

Abstract
Nat/Ivy is a diverse and ubiquitous CoA‐binding evolutionary lineage that catalyzes acyltransferase reactions, primarily converting thioesters into amides. At the heart of the Nat/Ivy fold is a phosphate‐binding loop that bears a striking resemblance to that of P‐loop NTPases—both are extended, glycine‐rich loops situated between a β‐strand and an α‐helix. Nat/Ivy, therefore, represents an intriguing intersection between thioester chemistry, a putative primitive energy currency, and an ancient mode of phospho‐ligand binding. Current evidence suggests that Nat/Ivy emerged independently of other cofactor‐utilizing enzymes, and that the observed structural similarity—particularly of the cofactor binding site—is the product of shared constraints instead of shared ancestry. The reliance of Nat/Ivy on a β‐α‐β motif for CoA‐binding highlights the extent to which this simple structural motif may have been a fundamental evolutionary “nucleus” around which modern cofactor‐binding domains condensed, as has been suggested for HUP domains, Rossmanns, and P‐loop NTPases. Finally, by dissecting the patterns of conserved interactions between Nat/Ivy families and CoA, the coevolution of the enzyme and the cofactor was analyzed. As with the Rossmann, it appears that the pyrophosphate moiety at the center of the cofactor predates the enzyme, suggesting that Nat/Ivy emerged sometime after the metabolite dephospho‐CoA.
Keywords: acetyltransferase, coenzyme A, GNAT, nucleotide cofactor, phosphate binding loop, P‐loop, Rossmann
1. INTRODUCTION
Nat/Ivy is an enzyme evolutionary lineage that primarily catalyzes acyl transfer reactions from a coenzyme A (CoA) thioester or a charged tRNA. Although somewhat limited in enzymatic breadth, Nat/Ivy is diverse both in sequence 1 and in the biological processes that it mediates, including eukaryotic pre‐ribosome maturation, 2 quorum sensing, 3 autoimmunity, 4 , 5 antibiotic resistance, 6 d‐amino acid detoxification, 7 and carbon metabolism (i.e., KEGG reaction R00233). At the heart of the Nat/Ivy fold is a phosphate‐binding loop that bears a striking resemblance to the phosphate binding loops of P‐loop NTPases and Rossmann enzymes—a fact noted over 20 years ago in the very first reports describing Nat/Ivy structures. 8 , 9 Indeed, phosphate binding loops are among the most ancient structures in biology 10 , 11 and it is believed that they may have been precursors to modern cofactor‐binding domains. 12 , 13 , 14 Continuity between the most ancient globular domains and short functional peptides was first proposed by Margaret Dayhoff, 15 and this realization inspired a generation of scientists, including Dan Tawfik. 16 Moreover, the observation that protein structures can be decomposed into closed loops 17 associated with elementary functions 11 , 18 supports this view. Given the importance of phosphate binding loops, the development of sensitive approaches to detect distant evolutionary relationships at the level of short fragments, 14 , 19 , 20 , 21 , 22 and interest in the role of thioesters as a primordial energy currency, 23 , 24 , 25 , 26 , 27 Nat/Ivy enzymes are a timely case study of protein evolution. Our intention is to revisit the Nat/Ivy enzyme lineage while taking inspiration from the work of the late Dan Tawfik.
2. NAT/IVY LIKELY EMERGED AS A COA‐BINDING PROTEIN
For the present analysis, evolutionary lineages are defined according to the Evolutionary Classification of Domains (ECOD) database 28 , 29 , 30 (version Develop283). The ECOD hierarchy consists of four levels, in which X‐groups (the broadest classification) correspond to evolutionary lineages and F‐groups (the narrowest level of classification) correspond to protein families. ECOD annotates 2,362 domains spread across 55 families (F‐groups) as belonging to the Nat/Ivy evolutionary lineage (X‐group 213) in the Protein Data Bank (PDB). From this set of domains, representative domains were selected, with preference going to liganded domains and high‐resolution structures (see Supplementary File 1, Representatives for the complete list with annotations). ECOD domain identifiers are provided throughout the text and are denoted by a leading “e” (e.g., e5kf9A2). Also note that while it is common for authors to refer to the phosphate‐binding loop in the Nat/Ivy fold as a P‐loop, 31 we will reserve the name P‐loop to refer to the evolutionary lineage of P‐loop NTPases (i.e., X‐group 2004).
Of the 2,433 protein evolutionary lineages in the ECOD database with an assigned F‐group, 61 bind either CoA or acetyl‐CoA (PDB ligand codes COA and ACO, respectively; interaction cutoff = 4 Å). To determine which of these X‐groups are most likely to have emerged as CoA binders, the fraction of F‐groups with at least one CoA binding event was calculated and plotted against the total number of F‐groups associated with that X‐group (Figure 1a). F‐group count reports the extent to which an X‐group has diverged, with a large number of F‐groups suggesting that a family is either evolutionarily diverse, ancient, or both. With 55 F‐groups, Nat/Ivy is in the top 2.1% of the most diverged evolutionary lineages in ECOD (median family count = 1; average family count = 6.25).
FIGURE 1.
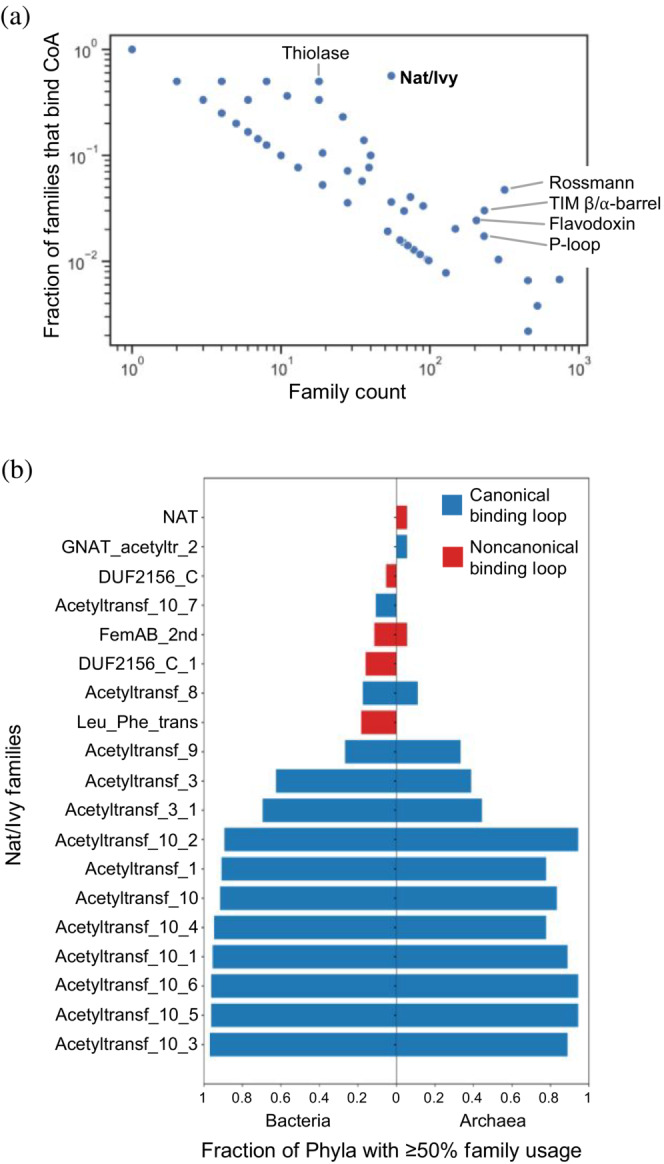
Nat/Ivy is a fundamental CoA‐binding domain. (a) Fraction of families (i.e., ECOD F‐groups) with a CoA or acetyl‐CoA binding event as a function of total family count. Nat/Ivy is the most diverged protein for which >50% of families bind CoA. (b) Distribution of Nat/Ivy domains in bacteria and archaea. Nat/Ivy proteins bearing a canonical CoA‐binding loop are essentially ubiquitous among microbes
From a parsimony perspective, the higher the fraction of families that bind CoA, the more likely CoA binding was a feature of the founding member of that family. Of the 55 Nat/Ivy families, 31 (56%) have a domain that binds to either CoA or acetyl‐CoA, making Nat/Ivy associated with the highest family fraction of CoA binding of any known evolutionary lineage, except for 3 X‐groups that are associated with just a single F‐group (and thus 100% of the families bind CoA; X‐groups 1119, 7531, and 7571). Furthermore, binding of CoA by Nat/Ivy is not generally associated with multiple participating domains: In 88.5% of cases, Nat/Ivy‐bound CoA molecules are associated with just a single domain. The next most specific evolutionary lineage, Thiolase (X‐group 7581), has only 18 F‐groups, 50% of which bind CoA or acetyl‐CoA.
Unlike other common cofactor‐binding proteins 32 , 33 —such as Rossmanns, P‐loops, or TIM barrels—Nat/Ivy is not strongly associated with binding to other common nucleotide‐containing cofactors. Compared to the 348 and 332 binding events in the PDB for CoA and acetyl‐CoA, respectively, AMP, UDP, SAM, and SAH are associated with no more than two binding events each (Supplemental File 1, Other Cofactors). In these cases, the non‐CoA cofactor is either a moiety of the natural substrate or the substrate itself. AMP (bound in e3r96A1), for example, is a fragment of the natural ligand, the antibiotic microcin C7, which inhibits aspartyl‐tRNA synthetase and can be inactivated by acetylation. 4 There were no observed binding events to any of the common redox cofactors FMN, FAD, NAD, or NADP. Thus, while Nat/Ivy has significantly diversified with respect to family counts—likely a consequence of the diverse substrates that Nat/Ivy operates on—the cofactor utilization of this family is apparently specialized and enduring.
3. NAT/IVY IS UBIQUITOUS ACROSS THE MICROBIAL TREE OF LIFE
Although phyletic distribution is an imperfect measure of the antiquity of a protein evolutionary lineage, ubiquity does suggest antiquity. To assess the phyletic distribution of Nat/Ivy domains across the microbial tree of life, HMM profiles from the ECOD database were searched against the Genome Taxonomy Database (GTDB) phylogenetic trees 34 for bacteria and archaea (version 95, randomly subsampled at a single proteome per genus) using HMMER 35 (version 3.3.2) with an i‐Evalue cutoff of 1 × 10−10, an HMM profile coverage cutoff of 75%, and a search space set to 106,052,079 targets using the ‐Z option (Figure 1b). We find that the majority of bacterial and archaeal phyla contain a Nat/Ivy domain in at least 50% of their respective proteomes, consistent with the view that this is an ancient evolutionary lineage. Furthermore, when families are classified by the presence of a canonical CoA‐binding loop (described in detail below), those families bearing the canonical loop are significantly more distributed across the microbial tree of life. This result echoes the family analysis in Figure 1a and supports the view that Nat/Ivy emerged as a CoA‐binding protein.
4. THE CHEMISTRY OF NAT/IVY: ACYL TRANSFER
To get a fingerprint of the chemical reactivity associated with Nat/Ivy, enzymes with a Nat/Ivy domain in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database 36 were identified. Briefly, the genes within each KEGG Orthology were clustered using an 80% identity cutoff by CD‐HIT. 37 Nat/Ivy families were then mapped onto each of the representative genes using HMMER, as described above. Those KEGG Orthologies for which ≥75% of the gene representatives contained a Nat/Ivy domain were collected (61 in total; see Supplemental File 1, Orthologies) and linked to enzyme commission (EC) numbers via the KEGG REST API. In total, Nat/Ivy domains were associated with 34 unique EC numbers (Supplemental File 1, Reactions).
A breakdown of the activities associated with the Nat/Ivy family confirms the dominance of acyltransferase activity, with 30 of the 34 enzymes classified as such (EC class 2.3) (Figure 2a). The acylating agents of these reactions fall into two groups: thioesters of CoA (EC 2.3.1) and charged tRNAs (EC 2.3.2), in which the attachment is via an ester not a thioester. The former group employs canonical CoA‐binding loops whereas the latter group does not. In the majority of cases, the substrate nucleophile is a primary amine, resulting in an amide product. In the case of lysine/arginine leucyltransferase (EC 2.3.2.6) and arginyltransferase (EC 2.3.2.8), the product is a bona fide peptide bond—though these reactions use charged tRNAs and not CoA. Ester formation, from attack by a hydroxyl group, also occurs. Less common reactions include oxidation coupled to phosphorylation (EC 1.2.1.38) and two phosphotransferase reactions (EC 2.7). The Nat/Ivy domain is apparently not required for either EC 1.2.1.38 or EC 2.7 activity, as alternative KEGG Orthologies with similar overall domain structure but lacking the Nat/Ivy domain can also perform these reactions. Finally, a conserved Nat/Ivy domain was identified in malonyl‐CoA decarboxylase (K01578). Despite limited chemistry, the Nat/Ivy domain is impressive in its ability to catalyze the acylation of a wide range of substrates, ranging in size from small molecules (such as polyamines) to proteins.
FIGURE 2.
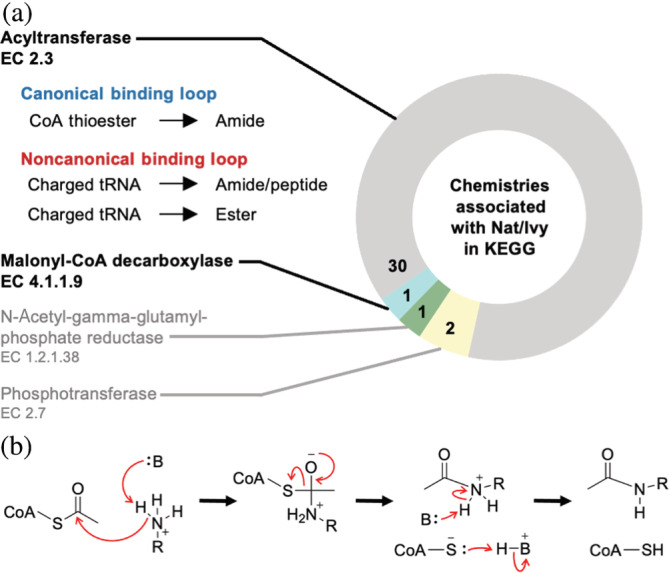
(a) Chemistry associated with Nat/Ivy domains in KEGG. Nat/Ivy primarily catalyzes acyl transfer chemistry. (b) The Nat/Ivy reaction mechanism. Although there is some debate, the curated M‐CSA database favors only mechanisms without an acyl enzyme intermediate
Cross‐referencing all PDB structures containing a Nat/Ivy domain with the Mechanism and Catalytic Site Atlas (M‐CSA) database 38 returns just six entries (Supplemental File 1, Mechanisms) associated with acyl transferase chemistry. In each case, the preferred catalytic mechanism involves direct attack of the CoA thioester by a substrate nucleophile, without an acyl‐enzyme intermediate (Figure 2b for an example of this mechanism involving a primary amine, the most common substrate nucleophile). Although it was previously reported that the Nat/Ivy enzyme ESA1 can employ a ping‐pong mechanism 39 —in which a protein Cys residue attacks the CoA thioester first to form a thioester acyl‐enzyme intermediate that is then resolved by nucleophilic attack of the substrate—subsequent mutagenesis studies have called this mechanism into question, in favor of direct nucleophilic attack by the substrate. 40
Christian de Duve proposed that thioesters were an essential component of primordial metabolism, 23 arguing that it was thioester derivatives of amino acids that gave rise to the first short peptides (which he dubbed “multimers”). In such a world, acyl transfer would no doubt be an important function as it could, for example, promote the formation of peptides. Indeed, Nat/Ivy enzymes can catalyze the formation of amide bonds, including peptide bonds. However, in the case of peptide bond formation, the acylating agent is a charged tRNA and the families associated with this activity (e.g., Leu_Phe_trans) lack a CoA‐binding loop and are not as highly distributed across the microbial tree of life. Nevertheless, the connection between acyl transfer activity and early peptide bond formation remains tantalizing. We also note that Nat/Ivy is a second family, like AARSs, that catalyzes peptide bond formation by charged tRNAs and performs thioester chemistry. 24 It has also been suggested that transthioesterases would be useful in a primitive thioester‐based metabolism, 26 but the KEGG analysis did not turn up any Nat/Ivy enzymes that perform this reaction, despite several such reactions being present in the KEGG database (namely, the CoA transferases of EC 2.8.3).
5. THE STRUCTURE OF NAT/IVY: AN Α + Β PROTEIN WITH AN Α/Β CORE
Nat/Ivy adopts a 3‐layer α + β sandwich architecture (Figure 3) comprised of a mixture of β‐β, α‐α, and β‐α motifs. In the middle of the fold is a conserved β‐α‐β‐α motif situated between two conserved β‐β hairpins, and so the Nat/Ivy structure may be described as an α + β protein with an α/β core. The α/β core is responsible for CoA binding: The loop between β4‐α3 forms an extended phosphate‐binding loop that wraps around the pyrophosphate moiety (what we refer to as the canonical phosphate‐binding loop of this fold); binding of the pantetheine moiety is achieved by a crevice between β4 and β5. The phosphate‐binding loop of Nat/Ivy is strikingly similar to that of the P‐loop (X‐group 2004). As such, it is worth noting that while Nat/Ivy and P‐loop proteins are grouped in the same Class and Architecture by the CATH protein structure database 41 —specifically, the 3‐layer α‐β‐α Sandwich (3.40)—this classification is somewhat misleading: P‐loop proteins are comprised exclusively of β‐α units, whereas Nat/Ivy proteins are not a repetitive fold outside the β/α core. That said, in all three evolutionary lineages—Rossmann, P‐loop, and Nat/Ivy—the phosphate binding loop is located within the very first β‐α motif of the fold. Assuming that the cofactor‐binding site is the most ancient part of Nat/Ivy, the evolution of the fold involved both N‐terminal and C‐terminal extensions, as opposed to the Rossmans, which are thought to have grown primarily by C‐terminal extensions. 14
FIGURE 3.
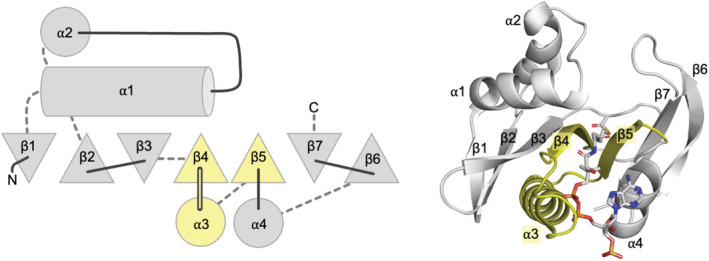
The conserved core of Nat/Ivy domains. β‐strands are represented as triangles; triangles pointing up denote β‐stands that are coming out of the plane of the paper (toward the reader) whereas triangles pointing down denote β‐strands that are going into the plane of the paper (away from the reader). Solid lines indicate loops at the top of the fold and dashed lines indicate loops at the bottom of the fold. Structural elements shaded yellow comprise the majority of the CoA binding site. Domain e2jddA1 pictured. All structure figures were generated in PyMOL (www.pymol.org)
There are several notable variations on the Nat/Ivy architecture. Foremost, some enzymes form a domain‐swapped arrangement in which β7 from one domain is replaced with β6 of an adjacent domain, resulting in two domains facing opposite directions (e.g., see e5kf9A1 and e5kf9A2). The lengths of both α1 and α2 are also variable, and rarely, one of the α‐helices will be absent. Finally, several Nat/Ivy proteins have N‐ and C‐terminal extensions beyond the conserved structural core depicted in Figure 3.
6. THE PHOSPHATE BINDING LOOP OF NAT/IVY
The features of the canonical CoA‐binding loop are now described in turn, and compared to those of common phosphate binding loops in other folds (Figure 4).
FIGURE 4.
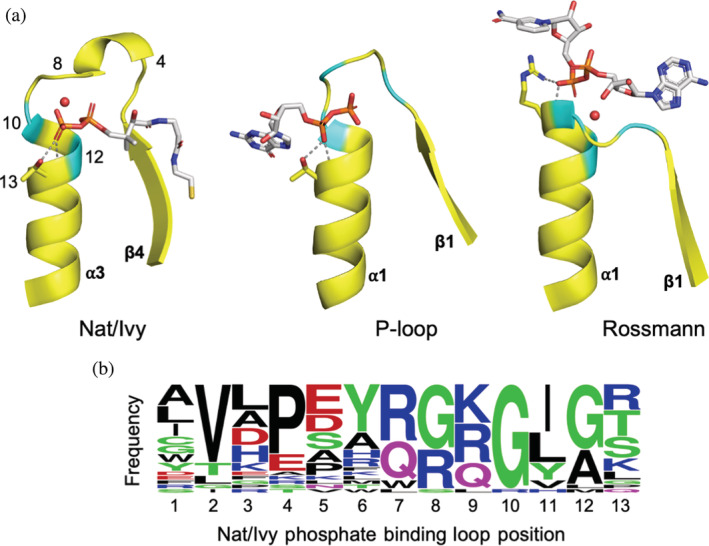
The canonical phosphate‐binding loop of Nat/Ivy strongly resembles those of other ancient enzymes. (a) A comparison between the phosphate binding loops of Nat/Ivy (e5kf9A2), P‐loop (e1yrbA1), and Rossmann (e1lssA1) enzymes. Conserved glycine residues are colored cyan. Conserved water molecules are rendered as red spheres. Residues forming bidentate interactions at the N‐terminus of an α‐helix are shown as sticks, with hydrogen bonds shown as dashed grey lines. For Nat/Ivy, numbers correspond to the positions in panel b. The 3′‐phosphoadenosine moiety of CoA is omitted for clarity. (b) Frequency plot derived from the consensus sequence of each Nat/Ivy family with a canonical phosphate binding loop (figure generated using WebLogo 63 )
6.1. N‐terminus of an α‐helix
Perhaps the most apparent feature of the CoA binding mode is that the pyrophosphate moiety is seated atop the N‐terminus of an α‐helix. Indeed, the preference for binding phosphate at the N‐terminus of an α‐helix has been recognized at least since the late 1970s. 42 We have argued that the N‐helix binding mode is a consequence of ancient constraints on protein function; specifically, a dearth of basic amino acids and the requirement for short, contiguous binding sites. 32 Binding at the N‐terminus of an α‐helix alone, therefore, should not be taken as a strong signature of homology; and we note that this feature is observed in TIM barrels, HUPs, flavodoxins, Rossmanns and P‐loops, 32 among others.
6.2. A glycine‐rich motif
Glycine‐rich loops are a common feature of nucleotide binding sites, 43 with P‐loop and Rossmann enzymes containing a GxxGxG or GxGxxG motif, respectively. Using sequence alignments taken from ConSurf‐DB 44 for each of the representative structures, the consensus phosphate binding loop sequence for each family was collected (Supplemental File 1, Representatives; Figure 4b). The resulting sequence profile closely resembles that reported in the Nucleotide Binding DataBase. 43 In most Nat/Ivy families, conserved glycine residues facilitate binding to the pyrophosphate moiety, specifically at positions 8, 10, and 12 of the phosphate binding loop (Figure 4)—forming a GxGxG motif. These glycine residues do not just reside in the extended portion of the loop, but also at the N‐terminus of the α‐helix, as is common in other phosphate binding loops.
6.3. Bidentate interactions
Previously, we have noted the importance of bidentate interactions, 32 in which both the backbone amide and the sidechain form hydrogen bonds with the same ligand molecule. Bidentate interactions are useful because they allow multiple, reinforcing interactions to be encoded by just a single residue. Inspection of the representative structures with a cofactor bound reveals that 14/26 structures make a bidentate interaction with either Ser, Thr, Arg, or Lys at the tip of α3 (position 13 of the phosphate binding loop; Figure 4). The consensus sequence for each family (Supplemental File 1, Representatives [Canonical]; Figure 4b) likewise indicates a preference for these residues at that site.
6.4. Conserved bridging water
Bottoms et al. 45 noted a conserved water in the Rossmann dinucleotide binding site. Of the 26 representative structures with a canonical binding loop bound to CoA, all but one (e2zpaB3) have a bridging water molecule that interacts with both the pyrophosphate moiety of CoA and the protein. Note, however, that some crystallographers have modeled the electron density at this site as a Mg2+ ion—for example, in e5ls7B1 and e4c7hA2—raising the possibility that this site can promiscuity bind Mg2+ as well. Dication binding at or near a phosphate‐binding loop is common (e.g., in P‐loop NTPases or in the Rossmann family of GTPases tubulin) and can signify a functional transition. The existence of nearby hydrogen bond donors, however, would seem to favor assignment as a water, as in the majority of structures. The CheckMyMetal server 46 supports this assessment, with the B‐factor of the putative metals relative to the surrounding protein atoms ranked as “borderline” and the associated ligands and binding geometry ranked as an “Outlier” for Mg2+. Taken together, Nat/Ivy is another enzyme, like the Rossmann, in which a water molecule has been recruited to facilitate interactions between protein and cofactor.
6.5. Is Nat/Ivy a distant relative of P‐loop NTPases and/or Rossmans?
Previously, the Tawfik Lab argued that Rossmanns and P‐loop NTPases may have a shared evolutionary origin based on various “bridge” structures with intermediate properties, 14 , 19 raising the question: Given the similarity of their phosphate‐binding loops (Figure 4), could Nat/Ivy be distantly related to Rossmanns and/or P‐loop NTPases? At present, there is little hard evidence in support of this hypothesis. Foremost, analyses of “bridging themes”—short sequence fragments shared across protein evolutionary lineages—do not return any evidence of sequence sharing involving Nat/Ivy and any other protein lineage, 19 , 22 suggesting that this family is something of an island in sequence space. Additionally, if Nat/Ivy emerged from a multi‐functional phosphate‐binding peptide, one might expect more variability in cofactor preference or the existence of bridge proteins like the Rossmann GTPase tubulin, which has properties intermediate between Rossmanns and P‐loops. 14 However, no such protein—for example, a Nat/Ivy NTPase—could be identified. Finally, we note that several features of the Nat/Ivy phosphate‐binding loop, such as an N‐helix binding site 32 or the glycine rich motifs, are known to be convergent features common to several ancient but unrelated proteins. Glycine rich motifs, for example, are also present in the helix‐hairpin‐helix motif (X‐group 102) 47 and in Ribonucleases (X‐group 2484). 48 Nevertheless, the deep evolutionary history of Nat/Ivy is yet to be fully understood and the search for bridging proteins should continue.
7. PANTETHEINE BINDING
The pantetheine moiety of CoA is largely situated between β4 and the middle or tip of β5 (Figure 3). An overlay of the representative structures with canonical‐binding loops (Figure 5b) reveals that the conformation of the 4′‐diphosphopantetheine moiety is consistent across families. In order to interact with both the N‐terminus of α3 and the edge of β4, the CoA moiety must adopt a bent conformation with an angle of about 107 ± 14° (calculated from atoms P1A, CP9, and S1P for all Nat/Ivy‐bound CoA molecules in the PDB with a resolution higher than 3.0 Å). This binding angle is a hallmark of the Nat/Ivy family, and distinguishes it from several other CoA‐binding protein families (Figure 5b). The phosphate binding loop also contributes to the binding of the pantetheine moiety: Position 7 acts as a sort of latch residue, resting atop the ligand (Figure 5c). Finally, the pantetheine moiety is anchored by two hydrogen bonds to the edge of β4, which can be expanded to three hydrogen bonds in the case of acetyl‐CoA (Figure 5d).
FIGURE 5.
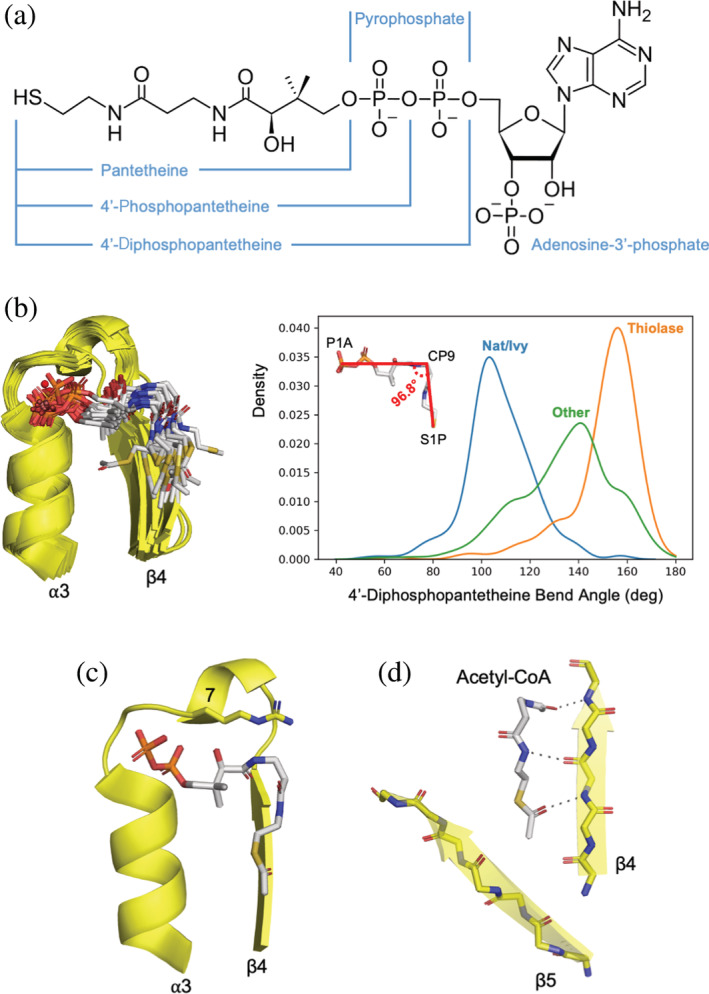
Binding of pantetheine by Nat/Ivy. (a) The structure of CoA. (b) Overlay of the representative structures with a canonical binding loop (see Supplemental File 1 for full list). The cofactor adopts a bent conformation. (c) A “latch residue” at position 7 of the phosphate‐binding loop lays across the top of the pantetheine moiety of the bound CoA (e4jxrB2). (d) Binding in a crevice formed by β4 and β5. Note how the cofactor extends the β‐sheet by binding the edge of β4 (e4jxrB2)
It has been suggested previously that the β1‐phosphate binding loop‐α1 of Rossmann and P‐loop proteins were part of the ancient peptide vocabulary. 10 In our analysis of Rossmanns, 49 P‐loops, 14 and HUP domains, 50 we instead favored a longer β‐α‐β motif as the evolutionary nucleus of these ancient folds. Three secondary structure elements, we note, is the approximate size of a cooperative, quasi‐independently foldable protein unit (a so‐called “foldon”) 51 , 52 or a mini‐protein. 53 And, although the exact nature of the crevice between β4 and β5 differs between families (specifically with respect to β5), the splayed β‐strands are highly conserved and present even in families that have lost the canonical phosphate binding loop. We therefore conclude that Nat/Ivy represents yet another ancient enzyme family that arose from a β‐α‐β peptide binding to a nucleotide‐containing cofactor (Figure 6).
FIGURE 6.
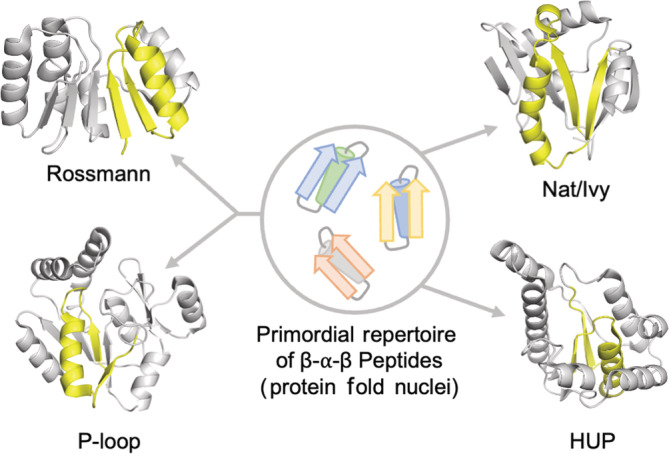
β‐α‐β peptides as nuclei for the emergence cofactor binding domains. At the heart of several ancient enzymes—including Rossmanns, P‐loops, and HUPs (the evolutionary lineage that encompasses Class I aminoacyl‐tRNA synthetases)—is a β‐α‐β motif that mediates ligand binding
8. NUCLEOSIDE BINDING AND A PEEK AT THE HISTORY OF NAT/IVY‐COA COEVOLUTION
An overlay of the representative domains reveals that the nucleoside and 3′‐phosphate moieties adopt a fairly wide range of conformations (Figure 7a). Furthermore, these parts of CoA make interactions predominantly with amino acid sidechains (Figure 7b and c) and, unlike the pyrophosphate and pantetheine moieties (Figures 4, 5, and 7), lack conserved backbone interactions. We have previously argued that sidechain binding interactions are likely more recent than backbone interactions, 32 especially in the context of ancient enzymes. And, for many structures, the 3′‐phosphate moiety is effectively pointing out into solvent. One notable binding interaction, however, is presented in Figure 7b, in which an Arg residue at position 8 of the phosphate binding loop stacks on top of the adenine ring while making hydrogen bonds with the 3′ phosphate. Arg at this site is preferred in about 50% of families, with Gly preferred otherwise (Figure 4b). To better quantify the distribution of backbone and sidechain binding, relatively short hydrogen bonds (estimated as a distance ≤3.2 Å) were identified for each family (Figure 7c). This analysis highlights the backbone interactions presented above as being conserved throughout the Nat/Ivy evolutionary lineage, and confirms the observation that binding of the nucleoside and 3′‐phosphate is mediated almost exclusively by sidechain interactions.
FIGURE 7.
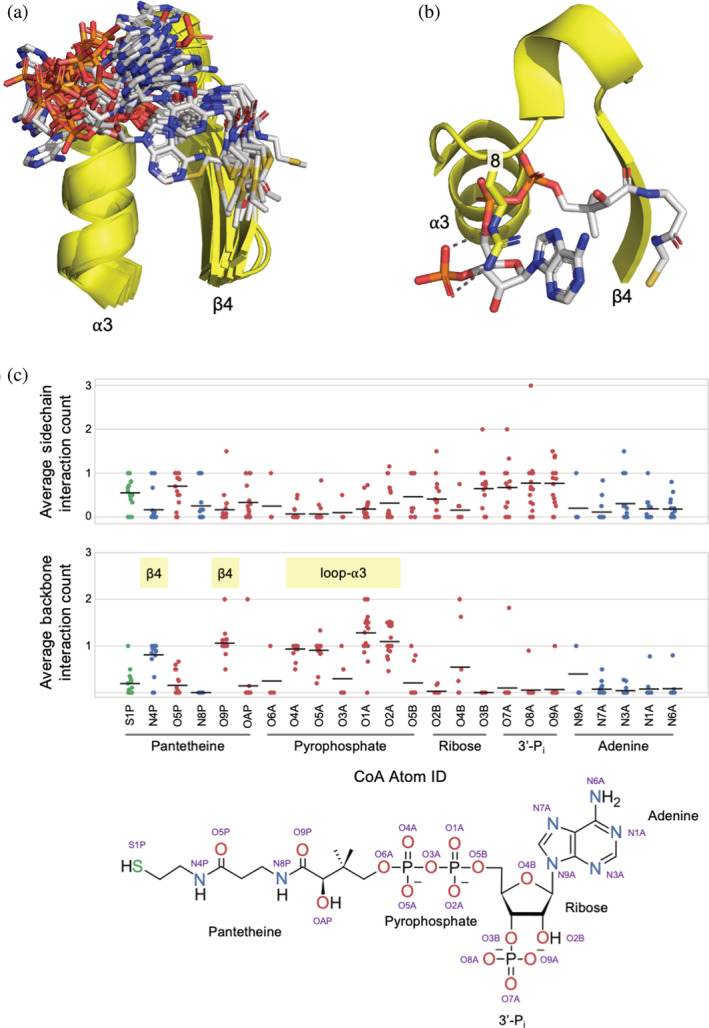
Binding to adenosine and the 3′‐phosphate. (a) An overlay of the representative domains reveals that the adenine moiety and the 3′‐phosphate adopt a range of conformations (c.f. Figure 6a, showing the comparatively tight conformational ensemble of the 4′‐diphosphopantetheine moiety). (b) An Arg residue at position 8 stacking on top of adenine and forming hydrogen bonds with the 3′‐phosphate (e3dddA2). (c) Family‐normalized interaction analysis between CoA and Nat/Ivy where each point corresponds to an F‐group in the Nat/Ivy evolutionary lineage. Interactions between O, N, and S atoms (3.2 Å distance cutoff) between CoA and protein for each family with a canonical phosphate‐binding loop were identified and classified as either a sidechain or backbone interaction. Each point corresponds to a family average and the bar indicates the average of all family averages. Whereas binding to the pyrophosphate and pantetheine moieties of CoA are both associated with conserved backbone interactions (indicated with yellow bars), binding to the nucleoside and the 3′‐phosphate are not. There is no case where a sidechain interaction is completely conserved
Given the comparatively loose binding to the adenosine moiety, and a lack of backbone interactions, is it possible that the Nat/Ivy fold predates CoA? After all, only the sulfhydryl at the end of CoA is required for chemical reactivity; the rest of the CoA molecule is (apparently) a handle for protein binding. Perhaps the evolutionary history of CoA is preserved within its biosynthetic pathway, 54 which involves two key intermediates prior to the formation of the contemporary cofactor 55 (Figure 8). These steps are the attachment of an AMP moiety to 4′‐phosphopantetheine to form dephospho‐CoA and, in the final step, the attachment of a 3′‐phosphate to form CoA. The role of the 3′‐phosphate is presumably to discriminate between binding of CoA, NADP (ribose phosphorylated on the 2′ hydroxyl) and NAD (ribose not phosphorylated on either the 2′ or the 3′ hydroxyl). Nat/Ivy domains, however, interact extensively with the CoA pyrophosphate moiety (Figures 4 and 7c), making 4′‐diphosphopantetheine the primary handle for binding. Indeed, there are structures in the PDB that involve binding to a fragment of CoA that lacks the adenosine moiety (e6pauA2) or the 3′ discriminating phosphate (e7ak9A1/B1). However, to the best of current knowledge, free 4′‐diphosphopantetheine is not only absent from the biosynthetic pathway of CoA but from essentially all of contemporary biology. A SMILES string 56 search for 4'‐diphosphopantetheine reveals that this metabolite is not present in KEGG. Thus, absent compelling evidence for the utilization of 4′‐diphosphopantetheine in ancient metabolism, it would seem that the Nat/Ivy phosphate binding loop emerged to bind either 4′‐phosphopanthetheine, dephospho‐CoA, or CoA—with only the latter two containing a full pyrophosphate moiety.
FIGURE 8.
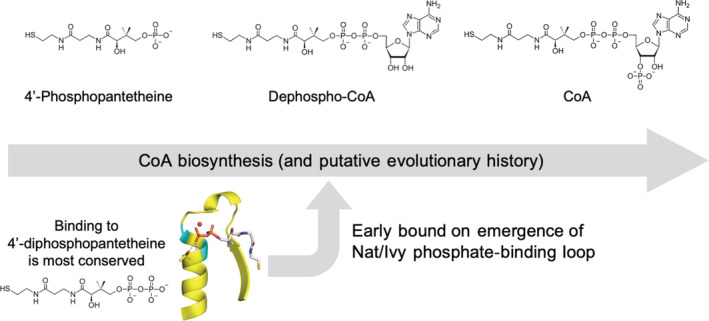
Coevolution of the Nat/Ivy phosphate‐binding loop and the CoA cofactor. The most conserved binding interactions—and those interactions that are mediated by the protein backbone—are centered on the 4′‐diphosphopantetheine moiety of CoA. However, the biosynthesis of CoA, which may report on its evolutionary history, does not involve a stage with a free 4′‐diphosphopantetheine metabolite. Instead, 4′‐diphosphopantetheine is, to the best of current knowledge, absent from contemporary metabolism. Assuming there was no stage during which 4′‐diphosphopantetheine was the major form of CoA, the emergence of the phosphate‐binding loop of Nat/Ivy seems to have occurred after the emergence of dephospho‐CoA (see main text for a more detailed discussion). This is in keeping with the Rossmann fold, which binds the pyrophosphate moiety of the dinucleotide cofactor NAD/P and therefore seems to have emerged after the appearance of the full dinucleotide cofactor
Given that fragments of pantetheine without a pyrophosphate group have been used as the basis of Nat/Ivy inhibitors, 57 we deem it very likely that Nat/Ivy can bind to free 4′‐phosphopantetheine (although no such structure could be identified in the PDB, nor could any such chemistry be found in KEGG). The Nat/Ivy family HlyC can accept a holo‐ACP that includes a covalently attached 4′‐phosphopantetheine moiety, though does so without a canonical‐binding loop. 58 Moreover, covalently‐attached 4′‐phosphopantetheine is primarily derived from CoA by the action of a holo‐[acyl‐carrier‐protein] synthase, 59 suggesting that CoA (a small molecule metabolite) predates holo‐ACP (a translated protein with a covalently attached cofactor).
Perhaps the binding of orthophosphate can provide insight into the preferences of the Nat/Ivy phosphate‐binding loop. There are six structures in which the canonical Nat/Ivy phosphate‐binding loop is bound only to orthophosphate. In one case, two orthophosphate molecules ghost the pyrophosphate binding site, with the P atoms positioned ~1 Å further apart from each other than in a molecule of pyrophosphate (PDB code: 1y7r). The observation of two orthophosphates in close proximity suggests a relatively strong preference for binding pyrophosphate, given that the orthophosphate groups will be negatively charged and thus mutually repulsive. In five other structures, where only one orthophosphate molecule is bound, the position of the phosphate ranges from being intermediate the two canonical binding sites (PDB codes: 2dxq, 6cx8, and 7kwh), to being approximately overlayed with the phosphate moiety on the pantetheine side (PDB code: 4mbu), to being shifted even further off the edge of the α‐helix and closer to β4 than in the canonical binding mode (PDB code: 5i0c). The presence of several degenerate binding modes for orthophosphate is consistent with a pyrophosphate binding element.
Taken together, we suggest that the emergence of Nat/Ivy phosphate binding loop took place after the emergence of the pyrophosphate moiety of CoA. This assessment is based on (a) the pattern of conserved interactions; (b) the general absence of structures or chemistry involving a free 4′‐phosphopanthetheine metabolite; and (c) the degenerate binding modes of orthophosphate to the Nat/Ivy phosphate binding loop, plus the observation of a ternary structure with two orthophosphate molecules simultaneously being bound. Once again, Nat/Ivy resembles the pyrophosphate‐binding Rossmann fold, which, in its present form, also seems to have emerged concurrent with or sometime after the establishment of the full dinucleotide cofactor.
9. CONCLUSIONS
The Tawfik laboratory was a pioneer in early protein evolution, showing the functional potential of simple peptides experimentally 12 , 13 , 60 , 61 , 62 and dissecting their histories through structural bioinformatics and state‐of‐the‐art sequence analysis. 14 , 19 , 32 , 49 Indeed, many of the features discussed within these works are relevant to Nat/Ivy as well, where a nucleotide cofactor is bound at the N‐terminus of an α‐helix, forms bidentate interactions, and interacts with a glycine‐rich motif. Prof. Tawfik was particularly interested in the potential of β‐α‐β fragments to serve as the evolutionary nuclei around which larger, structured domains could emerge—a feature we proposed for the P‐loop and the Rossmann 14 , 49 as well as the HUP evolutionary lineage. 50 Although Nat/Ivy is technically an α + β protein, the existence of an interior β‐α‐β motif that bears the phosphate binding loop and pantetheine binding cleft is in keeping with Tawfik's hypothesis. However, unlike Rossmanns and P‐loops, current evidence favors the assignment of Nat/Ivy as an independent evolutionary lineage, and its similarity to other phosphate binding loops seems to be the result of convergence not shared ancestry. By dissecting the binding site of Nat/Ivy, we now suggest a relative history between the evolution of CoA and the Nat/Ivy phosphate binding loop, a proposal that echoes the evolutionary history of Rossmann enzymes. Experimental characterization of simple Nat/Ivy‐derived peptides may offer an interesting look at the emergence of this acyl transferase enzyme lineage and may shed new light on the question of its independent origins and cofactor preferences. We close by noting that Prof. Tawfik himself was well aware of the Nat/Ivy family and its striking similarity to P‐loop proteins; during the preparation of Ref. [32], he instructed author L.M.L to “save it for a rainy day.”
AUTHOR CONTRIBUTIONS
Liam M Longo: Conceptualization (lead); data curation (lead); investigation (lead); visualization (lead); supervision (lead); writing – original draft (lead); writing – review and editing (lead). Hayate Hirai: Data curation (supporting); investigation (supporting); visualization (supporting); writing – review and editing (supporting). Shawn Erin McGlynn: Conceptualization (supporting); funding aquisition; supervision (supporting); project administration; resources; writing ‐ review and editing (supporting).
Supporting information
File S1:
ACKNOWLEDGMENTS
The authors are grateful to Dr. Dragana Despotović and Prof. Rachel Kolodny for helpful discussions during the preparation of this manuscript. Shawn E. McGlynn acknowledges support by NSF (Award No. 1724300) “Collaborative Research: Biochemical, Genetic, Metabolic, and Isotopic Constraints on an Ancient Thiobiosphere” and JSPS KAKENHI (Grant No. 22H01343).
Longo LM, Hirai H, McGlynn SE. An evolutionary history of the CoA‐binding protein Nat/Ivy. Protein Science. 2022;31(12):e4463. 10.1002/pro.4463
Review Editor: John Kuriyan
Funding information Japan Society for the Promotion of Science, Grant/Award Number: 22H01343; National Science Foundation, Grant/Award Number: 1724300
Contributor Information
Liam M. Longo, Email: llongo@elsi.jp.
Shawn Erin McGlynn, Email: mcglynn@elsi.jp.
REFERENCES
- 1. Krtenic B, Drazic A, Arnesen T, Reuter N. Classification and phylogeny for the annotation of novel eukaryotic GNAT acetyltransferases. PLoS Comput Biol. 2020;16:e1007988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kornprobst M, Turk M, Kellner N, et al. Architecture of the 90S pre‐ribosome: A structural view on the birth of the eukaryotic ribosome. Cell. 2016;166:380–393. [DOI] [PubMed] [Google Scholar]
- 3. Dong SH, Frane ND, Christensen QH, Greenberg EP, Nagarajan R, Nair SK. Molecular basis for the substrate specificity of quorum signal synthases. Proc Natl Acad Sci U S A. 2017;114:9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agarwal V, Metlitskaya A, Severinov K, Nair SK. Structural basis for microcin C7 inactivation by the MccE acetyltransferase. J Biol Chem. 2011;286:21295–21303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oda K, Matoba Y, Noda M, Kumagai T, Sugiyama M. Catalytic mechanism of bleomycin N‐acetyltransferase proposed on the basis of its crystal structure. J Biol Chem. 2010;285:1446–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kim KH, An DR, Song J, et al. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP‐7. Proc Natl Acad Sci U S A. 2012;109:7729–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yow GY, Uo T, Yoshimura T, Esaki N. Physiological role of D‐amino acid‐N‐acetyltransferase of Saccharomyces cerevisiae: Detoxification of D‐amino acids. Arch Microbiol. 2006;185:39–46. [DOI] [PubMed] [Google Scholar]
- 8. Weston SA, Camble R, Colls J, et al. Crystal structure of the anti‐fungal target N‐myristoyl transferase. Nat Struct Biol. 1998;5:213–221. [DOI] [PubMed] [Google Scholar]
- 9. Dutnall RN, Tafrov ST, Sternglanz R, Ramakrishnan V. Structure of the histone acetyltransferase Hat1: A paradigm for the GCN5‐related N‐acetyltransferase superfamily. Cell. 1998;94:427–438. [DOI] [PubMed] [Google Scholar]
- 10. Alva V, Söding J, Lupas AN. A vocabulary of ancient peptides at the origin of folded proteins. Elife. 2015;4:e09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goncearenco A, Berezovsky IN. Prototypes of elementary functional loops unravel evolutionary connections between protein functions. Bioinformatics. 2010;26:i497–i503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Romero Romero ML, Yang F, Lin Y‐R, et al. Simple yet functional phosphate‐loop proteins. Proc Natl Acad Sci. 2018;115:E11943–E11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vyas P, Trofimyuk O, Longo LM, Deshmukh FK, Sharon M, Tawfik DS. Helicase‐like functions in phosphate loop containing beta‐alpha polypeptides. Proc Natl Acad Sci U S A. 2021;118:e2016131118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Longo LM, Jabłońska J, Vyas P, et al. On the emergence of P‐loop NTPase and Rossmann enzymes from a Beta‐alpha‐Beta ancestral fragment. Elife. 2020;9:e64415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eck RV, Dayhoff MO. Evolution of the structure of ferredoxin based on living relics of primitive amino acid sequences. Science. 1966;1979(152):363–366. [DOI] [PubMed] [Google Scholar]
- 16. Romero Romero ML, Rabin A, Tawfik DS. Functional proteins from short peptides: Dayhoff's hypothesis turns 50. Angew Chem Int Ed. 2016;55:15966–15971. [DOI] [PubMed] [Google Scholar]
- 17. Berezovsky IN, Grosberg AY, Trifonov EN. Closed loops of nearly standard size: Common basic element of protein structure. FEBS Lett. 2000;466:283–286. [DOI] [PubMed] [Google Scholar]
- 18. Goncearenco A, Berezovsky IN. Protein function from its emergence to diversity in contemporary proteins. Phys Biol. 2015;12:045002. [DOI] [PubMed] [Google Scholar]
- 19. Kolodny R, Nepomnyachiy S, Tawfik DS, Ben‐Tal N. Bridging themes: Short protein segments found in different architectures. Mol Biol Evol. 2021;38:2191–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kolodny R. Searching protein space for ancient sub‐domain segments. Curr Opin Struct Biol. 2021;68:105–112. [DOI] [PubMed] [Google Scholar]
- 21. Longo LM, Kolodny R, McGlynn SE. Evidence for the emergence of β‐trefoils by “peptide budding” from an IgG‐like β‐sandwich. PLoS Comput Biol. 2022;18:e1009833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qiu K, Ben‐Tal N, Kolodny R. Similar protein segments shared between domains of different evolutionary lineages. Protein Sci. 2022;31:e4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brack A, editor. Duve C de clues from present‐day biology: The thioester world. The molecular origins of life: Assembling pieces of the puzzle. Cambridge: Cambridge University Press, 1998; p. 219–236. [Google Scholar]
- 24. Jakubowski H. Aminoacyl‐tRNA synthetases and the evolution of coded peptide synthesis: The thioester world. FEBS Lett. 2016;590:469–481. [DOI] [PubMed] [Google Scholar]
- 25. Goldford JE, Hartman H, Smith TF, Segrè D. Remnants of an ancient metabolism without phosphate. Cell. 2017;168:1126–1134. [DOI] [PubMed] [Google Scholar]
- 26. Goldford JE, Hartman H, Marsland R, Segrè D. Environmental boundary conditions for the origin of life converge to an organo‐sulfur metabolism. Nat Ecol Evol. 2019;3:1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanden SA, Yi R, Hara M, McGlynn SE. Simultaneous synthesis of thioesters and iron‐sulfur clusters in water: Two universal components of energy metabolism. Chem Commun. 2020;56:11989–11992. [DOI] [PubMed] [Google Scholar]
- 28. Cheng H, Schaeffer RD, Liao Y, et al. ECOD: An evolutionary classification of protein domains. PLoS Comput Biol. 2014;10:e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng H, Liao Y, Schaeffer RD, Grishin NV. Manual classification strategies in the ECOD database. Proteins. 2015;83:1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dustin Schaeffer R, Liao Y, Cheng H, Grishin NV. ECOD: New developments in the evolutionary classification of domains. Nucleic Acids Res. 2017;45:D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ud‐Din AIMS, Tikhomirova A, Roujeinikova A. Structure and functional diversity of GCN5‐related n‐acetyltransferases (GNAT). Int J Mol Sci. 2016;17:1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Longo LM, Petrović D, Kamerlin SCL, Tawfik DS. Short and simple sequences favored the emergence of N‐helix phospho‐ligand binding sites in the first enzymes. Proc Natl Acad Sci U S A. 2020;117:5310–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Medvedev KE, Kinch LN, Schaeffer RD, Grishin NV. Functional analysis of Rossmann‐like domains reveals convergent evolution of topology and reaction pathways. PLoS Comput Biol. 2019;15:e1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Parks DH, Chuvochina M, Rinke C, Mussig AJ, Chaumeil PA, Hugenholtz P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome‐based taxonomy. Nucleic Acids Res. 2022;50:D785–D794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46:W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fu L, Niu B, Zhu Z, Wu S, Li W. CD‐HIT: Accelerated for clustering the next‐generation sequencing data. Bioinformatics. 2012;28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ribeiro AJM, Holliday GL, Furnham N, Tyzack JD, Ferris K, Thornton JM. Mechanism and catalytic site atlas (M‐CSA): A database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 2018;46:D618–D623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yan Y, Harper S, Speicher DW, Marmorstein R. The catalytic mechanism of the ESA1 histone acetyltransferase involves a self‐acetylated intermediate. Nat Struct Biol. 2002;9:862–869. [DOI] [PubMed] [Google Scholar]
- 40. Berndsen CE, Albaugh BN, Tan S, Denu JM. Catalytic mechanism of a MYST family histone acetyltransferase. Biochemistry. 2007;46:623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sillitoe I, Dawson N, Lewis TE, et al. CATH: Expanding the horizons of structure‐based functional annotations for genome sequences. Nucleic Acids Res. 2019;47:D280–D284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hol WGJ, van Duijnen PT, Berendsen HJC. The α‐helix dipole and the properties of proteins. Nature. 1978;273:443–446. [DOI] [PubMed] [Google Scholar]
- 43. Zheng Z, Goncearenco A, Berezovsky IN. Nucleotide binding database NBDB ‐ a collection of sequence motifs with specific protein‐ligand interactions. Nucleic Acids Res. 2016;44:D301–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. ben Chorin A, Masrati G, Kessel A, et al. ConSurf‐DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins. Protein Sci. 2020;29:258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bottoms CA, Smith PE, Tanner JJ. A structurally conserved water molecule in Rossmann dinucleotide‐binding domains. Protein Sci. 2002;11:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W. CheckMyMetal: A macromolecular metal‐binding validation tool. Acta Crystallogr D Struct Biol. 2017;73:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Doherty AJ, Serpell LC, Ponting CP. The helix‐hairpin‐helix DNA‐binding motif: A structural basis for non‐sequence‐specific recognition of DNA. Nucleic Acids Res. 1996;24:2488–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bork P, Sander C, Valencia A. An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, Actin, and hsp70 heat shock proteins. Proc Natl Acad Sci U S A. 1992;89:7290–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Laurino P, Tóth‐Petróczy Á, Meana‐Pañeda R, Lin W, Truhlar DG, Tawfik DS. An ancient fingerprint indicates the common ancestry of Rossmann‐fold enzymes utilizing different ribose‐based cofactors. PLoS Biol. 2016;14:e1002396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gruic‐Sovulj I, Longo LM, Jabłońska J, Tawfik DS. The evolutionary history of the HUP domain. Crit Rev Biochem Mol Biol. 2022;57:1–15. [DOI] [PubMed] [Google Scholar]
- 51. Panchenko AR, Luthey‐Schulten Z, Wolynes PG. Foldons, protein structural modules, and exons. Proc Natl Acad Sci U S A. 1996;93:2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haglund E, Lindberg MO, Oliveberg M. Changes of protein folding pathways by circular permutation. J Biol Chem. 2008;283:27904–27915. [DOI] [PubMed] [Google Scholar]
- 53. Baker EG, Bartlett GJ, Porter Goff KL, Woolfson DN. Miniprotein design: Past, present, and prospects. Acc Chem Res. 2017;50:2085–2092. [DOI] [PubMed] [Google Scholar]
- 54. Noda‐Garcia L, Liebermeister W, Tawfik DS. Metabolite–enzyme coevolution: From single enzymes to metabolic pathways and networks. Annu Rev Biochem. 2018;87:187–216. [DOI] [PubMed] [Google Scholar]
- 55. Strauss E. Coenzyme a biosynthesis and enzymology. In: Mander L, Liu H‐W, editors. Comprehensive natural products II: Chemistry and biology. Oxford: Elsevier, 2010; p. 351–410. [Google Scholar]
- 56. Weininger D. SMILES, a chemical language and information system: 1: Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36. [Google Scholar]
- 57. Kung PP, Bingham P, Burke BJ, et al. Characterization of specific N‐α‐acetyltransferase 50 (Naa50) inhibitors identified using a DNA encoded library. ACS Med Chem Lett. 2020;11:1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Greene NP, Crow A, Hughes C, Koronakis V. Structure of a bacterial toxin‐activating acyltransferase. Proc Natl Acad Sci U S A. 2015;112:E3058–E3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rock CO, Jackowski S. Forty years of bacterial fatty acid synthesis. Biochem Biophys Res Commun. 2002;292:1155–1166. [DOI] [PubMed] [Google Scholar]
- 60. Smock RG, Yadid I, Dym O, Clarke J, Tawfik DS. De novo evolutionary emergence of a symmetrical protein is shaped by folding constraints. Cell. 2016;164:476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Longo L, Despotović D, Weil‐Ktorza O, et al. Primordial emergence of a nucleic acid binding protein via phase separation and statistical ornithine to arginine conversion. Proc Natl Acad Sci U S A. 2020;117:15731–15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Seal M, Weil‐Ktorza O, Despotović D, et al. Peptide‐RNA coacervates as a cradle for the evolution of folded domains. J Am Chem Soc. 2022;144:14150–14160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1: