

Abstract
Purpose:
Penetrance estimates of Birt-Hogg-Dubé (BHD)-associated cutaneous, pulmonary, and kidney manifestations are based on clinically ascertained families. In a healthcare system population, we used a genetics-first approach to estimate the prevalence of pathogenic/likely pathogenic (P/LP) truncating variants in FLCN, which cause BHD, and the penetrance of BHD-related phenotypes.
Methods:
Exomes from 135,990 patient-participants in Geisinger’s MyCode® cohort were assessed for P/LP truncating FLCN variants. BHD-related phenotypes were evaluated from electronic health records. Association between P/LP FLCN variants and BHD-related phenotypes was assessed by Firth’s logistic regression.
Results:
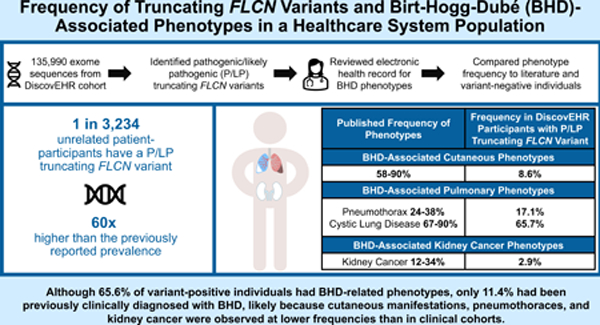
P/LP truncating FLCN variants were identified in 35 individuals (1 in 3,234 unrelated individuals), 68.6% of whom had BHD-related phenotype(s) including: cystic lung disease (65.7%), pneumothoraces (17.1%), cutaneous manifestations (8.6%), and kidney cancer (2.9%). Four (11.4%) had prior clinical BHD diagnoses.
Conclusion:
In this healthcare population, the frequency of P/LP truncating FLCN variants is 60 times higher than the previously reported prevalence. Although most variant-positive individuals had BHD-related phenotypes, a minority were previously clinically diagnosed, likely because cutaneous manifestations, pneumothoraces, and kidney cancer were observed at lower frequencies than in clinical cohorts. Improved clinical recognition of cystic lung disease and education concerning its association with FLCN variants could prompt evaluation for BHD.
Keywords: Birt-Hogg-Dubé syndrome, FLCN, healthcare system population, variant prevalence, basilar cystic lung disease
Graphical Abstract
Introduction
Birt-Hogg-Dubé syndrome (BHD; OMIM 135150) is an autosomal dominant condition associated with benign cutaneous manifestations, pulmonary cysts/blebs/bullae (hereafter referred to as “cystic lung disease”), spontaneous pneumothoraces, and kidney neoplasms.1,2 BHD is caused by germline variants in the folliculin (FLCN) gene, a tumor suppressor on chromosome 17p11.2 (OMIM 607273), that encodes the folliculin protein that is involved in numerous signaling pathways.3–6 Variants resulting in truncation of the protein (e.g., nonsense, frameshift, canonical splice site variants) are most common in individuals with BHD.2
BHD has an estimated prevalence of 1:200,0007, and, as of 2017, over 600 families with BHD had been described.8 The most common phenotypes in individuals with BHD are benign cutaneous manifestations, including fibrofolliculomas and trichodiscomas. Such findings have been identified in 58–90% of clinically ascertained patients and typically appear after age 25.9 Macroscopically, trichodiscomas and fibrofolliculomas are indistinguishable from one another and are believed to be on the same morphological spectrum as perifollicular fibromas.10
Patients with BHD also have increased risk of developing cystic lung disease and spontaneous pneumothoraces. Pulmonary cysts occur in 67–90% of patients with BHD, vary in number, and are typically bilateral and located in the lower basal zones2,9,11 Between 24–38% of patients with BHD experience a spontaneous pneumothorax at a median age of 38 years and earliest age of seven.2,11
Additionally, patients with P/LP FLCN variants are at an increased risk for benign and malignant kidney findings. Bilateral, multifocal renal masses are typical and are histologically diverse with most being hybrid oncocytic masses, chromophobe renal cell carcinoma, and clear cell carcinoma.2 It is estimated that 12 to 34% of individuals with BHD develop kidney cancers2,9,11 with a mean age of diagnosis of 50.7 years and earliest diagnosis at age 20.9 Additionally, individuals with BHD are at risk of developing oncocytosis, microscopic foci of oncocytic cells in the renal parenchyma, that is a precursor to kidney cancer.2 Other phenotypes have been reported in patients with BHD including lipomas, parathyroid adenomas, thyroid nodules, thyroid cancer, and parotid oncocytomas, but additional data are needed to clarify their association with BHD.2
To date, penetrance estimates of BHD-related features in individuals with P/LP FLCN variants have been based on families clinically ascertained due to a personal and/or family history. As genetic testing becomes widely integrated into clinical care, understanding variant prevalence and phenotypic presentation in broader populations will be necessary to enable accurate counseling and appropriate management. Population-based testing for pathogenic variants in other disease-associated genes has suggested that clinical penetrance estimates might be higher than penetrance in variant-positive individuals from broader cohorts due to ascertainment bias in clinical populations.12,13 Here, we explore the prevalence of P/LP truncating FLCN variants in a healthcare system-based population and utilize electronic health record (EHR) data to investigate the frequency of BHD-related phenotypes and prior clinical identification of the FLCN variant.
Materials and Methods
MyCode Participants
The Geisinger MyCode® Community Health Initiative (MyCode) serves as a biobank of blood and other samples from over 300,000 patient-participants who consent to health-related research.14–16 Participants are recruited throughout Geisinger regardless of disease or phenotype.14–17 The population for this study includes a subset of MyCode patient-participants, referred to as the DiscovEHR cohort15, with available exome sequencing and linked EHR data. Most patient-participants also have available SNP genotyping data. MyCode participants have higher rates of self-reported White race and non-Hispanic ethnicity, older median age, and higher comorbidity index compared to the overall Geisinger population.17 MyCode and the research outlined are governed by the Geisinger Institutional Review Board; all participants provided written informed consent.
Exome Sequencing and Intragenic CNV Calling
Exome sequencing for 135,990 adult MyCode patient-participants was performed in collaboration with the Regeneron Genetics Center, as described previously.14,15 Exome sequence data were aligned to human reference GRCh38 with BWA-MEM, and the resultant BAM files were processed using the Picard MarkDuplicates tool (http://broadinstitute.github.io/picard) to flag duplicate reads. After exome data were aligned, variants were called using weCall.18 Joint genotyping was performed across the cohort using GLnexus (Lin et al., 2018, unpublished manuscript, https://www.biorxiv.org/content/10.1101/343970v1.full.pdf) . CNV calling from exome data was carried out using Copy number estimation using the Lattice-Aligned Mixture ModelS (CLAMMS) algorithm19 that demonstrates high sensitivity and specificity down to single-exon level CNVs (Maxwell et al., 2017, unpublished manuscript, https://www.biorxiv.org/content/biorxiv/early/2017/03/22/119461.full.pdf). CNV calls were validated with SNP-array CNV calls using Penn CNV.20
Identification of P/LP Truncating FLCN Variants
Exome sequencing data (n=135,990) were reviewed for truncating FLCN variants. Variants in VCF files were annotated with Ensembl Variant Effect Predictor (VEP) version 96 using RefSeq transcripts. High impact variants (nonsense, frameshift, +/− 1,2 splice site) based on the FLCN transcript most expressed in adult tissues (transcript variant 1, GenBank: NM_144997.7) in genome build GRCh38.5 Sequence variants with an allele balance greater or equal to 0.20, genotype quality greater than or equal to 80, coverage greater than or equal to 20, and maximum gnomAD subpopulation allele frequency of less than 0.001 were identified. Two FLCN variants that nearly met our quality thresholds were included in our analysis upon confirmation of the FLCN variant from a clinical genetic testing report documented in the EHR (n=1) and validation of a variant by evaluation of IGV plots (n=1). Truncating variants were then reviewed (JMS and HS) using the American College of Medical and Genomics and Association for Molecular Pathology variant interpretation guidelines and classifications were confirmed by a third reviewer (NTS), who is board certified in laboratory genetics and genomics (Supplemental Table 2).21–23 Truncating variants with P/LP classifications were included in the analysis.
While this study focuses the most common BHD-causative variants (truncating FLCN variants), a secondary analysis of non-truncating variants reported in the National Center for Biotechnology Information ClinVar24 database as P/LP was conducted (Data not incorporated into further analyses, Supplementary Materials and Methods, Supplemental Tables 1, 2, 6).
Determination of Relatedness
To report on the number of unique families with P/LP truncating FLCN variants, the frequency of phenotypes within families, and P/LP truncating variant prevalence in an unrelated subset of the MyCode cohort, first- and second-degree familial relationships were identified from genotype data using Pedigree Reconstruction and Identification of the Maximally Unrelated Set (PRIMUS).25
Phenotype Evaluation – EHR and Imaging Review
BHD-related pulmonary, cutaneous, and kidney phenotypes were characterized in variant-positive individuals via manual review of patients’ EHRs. A chart review guide was developed to collect variables of interest and independent, double reviews were completed by a genetic counselor and board-certified neurodevelopmental pediatrician (JMS, SMM) between March and November 2020 (Supplemental Materials and Methods). Discrepancies were resolved through joint review and consensus.
Cystic lung disease and renal masses might be incidentally identified on imaging studies of adjacent body parts (e.g., Computed Tomography (CT) of the abdomen includes the lung bases and lumbar spine Magnetic Resonance Imaging (MRI) includes part of the kidneys). Additionally, cystic lung disease may be identified, but sometimes not mentioned, in radiology reports or elsewhere in the EHR. Because of this, dedicated review of imaging studies including all or part of the kidneys and/or lungs was completed by a board-certified radiologist (AMDL) for all patients with available imaging (Supplemental Materials and Methods).
Phenotype Evaluation – ICD-9/10 Diagnostic Codes
While chart review provides more accurate phenotyping, it is low throughput and not feasible for large sample sizes. As such, clinical manifestations of BHD-related phenotypes were also evaluated in variant-positive and negative patient-participants by extracting relevant International Statistical Classification of Diseases and Related Health Problems 9 and 10 codes (ICD-9/10) from the EHR in February 2021. Selected ICD-9/10 diagnostic codes were defined prior to manual chart review to capture BHD-related pulmonary, cutaneous, and kidney phenotypes (Supplemental Table 3). While ICD-9/10 codes can provide an estimate of BHD-related phenotypes, some codes that correspond to BHD-features are non-specific and could be used for other, unrelated clinical findings (e.g., ICD-10 codes for trichodiscoma and fibrofolliculoma are broadly used for benign neoplasms of the skin), and others might not be applied even when such a feature is identified due to its indolent or benign nature (e.g., ICD-9/10 codes for pulmonary cyst). Since use of ICD-9/10 codes alone might not correctly reflect patient phenotypes,26 we evaluated the ability of ICD-9/10 codes in the EHR to accurately identify BHD-related phenotypes by comparing diagnoses captured using diagnostic codes to those captured with manual review of EHR data in variant-positive individuals.
Statistical Analysis
Continuous variables are summarized as median and interquartile range and categorical variables are described as frequency and percentage. Two-tailed Fisher’s exact and Wilcoxon rank-sum tests were performed to test significant differences in demographics between variant-positive and variant-negative individuals. Associations between P/LP truncating FLCN variants and BHD phenotype were assessed by comparing the frequency of BHD-related phenotypes in variant-positive individuals with the variant-negative group using relevant ICD-9/10 diagnostic codes. The statistical significance of associations between P/LP truncating FLCN variants and BHD-related phenotypes was calculated using Firth logistic regression model adjusting for age, sex and the first four principal components of ancestry. To reduce the impact of population stratification on the results, the European subset, as determined by genetic ancestry, (121,876) was used for the association analysis. The European subset used in the association analysis was identified by principal component analysis and clustering with the 1000 Genomes reference population using the same protocol applied to the UK Biobank.27 Bonferroni correction was used to account for multiple testing (α = 0.05). All analyses were conducted using R version 4.0.1.
Results
Prevalence of P/LP Truncating FLCN Variants
Of the 135,990 patient-participants, 35 (0.03%, 1 in 3,885) had a P/LP truncating variant in FLCN (1 patient-participant not included in analyses had a non-truncating P/LP variant - Supplemental Tables 1, 6). Based on genetic relatedness data,25 we determined that these 35 individuals are from 28 families, representing 0.03%(1 in 3,234) of the 90,563 first and second-degree unrelated subset of the cohort. Thirteen unique truncating FLCN variants were identified, including five frameshift variants in 17 individuals, five nonsense variants in six patient-participants, two canonical splice site variants in 12 individuals, and a single intragenic deletion of exons 4–11 in one participant (Figure 1). The individual with the intragenic deletion also had a frameshift variant in cis. Nine of the thirteen variants identified in 32 of the 35 participants were previously reported in the literature and/or submitted to the ClinVar24 database. Table 1 summarizes demographics of variant-positive and variant-negative individuals in the MyCode cohort. Variant-positive participants were 62.9% female, 97.1% self-reported their race as White, 100% self-reported non-Hispanic ethnicity, and had a median age of 62 years. Collection of patient-reported categorical race and ethnicity data occurs as part of participants’ interaction with the healthcare system. Eighty-nine percent of variant-positive patient-participants were alive, and the median length of EHR data was 17.43 years at the time of manual chart review. No statistically significant demographic differences were identified between the variant-positive and variant-negative individuals.
Figure 1. Pathogenic/Likely Pathogenic truncating variants in FLCN identified in the MyCode cohort.
Based on build GRCh38 (NC_000017.11) and FLCN transcript most expressed in adult tissues (transcript variant 1, GenBank: NM_144997.7, 14 exons total with 11 coding exons, 579 amino acid residues) P=number of patient-participants with that variant and F=number of Families with that variant. ∓denotes variants that were previously reported in the literature and/or submitted to ClinVar. a denotes two variants identified in the same participant in cis.
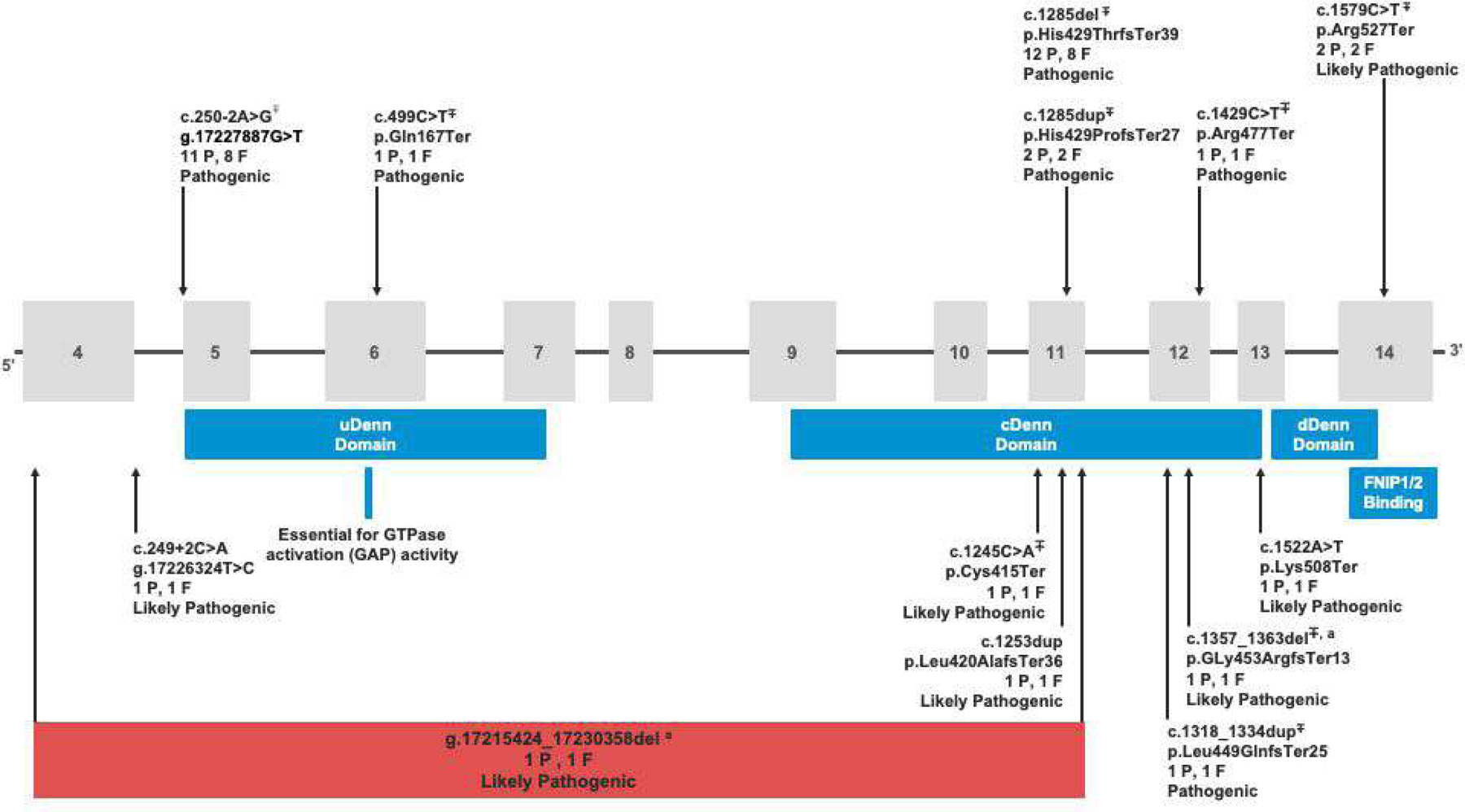
Table 1.
Demographic Details in Truncating FLCN Variant-Positive Patient-Participants and the Overall MyCode Cohort
| Demographics | P/LP Truncating FLCN Variant-Positive Patient-participants (n=35) |
Remaining MyCode Cohort (n=135,955) |
P-value |
|---|---|---|---|
| Median Age (IQR) | 62 years (54–76) | 61 years (45–72) | 0.20 |
|
Sex Female |
22 (62.9%) |
82,958 (61.0%) |
0.86 |
|
Racea American Indian or Alaska Native Asian Black or African American Native Hawaiian or Pacific Islander Other Unknown White |
1 (2.9%) -- -- -- -- -- 34 (97.1%) |
161 (0.1%) 487 (0.4%) 2,689 (2.0%) 231 (0.1%) 3 (0.0%) 325 (0.2%) 132,059 (97.1%) |
-- -- -- -- -- -- 0.99 |
|
Ethnicity
Hispanic or Latino Not Hispanic or Latino Unknown |
-- 35 (100%) -- |
2,978 (2.2%) 131,090 (96.4%) 1,887 (1.4%) |
-- 0.99 -- |
| Alive During Study Period | 31 (88.6%) | 125,455 (92.3%) | 0.34 |
|
Smoking Status Current Smoker |
4 (11.4%) |
23,094 (17.0%) |
0.50 |
|
Median Length of EHR (Range)
Number with <2 years |
17.43 years (0.05–22.61) 2 (5.7%) |
14.68 years (0.01–22.58) 5,301 (3.9%) |
-- 0.40 |
Collection of race and ethnicity data occurs as part of participants’ interaction with the healthcare system. Race and ethnicity are participant-provided and categories were defined based on what is included in the EHR. “Other” is a category available within the EHR.
BHD-related Phenotypes – Chart Review of Patient-participants with Truncating FLCN Variants
The frequencies of major BHD-related phenotypes were evaluated by manual chart review of each variant-positive patient-participant. We found that 68.6% (n=24/35) of variant-positive individuals had EHR-documentation of a BHD-related phenotype (Figure 2 and Table 2).
Figure 2. Frequency of BHD-related phenotypes in the literature compared to that in MyCode patient-participants with truncating P/LP FLCN variants and in unique MyCode families.
a Frequency of BHD-associated cutaneous manifestations is presented in all individuals with truncating P/LP FLCN variants. When restricting to those who were examined by a dermatologist and/or had a skin biopsy, 14.3% (n=3/21) had a BHD-related cutaneous finding. b An additional three participants had renal masses identified through manual EHR review. If we assume the three participants with a renal mass noted on imaging have an undiagnosed kidney cancer and only consider those with bilateral kidney imaging as being assessed for kidney cancer, an upper limit of potential kidney cancer in variant-positive individuals is 18.2% (n=4/22). Exome data was used to identify first-second degree
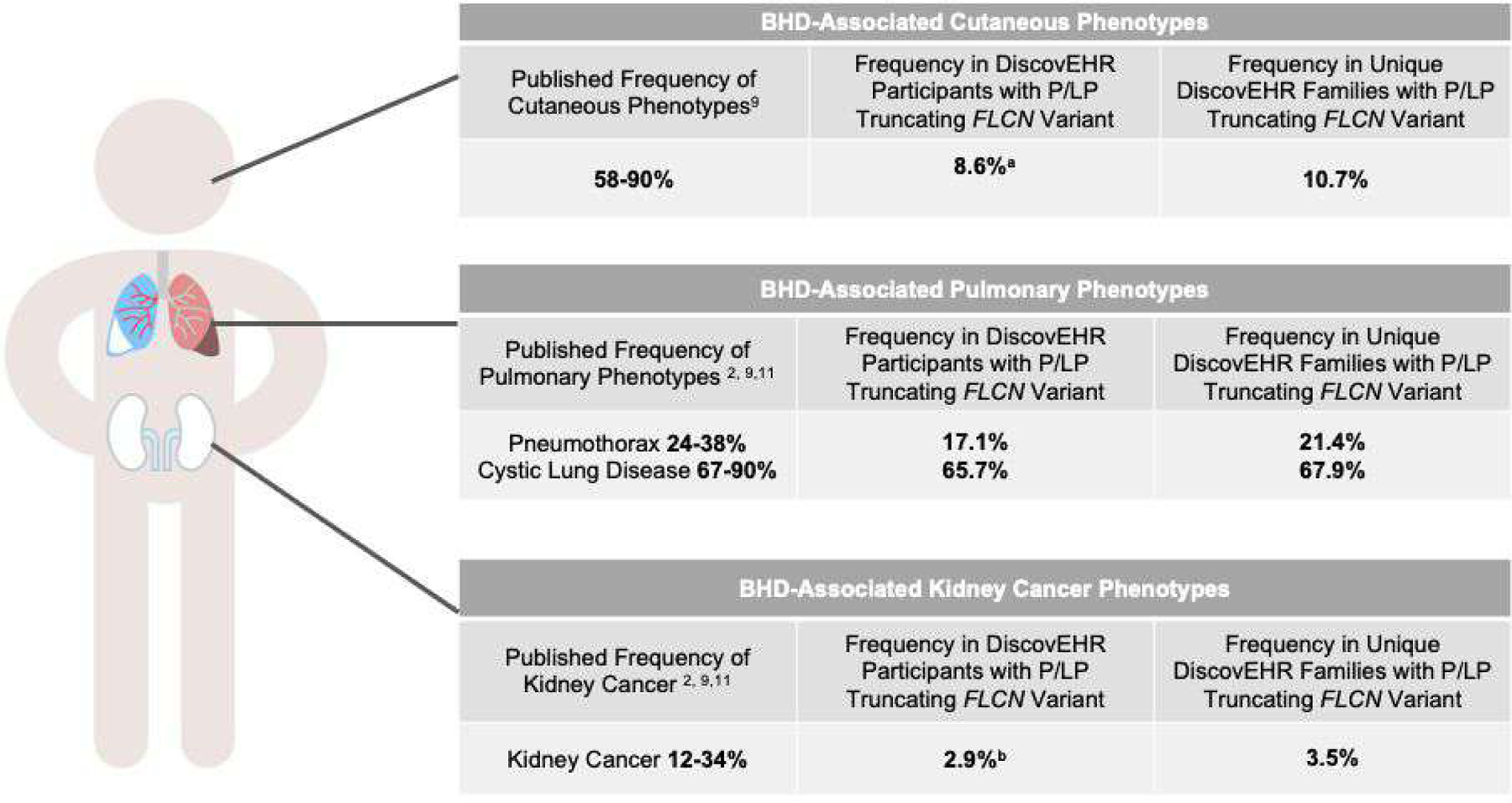
Table 2.
EHR Review for BHD-related Phenotypic Features in Truncating Variant-Positive Patient-Participants
| Phenotypic Feature | Frequency in Participants with P/LP Truncating FLCN Variant (n=35) |
|---|---|
| BHD Diagnosis and/or Clinical Identification of FLCN Variant | 4/35 (11.4%) |
| Any Personal History of BHD Cutaneous, Pulmonary, and/or Renal Phenotypes | 24/35 (68.6%) |
|
Any Cutaneous Phenotype Dermatologic Assessment (Dermatologist Exam and/or Skin Biopsy) Fibrofolliculoma, Perifollicular Fibroma, Trichodiscoma |
3/35 (8.6%)
21/35 (60.0%) 3/21(14.3%) |
|
Any Pulmonary Phenotype Basilar Cystic Lung Disease Single Spontaneous Pneumothorax Multiple Spontaneous Pneumothoraces Lung Imaging (Adequate Visualization and Any CT/MRI Imaging) Basilar Cystic Lung Disease |
23/35 (65.7%) 23/35 (65.7%) 2/35 (5.7%) 4/35 (11.4%) 13/35 (37.1%) and 29/35 (82.9%) 13/13 (100%) and 23/29 (79.3%) |
|
Any Kidney Phenotype
Kidney Cancer Renal Massa Renal Imaging (Adequate Visualization and Any Imaging)a Renal Massa |
4/35 (11.4%)
1/35 (2.9%) 3/34 (8.8%) 23/34 (67.6%) and 27/34 (79.4%) 3/23 (13.0%) and 3/27 (11.1%) |
|
Documented Family History of BHD-related Phenotypic Feature
Family History of Renal Cancer Family History of Pneumothorax Family History of Fibrofolliculomas/Papules Documented Family History of Clinical BHD or FLCN Variant |
7/35 (20.0%) 2/35 (5.7%) 3/35 (8.6%) 2/35 (5.7%) 2/35 (5.7%) |
excludes individual with renal cancer diagnosed
BHD-related cutaneous findings including fibrofolliculomas, perifollicular fibromas, and trichodiscomas were identified in three patient-participants (8.6%). Among all variant-positive participants, 60% (n=21/35) had documentation of an evaluation with a dermatologist and/or at least one skin biopsy. When restricting to those who were examined by a dermatologist and/or had a skin biopsy 14.3% (n=3/21) had a BHD-related cutaneous finding. BHD-related cutaneous findings were identified in 10.7% (n=3/28) of unique families with FLCN variants.
Pulmonary phenotypes, including basilar cystic lung disease and/or pneumothorax, were identified in 65.7% (n=23/35) of patient-participants. Of the 23 individuals with pulmonary phenotypes, six had at least one spontaneous pneumothorax; two had exactly one and four had more than one. Participants experienced their first pneumothorax between the ages of 21 and 79 years (median 27.5 years). The individual that experienced their first pneumothorax at age 79 had a diagnosis of chronic obstructive pulmonary disease. All 23 individuals with a pulmonary phenotype had basilar cystic lung disease. Eighty-three percent (n=29/35) of variant-positive patient-participants had CT or MRI imaging that included at least a portion of the lungs (e.g., abdomen CT includes lung bases) and 37.1% (n=13/35) had adequate visualization of the entire lungs. Among those with any lung imaging and those with adequate imaging reviewed, 79.3% (n=23/29) and 100% (n=13/13) had basilar cystic lung disease, respectively. Among unique families with FLCN variants, 21.4% (n=6/28) had pneumothorax and 67.9% (n=19/28) had cystic lung disease in at least one family member (Figure 2).
Kidney cancer was identified in one patient-participant (2.9%) with a chromophobe renal cancer diagnosed at age 71. Among the remaining 34 patient-participants without kidney cancer, 79.4% (n=27/34) had imaging that included the kidneys and 67.6% (n=23/34) had adequate visualization of both kidneys in their entirety. Renal masses were identified in 11.1 % (n=3/27) and 13.0% (n=3/23) of those who had any kidney imaging or bilateral imaging of the entire kidneys, respectively. These renal masses had not been pathologically confirmed to be kidney cancer or benign BHD-associated renal masses. In one case, follow-up imaging suggested the mass was likely a cyst while, in the other two cases, unrelated, benign renal cysts had not been ruled out.
In addition to core BHD-related phenotypes, the EHR review included evaluation of other phenotypes that have been reported in patients with BHD including thyroid cancer and parotid oncocytomas. Bilateral parotid oncocytosis was noted in a single participant with bilateral cystic lung disease. Thyroid cancer was not noted in any individuals.
Four individuals (11.4%) from three families had a diagnosis of BHD or documentation of a germline FLCN variant in their EHR. Two of those individuals had EHR documentation of multiple spontaneous pneumothoraces and bilateral blebs; one had a fibrofolliculoma, bilateral blebs, and renal mass; and one had a perifollicular fibroma.
BHD-related Phenotypes – Diagnostic Code Comparison
Comparison of BHD-related phenotypes captured by ICD codes with manual chart review (Supplemental Table 4) revealed that ICD-9/10 codes for BHD-related cutaneous features lack specificity and, as such, estimate the rates of such findings in variant-positive individuals at a higher rate than observed via manual review. We found that 42.9% (n=15/35) versus 8.6% (n=3/35) had BHD-related cutaneous features based on the ICD code approach and chart review respectively. ICD codes underestimated the frequency of cystic lung disease [20.0% (n=7/35) based on ICD codes versus 65.7% (n=23/35) chart and radiology review], likely due to the benign nature and incidental identification on imaging. Diagnostic codes for pneumothorax were found to accurately identify this phenotype in variant-positive individuals [17.1% (n=6/35) ICD codes versus 17.1% (n=6/35) chart and radiology review]. All six individuals with pneumothoraces identified on manual chart review had an appropriate ICD-9/10 code in their EHR. ICD diagnostic codes for kidney cancer appropriately identified the single individual with kidney cancer. Diagnostic codes associated with a renal mass underestimated this phenotype [2.9% (n=1/34) ICD codes versus 8.8% (n=3/34) chart review].
The associations between FLCN P/LP variants and ICD-9/10 based BHD-related phenotypes were assessed by Firth logistic regression in the unrelated, European subset of the cohort (Table 3). P/LP variants in FLCN were associated with a higher prevalence of pulmonary phenotypes (OR=4.16; 95% CI:11.65–9.43; p=0.026), and, more specifically, spontaneous pneumothorax (OR=15.63; 95% CI: 5.96–35.69; p=1.92×10−5) in variant-positive patient-participants. Association of P/LP truncating FLCN variants with other pulmonary, cutaneous, and kidney phenotypes were not statistically significant when correcting for multiple testing. We performed the association analysis in the full unrelated subset of the cohort including European and non-European minority populations. The results of the full unrelated cohort were consistent with those of the European subset (Supplemental Table 5).
Table 3.
Associations Between P/LP Truncating FLCN Variants and Diagnosis of BHD-related Phenotypesa in the First- and Second-Degree Unrelated European Subset of the MyCode Cohort
| Phenotype | Patient-Participants with P/LP Truncating FLCN Variant N=27 |
Remaining MyCode Cohort N=79496 |
Odds Ratio (95% CI) | p-valueb |
|---|---|---|---|---|
| Any Pulmonary Phenotype | 7 (25.9%) | 6,422 (8.1%) | 4.16 (1.65–9.43) | 0.026* |
| Spontaneous Pneumothorax | 6 (22.2%) | 1,523 (1.9%) | 15.63 (5.96–35.69) | 1.92×10−5* |
| Pulmonary Cyst | 5 (18.5%) | 5,245 (18.5%) | 3.40 (1.17–8.32) | 0.185 |
| Cutaneous Phenotype | 10 (37.0%) | 16,243 (20.4%) | 2.19 (0.97–4.70) | 0.405 |
| Any Kidney Phenotype | 1 (3.7%) | 2,200 (2.8%) | 1.93 (0.21–7.43) | >0.99 |
| Renal Mass | 0 (0%) | 1,693 (2.1%) | 0.81 (0.01–5.81) | >0.99 |
| Kidney Cancer | 1 (3.7%) | 852 (1.1%) | 4.94 (0.55–19.37) | 0.891 |
Ability of ICD-9/10 codes in the EHR to accurately identify BHD-related phenotypes varied between phenotypes.
Bonferroni corrected for multiple testing (7-tests).
Discussion
Frequency of P/LP Truncating FLCN Variants
BHD is considered a rare disorder with an estimated prevalence of 1 in 200,000.7 BHD prevalence and penetrance estimates have been based on clinically ascertained families to date. This study examined exome sequencing data from a large healthcare population to estimate the prevalence of P/LP truncating FLCN variants and frequency of BHD-related phenotypes. Truncating FLCN variants were identified in 28 out of 90,563 first- and second-degree unrelated individuals (1 in 3,234 in unrelated cohort) suggesting that such variants are significantly more common than previously reported. This finding is consistent with the increased frequency of variants associated with other genetic syndromes identified in broader, population cohorts via genome-first approaches.28–30
Prevalence of BHD-related Phenotypes
In participants with P/LP truncating FLCN variants from this healthcare cohort, BHD-related lung findings were the most common phenotype (65.7%, n=23/35). The frequency of basilar pulmonary cystic lung disease in those with CT and MRI lung imaging (79.3%, n=23/29) overlapped with the reported frequency in clinically ascertained individuals (67–90%)2,9,11 and was even higher than the reported frequency in those with adequate visualization of the lungs (100%). The frequency of pneumothorax (17.1%, n=6/35) was lower than that reported in the literature (24–38%). Pneumothorax-associated ICD-9/10 codes were present in the EHR of all patient-participants with pneumothoraces identified by chart review, suggesting that these codes capture the phenotype with high sensitivity. When we assessed the association between P/LP truncating FLCN variants and pneumothorax, we identified a higher prevalence of spontaneous pneumothorax diagnosis (OR=15.63 95%CI (5.96–35.69), p=1.92×10−5) in variant-positive individuals. Together, these data suggest that while patient-participants with truncating FLCN variants in this healthcare-based cohort may have lower rates of pneumothorax compared to the clinically ascertained BHD population and higher rates compared to the variant-negative population. The prevalence of diagnosis codes associated with cystic lung disease was not statistically different between the variant-positive and variant-negative patient-participants. However, this could be due to inability of the selected ICD-9/10 codes to identify all individuals with the phenotype, as a minority of variant-positive participants with this phenotype based on manual chart review had the relevant ICD codes in the EHR. Therefore, this pulmonary finding, which can be a precursor to spontaneous pneumothorax, may also occur at higher rates in individuals with P/LP truncating FLCN variants. Additional research is needed to determine whether P/LP FLCN variants are associated with increased prevalence of cystic lung disease.
Other BHD-related phenotypes, including cutaneous and kidney manifestations appeared to be less common in the variant-positive individuals in this cohort compared with BHD probands in the reported literature. BHD-related dermatologic findings were identified in only 8.6% (n=3/35) of FLCN variant-positive individuals compared to 58–90% of patients with BHD in the literature.9 When restricting the analysis to patient-participants who were examined by a dermatologist and/or had a skin biopsy that would have likely identified these benign phenotypes, only 14.3% (n=3/21) had BHD-related skin findings. Since lack of clinical assessment does not alone account for differences in frequency, this finding is consistent with a lower penetrance of P/LP truncating FLCN variants than previously reported. P/LP truncating FLCN variants were not associated with an increased rate of cutaneous diagnosis, based on ICD-9/10 codes. Given the non-specific nature of cutaneous ICD-9/10 codes and their overestimation of BHD-related skin-findings in variant-positive individuals, however, additional assessment comparing the frequency of BHD-related cutaneous findings between variant-positive and variant-negative populations is needed.
Only one participant with a FLCN variant was identified to have kidney cancer (2.9%, n=1/35). This is a lower frequency than reported in the literature (12–34%).2,9,11 Although some participants are younger (e.g., five variant-positive participants <40 years of age) and could go on to develop a kidney cancer in their lifetime, the majority of FLCN variant-positive individuals in the cohort are older than 50.7 years (74.3%, n=26/35) which is the median age of kidney cancer diagnosis reported in individuals with BHD. Due to the indolent nature of BHD-related kidney cancers,2 some variant-positive patient-participants might have yet to come to clinical attention. If we assume the three participants with a renal mass noted on imaging have an undiagnosed kidney cancer and only consider those with bilateral renal imaging as being assessed for a kidney cancer, an upper limit of potential kidney cancer in variant-positive individuals is 18.2% (n=4/22), which overlaps with the frequency of kidney cancer reported in the literature. P/LP truncating FLCN variants were not associated with increased prevalence of kidney cancer in the MyCode cohort based on EHR data. Although additional studies are needed due to small numbers, this suggests the rates of kidney cancer among individuals with truncating FLCN variants may not be as high as previously estimated.
These data suggest that, among individuals in a healthcare-system population who volunteered to participate in genetic research but were not selected for any disease state, the majority of individuals with P/LP truncating FLCN variants have a phenotype consistent with BHD. Of all MyCode participants without considering relatedness, 1 in 5,663 had a P/LP truncating FLCN variant and an associated phenotype. Individual BHD-related phenotypes including pneumothorax, cutaneous findings, and kidney cancer may be less common among individuals with P/LP truncating FLCN variants than previously reported, a finding that is consistent with other studies examining frequency of disease-associated phenotypes in population-based cohorts.12,13 Although we are not aware of any prior population-based studies of FLCN variants, the FLCN gene is located within the recurrent copy number variant region on 17p11.2 that is deleted in 90% of individuals with Smith-Magenis syndrome (SMS), hence these individuals are haploinsufficient for FLCN; however, there are few published reports of BHD-related phenotypes in individuals with SMS.31-34Although SMS cohorts may be younger in age and not all individuals with SMS have undergone clinical evaluation for BHD, these data also suggest that published penetrance of BHD-related phenotypes may be overestimated.
Clinical Identification of Truncating FLCN Variants
Even though most participants with P/LP truncating FLCN variants had features consistent with BHD documented in the EHR, the majority of variant-positive patient-participants (88.6%, n=31/35) did not have a clinical diagnosis of BHD. The majority of those with BHD-related features had cystic lung disease (95.8%, n=23/24 with phenotype). Although cystic lung disease is included in the diagnostic criteria for BHD,2,35 these findings did not prompt a referral to genetics or clinical genetic testing in any variant-positive patient-participants. The four individuals with clinically identified FLCN variants or BHD diagnoses all had cystic lung disease but were evaluated due to other phenotypes (fibrofolliculoma, perifollicular fibroma, pneumothoraces, family history of BHD). Although cystic lung disease was identified in 23 individuals in total, nine patient-participants were only identified on radiologist review of imaging data, meaning these findings were not documented in the EHR and were not commented on in the initial radiology report. Improved clinical recognition of these benign findings and education concerning their potential association with truncating FLCN variants could prompt additional evaluation for BHD. Identifying patients at risk for BHD-phenotypes could inform appropriate management. For example, in patients with BHD, a first pneumothorax might be treated more aggressively with pleurectomy/pleurodesis given the risk for recurrent pneumothoraces36,37 and identification of individuals with FLCN variants could enable earlier screening for kidney cancer, potentially allowing detection of cancers at earlier stages.38
Limitations
This study has several limitations. Because the MyCode population is primarily of European ancestry, from a single healthcare system participating in genetics research, it is unclear if these findings are generalizable to more diverse patient populations. Future work that combines data from multiple, more diverse cohorts are needed. Our estimated phenotype frequency relies on data within the EHR; as such, not all patient-participants have had assessments that would identify BHD-related features and imaging studies reviewed differ in their ability to detect lung and kidney phenotypes. Finally, this study focused on truncating variants since they are most common in individuals with BHD; additional variant types could be considered in future work.
Conclusions
P/LP truncating FLCN variants were identified in 1 in 3,234 individuals unrelated cohort in a healthcare population. Of those with such variants, 65.6% were identified to have BHD-related phenotypes (1 in 5,663 individuals with a truncating FLCN variant and an associated phenotype); however, only 11.4% had been clinically diagnosed with BHD. This limited ascertainment is likely because classic BHD-related phenotypes, such as cutaneous manifestations, pneumothorax, and kidney cancer, were observed at lower frequencies. Cystic lung disease was the most common phenotype in our cohort and could be used to prompt evaluation for BHD. This finding could inform future screening strategies for BHD in broad patient populations.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Mental Health of the National Institutes of Health (NIH) under award number: R01 MH074090. The authors would like to acknowledge the MyCode patient-participants that make this work possible. We would also like to acknowledge those involved in the Geisinger-Regeneron DiscovEHR Collaboration who have enabled the generation of the genomics data used in this study and Dr. H. Les Kirchner for his thoughtful review and comments that helped shape the statistical analysis.
Footnotes
Ethics Declaration
The Geisinger MyCode® Community Health Initiative (MyCode) serves as a biobank of blood and other samples from over 300,000 patient-participants who consent to health-related research (https://www.geisinger.org/precision-health/mycode). MyCode and the research outlined are approved by the Geisinger Institutional Review Board. MyCode participants or their parent/legal guardian provide written consent and HIPAA authorization. As part of participation, they agree to provide samples for broad research use and permit access to data in their electronic health record for research use. 14 Individual data included in this manuscript have been de-identified and presented in aggregate.
Conflict of Interest Statement
DHL: Employee of Unified Patient Network, Inc., and scientific consultant for Natera, Inc., MyOme, Inc., and Seven Bridges Genomics, Inc. relationships and to group individuals into unique families. Exome data was used to identify first-second degree relationships and to group individuals into unique families.
Data Availability
The data that support the findings of this study are available within the article and/or are available on request from the corresponding author, JMS.
References
- 1.Birt AR, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 1977;113(12):1674–1677. [PubMed] [Google Scholar]
- 2.Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol 2015;12(10):558–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasumi Y, Baba M, Ajima R, et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A 2009;106(44):18722–18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baba M, Hong SB, Sharma N, et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A 2006;103(42):15552–15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickerson ML, Warren MB, Toro JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé syndrome. Cancer Cell 2002;2(2):157–164. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt LS, Linehan WM. FLCN: The causative gene for Birt-Hogg-Dubé syndrome. Gene 2018;640:28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=122. . Accessed December 14, 2020.
- 8.Foundation B-H-DB, 2017. https://www.bhdsyndrome.org/for-researchers/what-is-bhd/introduction/published-bhd-families/ December 18, 2020.
- 9.Jensen DK, Villumsen A, Skytte AB, Madsen MG, Sommerlund M, Bendstrup E. Birt-Hogg-Dubé syndrome: a case report and a review of the literature. Eur Clin Respir J 2017;4(1):1292378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Garcia DR, Teague D, Landis ET, Sangueza OP. Morphological diversity of trichodiscomas and fibrofolliculomas. Am J Dermatopathol 2014;36(9):734–740. [DOI] [PubMed] [Google Scholar]
- 11.Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé Syndrome. Clin Chest Med 2016;37(3):475–486. [DOI] [PubMed] [Google Scholar]
- 12.van Rooij J, Arp P, Broer L, et al. Reduced penetrance of pathogenic ACMG variants in a deeply phenotyped cohort study and evaluation of ClinVar classification over time. Genetics in Medicine 2020;22(11):1812–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tuke MA, Ruth KS, Wood AR, et al. Mosaic Turner syndrome shows reduced penetrance in an adult population study. Genet Med 2019;21(4):877–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carey DJ, Fetterolf SN, Davis FD, et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med 2016;18(9):906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354(6319):aaf6814. [DOI] [PubMed] [Google Scholar]
- 16.https://www.geisinger.org/-/media/OneGeisinger/pdfs/ghs/research/mycode/mycode-scorecard.pdf?la=en. Accessed March 12, 2022 2020.
- 17.Buchanan AH, Lester Kirchner H, Schwartz MLB, et al. Clinical outcomes of a genomic screening program for actionable genetic conditions. Genetics in Medicine 2020;22(11):1874–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yun T, Li H, Chang P-C, Lin MF, Carroll A, McLean CY. Accurate, scalable cohort variant calls using DeepVariant and GLnexus. Bioinformatics 2021;36(24):5582–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Packer JS, Maxwell EK, O’Dushlaine C, et al. CLAMMS: a scalable algorithm for calling common and rare copy number variants from exome sequencing data. Bioinformatics 2016;32(1):133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007;17(11):1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abou Tayoun AN, Pesaran T, DiStefano MT, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat 2018;39(11):1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015;17(5):405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 2020;22(2):245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–d1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Staples J, Qiao D, Cho MH, Silverman EK, Nickerson DA, Below JE. PRIMUS: rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. Am J Hum Genet 2014;95(5):553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quan H, Li B, Saunders LD, et al. Assessing validity of ICD-9-CM and ICD-10 administrative data in recording clinical conditions in a unique dually coded database. Health services research 2008;43 4:1424–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manickam K, Buchanan AH, Schwartz MLB, et al. Exome Sequencing-Based Screening for BRCA1/2 Expected Pathogenic Variants Among Adult Biobank Participants. JAMA Netw Open 2018;1(5):e182140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho BPH, Nannoni S, Harshfield EL, et al. NOTCH3 variants are more common than expected in the general population and associated with stroke and vascular dementia: an analysis of 200 000 participants. J Neurol Neurosurg Psychiatry 2021;92(7):694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dershem R, Gorvin CM, Metpally RPR, et al. Familial Hypocalciuric Hypercalcemia Type 1 and Autosomal-Dominant Hypocalcemia Type 1: Prevalence in a Large Healthcare Population. Am J Hum Genet 2020;106(6):734–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dardour L, Verleyen P, Lesage K, Holvoet M, Devriendt K. Bilateral renal tumors in an adult man with Smith-Magenis syndrome: The role of the FLCN gene. Eur J Med Genet 2016;59(10):499–501. [DOI] [PubMed] [Google Scholar]
- 32.Truong HT, Dudding T, Blanchard CL, Elsea SH. Frameshift mutation hotspot identified in Smith-Magenis syndrome: case report and review of literature. BMC Med Genet 2010;11:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finucane B, Savatt JM, Shimelis H, Girirajan S, Myers SM. Birt-Hogg-Dubé symptoms in Smith-Magenis syndrome include pediatric-onset pneumothorax. Am J Med Genet A 2021;185(6):1922–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith ACM, Fleming LR, Piskorski AM, Amin A, Phorphutkul C, de la Monte S, Stopa E, Introne W, Vilboux T, Duncan F, Pellegrino J, Braddock B, Middleton LA, Vocke C, Lonehan WM Deletion of 17p11.2 encompasses FLCN with increased risk of Birt-Hogg-Dubé in Smith Magenis Syndrome: Recommendation for Cancer Screening. . Annual conference of the American Society of Human Genetics San Diego, CA, United States, October 18–22, 2014. [Google Scholar]
- 35.Menko FH, van Steensel MA, Giraud S, et al. Birt-Hogg-Dubé syndrome: diagnosis and management. Lancet Oncol 2009;10(12):1199–1206. [DOI] [PubMed] [Google Scholar]
- 36.Sattler EC, Syunyaeva Z, Mansmann U, Steinlein OK. Genetic Risk Factors for Spontaneous Pneumothorax in Birt-Hogg-Dubé Syndrome. Chest 2020;157(5):1199–1206. [DOI] [PubMed] [Google Scholar]
- 37.Gupta N, Kopras EJ, Henske EP, et al. Spontaneous Pneumothoraces in Patients with Birt-Hogg-Dubé Syndrome. Ann Am Thorac Soc 2017;14(5):706–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johannesma PC, Reinhard R, Kon Y, et al. Prevalence of Birt-Hogg-Dubé syndrome in patients with apparently primary spontaneous pneumothorax. Eur Respir J 2015;45(4):1191–1194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the article and/or are available on request from the corresponding author, JMS.