Patients with acromegaly, which is characterized by excessive circulating levels of GH, have increased cardiovascular mortality that is associated with hypertension and heart failure.
Patients with GH deficiency have an increased risk of cardiovascular morbidity and mortality that is associated with cardiovascular risk factors and premature atherosclerosis.
GH actions in cancer are particularly implicated in mechanisms of therapy resistance; for example, active drug efflux, the epithelial-to-mesenchymal transition, apoptosis inhibition and development of a tumour-supportive microenvironment.
GH has a ‘Goldilocks effect’, where too little or too much can lead to poor clinical outcomes.
Acknowledgements
J.J.K. acknowledges the support of the State of Ohio’s Eminent Scholar Program that includes a gift from Milton and Lawrence Goll, NIH-R01AG059779, the AMVETS and the Edison Biotechnology Institute at Ohio University. D.E.B. acknowledges the support of ASPIRE funding from Pfizer, NIH-R01AG059779 and the Heritage College of Osteopathic Medicine at Ohio University. V.P. acknowledges the support of funds from Osteopathic Heritage Foundation’s Vision 2020 to Heritage College of Osteopathic Medicine at Ohio University, R01HL140836, R01MD012579 and RO1DK124126. The authors acknowledge J. Young, L. Householder, S. Zhu and A. Jara (Ohio University) for their assistance with the original version of Fig. 1.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Endocrinology thanks César Boguszewski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Jorgensen JOL, Johannsson G & Barkan A Should patients with adult GH deficiency receive GH replacement? Eur. J. Endocrinol. 186, D1–D15 (2021). [DOI] [PubMed] [Google Scholar]
- 2.Ranke MB & Wit JM Growth hormone–past, present and future. Nat. Rev. Endocrinol. 14, 285–300 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Bernardo A & Houssay MD The hypophysis and metabolism. N. Engl. J. Med. 214, 961–971 (1936). [Google Scholar]
- 4.Guevara-Aguirre J et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl Med. 3, 70ra13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguiar-Oliveira MH & Bartke A Growth hormone deficiency: health and longevity. Endocr. Rev. 40, 575–601 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milman S, Huffman DM & Barzilai N The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab. 23, 980–989 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wynn TA Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4, 583–594 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Householder LA et al. Increased fibrosis: a novel means by which GH influences white adipose tissue function. Growth Horm. IGF Res. 39, 45–53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fruchtman S et al. Suppressor of cytokine signaling-2 modulates the fibrogenic actions of GH and IGF-I in intestinal mesenchymal cells. Am. J. Physiol. Gastrointest. Liver Physiol 289, G342–G350 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Ong LK et al. Growth hormone improves cognitive function after experimental stroke. Stroke 49, 1257–1266 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Yang CW, Striker GE, Chen WY, Kopchick JJ & Striker LJ Differential expression of glomerular extracellular matrix and growth factor mRNA in rapid and slowly progressive glomerulosclerosis: studies in mice transgenic for native or mutated growth hormone. Lab. Invest. 76, 467–476 (1997). [PubMed] [Google Scholar]
- 12.Nielsen RH et al. Chronic alterations in growth hormone/insulin-like growth factor-I signaling lead to changes in mouse tendon structure. Matrix Biol. 34, 96–104 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doessing S et al. Growth hormone stimulates the collagen synthesis in human tendon and skeletal muscle without affecting myofibrillar protein synthesis. J. Physiol. 588, 341–351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gadelha MR, Kasuki L, Lim DST & Fleseriu M Systemic complications of acromegaly and the impact of the current treatment landscape: an update. Endocr. Rev. 40, 268–332 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Constantin T et al. Calcium and bone turnover markers in acromegaly: a prospective, controlled study. J. Clin. Endocrinol. Metab. 102, 2416–2424 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Arlien-Soborg MC et al. Fibroblast activation protein is a GH target: a prospective study of patients with acromegaly before and after treatment. J. Clin. Endocrinol. Metab. 105, dgz033 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Nielsen RH et al. GH receptor blocker administration and muscle-tendon collagen synthesis in humans. Growth Horm. IGF Res. 21, 140–145 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Holt RIG & Ho KKY The use and abuse of growth hormone in sports. Endocr. Rev. 40, 1163–1185 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Coghlan RF et al. A degradation fragment of type X collagen is a real-time marker for bone growth velocity. Sci. Transl Med. 9, eaan4669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antoniazzi F et al. GH in combination with bisphosphonate treatment in osteogenesis imperfecta. Eur. J. Endocrinol. 163, 479–487 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Randeva HS et al. Growth hormone replacement decreases plasma levels of matrix metalloproteinases (2 and 9) and vascular endothelial growth factor in growth hormone-deficient individuals. Circulation 109, 2405–2410 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Karci AC, Canturk Z, Tarkun I & Cetinarslan B Matrix metalloproteinase 2 (MMP-2) levels are increased in active acromegaly patients. Endocrine 57, 148–155 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Imanishi R et al. GH suppresses TGF-β-mediated fibrosis and retains cardiac diastolic function. Mol. Cell Endocrinol. 218, 137–146 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y et al. TIMP3 modulates GHR abundance and GH sensitivity. Mol. Endocrinol. 30, 587–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amor C et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 583, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu RM & Liu G Cell senescence and fibrotic lung diseases. Exp. Gerontol. 132, 110836 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker DJ et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stout MB et al. Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging 6, 575–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumoto R et al. Accelerated telomere shortening in acromegaly; IGF-I induces telomere shortening and cellular senescence. PLoS ONE 10, e0140189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran D et al. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 13, 669–678 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chesnokova V & Melmed S GH and senescence: a new understanding of adult GH action. J. Endocr. Soc 6, bvab177 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thum T et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ. Res. 100, 434–443 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Luo X et al. Insulin-like growth factor-1 attenuates oxidative stress-induced hepatocyte premature senescence in liver fibrogenesis via regulating nuclear p53-progerin interaction. Cell Death Dis. 10, 451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henderson NC, Rieder F & Wynn TA Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freeth JS et al. Human skin fibroblasts as a model of growth hormone (GH) action in GH receptor-positive Laron’s syndrome. Endocrinology 138, 55–61 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Guller S, Sonenberg M, Wu KY, Szabo P & Corin RE Growth hormone-dependent events in the adipose differentiation of 3T3-F442A fibroblasts: modulation of macromolecular synthesis. Endocrinology 125, 2360–2367 (1989). [DOI] [PubMed] [Google Scholar]
- 37.Thorey IS et al. Transgenic mice reveal novel activities of growth hormone in wound repair, angiogenesis, and myofibroblast differentiation. J. Biol. Chem. 279, 26674–26684 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Fan MH et al. Fibroblast activation protein (FAP) accelerates collagen degradation and clearance from lungs in mice. J. Biol. Chem. 291, 8070–8089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang XM et al. Fibroblast activation protein and chronic liver disease. Front. Biosci. 13, 3168–3180 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Ben-Shlomo A & Melmed S Skin manifestations in acromegaly. Clin. Dermatol. 24, 256–259 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Lie JT Pathology of the heart in acromegaly: anatomic findings in 27 autopsied patients. Am. Heart J. 100, 41–52 (1980). [DOI] [PubMed] [Google Scholar]
- 42.dos Santos Silva CM et al. Low frequency of cardiomyopathy using cardiac magnetic resonance imaging in an acromegaly contemporary cohort. J. Clin. Endocrinol. Metab. 100, 4447–4455 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Koutsou-Tassopoulou A et al. Hepatic steatosis in patients with acromegaly. Endocrinol. Diabetes Metab. 2, e00090 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrioli M et al. Thyroid nodules in acromegaly: the role of elastography. J. Ultrasound 13, 90–97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lai NB, Garg D, Heaney AP, Bergsneider M & Leung AM No benefit of dedicated thyroid nodule screening in patients with acromegaly. Endocr. Pract. 26, 16–21 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Jara A et al. Elevated systolic blood pressure in male GH transgenic mice is age dependent. Endocrinology 155, 975–986 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munoz MC et al. Downregulation of the ACE2/Ang-(1–7)/Mas axis in transgenic mice overexpressing GH. J. Endocrinol. 221, 215–227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jensen EA et al. Growth hormone alters gross anatomy and morphology of the small and large intestines in age- and sex-dependent manners. Pituitary 25, 116–130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.List EO et al. Adipocyte-specific GH receptor-null (AdGHRKO) mice have enhanced insulin sensitivity with reduced liver triglycerides. Endocrinology 160, 68–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.List EO et al. GH knockout mice have increased subcutaneous adipose tissue with decreased fibrosis and enhanced insulin sensitivity. Endocrinology 160, 1743–1756 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yakar S & Isaksson O Regulation of skeletal growth and mineral acquisition by the GH/IGF-1 axis: lessons from mouse models. Growth Horm. IGF Res. 28, 26–42 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dichtel LE et al. The association between IGF-1 levels and the histologic severity of nonalcoholic fatty liver disease. Clin. Transl Gastroenterol. 8, e217 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Polyzos SA et al. Targeted analysis of three hormonal systems identifies molecules associated with the presence and severity of NAFLD. J. Clin. Endocrinol. Metab. 105, e390–e400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nishizawa H et al. Nonalcoholic fatty liver disease in adult hypopituitary patients with GH deficiency and the impact of GH replacement therapy. Eur. J. Endocrinol. 167, 67–74 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Theiss AL et al. Growth hormone reduces the severity of fibrosis associated with chronic intestinal inflammation. Gastroenterology 129, 204–219 (2005). [DOI] [PubMed] [Google Scholar]
- 56.de Oliveira GV et al. Growth hormone effects on hypertrophic scar formation: a randomized controlled trial of 62 burned children. Wound Repair Regen. 12, 404–411 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Breederveld RS & Tuinebreijer WE Recombinant human growth hormone for treating burns and donor sites. Cochrane Database Syst. Rev. 2014, CD008990 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu ZC et al. Expression of insulin-like growth factor-1 receptor in keloid and hypertrophic scar. Clin. Exp. Dermatol. 39, 822–828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Doi T et al. Progressive glomerulosclerosis develops in transgenic mice chronically expressing growth hormone and growth hormone releasing factor but not in those expressing insulinlike growth factor-1. Am. J. Pathol. 131, 398–403 (1988). [PMC free article] [PubMed] [Google Scholar]
- 60.Bengtsson BA, Eden S, Ernest I, Oden A & Sjogren B Epidemiology and long-term survival in acromegaly. A study of 166 cases diagnosed between 1955 and 1984. Acta Med. Scand. 223, 327–335 (1988). [DOI] [PubMed] [Google Scholar]
- 61.Dal J et al. Acromegaly incidence, prevalence, complications and long-term prognosis: a nationwide cohort study. Eur. J. Endocrinol. 175, 181–190 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Colao A, Grasso LFS, Di Somma C & Pivonello R Acromegaly and heart failure. Heart Fail. Clin. 15, 399–408 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Thuesen L et al. Increased myocardial contractility following growth hormone administration in normal man. An echocardiographic study. Dan. Med. Bull. 35, 193–196 (1988). [PubMed] [Google Scholar]
- 64.Thuesen L et al. Short and long-term cardiovascular effects of growth hormone therapy in growth hormone deficient adults. Clin. Endocrinol. 41, 615–620 (1994). [DOI] [PubMed] [Google Scholar]
- 65.Napoli R et al. Acute effects of growth hormone on vascular function in human subjects. J. Clin. Endocrinol. Metab. 88, 2817–2820 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Kamenicky P, Mazziotti G, Lombes M, Giustina A & Chanson P Growth hormone, insulin-like growth factor-1, and the kidney: pathophysiological and clinical implications. Endocr. Rev. 35, 234–281 (2014). [DOI] [PubMed] [Google Scholar]
- 67.Moller J, Jorgensen JO, Frandsen E, Laursen T & Christiansen JS Body fluids, circadian blood pressure and plasma renin during growth hormone administration: a placebo-controlled study with two growth hormone doses in healthy adults. Scand. J. Clin. Lab. Invest. 55, 663–669 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Thuesen L, Christensen SE, Weeke J, Orskov H & Henningsen P A hyperkinetic heart in uncomplicated active acromegaly. Explanation of hypertension in acromegalic patients? Acta Med. Scand. 223, 337–343 (1988). [DOI] [PubMed] [Google Scholar]
- 69.Ikkos D, Luft R & Sjogren B Body water and sodium in patients with acromegaly. J. Clin. Invest. 33, 989–994 (1954). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moller N et al. Basal- and insulin-stimulated substrate metabolism in patients with active acromegaly before and after adenomectomy. J. Clin. Endocrinol. Metab. 74, 1012–1019 (1992). [DOI] [PubMed] [Google Scholar]
- 71.Bolfi F, Neves AF, Boguszewski CL & Nunes-Nogueira VS Mortality in acromegaly decreased in the last decade: a systematic review and meta-analysis. Eur. J. Endocrinol. 181, L5–L6 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Jaffrain-Rea ML et al. Impact of successful transsphenoidal surgery on cardiovascular risk factors in acromegaly. Eur. J. Endocrinol. 148, 193–201 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Maison P, Tropeano AI, Macquin-Mavier I, Giustina A & Chanson P Impact of somatostatin analogs on the heart in acromegaly: a metaanalysis. J. Clin. Endocrinol. Metab. 92, 1743–1747 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Yuen KCJ et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr. Pract. 25, 1191–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Hammarstrand C et al. Higher glucocorticoid replacement doses are associated with increased mortality in patients with pituitary adenoma. Eur. J. Endocrinol. 177, 251–256 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Klose M et al. Central hypothyroidism and its replacement have a significant influence on cardiovascular risk factors in adult hypopituitary patients. J. Clin. Endocrinol. Metab. 98, 3802–3810 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Tomlinson JW et al. Association between premature mortality and hypopituitarism. West Midlands Prospective Hypopituitary Study Group. Lancet 357, 425–431 (2001). [DOI] [PubMed] [Google Scholar]
- 78.Abe SY et al. Metabolic syndrome and its components in adult hypopituitary patients. Pituitary 23, 409–416 (2020). [DOI] [PubMed] [Google Scholar]
- 79.Rosen T & Bengtsson BA Premature mortality due to cardiovascular disease in hypopituitarism. Lancet 336, 285–288 (1990). [DOI] [PubMed] [Google Scholar]
- 80.Isgaard J, Wahlander H, Adams MA & Friberg P Increased expression of growth hormone receptor mRNA and insulin-like growth factor-I mRNA in volume-overloaded hearts. Hypertension 23, 884–888 (1994). [DOI] [PubMed] [Google Scholar]
- 81.Ratku B et al. Effects of adult growth hormone deficiency and replacement therapy on the cardiometabolic risk profile. Pituitary 25, 211–228 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johannsson G & Ragnarsson O Growth hormone deficiency in adults with hypopituitarism — What are the risks and can they be eliminated by therapy? J. Intern. Med. 290, 1180–1193 (2021). [DOI] [PubMed] [Google Scholar]
- 83.Moller N & Jorgensen JO Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 30, 152–177 (2009). [DOI] [PubMed] [Google Scholar]
- 84.Zierler KL & Rabinowitz D Roles of insulin and growth hormone, based on studies of forearm metabolism in man. Medicine 42, 385–402 (1963). [DOI] [PubMed] [Google Scholar]
- 85.Press M, Notarfrancesco A & Genel M Risk of hypoglycaemia with alternate-day growth hormone injections. Lancet 1, 1002–1004 (1987). [DOI] [PubMed] [Google Scholar]
- 86.Johansson JO, Fowelin J, Landin K, Lager I & Bengtsson BA Growth hormone-deficient adults are insulin-resistant. Metabolism 44, 1126–1129 (1995). [DOI] [PubMed] [Google Scholar]
- 87.Hew FL et al. Insulin resistance in growth hormone-deficient adults: defects in glucose utilization and glycogen synthase activity. J. Clin. Endocrinol. Metab. 81, 555–564 (1996). [DOI] [PubMed] [Google Scholar]
- 88.Berryman DE, Glad CA, List EO & Johannsson G The GH/IGF-1 axis in obesity: pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 9, 346–356 (2013). [DOI] [PubMed] [Google Scholar]
- 89.Boger RH et al. Nitric oxide may mediate the hemodynamic effects of recombinant growth hormone in patients with acquired growth hormone deficiency. A double-blind, placebo-controlled study. J. Clin. Invest. 98, 2706–2713 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sverrisdottir YB, Elam M, Herlitz H, Bengtsson BA & Johannsson G Intense sympathetic nerve activity in adults with hypopituitarism and untreated growth hormone deficiency. J. Clin. Endocrinol. Metab. 83, 1881–1885 (1998). [DOI] [PubMed] [Google Scholar]
- 91.Moller J, Frandsen E, Fisker S, Jorgensen JO & Christiansen JS Decreased plasma and extracellular volume in growth hormone deficient adults and the acute and prolonged effects of GH administration: a controlled experimental study. Clin. Endocrinol. 44, 533–539 (1996). [DOI] [PubMed] [Google Scholar]
- 92.Maison P et al. Impact of growth hormone (GH) treatment on cardiovascular risk factors in GH-deficient adults: a metaanalysis of blinded, randomized, placebo-controlled trials. J. Clin. Endocrinol. Metab. 89, 2192–2199 (2004). [DOI] [PubMed] [Google Scholar]
- 93.Sverrisdottir YB et al. The effect of growth hormone (GH) replacement therapy on sympathetic nerve hyperactivity in hypopituitary adults: a double-blind, placebo-controlled, crossover, short-term trial followed by long-term open GH replacement in hypopituitary adults. J. Hypertens. 21, 1905–1914 (2003). [DOI] [PubMed] [Google Scholar]
- 94.Evans LM et al. The effect of GH replacement therapy on endothelial function and oxidative stress in adult growth hormone deficiency. Eur. J. Endocrinol. 142, 254–262 (2000). [DOI] [PubMed] [Google Scholar]
- 95.Aguiar-Oliveira MH & Salvatori R Disruption of the GHRH receptor and its impact on children and adults: the Itabaianinha syndrome. Rev. Endocr. Metab. Disord. 22, 81–89 (2021). [DOI] [PubMed] [Google Scholar]
- 96.Johannsson G, Sverrisdottir YB, Ellegard L, Lundberg PA & Herlitz H GH increases extracellular volume by stimulating sodium reabsorption in the distal nephron and preventing pressure natriuresis. J. Clin. Endocrinol. Metab. 87, 1743–1749 (2002). [DOI] [PubMed] [Google Scholar]
- 97.Kamenicky P et al. Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity. J. Clin. Endocrinol. Metab. 96, 2127–2135 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Moller J et al. Blockade of the renin-angiotensin-aldosterone system prevents growth hormone-induced fluid retention in humans. Am. J. Physiol. 272, E803–E808 (1997). [DOI] [PubMed] [Google Scholar]
- 99.Yokota N, Bruneau BG, Kuroski de Bold ML & de Bold AJ Atrial natriuretic factor significantly contributes to the mineralocorticoid escape phenomenon. Evidence for a guanylate cyclase-mediated pathway. J. Clin. Invest. 94, 1938–1946 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Moller J, Jorgensen JO, Marqversen J, Frandsen E & Christiansen JS Insulin-like growth factor I administration induces fluid and sodium retention in healthy adults: possible involvement of renin and atrial natriuretic factor. Clin. Endocrinol. 52, 181–186 (2000). [DOI] [PubMed] [Google Scholar]
- 101.Widdowson WM & Gibney J The effect of growth hormone replacement on exercise capacity in patients with GH deficiency: a metaanalysis. J. Clin. Endocrinol. Metab. 93, 4413–4417 (2008). [DOI] [PubMed] [Google Scholar]
- 102.Jorgensen JO et al. Beneficial effects of growth hormone treatment in GH-deficient adults. Lancet 1, 1221–1225 (1989). [DOI] [PubMed] [Google Scholar]
- 103.Johannsson G, Grimby G, Sunnerhagen KS & Bengtsson BA Two years of growth hormone (GH) treatment increase isometric and isokinetic muscle strength in GH-deficient adults. J. Clin. Endocrinol. Metab. 82, 2877–2884 (1997). [DOI] [PubMed] [Google Scholar]
- 104.Amato G et al. Body composition, bone metabolism, and heart structure and function in growth hormone (GH)-deficient adults before and after GH replacement therapy at low doses. J. Clin. Endocrinol. Metab. 77, 1671–1676 (1993). [DOI] [PubMed] [Google Scholar]
- 105.Hansson GK Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352, 1685–1695 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Rudling M et al. Importance of growth hormone for the induction of hepatic low density lipoprotein receptors. Proc. Natl Acad. Sci. USA 89, 6983–6987 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sharma VM et al. Growth hormone acts along the PPARγ-FSP27 axis to stimulate lipolysis in human adipocytes. Am. J. Physiol. Endocrinol. Metab 316, E34–E42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Elam MB, Wilcox HG, Solomon SS & Heimberg M In vivo growth hormone treatment stimulates secretion of very low density lipoproteins by the perfused rat liver. Endocrinology 131, 2717–2722 (1992). [DOI] [PubMed] [Google Scholar]
- 109.Beentjes JA, van Tol A, Sluiter WJ & Dullaart RP Effect of growth hormone replacement therapy on plasma lecithin:cholesterol acyltransferase and lipid transfer protein activities in growth hormone-deficient adults. J. Lipid Res. 41, 925–932 (2000). [PubMed] [Google Scholar]
- 110.Rosen T, Bosaeus I, Tolli J, Lindstedt G & Bengtsson B-Å Increased body fat mass and decreased extracellular fluid volume in adults with growth hormone deficiency. Clin. Endocrinol. 38, 63–71 (1993). [DOI] [PubMed] [Google Scholar]
- 111.Cuneo RC, Salomon F, Wiles CM, Hesp R & Sönksen PH Growth hormone treatment in growth hormone-deficient adults. II. Effects on exercise performance. J. Appl. Physiol. 70, 695–700 (1991). [DOI] [PubMed] [Google Scholar]
- 112.Olivecrona H, Ericsson S, Berglund L & Angelin B Increased concentrations of serum lipoprotein (a) in response to growth hormone treatment. BMJ 306, 1726–1727 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ridker PM, Hennekens CH, Buring JE & Rifai N C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N. Engl. J. Med. 342, 836–843 (2000). [DOI] [PubMed] [Google Scholar]
- 114.Leonsson M et al. Increased Interleukin-6 levels in pituitary-deficient patients are independently related to their carotid intima-media thickness. Clin. Endocrinol. 59, 242–250 (2003). [DOI] [PubMed] [Google Scholar]
- 115.Sesmilo G et al. Effects of growth hormone administration on inflammatory and other cardiovascular risk markers in men with growth hormone deficiency. A randomized, controlled clinical trial. Ann. Intern. Med. 133, 111–122 (2000). [DOI] [PubMed] [Google Scholar]
- 116.Franco C et al. Growth hormone treatment reduces abdominal visceral fat in postmenopausal women with abdominal obesity: a 12-month placebo-controlled trial. J. Clin. Endocrinol. Metab. 90, 1466–1474 (2005). [DOI] [PubMed] [Google Scholar]
- 117.Pappachan JM, Raskauskiene D, Kutty VR & Clayton RN Excess mortality associated with hypopituitarism in adults: a meta-analysis of observational studies. J. Clin. Endocrinol. Metab. 100, 1405–1411 (2015). [DOI] [PubMed] [Google Scholar]
- 118.Drake CJ Embryonic and adult vasculogenesis. Birth Defects Res. C. Embryo Today 69, 73–82 (2003). [DOI] [PubMed] [Google Scholar]
- 119.Bouloumie A, Schini-Kerth VB & Busse R Vascular endothelial growth factor up-regulates nitric oxide synthase expression in endothelial cells. Cardiovasc. Res. 41, 773–780 (1999). [DOI] [PubMed] [Google Scholar]
- 120.Brunet-Dunand SE et al. Autocrine human growth hormone promotes tumor angiogenesis in mammary carcinoma. Endocrinology 150, 1341–1352 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Walsh MF et al. Insulin-like growth factor I diminishes in vivo and in vitro vascular contractility: role of vascular nitric oxide. Endocrinology 137, 1798–1803 (1996). [DOI] [PubMed] [Google Scholar]
- 122.Setola E et al. Effects of growth hormone treatment on arginine to asymmetric dimethylarginine ratio and endothelial function in patients with growth hormone deficiency. Metabolism 57, 1685–1690 (2008). [DOI] [PubMed] [Google Scholar]
- 123.Lilien MR, Schroder CH, Levtchenko EN & Koomans HA Growth hormone therapy influences endothelial function in children with renal failure. Pediatr. Nephrol. 19, 785–789 (2004). [DOI] [PubMed] [Google Scholar]
- 124.Lanes R et al. Endothelial function, carotid artery intima-media thickness, epicardial adipose tissue, and left ventricular mass and function in growth hormone-deficient adolescents: apparent effects of growth hormone treatment on these parameters. J. Clin. Endocrinol. Metab. 90, 3978–3982 (2005). [DOI] [PubMed] [Google Scholar]
- 125.Ishikawa M et al. Role of growth hormone signaling pathways in the development of atherosclerosis. Growth Horm. IGF Res. 53–54, 101334 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Maffei P, Dassie F, Wennberg A, Parolin M & Vettor R The endothelium in acromegaly. Front. Endocrinol. 10, 437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Messias de Lima CF, Dos Santos Reis MD, da Silva Ramos FW, Ayres-Martins S & Smaniotto S Growth hormone modulates in vitro endothelial cell migration and formation of capillary-like structures. Cell Biol. Int. 41, 577–584 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Rymaszewski Z, Cohen RM & Chomczynski P Human growth hormone stimulates proliferation of human retinal microvascular endothelial cells in vitro. Proc. Natl Acad. Sci. USA 88, 617–621 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Speakman JS et al. Pituitary ablation for diabetic retinopathy. Can. Med. Assoc. J. 94, 627–635 (1966). [PMC free article] [PubMed] [Google Scholar]
- 130.Smith LE et al. Essential role of growth hormone in ischemia-induced retinal neovascularization. Science 276, 1706–1709 (1997). [DOI] [PubMed] [Google Scholar]
- 131.Steenfos HH & Jansson JO Growth hormone stimulates granulation tissue formation and insulinlike growth factor-I gene expression in wound chambers in the rat. J. Endocrinol. 132, 293–298 (1992). [DOI] [PubMed] [Google Scholar]
- 132.Isgaard J, Arcopinto M, Karason K & Cittadini A GH and the cardiovascular system: an update on a topic at heart. Endocrine 48, 25–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Santini MP et al. Enhancing repair of the mammalian heart. Circ. Res. 100, 1732–1740 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jara A et al. Cardiac-specific disruption of GH receptor alters glucose homeostasis while maintaining normal cardiac performance in adult male mice. Endocrinology 157, 1929–1941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim J et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 22, 2531–2543 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Imrie H et al. Novel role of the IGF-1 receptor in endothelial function and repair: studies in endothelium-targeted IGF-1 receptor transgenic mice. Diabetes 61, 2359–2368 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abbas A et al. The insulin-like growth factor-1 receptor is a negative regulator of nitric oxide bioavailability and insulin sensitivity in the endothelium. Diabetes 60, 2169–2178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liang M et al. Protective role of insulin-like growth factor-1 receptor in endothelial cells against unilateral ureteral obstruction-induced renal fibrosis. Am. J. Pathol. 185, 1234–1250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Higashi Y et al. Insulin-like growth factor-1 receptor deficiency in macrophages accelerates atherosclerosis and induces an unstable plaque phenotype in apolipoprotein E-deficient mice. Circulation 133, 2263–2278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Spadaro O et al. Growth hormone receptor deficiency protects against age-related NLRP3 inflammasome activation and immune senescence. Cell Rep. 14, 1571–1580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nehra S, Gumina RJ & Bansal SS Immune cell dilemma in ischemic cardiomyopathy: to heal or not to heal. Curr. Opin. Physiol. 19, 39–46 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Moon HD, Simpson ME, Li CH & Evans HM Neoplasms in rats treated with pituitary growth hormone; pulmonary and lymphatic tissues. Cancer Res. 10, 297–308 (1950). [PubMed] [Google Scholar]
- 143.Chesnokova V et al. Local non-pituitary growth hormone is induced with aging and facilitates epithelial damage. Cell Rep. 37, 110068 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harvey S, Aramburo C & Sanders EJ Extrapituitary production of anterior pituitary hormones: an overview. Endocrine 41, 19–30 (2012). [DOI] [PubMed] [Google Scholar]
- 145.Basu R & Kopchick JJ The effects of growth hormone on therapy resistance in cancer. Cancer Drug Resist. 2, 827–846 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Waters MJ & Conway-Campbell BL The oncogenic potential of autocrine human growth hormone in breast cancer. Proc. Natl Acad. Sci. USA 101, 14992–14993 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chesnokova V & Melmed S Growth hormone in the tumor microenvironment. Arch. Endocrinol. Metab 63, 568–575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lincoln DT, Singal PK & Al-Banaw A Growth hormone in vascular pathology: neovascularization and expression of receptors is associated with cellular proliferation. Anticancer Res. 27, 4201–4218 (2007). [PubMed] [Google Scholar]
- 149.Dos Santos Reis MD, Dos Santos YMO, de Menezes CA, Borbely KSC & Smaniotto S Resident murine macrophage migration and phagocytosis are modulated by growth hormone. Cell Biol. Int. 42, 615–623 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Kopchick JJ, Berryman DE, Puri V, Lee KY & Jorgensen JOL The effects of growth hormone on adipose tissue: old observations, new mechanisms. Nat. Rev. Endocrinol. 16, 135–146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lee SW, Kim SH, Kim JY & Lee Y The effect of growth hormone on fibroblast proliferation and keratinocyte migration. J. Plast. Reconstr. Aesthet. Surg. 63, e364–e369 (2010). [DOI] [PubMed] [Google Scholar]
- 152.Hua H, Kong Q, Yin J, Zhang J & Jiang Y Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: a challenge for cancer therapy. J. Hematol. Oncol. 13, 64 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pollak M Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928 (2008). [DOI] [PubMed] [Google Scholar]
- 154.Basu R et al. Growth hormone upregulates melanocyte-inducing transcription factor expression and activity via JAK2-STAT5 and SRC signaling in GH receptor-positive human melanoma. Cancers 11, 1352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chesnokova V et al. Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc. Natl Acad. Sci. USA 110, E3331–E3339 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ben-Shlomo A et al. DNA damage and growth hormone hypersecretion in pituitary somatotroph adenomas. J. Clin. Invest. 130, 5738–5755 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chesnokova V et al. Excess growth hormone suppresses DNA damage repair in epithelial cells. JCI Insight 4, e125762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chesnokova V et al. Growth hormone is permissive for neoplastic colon growth. Proc. Natl Acad. Sci. USA 113, E3250–E3259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Podlutsky A et al. The GH/IGF-1 axis in a critical period early in life determines cellular DNA repair capacity by altering transcriptional regulation of DNA repair-related genes: implications for the developmental origins of cancer. Geroscience 39, 147–160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dominick G, Bowman J, Li X, Miller RA & Garcia GG mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell 16, 52–60 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Menashe I et al. Pathway analysis of breast cancer genome-wide association study highlights three pathways and one canonical signaling cascade. Cancer Res. 70, 4453–4459 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kleinberg DL & Barcellos-Hoff MH The pivotal role of insulin-like growth factor I in normal mammary development. Endocrinol. Metab. Clin. North Am. 40, 461–471 (2011). [DOI] [PubMed] [Google Scholar]
- 163.Lombardi S et al. Growth hormone is secreted by normal breast epithelium upon progesterone stimulation and increases proliferation of stem/progenitor cells. Stem Cell Rep. 2, 780–793 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Boulter L, Bullock E, Mabruk Z & Brunton VG The fibrotic and immune microenvironments as targetable drivers of metastasis. Br. J. Cancer 124, 27–36 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Piersma B, Hayward MK & Weaver VM Fibrosis and cancer: a strained relationship. Biochim. Biophys. Acta Rev. Cancer 1873, 188356 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chakraborty D et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 8, 1130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tandon M et al. Prolactin promotes fibrosis and pancreatic cancer progression. Cancer Res. 79, 5316–5327 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sun W et al. Regulation of the IGF1 signaling pathway is involved in idiopathic pulmonary fibrosis induced by alveolar epithelial cell senescence and core fucosylation. Aging 13, 18852–18869 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Duran-Ortiz S et al. Growth hormone receptor gene disruption in mature-adult mice improves male insulin sensitivity and extends female lifespan. Aging Cell 20, e13506 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ikeno Y et al. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 64, 522–529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bougen NM et al. Autocrine human GH promotes radioresistance in mammary and endometrial carcinoma cells. Endocr. Relat. Cancer 19, 625–644 (2012). [DOI] [PubMed] [Google Scholar]
- 172.Basu R, Baumgaertel N, Wu S & Kopchick JJ Growth hormone receptor knockdown sensitizes human melanoma cells to chemotherapy by attenuating expression of ABC drug efflux pumps. Horm. Cancer 8, 143–156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Qian Y et al. Growth hormone upregulates mediators of melanoma drug efflux and epithelial-to-mesenchymal transition in vitro and in vivo. Cancers 12, 3640 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Arumugam A et al. Silencing growth hormone receptor inhibits estrogen receptor negative breast cancer through ATP-binding cassette sub-family G member 2. Exp. Mol. Med. 51, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lantvit DD et al. Mammary tumors growing in the absence of growth hormone are more sensitive to doxorubicin than wild-type tumors. Endocrinology 162, bqab013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Begicevic RR & Falasca M ABC transporters in cancer stem cells: beyond chemoresistance. Int. J. Mol. Sci. 18, 2362 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chen YJ et al. Autocrine human growth hormone promotes invasive and cancer stem cell-like behavior of hepatocellular carcinoma cells by STAT3 dependent inhibition of CLAUDIN-1 expression. Int. J. Mol. Sci. 18, 1274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wang JJ et al. Autocrine hGH stimulates oncogenicity, epithelial-mesenchymal transition and cancer stem cell-like behavior in human colorectal carcinoma. Oncotarget 8, 103900–103918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Subramani R, Nandy SB, Pedroza DA & Lakshmanaswamy R Role of growth hormone in breast cancer. Endocrinology 158, 1543–1555 (2017). [DOI] [PubMed] [Google Scholar]
- 180.Subramani R et al. Growth hormone receptor inhibition decreases the growth and metastasis of pancreatic ductal adenocarcinoma. Exp. Mol. Med. 46, e117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Basu R, Wu S & Kopchick JJ Targeting growth hormone receptor in human melanoma cells attenuates tumor progression and epithelial mesenchymal transition via suppression of multiple oncogenic pathways. Oncotarget 8, 21579–21598 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang J et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 21, 341–352 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brittain AL, Basu R, Qian Y & Kopchick JJ Growth hormone and the epithelial-to-mesenchymal transition. J. Clin. Endocrinol. Metab. 102, 3662–3673 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Mukhina S et al. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc. Natl Acad. Sci. USA 101, 15166–15171 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Shafiei F et al. DNMT3A and DNMT3B mediate autocrine hGH repression of plakoglobin gene transcription and consequent phenotypic conversion of mammary carcinoma cells. Oncogene 27, 2602–2612 (2008). [DOI] [PubMed] [Google Scholar]
- 186.Zhang W et al. Autocrine/paracrine human growth hormone-stimulated microRNA 96–182-183 cluster promotes epithelial-mesenchymal transition and invasion in breast cancer. J. Biol. Chem. 290, 13812–13829 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chien CH, Lee MJ, Liou HC, Liou HH & Fu WM Growth hormone is increased in the lungs and enhances experimental lung metastasis of melanoma in DJ-1 KO mice. BMC Cancer 16, 871 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chandler C, Liu T, Buckanovich R & Coffman LG The double edge sword of fibrosis in cancer. Transl Res. 209, 55–67 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Leslie M Genetics and disease. Growth defect blocks cancer and diabetes. Science 331, 837 (2011). [DOI] [PubMed] [Google Scholar]
- 190.Steuerman R, Shevah O & Laron Z Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 164, 485–489 (2011). [DOI] [PubMed] [Google Scholar]
- 191.Boguszewski CL & Boguszewski M Growth hormone’s links to cancer. Endocr. Rev. 40, 558–574 (2019). [DOI] [PubMed] [Google Scholar]
- 192.Lu M, Flanagan JU, Langley RJ, Hay MP & Perry JK Targeting growth hormone function: strategies and therapeutic applications. Signal Transduct. Target. Ther. 4, 3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Basu R et al. A novel peptide antagonist of the human growth hormone receptor. J. Biol. Chem. 296, 100588 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang H et al. Inhibition of experimental small-cell and non-small-cell lung cancers by novel antagonists of growth hormone-releasing hormone. Int. J. Cancer 142, 2394–2404 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Paisley AN, Trainer P & Drake W Pegvisomant: a novel pharmacotherapy for the treatment of acromegaly. Expert Opin. Biol. Ther. 4, 421–425 (2004). [DOI] [PubMed] [Google Scholar]
- 196.Basu R, Qian Y & Kopchick JJ Mechanisms in endocrinology: lessons from growth hormone receptor gene-disrupted mice: are there benefits of endocrine defects? Eur. J. Endocrinol. 178, R155–R181 (2018). [DOI] [PubMed] [Google Scholar]
- 197.Cohen P, Clemmons DR & Rosenfeld RG Does the GH-IGF axis play a role in cancer pathogenesis? Growth Horm. IGF Res. 10, 297–305 (2000). [DOI] [PubMed] [Google Scholar]
- 198.Chesnokova V, Zonis S, Barrett RJ, Gleeson JP & Melmed S Growth hormone induces colon DNA damage independent of IGF-1. Endocrinology 160, 1439–1447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00976508 (2013). [DOI] [PubMed]
- 200.Hermansen K, Bengtsen M, Kjaer M, Vestergaard P & Jorgensen JOL Impact of GH administration on athletic performance in healthy young adults: a systematic review and meta-analysis of placebo-controlled trials. Growth Horm. IGF Res. 34, 38–44 (2017). [DOI] [PubMed] [Google Scholar]
- 201.Lupu F, Terwilliger JD, Lee K, Segre GV & Efstratiadis A Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 229, 141–162 (2001). [DOI] [PubMed] [Google Scholar]
- 202.Wang Z et al. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology 146, 5188–5196 (2005). [DOI] [PubMed] [Google Scholar]
- 203.Zhang X et al. Inhibition of estrogen-independent mammary carcinogenesis by disruption of growth hormone signaling. Carcinogenesis 28, 143–150 (2007). [DOI] [PubMed] [Google Scholar]
- 204.Swanson SM & Unterman TG The growth hormone-deficient Spontaneous Dwarf rat is resistant to chemically induced mammary carcinogenesis. Carcinogenesis 23, 977–982 (2002). [DOI] [PubMed] [Google Scholar]
- 205.Bugni JM, Poole TM & Drinkwater NR The little mutation suppresses DEN-induced hepatocarcinogenesis in mice and abrogates genetic and hormonal modulation of susceptibility. Carcinogenesis 22, 1853–1862 (2001). [DOI] [PubMed] [Google Scholar]
- 206.Snibson KJ, Bhathal PS & Adams TE Overexpressed growth hormone (GH) synergistically promotes carcinogen-initiated liver tumour growth by promoting cellular proliferation in emerging hepatocellular neoplasms in female and male GH-transgenic mice. Liver 21, 149–158 (2001). [DOI] [PubMed] [Google Scholar]
- 207.Emerald BS et al. αCP1 mediates stabilization of hTERT mRNA by autocrine human growth hormone. J. Biol. Chem. 282, 680–690 (2007). [DOI] [PubMed] [Google Scholar]
- 208.Snibson KJ, Bhathal PS, Hardy CL, Brandon MR & Adams TE High, persistent hepatocellular proliferation and apoptosis precede hepatocarcinogenesis in growth hormone transgenic mice. Liver 19, 242–252 (1999). [DOI] [PubMed] [Google Scholar]
- 209.Zhang X et al. Human growth hormone-regulated HOXA1 is a human mammary epithelial oncogene. J. Biol. Chem. 278, 7580–7590 (2003). [DOI] [PubMed] [Google Scholar]
- 210.Zhu T et al. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 65, 317–324 (2005). [PubMed] [Google Scholar]
- 211.Ma Y et al. Dysregulation and functional roles of miR-183–96-182 cluster in cancer cell proliferation, invasion and metastasis. Oncotarget 7, 42805–42825 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mertani HC et al. Autocrine human growth hormone (hGH) regulation of human mammary carcinoma cell gene expression. Identification of CHOP as a mediator of hGH-stimulated human mammary carcinoma cell survival. J. Biol. Chem. 276, 21464–21475 (2001). [DOI] [PubMed] [Google Scholar]
- 213.Chen YJ et al. Autocrine human growth hormone stimulates the tumor initiating capacity and metastasis of estrogen receptor-negative mammary carcinoma cells. Cancer Lett. 365, 182–189 (2015). [DOI] [PubMed] [Google Scholar]
- 214.Divisova J et al. The growth hormone receptor antagonist pegvisomant blocks both mammary gland development and MCF-7 breast cancer xenograft growth. Breast Cancer Res. Treat. 98, 315–327 (2006). [DOI] [PubMed] [Google Scholar]
- 215.Kaulsay KK et al. Autocrine stimulation of human mammary carcinoma cell proliferation by human growth hormone. Exp. Cell Res. 250, 35–50 (1999). [DOI] [PubMed] [Google Scholar]
- 216.Takahara K et al. Human prostate cancer xenografts in lit/lit mice exhibit reduced growth and androgen-independent progression. Prostate 71, 525–537 (2011). [DOI] [PubMed] [Google Scholar]
- 217.Recouvreux MV et al. Androgen receptor regulation of local growth hormone in prostate cancer cells. Endocrinology 158, 2255–2268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Friend KE, Radinsky R & McCutcheon IE Growth hormone receptor expression and function in meningiomas: effect of a specific receptor antagonist. J. Neurosurg. 91, 93–99 (1999). [DOI] [PubMed] [Google Scholar]
- 219.McCutcheon IE et al. Antitumor activity of the growth hormone receptor antagonist pegvisomant against human meningiomas in nude mice. J. Neurosurg. 94, 487–492 (2001). [DOI] [PubMed] [Google Scholar]
- 220.Yan HZ et al. GHR is involved in gastric cell growth and apoptosis via PI3K/AKT signalling. J. Cell Mol. Med. 25, 2450–2458 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Evans A, Jamieson SM, Liu DX, Wilson WR & Perry JK Growth hormone receptor antagonism suppresses tumour regrowth after radiotherapy in an endometrial cancer xenograft model. Cancer Lett. 379, 117–123 (2016). [DOI] [PubMed] [Google Scholar]
- 222.Wu X et al. Growth hormone receptor overexpression predicts response of rectal cancers to pre-operative radiotherapy. Eur. J. Cancer 42, 888–894 (2006). [DOI] [PubMed] [Google Scholar]
- 223.Wu XY et al. Growth hormone protects colorectal cancer cells from radiation by improving the ability of DNA damage repair. Mol. Med. Rep. 10, 486–490 (2014). [DOI] [PubMed] [Google Scholar]
- 224.Minoia M et al. Growth hormone receptor blockade inhibits growth hormone-induced chemoresistance by restoring cytotoxic-induced apoptosis in breast cancer cells independently of estrogen receptor expression. J. Clin. Endocrinol. Metab. 97, E907–E916 (2012). [DOI] [PubMed] [Google Scholar]
- 225.Zatelli MC et al. Growth hormone excess promotes breast cancer chemoresistance. J. Clin. Endocrinol. Metab. 94, 3931–3938 (2009). [DOI] [PubMed] [Google Scholar]
- 226.Bougen NM, Yang T, Chen H, Lobie PE & Perry JK Autocrine human growth hormone reduces mammary and endometrial carcinoma cell sensitivity to mitomycin C. Oncol. Rep. 26, 487–493 (2011). [DOI] [PubMed] [Google Scholar]
- 227.Gentilin E et al. Growth hormone differentially modulates chemoresistance in human endometrial adenocarcinoma cell lines. Endocrine 56, 621–632 (2017). [DOI] [PubMed] [Google Scholar]
- 228.Bogazzi F et al. Growth hormone inhibits apoptosis in human colonic cancer cell lines: antagonistic effects of peroxisome proliferator activated receptor-gamma ligands. Endocrinology 145, 3353–3362 (2004). [DOI] [PubMed] [Google Scholar]
- 229.Arnold RE & Weigent DA The inhibition of apoptosis in EL4 lymphoma cells overexpressing growth hormone. Neuroimmunomodulation 11, 149–159 (2004). [DOI] [PubMed] [Google Scholar]