

Abstract
Young women represent a target of E-cigarette (E-cig) companies, raising concern for potential connections with breast cancer (BC) that have not yet been elucidated. We hypothesized that E-cig promotes BC development and lung metastasis possibly through BC-monocyte/tumor-associated macrophage (TAM) crosstalk via CCL5 and V-CAM-1 axes. We demonstrated that E-cig promoted the infiltration of circulating monocytes in mammary fat pad (MFP) model. Furthermore, E-cig exposure significantly enhanced BC cell growth in MFP tumor and metastatic lung colonization; immunohistochemical stains illustrated the increase of TAMs infiltration, reduced BC cell apoptosis and increased proliferation index after E-cig exposure. In vitro studies show E-cig vapor condensate (EVC) treatment upregulated protein expressions of CCL5, V-CAM-1, and other pro-tumorigenic factors in BC cells. Mechanistically, co-culture system demonstrated both EVC and macrophages independently stimulated BC cell growth and the migration via CCL5/CCR1/CCR5 axis. During metastasis, E-Cig exposure stimulated BC cell survival via direct interaction with infiltrated macrophages, regulated by VCAM-1 and integrin α4β1. Our findings, for the first time, showed that E-cig promotes BC growth and metastasis. This study highlights the critical role of TAMs via CCL5 and VCAM-1 pathways in E-cig promoted BC tumor development.
Keywords: E-Cigarette, Breast cancer, Lung metastasis, Macrophages, CCL5, V-CAM-1
1. Introduction
Breast cancer (BC) is the most commonly diagnosed cancer among women in the United States, accounting for nearly one-third of all female cancer diagnoses and over 20% of female cancer deaths [1,2]. Metastasis remains the primary cause of morbidity and mortality from BC [3]. Approximately 16% of all US adults regularly engage in cigarette smoking (CS) [4,5], representing the largest preventable cause of death in the USA [6]. In 2009, the Canadian Expert Panel on Tobacco Smoke and Breast Cancer Risk concluded that the relationship between active smoking and BC was consistent with causality [7]. A slight increase in BC incidence was seen with every increase of 20 pack-years, and hazard ratio would increase relative to the duration, quantity, and starting age of smoking [8]. Pierce et al. found that BC patients who were former smokers (>a 30 pack-year history) had a 54% increased risk of overall BC mortality than nonsmoking patients [9]. In contrast to the well-established epidemiological relationship, mechanisms linking smoking and BC remain incompletely described. Combustion of a cigarette during smoking generates thousands of agents [6]. Exposure to those agents increases motility and epithelial mesenchymal transition (EMT) of BC cells [10,11]. Nicotine, a major component in CS, damages the cellular genome, disrupts cellular metabolic processes and facilitates the proliferation of transformed cells [12]. The nicotinic acetylcholine receptors, which are activated by nicotine, can stimulate several signaling pathways leading to tumorigenic effects. Clinically, smoking-related DNA changes in epithelial cells are detected in the breast milk of smokers [13,14].
In the past decade, an alternative to CS known as the E-Cigarette (E-Cig) was introduced into the US market, initially as an effort to reduce the harm associated with tobacco use. E-Cig liquid contains vehicle solvents such as vegetable glycerin (VG) and propylene glycol (PG), nicotine and usually agents associated with specific flavor [15]. Since its introduction, E-Cig use has experienced widespread acceptance among smokers and nonsmokers alike, particularly among young people [16]. Emerging evidence, including results from our lab, have suggested that E-Cig use induces systematic inflammation and immune cell activation. E-Cig vapor condensate (EVC) significantly increases production of IL-6, tumor necrosis factor-α (TNF-α), and MMP-9 from cultured alveolar macrophages [17] and suppresses cellular antioxidant defenses in cultured epithelial cells [18]. Chronic vaping induces protease release from pulmonary immune cells [19]. The impact of E-Cig on BC development and metastasis, however, remains largely unknown. The limited literature, primarily focused on bronchial epithelial and urothelial cells, does suggest E-Cig exert a powerful stimulatory effect on phase-I carcinogen-bioactivating enzymes [20], induce DNA damage after short term exposure and increase the rate of spontaneous lung adenocarcinoma in FVB/N mice [21,22].
In a physiological state, the immune system represents a major barrier to tumor formation. However, chronic inflammation, caused by various triggers, can result in a tumor-promoting microenvironment (TME) [23]. The TME is composed of extracellular matrix (ECM), stromal cells and immune cells, including tumor-associate macrophages (TAMs). It has become increasingly clear that the TME has a fundamental role in tumor progression, metastasis and immunosuppression [24]. TAM infiltration in human BC, for example, is associated with poor prognosis and metastatic disease [25]. The macrophage targeting chemotherapeutic agent Trabectedin, a CSF1 inhibitor, decreases TAM infiltration and reduces tumor growth and metastasis in a BC xenograft mouse model [26,27]. TAMs constitute up to 50% of cells by number within the BC TME [28]. Most TAMs are recruited to the TME by cytokines released from cancer and stromal cells. These cytokines include CSF1, CCL2 and CCL5 [29,30]. Recruited macrophages develop into non-polarized (M0) macrophages by CSF1 stimulation [31]. M0 macrophages are highly plastic, exhibiting dynamic responses to environmental signals. The resulting TAMs can be classified along a functional spectrum. M1-like and M2-like macrophages represent opposite extremes of this continuum [32,33]. M1-like macrophages are stimulated by the Th1-associated cytokines and exhibit antitumor effects by releasing pro-inflammatory cytokines as well as reactive nitrogen and oxygen intermediates [31,34]. M2-like macrophages are stimulated by the Th2-associated cytokines and typically demonstrate pro-tumor characteristics. Depending on subsequent stimuli, M2-like macrophages can further differentiate into three subtypes: M2a, M2b and M2c macrophages, all of which play a role in the metastatic process [35,36]. Most TAMs in the BC microenvironment are closely related to the M2-like phenotype [33]. No study in the literature addresses the effects of E-Cig exposure on lung metastasis in BC, though recent experimental and epidemiological studies suggest CS enhances lung metastasis of BC [37,38]. It is reported that CS promotes cell growth and invasiveness of BC cell lines; triggers a subpopulation of CD44+,CD49f + tumor stem-cells and stimulates metastasis from primary injection sites [39]. These studies on BC metastasis focus on the tumor cell itself, effects of CS or E-Cig exposure on TAM recruitment and function, as well as cross-talk between BC cells and TAMs, is lacking.
This study is designed to elucidate the effects of E-Cig exposure on BC development and lung metastasis along with associated underlying mechanisms. We hypothesize that E-Cig exposure activates circulating monocytes and alters the cross-talk between BC cancer cells and TAMs to 1) increase the development and migration of BC cells via CCL5/CCR1/CCR5 axis and 2) promote the binding of TAMs to BC cells and the survival of disseminated BC cells in the lung via a VCAM-1-mediated signaling pathway, ultimately leading to pulmonary metastasis. Our results highlight the potential impact of E-Cig use on the development and metastasis of BC, representing a significant contribution to our understanding of the risk associated with widespread E-Cig adoption.
2. Materials and methods
2.1. Animal studies
All procedures were carried out in accordance to a protocol approved by Rutgers Institutional Animal Care and Use Committee (IACUC, protocol #PROTO201900009). Female 5–7 weeks old BALB/C were purchased from Charles River Laboratories. In a series of experiments, for each cell line, 16 mice were equally divided into 2 groups, air and E-Cig exposure. In the E-Cig exposure group, mice were exposed to E-Cig vapor using the InExpose system, specifically customized by Scientific Respiratory Equipment Inc. (SCIREQ, Montreal, Canada) for 2 h/day, 5 days/week, for 6 weeks. The E-Cig formula (24 mg/mL nicotine, 50% PG and 50% VG) and exposure setting were optimized according to the American E-Cig Liquid Manufacturing Standard and American Vaping Standard (70 mL/1 puff/min). The air exposure group was kept in separate animal room with ambient filtered air. After 1 week of air or E-Cig exposure in mice, BC cells were injected orthotopically to establish BC and/or lung metastasis as previously described [40]. Briefly, 1 × 106 GFP-labeled EpRAS cells in 50 μL of 1:1 mix of PBS/growth factor reduced Matrigel Matrix (BD Biosciences, San Jose, CA) or 1 × 106 GFP/luciferase-labeled 4T07 cells resuspended in 50 μl of PBS were injected into the 4th right mammary fat pad or the tail vein of mice, respectively. Tumor growth and lung metastasis were monitored weekly with the Spectrum In Vivo Imaging System (IVIS, PerkinElmer, MA, USA). After the experimental endpoint (4 weeks after air or E-Cig exposure), all mice were euthanized with CO2 inhalation, followed by cervical dislocation, at the same day. Blood samples were collected from the vena cava and subjected to flow cytometric analysis; the tumors in the fat pad and bilateral lungs were collected, weighed, and the number of metastatic tumor nodules on the surface of the lung were counted; the tumor tissue was then embedded in paraffin for histological and immunohistochemical analysis. Harvested primary MFP tissues were fixed with 10% neutralized formalin for 1 day and thereafter kept in 70% ethanol; lung tissues were perfusion-fixed with 10% neutralized formalin at 200 mm H2O pressure for 10 min, harvested and fixed in the same solution for 1 day and thereafter kept in 70% ethanol. MFP or lung tissues were then bi-halved to reveal the largest cross sections, embedded in paraffin and sectioned at 5-μm before H&E stained or GFP immunostained (2.4, immunohistochemical analysis). The stained tissue sections were scanned, tumor areas were outlined by 2 board-certified pathologists (blind for the animal treatment conditions), and tumor area quantified by Imagescope software (Apeiro).
For inhibition study, a total of 28 female BALB/C mice (6 weeks old) were placed into 4 groups, each group 7 mice (n = 7): Electronic cigarette smoke (ECS) exposure group (E-Group), inhibitor treated ECS exposure group (iE-Group), Air exposure group (A-Group) and inhibitor treated air exposure group (iA-Group). Two groups included E-Group and iE-Group were exposed to ECS generated from E-liquid (24 mg/mL of nicotine dissolved in the mixture of Propylene glycol and vegetable glycerin at a 1:1 ratio) using InExpose system (SCIREQ, Montreal, Canada). Mice were exposed in whole-body exposure with ECS, 70 mL puff volume, 1 puff/min, 2 h per day, 5 days per week for 7 weeks. The consistence of ECS exposure was monitored by controlling particulate concentration and gravimetric measurement. The last two groups A-Group and iA-Group, remained housed in the animal room, expose to the ambient filtered air. After 1 week of exposure in mice, 2 × 106 of EpRAS breast cancer cells (in the mixture of PBS and high concentration Matrigel at 1:1 ratio) were injected into the 4th right mammary fat pad of mice (4 days later, 1 mouse in E-Group died). One week after injection, all mice in iE-Group and iA-Group were treated with 20 mg/ kg/day of Maraviroc (Selleckchem, Cat. #S2003) by intraperitoneal (IP) injection, 5 days per week for 5 weeks. The mice in E-group and A-group were provided with 100 μl of vehicle, one a day, 5 days per week for 5 weeks. After 5 weeks of Maraviroc treatment, all mice were euthanized with CO2 followed by cervical dislocation, at the same day. Blood samples were collected from the vena cava and subjected to flow cytometric analysis; urine was collected for cotinine ELISA assay; the tumors in the fat pad and bilateral lungs were collected, and the number of metastatic tumor nodules on the surface of the lung were counted; the tumor tissue was then embedded in paraffin for histological and immunohistochemical analysis.
2.2. Cell culture
GFP-labeled EpRAS, GFP/luciferase-labeled 4T07 (gifts from Dr. Kang YB from Princeton University) and RAW267.4 (ATCC) cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, Gaithersburg, MD) supplemented with 10% fetal bovine serum, 2 mM Glutamine, and 1% Penicillin/Streptomycin. Cells were grown in a humidified incubator at 37 °C with 5% CO2. All supplements were purchased from Life Technologies (Carlsbad, CA, USA).
2.3. E-cig vapor condensate (EVC or E-Cig) preparation for in vitro studies
ECV aerosol was be generated from 30.3 μl in-house-prepared E-liquid (50% PG, 50% VG, and 18 mg/mL nicotine) using a Kangertech EVOD Mega pen operated at a fixed voltage of 3.7V with heating temperature ranging between 100 °C and 300 °C. The aerosol will be drawn into 12.5 mL DMEM with peristaltic pump set at an optimum speed to allow the E-liquid to burn in approximately 15 min and the resulting solution will be considered 100% ECV extract. This solution will be filtered through a 0.22 μm pore acrodisc syringe filter and aliquot into 1 mL/tube and stored at −80 °C. Nicotine concentrations from each batch of preparation will be measured at the lab of Matthew S. Halquist (Bioanalytical Shared Resource Laboratory, University School of Pharmacy).
2.4. Immunohistochemical analysis
Upon termination of animal experiments, tumors and lungs were removed and fixed in 10% neutral-buffered formalin before processing into paraffin blocks. Tissue sections (5-μm thick) were cut from the blocks and subjected to H&E or immunohistochemical (IHC) staining. Briefly, paraffin sections were rehydrated and processed using the standard protocol (Rutgers CINJ Histology Core Facility) for IHC staining. The following antibodies were used: GFP (Ab290, 1:2,000, Abcam, USA), F4/80 (MCA4976A, 1:50, Serotech, USA), Ki-67 (Ab16667, 1:500, Abcam, USA), cleaved Caspase-3 (9661, 1:200, Cell Signaling, USA). Both H&E and immunohistochemically stained slides were scanned on an Ariol SL-50 (version 3.0.70) Automatic Scanning System from Applied Imaging. Tumor sections were scanned at 20 × magnification and the regions of interest were identified and outlined by a pathologist. The tumor sections with immune-positive regions from control and treated mice were quantified for intensity, size and area using Ariol software Review Station. P-value was determined for statistical significance using the student’s t-test.
2.5. Reverse transcriptase polymerase chain reaction (RT-PCR)
TRIzol™ reagent (ThermoFisher Scientific, Waltham MA) was used to extract total RNA from cells. 1 μg of RNA was reverse-transcribed using Maxima First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Waltham MA). PCR amplification was performed using Applied Biosystems PowerUp™ SYRB™ Green Master Mix (ThermoFisher Scientific, Waltham MA) for quantitative real-time PCR. The following PCR primers were used: murine GAPDH: 5′- ACAACTTTGGTATCGTGGAAGG-3’ (forward), 5′- GCCATCACGCCACAGTTTC-3’ (reverse); murine CCL5: 5′- CTCACCATATGGCTCGGA-3’ (forward), 5′-CGAGTGACAAACACGACTG-3’ (reverse); murine CCR1: 5′-GTGGTG GGCAATGTCCTAGT-3’ (forward), 5′-TCAGATTGTAGGGGG TCCAG-3’ (reverse); murine CCR5: 5′-GGAGAGAAGTTCCGGAGTTA -3’ (forward), 5′-CTCCTGTGGATCGGGTATAG -3’ (reverse); murine VCAM-1: 5′-CCCGTCATTGAGGATATTGG-3’ (forward), 5′-CCTGGGAGA-GATGTAGACTT-3’ (reverse); murine ITGB1: 5′-CCCAGAGGCTCTCAAACTA-3’ (forward), 5′-TCTGTG GTT CTC CTG ATC TC-3’ (reverse); murine ITGA4: 5′-GGCTCTATCGTGACTTGTG-3’ (forward), 5′-CTCAGTTCTGTCCGCAAA-3’ (reverse). The relative expression of gene of interest was quantified using the relative quantitation method. Each experiment was independently repeated at least two times.
2.6. Flow cytometry with whole blood samples
At the experimental endpoint, up to 1 mL of whole blood from each animal was collected into 3 mL K2EDTA vacutainers (BD Biosciences, San Jose, CA) and subjected to red blood cell lysis with BD Pharm Lyse Lysing Buffer (BD Biosciences, San Jose, CA), according to the manufacturer’s protocol. After red blood cell lysis, the remaining cells were stained with 16 fluorochrome-conjugated anti-mouse antibodies listed in the biomarker panel for murine monocytes (Table 1). Panel design was based on fluorochrome brightness, antigen density and co-expression, fluorochrome spillover of interested immune-cell subsets and reagent availability in each panel for the available flow cytometer. These markers were designed to assess the immune profile of circulating monocytes and interested targets. A fluorescence minus one (FMO) and unstained cells were included for each antibody as a control. Live cells were identified with Live/Dead Fixable Blue Dead Cell Stain Kit (Thermo Fisher, Waltham, MA). Stained cells were subjected to flow cytometric analysis with a BD LSR Fortessa X-20 cytometer equipped with 5 lasers (355 nm, 405 nm, 488 nm, 561 nm, and 642 nm), performed by Rutgers NJMS Cytometry and Immunology Core Laboratory.
Table 1.
List of used antibodies for flow cytometry and histology analyses.
| CD115 | BioLegend #135526 | CD43 | BD Biosciences #562866 |
|---|---|---|---|
|
| |||
| CCR1 | BioLegend #152506 | CX3CR1 | BioLegend #149013 |
| Ly6C | BioLegend #128025 | TLR3 | BioLegend #141903 |
| CCR2 | R&D Systems #FAB5538 N | F4/80 | Serotech #MCA4976A |
| TLR9 | R&D Systems #FAB7960R | GFP | Abcam #Ab290 |
| CD49d | BD Biosciences #740341 | Ki67 | Abcam #Ab16667 |
| MHC II | BD Biosciences #743874 | Caspase-3 | Cell Signaling #9661 |
| CCR5 | BD Biosciences #743697 | ||
| CD62L | BD Biosciences #563117 | ||
| CD45 | BD Biosciences #563890 | ||
| CD3 | BD Biosciences #563565 | ||
| CD19 | BD Biosciences #563557 | ||
| NK1.1 | BD Biosciences #564144 | ||
2.7. Wound healing assay
In vitro cell migration was evaluated with a CytoSelect would healing assay (Cell Biolabs Inc., San Diego CA). Briefly, EpRAS cells were seeded at a density of 350,000 cells/mL in 2 mL/well of a 6-well plate containing a wound healing insert placed in the center of each well. Upon reaching confluency, the inserts were removed and cells were washed with PBS to remove non-adherent cells. Then the cells were treated with either 0.5% E-Cig or conditioned medium of RAW267.4 pretreated with 0.5% E-Cig for 24 h. Images were taken every 2 h from the starting point (T0) with Keyence automated microscope (Keyence, Osaka, Japan). The effect on migration was quantified by the unoccupied area, measured in square pixels using ImageJ software. Each experiment was performed in triplicate and repeated at least two times using different cell preparations.
2.8. Cytokine protein array
To prepare supernatant samples for proteome profiler array, EpRAS cells were seeded in a 35-mm cell culture treated dishes at a density of 0.28 × 106 cells/cm2 and treated with 0.5% E-Cig. Non-treated condition was included as a control. After 24 h, the supernatant was collected and analyzed with mouse cytokine antibody array (R&D System, Minneapolis, MN) per manufacturer’s instruction. Briefly, the membrane was treated with blocking buffer for 30 min then incubated with 1 mL of EpRAS supernatant overnight at 4 °C. After this incubation period, the membranes were washed five times with a washing buffer and incubated for 1 h at room temperature with biotin-conjugated antibodies against murine cytokines (1:250 dilution). Thereafter, the membranes were washed five times, incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated streptavidin (1:1000 dilution) and washed five times again. Finally, the reaction was developed in a mixture of SuperSignal West Pico luminol/enhancer and stable peroxide solutions (Thermo Scientific, Rockford, IL) and exposed to BioRad imaging system. The pixel density of each spot was subtracted to that of the correlated negative control (neg)/background and then analyzed with ImageJ software (NIH). The experiment was performed in duplicate.
2.9. Enzyme-linked immunosorbent assay (ELISA)
After the treatment with 0.5% E-Cig, the concentration of mouse CCL5 and VEGF in the supernatant of the cultured EpRAS cells were quantified, using the Mouse/Rat CCL5 or VEGF Quantikine ELISA kit ((R&D Systems, Minneapolis, MN), according to the manufacturer’s instruction. Briefly, 50 μl EpRAS supernatant was added into each well of the 96-well strip plate precoated with associated capturing antibodies and incubated at 4 °C overnight. After washing with assay buffer, the corresponding conjugations were added to each well and incubated for 2 h at room temperature. After 3 washes, 100 μl substrate was added to the plate and incubated for 30 min at room temperature in dark. The reaction was stopped by adding the provided stopping reagent. Optical densities at 450 nm and 570 nm were read in an ELISA reader (Bio-Tek, Winooski, VT). The actual concentrations were determined by comparing to the standard curves and analyzed with GraphPad Prism software. All experiments were performed in triplicate.
2.10. Cell-cell binding and survival assays
For cell-cell binding assays, RAW267.4 monocytes pre-labeled with Cell Tracker Red CMTPX Dye (Invitrogen, Carlsbad, CA) were co-cultured with confluent EpRAS cancer cells pre-treated with E-Cig 0.5% at a density of 2.5 × 105 cells/cm2 for 75 min to allow cell-cell binding. After the incubation, the co-culture was washed 3 times with 1X PBS to remove unbound monocytes, and fixed with 4% paraformaldehyde, followed by Hoechst 33342 staining. After three-washes, cells were visualized on fluorescent microscope. For inhibition study, RAW267.4 cells were pre-treated with 25 nM BIO 5192 (Tocris, Bristol, UK) for 4 h before co-cultured with EpRAS cells. For survival assays, the co-culture of non-labeled RAW267.4 monocytes and EpRAS cancer cells was further incubated with 100 ng/mL recombinant TRAIL (PeproTech Inc., Rocky Hill, NJ) to trigger apoptosis. After overnight treatment, the cells were washed 3 times with 1X PBS and harvested by Accutase solution. The single cell suspension was stained with anti-mouse CD45 antibody (BD Biosciences, San Jose, CA) and Annexin V apoptosis kit (Invitrogen, Carlsbad, CA) as per manufacturer’s instructions. The stained cells were analyzed with the LSR II flow cytometer (BD Biosciences, San Jose, CA). All experiments were independently repeated at least 2 times.
2.11. Statistical analysis
Statistical analyses were performed with GraphPad Prism software version 6. Data are presented as means and standard error of the mean (SEM). Differences between groups in in vitro analyses were calculated with the Student’s t-test or two-way ANOVA followed by the Student’s t-test. The animal number used in the in vivo study was justified with Power and Sample Size Calculation software version 3.1.6 (Vanderbilt University) with a power of 90% at a significance level of 5%. For all experiments, significance is denoted by *P < 0.05; **P < 0.01; ***P < 0.001, or ****P < 0.0001 as indicated in the figures and legends.
3. Results
3.1. E-cig exposure enhances tumorigenicity of primary and metastatic breast cancer
Traditional smoking is well established as a risk factor for carcinogenesis. The impact of E-Cig exposure on cancer progression, however, has not been reported. In this study, we aimed to evaluate the direct effect of E-Cig exposure on tumor growth, using an immunocompetent mouse model with orthotopic implantation of EpRAS cells into the mammary fat pad. After 4 weeks of E-Cig exposure, tumor growth was significantly faster (2.27-week doubling time) than that in the animals exposed to filtered air (1.24 week doubling time). The tumor area increased from 35.46% (air control) to 69.24% (E-Cig) (Fig. 1A and B). In addition, E-Cig exposure was associated with 100% (6/6) tumor development, while only 33.3% (2/6) of tumors formed in the air control group. Consistent with this observation, the immunohistochemical analysis of the primary tumor revealed an increase in proliferation by 15.74% and decrease in apoptosis by 14.98%, represented by Ki67 and cleaved Caspase-3 staining, respectively, in E-Cig exposed mice (Fig. 1C and D). These observations strongly suggest a significant impact of E-Cig exposure on tumorigenesis at the primary site of breast cancer.
Fig. 1. The impact of E-Cig exposure on mammary fat pad EpRAS breast cancer progression.

(A) Site of MFP primary tumor and tumor volume with and without E-Cigarette exposure. (B) Histological measurement of MFP primary tumor areas with and without E-Cigarette exposure. (C) Measurements of tumor cell proliferation (Ki-67 stain) and tumor cell apoptosis (cleaved caspas-3 stain) with and without E-Cigarette exposure. (D) H&E stain of MFP tumor cells (upper column); Stains of Ki67 (middle column); and cleaved Caspase-3 (lower column). *P < 0.05, **P < 0.01, n = 8 for each experimental group. Bar scale = 0.02 mm.
The lung represents one of the most common sites of metastasis associated with breast cancer. We therefore investigated the impact of E-Cig exposure on the process of lung metastasis from EpRAS mammary breast cancer cells. In the same orthotopic model, E-Cig exposure significantly promoted the development of lung metastasis in all animals (6/6) compared to the control cohort (2/6) (Fig. 2A and B). Similar to our observations in primary tumors, the incidence of metastasis also correlated with increased proliferation by 20.71% and decreased apoptosis by 27.8%, quantified by Ki67 and cleaved Caspase-3 staining, respectively, in the lungs of animals exposed to E-Cig.
Fig. 2. The impact of E-Cig exposure on lung metastasis of EpRAS breast cancer.

(A) Site of lung metastasis of breast cancer and number of metastatic nodules with and without E-Cigarette exposure. (B) Histological measurement of metastatic tumor areas with and without E-Cigarette exposure. (C) Measurements of metastatic tumor cell proliferation (Ki-67 stain) and tumor cell apoptosis (cleaved caspase-3 stain) with and without E-Cigarette exposure. (D) H&E stain of metastatic tumor nodules (upper column); Ki67 stain (middle column); cleaved Caspase-3 staining (lower column). *P < 0.05, ***P < 0.001, n = 8 for each experimental group. Bar scale = 0.02 mm.
The enhancement of lung metastasis during E-Cig exposure was also demonstrated in another murine breast cancer model intravenously injected with 4T07 cells. This cell line which was isolated from line 44FT0 after 7 in vivo passages through the lungs of syngeneic mice [42] is known to carry a higher metastatic characteristic than EpRAS line. Consistent with the in vivo model using the mammary fat pad implantation of EpRAS cells, our data showed that E-Cig also enhanced the metastatic potential in the lung of mice injected with 4T07 cells (Fig. 6A). The tumor nodules and GFP-labeled 4T07 cells increased 60.20% (p = 0.024) and 77.50% (p = 0.0036), respectively, in the lungs in E-Cig exposure group, when compared to mice exposed to air (Fig. 6B). In addition, the immunohistochemical analysis of the 4T07 lung metastasis revealed an significant decrease in apoptosis by 84.33% (p = 0.0007) represented cleaved Caspase-3 staining in E-Cig exposed mice (Fig. 6C, bottom). The consistent findings established on both EpRAS and 4T07 murine breast cancer model strongly suggest that E-Cig exposure plays an important role in the progression of primary tumor and the development of distant metastasis.
Fig. 6. The in vivo and in vitro impact of E-Cig exposure in 4T07 breast cancer.

(A) Site of lung metastasis of developed by intravenous injection of GFP/luciferase-labeled 4T07 cells showed by Spectrum In Vivo Imaging System. (B) Histological measurement of metastatic tumor nodules and areas with and without E-Cigarette exposure. (C) Measurements of tumor infiltrated macrophages (F4/80 as DAB, GFP as purple, top) and tumor cell apoptosis (cleaved caspase-3 as DAB, GFP as purple, bottom) with and without E-Cigarette exposure. The bar graphs indicate corresponding quantitative measurements. (D) The bar graph represents the quantification of unoccupied areas measured in the wound healing assay of 4T07 cells treated with 0.5% E-Cig, with or without Met-CCL5. Negative control (NC) was included as a control. (E) Quantitative analysis of the number of RAW267.4 cells binding to 4T07 breast cancer cells in the presence or absence of E-Cig in cell-cell binding assay. *P < 0.05, **P < 0.01, ***P < 0.001. n = 8 for each in vivo experimental group. Bar scale = 0.02 mm. Each in vitro experiment was performed in triplicates and repeated at least 2 times independently. Data is the representative of independent experiments (n = 3).
3.2. E-cig exposure increased the number of circulating monocytes and infiltration of tumor-associated macrophages in the primary and metastatic tumor microenvironment
Monocytes/macrophages are well documented for their putative roles in tumorigenicity. We hypothesized that the enhancement of breast cancer progression and metastasis by E-Cig exposure could be regulated via the crosstalk between monocytes/macrophages and tumor cells in the tumor microenvironment. The advantage of using an immunocompetent Balb/c mouse with Balb/c-derived EpRAS cancer cells allowed us to examine this hypothesis. The strategy used to identify circulating monocyte population in the whole blood was described (Fig. 3A). Flow cytometric analysis revealed a significant increase in circulating monocytes in E-Cig exposed animals (3.49%, compared to 1.15% in the air control group) (Fig. 3B). This population was further remarkable for upregulation of the chemokine receptor, CCR5 (P value = 0.05), a biomarker known to facilitate monocyte/macrophage infiltration and tumor crosstalk. We also observed a trend toward significance in the expression of CX3CR1 (P value = 0.08), marker of monocyte differentiation (Fig. 3C). Interestingly, the number of F4/80-labeled resident monocytes increased by a factor of 4 and 2.5, respectively, in primary and metastatic sites of E-Cig exposed mice (Fig. 3D). Consistent with the observation seen in EpRAS tumor model, the immunohistochemical analysis of the 4T07-derived lung metastasis in E-Cig exposed mice also revealed an significant increase in the infiltration of F4/80-labeled macrophages by over 400% (p = 0.023), when compared to that in the tumor bearing mice exposed to air (Fig. 6C, top). The findings suggest a contribution of E-Cig exposure to monocyte activation and infiltration into the tumor microenvironment at both primary and metastatic sites, potentially through the CCR5/CCL5 axis.
Fig. 3. E-Cig exposure increases the circulating monocytes and infiltrated TAMs in EpRAS primary and metastatic tumor areas.
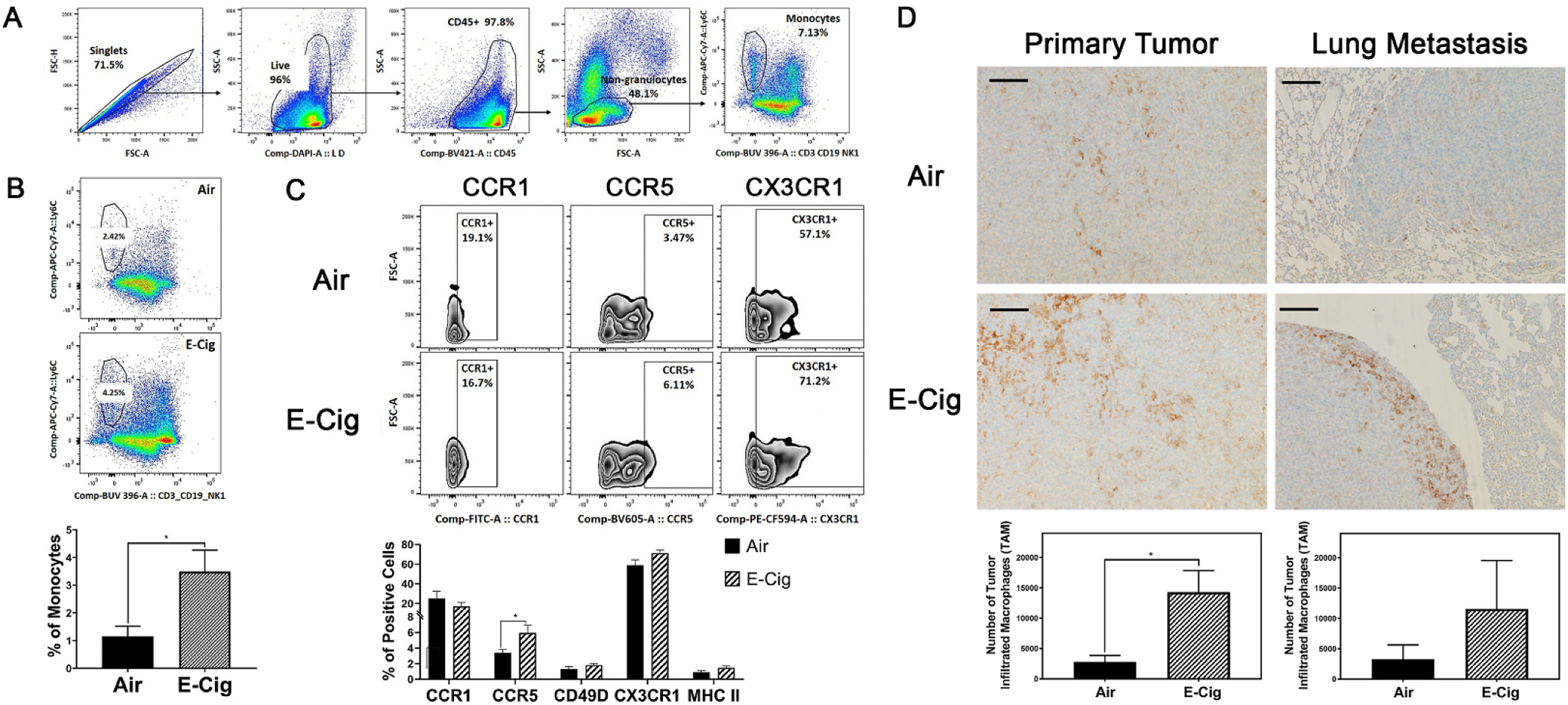
(A) Flow cytometry gating strategy for monocyte surface markers. (B) Gating strategy to measure circulating monocyte number with and without E-Cigarette exposure, corresponding quantitative measurement. (C) Measurement of monocyte surface markers, CCR1, CCR5, CX3CR1 using flow cytometry. (D) Immunostaining of macrophages associated to primary or metastatic breast cancers with and without E-Cigarette exposure (F4/80) and corresponding quantitative measurements. *P < 0.05, n = 8 for each experimental group. Bar scale = 0.02 mm.
3.3. E-cig exposure stimulates the migration of breast cancer cells in both dependent and independent manner of monocytes
The E-Cig enhanced infiltration of monocytes/macrophages into the tumor microenvironment in the immunocompetent mouse model suggests an important role of E-Cig and TAMs in the tumorigenicity of BC. In order to gain a more complete picture of E-Cig exposure in the tumor microenvironment, we next probed the aggressive migratory phenotype of BC cells using an in vitro wound healing assay. Under both direct stimulation via EVC or conditioned medium of RAW267.4 monocytes that were exposed to EVC (CME), a significant increase was observed in the migration of murine mammary carcinoma EpRAS cells, when compared to the non-treated (NC) or conditioned medium-treated cells (CM, no EVC treatment), respectively. This observation suggests that E-Cig may contribute to the migration of EpRAS cells both directly and indirectly through monocyte crosstalk. Further, the migratory effect of EVC on EpRAS can be effectively inhibited by Met-CCL5, a well-known inhibitor of CCR1 and CCR5, suggesting that this phenotype may be controlled by CCL5 and its receptors CCR1/CCR5 (Fig. 4A and B). Consistent with EpRAS model, the impact of E-Cig on CCL5-mediated cell migration was also demonstrated in 4T07 cell line (Fig. 6D).
Fig. 4. Effect of E-Cig on the in vitro migration and the expression of CCL5/ CCR1/CCR5.

(A) and (B), the wound healing assay of EpRAS cells treated with (A) 0.5% E-Cig or (B) conditioned medium from RAW264.7 pre-treated with 0.5% E-Cig (CME), with or without Met-CCL5. Negative control (NC) or treatment with RAW267.4 conditioned medium (CM) was included as a control. The gap area was quantitated in the right bar graphs. (C) mRNA expression of CCL5, CCR1, and CCR5 in EpRAS (left) and RAW264.7 (right) cells was detected by RT-qPCR. (D) E-Cigarette enhanced the secretion of CCL5 and other cytokines from EpRAS cells. Secreted cytokines in EpRAS conditioned medium, with (bottom) or without (top) 0.5% E-Cig treatment, were analyzed using Mouse XL Cytokine array kit. The blot displays the pixel density from selected cytokines (present in duplicates). Positive (Pos) and negative (Neg) reference spots of each membrane blot were included for quality control purpose. The upper right bar graphs represented the spot pixel density quantification of selected cytokines in the nontreated (black) and E-Cig-treated EpRAS conditioned medium (green). (E) Quantitative analysis of CCL5 (left Y axis) and VEGF (right Y axis) concentrations detected in the supernatant of EpRAS with (E-Cig) or without (NC) treatment, using Mouse/Rat Quantikine ELISA kits. *P < 0.05; **P < 0.01; ***P < 0.001, or ****P < 0.0001. Each experiment was performed in triplicates (duplicates, for protein array) and repeated at least 2 times independently. Data is the representative of independent experiments (n = 3).
To evaluate the correlation of this chemokine/chemokine receptor axis to the migratory phenotype, the CCL5, CCR1, and CCR5 expression in EpRAS and RAW267.4 cells was analyzed using real-time polymerase chain reaction. In EpRAS cancer cells, CCL5 was significantly upregulated when stimulated with RAW267.4 conditioned medium in an E-Cig independent manner. While contributing little to no effect on CCL5 mRNA expression, direct EVC treatment enhanced the expression of both CCR1 and CCR5 receptors by 5.5 fold and 3.3 fold, respectively, when compared to non-treated EpRAS cells (Fig. 4C, left). In contrast to EpRAS cells, EVC exposure did not have any impact on the mRNA expression of these ligands and receptors in RAW267.4 monocytes (Fig. 4C, right).
Although the change of CCL5 was not detected at mRNA level, the proteomic analysis with Mouse XL Cytokine Array showed a significantly increase in the secretion of CCL5 in the conditioned medium of EpRAS cells after EVC treatment, when compared to non-treated sample (p = 0.0071). In addition to CCL5, EVC also stimulated the secretion of other key factors that contribute to immune cell recruitment and metastasis, such as CX3CL1 (p = 0.0254), CXCL1 (p = 0.0039), CXCL16 (p = 0.0013), Endostatin (p = 0.0065), and VEGF (p = 0.01) from EpRAS cells (Fig. 4D). Consistent with the proteomic analysis, quantitative analysis with Mouse/Rat Quantikine ELISA kit for CCL5 and VEGF also confirmed the effect of E-Cig on secretion of CCL5 (p = 0.0397) and VEGF (p = 0.0033) from EpRAS cells (Fig. 4E). These data together strongly support the contribution of E-Cig exposure in stimulating cancer-immune cells communication and metastatic potential in breast cancer cells.
3.4. E-cig enhances the binding of monocytes to breast cancer cells and supports apoptotic resistance
Our in vivo studies clearly demonstrated the enhancement of tumor-infiltrated macrophages at pulmonary metastatic sites. To understand the impact of macrophage filtration on metastatic development, we studied the interaction of tumor cells and monocytes in an in vitro cell-cell binding assay [43,44]. This analysis demonstrated that E-Cig exposure supported direct binding of monocytes with BC cells. Mechanistically, EVC treatment upregulated VCAM-1 mRNA expression >5 folds in EpRAS BC cells; while there is no change in mRNA level of IGA4 and IGB1 genes encoding integrin subunits α4 and β1 (Fig. 5B); monocyte/EpRAS binding can be effectively inhibited by BIO 5192 (Fig. 5A, top and bottom), a highly potent and selective inhibitor of integrin α4β1 (VLA-4). This phenomenon was also confirmed in 4T07 cells (Fig. 6E). Altogether, the data established in two different models of murine breast cancer suggested that this receptor and its ligand VCAM-1 play a significant role in mediating myeloid-tumor crosstalk in metastasis.
Fig. 5. E-Cig enhances the binding of monocytes to breast cancer cells and support the apoptosis resistance.

(A) Cell-cell binding assay between 4T07 breast cancer cells, with and without EVC treatment and RAW246.7 cells labeled with cell tracker Red CMPTX dye in presence of 25 nM VLA4 inhibitor BIO5192 (top) and corresponding quantitative measurement (bottom). (B) Percentage of apoptotic tumor cells after 24 h 100 ng/mL TRAIL treatment with and without 25 nM VLA4 inhibitor BIO5192. (C) mRNA expression of VCAM-1 in EpRAS breast cancer (left) and ITGA4 and ITGB1 in RAW246.7 monocytes (right) with and without E-Cig treatment were measure by qPCR. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two-tailed Student’s t-test). Each experiment was performed in triplicates and repeated 3 times independently. Data is the representative of independent experiments (n = 3).
It has been reported that the direct interaction of TAM and tumor cells through VCAM-1 and its receptor α4β1 is critical for apoptotic resistance of tumor cells [45]. Consistent with this finding, we showed that the percentage of TRAIL-induced apoptotic EpRAS cells inversely correlates with the number of EpRAS-bound monocytes under EVC stimulation. This protective effect can be inhibited by treatment with BIO 5192 (Fig. 5C). Taken together, these data strongly support the role of E-Cig in regulating the VCAM-1-dependent crosstalk between monocytes/macrophages and tumor cells, resulting in apoptotic protection and seeding enhancement of breast cancer cells in the pulmonary microenvironment.
3.5. The CCR5 inhibitor, maraviroc, significantly inhibit the effect of E-Cig on lung metastasis
For further confirmation of E-Cig effect on lung metastasis driven by CCL5/CCR5 axis, we performed an in vivo inhibition study using Maraviroc, a specific small molecule inhibitor of CCR5 chemokine receptor that had received FDA approval in 2007 for the treatment of patients with human immunodeficiency virus (HIV) infection [46]. Acting as an antagonist at CCR5 coreceptor, this inhibitor is now being repositioned in clinical trials as a new cancer therapeutic approach [47,48]. At the experimental end point (5 weeks after Maraviroc treatment), the histological analysis revealed that Maraviroc significantly reduced the tumor area in lung metastasis in mice exposed to E-Cig by 46.56% (p < 0.0001), when compared to the vehicle group (Fig. 7A). Of interest, this CCR5 antagonist also attenuated the filtration of F4/80-labeled macrophages into the tumor site of E-Cig exposed mice by 28.83% (p = 0.0003) (Fig. 7B). These in vivo observations provide a strong evidence for the important role of CCL5/CCR5 axis in regulating E-Cig induced lung metastasis in the murine breast cancer model.
Fig. 7. The CCR5 inhibitor, Maraviroe, significantly inhibit the effect of E-Cig on lung metastasis.

(A) Histological measurement of metastatic tumor areas in the lung of mice treated with Maraviroc or vehicle, with or without E-Cigarette exposure. (B) Co-immunostaining of GFP (purple) and F4/80 (DAB)-labeled macrophages associated to primary or metastatic breast cancers, with and without E-Cigarette exposure. The bar graphs indicate corresponding quantitative measurements. *P < 0.05, ***p < 0.001. n = 7 for each experimental group. Bar scale = 0.02 mm.
4. Discussion
According to a recent survey on vaping usage, an estimated 2.1 million middle school and high school students reported using E-Cigarettes in 2017 and this number jumped to 4.9 million in 2018 [55]. The increasing popularity of vaping, especially in teenagers, raises an alarm in the public health community. Early evidence has revealed the direct impact of E-Cig on overall health, the development of the adolescent brain, and the potential for addiction [56]. Its contribution to cancer, however, has rarely been reported or discussed. In this study using both animal models and cell-based systems, we uncover versatile roles of E-Cig exposure in BC progression and lung metastasis. As summarized in Fig. 8, our data revealed that E-Cig exposure promoted the infiltration of circulating monocytes into the tumor microenvironment through the expression of chemokine receptors CCR1/CCR5 and the secretion of their ligand CCL5 by monocytes and cancer cells, respectively. In the primary tumor, both E-Cig exposure and infiltrating macrophages can induce growth and migration of BC cells through the regulation of CCL5/CCR1/CCR5 axis and possibly other factors related to tumor proliferation and metastasis. During the stage of metastasis, E-Cig exposure supports the survival of disseminated BC cells and colonization via interaction with infiltrated macrophages, regulated in part by VCAM-1 and integrin α4β1. Our findings provide insight into the potential mechanism by which E-Cig exposure modulates progression and pulmonary metastasis of BC. Further, these findings contribute timely scientific evidence to stress public awareness regarding E-Cig regulation, particularly in the younger population.
Fig. 8. The schematic mechanism of E-Cigarette promotes BC growth and lung metastasis.

E-Cig treatment drives the infiltration of monocytes to tumor areas both in primary tumor and lung colonized tumor via CCR5 upregulation on tumor associated macrophage (TAMs) surface and VCAM-1 on BC cells. CCL5-CCR1/CCR5 axis maintains the crosstalk between BC cells and TAMs while VCAM-1 upregulation increases the binding of TAMs and BC cells during infiltration and enhances the survival rate of metastatic BC cells in lung colonization progress. In addition, TAMs also trigger the secretion of CCL5 derived from BC cells (dash arrow), assist the migration of BC cells to lung. Met-CCL5 inhibitor prevents effectively the contribution of CCL5 to BC cells migration. Furthermore, other cytokines such as CXCL5/10/16, MMPs, Osteopontin, Proliferin, VEGF, and TNFα are also secreted from BC cells after Electronic-cigarettes treatment, prompting tumor progression and metastasis.
BC is the second most common cancer and is the second leading cause of cancer mortality among women [57,58]. In contrast to the well-established causal relationship between traditional CS and BC, no study in the literature directly evaluates the effect of E-Cig exposure on BC progression and lung metastasis. Pioneering work by Tang MS and his group demonstrated the effect of E-Cig on malignant/premalignant cells directly: exposure for 4 months induced mutagenic O6-methyl-deoxyguanosine and γ-hydroxy-1,N2-propano-deoxyguanosine formation in the DNA of lung and bladder of murine epithelial cells, and exposure for 52 weeks induced spontaneous lung adenocarcinoma in FEBV mice [21,22]. In contrast to Tang’s studies, our investigation evaluates the critical role of myeloid cells and related signal transduction pathways in the setting of E-Cig enhanced BC growth and metastasis. In a physiological state, the microenvironment of any organ in the body is generally tumor suppressive. However, chronic inflammation, caused by various triggers, can result in a tumor-promoting microenvironment (TME) [23]. The TME is composed of altered extracellular matrix (ECM), stromal cells and immune cells, including TAMs. It has become increasingly clear that the TME has a fundamental role in tumor progression, metastasis and immunosuppression [24]. TAM infiltration in human BC, for example, is associated with poor prognosis and metastatic disease [25]. Macrophage targeting agents, such as trabectedin or CSF1 inhibitors, decrease TAM infiltration and reduce tumor growth and metastasis formation in BC xenograft mouse models [26,27]. TAMs constitute up to 50% of the cell number within BC [28]. Most TAMs derive from macrophages recruited by soluble factors released from cancer cells and other TME-associated stromal cells. These cytokines/chemokines include CSF1, CCL2 and CCL5 [29]. TAMs have demonstrated a role in nearly all metastatic processes, including local invasion, blood vessel intravasation, extravasation at distant sites and metastatic tumor growth [29,45,59,60]. Flow cytometry analysis in the current study demonstrates that E-Cig exposure significantly increases the number of monocytes in circulation and upregulates the immune/inflammatory marker CCR5 in circulating monocytes. The increased number of activated monocytes, together with increased monocyte chemokine expression in EpRAS cells after E-Cig exposure, including VCAM-1, CCR1, CCR5, can be reasonably speculated to accelerate monocyte infiltration into the tumor microenvironment. Our results therefore suggest that E-Cig exposure stimulates TAM infiltration in both mammary orthotopic and lung metastatic tumors. This increase in TAM infiltration is associated with accelerated BC tumor growth in the mammary fat pad and increased nodule number and total area of lung metastatic BC tumors.
Our in vitro assays show E-Cig treatment significantly upregulated secreted CCL5 protein levels from EpRAS and RAW267.4 cells. Further, E-Cig exposure enhances the expression of the CCL5 receptor in circulating monocytes by flow cytometry analysis. The data suggest a critical role for the CCL5/CCR5 pathway in regulating E-Cig enhanced BC growth and metastasis. CCL5, a member of the CC family, promotes metastasis by triggering MMP production and improving cancer cell motility through a complex network of interacting factors and positive feedback loops [61]. Our observation that the migratory stimulation of EpRAS cells by E-Cig exposure can be effectively inhibited by Met-CCL5, a known inhibitor of CCR1 and CCR5, suggests that this phenotype is also regulated by CCL5 signaling pathway. Related to this finding, CCL5 has been previously reported to promote macrophage and lymphocyte infiltration in various types of human cancers including BC [62–64]. CCL5 binds to its receptors, CCR5 or CCR1, on macrophages and activates AKT signaling to recruit and repolarize TAMs. While minimally expressed by normal breast epithelial duct cells, CCL5 is aberrantly expressed by breast cancer cells at primary tumor sites. Previous studies suggest CCL5 play important role in the cross talk between BC cells and other cell types within the tumor microenvironment by: 1) shifting the balance of infiltrating immune cells, increasing the presence of deleterious TAMs and inhibiting potential antitumor T cell activities; 2) enhancing metastatic processes; 3) promoting migratory and invasion-related properties of BCs [65,66].
Our study also shows that direct E-Cig treatment upregulates VCAM-1 mRNA expression >5 folds in EpRAS BC cells. Further, monocyte/EpRAS binding can be effectively inhibited by BIO 5192, a highly potent and selective inhibitor of integrin α4β1 (VLA-4), suggesting that this receptor and its ligand, VCAM-1, play a significant role in mediating crosstalk between BC cells and TAMs. VCAM-1, an immunoglobulin (Ig)-like adhesion molecule with seven extracellular Ig domains, is aberrantly expressed in BC cells and binds to its receptor, α4β1 integrin [67, 68]. Most early studies on the crosstalk between BC cells and TAMs focus on soluble factors [69]. However, recent studies have shown that depleting lung macrophages or preventing the recruitment of macrophages into the lung significantly decreases lung metastasis via unknown molecular mechanisms [69,70]. Our data regarding VCAM-1 induction provide one such mechanism [45]. Tumor cells entering the lung parenchyma are typically surrounded by macrophages, possibly related to an underlying innate immune response. The close proximity of macrophage and tumor cells subsequently facilitates contact between the α4 integrins and VCAM-1, recruits Ezrin, a cytoplasmic adaptor protein that links the actin cytoskeleton to the VCAM-1 cytoplasmic tail, leading to phosphorylation of Ezrin [65]. Once activated, Ezrin serves as an adaptor that binds both PI3K and its downstream mediator, AKT, leading to activation of AKT-mediated pro-survival signaling [45]. Therefore, α4 integrin-expressing TAMs create a favorable microenvironment for high VCAM-1 expressing BC cells in the lungs as well as primary tumors. We provide support of this proposed interaction, demonstrating that BC-TAM binding is associated with reduced apoptosis of BC cells in vitro; this apoptotic resistance is negated after BC-TAM binding is hindered by an α4 integrin inhibitor.
Both in vitro and in vivo evidences provided in this study help us gain better understanding on the impact of E-Cig during breast cancer progression and metastasis. The active component(s) of E-Cig that drives these biological effects, however, remained unknown. The raw E-liquid composes of 3 main components: nicotine, vegetable glycerin (VG), and polyethylene glycol (PG). After aerosolization that involves heating of the E-liquid at high temperature, many by-products are detected, besides the main ones. This may account for the application of heat that can cause chemical changes or chemical reactions, in which new substances are formed, with different properties from the original. In fact, the additional components that have been reported include formaldehyde, acetaldehyde, acroleine, propanal, acetone, reactive oxygen species, heavy metals, nicotyrine, o-methyl-benzaldehyde, carcinogenic nitrosamines and many more, due to the chemical nature of PG, VG, and heating coil [71,72]. More importantly, secondary and tertiary chemical reaction products between E-liquid and E-Cig constituents have been reported [73]. In an effort to identify the active component(s), we tested the effect of nicotine, VG, PG, and their aerosol condensate generated by heating on the expression of CCR5 and VCAM-1 in EpRAS cells. Although it is very preliminary, our data showed that the mRNA level of both CCR5 and VCAM-1 was significantly upregulated by nicotine vapor, while it remained unchanged under the treatment of unheated nicotine. Neither VG nor PG (in original or aerosol form) produced any effects on the expression of these genes (data not shown). It has to be mentioned that the nicotine concentration in our EVC is approximately 2 nM, which is much lower than the literature reported biological effective dose of nicotine on endothelial cells or animal in vivo studies [74,75]. With some degree of certainty, our observation indicated that nicotine aerosol and/or its derivatives generated from the aerosolization may be a potential active component responsible for the biological effect of EVC in EpRAS cells. To further identify specific component(s) in the complex composition of EVC after the aerosolization, an extensive study involving the use of highly throughput technique in analytical chemistry, such as High-Performance Liquid Chromatography (HPLC), will be required.
In summary, we provide the first scientific evidence for the involvement of E-Cig exposure in enhancing BC progression and pulmonary metastasis in both cellular and animal models. Mechanistically, E-Cig exposure appears to be a central component in both tumor-immune cell crosstalk and tumor metastasis. Exposure to E-Cig led to an increase in circulating monocytes. In our proposed mechanism, monocytes are recruited to tumor sites via CCL5, secreted by breast cancer cells, and its chemokine receptors CCR1/CCR5, increased in E-Cig treated monocytes. This chemokine signaling axis also enhances migration of BC cells when stimulated directly with E-Cig or monocyte conditioned medium pretreated with E-Cig. In addition, the direct binding of macrophages to cancer cells, via E-Cig-regulated VCAM-1 expression by BC cells, provides a protective effect against apoptosis, allowing metastatic seeding in the new microenvironment. Consistently established in both animal- and cell-based systems, with a combination of agonist and antagonist approaches, our findings highlight potentially serious and avoidable risks associated with vaping. These microenvironmental observations also inform therapeutic approaches in breast cancers with high risk for metastasis.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].DeSantis C, Siegel R, Bandi P, Jemal A, Breast cancer statistics, Ca - Cancer J. Clin. 61 (2011) (2011) 409–418. [DOI] [PubMed] [Google Scholar]
- [2].Luo J, Margolis KL, Wactawski-Wende J, Horn K, Messina C, Stefanick ML, Tindle HA, Tong E, Rohan TE, Association of active and passive smoking with risk of breast cancer among postmenopausal women: a prospective cohort study, BMJ 342 (2011) d1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Redig AJ, McAllister SS, Breast cancer as a systemic disease: a view of metastasis, J. Intern. Med. 274 (2013) 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].U.S. Department of Health and Human Services, The Health Consequences of Smoking: 50 Years of Progress. A Report of the Surgeon General, U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, Atlanta, GA, 2014. [Google Scholar]
- [5].A J, Dm H, E OC, Sd B, Rs C, T S, Ss H, Ba K, Current Cigarette Smoking Among Adults - United States, 2005–2014, MMWR. Morbidity and Mortality Weekly Report, 2015, p. 64. [DOI] [PubMed] [Google Scholar]
- [6].Secretan B, Straif K, Baan R, Grosse Y, Ghissassi FE, Bouvard V, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V, A review of human carcinogens—Part E: tobacco, areca nut, alcohol, coal smoke, and salted fish, Lancet Oncol. 10 (2009) 1033–1034. [DOI] [PubMed] [Google Scholar]
- [7].Kc J, Ab M, Ne C, JR P, Sk H, Ag S, Kp C, Md M, Nf B, J M, F T, Active smoking and secondhand smoke increase breast cancer risk: the report of the Canadian Expert panel on tobacco smoke and breast cancer risk, Tobac. Contr. 2011 (2009) 20. [DOI] [PubMed] [Google Scholar]
- [8].F X, Wc W, Ba R, Se H, Kb M, Cigarette Smoking and the Incidence of Breast Cancer, Archives of internal medicine, 2011, p. 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jp P, Re P, Cm S, Sw F, Bj C, L N, Sj N, Em P, Xo S, Wy C, Lifetime cigarette smoking and breast cancer prognosis in the after breast cancer pooling project, J. Natl. Cancer Inst. 106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].S K, J M, J M, Cigarette smoke induces cell motility via platelet-activating factor Accumulation in breast cancer cells: a potential mechanism for metastatic disease, Physiological reports 3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].P D, W R, S P, R K, M K, S R, S B, M C, E K, D C, E H, S C, Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines, Int. J. Canc. (2009) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sa G, Connections of nicotine to cancer, Nat. Rev. Canc. 14 (2014). [DOI] [PubMed] [Google Scholar]
- [13].Nl P, Ld G, Tc B, N C, Jc C, Nicotine in breast fluid of nonlactating women, Science (1978) 199. New York, N.Y. [DOI] [PubMed] [Google Scholar]
- [14].Pa T, Dm D, Ff K, Gy M, Lr B, Bl G, My F, A S, Pd J, Cb A, Evidence for the presence of mutagenic arylamines in human breast milk and DNA adducts in exfoliated breast ductal epithelial cells, Environ. Mol. Mutagen. 39 (2002). [DOI] [PubMed] [Google Scholar]
- [15].Ee D, S K-S, E-cigarettes: impact of E-liquid components and device characteristics on nicotine exposure, Curr. Neuropharmacol. 16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].G S, A J, Ba K, Jm M, Electronic Cigarette Use Among Working Adults - United States, 2014, MMWR. Morbidity and Mortality Weekly Report, 2016, p. 65. [DOI] [PubMed] [Google Scholar]
- [17].Scott A, Lugg ST, Aldridge K, Lewis KE, Bowden A, Mahida RY, Grudzinska FS, Dosanjh D, Parekh D, Foronjy R, Sapey E, Naidu B, Thickett DR, Pro-inflammatory effects of e-cigarette vapour condensate on human alveolar macrophages, Thorax 73 (2018) 1161–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ganapathy V, Manyanga J, Brame L, McGuire D, Sadhasivam B, Floyd E, Rubenstein DA, Ramachandran I, Wagener T, Queimado L, Electronic cigarette aerosols suppress cellular antioxidant defenses and induce significant oxidative DNA damage, PloS One 12 (2017), e0177780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghosh A, Coakley RD, Ghio AJ, Muhlebach MS, Esther CR Jr., Alexis NE, Tarran R, Chronic E-cigarette use increases neutrophil elastase and matrix metalloprotease levels in the lung, Am. J. Respir. Crit. Care Med. 200 (2019) 1392–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Canistro D, Vivarelli F, Cirillo S, Babot Marquillas C, Buschini A, Lazzaretti M, Marchi L, Cardenia V, Rodriguez-Estrada MT, Lodovici M, Cipriani C, Lorenzini A, Croco E, Marchionni S, Franchi P, Lucarini M, Longo V, Della Croce CM, Vornoli A, Colacci A, Vaccari M, Sapone A, Paolini M, E-cigarettes induce toxicological effects that can raise the cancer risk, Sci. Rep. 7 (2017) 2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hw L, Sh P, Mw W, Ht W, Wc H, H L, Xr W, Lc C, Ms T, E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 115, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ms T, Xr W, Hw L, Y X, Fm D, AL M, Lc C, Wc H, H L, Electronic-cigarette smoke induces lung adenocarcinoma and bladder urothelial hyperplasia in mice, in: Proceedings of the National Academy of Sciences of the United States of America, vol. 116, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].X Z, K T, J M, B B, W S, A W, R S, Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy, Oncotarget 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bs H, A S, G DA, A Z, Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer, Semin. Canc. Biol. (2019). [DOI] [PubMed] [Google Scholar]
- [25].Ta K, Y C, H A, E W, la A, Tumor-associated macrophages are strongly related to vascular invasion, non-luminal subtypes, and interval breast cancer, Hum. Pathol. (2017) 69. [DOI] [PubMed] [Google Scholar]
- [26].S A, P P, M S, M H, K Z, R S, Er S, D A, Colony-stimulating factor-1 blockade by antisense oligonucleotides and small interfering RNAs suppresses growth of human mammary tumor xenografts in mice, Canc. Res. (2004) 64. [DOI] [PubMed] [Google Scholar]
- [27].G G, R F, C B, A A, S P, M L, E E, S U, M Z, F P, M N, N v.R., R M, L B, S M, I FN, R S, Pg C, S P, Cm G, A A, A M, M DI, P A, Role of macrophage targeting in the antitumor activity of trabectedin, Canc. Cell 23 (2013). [DOI] [PubMed] [Google Scholar]
- [28].Sq Q, Sjh W, Mc Z, Ege d.V., B v.d.V., Cp S, Tumor-associated macrophages in breast cancer: innocent bystander or important player? Canc. Treat Rev. (2018) 70. [DOI] [PubMed] [Google Scholar]
- [29].K S-A, E P, K B, D S, Y P, Cd M, Dj L, Wp S, Ef P, Kindlin-2 regulates the growth of breast cancer tumors by activating CSF-1-Mediated macrophage infiltration, Canc. Res. (2017) 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nagarsheth N, Wicha MS, Zou W, Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy, Nat. Rev. Immunol. 17 (2017) 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fo M, S G, The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment, F1000prime reports, 2014, p. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dm M, Jp E, Exploring the full spectrum of macrophage activation, Nat. Rev. Immunol. 8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].A M, S S, M L, P A, A S, Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes, Trends Immunol. (2002) 23. [DOI] [PubMed] [Google Scholar]
- [34].Sk B, A M, Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm, Nat. Immunol. 11 (2010). [DOI] [PubMed] [Google Scholar]
- [35].A M, A S, Macrophages, innate immunity and cancer: balance, tolerance, and diversity, Curr. Opin. Immunol. (2010) 22. [DOI] [PubMed] [Google Scholar]
- [36].Yao Y, Xu XH, Jin L, Macrophage polarization in physiological and pathological pregnancy, Front. Immunol. 10 (2019) 792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].S M, Ke P, Ne H, K E, The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer, Chest (2004) 125. [DOI] [PubMed] [Google Scholar]
- [38].S S, S D, A N, Cn K, SURVIVIN as a marker for quiescent-breast cancer stem cells-an intermediate, adherent, pre-requisite phase of breast cancer metastasis, Clin. Exp. Metastasis 33 (2016). [DOI] [PubMed] [Google Scholar]
- [39].F DC, Vl F, H L, B V-P, B G, K H, J S, R B, W W, C B, Sb B, Ca Z, Cigarette smoke induces epithelial to mesenchymal transition and increases the metastatic ability of breast cancer cells, Mol. Canc. (2013) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J, TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4, Cell 133 (2008) 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Be M, Fr M, Dj W, Gh H, Analysis of tumour cell composition in tumours composed of paired mixtures of mammary tumour cell lines, Br. J. Canc. (1987) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen Q, Zhang XH, Massague J, Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs, Canc. Cell 20 (2011) 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen Q, Massague J, Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis, Clin. Canc. Res. 18 (2012) 5520–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Q C, Xh Z, J M, Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs, Canc. Cell (2011) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].S S, H K, Maraviroc: A New CCR5 Antagonist, Expert Review of Anti-infective Therapy, vol. 7, 2009. [DOI] [PubMed] [Google Scholar]
- [47].X J, O N, T P, Av K, N H, D J, Rg P, Recent advances targeting CCR5 for cancer and its role in immuno-oncology, Canc. Res. 79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].A P, M Z, S M, Dm A, Mr B, H A, CCR5 blockage by Maraviroc: a potential therapeutic option for metastatic breast cancer, Cell. Oncol. (2019) 42. Dordrecht. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].C.f.D.C.a. Prevention, Progress Erased: Youth Tobacco Use Increased during 2017–2018 | CDC Online Newsroom, CDC, 2019. [Google Scholar]
- [56].E-Cigarette Use Among Youth and Young Adults: A Report of the Surgeon General, 2016. Atlanta (GA). [PubMed] [Google Scholar]
- [57].C D, R S, P B, A J, Breast cancer statistics, Ca - Cancer J. Clin. 2011 (2011) 61. [Google Scholar]
- [58].Luo J, Margolis KL, Wactawski-Wende J, Horn K, Messina C, Stefanick ML, Tindle HA, Tong E, Rohan TE, Association of Active and Passive Smoking with Risk of Breast Cancer Among Postmenopausal Women: a Prospective Cohort Study, BMJ 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ja J, Jw P, Microenvironmental regulation of metastasis, Nat. Rev. Canc. 9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].As H, En A, D E, Y W, P G, Bz Q, Mh O, Jw P, Jg J, Js C, Real-time imaging reveals local, transient vascular permeability, and tumor cell intravasation stimulated by TIE2hi macrophage-derived VEGFA, Canc. Discov. 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Khalid A, Wolfram J, Ferrari I, Mu C, Mai J, Yang Z, Zhao Y, Ferrari M, Ma X, Shen H, Recent advances in discovering the role of CCL5 in metastatic breast cancer, Mini Rev. Med. Chem. 15 (2015) 1063–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].K P, D L, C L, Jk H, CCL5, CCR1 and CCR5 in murine glioblastoma: immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5, J. Neuroimmunol. (2012) 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sc R, Ka S, Jl W, Rg T, Ae P, Fr B, A chemokine receptor antagonist inhibits experimental breast tumor growth, Canc. Res. (2003) 63. [PubMed] [Google Scholar]
- [64].G S, A B-B, The inflammatory chemokines CCL2 and CCL5 in breast cancer, Canc. Lett. (2008) 267. [DOI] [PubMed] [Google Scholar]
- [65].Rg F, Ai M, A B, Organizing the cell cortex: the role of ERM proteins, Nat. Rev. Mol. Cell Biol. 11 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Jm A, Ac G, A A, R S, Jm B, L B, F D, D B, Z M, C F, Hl G, Ja P, Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer, Sci. Rep. 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Q C, J M, Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis, Clin. Canc. Res.: an official journal of the American Association for Cancer Research 18 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].S A, J K, Kd P, Intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and regulated on activation normal T cell expressed and secreted are expressed by human breast carcinoma cells and support eosinophil adhesion and activation, Am. J. Pathol. 157 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].B Q, Y D, Jh I, Rj M, Y Z, J L, Ra L, Jw P, A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth, PloS One (2009) 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bz Q, J L, H Z, T K, J Z, Lr C, Ea K, la S, Jw P, CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis, Nature 475 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Jankowski M BG, Lawson J, Skoczyński S, Zejda JE, E-smoking: emerging public health problem, Int. J. Occup. Med. Environ. Health 30 (2017) 329–344. [DOI] [PubMed] [Google Scholar]
- [72].Erythropel HC, Jabba SV, DeWinter TM, Mendizabal M, Anastas PT, Jordt SE, Zimmerman JB, Formation of flavorant–propylene glycol adducts with novel toxicological properties in chemically unstable E-cigarette liquids, Nicotine Tob. Res. 21 (2019) 1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rp J, Rm S, Dh P, Solvent chemistry in the electronic cigarette reaction vessel, Sci. Rep. 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hs Y, Yj L, Za J, Sy L, By G, T W, Nicotine-induced ICAM-1 and VCAM-1 expression in mouse cardiac vascular endothelial cell via p38 MAPK signaling pathway, Analytical and quantitative cytopathology and histopathology (2014) 36. [PubMed] [Google Scholar]
- [75].Bradford ST, Stamatovic SM, Dondeti RS, Keep RF, Andjelkovic AV, Nicotine aggravates the brain postischemic inflammatory response, Am. J. Physiol. Heart Circ. Physiol. 300 (2011) H1518–H1529. [DOI] [PMC free article] [PubMed] [Google Scholar]