

Abstract
TLR9 is a critical nucleic acid sensing receptor in mediating periodontitis and periodontitis-associated comorbidities. Emerging evidence implicates TLR9 as a key sensor during aging, although its participation in periodontal aging is unexplored. Here, we investigated whether TLR9-mediated host responses can promote key hallmarks of aging, inflammaging, and senescence, in the course of periodontitis using a multipronged approach comprising clinical and preclinical studies. In a case-control model, we found increased TLR9 gene expression in gingival tissues of older (≥55 y) subjects with periodontitis compared to older healthy subjects as well as those who are younger (<55 y old) with and without the disease. Mechanistically, this finding was supported by an in vivo model in which wild-type (WT) and TLR9–/– mice were followed for 8 to 10 wk (young) and 18 to 22 mo (aged). In this longitudinal model, aged WT mice developed severe alveolar bone resorption when compared to their younger counterpart, whereas aged TLR9–/– animals presented insignificant bone loss when compared to the younger groups. In parallel, a boosted inflammaging milieu exhibiting higher expression of inflammatory/osteoclast mediators (Il-6, Rankl, Cxcl8) and danger signals (S100A8, S100A9) was noted in gingival tissues of aged WT mice compared to the those of aged TLR9–/– mice. Consistently, WT aged mice displayed an increase in prosenescence balance as measured by p16INK4a/p19ARF ratio compared to the younger groups and aged TLR9–/– animals. Ex vivo experiments with bone marrow–derived macrophages primed by TLR9 ligand (ODN 1668) further corroborated in vivo and clinical data and showed enhanced inflammatory-senescence circuit followed by increased osteoclast differentiation. Together, these findings reveal first systematic evidence implicating TLR9 as one of the drivers of periodontitis during aging and functioning by boosting a deleterious inflammaging/senescence environment. This finding calls for further investigations to determine whether targeting TLR9 will improve periodontal health in an aging population.
Keywords: periodontitis, toll-like receptor 9, nucleic acids, cellular senescence, macrophages, S100 proteins
Introduction
Aging is defined as a time-dependent functional decline phenomenon that results in the progressive loss of tissue integrity, which increases susceptibility to chronic diseases, including periodontitis (Ebersole et al. 2016). Given its cumulative nature (i.e., lifetime periodontal attachment loss), the prevalence of periodontitis rises as the population ages. Nonetheless, there is no definitive evidence that periodontitis pathophysiology is affected by age per se, although it is likely that the dysregulated host response due to aging may lead to increased periodontitis progression and/or susceptibility (Preshaw et al. 2017).
Cellular senescence is one the most widely studied hallmarks of aging, and it is characterized by mitotic cycle arrest where cells are no longer able to replicate (Di Micco et al. 2021). Two key features of a senescent cell (SC) are increased ratio between p16INK4a and p19ARF proteins encoded at the Cdkn2a locus, which favor cell cycle inhibition, and elevated activity of the β-galactosidase enzyme (Baker, Jin, and van Deursen 2008; Baker, Perez-Terzic, et al. 2008; Di Micco et al. 2021). Notably, SCs within aged tissues have a senescence-associated secretory phenotype, which includes increased basal levels of mediators like the receptor activator of nuclear factor κΒ ligand (Rankl), interleukin-6 (Il-6), and C-X-C-motif chemokine ligand 8 (Cxcl8) (Kumari and Jat 2021). Such shift in cell phenotype during aging results in an impaired tissue turnover followed by the development of a chronic inflammatory milieu termed inflammaging. Cooperatively, these changes can drive age-related pathologies (Franceschi et al. 2018; Di Micco et al. 2021). The factors and underlying mechanisms mediating these biological events in periodontal tissues are still poorly understood.
Continued activation of innate sensors (e.g., Toll-like receptors [TLRs]) by exogenous and endogenous danger signals (MAMPs/DAMPs) (e.g., unmethylated CpG-motifs from bacterial DNA [CpG-ODNs], nucleic acids released by dead cells, and storage of S100 proteins into SCs) can trigger an inflammaging state (Huang et al. 2015; Franceschi et al. 2018). These biological pathways driving aging can be operant in the oral cavity and likely contribute to periodontal diseases (Preshaw et al. 2017). As such, TLR9 is a DNA sensing receptor expressed in immune and resident cells of the periodontium which is implicated in the pathogenesis of periodontitis and periodontitis-related comorbidities. (Zou et al. 2002; Sahingur et al. 2013; Chen et al. 2014; Crump et al. 2016; Gonçalves-Anjo et al. 2019; Lyu et al. 2019; Nishimoto et al. 2020). Remarkably, the activation of TLR9 can also trigger the synthesis of DAMPs like S100 proteins (S100A8, S100A9), which are calcium-binding molecules implicated in aging and periodontal inflammation (Swindell et al. 2013; Hsu et al. 2014; Maekawa et al. 2019; Kim et al. 2020). Furthermore, TLR9-mediated responses can lead to the development of various comorbidities such as skeletal muscle fibrosis, opportunistic infections, and brain deposition of amyloid plaque in aged organisms (Frank et al. 2009; Lyu et al. 2019; Sato et al. 2020). Thus, collectively emerging evidence underscores the significance of TLR9 in the pathophysiology of aging-related disorders. Yet there is still limited information about the pathobiological crosstalk between TLR9 and MAMP/DAMP signaling within the aging oral mucosa.
Given the lifetime exposure of periodontal tissues to nucleic acids from different sources and ability of TLR9 to sense MAMPs/DAMPs involved in the aging process, we hypothesized that TLR9 exerts an essential function in the pathogenesis of periodontitis during aging. For this, we configured a study in 3 fronts: 1) clinical, by evaluating gingival expression of TLR9 in periodontitis/healthy and young/aged individuals; 2) in vivo, by tracing lifetime developed alveolar bone loss and inflammaging/senescence in gingival tissues from WT and TLR9–/– mice; and 3) ex vivo, by investigating the aging-related mechanisms triggered by TLR9 activation.
Materials and Methods
Clinical Study
Study population and sample collection
A case-control study following the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) guidelines (Appendix file) was approved by the Institutional Review Board of Virginia Commonwealth University (Protocol No. 11025) and University of Pennsylvania (Protocol No. 844212) and conducted in accordance with the Helsinki Declaration of 1975, as revised in 2000. The case-control design was used to compare individuals with (case) or without (control) periodontitis taking 30-y (lower) and 55-y (upper) age cutoffs: 30 to 55 y (young/middle) and >55 y (aged) (Billings et al. 2018). The criteria for periodontitis diagnosis comprised stage II and III in its generalized form and periodontally healthy individuals defined as described (Papapanou et al. 2018). In both groups, the presence of at least 20 natural teeth was required. Gingival tissue was harvested from the site with the deepest periodontal probing depth in periodontitis group. Healthy biopsy samples were obtained during crown lengthening surgery. Inclusion and exclusion criteria and further details for sample collection are in Sahingur et al. (2013).
Quantitative real-time polymerase chain reaction
Total RNA was isolated from gingival specimens using the RNeasy Plus Mini kit (Qiagen) following the manufacturer’s recommendations. Quality/concentration was determined at A260/A280 absorbance in a NanoDrop One Spectrophotometer (Thermo Scientific). Complementary DNA (cDNA) was synthesized using High-Capacity cDNA reverse-transcription kit (Applied Biosystems). Quantitative real-time polymerase chain reaction (qPCR) was determined using specific SYBR Green primers for TLR9 and Gapdh (sequences in the Appendix) and SYBR Green Master Mix (SABiosciences) in the StepOne Plus System (Applied Biosystems). Relative expression analysis was performed by the ΔΔCT method using Gapdh as endogenous control.
Animal Model
All animal procedures were approved by the Institutional Animal Care and Use Committee at University of Pennsylvania (Protocol No. 806809) and Virginia Commonwealth University (Protocol No. 10000566). This animal study followed the ARRIVE 2.0 guidelines (Appendix file). TLR9–/– mice (BALB/c) (Dr. Shizuo Akira, Osaka University) and wild-type (WT) counterparts (Jackson Laboratory) were bred in house. Mice were genotyped by tail snip using specific primers (sequences in the Appendix). We used a murine longitudinal aging model where mice were kept in a specific-pathogen-free environment until the time points for biological sample harvesting were reached: 8 to 10 wk (young) and 18 to 22 mo (aged).
Micro–computed tomography (µCT)
Periodontal bone loss around the maxillary molars was determined using micro–computed tomography (µCT) analysis as described (Crump et al. 2016). Details are in the Appendix.
qPCR
Total RNA was extracted from mouse gingival tissues around maxillary teeth and qPCR was performed according to the protocol described earlier. Messenger RNA (mRNA) levels for Rankl, Cxcl8, Il-6, S100A8, and S100A9 and for the encoded exons p16INK4a and p19ARF in the Cdkn2a locus were assessed (sequences in the Appendix).
Immunofluorescence
Immunofluorescence staining for p16INK4a and p19ARF was performed on gingival tissues following the previous protocol (Zaqout et al. 2020). Details are in the Appendix.
Ex Vivo Model
BMDM isolation and ex vivo stimulation
Bone marrow–derived macrophages (BMDMs) were differentiated from bone marrow (BM) cells harvested from femurs and tibias of 8- to 10-wk-old WT and TLR9−/− mice as described (Kim et al. 2015). BMDMs were challenged with different concentrations of TLR9 ligand ODN1668 (6 µg/mL and 60 µg/mL) (InVivogen) for 12 h, 24 h, and 72 h. Unstimulated cells were used as negative controls. Culture supernatants and cell lysates were collected for further analyses. Details are in the Appendix.
Enzyme-linked immunosorbent assay
Supernatant and lysates of BMDMs were assayed by enzyme-linked immunosorbent assay (ELISA) for the following markers: senescence-β-galactosidase enzyme (LifeSpan BioSciences), Il-6 (Thermo Fisher), and S100A8/A9 heterodimer (R&D Systems). All procedures were performed following the manufacturer’s recommendation.
Immunofluorescence
In this set of experiments, BMDMs isolated from WT and TLR9–/– described earlier were seeded in chamber polystyrene vessels (BD Falcon). After a 72-h stimulation with ODN as aforementioned, immunofluorescence staining for p16INK4a and p19ARF was performed. Details are in the Appendix.
BMDM-osteoclast differentiation and tartrate-resistant acid phosphate staining assay
To evaluate osteoclast differentiation, BMDMs were stimulated as outlined earlier and culture media were refreshed every 2-days for 6-days treatment. Cells treated with Rankl-mouse recombinant protein (20 ng/mL) (BioLegend) were used as positive controls. Tartrate-resistant acid phosphate (TRAP) staining was performed to quantify osteoclasts (TRAP+ cells) following the manufacturer’s recommendations (Sigma-Aldrich). Details are in the Appendix.
Statistical Analysis
Sample size calculation for the clinical study was based on 4 samples from each group randomly selected from our previous published study (Sahingur et al. 2013), taking a variation of at least 2-fold changes in TLR9 gene expression as the main variable. Considering a power of 80% and significance level of 5%, a minimum of 10 individuals per group would be required. Sample size for the animal study (5 animals per group) was based on published data, taking age-related alveolar bone loss as the main variable (Clark et al. 2021).
In the clinical study, the patient was maintained as the unit of measurement, and normality distribution was evaluated by Kolmogorov–Smirnov test with Lilliefors correction. Kruskal–Wallis with post hoc Dunn’s test and Welch’s analysis of variance (ANOVA) with post hoc Games–Howell test were used to evaluate intergroup differences. In the in vivo study, the animal was maintained as the unit of measurement, and 1-way ANOVA with post hoc Tukey’s test was applied. All tests were performed at the significance level of 0.05 using the GraphPad Prism software (GraphPad Software).
Results
Increased Gingival TLR9 Expression Was a Hallmark of Periodontitis in Aged Subjects
A total of 62 participants were included into 4 groups based on their age and diagnoses as follows: 17 young healthy, 10 aged healthy, 10 young periodontitis, and 25 aged periodontitis. All the subjects were nonsmokers and systemically healthy, and there was similar gender distribution among groups in order to avoid potential confounders (see Appendix). Gingival biopsies from older periodontitis subjects (≥55 y) exhibited a marked increase in TLR9 expression compared to those obtained from both young and aged healthy groups (mean fold-change of 17.84 vs. 2.09 and 1.28, respectively) (Fig. 1). This finding established nucleic acid sensing by TLR9 as a potential instigator of periodontal pathology during aging.
Figure 1.
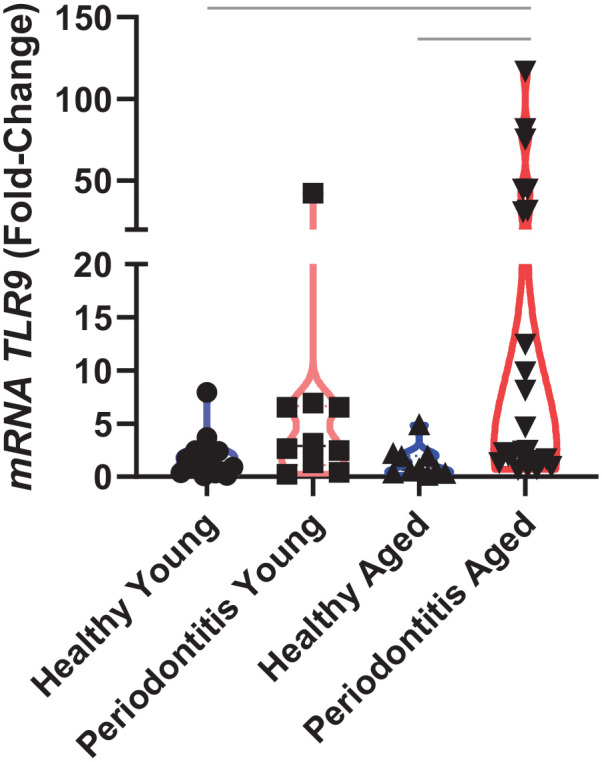
TLR9 gene expression in the healthy gingival tissues of younger (n = 17) and older (n = 10) individuals (control groups), and younger (n = 10) and older (n = 25) subjects with periodontitis (case groups). Data are presented in fold change, and each dot represents a biopsied site per individual. Gapdh was used as endogenous control. Bars indicate intergroup significant difference using Kruskal–Wallis with post hoc Dunn’s test and Welch’s analysis of variance with post hoc Games–Howell test at the significance level of 5%.
Lack of TLR9 Reduced Aging-Associated Physiological Alveolar Bone Resorption and Inflammaging Markers
Since TLR9 was highly expressed in gingival tissues from older periodontitis subjects, we then tested in a longitudinal animal study whether TLR9 could have an effect on the lifetime physiological development of periodontitis (Fig. 2A). No animal during this longitudinal study was lost due to health problems or signs of illness (e.g., diarrhea, spontaneous tumor, bleeding). As expected and consistent with our clinical data, aged WT mice presented significantly increased alveolarbone loss (−39.24 µm) compared to young WT counterparts(P < 0.01), whereas lack of TLR9 resulted in minimal and insignificant physiological bone loss (−8.04 µm) in aged TLR9–/– mice (P > 0.05 vs. young WT or young TLR9–/– groups) (Fig. 2B, C). Furthermore, gingival tissues from aged WT mice presented higher expression of Rankl and Cxcl8 (~1.83- and ~2.41-fold change vs. young WT, respectively). On the contrary, the transcripts for Rankl were reduced (by ~0.31-fold change vs. aged WT), and the expression of Cxcl8 and Il-6 was practically abolished in aged TLR9–/– animals when compared to all experimental groups (Fig. 2D). Similar to the changes observed in inflammaging markers, the transcripts for DAMPs signals (S100A8 and S100A9) were also upregulated in aged WT mice compared to its young counterparts. On the contrary, the expression of these S100 proteins was reduced in aged TLR9–/– animals when compared to all the other experimental groups (Fig. 2E, F).
Figure 2.

In vivo murine physiological aging model using wild-type (WT) (n = 5) and TLR9–/– (n = 5) mice. (A) Study model displaying the longitudinal timeline for sample collection 8 to 10 wk (young mice) to 18 to 22 mo (aged mice). (B) Representative images of alveolar bone levels used in micro–computed tomography analysis for each group. (C) Mean (in µm) of the distance from the cementoenamel junction (CEJ) to the alveolar bone crest (ABC) for each animal. Negative values represent bone resorption in relation to the mean bone level of the control group (young WT mice). (D) Heatmap for messenger RNA (mRNA) levels of inflammatory markers (Cxcl8, Il-6, and Rankl) in gingival tissues of young and aged mice. (E, F) mRNA levels of the DAMPs (S100A8 and S100A9) in the gingival tissue of each experimental group. All mRNA data are presented as fold change, and Gapdh was used as an endogenous control. In the heatmap, (*) indicates significant difference in the group in relation to all other experimental groups and (Ø) indicates significant difference in relation to aged WT mice. Bars indicate intergroup significant difference. One-way ANOVA with post hoc Tukey’s test was used at the significance level of 5%.
Lack of TLR9 Improves Senescence Phenotype by Instigating a Balanced p16INK4A:p19ARF Ratio in Aged Periodontal Tissues
Next, in order to evaluate whether TLR9 could alter the aging phenotype of periodontal tissues, we assessed the expression of p16INK4a and p19ARF proteins, which are widely studied markers of senescence (Baker, Jin, and van Deursen 2008; Baker, Perez-Terzic, et al. 2008). At the gene level, both aged WT and aged TLR9–/– mice (18–22 mo old) had higher expression of p16INK4a when compared to their young counterparts (8–10 wk old). However, there was significantly reduced expression of p16INK4a in aged TLR9–/– mice compared to the aged WT mice, which further supports the notion that lack of TLR9 can improve senescence phenotype (Fig. 3A). Likewise, higher levels of p19ARF transcripts were found in aged groups when compared to younger mice (Fig. 3B). At the protein level, there was reduced frequency of p16INK4a+ cells in gingival specimens from aged TLR9–/– compared to all other groups, further confirming that nucleic acid sensing can trigger senescence in gingival tissues (Fig. 3D, G). Furthermore, aged WT mice displayed decreased number of p19ARF+ cells compared to other groups (Fig. 3E, G). Previous evidence implicates increased p16INK4a:p19ARF ratio as one of the key features of a deleterious senescence (Baker, Jin, and van Deursen. 2008; Baker et al. 2016; Zhang et al. 2019). Consistently, aged WT mice had a higher p16INK4a:p19ARF ratio at both gene and protein levels when compared to all experimental groups, which was not observed in aged TLR9–/– mice (Fig. 3C, F).
Figure 3.

In vivo evaluation of the Cdkn2a locus through quantitative real-time polymerase chain reaction and immunofluorescence (IF) of the exons for p16INK4a and p19ARF in gingival tissues from young and aged wild-type (WT) and TLR9–/– mice. (A) Transcription levels of p16INK4a. (B) Transcription levels of p19ARF. (C) Balance of the prosenescence messenger RNA p16INK4a/p19ARF ratio. (D) Percentage of p16INK4a+ cells. (E) Percentage of p19ARF+ cells. (F) Ratio between p16INK4a+ and p19ARF+ cells. (G) Representative IF images taken at 40× magnification. Ct, connective tissue; Ge, gingival epithelium. Messenger RNA data are presented as fold change, and Gapdh was used as endogenous control. The number of marked cells for each antibody was calculated taking DAPI as endogenous control. Bars indicate intergroup significant difference using 1-way analysis of variance with post hoc Tukey’s test at the significance level of 5%.
TLR9-Mediated Responses Trigger Senescence and Inflammaging Followed by Increased Osteoclastic Differentiation
Last, we evaluated whether TLR9 activation triggers an inflammaging response followed by premature senescence and osteoclastic differentiation ex vivo. Priming of TLR9 with ODN1668 in BMDMs derived from WT mice increased synthesis of S100A8/A9, Il-6, and senescence-β-galactosidase enzyme (Fig. 4A–C). Immunofluorescence assays revealed increased p16INK4a expression over p19ARF (Fig. 4D, E). Moreover, this boosted prosenescence/inflammaging milieu under TLR9 priming was accompanied by increased osteoclast differentiation (Fig. 4F, G). Confirming the specificity, BMDMs obtained from TLR9–/– mice did not show any of the reported outcomes.
Figure 4.

Ex vivo evaluation of inflammaging and senescence markers in bone marrow–derived macrophages (BMDMs) isolated from wild-type (WT) and TLR9–/– that were stimulated with varying concentrations of TLR9 ligand (ODN 1668) (6 µg/mL and 60 µg/mL) or left unstimulated over different time points (12 h, 24 h, and 72 h). Enzyme-linked immunosorbent assay was used to measure (A) S100A8/A9 heterodimer (calprotectin) accumulation in BMDM cell lysate and (B, C) levels of β-galactosidase and interleukin-6 (Il-6) in BMDM cell supernatant, respectively. (D, E) Immunofluorescence (IF) for p16INK4a, p19ARF, and DAPI was performed at the 72-h time point. (D) Representative IF images taken at 40× magnification. (E) Balance of the p16INK4a/p19ARF expression in relation to DAPI. (F, G) Evaluation of osteoclast differentiation (purple/TRAP+ cells) over 6-d stimulation with ODN. Treatment with RANKL (20 ng/mL) was used as positive control for osteoclast differentiation. (F) Number of osteoclasts (TRAP+ cells) per field at 40× magnification. (G) Representative images for TRAP staining. (*) indicates significant difference in relation to BMDMs isolated from WT mice not stimulated with ODN (negative control). One-way analysis of variance with post hoc Tukey’s test was used at the significance level of 5%. TRAP, tartrate-resistant acid phosphate.
Discussion
TLR9 is implicated in the pathogenesis of periodontitis, and evidence suggests its critical role in aging (Sahingur et al. 2013; Huang et al. 2015; Kim et al. 2015; Crump and Sahingur 2016; Crump et al. 2016; Gonçalves-Anjo et al. 2019). Herein, using a systematic approach including clinical and preclinical studies, we uncovered several lines of novel evidence that collectively reveal TLR9 as one of the drivers of aging in periodontal tissues possibly through its tight control on DAMP signaling and senescence/inflammaging axis.
According to the “DAMP hypothesis,” inflammaging arises from the accumulation of danger signals like S100 proteins by SCs, which could then mediate aging-related disorders through TLR signaling (Huang et al. 2015). Importantly, the lifetime tissue exposure of endogenous and microbial nucleic acids like CpG-ODNs could favor this response via TLR9 activation (Huang et al. 2015). In this context, Rankl, Cxcl8, and Il-6 exist as part of a typical proinflammatory response that leads to periodontal tissue destruction, but they are also elevated during inflammaging (Franceschi et al. 2018; Di Micco et al. 2021; Kumari and Jat 2021). These mediators are produced as part of TLR9 signaling and affect osteoclastogenesis (Souza and Lerner 2019). In fact, TLR9-mediated responses can be context dependent and exert anti- or pro-osteoclastogenic effects based on the type of extracellular stimulus and stress (Zou et al. 2002). Here, we reported enhanced inflammaging milieu, senescence-β-galactosidase enzyme activity, and osteoclast differentiation upon TLR9 activation with ODN 1668. Our data indicate that this pathobiological circuit is operant in periodontal tissues and can explain the reduced physiological bone loss and gingival expression of inflammatory/osteoclast markers noted in aged mice lacking this receptor.
Furthermore, we also noted lack of S100A8 and S100A9 expression in the gingival tissues of aged TLR9–/– mice where, in contrast, aged WT mice exhibited overexpression of these proteins. This observation was also mechanistically confirmed by our ex vivo data showing higher levels of these S100 proteins (DAMPs) in macrophages upon TLR9 activation with CpG-ODNs (DAMPs/MAMPs), which was also consistent with previous reports in other cells (Hsu et al. 2014; Singh et al. 2016). S100A8 and S100A9 proteins have been previously implicated in periodontal disease pathogenesis where they can possibly act as danger signals (Maekawa et al. 2019; Kim et al. 2020). Similar to the periodontium, a robust shift in S100A8 and S100A9 expression has been noted in other aged tissues, suggesting their likely contribution to senescence phenomenon as DAMPs (Swindell et al. 2013). It has been shown that S100A8 could induce cellular senescence-like changes in bovine oviduct epithelial cells (Nakamura et al. 2019). Similarly, S100A8/A9 promoted aging-related cardiac fibrosis in myeloid-derived suppressor cells (Sun et al. 2021). At the organ level, in lungs, for example, aging per se was followed by CS and concomitant upregulation of S100A8 and increased severity of lung inflammation (Rashid et al. 2018). Likewise, higher S100A9 expression was associated with increased rates of neurodegeneration and cognitive deficits in an Alzheimer’s disease model (Kim et al. 2014). Hence, our results in periodontal tissues are consistent with the proposed role of these danger signals in other organs and reveal the first evidence of a possible crosstalk between TLR9 and DAMPs like S100 proteins in periodontal senescence. However, given that S100 proteins have either proinflammatory effect or antimicrobial activity (Wang et al. 2018), future studies targeting the TLR9/S100 protein axis also need to assess its consequences in terms of modulate host susceptibility to infectious diseases.
Another important feature of aging that is altered by TLR9 activation seems to be the balance between p16INK4a and p19ARF, which are 2 proteins encoded in the Cdkn2a locus with opposing functions (Baker, Jin, and van Deursen 2008; Baker, Perez-Terzic, et al. 2008; Baker et al. 2016). p16INK is a well-recognized prosenescence protein that induces an irreversible cell cycle arrest and, consequently, leads to an impaired tissue homeostasis due to the loss of cellular replicative capacity (Baker et al. 2016; Kumari and Jat 2021). On the contrary, p19ARF has antisenescence effects by inducing a reversible arrest that allows cells to replicate under minor stress-related damage and by guaranteeing long-term genomic stability and repair DNA damage (Baker, Perez-Terzic, et al. 2008; Bieging-Rolett et al. 2016). Such opposite functions in the Cdkn2a locus were recently corroborated by a study highlighting that p19ARF mediates repression of p16INK4a via long-range chromatin interactions (Zhang et al. 2019). Thus, analysis of the p16INK4a/p19ARF balance is used as a marker to assess age-related phenotypes resulting from the deleterious effect of a prosenescent environment (↑p16) or from the protective action of an antisenescence response (↑p19) (Baker, Perez-Terzic, et al. 2008; Baker et al. 2016). This is the first study that evaluated this balance in periodontal aging and revealed pronouncedly increased p16INK4a/p19ARF ratio in gingival tissues at both gene and protein levels in aged mice expressing TLR9. This finding was verified through ex vivo studies demonstrating an elevated p16INK4a/p19ARF balance upon TLR9 activation. In addition, despite increased number of transcripts for p19ARF, the number of p19ARF+ cells was practically abolished in the gingival epithelium of aged WT mice compared to the TLR9–/– counterpart. Thus, the increased p16INK4a/p19ARF ratio came at the expense of a lifetime reduction in p19ARF rather than an increase in the number of cells expressing p16INK4a. This finding indicates that an increase in p16INK4a/p19ARF ratio boosts the deleterious senescence effects of aging (Baker, Jin, and van Deursen 2008; Bieging-Rolett et al. 2016). Since p16INK4a expression during aging is organ dependent (Idda et al. 2020) and works in counterbalance with p19ARF (Baker, Perez-Terzic, et al. 2008; Bieging-Rolett et al. 2016), it is plausible that determining the expression of both markers and their interaction may be a better measure of the aging phenomenon.
The complexity of this balance also appears to have an interaction with TLR9, as the activation of this receptor is required for posttranslational stabilization of p16INK4a (Parroche et al. 2016). Interestingly, no significant change in p16INK4a expression was observed in young animals lacking TLR9, whereas aged mice lacking the receptor had increased transcription of p16INK4a but reduced number of p16INK4a-expressing cells, especially at the basal layer of the gingival epithelium, where a higher pattern of TLR9 expression was reported in a previous study (Chen et al. 2014). These findings are consistent with the notion that the absence of this prosenescence signaling may exert more profound effect in aging periodontal tissues possibly due to the lifetime accumulation of stressors (e.g., DAMPs) that engage TLR9. Thus, it is likely that in animals lacking TLR9, destabilization of p16INK4a may occur throughout life, eventually rescuing the expression of the p19ARF and favoring healthier periodontium. Our findings are also in line with the previous reports that reported that the clearance of p16INK4a+ cells improves life span and promotes healthy aging in several organs (Baker et al. 2016), which seems to naturally occur in the absence of TLR9 signaling. These data also draw further attention to the divergent responses between the expression of genes and proteins in aging tissues, which likely results from posttranslational modifications, epigenetic alterations, and changes in cellular metabolism that emerge with senescence (Santos and Lindner 2017; Di Micco et al. 2021).
Our knowledge about the host factors dictating the development and progression of periodontitis in aged tissues is still in its infancy. In this study, using clinical analyses and mechanistically using longitudinal in vivo model and ex vivo studies, we established that TLR9 induces periodontal aging-associated deleterious effects. This novel finding indicates that targeting TLR9 can be a promising strategy to improve clinical outcomes, especially in aging population. In the light of this evidence, future investigations are warranted to identify specific host cells within the periodontium exhibiting higher TLR9 expression with a concomitant shift in the senescence balance and characterize cellular and molecular pathways in aging tissues to develop targeted therapies. It will also be important to design population-based cohorts to prove a relation of causality between TLR9 and periodontitis during aging.
Conclusion
In summary, our data support the hypothesis that lifetime TLR9 activation due to the presence of MAMPs/DAMPs within the periodontium is one of the drivers of periodontitis possibly through its effect on boosting inflammaging/senescence (Fig. 5). Thus, targeting TLR9 has the potential to become an effective strategy to manage periodontal diseases, especially in an aging population.
Figure 5.
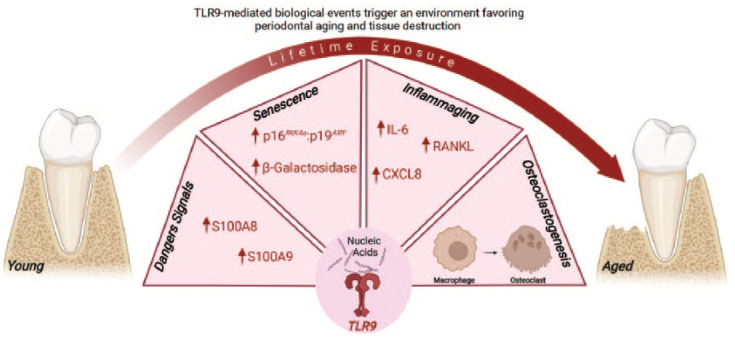
Model illustrating how TLR9-mediated responses can govern periodontitis pathophysiology during aging. The lifetime activation of TLR9 by nucleic acids triggers the synthesis of inflammatory mediators and DAMPs-like S100 proteins and fosters a deleterious senescence environment characterized by an elevated activity of the β-galactosidase enzyme and p16INK4a/p19ARF balance, as well as a boosted inflammaging milieu with higher expression of inflammatory/osteoclast mediators like Cxcl8, Il-6, and Rankl. As a result, macrophages undergo osteoclast differentiation, which leads to alveolar bone resorption and periodontitis progression during aging.
Author Contributions
E. Albuquerque-Souza, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; K.E. Crump, contributed to design, data acquisition, analysis, and interpretation, critically revised the manuscript; K. Rattanaprukskul, contributed to design, data acquisition and analysis, critically revised the manuscript; Y. Li, B. Shelling, contributed to data acquisition, critically revised the manuscript; X. Xia-Juan, M. Jiang, contributed to data acquisition and analysis, critically revised the manuscript; S.E. Sahingur, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-docx-1-jdr-10.1177_00220345221110108 for TLR9 Mediates Periodontal Aging by Fostering Senescence and Inflammaging by E. Albuquerque-Souza, K.E. Crump, K. Rattanaprukskul, Y. Li, B. Shelling, X. Xia-Juan, M. Jiang and S.E. Sahingur in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by US Public Health Service grants R01DE025037 and R01DE027374 to S.E. Sahingur from National Institute of Dental and Craniofacial Research/National Institute of Health.
Data Availability Statement: The data that support the findings of this study are available from the corresponding author upon request.
References
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. 2016. Naturally occurring p16INK4a-positive cells shorten healthy lifespan. Nature. 530(7589):184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jin F, van Deursen JM. 2008. The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle. 7(18):2795–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, Yamada S, Reyes S, Rowe L, Hiddinga HJ, et al. 2008. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 10(7):825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging-Rolett KT, Johnson TM, Brady CA, Beaudry VG, Pathak N, Han S, Attardi LD. 2016. p19ARF is required for the cellular response to chronic DNA damage. Oncogene. 35(33):4414–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings M, Holtfreter B, Papapanou PN, Mitnik GL, Kocher T, Dye BA. 2018. Age-dependent distribution of periodontitis in two countries: findings from NHANES 2009 to 2014 and SHIP-TREND 2008 to 2012. J Clin Periodontol. 45(Suppl 20):S130–S148. [DOI] [PubMed] [Google Scholar]
- Chen YC, Liu CM, Jeng JH, Ku CC. 2014. Association of pocket epithelial cell proliferation in periodontitis with TLR9 expression and inflammatory response. J Formos Med Assoc. 113(8):549–556. [DOI] [PubMed] [Google Scholar]
- Clark D, Halpern B, Miclau T, Nakamura M, Kapila Y, Marcucio R. 2021. The contribution of macrophages in old mice to periodontal disease. J Dent Res. 100(12):1397–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump KE, Oakley JC, Xia-Juan X, Madu TC, Devaki S, Mooney EC, Sahingur SE. 2016. Interplay of toll-like receptor 9, myeloid cells, and deubiquitinase A20 in periodontal inflammation. Infect Immun. 85(1):e00814-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump KE, Sahingur SE. 2016. Microbial nucleic acid sensing in oral and systemic diseases. J Dent Res. 95(1):17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Krizhanovsky V, Baker D, d’Adda di Fagagna F. 2021. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 22(2):75–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole JL, Graves CL, Gonzalez OA, Dawson D, 3rd, Morford LA, Huja PE, Hartsfield JK, Jr, Huja SS, Pandruvada S, Wallet SM. 2016. Aging, inflammation, immunity and periodontal disease. Periodontol 2000. 72(1):54–75. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. 2018. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 14(10):576–590. [DOI] [PubMed] [Google Scholar]
- Frank S, Copanaki E, Burbach GJ, Müller UC, Deller T. 2009. Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett. 453(1):41–44. [DOI] [PubMed] [Google Scholar]
- Gonçalves-Anjo N, Leite-Pinheiro F, Ribeiro R, Requicha JF, Lourenço AL, Dias I, Viegas C, Bastos E. 2019. Toll-like receptor 9 gene in periodontal disease—a promising biomarker. Gene. 687:207–211. [DOI] [PubMed] [Google Scholar]
- Hsu K, Chung YM, Endoh Y, Geczy CL. 2014. TLR9 ligands induce S100A8 in macrophages via a STAT3-dependent pathway which requires IL-10 and PGE2. PLoS One. 9(8):e103629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Xie Y, Sun X, Zeh HJ, III, Kang R, Lotze MT, Tang D. 2015. Damps, ageing, and cancer: the ‘damp hypothesis’. Ageing Res Rev. 24(Pt A):3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idda ML, McClusky WG, Lodde V, Munk R, Abdelmohsen K, Rossi M, Gorospe M. 2020. Survey of senescent cell markers with age in human tissues. Aging (Albany NY). 12(5):4052–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Kim S, Jeon S, Kim SJ, Cho HJ, Choi YN. 2020. Diagnostic and prognostic ability of salivary MMP-9 and S100A8 for periodontitis. J Clin Periodontol. 47(10):1191–1200. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Chang KA, Ha TY, Kim J, Ha S, Shin KY, Moon C, Nacken W, Kim HS, Suh YH. 2014. S100A9 knockout decreases the memory impairment and neuropathology in crossbreed mice of Tg2576 and S100A9 knockout mice model. PLoS One. 9(2):e88924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PD, Xia-Juan X, Crump KE, Abe T, Hajishengallis G, Sahingur SE. 2015. Toll-like receptor 9-mediated inflammation triggers alveolar bone loss in experimental murine periodontitis. Infect Immun. 83(7):2992–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R, Jat P. 2021. Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype. Front Cell Dev Biol. 9:645593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu AK, Zhu SY, Chen JL, Zhao YX, Pu D, Luo C, Lyu Q, Fan Z, Sun Y, Wu J, et al. 2019. Inhibition of TLR9 attenuates skeletal muscle fibrosis in aged sarcopenic mice via the p53/SIRT1 pathway. Exp Gerontol. 122:25–33. [DOI] [PubMed] [Google Scholar]
- Maekawa S, Onizuka S, Katagiri S, Hatasa M, Ohsugi Y, Sasaki N, Watanabe K, Ohtsu A, Komazaki R, Ogura K, et al. 2019. RNA sequencing for ligature induced periodontitis in mice revealed important role of S100A8 and S100A9 for periodontal destruction. Sci Rep. 9(1):14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Iwata H, Kuwayama T, Shirasuna K. 2019. S100A8, which increases with age, induces cellular senescence-like changes in bovine oviduct epithelial cells. Am J Reprod Immunol. 82(3):e13163. [DOI] [PubMed] [Google Scholar]
- Nishimoto S, Fukuda D, Sata M. 2020. Emerging roles of toll-like receptor 9 in cardiometabolic disorders. Inflamm Regen. 40:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapanou PN, Sanz M, Buduneli N, Dietrich T, Feres M, Fine DH, Flemmig TF, Garcia R, Giannobile WV, Graziani F, et al. 2018. Periodontitis: consensus report of Workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-implant Diseases and Conditions.J Periodontol. 89 Suppl 1:S173–S182. [DOI] [PubMed] [Google Scholar]
- Parroche P, Roblot G, Le Calvez-Kelm F, Tout I, Marotel M, Malfroy M, Durand G, McKay J, Ainouze M, Carreira C, et al. 2016. TLR9 re-expression in cancer cells extends the S-phase and stabilizes p16INK4a protein expression. Oncogenesis. 5(7):e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preshaw PM, Henne K, Taylor JJ, Valentine RA, Conrads G. 2017. Age-related changes in immune function (immune senescence) in caries and periodontal diseases: a systematic review. J Clin Periodontol. 44(Suppl 18):S153–S177. [DOI] [PubMed] [Google Scholar]
- Rashid K, Sundar IK, Gerloff J, Li D, Rahman I. 2018. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci Rep. 8(1):9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahingur SE, Xia XJ, Voth SC, Yeudall WA, Gunsolley JC. 2013. Increased nucleic acid receptor expression in chronic periodontitis. J Periodontol. 84(10):e48–e57. [DOI] [PubMed] [Google Scholar]
- Santos AL, Lindner AB. 2017. Protein posttranslational modifications: roles in aging and age-related disease. Oxid Med Cell Longev. 2017:5716409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Tansho-Nagakawa S, Ubagai T, Ono Y. 2020. Analysis of immune responses in Acinetobacter baumannii-infected klotho knockout mice: a mouse model of Acinetobacter baumannii infection in aged hosts. Front Immunol. 11:601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Agrawal NK, Gupta SK, Sinha P, Singh K. 2016. Increased expression of TLR9 associated with pro-inflammatory S100A8 and IL-8 in diabetic wounds could lead to unresolved inflammation in type 2 diabetes mellitus (T2DM) cases with impaired wound healing. J Diabetes Complications. 30(1):99–108. [DOI] [PubMed] [Google Scholar]
- Souza PPC, Lerner UH. 2019. Finding a toll on the route: the fate of osteoclast progenitors after toll-like receptor activation. Front Immunol. 10:1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SN, Ni SH, Li Y, Liu X, Deng JP, Chen ZX, Li H, Feng WJ, Huang YS, Li DN, et al. 2021. G-MDSCs promote aging-related cardiac fibrosis by activating myofibroblasts and preventing senescence. Cell Death Dis. 12(6):594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swindell WR, Johnston A, Xing X, Little A, Robichaud P, Voorhees JJ, Fisher G, Gudjonsson JE. 2013. Robust shifts in S100a9 expression with aging: a novel mechanism for chronic inflammation. Sci Rep. 3:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. 2018. S100A8/A9 in inflammation. Front Immunol. 9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaqout S, Becker LL, Kaindl AM. 2020. Immunofluorescence staining of paraffin sections step by step. Front Neuroanat. 14:582218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hyle J, Wright S, Shao Y, Zhao X, Zhang H, Li C. 2019. A cis-element within the ARF locus mediates repression of p16INK4a expression via long-range chromatin interactions. Proc Natl Acad Sci U S A. 116(52):26644–26652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Schwartz H, Endres S, Hartmann G, Bar-Shavit Z. 2002. CpG oligonucleotides: novel regulators of osteoclast differentiation. FASEB J. 16(3):274–282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-jdr-10.1177_00220345221110108 for TLR9 Mediates Periodontal Aging by Fostering Senescence and Inflammaging by E. Albuquerque-Souza, K.E. Crump, K. Rattanaprukskul, Y. Li, B. Shelling, X. Xia-Juan, M. Jiang and S.E. Sahingur in Journal of Dental Research