

Abstract
Labrune syndrome is an extremely rare disorder characterized by a radiological triad of leukoencephalopathy, cerebral calcifications, and cysts. The condition is the result of an autosomal mutation in the SNORD118 gene, a non-protein encoding gene that mediates rRNA synthesis. The mutation results selectively in cerebral microangiopathy through an unknown mechanism. Radiological imaging is central to diagnosing the condition, but, because the condition is so rare, there is no standard treatment paradigm. We describe the longitudinal progression of a case of Labrune syndrome, including the radiological diagnosis and imaging and surgical management.
Keywords: Leukoencephalopathy, Cerebral calcifications, Cerebral cysts, Microangiopathy, Ribosomopathy, White matter
Introduction
In 1996, Labrune et al. described a series of 3 cases defined by a radiological triad of leukoencephalopathy, brain calcifications, and cysts secondary to microangiopathic changes of the cerebral vasculature; the condition was subsequently termed “Labrune Syndrome,” otherwise known as “leukoencephalopathy with calcifications and cysts (LCC).” This syndrome is symptomatically defined by seizures, pyramidal, extrapyramidal, and cerebellar signs [1]. Jenkinson et al. have recently shown that this syndrome results from autosomal recessive mutations in the SNORD118 gene, a non-protein encoding RNA that assists in ribosome synthesis [2]. The mechanism by which a ribosomopathy specifically induces cerebral microangiopathy is unknown. Nevertheless, the microangiopathic changes are defined by increased small vessel tortuosity and calcification, leading to increased focal edema and increased water content of the white matter; additional histopathological findings include Rosenthal fibers, hyaline deposition and perivascular calcifications [3].
This condition is extremely rare, with less than 100 reported cases since its identification [4]. Consequently, there is no established paradigm for the diagnosis or treatment of LCC. Prior to the identification of SNORD118, Wang et al. proposed a diagnostic algorithm to work through the differential [5]. Several other diseases must be ruled out, including other leukodystrophies, parasitic infections and astrocytoma [5]. After working through the differential, genetic analysis allows for the definitive diagnosis of LCC. These different diseases dictate different treatments, so it is imperative to arrive at a correct diagnosis. Treatments are aimed at providing symptomatic relief, including surgical removal of cysts, antiepileptics, and antipsychotics [4]. Interestingly, vascular endothelial growth factor (VEGF) has been used experimentally with some success in a few cases [6,7].
Case section
We present a longitudinal case of LCC in a 23-year-old male that displayed marked disease progression on imaging over a course of approximately 9 years. The patient initially presented at the age of 14 for new onset seizures. Initial computed tomographic (CT) and magnetic resonance imaging (MRI) revealed extensive subcortical calcification and calcifications in the pons and left dentate nucleus; several cysts within the right frontal and temporal lobes; and white matter disease encompassing the centrum semiovale with involvement of the midbrain, pons, and middle cerebellar peduncle (Fig. 1). An initial diagnosis of Aicardi-Gouteire's disease was made, but addended to LCC after LCC was identified following a brief literature review. There was a striking resemblance to the imaging observed in the literature and the patient's films. The patient's seizures were controlled medically and no surgical intervention was recommended.
Fig. 1.

Initial imaging at age 14, with CT imaging demonstrating extensive calcifications in a subcortical distribution (A), and MR imaging demonstrating extensive white matter disease (B).
Follow-up imaging 2.5 years later demonstrated bilateral hemispheric cysts with an increase in size of the right temporal cyst from 1 to 2.7 cm, increased white matter FLAIR intensity in both cerebral hemispheres, and spread of calcifications with some expressing peripheral enhancement (Fig. 2).
Fig. 2.

Follow-up imaging 2.5 years after initial imaging. MR imaging demonstrates several bilateral cysts in the subcortical white matter accompanied with white matter disease (A). CT imaging demonstrates increased calcification compared to prior CT (B).
Follow-up imaging 3 years later (5.5 years from initial image) demonstrated further progression of the disease. Several cysts demonstrated dense calcifications with associated microhemorrhages. Several cysts grew significantly, including the cyst in the right temporal lobe, which measured 4.2 cm with blood fluid level association. Increased white matter involvement was demonstrated, most notably including new onset involvement of the left cerebral peduncle, with extensions into the pons and bilaterally into the cerebellum through the cerebellar peduncles.
Subsequent follow-up MR imaging 2 years later (7.5 years from initial imaging) demonstrated a marked increase in the size and number of intracranial cysts. The largest cyst, located in the right temporal lobe, was measured at 5.7 cm. A new prominent cyst emerged in the right periventricular white matter. These 2 large right-sided cerebral cysts began to exert mass effects, causing a 7 mm leftward midline shift and uncal compression of the right cerebral peduncle. The cyst in the posterior pons progressed to the point of nearly filling the fourth ventricle (Fig. 3). Gradient echo imaging demonstrated extensive calcification; however, not much progression of calcification was noted from 2 year's prior images.
Fig. 3.

Pre-operative imaging prior to the first surgery (7.5 years after initial imaging). MR imaging demonstrates significant cyst progression, including the right temporal lobe (A) and throughout the rest of the cerebrum (B). White matter disease and calcifications are also visualized.
After disease progression over the prior 7.5 years, concern arose regarding the possibility of herniation or ventricular obstruction, prompting surgical intervention. Cyst aspiration to relieve mass effect was completed during one procedure, in 3 separate stages. “Stage one” was a right-sided stereotactic, image-guided burr hole aspiration of the 2 largest right-sided cysts responsible for the demonstrated left midline shift and uncal herniation. “Stage two” was a left-sided stereotactic, image-guided burr hole aspiration of a left parietal cyst. “Stage three” was an open, image-guided suboccipital craniotomy for open fenestration of the brain stem cyst in the floor of the fourth ventricle. Later that day, the patient complained of headaches and experienced projectile vomiting; a stat CT demonstrated a left frontoparietal epidural hematoma (Fig. 4). An emergency craniotomy was performed to evacuate the hematoma. CT imaging performed the next day demonstrated successful hematoma removal (Fig. 5). Beyond mild nystagmus, nausea, and vomiting, the post-op course was unremarkable and the patient was discharged 3 days later.
Fig. 4.
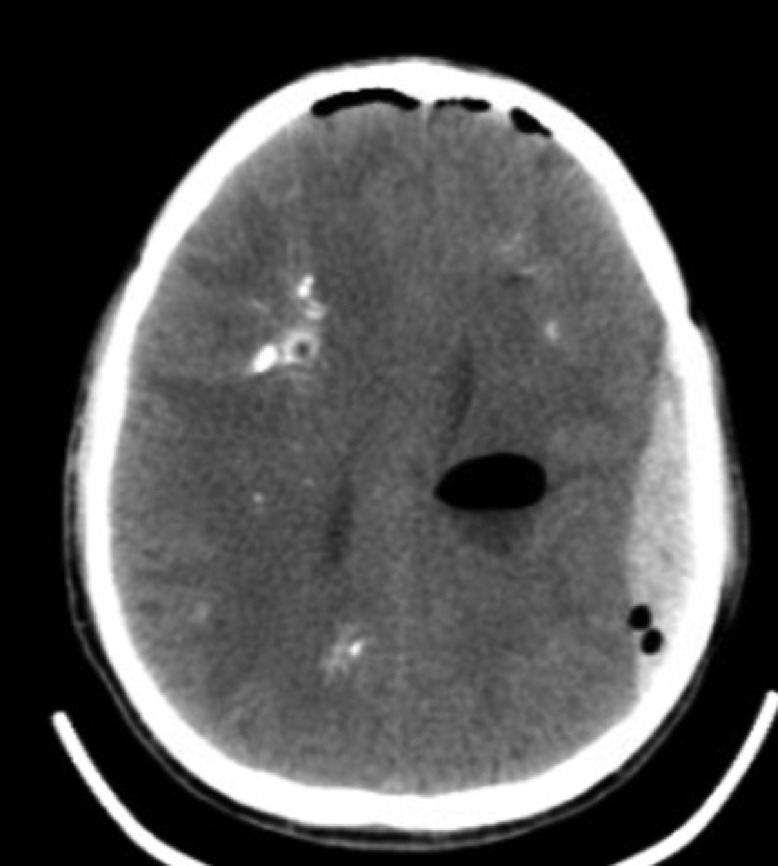
Post-operative CT image the day of first surgery complicated by a left-sided epidural hematoma. Post-operative pneumocephalus is also seen.
Fig. 5.
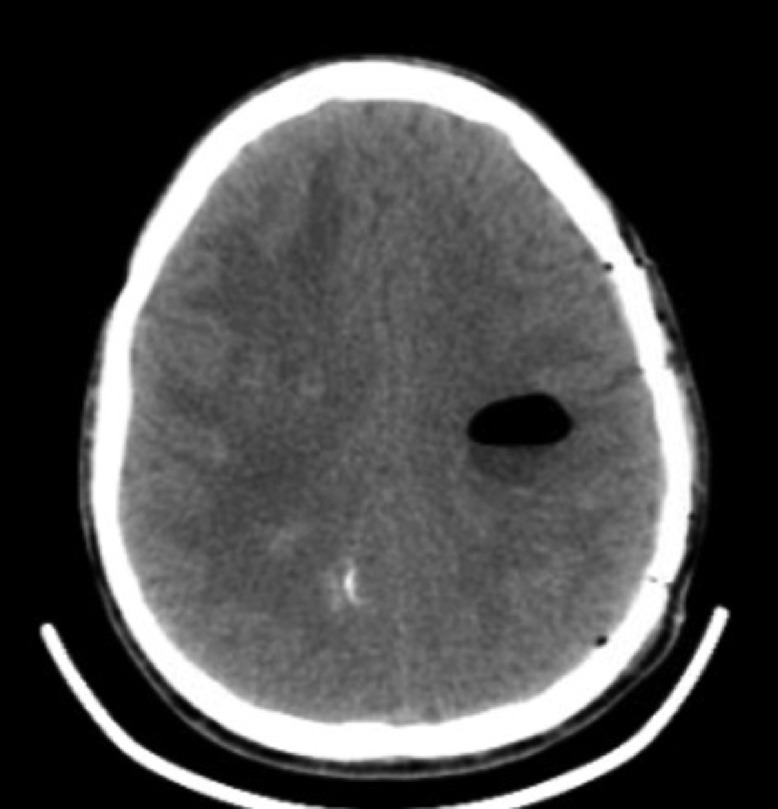
Post-evacuation CT image the day following the initial surgery and epidural hematoma evacuation.
MR imaging 3 weeks later demonstrated resolution of midline shift. Furthermore, the ring-enhancing cystic lesions appeared to resolve. The brain still contained numerous cysts and calcifications. MR imaging 6 months later revealed interval cyst growth. The large cyst in the right temporal lobe that had been aspirated increased in size to 7.7 cm, causing both a 5 mm leftward midline shift and right uncal compression of the midbrain. All other cysts and calcifications remained stable. The patient remained minimally symptomatic and observation was continued. Imaging performed another 6 months later (12 months post-op) showed mild interval cyst progression, most notably in the right temporoparietal cyst, now measuring 8.1 cm and exerting a 5 mm leftward midline shift. There was a marked increase in left cerebellar leukodystrophy as compared to prior imaging. Also noted were areas of gliosis surrounding multiple sites of infarction. The patient remained stably symptomatic throughout, complaining of complex partial seizures, auras, dizziness, tunnel vision, left-sided paresthesia, tinnitus, and fear of impending doom. The patient had no outward deficits and appeared to be fully intact of cognitive function. The patient was maintained on levetiracetam, lorazepam, and meclizine.
Follow-up imaging another 3 months later (15 months post-op) revealed significant enough cyst growth and leukoencephalopathy progression to prompt a second surgery (Fig. 6). One month later, the patient underwent Wada testing in preparation for cyst aspiration. The results of the Wada demonstrated the right-handed patient to have speech localization in the left hemisphere. Also, the left side hippocampal complex was able to independently support memory, suggesting it was safe to operate on the right side of the brain. Approximately 1 month later, the patient underwent a craniotomy to fenestrate the right temporoparietal cyst cavity. Postoperative imaging demonstrated significant relief of mass effect (Fig. 7). The post-op course has remained uneventful, and the patient will continue to be monitored.
Fig. 6.

Pre-operative MR imaging for the second surgery, approximately 15 months after the first surgery. Significant cyst growth and progressive leukodystrophy are demonstrated. Notably, marked growth in the previously fenestrated right temporal lobe cyst is demonstrated (B).
Fig. 7.

Post-operative MR image demonstrating significant reduction in the size of the right temporal lobe cyst with consequent relief of mass effect.
Discussion
LCC is defined by a radiological triad of leukoencephalopathy, brain calcifications, and cysts [1]. Typical symptoms include seizures, pyramidal, extrapyramidal, and cerebellar signs [1].
Jenkinson et al. demonstrate that LCC is an autosomal recessive condition in the small nucleolar RNA, C/D box 118 (“SNORD118”) gene [2]. This gene encodes for the snoRNA U8, a non-protein encoding gene which aids in rRNA processing as part of ribosomal biogenesis [8]. SNORD118 displays significant polymorphism. In an analysis of 40 LCC patients from 33 families, Jenkinson et al. demonstrate rare or novel mutations in SNORD118 in all 40 patients; furthermore, only 2 out of 33 pedigrees display consanguinity, while the remaining 31 pedigrees display compound heterozygosity [2]. Taken together, these findings imply LCC results from one null mutation paired with one rare hypomorphic mutation, allowing for embryogenesis but not proper functioning, while homozygous null mutations must be embryologically lethal [2]. Indeed, in a zebrafish model, Badrock et al. demonstrate U8-3 null mutations are embryologically lethal [9]. However, Crow et al. were unable to identify any specific genotype-phenotype relationship to explain symptom variability, including age of onset, implying additive and environmental factors likely play a role in phenotypic expression as well [10]. As ribosomes are central to translation, a process central to protein production to cells throughout the body, a ribosomopathy should be expected to exert ubiquitous deleterious effects; however, the only demonstrated pathology in LCC is cerebral microangiopathy [3]. The mechanism by which a ribosomopathy selectively causes cerebral microangiopathy is unknown. The mechanism is especially difficult to elucidate, as it is a non-protein coding gene, so in silico algorithms cannot be used to predict loss of function [10].
LCC is histopathologically defined by cerebral microangiopathy with angiomatous-like microvasculature changes, leading to edematous changes throughout the brain. Rosenthal fibers, proteinaceous aggregates of glial fibrillary acidic protein and ubiquitin found in fibrous astrocytes under inflammatory conditions, are also observed, as are blood vessel calcifications and gliosis [3]. Other case reports demonstrate decreased N-acetylaspartate (NAA) and choline (Cho) peaks on magnetic resonance spectrometry (MRS), indicating the leukoencephalopathy observed in LCC is due to increased water content in myelin, not demyelination [5,11]. Although there is much left to be understood about the pathogenesis of LCC, the disease can be well defined genetically, radiologically, and histopathologically.
Due to its extreme rarity and similarity to several diseases, LCC may evade timely diagnosis and treatment. Furthermore, the radiological triad defining LCC is not pathognomonic for LCC; other diseases display similar radiological findings [10]. There are no established diagnostic criteria, and most cases of LCC are diagnosed later in the disease course. Although there is no cure, symptomatic relief can be achieved through surgery, anti-psychotics, and anti-epileptics. Even though LCC is extremely rare, it should be considered in the differential of leukoencephalopathy with calcifications and cysts. Prior to the discovery of SNORD118 mutations underlying LCC, Wang et al. proposed an algorithm for navigating the differential of associated neurological conditions [5]. Although SNORD118 genotyping alone should now be sufficient for ultimate diagnosis of LCC, it is worthwhile to consider each disease in the differential. Several of these different diseases are treatable with the appropriate indication; thus, arriving at a correct diagnosis may yield clinically beneficial results. The differential includes parasitic infections, astrocytomas, and other leukodystrophies, including Coats Plus syndrome and Aicardi-Goutieres syndrome.
Parasitic infections can be differentiated from LCC based on the presence or absence of anti-parasitic antibodies [5]. Several parasites can cause brain infections, such as Taenia solium, the most common parasitic infection in the world. Taenia solium is found in endemic areas where pigs and humans cohabitate under conditions of poor sanitation. These parasites cause neurocysticercosis, in which the secondary larval form creates cysts in the brain that subsequently calcificy [12]. The diagnosis of an intracranial parasitic infection is made through the detection of anti-parasitic antibodies/antigens in serology. These infections can subsequently be treated with the appropriate anti-parasitic medication [5].
After ruling out parasitic infections, Wang et al. recommend performing nuclear MRS to rule out astrocytoma [5]. Astrocytomas generally present with elevated Cho and decreased NAA on MRS [13]. For LCC, the majority of the cases reviewed by Wang et al. demonstrate reduced NAA and Cho levels within the cysts, as well as the absence of metabolites, suggesting water content within the cyst [5]; furthermore, they report reduced NAA and Cho in the leukoencephalic lesions, which suggests water content in the white matter [5]. Astrocytomas have a treatment protocol, so it is important to differentiate these lesions from LCC. After ruling out astrocytoma, Wang et al. recommend using genetic analysis to differentiate between several radiologically similar diseases, including Aicardi-Goutieres and Coats Plus [5].
Aicardi-Goutieres syndrome (AGS) is a “type 1 interferonopathy” that may present similarly to LCC. AGS presents as a progressively worsening neurological disorder that presents early in life and frequently involves the skin, with characteristic “chilblains” [14]. Mutations of several different genes have been implicated in AGS, including TREX1, RNASE H2 proteins, SAMHD1, ADAR, and MDA5; mutations in these genes lead to increased levels of type 1 interferon (IFN-1) [14]. AGC is radiologically similar to LCC, as it also presents with intracranial calcification, cysts and leukodystrophy, although AGS also frequently demonstrates cortical atrophy [15]. Additionally, IFN-1 is elevated in cerebrospinal fluid and serum in AGS [16]. There is currently no established treatment for AGS, although therapies targeting the IFN-1 pathway, reverse transcriptase inhibitors and JAK inhibitors have all shown promise as a potential therapy for AGS [17]. Nevertheless, it is important to distinguish between AGS and LCC, so that patients receive adequate treatment when therapies are approved for each condition.
Coats Plus syndrome (CPS) is radiologically indistinguishable from LCC, presenting with the classic triad of leukoencephalopathy, cysts, and calcifications [18]. However similar on imaging, LCC and CPS can be distinguished as separate diseases both genetically and clinically. CPS is an autosomal recessive disorder of the conserved telomere maintenance component 1 (CTC1) gene, which encodes the CTC1 protein, a component of the CTC-1, STN-1, TEN-1 (CST) complex, which is responsible for maintaining the structural integrity of the telomere [18]. CTC1 mutations also decrease protein STN1’s (another component of CST) ability to bind DNA alpha polymerase during DNA replication [19]. Clinically, CPS displays the intracranial findings typical of LCC, with all the associated symptoms; in addition, extracranial symptoms are demonstrated, including osteopenia, gastrointestinal bleeding, portal hypertension, and, most notably, bilateral retinal telangiectasia with exudative retinopathy [18]. CPS is most often identified clinically via retinal telangiectasia.
There is currently no established paradigm for the management of LCC, and most patients are managed on a case-by-case basis. Surgery is often employed to remove mass effect, with concomitant medical management of antiepileptics and antipsychotics. Interestingly, bevacizumab, an anti-VEGF biologic, has yielded promising results in 2 cases of LCC. Bevacizumab is used in diabetic retinopathy and Coats Disease (Coats Plus without extraocular features) to reduce VEGF-induced vascular permeability and thus reduce retinal edema. The goal of bevacizumab in LCC is to reduce cyst volume by decreasing vascular leakage secondary to microangiopathy. Fay et al. administered bevacizumab biweekly for 1 year to an 18-year-old with LCC; both clinical and radiological improvements were noted, including restoration of lost range of motion, decreased bradykinesia and shrinkage in cyst volume [7]. Similarly, Martinez-Matilla attempted bevacizumab treatment in a 19-month-old with LCC. Radiological improvement was noted, but there were no clinical improvements [6]. Nevertheless, there is no cure for LCC, but symptoms can at least be managed by controlling mass effect.
LCC is an extremely rare neurological condition defined by cerebral microangiopathic changes inducing leukoencephalopathy, cyst formation, and calcifications [1]. There are currently no established diagnostic or treatment algorithms, although the disease displays a characteristic imaging pattern and genetic testing can confirm the SNORD118 gene mutation. Diagnosis can be achieved by excluding similar pathologies, and definitive diagnosis can be achieved through genetic analysis of SNORD118. Symptomatic treatments involve surgery to remove mass effect and medical management to control seizures and mental status changes. A disease modifying or curative treatment remains elusive and merits further research despite the rarity of the condition.
Patient consent
Informed consent was obtained for publication of this case report.
Footnotes
Competing Interests: The authors report no conflict of interest.
References
- 1.Labrune P, Lacroix C, Goutières F, de Laveaucoupet J, Chevalier P, Zerah M, et al. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: a new progressive disorder due to diffuse cerebral microangiopathy. Neurology. 1996;46(5):1297–1301. doi: 10.1212/wnl.46.5.1297. [DOI] [PubMed] [Google Scholar]
- 2.Jenkinson EM, Rodero MP, Kasher PR, Uggenti C, Oojageer A, Goosey LC, et al. Mutations in SNORD118 cause the cerebral microangiopathy leukoencephalopathy with calcifications and cysts. Nat Genet. 2016;48(10):1185–1192. doi: 10.1038/ng.3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sener U, Zorlu Y, Men S, Bayol U, Zanapalioglu U. Leukoencephalopathy, cerebral calcifications, and cysts. AJNR Am J Neuroradiol. 2006;27(1):200–203. [PMC free article] [PubMed] [Google Scholar]
- 4.Sim CY, Mukari SAM, Ngu LH, Loh CY, Remli R, Ibrahim NM. Labrune's syndrome presenting with stereotypy-like movements and psychosis: a case report and review. J Mov Disord. 2022;15(2):162–166. doi: 10.14802/jmd.21120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang M, Zhang M, Wu L, Dong Z, Yu S. Leukoencephalopathy with cerebral calcification and cysts: cases report and literature review. J Neurol Sci. 2016;370:173–179. doi: 10.1016/j.jns.2016.09.048. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Matilla M, Ferre-Fernandez JJ, Aparisi MJ, Marco-Hernandez AV, Ceron JA, Crow YJ, et al. Apparent radiological improvement in an infant with Labrune syndrome treated with bevacizumab. Pediatr Neurol. 2020;112:53–55. doi: 10.1016/j.pediatrneurol.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Fay AJ, King AA, Shimony JS, Crow YJ, Brunstrom-Hernandez JE. Treatment of leukoencephalopathy with calcifications and cysts with bevacizumab. Pediatr Neurol. 2017;71:56–59. doi: 10.1016/j.pediatrneurol.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3(3):397–414. doi: 10.1002/wrna.117. [DOI] [PubMed] [Google Scholar]
- 9.Badrock AP, Uggenti C, Wacheul L, Crilly S, Jenkinson EM, Rice GI, et al. Analysis of U8 snoRNA variants in zebrafish reveals how bi-allelic variants cause leukoencephalopathy with calcifications and cysts. Am J Hum Genet. 2020;106(5):694–706. doi: 10.1016/j.ajhg.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crow YJ, Marshall H, Rice GI, Seabra L, Jenkinson EM, Baranano K, et al. Leukoencephalopathy with calcifications and cysts: genetic and phenotypic spectrum. Am J Med Genet A. 2021;185(1):15–25. doi: 10.1002/ajmg.a.61907. [DOI] [PubMed] [Google Scholar]
- 11.Murphy S, Grima G, Mankad K, Aquilina K. Pediatric neurosurgical implications of a ribosomopathy: illustrative case and literature review. Child's Nervous System. 2022;38(3):643–648. doi: 10.1007/s00381-021-05208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osborn AG. First. Amirsys; 2013. Osborn's brain: imaging, pathology and anatomy. [Google Scholar]
- 13.Jaskolski DJ, Fortuniak J, Majos A, Gajewicz W, Papierz W, Liberski PP, et al. Magnetic resonance spectroscopy in intracranial tumours of glial origin. Neurol Neurochir Pol. 2013;47(5):438–449. doi: 10.5114/ninp.2013.32999. [DOI] [PubMed] [Google Scholar]
- 14.Crow YJ, Manel N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15(7):429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 15.Uggetti C, La Piana R, Orcesi S, Egitto MG, Crow YJ, Fazzi E. Aicardi-Goutières syndrome: neuroradiologic findings and follow-up. Am J Neuroradiol. 2009;30(10):1971–1976. doi: 10.3174/ajnr.A1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuang SY, Li Y, Yang SL, Han X. Child neurology: Aicardi-Goutieres syndrome presenting as recurrent ischemic stroke. Neurology. 2022;99(9):393–398. doi: 10.1212/WNL.0000000000200952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu D, Fang L, Huang T, Ying S. Case report: Aicardi-Goutières syndrome caused by novel TREX1 variants. Front Pediatr. 2021;9 doi: 10.3389/fped.2021.634281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44(3):338–342. doi: 10.1038/ng.1084. [DOI] [PubMed] [Google Scholar]
- 19.Gu P, Chang S. Functional characterization of human CTC1 mutations reveals novel mechanisms responsible for the pathogenesis of the telomere disease Coats plus. Aging Cell. 2013;12(6):1100–1109. doi: 10.1111/acel.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]