

Abstract
Vitamin D exerts anti-cancer effects in recent clinical trials and preclinical models. The actions of vitamin D are primarily mediated through its hormonal form, 1,25-dihydroxyvitamin D (1,25(OH)2D). Previous literature describing in vitro studies has predominantly focused on the anti-tumourigenic effects of the hormone, such as proliferation and apoptosis. However, recent evidence has identified 1,25(OH)2D as a regulator of energy metabolism in cancer cells, where requirements for specific energy sources at different stages of progression are dramatically altered. The literature suggests that 1,25(OH)2D regulates energy metabolism, including glucose, glutamine and lipid metabolism during cancer progression, as well as oxidative stress protection, as it is closely associated with energy metabolism. Mechanisms involved in energy metabolism regulation are an emerging area in which vitamin D may inhibit multiple stages of cancer progression.
Keywords: cancer, glucose, glutamine, lipids, metabolism, oxidative Stress, Vitamin D
1 |. INTRODUCTION
It is estimated that over 1.8 million new cancer cases will be diagnosed in the United States in 2021 (Siegel et al., 2021). In addition, cancer is the second-leading cause of death in the United States, with the majority of cancer-related deaths due to metastatic disease (Chaffer & Weinberg, 2011; Dillekås et al., 2019). These data highlight the critical need for the prevention of not only disease development but also cancer progression through metastasis. Because metastatic disease is the primary cause of mortality in cancer patients, identifying compounds, such as vitamin D, to prevent progression through metastasis has the potential to improve the overall survival and quality of life of cancer patients.
Mounting epidemiological evidence suggests that vitamin D may prevent cancer and its progression. For example, epidemiological studies demonstrate an inverse association between incidence of cancers and ultraviolet B (UVB) exposure (Garland et al., 2006; Garland & Garland, 1980; Hanchette & Schwartz, 1992). Because of the body’s ability to obtain vitamin D not only through the diet but also through sunlight exposure, these studies led to the hypothesis that UVB exposure decreases cancer risk through the production of vitamin D. In support of this hypothesis, increased UVB exposure is associated with increased vitamin D status (Grant, 2018) and higher vitamin D status is associated with decreased cancer risk (Garland et al., 2006). In addition, a recent meta-analysis identified a significant linear dose-dependent relationship between vitamin D status and overall survival in breast cancer patients (Hu et al., 2018). Since the majority of cancer-related deaths are the consequence of metastatic disease, these results suggest that vitamin D may also prevent metastasis.
Results from animal models assessing the effect of vitamin D also support a chemopreventive role of vitamin D. Preclinical rodent models, for example, indicate that vitamin D metabolites, analogues or dietary vitamin D alone or in combination with other compounds inhibit tumour formation, tumour volume, cancer cell proliferation and reduce overall tumour burden (Feldman et al., 2014; Krishnan et al., 2010). Furthermore, preclinical studies demonstrate that vitamin D significantly reduces cancer metastasis (Horas et al., 2019; Y. Zhang et al., 2014) and, conversely, that vitamin D deficiency increases tumour growth and metastasis (Williams et al., 2016). Collectively, these results suggest that vitamin D may not only reduce cancer initiation but also improve cancer prognosis in animal models.
It is important to determine the vitamin D status, acquired through diet, supplements or UVB exposure, needed to achieve anti-cancer effects in order to translate research to recommendations. In the United States, the current recommended daily allowance for vitamin D is 600 IU day−1 for individuals aged 1–70 years, which is sufficient to maintain adequate bone health among healthy people (Kimball & Holick, 2020; Newberry et al., 2014). Other evidence suggests that higher daily intakes of vitamin D (up to 2,000 IU day−1 for normal-weight individuals) may prevent the development of chronic diseases, including cancer, without adverse effects on calcium homeostasis (Kimball & Holick, 2020; Shirvani et al., 2020). In addition, higher daily intake of vitamin D may be required to increase the vitamin D status for individuals with obesity (body mass index [BMI] > 30). Obesity is associated with lower serum vitamin D status compared to those with lower BMIs, which is purported to be due to the sequestering of vitamin D within the adipose tissue (Migliaccio et al., 2019). In addition to obesity, several other factors including increasing age, darker skin colour and reduced sunlight exposure negatively affect the vitamin D status and may increase the dietary vitamin D requirements at an individual level. Therefore, it is important to consider not only intake levels but also other influential factors, to achieve an appropriate vitamin D status that may prevent cancer progression.
Clinical trials examining the effect of dietary vitamin D interventions on cancer incidence and progression have shown mixed results. The Women’s Health Initiative Study, which provided participants with a relatively low dose (400 IU vitamin D per day) for an average of 7 years, demonstrated no effect of supplementation on colorectal cancer incidence (Prentice et al., 2013). Other trials testing a wide range of vitamin D doses have similarly shown null effects on total cancer incidence (Grasset et al., 2012; Jorde et al., 2016; Scragg et al., 2018). In contrast, a higher dose (1,100 IU day−1 of vitamin D in combination with 1,400–1,500 mg day−1 of calcium) significantly decreased the total cancer incidence in healthy postmenopausal women when the analyses excluded the first year of follow-up (Lappe et al., 2007). In this study, no significant effect on cancer incidence was observed in the calcium-alone group, suggesting that vitamin D was critical for the reduction in cancer incidence. The recent Vitamin D and Omega-3 Trial (VITAL) study, which assessed the effect of 2,000 IU vitamin D per day on cancer outcomes, reported no effect of supplementation on cancer incidence 5 years after follow-up (Manson et al., 2019). Interestingly, the supplementation reduced cancer deaths by 25% when the first 2 years of the trial were excluded from the analysis (Manson et al., 2019), suggesting that vitamin D reduces cancer progression. Additionally, a secondary analysis of the VITAL study demonstrated that vitamin D supplementation reduced the incidence of metastatic or fatal cancer in normal-weight participants only, whereas there was no effect in participants with overweight or obesity (Chandler et al., 2020). These results highlight the heterogeneous response to vitamin D supplementation and necessitate further research on doses required to achieve anti-cancer effects of vitamin D in a diverse population. Finally, results of a recent meta-analysis show that vitamin D supplementation had no effect on cancer incidence but significantly reduced cancer mortality (Keum et al., 2019). Collectively, the results from epidemiological, preclinical and clinical studies support a role for vitamin D in preventing cancer progression.
2 |. VITAMIN D METABOLISM
Vitamin D must be metabolized in the body to produce its most active metabolite, 1,25-dihydroxyvitamin D (1,25(OH)2D) (Figure 1). Vitamin D is obtained from dietary sources as well as through cutaneous synthesis in the skin from its precursor, 7-dehydrocholesterol, following UVB exposure (Holick et al., 2007; Webb et al., 1989). The prohormone vitamin D, whether from the diet or skin, is hydroxylated in the liver to 25-hydroxyvitamin D (25(OH)D/calciferol) by the enzyme 25-hydroxylase. Because of the long half-life (15 days) of 25(OH)D and the lack of regulation of the activity of the hepatic enzyme 25-hydroxylase, 25(OH)D blood concentration serves as an indicator of vitamin D status.
FIGURE 1.

Overview of vitamin D metabolism and transcriptional effects within cancer cells. The most active vitamin D metabolite, 1,25 (OH)2D, is synthesized in the body following two hydroxylation events. This metabolite binds the vitamin D receptor to induce heterodimerization to retinoid X receptor, which subsequently binds to the vitamin D response element within the promoter region of vitamin D target genes. A number of different gene families are regulated by 1,25(OH)2D, including those involved in apoptosis and cell cycle arrest, as well as cell metabolism. Generally, 1,25(OH)2D downregulates a variety of genes involved in glucose, glutamine and lipid metabolism, as well as oxidative stress protection. Additional studies are needed (indicated by “?”) to connect these mechanisms to cancer-related outcomes involved in cancer progression. Abbreviations: 25(OH)D-25—hydroxyvitamin D; 1,25(OH)2D-1α,25—dihydroxyvitamin D; VDR—vitamin D receptor; RXR—retinoid X receptor; VDRE—vitamin D response element
Circulating 25(OH)D is further hydroxylated in renal cells by 1α-hydroxylase to form the active metabolite and hormonal form, 1,25(OH)2D (Figure 1). In the kidney, 1α-hydroxylase expression is highly regulated by factors that control serum calcium levels. For example, parathyroid hormone (PTH) increases 1α-hydroxylase activity and FGF-23 decreases its activity (Murayama et al., 1998). Importantly, 1α-hydroxylase is also expressed in other tissues, including cancer cells, where it is differentially regulated compared to the renal form (Bikle, 2009; Friedrich et al., 2006; Li et al., 2008; Zehnder et al., 2001). Expression of 1α-hydroxylase in extrarenal tissue permits the local production of 1,25(OH)2D, which may exert direct action within the host cell (Bikle, 2014) (Figure 1). Therefore, high 25(OH)D levels may lead to higher local intracellular levels of the active metabolite, 1,25(OH)2D. Levels of 1,25(OH)2D are maintained at appropriate physiological levels by 24-hydroxylase-mediated hydroxylation, which targets the metabolite for excretion. In certain cancer types, evidence suggests that expression of 24-hydroxylase, 1α-hydroxylase and the vitamin D receptor (NR1I1) are reduced compared to untransformed tissue, which may suggest that there is reduced availability and action of the active form of vitamin D in these cancers (Francis et al., 2019; Slominski et al., 2018). Nevertheless, results from preclinical and clinical trials demonstrate vitamin D is effective in preventing cancer, including metastasis, which suggests sufficient 1,25(OH)2D action remains to mediate anti-cancer effects despite the downregulation of components of vitamin D metabolism.
The hormone 1,25(OH)2D regulates cellular processes by two mechanisms. First, similar to other steroid hormones, 1,25(OH)2D regulates gene expression by binding to its cognate steroid hormone receptor, the vitamin D receptor. The vitamin D receptor subsequently heterodimerizes with its binding partner, retinoid X receptor (RXR/NR2B1–3), which associates with vitamin D response elements in the promoter region of target genes (Deeb et al., 2007; Jin & Pike, 1996; Kerner et al., 1989) (Figure 1). Vitamin D response elements consist of two direct repeat sequences with three spacer nucleotides, although there is significant variation in the sequence (Carlberg & Seuter, 2009; Christakos et al., 2016). The vitamin D receptor/retinoid X receptor liganded complex binds with high affinity to the vitamin D response element, recruits the transcriptional co-activators steroid receptor activator 1, 2 and 3, which have histone acetylase activity and initiates the regulation of gene transcription (Figure 1). Binding of the vitamin D receptor/retinoid X receptor complex to vitamin D response elements induces or suppresses transcription of a wide variety of target genes involved in several cellular processes. In addition to transcriptional regulation by 1,25 (OH)2D, evidence also suggests that 1,25(OH)2D or the vitamin D receptor regulate intracellular pathways and mitochondrial function through nongenomic mechanisms (Blajszczak & Nonn, 2019; Hii & Ferrante, 2016; Nemere et al., 2012; Norman, 1998; Zmijewski & Carlberg, 2020). Notably, the vitamin D receptor and other proteins involved in vitamin D metabolism localize to the mitochondria in untransformed and transformed models, suggesting a potential direct regulation of mitochondrial processes (Ricca et al., 2018; Silvagno et al., 2010). In addition, the genetic depletion of vitamin D receptor in the absence of 1,25(OH)2D reduced the components of the mitochondrial respiratory chain and increased the mitochondrial membrane potential, supporting the connection between vitamin D, mitochondrial function and energy metabolism (Consiglio et al., 2014). Thus, 1,25(OH)2D regulates cell function via both transcriptional regulation and nongenomic pathways.
Classical pathways regulated by 1,25(OH)2D signalling include those involved in calcium and phosphorous homeostasis (Holick, 1996); however, 1,25(OH)2D has also been identified as a promising anti-cancer and anti-metastatic agent (El-Sharkawy & Malki, 2020). Several studies have assessed the effect of 1,25(OH)2D or its analogues, which have traditionally been used to mitigate hypercalcaemic effects of 1,25(OH)2D, using microarray analyses, and have identified over 1,000 vitamin D target genes involved in a wide variety of cell functions (Carlberg & Muñoz, 2020; Feldman et al., 2014; Fleet et al., 2012; Maestro et al., 2019; Mazzaferro et al., 2014; Slominski et al., 2020; Welsh, 2017). Interestingly, some vitamin D target genes identified in untransformed models are critical to cell metabolism, such as the fructose-bisphosphatase 1 gene (PFKFB3) identified from a microarray analysis in human monocytes (Nurminen et al., 2019). In transformed models, microarray datasets measuring the effects of 1,25(OH)2D have reported significant expression changes within a wide variety of gene families (Krishnan et al., 2004; Milani et al., 2013; Shan et al., 2020; Swami et al., 2003; Vanoirbeek et al., 2009) such as apoptosis (X. Zhang et al., 2005) and proliferation (Guzey et al., 2004). However, the effects of 1,25(OH)2D on metabolism-related genes were not highlighted in these reports. Nonetheless, numerous studies have identified a 1,25(OH)2D-mediated regulation of individual genes involved in cell metabolism.
The growth of cancer cells in the primary tumour and progression of cancer to metastatic disease require both metabolic plasticity and metabolic flexibility in order to respond to microenvironmental changes (Bergers & Fendt, 2021; McGuirk et al., 2020; Sullivan et al., 2019). Thus, metastatic breast cancer cells demonstrate metabolic plasticity, or the ability to utilize metabolic substrates to drive a multitude of cellular processes, through differential use of the metabolite pyruvate. For example, breast cancer cells that preferentially metastasize to the lung or bone metabolized pyruvate through the tricarboxylic acid (TCA) cycle, whereas breast cancer cells that had metastasized to the liver preferentially metabolize pyruvate to lactate (Döppler & Storz, 2015). These observations suggest that breast cancer cells that differentially regulate genes involved in pyruvate metabolism, thus conferring metabolic plasticity, may be more successful at site-specific metastatic outgrowth. In addition, cancer cells often display metabolic flexibility, or the ability to utilize different nutrients for energy production or anabolic reactions depending on the cellular context, thereby reducing dependence on single nutrients and improving survival in variable nutrient environments. For example, glioblastoma cells and cells that metastasize to the brain display increased utilization of acetate, since glucose is preferentially consumed by resident glial cells (Mashimo et al., 2014). Thus, metabolic plasticity and flexibility are critical mechanisms by which cancer cells are able to survive in changing environmental circumstances throughout progression.
Previous research suggests that 1,25(OH)2D regulates genes involved in cell metabolism in both untransformed and transformed models. For example, vitamin D receptor knockout mice are resistant to high-fat diet-induced weight gain, demonstrating that vitamin D is a crucial regulator of whole-body energy metabolism (Fraser, 2015). The mechanism underlying the resistance to obesity is suggested to be increased energy expenditure, as the vitamin D receptor knockout mice exhibit increased fatty acid oxidation and uncoupling of ATP formation in white adipose tissue (Narvaez et al., 2009). The vitamin D regulation of whole-body energy metabolism may be evolutionarily advantageous in colder climates with reduced UVB exposure, where vitamin D deficiency may have led to increased energy release as heat as opposed to storing fatty acids (Fraser, 2015). In addition, the vitamin D-mediated regulation of energy metabolism may be advantageous directly in skin cells. While UVB exposure causes DNA damage in skin cells (Jagoda & Dixon, 2020), this damage is countered by vitamin D-mediated increases in glycolysis and energy production, which result in an improved cellular ability to repair damaged DNA (Rybchyn et al., 2018). These results suggest an evolutionary advantage to cutaneous synthesis and the regulation of gene activity by vitamin D in response to UVB exposure as well as adaptations to colder climates.
In addition, vitamin D and its metabolite 1,25(OH)2D have been proposed to regulate glucose, glutamine and lipid metabolism, all of which are increased at different stages of cancer progression and contribute to overall metabolic plasticity. Modulation of energy metabolizing pathways in cancer cells by 1,25(OH)2D may reduce viability when the cells are metabolically stressed or in nutrient-depleted environments, providing a mechanism by which vitamin D inhibits progression through the stages of carcinogenesis. This review highlights the current state of the evidence for the regulation of glucose, glutamine and lipid metabolism, as well as oxidative stress, by 1,25(OH)2D in cancer cells and emphasizes the need for future studies to investigate the mechanistic link between the regulation of energy metabolism and the chemopreventive effects of vitamin D.
3 |. VITAMIN D REGULATION OF GLUCOSE METABOLISM
Altered glucose energy metabolism is a common characteristic in cancer cells (Ghanavat et al., 2021). Otto Warburg was the first to observe that cancer cells have altered glucose metabolism (Warburg et al., 1927). In this phenomenon, termed the “Warburg effect”, cancer cells increased glucose uptake and glycolysis, and decreased glucose metabolism via oxidative phosphorylation, even in the presence of oxygen (Liberti & Locasale, 2016) (Figure 2). Although not all cancer cells display the Warburg effect, most cancer cells have altered glucose metabolism to support growth (Bose & Le, 2018). For example, increased flux of glucose through glycolysis increases the production of carbon-based intermediates which may be used for biosynthetic reactions to support growth, as well as the production of lactate which may be an important fuel source for cancer-associated fibroblasts or cancer cells in select contexts (Sazeides & Le, 2018; Wilde et al., 2017). Additionally, glucose provides substrates for the pentose phosphate pathway which results in increased synthesis of nucleic acids and reducing equivalents through NADPH production, both of which are required to sustain the rapid cell proliferation observed in cancer (Scheel & Weinberg, 2012). Current therapies targeting glucose metabolism, while promising as anti-cancer agents, pose serious concerns for their off-target effects demonstrated in clinical trials (Abdel-Wahab et al., 2019). Given the prevalence of altered glucose metabolism in cancer and its critical contribution to cell proliferation, the identification of agents that target this characteristic with minimal effects on benign tissue, such as vitamin D, may provide a promising strategy for preventing cancer and its progression.
FIGURE 2.
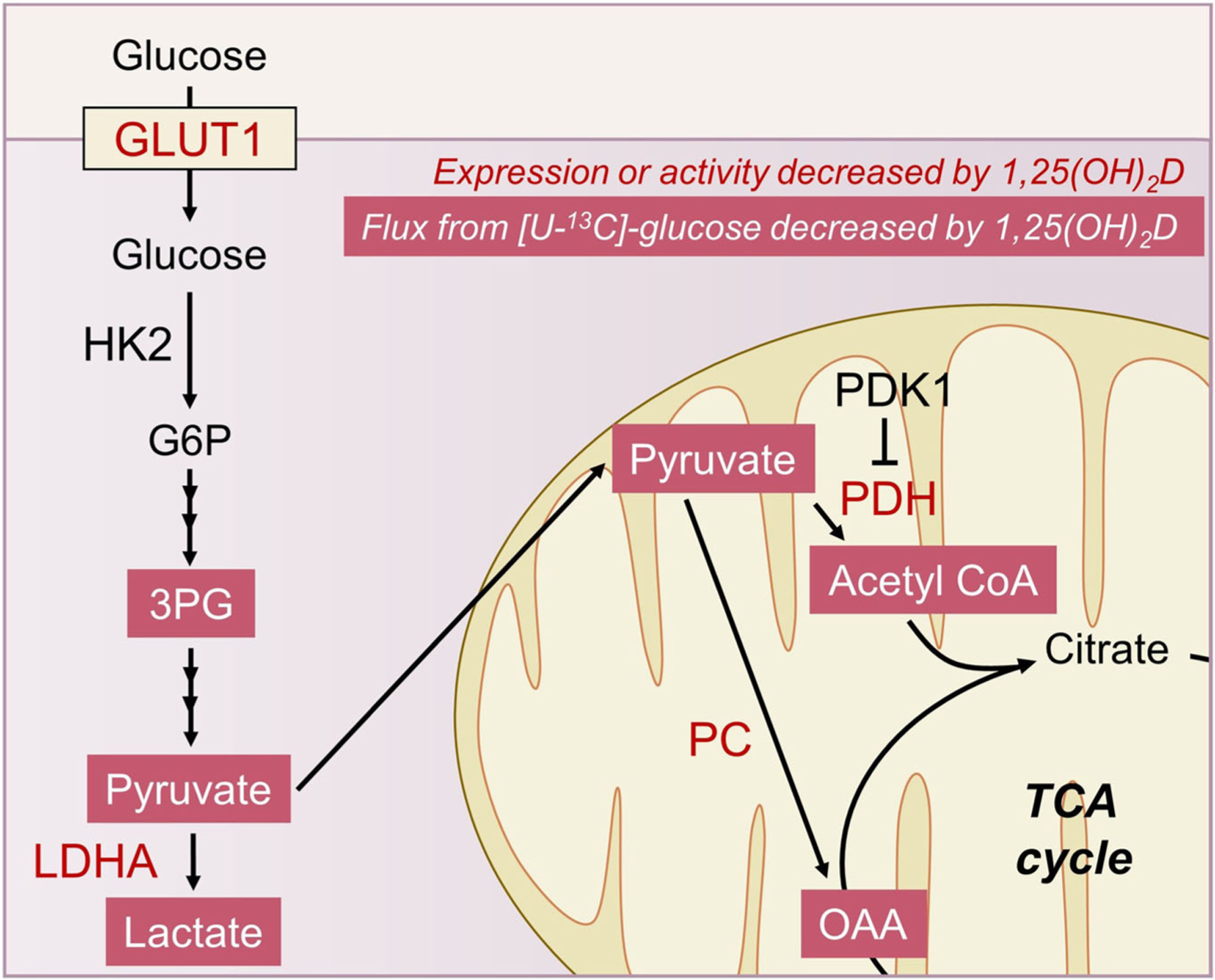
Decreased glucose metabolism mediated by 1,25(OH)2D. An overall reduction in glycolysis and glucose entry into the TCA cycle is observed in cancer cells treated with the active metabolite of vitamin D, 1,25 (OH)2D. Treatment of cancer cells in vitro with 1,25(OH)2D results in reduced enzyme expression or activity (red font) and [U-13C]-glucose flux to metabolites (red shaded boxes). Abbreviations: 1,25 (OH)2D-1,25—dihydroxyvitamin D; [U-13C]—universally labelled carbon 13; GLUT1—glucose transporter 1; HK2—hexokinase 2; G6P—glucose-6-phosphate; 3PG—3-phosphoglycerate; LDHA—lactate dehydrogenase A; PDK1—pyruvate dehydrogenase kinase 1; PDH—pyruvate dehydrogenase; PC—pyruvate carboxylase; OAA—oxaloacetate; TCA—tricarboxylic acid
One potential mechanism by which vitamin D may inhibit cancer progression is through inhibition or reversal of altered glucose metabolism, including reducing glucose uptake into cancer cells. Glucose enters cells via different isoforms of the glucose transporter (GLUT1–7) and increased expression of GLUT1 in breast cancer is associated with a worse prognosis (Kang et al., 2002). In agreement with this, GLUT1 expression is associated with increased invasiveness in various human breast cancer cell lines, while the expression of GLUT2–5 is not associated with invasive capabilities (Grover-McKay et al., 1998). Treatment with 1,25(OH)2D decreased GLUT1 mRNA and protein expression in cell models of prostate cancer (Abu El Maaty et al., 2017). Additionally, 1,25(OH)2D decreased GLUT1 mRNA and protein expression and glucose uptake in MCF-7 and MDA-MB-231 breast cancer cells (Abu El Maaty et al., 2018; Santos et al., 2018). However, the impact of 1,25(OH)2D on GLUT1 expression may be dependent on stage or type of cancer, as 1,25(OH)2D treatment decreased glucose uptake without affecting GLUT1 mRNA levels in an in vitro model of early-stage breast cancer (Zheng et al., 2013). Further, in 1-methyl-1-nitrosourea-induced breast cancer in Sprague–Dawley rats, supplementation with vitamin D or the vitamin D analogue seocalcitol reduced glucose uptake in tumour tissue, measured using flouro-2-deoxy-glucose (Macejová et al., 2011). Taken together, these results suggest that 1,25(OH)2D suppresses glucose uptake in models of prostate and breast cancer.
In addition to glucose uptake, 1,25(OH)2D also regulates glycolysis in in vitro cancer models, although the degree and direction of regulation varies between models. For example, 1,25(OH)2D decreased protein levels of hexokinase 2, the enzyme responsible for the first step of glycolysis, in MCF-7 breast cancer cells, but increased hexokinase 2 protein expression in MDA-MB-231 cells (Abu El Maaty et al., 2018) (Figure 2). Treatment with 1,25(OH)2D suppressed mRNA levels of hexokinase 2 in both cell lines (Santos et al., 2018). In addition, 1,25(OH)2D decreased the expression of lactate dehydrogenase, the enzyme that reduces pyruvate to lactate, in breast cancer and prostate cancer cells (Abu El Maaty et al., 2017; Santos et al., 2018). In nonmetastatic breast cancer cells, 1,25(OH)2D had no effect on mRNA expression of the glycolytic enzymes hexokinase 2 and phosphoglycerate kinase 1 but decreased glycolysis, lactate production and flux of universally labelled 13C-glucose into 3-phosphoglycerate (3PG), pyruvate and lactate (Zheng et al., 2013). Additionally, 1,25 (OH)2D suppressed glycolysis as measured by a biosensor chip system, which uses extracellular acidification as an indicator of glycolysis (Abu El Maaty et al., 2017). Decreased acidification was detected over time to a greater extent in less progressed cell lines versus highly metastatic cells, suggesting decreased glycolysis (Abu El Maaty et al., 2017). In contrast, 1,25(OH)2D treatment did not affect lactate production in MCF-7 cells, while a high 1 μM dose of 1,25(OH)2D reduced the lactate production in the MDA-MB-231 cells (Santos et al., 2018). Together, these results demonstrate that 1,25(OH)2D targets glucose metabolism via inhibition of glycolysis; however, the extent of this regulation may depend on the cancer model.
In addition to glycolysis, 1,25(OH)2D is also proposed to regulate the flux of glucose into the TCA cycle (Figure 2). Pyruvate, the end-product of glycolysis, enters the TCA cycle as acetyl-CoA through the activity of pyruvate dehydrogenase and oxaloacetate via pyruvate carboxylase activity. Treatment with 1,25(OH)2D decreased mRNA abundance and protein expression of the negative regulator of pyruvate dehydrogenase, pyruvate dehydrogenase kinase 1, in various stages of prostate cancer (Abu El Maaty et al., 2017), suggesting that 1,25(OH)2D may promote flux of pyruvate into the TCA cycle. However, in MCF10A-ras transfected breast cancer cells, 1,25(OH)2D treatment reduced the pyruvate dehydrogenase activity although, in contrast, had no effect on pyruvate dehydrogenase kinase 1 mRNA expression (Zheng et al., 2013). Additionally, treatment with 1,25 (OH)2D reduced pyruvate carboxylase expression in MCF10A-ras transfected breast cancer cells, through the direct regulation of a vitamin D response element in the promoter of the pyruvate carboxylase gene (Wilmanski, Buhman, et al., 2017; Wilmanski, Zhou, et al., 2017). In agreement with these results, 1,25(OH)2D reduced flux from universally labelled 13C-glucose to acetyl-CoA and oxaloacetate and decreased the pool size of succinate, a TCA cycle intermediate, demonstrating that 1,25(OH)2D reduces glucose incorporation into the TCA cycle (Zheng et al., 2013). Following pyruvate entry into the TCA cycle, acetyl-CoA and oxaloacetate condense into citrate. Replenishing the citrate pool provides substrate for energy metabolism through the production of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) in the TCA cycle, as well as fueling fatty acid synthesis, both of which are utilized by cancer cells. MCF-7 breast cancer cells had reduced levels of citrate and downstream TCA cycle intermediates when treated with 1,25 (OH)2D, while 1,25(OH)2D-treated MDA-MB-231 breast cancer cells had increased citrate levels (Abu El Maaty et al., 2018). Further, 1,25 (OH)2D treatment of prostate cancer cells decreased mRNA expression of the TCA cycle enzyme isocitrate dehydrogenase 1 (IDH1), which converts isocitrate to α-ketoglutarate (Abu El Maaty et al., 2017), a metabolite which is pivotal in supporting both oxidative and reductive metabolism in cancer (Abla et al., 2020). Collectively, these results suggest 1,25(OH)2D treatment decreases glucose utilization in the TCA cycle in cancer cells.
Oxidative phosphorylation is used by cells to produce ATP from the NADH and FAD generated in the TCA cycle. In the context of cancer, oxidative phosphorylation is utilized in oxygenated environments including breast cancer and Hodgkin lymphoma (Ashton et al., 2018; Birkenmeier et al., 2016; Whitaker-Menezes et al., 2011). Evidence suggests that the effects of vitamin D on oxidative phosphorylation may also have differential effects depending on stage and type of cancer. For example, 1,25(OH)2D decreased oxygen consumption rate, a measurement of oxidative phosphorylation in both nonmetastatic and metastatic prostate cancer cell lines, although the metastatic cells were more resistant to 1,25(OH)2D-mediated decreases compared to nonmetastatic cells (Abu El Maaty et al., 2017). However, the opposite effect was observed in breast cancer cells, as 1,25(OH)2D had no effect on oxygen consumption rate in nonmetastatic cells, but decreased oxygen consumption rate of metastatic cells (Abu El Maaty et al., 2018). Further investigation into understanding the conditions that influence the regulation of oxidative phosphorylation by 1,25(OH)2D is necessary in order to develop strategies for the use of vitamin D as a means of cancer prevention or treatment through targeting oxidative phosphorylation.
With increasing evidence of vitamin D’s negative regulation of glucose metabolism, investigations have focused on elucidating the mechanism underlying this effect. Evidence shows that 1,25(OH)2D increases the activation of adenosine monophosphate-activated protein kinase (AMPK) in breast cancer cells (Abu El Maaty et al., 2018; Santos et al., 2018). AMPK activity is regulated by the energy status of the cell and is increased in energy-deplete conditions. Phosphorylation targets of AMPK are proteins involved in increasing the bioenergetic status of the cell (Cork et al., 2018). For example, one AMPK target is the mammalian target of rapamycin (mTOR) pathway where AMPK activity results in inhibition of mTOR activity through direct and indirect mechanisms, thus suppressing anabolic reactions and conserving energy in the cell (Luo et al., 2010). Indeed, 1,25(OH)2D treatment in breast cancer cells increased levels of the active form of AMPK and thereby decreased levels of the active form of mTOR (Santos et al., 2018). In addition, in an in vitro colorectal cancer model, 1,25(OH)2D treatment reduced glycolysis and lactate production through induction of the long noncoding RNA (lncRNA) maternally expressed gene 3 (Zuo et al., 2020). Further, maternally expressed gene 3 and 25(OH)D serum levels were positively correlated in cancer patients, suggesting that 1,25(OH)2D may increase the expression of maternally expressed gene 3, which then may exert its effects to suppress glucose metabolism. Thus, the regulation of glycolysis by 1,25(OH)2D may be through the regulation of key players in energy metabolism such as AMPK/mTOR signalling or by increasing the expression of maternally expressed gene 3 lncRNA.
Collectively, evidence suggests that 1,25(OH)2D regulates glycolysis, the TCA cycle and oxidative phosphorylation, with differential responses that may be dependent on cancer type and stage of progression (Figure 2). In some types of cancer, including breast and prostate cancer, the regulation of glucose metabolism by 1,25(OH)2D is most prominent in nonmetastatic cancer cells with lesser effects in metastatic cells (Abu El Maaty et al., 2017; Abu El Maaty et al., 2018), suggesting that 1,25(OH)2D may play an important role in regulating glucose metabolism in early stage tumours. However, models of breast cancer show that 1,25(OH)2D treatment suppresses oxidative phosphorylation to a greater extent in metastatic cells with less regulation in the TCA cycle (Abu El Maaty et al., 2018). This is particularly interesting as the TCA cycle synthesizes the substrates used in oxidative phosphorylation. Further investigation into this differential response may lead to better application of vitamin D as a chemopreventive agent in the treatment of metastatic cancer. In addition, future research addressing the effect of 1,25(OH)2D-mediated changes in glucose metabolism as it relates to cellular processes associated with metastatic progression is needed to provide mechanistic links between 1,25(OH)2D treatment, alterations in glucose metabolism and metastasis.
4 |. VITAMIN D REGULATION OF GLUTAMINE METABOLISM
Increased dependence on glutamine metabolism is commonly observed in cancer cells and in certain circumstances is required for cancer cell survival (Cluntun et al., 2017; Hensley et al., 2013). Tumour cells use glutamine for several purposes including energy production via entry of carbon into the TCA cycle (Figure 3), oxidative stress protection through the production of the anti-oxidant GSH and the synthesis of nonessential amino acids such as alanine, serine, aspartate and glycine (Hensley et al., 2013). Given the importance of glutamine metabolism for cell proliferation, identifying strategies to inhibit glutamine metabolism to suppress tumour growth is a promising area of research. Interestingly, 1,25(OH)2D treatment is demonstrated to regulate glutamine metabolism in in vitro models of breast cancer (Abu El Maaty et al., 2017; Beaudin & Welsh, 2017; Zhou et al., 2016) through several mechanisms.
FIGURE 3.

Decreased glutamine metabolism mediated by 1,25 (OH)2D. A decrease in glutamine metabolism and replenishment of the TCA cycle by glutamine is observed in 1,25(OH)2D-treated cancer cells. Treatment of cancer cells in vitro with 1,25(OH)2D reduces enzyme expression or activity (red font) and [U-13C]-glutamine flux to metabolites (red shaded boxes). Abbreviations: 1,25 (OH)2D-1,25—dihydroxyvitamin D; SLC1A5—solute carrier family 1 member 5; GLS—glutaminase; GLUL—glutamate ammonia ligase/glutamine synthetase; αKG-α—ketoglutarate; TCA—tricarboxylic acid
First, evidence suggests that treatment with 1,25(OH)2D suppresses glutamine uptake in cancer cells (Figure 3). For example, following 1,25(OH)2D treatment, expression of the major glutamine transporter solute carrier family 1 member 5 (SLC1A5/ASCT2) is decreased at the mRNA and protein level in breast cancer cells (Zhou et al., 2016). A vitamin D response element was identified in the promoter of the SLC1A5 gene and verified using site-directed mutagenesis, suggesting that 1,25(OH)2D downregulates SLC1A5 directly (Zhou et al., 2016). In agreement with these results, glutamine uptake was decreased by 1,25(OH)2D treatment, with no further decrease in uptake observed with the combination of 1,25 (OH)2D treatment and the SLC1A5 inhibitor L-γ-glutamyl-p-nitroanilide. In contrast, SLC1A5 mRNA and protein expression were unaffected by 1,25(OH)2D treatment in untransformed breast epithelial cells (Beaudin & Welsh, 2017; Zhou et al., 2016), suggesting that the 1,25(OH)2D-mediated regulation of glutamine uptake may occur specifically in cancer cells. This is particularly interesting, as 1,25(OH)2D’s regulation of SLC1A5 in cancer cells may have broader effects on cell metabolism apart from directly inhibiting glutamine uptake. For example, glutamine uptake is linked to the regulation of mTOR signalling, where inhibiting glutamine uptake prevents mTOR activation (Yoshida, 2021). Given this connection, the 1,25(OH)2D-mediated downregulation of SLC1A5 may also lead to reduced mTOR signalling and therefore a further reduction of cell growth. These data support that 1,25(OH)2D may specifically target cancer cells to reduce glutamine, with minimal effects on this process in untransformed cells.
Evidence also supports that 1,25(OH)2D regulates intracellular glutamine metabolism (Figure 3). After uptake into the cell, glutamine is metabolized by glutaminase to glutamate, which can be used for protein synthesis or metabolized into α-ketoglutarate and enter the TCA cycle. In in vitro models of breast and prostate cancer, glutaminase is suppressed by 1,25(OH)2D treatment (Abu El Maaty et al., 2017; Beaudin & Welsh, 2017; Narvaez et al., 2020). In accord with this, several studies show that 1,25(OH)2D inhibits conversion of glutamine to its downstream metabolites, including glutamate and methionine (Saracligil et al., 2017; Zhou et al., 2016). As a result, glutamine oxidation is reduced following 1,25(OH)2D treatment in breast cancer cells (Beaudin & Welsh, 2017), suggesting decreased utilization of glutamine for energy production via oxidative phosphorylation in 1,25(OH)2D-treated cells. These results indicate that 1,25 (OH)2D treatment reduces glutamine metabolism into glutamate and subsequent utilization in the TCA cycle.
Synthesis of glutamine is also decreased by 1,25(OH)2D in cancer cell models (Figure 3). Synthesis of glutamine from glutamate is mediated by glutamine synthetase, and glutamine synthetase expression in cancer cells is associated with increased metabolic flexibility, as it supports glutamine independence (Hensley et al., 2013). For example, breast cancer cells that synthesize glutamine through glutamine synthetase are less sensitive to glutamine deprivation (Beaudin & Welsh, 2017). Interestingly, 1,25(OH)2D treatment decreased glutamine synthetase mRNA and protein expression in these cell models, with a subsequent decrease in glutamine synthetase activity (Beaudin & Welsh, 2017). In agreement with these results, 1,25(OH)2D-mediated inhibition of glutamine synthetase causes increased dependence on extracellular glutamine (Beaudin & Welsh, 2017). These results suggest that inhibition of glutamine synthesis by 1,25(OH)2D may increase the cellular requirement for extracellular glutamine and result in inhibition of cancer progression.
The regulation of glutamine metabolism in cancer cells by 1,25 (OH)2D may be of particular interest as a chemopreventive compound and in therapeutic applications, particularly due to the differential effect of 1,25(OH)2D across cancer models. For example, 1,25(OH)2D treatment had little effect on glutamine metabolism in untransformed cells, with more prominent effects observed in transformed MCF10A-ras transfected breast cancer cells (Zhou et al., 2016), suggesting that 1,25(OH)2D may alter glutamine metabolism with minimal cytotoxic effects on normal tissue. Treatment with 1,25(OH)2D suppressed glutamine uptake as well as glutamine synthesis (Figure 3), suggesting that 1,25(OH)2D’s role in targeting glutamine metabolism may be clinically relevant particularly in cancers that display glutamine independence, such as glioblastoma and non-small-cell lung cancer (Cluntun et al., 2017). This specificity emphasizes the potential for glutamine metabolism inhibitors such as 1,25(OH)2D to increase cancer cell death in nutrient restricted environments, thereby inhibiting cancer progression. Further research is needed to determine the impact of 1,25(OH)2D’s regulation of glutamine metabolism on specific outcomes in cancer progression, such as migration or invasion, in order to determine the mechanisms underlying its anti-cancer effects.
5 |. VITAMIN D REGULATION OF LIPID METABOLISM
Cancer cells have dysregulated lipid metabolism, including abnormal accumulation of neutral lipids in cytoplasmic lipid droplets. Notably, lipid-rich tumours are linked to more aggressive phenotypes and poorer clinical outcomes in cancer patients and in animal models (Abourmad et al., 1963; Ramos & Taylor, 1974). In accordance with this abnormal lipid accumulation, evidence supports that cancer cells have increased the expression of several enzymes involved in fatty acid synthesis and cholesterol synthetic pathways (Cruz et al., 2020; Petan et al., 2018) (Figure 4). Multiple hypotheses exist regarding the role of lipid accumulation to potentially promote cancer progression, including protection from lipotoxicity, and serving as a storage pool for fatty acids that can be used for energy production, biosynthetic processes, or intracellular signalling (Petan et al., 2018; Saulnier et al., 1988; Shyu et al., 2018). Interestingly, enzymes involved in fatty acid oxidation are also often overexpressed in cancer cells, supporting that cancer cells may have increased oxidation of fatty acids concurrently with increased lipid storage (Koundouros & Poulogiannis, 2020). Due to this information highlighting the increased metabolism of lipids in cancer cells, several inhibitors of lipid metabolism are currently being tested in preclinical and clinical trials in efforts to prevent cancer progression (Beloribi-Djefaflia et al., 2016). Evidence also suggests that vitamin D metabolites modulate lipid metabolism in a variety of cell and animal models (Abu El Maaty & Wölfl, 2017); thus, vitamin D may exert chemopreventive effects through targeting lipid metabolism pathways in cancer cells.
FIGURE 4.

The regulation of lipid metabolism by 1,25(OH)2D. A decrease in palmitate synthesis (red shaded box) from glucose, through the downregulation of pyruvate carboxylase (red font), is observed in 1,25 (OH)2D treated breast cancer cells. Other in vitro studies indicate that 1,25(OH)2D reduces malonyl-CoA (red shaded box) and differentially affects (black and red arrows) fatty acid synthase as well as neutral lipid accumulation, depending on the type and stage of the cancer model. Abbreviations: 1,25(OH)2D-1α,25—dihydroxyvitamin D; CD36—cluster of differentiation 36; FATP—fatty acid transport protein; ACC—acetyl-CoA carboxylase; FASN—fatty acid synthase; PC—pyruvate carboxylase; OAA—oxaloacetate; TCA—tricarboxylic acid
Studies suggest that 1,25(OH)2D decreases lipid accumulation in cancer cells, highlighting the regulation of lipid metabolism as a potential mechanism underlying vitamin D’s anti-cancer effect (Figure 4). For example, 1,25(OH)2D treatment significantly reduced de novo fatty acid synthesis and lipid accumulation through the downregulation of the anaplerotic enzyme pyruvate carboxylase in metastatic breast cancer cells (Wilmanski, Buhman, et al., 2017). In addition, 1,25(OH)2D and its analogues may suppress fatty acid synthesis through inhibition of fatty acid synthase. For example, in prostate cancer cells, 1,25(OH)2D treatment significantly down-regulated fatty acid synthase at mRNA and protein levels through the upregulation of long-chain fatty-acid-CoA ligase 3 (Qiao et al., 2003; Qiao & Tuohimaa, 2004a, 2004b). Similarly, the vitamin D analogue MT19c suppressed fatty acid synthase expression and activity in a xenograft model of ovarian cancer (Moore et al., 2012). In an in vitro model of ovarian cancer, the MT19c analogue reduced cellular levels of malonyl-CoA, a precursor of fatty acid synthesis (Moore et al., 2012), suggesting that the treatment may also target other lipogenic enzymes upstream of fatty acid synthase. Similarly, 1,25 (OH)2D and its analogues decreased the mRNA abundance of lipogenic enzymes in various colon cancer cell lines, including acetyl-CoA carboxylase, fatty acid synthase and fatty acid elongase, enzymes which mediate fatty acid synthesis and elongation (Leyssens et al., 2015). Because acetyl-CoA carboxylase produces malonyl-CoA and fatty acid synthase catalyses the synthesis of saturated fatty acids, their suppression may result in reduced fatty acid synthesis (Leyssens et al., 2015). Finally, vitamin D receptor activators can inhibit the AMPK/mTOR/thioredoxin interacting protein, thereby blocking synthesis of fatty acids (Abu El Maaty et al., 2017; Abu El Maaty & Wölfl, 2017). These results indicate that 1,25(OH)2D may decrease lipid synthesis more globally through modulation of signalling pathways, such as mTOR, in addition to its direct transcriptional regulation of lipid metabolism genes.
In contrast, other studies indicate that 1,25(OH)2D increases lipid accumulation (Figure 4). For example, 1,25(OH)2D and its analogue 1-hydroxyvitamin D5 increased lipid accumulation in several human breast cancer cell lines (Lazzaro et al., 2000; Mehta et al., 2000; Punj et al., 2004). These results are in agreement with a study which shows that 1,25(OH)2D, either alone or in combination with testosterone, increased neutral lipid accumulation in C4–2B prostate cancer cells, although 1,25(OH)2D treatment alone was insufficient to mediate this effect in PC346C or LNCaP cells (P. Zhang et al., 2019). Increased lipid accumulation induced by 1,25(OH)2D may be driven by an increase in peroxisome-proliferator activated receptor alpha (PPARα) (Esquenet et al., 1997; Wang & Tenniswood, 2014; Wang et al., 2013; P. Zhang et al., 2019), a key activator of lipid synthesis (Knight et al., 2005). Dual treatment of 1,25(OH)2D and androgens induced PPARα protein levels by decreasing RNA expression of its negative regulator miR-17/92 cluster in prostate cancer cells (Wang & Tenniswood, 2014). In this model, increased lipid accumulation through 1,25(OH)2D signalling was associated with reduced tumour progression and growth (Wang & Tenniswood, 2014), although further research is required to determine if changes in lipid metabolism played a causal role in reducing tumour growth.
In order to develop vitamin D recommendations to inhibit tumour or metastatic growth, it is critical to clearly elucidate the mechanistic basis of 1,25(OH)2D’s regulation of lipid metabolism in specific circumstances and the link between these effects to cancer-specific outcomes involved in progression. First, additional research is required to determine the differential mechanisms driving either increased or decreased lipid accumulation in 1,25(OH)2D-treated cells. Further, current evidence from models of breast, colon and prostate cancer suggest that cancer stage, molecular subtype and tumour microenvironment may all contribute to the net effect of 1,25(OH)2D on lipid accumulation (Zhang et al., 2019). Finally, although several preclinical models indicate that vitamin D metabolites and analogues alter lipid metabolism, additional research is needed to causally link changes in lipid metabolism with cancer-related outcomes, such as migration and metastasis.
6 |. VITAMIN D REGULATION OF OXIDATIVE STRESS
High levels of ROS have been reported in many types of cancer (Liou & Storz, 2010). Because ROS is produced as a by-product of energy production in the electron transport chain in the mitochondria, the high metabolic rate observed in cancer cells may produce increased total cellular ROS and oxidative stress. Moderate levels of oxidative stress contribute to mutagenesis which promotes cancer progression; however, excess ROS can also result in cell death. In order to combat the accumulation of cytotoxic levels of ROS, cancer cells must maintain reductive–oxidative (redox) balance through synthesis of reducing equivalents such as NADPH. NADPH functions to regenerate the primary cellular anti-oxidant, GSH, from its oxidized form, GSSG (Liou & Storz, 2010). The metabolism of both glucose and glutamine may increase NADPH synthesis and cancer cells can maintain redox balance by increasing the activity of NADPH synthesizing enzymes. Thus, 1,25(OH)2D-mediated downregulation of glucose and glutamine metabolism, specifically by regulating enzymes that synthesize NADPH, may disrupt the redox balance in cancer cells, thereby contributing to 1,25(OH)2D-mediated cell death due to excessive ROS accumulation.
The regulation of intracellular ROS by 1,25(OH)2D in cancer cells is under current investigation. The regulation of ROS by 1,25(OH)2D is particularly interesting in the context of skin cancer where UVB exposure, which initiates vitamin D synthesis in the skin, may also induce cancer through DNA mutagenesis both directly and indirectly by increasing ROS production (Jagoda & Dixon, 2020). In untransformed keratinocytes, 1,25(OH)2D signalling protects cells from UVB-induced ROS (Rybchyn et al., 2018) and in non-malignant prostate epithelial cells, 1,25(OH)2D was protective against hydrogen peroxide (H2O2)-induced oxidative stress (Bao et al., 2008). However, in prostate cancer cells, 1,25(OH)2D increases the susceptibility of cells to oxidative stress, thereby increasing cancer cell death (Bao et al., 2008). Additionally, treatment of A375 human melanoma cells with either 1,25(OH)2D, 25(OH)D, or the vitamin D analogue calcipotriol decreased cell proliferation due to increased sensitivity to H2O2 (Piotrowska et al., 2019). In breast cancer cells, 1,25(OH)2D treatment increased ROS and decreased the ratio of GSH/GSSG (Abu El Maaty et al., 2018; Koren et al., 2001; Wilmanski, Zhou, et al., 2017). Further, 1,25(OH)2D treatment decreased the NADPH/NADP+ ratio in breast cancer cells, suggesting that 1,25(OH)2D treatment disrupts the regeneration of GSH from NADPH (Wilmanski, Zhou, et al., 2017). Overall, evidence suggests that 1,25(OH)2D alters the redox balance, increasing ROS or sensitivity to oxidative stress in cancer cells, and minimally or oppositely effecting non-malignant cells.
There are multiple enzymes involved in GSH regeneration, including GSH peroxidase, reductase and transferase, and many enzymes that generate NADPH such as glucose-6-phosphate dehydrogenase (G6PD), isocitrate dehydrogenase, malic enzyme and others (Gill et al., 2016). GSH peroxidase, reductase and transferase activities were not regulated by 1,25(OH)2D in breast cancer cells (Koren et al., 2001). However, changes in NADPH producing enzymes were observed with 1,25(OH)2D treatment. For example, G6PD levels and activity were increased following 1,25(OH)2D treatment in breast cancer cells (Abu El Maaty et al., 2018; Beaudin & Welsh, 2016; Koren et al., 2001; Noun et al., 1989; Wilmanski, Zhou, et al., 2017). Conversely, 1,25(OH)2D treatment reduced G6PD mRNA expression in prostate cancer cells (Abu El Maaty et al., 2017). A validated vitamin D response element in the promoter region of the G6PD gene was identified in malignant prostate cells (Bao et al., 2008). However, the 1,25(OH)2D-mediated increase in G6PD activity does not result in an overall increase in oxidative stress protection from increased NADPH synthesis, an expected result since 1,25(OH)2D treatment increased ROS in these models (Bao et al., 2008; Wilmanski, Zhou, et al., 2017). Further, 1,25(OH)2D treatment did not change malic enzyme 1 mRNA expression in breast cancer cells, another enzyme which produces NADPH (Wilmanski, Zhou, et al., 2017). Isocitrate dehydrogenase 1 mRNA abundance was decreased by 1,25(OH)2D in prostate cancer cells (Abu El Maaty et al., 2017), but isocitrate dehydrogenase 2 mRNA abundance was increased by 1,25(OH)2D treatment in untransformed breast epithelial cells (Beaudin & Welsh, 2016). Thus, treatment with 1,25(OH)2D does not affect mRNA levels of genes associated with de novo synthesis of reduced glutathione but may modulate the expression of genes involved in reducing NADP+ to NADPH.
The downregulation of pyruvate carboxylase by 1,25(OH)2D is identified as a mechanism by which 1,25(OH)2D impairs redox balance and mediates an increase in ROS in breast cancer cells (Wilmanski, Zhou, et al., 2017). In this model, 1,25(OH)2D treatment decreased the GSH/GSSG and NADPH/NADP+ ratios. Similarly, genetic depletion of pyruvate carboxylase decreased these ratios, with no further decrease in these ratios with the addition of 1,25(OH)2D treatment. A vitamin D response element was identified and validated by site-specific mutation in the promoter region of the pyruvate carboxylase gene (Wilmanski, Zhou, et al., 2017). Through this mechanism, pyruvate carboxylase may play a role in maintaining redox balance by regulating pyruvate cycling. In this pathway, pyruvate carboxylase converts pyruvate to oxaloacetate, which can be metabolized first to malate through malate dehydrogenase and subsequently to pyruvate through malic enzyme 1 (Ratledge, 2014). The malic enzyme 1 reaction synthesizes NADPH from NADP+ in tandem with conversion of malate to pyruvate; thus, the pyruvate cycling pathway contributes to redox balance and oxidative stress protection. Therefore, transcriptional repression of pyruvate carboxylase, which contributes to pyruvate cycling and the production of NADPH, is a unique mechanism by which 1,25(OH)2D may regulate oxidative stress protection in cancer.
Emerging evidence suggests that 1,25(OH)2D may also synergize with other small molecules and active compounds, including cytokines, antibiotics and drugs, to exert anti-cancer effects. For example, a synergistic effect was shown with 1,25(OH)2D and IL-6, IL1-β or TNFα to reduce breast cancer cell proliferation (Koren et al., 2001). This synergistic effect was abolished when cells were given GSH or N-acetylcysteine, an anti-oxidant that donates the amino acid cysteine used in GSH synthesis, suggesting that 1,25(OH)2D treatment may be useful to exacerbate ROS levels and increase inflammation produced by cancer treatments. Additionally, 1,25(OH)2D in combination with doxorubicin, an anti-tumour antibiotic, induced ROS in breast cancer cells (Ravid et al., 1999). Here, 1,25(OH)2D was shown to induce ROS through reduced mRNA abundance, protein expression and activity of the Cu/Zn superoxide dismutase, an enzyme involved in hydrogen peroxide neutralization. In agreement with these studies, 1,25(OH)2D and other active 1,25(OH)2D analogues, in conjunction with the anti-cancer drug dacarbazine, increased ROS and mitochondrial membrane potential and inhibited the proliferation of melanoma cells (Piotrowska et al., 2019). Collectively, these results demonstrate that co-treatment with 1,25(OH)2D may enhance the efficacy of cancer therapies which induce oxidative stress-related cell death.
In conclusion, 1,25(OH)2D treatment increases oxidative stress in cancer cells. This effect is likely advantageous, as cancer cells produce high levels of ROS (Gill et al., 2016) due to their increased metabolism. Treatments that further increase ROS levels, such as 1,25(OH)2D, induce cytotoxic effects (Gill et al., 2016). Further investigation into additional mechanisms by which 1,25(OH)2D increases oxidative stress, beyond inhibition of pyruvate carboxylase and superoxide dismutase, have yet to be identified. However, 1,25(OH)2D may be useful to amplify ROS-inducing treatments, as demonstrated in a breast cancer cell model with doxorubicin (Ravid et al., 1999). Collectively, these results highlight a novel mechanism by which 1,25 (OH)2D alters cell metabolism to reduce cancer cell viability.
7 |. CONCLUSIONS
A substantial body of literature using cell, preclinical and human models supports that vitamin D or its metabolites may prevent cancer progression (Feldman et al., 2014; Ferrer-Mayorga et al., 2019). Several mechanisms by which 1,25(OH)2D exerts its anti-cancer effects have been identified, including a recent focus on the critical area of metabolic plasticity (Figure 1). Metabolic plasticity is essential for cells to metastasize and highlights several pathways that may be targeted to reduce cancer progression. The active metabolite of vitamin D, 1,25(OH)2D, inhibits glucose, glutamine and fatty acid metabolism. In addition, 1,25(OH)2D also decreases the production of NADPH, thereby increasing oxidative stress and reducing viability of highly metabolic cancer cells.
Little is known regarding the dose or status of vitamin D that may be required to inhibit cancer progression, as the current dietary recommendations are based on bone health (Newberry et al., 2014). Thus, future experimental designs should consider the level of 25(OH) D, the vitamin D status marker and substrate for intracellular conversion to the active metabolite, needed to alter metabolism and inhibit specific steps in cancer. Information about the concentration of 25(OH)D can be used to infer, along with other literature on dose responses in varying conditions, the amount of dietary vitamin D needed to prevent cancer progression.
In addition to variability in doses, discrepancies in clinical trials may be due to the effects of vitamin D at different stages of cancer progression. As such, it is critical to recognize and utilize appropriate models that represent these specific stages. The nutrient needs of cells may vary significantly at different stages of cancer; therefore, changes in cellular energy processes need to be linked to specific outcomes, such as migration, invasion or tumour formation. Thus, it is critical to clearly identify specific cancer conditions where vitamin D is most effective, in order to design strategies to utilize vitamin D in conjunction with cancer treatments, or as a preventive agent.
To develop effective recommendations to prevent disease progression or enhance therapies, it is important to determine 1,25 (OH)2D’s regulation of overall cellular energy metabolism. Utilizing innovative techniques such as modelling stable isotope flux of specific metabolites (Antoniewicz, 2018), rather than only examining mRNA abundance or protein expression to infer changes in metabolism, can improve our understanding of the complexity of the metabolic status of cells in different steps of metastasis and varying genetic backgrounds. In addition, future studies focused on the effects these metabolic changes have on cancer outcomes measured through both in vitro and in vivo experimentation will contribute to a better understanding of vitamin D’s effects on cancer progression.
Overall, research on vitamin D to inhibit cancer progression is overwhelmingly promising. Future studies are needed to identify the necessary dose and specific stages of cancer where energy metabolism may be most greatly altered by vitamin D. Determining how vitamin D may regulate outcomes that inhibit progression will aid in designing appropriate strategies for vitamin D to prevent the devastating consequences of cancer progression, and potentially enhance cancer therapies.
7.1 |. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander, Cidlowski, et al., 2019; Alexander, Fabbro, et al., 2019).
ACKNOWLEDGEMENTS
This work was supported by the Purdue Center for Cancer Research NIH grant P30 CA023168, Indiana Clinical Translational Science Institute NIH/NCRR [#TR000006] and the National Institutes of Health [R01CA232589].
Funding information
Indiana Clinical Translational Science Institute NIH/NCRR, Grant/Award Number: #TR000006; National Institutes of Health, Grant/Award Number: R01CA232589; Purdue Center for Cancer Research NIH grant, Grant/Award Number: P30 CA023168
Abbreviations:
- 1,25(OH)2D
1,25-dihydroxyvitamin D
- 25(OH)D
25-hydroxyvitamin D
- AMPK
adenosine monophosphate-activated protein kinase
- GLUT
glucose transporter
- GSH
glutathione
- lncRNA
long noncoding RNA
- mTOR
mammalian target of rapamycin
- PPARα
peroxisome-proliferator activated receptor alpha
- SLC1A5
solute carrier family 1 member 5
- TCA
tricarboxylic acid
- UVB
ultraviolet B
- VITAL
Vitamin D and Omega-3 Trial
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article because no new data were created or analysed in this study.
REFERENCES
- Abdel-Wahab AF, Mahmoud W, & Al-Harizy RM (2019). Targeting glucose metabolism to suppress cancer progression: Prospective of anti-glycolytic cancer therapy. Pharmacological Research, 150, 104511. 10.1016/j.phrs.2019.104511 [DOI] [PubMed] [Google Scholar]
- Abla H, Sollazzo M, Gasparre G, Iommarini L, & Porcelli AM (2020). The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Seminars in Cell & Developmental Biology, 98, 26–33. 10.1016/j.semcdb.2019.05.031 [DOI] [PubMed] [Google Scholar]
- Abourmad MH, Horn RC, & Fine G (1963). Lipid-secreting mammary carcinoma. Report of a case associated with Paget’s disease of the nipple. Cancer, 16, 521–525. 10.1002/1097-0142(196304)16:4<521::aid-cncr2820160414>3.0.co;2-b [DOI] [PubMed] [Google Scholar]
- Abu El Maaty MA, Alborzinia H, Khan SJ, Büttner M, & Wölfl S (2017). 1,25(OH)2D3 disrupts glucose metabolism in prostate cancer cells leading to a truncation of the TCA cycle and inhibition of TXNIP expression. Biochimica et Biophysica Acta, Molecular Cell Research, 1864(10), 1618–1630. 10.1016/j.bbamcr.2017.06.019 [DOI] [PubMed] [Google Scholar]
- Abu El Maaty MA, Dabiri Y, Almouhanna F, Blagojevic B, Theobald J, Büttner M, & Wölfl S (2018). Activation of pro-survival metabolic networks by 1,25(OH)2D3 does not hamper the sensitivity of breast cancer cells to chemotherapeutics. Cancer & Metabolism, 6, 11. 10.1186/s40170-018-0183-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu El Maaty MA, & Wölfl S (2017). Vitamin D as a novel regulator of tumor metabolism: Insights on potential mechanisms and implications for anti-cancer therapy. International Journal of Molecular Sciences, 18(10), 2184. 10.3390/ijms18102184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA, & GTP collaborators. (2019). The Concise Guide to PHARMACOLOGY 2019/2020: Nuclear hormones receptors. British Journal of Pharmacology, 176, S229–S246. 10.1111/bph.14748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA, & GTP collaborators. (2019). The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniewicz MR (2018). A guide to 13C metabolic flux analysis for the cancer biologist. Experimental & Molecular Medicine, 50(4), 19–13. 10.1038/s12276-018-0060-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton TM, McKenna WG, Kunz-Schughart LA, & Higgins GS (2018). Oxidative phosphorylation as an emerging target in cancer therapy. Clinical Cancer Research, 24(11), 2482–2490. 10.1158/1078-0432.CCR-17-3070 [DOI] [PubMed] [Google Scholar]
- Bao BY, Ting HJ, Hsu JW, & Lee YF (2008). Protective role of 1α, 25-dihydroxyvitamin D3 against oxidative stress in nonmalignant human prostate epithelial cells. International Journal of Cancer, 122(12), 2699–2706. 10.1002/ijc.23460 [DOI] [PubMed] [Google Scholar]
- Beaudin S, & Welsh J (2016). 1,25-Dihydroxyvitamin D induces the glutamate transporter SLC1A1 and alters glutamate handling in non-transformed mammary cells. Molecular and Cellular Endocrinology, 424, 34–41. 10.1016/j.mce.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudin S, & Welsh J (2017). 1,25-Dihydroxyvitamin D regulation of glutamine synthetase and glutamine metabolism in human mammary epithelial cells. Endocrinology, 158(12), 4174–4188. 10.1210/en.2017-00238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloribi-Djefaflia S, Vasseur S, & Guillaumond F (2016). Lipid metabolic reprogramming in cancer cells. Oncogene, 5, e189. 10.1038/oncsis.2015.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, & Fendt SM (2021). The metabolism of cancer cells during metastasis. Nature Reviews. Cancer, 21, 162–180. 10.1038/s41568-020-00320-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikle DD (2009). Extra renal synthesis of 1,25-dihydroxyvitamin D and its health implications. Clinical Reviews in Bone and Mineral Metabolism, 7, 114–125. 10.1007/s12018-009-9033-y [DOI] [Google Scholar]
- Bikle DD (2014). Vitamin D metabolism, mechanism of action, and clinical applications. Chemistry & Biology, 21(3), 319–329. 10.1016/j.chembiol.2013.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeier K, Dröse S, Wittig I, Winkelmann R, Käfer V, Döring C, Hartmann S, Wenz T, Reichert AS, Brandt U, & Hansmann ML (2016). Hodgkin and Reed–Sternberg cells of classical Hodgkin lymphoma are highly dependent on oxidative phosphorylation. International Journal of Cancer, 138(9), 2231–2246. 10.1002/ijc.29934 [DOI] [PubMed] [Google Scholar]
- Blajszczak CC, & Nonn L (2019). Vitamin D regulates prostate cell metabolism via genomic and non-genomic mitochondrial redox-dependent mechanisms. The Journal of Steroid Biochemistry and Molecular Biology, 195, 105484. 10.1016/j.jsbmb.2019.105484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S, & Le A (2018). Glucose metabolism in cancer. Advances in Experimental Medicine and Biology, 1063, 3–12. 10.1007/978-3-319-77736-8_1 [DOI] [PubMed] [Google Scholar]
- Carlberg C, & Muñoz A (2020). An update on vitamin D signaling and cancer. Seminars in Cancer Biology. 10.1016/j.semcancer.2020.05.018 [DOI] [PubMed] [Google Scholar]
- Carlberg C, & Seuter S (2009). A genomic perspective on vitamin D signaling. Anticancer Research, 29(9), 3485–3493. [PubMed] [Google Scholar]
- Chaffer CL, & Weinberg RA (2011). A perspective on cancer cell metastasis. Science, 331(6024), 1559–1564. 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- Chandler PD, Chen WY, Ajala ON, Hazra A, Cook N, Bubes V, Lee IM, Giovannucci EL, Willett W, Buring JE, Manson JE, & VITAL Research Group. (2020). Effect of vitamin D3 supplements on development of advanced cancer: A secondary analysis of the VITAL randomized clinical trial. JAMA Network Open, 3(11), e2025850. 10.1001/jamanetworkopen.2020.25850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christakos S, Dhawan P, Verstuyf A, Verlinden L, & Carmeliet G (2016). Vitamin D: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiological Reviews, 96(1), 365–408. 10.1152/physrev.00014.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluntun AA, Lukey MJ, Cerione RA, & Locasale JW (2017). Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer, 3(3), 169–180. 10.1016/j.trecan.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consiglio M, Destefanis M, Morena D, Foglizzo V, Forneris M, Pescarmona G, & Silvagno F (2014). The vitamin D receptor inhibits the respiratory chain, contributing to the metabolic switch that is essential for cancer cell proliferation. PLoS One, 9(12), e115816. 10.1371/journal.pone.0115816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cork GK, Thompson J, & Slawson C (2018). Real talk: The inter-play between the mTOR, AMPK, and hexosamine biosynthetic pathways in cell signaling. Frontiers in Endocrinology, 9, 522. 10.3389/fendo.2018.00522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz ALS, Barreto EA, Fazolini NPB, Viola JPB, & Bozza PT (2020). Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death & Disease, 11(2), 105. 10.1038/s41419-020-2297-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb KK, Trump DL, & Johnson CS (2007). Vitamin D signalling pathways in cancer: Potential for anti-cancer therapeutics. Nature Reviews. Cancer, 7(9), 684–700. 10.1038/nrc2196 [DOI] [PubMed] [Google Scholar]
- Dillekås H, Rogers MS, & Straume O (2019). Are 90% of deaths from cancer caused by metastases? Cancer Medicine, 8(12), 5574–5576. 10.1002/cam4.2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döppler H, & Storz P (2015). Differences in metabolic programming define the site of breast cancer cell metastasis. Cell Metabolism, 22(4), 536–537. 10.1016/j.cmet.2015.09.022 [DOI] [PubMed] [Google Scholar]
- El-Sharkawy A, & Malki A (2020). Vitamin D signaling in inflammation and cancer: Molecular mechanisms and therapeutic implications. Molecules, 25(14), 3219. 10.3390/molecules25143219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquenet M, Swinnen JV, Van Veldhoven PP, Denef C, Heyns W, & Verhoeven G (1997). Retinoids stimulate lipid synthesis and accumulation in LNCaP prostatic adenocarcinoma cells. Molecular and Cellular Endocrinology, 136(1), 37–46. 10.1016/s0303-7207(97)00210-4 [DOI] [PubMed] [Google Scholar]
- Feldman D, Krishnan AV, Swami S, Giovannucci E, & Feldman BJ (2014). The role of vitamin D in reducing cancer risk and progression. Nature Reviews. Cancer, 14(5), 342–357. 10.1038/nrc3691 [DOI] [PubMed] [Google Scholar]
- Ferrer-Mayorga G, Larriba MJ, Crespo P, & Muñoz A (2019). Mechanisms of action of vitamin D in colon cancer. The Journal of Steroid Biochemistry and Molecular Biology, 185, 1–6. 10.1016/j.jsbmb.2018.07.002 [DOI] [PubMed] [Google Scholar]
- Fleet JC, DeSmet M, Johnson R, & Li Y (2012). Vitamin D and cancer: A review of molecular mechanisms. The Biochemical Journal, 441(1), 61–76. 10.1042/BJ20110744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis I, AlAbdali N, Kapila K, John B, & Al-Temaimi RA (2019). Vitamin D pathway related polymorphisms and vitamin D receptor expression in breast cancer. International Journal for Vitamin and Nutrition Research, 91, 1–9. 10.1024/0300-9831/a000615 [DOI] [PubMed] [Google Scholar]
- Fraser DR (2015). Vitamin D deficiency and energy metabolism. Endocrinology, 156(6), 1933–1935. 10.1210/en.2015-1298 [DOI] [PubMed] [Google Scholar]
- Friedrich M, Diesing D, Cordes T, Fischer D, Becker S, Chen TC, Flanagan JN, Tangpricha V, Gherson I, Holick MF, & Reichrath J (2006). Analysis of 25-hydroxyvitamin D3–1α-hydroxylase in normal and malignant breast tissue. Anticancer Research, 26(4A), 2615–2620. [PubMed] [Google Scholar]
- Garland CF, & Garland FC (1980). Do sunlight and vitamin D reduce the likelihood of colon cancer? International Journal of Epidemiology, 9(3), 227–231. [DOI] [PubMed] [Google Scholar]
- Garland CF, Garland FC, Gorham ED, Lipkin M, Newmark H, Mohr SB, & Holick MF (2006). The role of vitamin D in cancer prevention. American Journal of Public Health, 96(2), 252–261. 10.2105/AJPH.2004.045260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanavat M, Shahrouzian M, Deris Zayeri Z, Banihashemi S, Kazemi SM, & Saki N (2021). Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sciences, 264, 118603. 10.1016/j.lfs.2020.118603 [DOI] [PubMed] [Google Scholar]
- Gill JG, Piskounova E, & Morrison SJ (2016). Cancer, oxidative stress, and metastasis. Cold Spring Harbor Symposia on Quantitative Biology, 81, 163–175. 10.1101/sqb.2016.81.030791 [DOI] [PubMed] [Google Scholar]
- Grant WB (2018). A review of the evidence supporting the vitamin D-cancer prevention hypothesis in 2017. Anticancer Research, 38(2), 1121–1136. 10.21873/anticanres.12331 [DOI] [PubMed] [Google Scholar]
- Grasset W, Mercier N, Chaussard C, Carpentier E, Aldridge S, & Saragaglia D (2012). The surgical treatment of peroneal tendinopathy (excluding subluxations): A series of 17 patients. The Journal of Foot and Ankle Surgery, 51(1), 13–19. 10.1053/j.jfas.2011.10.010 [DOI] [PubMed] [Google Scholar]
- Grover-McKay M, Walsh SA, Seftor EA, Thomas PA, & Hendrix MJ (1998). Role for glucose transporter 1 protein in human breast cancer. Pathology Oncology Research, 4(2), 115–120. 10.1007/BF02904704 [DOI] [PubMed] [Google Scholar]
- Guzey M, Luo J, & Getzenberg RH (2004). Vitamin D3 modulated gene expression patterns in human primary normal and cancer prostate cells. Journal of Cellular Biochemistry, 93(2), 271–285. 10.1002/jcb.20182 [DOI] [PubMed] [Google Scholar]
- Hanchette CL, & Schwartz GG (1992). Geographic patterns of prostate cancer mortality. Evidence for a protective effect of ultraviolet radiation. Cancer, 70(12), 2861–2869. 10.1002/1097-0142(19921215)70:12<2861::aid-cncr2820701224>3.0.co;2-g [DOI] [PubMed] [Google Scholar]
- Hensley CT, Wasti AT, & DeBerardinis RJ (2013). Glutamine and cancer: Cell biology, physiology, and clinical opportunities. The Journal of Clinical Investigation, 123(9), 3678–3684. 10.1172/JCI69600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hii CS, & Ferrante A (2016). The non-genomic actions of vitamin D. Nutrients, 8(3), 135. 10.3390/nu8030135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holick MF (1996). Vitamin D and bone health. The Journal of Nutrition, 126(4 Suppl), 1159S–1164S. 10.1093/jn/126.suppl_4.1159S [DOI] [PubMed] [Google Scholar]
- Holick MF, Chen TC, Lu Z, & Sauter E (2007). Vitamin D and skin physiology: A D-lightful story. Journal of Bone and Mineral Research, 22 (Suppl 2), V28–V33. 10.1359/jbmr.07s211 [DOI] [PubMed] [Google Scholar]
- Horas K, Zheng Y, Fong-Yee C, Macfarlane E, Manibo J, Chen Y, Qiao J, Gao M, Haydar N, McDonald MM, Croucher PI, Zhou H, & Seibel MJ (2019). Loss of the vitamin D receptor in human breast cancer cells promotes epithelial to mesenchymal cell transition and skeletal colonization. Journal of Bone and Mineral Research, 34(9), 1721–1732. 10.1002/jbmr.3744 [DOI] [PubMed] [Google Scholar]
- Hu K, Callen DF, Li J, & Zheng H (2018). Circulating vitamin D and overall survival in breast cancer patients: A dose-response meta-analysis of cohort studies. Integrative Cancer Therapies, 17(2), 217–225. 10.1177/1534735417712007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagoda SV, & Dixon KM (2020). Protective effects of 1,25 dihydroxyvitamin D and its analogs on ultraviolet radiation-induced oxidative stress: A review. Redox Report, 25(1), 11–16. 10.1080/13510002.2020.1731261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin CH, & Pike JW (1996). Human vitamin D receptor-dependent transactivation in Saccharomyces cerevisiae requires retinoid X receptor. Molecular Endocrinology, 10(2), 196–205. 10.1210/mend.10.2.8825559 [DOI] [PubMed] [Google Scholar]
- Jorde R, Sollid ST, Svartberg J, Schirmer H, Joakimsen RM, Njølstad I, Fuskevåg OM, Figenschau Y, & Hutchinson MY (2016). Vitamin D 20,000 IU per week for five years does not prevent progression from prediabetes to diabetes. The Journal of Clinical Endocrinology and Metabolism, 101(4), 1647–1655. 10.1210/jc.2015-4013 [DOI] [PubMed] [Google Scholar]
- Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR, Lee JH, Lee SG, & Park YK (2002). Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Japanese Journal of Cancer Research, 93(10), 1123–1128. 10.1111/j.1349-7006.2002.tb01214.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerner SA, Scott RA, & Pike JW (1989). Sequence elements in the human osteocalcin gene confer basal activation and inducible response to hormonal vitamin D3. Proceedings of the National Academy of Sciences of the United States of America, 86(12), 4455–4459. 10.1073/pnas.86.12.4455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum N, Lee DH, Greenwood DC, Manson JE, & Giovannucci E (2019). Vitamin D supplementation and total cancer incidence and mortality: A meta-analysis of randomized controlled trials. Annals of Oncology, 30(5), 733–743. 10.1093/annonc/mdz059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball SM, & Holick MF (2020). Official recommendations for vitamin D through the life stages in developed countries. European Journal of Clinical Nutrition, 74(11), 1514–1518. 10.1038/s41430-020-00706-3 [DOI] [PubMed] [Google Scholar]
- Knight BL, Hebbachi A, Hauton D, Brown AM, Wiggins D, Patel DD, & Gibbons GF (2005). A role for PPARα in the control of SREBP activity and lipid synthesis in the liver. The Biochemical Journal, 389(Pt 2), 413–421. 10.1042/BJ20041896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren R, Hadari-Naor I, Zuck E, Rotem C, Liberman UA, & Ravid A (2001). Vitamin D is a prooxidant in breast cancer cells. Cancer Research, 61(4), 1439–1444. [PubMed] [Google Scholar]
- Koundouros N, & Poulogiannis G (2020). Reprogramming of fatty acid metabolism in cancer. British Journal of Cancer, 122(1), 4–22. 10.1038/s41416-019-0650-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan AV, Shinghal R, Raghavachari N, Brooks JD, Peehl DM, & Feldman D (2004). Analysis of vitamin D-regulated gene expression in LNCaP human prostate cancer cells using cDNA microarrays. Prostate, 59(3), 243–251. 10.1002/pros.20006 [DOI] [PubMed] [Google Scholar]
- Krishnan AV, Trump DL, Johnson CS, & Feldman D (2010). The role of vitamin D in cancer prevention and treatment. Endocrinology and Metabolism Clinics of North America, 39(2), 401–418. 10.1016/j.ecl.2010.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe JM, Travers-Gustafson D, Davies KM, Recker RR, & Heaney RP (2007). Vitamin D and calcium supplementation reduces cancer risk: Results of a randomized trial. The American Journal of Clinical Nutrition, 85(6), 1586–1591. 10.1093/ajcn/85.6.1586 [DOI] [PubMed] [Google Scholar]
- Lazzaro G, Agadir A, Qing W, Poria M, Mehta RR, Moriarty RM, Das Gupta TK, Zhang XK, & Mehta RG (2000). Induction of differentiation by 1α-hydroxyvitamin D5 in T47D human breast cancer cells and its interaction with vitamin D receptors. European Journal of Cancer, 36(6), 780–786. 10.1016/s0959-8049(00)00016-2 [DOI] [PubMed] [Google Scholar]
- Leyssens C, Marien E, Verlinden L, Derua R, Waelkens E, Swinnen JV, & Verstuyf A (2015). Remodeling of phospholipid composition in colon cancer cells by 1α,25(OH)2D3 and its analogs. The Journal of Steroid Biochemistry and Molecular Biology, 148, 172–178. 10.1016/j.jsbmb.2015.01.018 [DOI] [PubMed] [Google Scholar]
- Li J, Byrne ME, Chang E, Jiang Y, Donkin SS, Buhman KK, Burgess JR, & Teegarden D (2008). 1α,25-Dihydroxyvitamin D hydroxylase in adipocytes. The Journal of Steroid Biochemistry and Molecular Biology, 112(1–3), 122–126. 10.1016/j.jsbmb.2008.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, & Locasale JW (2016). The Warburg effect: How does it benefit cancer cells? Trends in Biochemical Sciences, 41(3), 211–218. 10.1016/j.tibs.2015.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou GY, & Storz P (2010). Reactive oxygen species in cancer. Free Radical Research, 44(5), 479–496. 10.3109/10715761003667554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Zang M, & Guo W (2010). AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncology, 6 (3), 457–470. 10.2217/fon.09.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macejová D, Ondková S, Jakubíková L, Mlynarčíková A, Scsuková S, Liška J, & Brtko J (2011). MNU-induced mammary gland carcinogenesis: Chemopreventive and therapeutic effects of vitamin D and Seocalcitol on selected regulatory vitamin D receptor pathways. Toxicology Letters, 207(1), 60–72. 10.1016/j.toxlet.2011.07.029 [DOI] [PubMed] [Google Scholar]
- Maestro MA, Molnár F, & Carlberg C (2019). Vitamin D and its synthetic analogs. Journal of Medicinal Chemistry, 62(15), 6854–6875. 10.1021/acs.jmedchem.9b00208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Gordon D, Copeland T, D’Agostino D, Friedenberg G, Ridge C, Bubes V, Giovannucci EL, Willett WC, Buring JE, & VITAL Research Group. (2019). Vitamin D supplements and prevention of cancer and cardiovascular disease. The New England Journal of Medicine, 380(1), 33–44. 10.1056/NEJMoa1809944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, & Bachoo RM (2014). Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell, 159(7), 1603–1614. 10.1016/j.cell.2014.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzaferro S, Goldsmith D, Larsson TE, Massy ZA, & Cozzolino M (2014). Vitamin D metabolites and/or analogs: Which D for which patient? Current Vascular Pharmacology, 12(2), 339–349. 10.2174/15701611113119990024 [DOI] [PubMed] [Google Scholar]
- McGuirk S, Audet-Delage Y, & St-Pierre J (2020). Metabolic fitness and plasticity in cancer progression. Trends Cancer, 6(1), 49–61. 10.1016/j.trecan.2019.11.009 [DOI] [PubMed] [Google Scholar]
- Mehta RR, Bratescu L, Graves JM, Green A, & Mehta RG (2000). Differentiation of human breast carcinoma cells by a novel vitamin D analog: 1α-hydroxyvitamin D5. International Journal of Oncology, 16(1), 65–73. 10.3892/ijo.16.1.65 [DOI] [PubMed] [Google Scholar]
- Migliaccio S, Di Nisio A, Mele C, Scappaticcio L, Savastano S, Colao A, & Obesity Programs of nutrition, E., R.search and Assessment (OPERA) Group. (2019). Obesity and hypovitaminosis D: Causality or casualty? International Journal of Obesity Supplements, 9 (1), 20–31. 10.1038/s41367-019-0010-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C, Katayama ML, de Lyra EC, Welsh J, Campos LT, Brentani MM, Maciel Mdo S, Roela RA, del Valle PR, Góes JC, Nonogaki S, Tamura RE, & Folgueira MA (2013). Transcriptional effects of 1,25 dihydroxyvitamin D3 physiological and supra-physiological concentrations in breast cancer organotypic culture. BMC Cancer, 13, 119. 10.1186/1471-2407-13-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RG, Lange TS, Robinson K, Kim KK, Uzun A, Horan TC, Kawar N, Yano N, Chu SR, Mao Q, Brard L, DePaepe ME, Padbury JF, Arnold LA, Brodsky A, Shen TL, & Singh RK (2012). Efficacy of a non-hypercalcemic vitamin-D2 derived anti-cancer agent (MT19c) and inhibition of fatty acid synthesis in an ovarian cancer xenograft model. PLoS One, 7(4), e34443. 10.1371/journal.pone.0034443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama A, Takeyama K, Kitanaka S, Kodera Y, Hosoya T, & Kato S (1998). The promoter of the human 25-hydroxyvitamin D31α-hydroxylase gene confers positive and negative responsiveness to PTH, calcitonin, and 1α,25(OH)2D3. Biochemical and Biophysical Research Communications, 249(1), 11–16. 10.1006/bbrc.1998.9098 [DOI] [PubMed] [Google Scholar]
- Narvaez CJ, Grebenc D, Balinth S, & Welsh JE (2020). Vitamin D regulation of HAS2, hyaluronan synthesis and metabolism in triple negative breast cancer cells. The Journal of Steroid Biochemistry and Molecular Biology, 201, 105688. 10.1016/j.jsbmb.2020.105688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaez CJ, Matthews D, Broun E, Chan M, & Welsh J (2009). Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology, 150(2), 651–661. 10.1210/en.2008-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemere I, Garbi N, Hammerling G, & Hintze KJ (2012). Role of the 1,25D3-MARRS receptor in the 1,25(OH)2D3-stimulated uptake of calcium and phosphate in intestinal cells. Steroids, 77(10), 897–902. 10.1016/j.steroids.2012.04.002 [DOI] [PubMed] [Google Scholar]
- Newberry SJ, Chung M, Shekelle PG, Booth MS, Liu JL, Maher AR, Motala A, Cui M, Perry T, Shanman R, & Balk EM (2014). Vitamin D and calcium: A systematic review of health outcomes (update). Evidence Report/Technology Assessment, 217, 1–929. 10.23970/AHRQEPCERTA217 [DOI] [PubMed] [Google Scholar]
- Norman AW (1998). Receptors for 1α,25(OH)2D3: Past, present, and future. Journal of Bone and Mineral Research, 13(9), 1360–1369. 10.1359/jbmr.1998.13.9.1360 [DOI] [PubMed] [Google Scholar]
- Noun A, Garabedian M, & Monet JD (1989). Stimulatory effect of 1,25-dihydroxyvitamin D3 on the glucose-6-phosphate dehydrogenase activity in the MCF-7 human breast cancer cell line. Cell Biochemistry and Function, 7(1), 1–6. 10.1002/cbf.290070102 [DOI] [PubMed] [Google Scholar]
- Nurminen V, Seuter S, & Carlberg C (2019). Primary vitamin D target genes of human monocytes. Frontiers in Physiology, 10, 194. 10.3389/fphys.2019.00194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petan T, Jarc E, & Jusovi c M (2018). Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules, 23(8), 1941. 10.3390/molecules23081941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowska A, Wierzbicka J, Rybarczyk A, Tuckey RC, Slominski AT, & Żmijewski MA (2019). Vitamin D and its low calcemic analogs modulate the anticancer properties of cisplatin and dacarbazine in the human melanoma A375 cell line. International Journal of Oncology, 54 (4), 1481–1495. 10.3892/ijo.2019.4725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice RL, Pettinger MB, Jackson RD, Wactawski-Wende J, Lacroix AZ, Anderson GL, Chlebowski RT, Manson JE, Van Horn L, Vitolins MZ, Datta M, LeBlanc ES, Cauley JA, & Rossouw JE (2013). Health risks and benefits from calcium and vitamin D supplementation: Women’s Health Initiative clinical trial and cohort study. Osteoporosis International, 24(2), 567–580. 10.1007/s00198-012-2224-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punj V, Graves JM, & Mehta RR (2004). Effect of vitamin D analog (1α hydroxy D5) immunoconjugated to Her-2 antibody on breast cancer. International Journal of Cancer, 108(6), 922–929. 10.1002/ijc.11590 [DOI] [PubMed] [Google Scholar]
- Qiao S, Pennanen P, Nazarova N, Lou YR, & Tuohimaa P (2003). Inhibition of fatty acid synthase expression by 1α,25-dihydroxyvitamin D3 in prostate cancer cells. The Journal of Steroid Biochemistry and Molecular Biology, 85(1), 1–8. 10.1016/s0960-0760(03)00142-0 [DOI] [PubMed] [Google Scholar]
- Qiao S, & Tuohimaa P (2004a). The role of long-chain fatty-acid-CoA ligase 3 in vitamin D3 and androgen control of prostate cancer LNCaP cell growth. Biochemical and Biophysical Research Communications, 319 (2), 358–368. 10.1016/j.bbrc.2004.05.014 [DOI] [PubMed] [Google Scholar]
- Qiao S, & Tuohimaa P (2004b). Vitamin D3 inhibits fatty acid synthase expression by stimulating the expression of long-chain fatty-acid-CoA ligase 3 in prostate cancer cells. FEBS Letters, 577(3), 451–454. 10.1016/j.febslet.2004.10.044 [DOI] [PubMed] [Google Scholar]
- Ramos CV, & Taylor HB (1974). Lipid-rich carcinoma of the breast. A clinicopathologic analysis of 13 examples. Cancer, 33(3), 812–819. 10.1002/1097-0142(197403)33:3<812::aidcncr2820330328>3.0.co;2-4 [DOI] [PubMed] [Google Scholar]
- Ratledge C (2014). The role of malic enzyme as the provider of NADPH in oleaginous microorganisms: A reappraisal and unsolved problems. Biotechnology Letters, 36(8), 1557–1568. 10.1007/s10529-014-1532-3 [DOI] [PubMed] [Google Scholar]
- Ravid A, Rocker D, Machlenkin A, Rotem C, Hochman A, Kessler-Icekson G, Liberman UA, & Koren R (1999). 1,25-Dihydroxyvitamin D3 enhances the susceptibility of breast cancer cells to doxorubicin-induced oxidative damage. Cancer Research, 59(4), 862–867. [PubMed] [Google Scholar]
- Ricca C, Aillon A, Bergandi L, Alotto D, Castagnoli C, & Silvagno F (2018). Vitamin D receptor is necessary for mitochondrial function and cell health. International Journal of Molecular Sciences, 19(6), 1672. 10.3390/ijms19061672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybchyn MS, De Silva WGM, Sequeira VB, McCarthy BY, Dilley AV, Dixon KM, Halliday GM, & Mason RS (2018). Enhanced repair of UV-induced DNA damage by 1,25-dihydroxyvitamin D. The Journal of Investigative Dermatology, 138(5), 1146–1156. 10.1016/j.jid.2017.11.037 [DOI] [PubMed] [Google Scholar]
- Santos JM, Khan ZS, Munir MT, Tarafdar K, Rahman SM, & Hussain F (2018). Vitamin D3 decreases glycolysis and invasiveness, and increases cellular stiffness in breast cancer cells. The Journal of Nutritional Biochemistry, 53, 111–120. 10.1016/j.jnutbio.2017.10.013 [DOI] [PubMed] [Google Scholar]
- Saracligil B, Ozturk B, Unlu A, Abusoglu S, & Tekin G (2017). The effect of vitamin D on MCF-7 breast cancer cell metabolism. Bratislavské Lekárske Listy, 118(2), 101–106. 10.4149/BLL_2017_021 [DOI] [PubMed] [Google Scholar]
- Saulnier J, Bostancioglu K, Favre-Bonvin G, & Wallach J (1988). Conductimetry: A new tool for studying inhibition of elastolysis. Biological Chemistry Hoppe-Seyler, 369(Suppl), 75–78. [PubMed] [Google Scholar]
- Sazeides C, & Le A (2018). Metabolic relationship between cancer-associated fibroblasts and cancer cells. Advances in Experimental Medicine and Biology, 1063, 149–165. 10.1007/978-3-319-77736-8_11 [DOI] [PubMed] [Google Scholar]
- Scheel C, & Weinberg RA (2012). Cancer stem cells and epithelialmesenchymal transition: Concepts and molecular links. Seminars in Cancer Biology, 22(5–6), 396–403. 10.1016/j.semcancer.2012.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scragg R, Khaw KT, Toop L, Sluyter J, Lawes CMM, Waayer D, Giovannucci E, & Camargo CA (2018). Monthly high-dose vitamin D supplementation and cancer risk: A post hoc analysis of the vitamin D assessment randomized clinical trial. JAMA Oncology, 4(11), e182178. 10.1001/jamaoncol.2018.2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan NL, Minden A, Furmanski P, Bak MJ, Cai L, Wernyj R, Sargsyan D, Cheng D, Wu R, Kuo HCD, Li SN, Fang M, Maehr H, Kong AN, & Suh N (2020). Analysis of the transcriptome: Regulation of cancer stemness in breast ductal carcinoma. Cancer Prevention Research (Philadelphia, Pa.), 13(8), 673–686. 10.1158/1940-6207.CAPR-19-0566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirvani A, Kalajian TA, Song A, Allen R, Charoenngam N, Lewanczuk R, & Holick MF (2020). Variable genomic and metabolomic responses to varying doses of vitamin D supplementation. Anticancer Research, 40(1), 535–543. 10.21873/anticanres.13982 [DOI] [PubMed] [Google Scholar]
- Shyu P, Wong XFA, Crasta K, & Thibault G (2018). Dropping in on lipid droplets: Insights into cellular stress and cancer. Bioscience Reports, 38(5), BSR20180764. 10.1042/BSR20180764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Fuchs HE, & Jemal A (2021). Cancer statistics, 2021. CA: A Cancer Journal for Clinicians, 71(1), 7–33. 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- Silvagno F, De Vivo E, Attanasio A, Gallo V, Mazzucco G, & Pescarmona G (2010). Mitochondrial localization of vitamin D receptor in human platelets and differentiated megakaryocytes. PLoS One, 5(1), e8670. 10.1371/journal.pone.0008670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski AT, Brożyna AA, Skobowiat C, Zmijewski MA, Kim TK, Janjetovic Z, Oak AS, Jozwicki W, Jetten AM, Mason RS, Elmets C, Li W, Hoffman RM, & Tuckey RC (2018). On the role of classical and novel forms of vitamin D in melanoma progression and management. The Journal of Steroid Biochemistry and Molecular Biology, 177, 159–170. 10.1016/j.jsbmb.2017.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slominski AT, Brożyna AA, Zmijewski MA, Janjetovic Z, Kim TK, Slominski RM, Tuckey RC, Mason RS, Jetten AM, Guroji P, Reichrath J, Elmets C, & Athar M (2020). The role of classical and novel forms of vitamin D in the pathogenesis and progression of nonmelanoma skin cancers. Advances in Experimental Medicine and Biology, 1268, 257–283. 10.1007/978-3-030-46227-7_13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, & Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife, 8, e4423. 10.7554/eLife.44235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swami S, Raghavachari N, Muller UR, Bao YP, & Feldman D (2003). Vitamin D growth inhibition of breast cancer cells: Gene expression patterns assessed by cDNA microarray. Breast Cancer Research and Treatment, 80(1), 49–62. 10.1023/A:1024487118457 [DOI] [PubMed] [Google Scholar]
- Vanoirbeek E, Eelen G, Verlinden L, Marchal K, Engelen K, De Moor B, Beullens I, Marcelis S, De Clercq P, Bouillon R, & Verstuyf A (2009). Microarray analysis of MCF-7 breast cancer cells treated with 1,25-dihydroxyvitamin D3 or a 17-methyl-D-ring analog. Anticancer Research, 29(9), 3585–3590. [PubMed] [Google Scholar]
- Wang WL, & Tenniswood M (2014). Vitamin D, intermediary metabolism and prostate cancer tumor progression. Frontiers in Physiology, 5, 183. 10.3389/fphys.2014.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WL, Welsh J, & Tenniswood M (2013). 1,25-Dihydroxyvitamin D3 modulates lipid metabolism in prostate cancer cells through miRNA mediated regulation of PPARA. The Journal of Steroid Biochemistry and Molecular Biology, 136, 247–251. 10.1016/j.jsbmb.2012.09.033 [DOI] [PubMed] [Google Scholar]
- Warburg O, Wind F, & Negelein E (1927). The metabolism of tumors in the body. The Journal of General Physiology, 8(6), 519–530. 10.1085/jgp.8.6.519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb AR, DeCosta BR, & Holick MF (1989). Sunlight regulates the cutaneous production of vitamin D3 by causing its photodegradation. The Journal of Clinical Endocrinology and Metabolism, 68(5), 882–887. 10.1210/jcem-68-5-882 [DOI] [PubMed] [Google Scholar]
- Welsh J (2017). Function of the vitamin D endocrine system in mammary gland and breast cancer. Molecular and Cellular Endocrinology, 453, 88–95. 10.1016/j.mce.2017.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Pavlides S, Tsirigos A, Ertel A, Pestell RG, Broda P, Minetti C, Lisanti MP, & Sotgia F (2011). Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle, 10(23), 4047–4064. 10.4161/cc.10.23.18151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde L, Roche M, Domingo-Vidal M, Tanson K, Philp N, Curry J, & Martinez-Outschoorn U (2017). Metabolic coupling and the reverse Warburg effect in cancer: Implications for novel biomarker and anti-cancer agent development. Seminars in Oncology, 44(3), 198–203. 10.1053/j.seminoncol.2017.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JD, Aggarwal A, Swami S, Krishnan AV, Ji L, Albertelli MA, & Feldman BJ (2016). Tumor autonomous effects of vitamin D deficiency promote breast cancer metastasis. Endocrinology, 157(4), 1341–1347. 10.1210/en.2015-2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmanski T, Buhman K, Donkin SS, Burgess JR, & Teegarden D (2017). 1α,25-dihydroxyvitamin D inhibits de novo fatty acid synthesis and lipid accumulation in metastatic breast cancer cells through downregulation of pyruvate carboxylase. The Journal of Nutritional Biochemistry, 40, 194–200. 10.1016/j.jnutbio.2016.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmanski T, Zhou X, Zheng W, Shinde A, Donkin SS, Wendt M, Burgess JR, & Teegarden D (2017). Inhibition of pyruvate carboxylase by 1α,25-dihydroxyvitamin D promotes oxidative stress in early breast cancer progression. Cancer Letters, 411, 171–181. 10.1016/j.canlet.2017.09.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida GJ (2021). The harmonious interplay of amino acid and monocarboxylate transporters induces the robustness of cancer cells. Metabolites, 11(1), 27. 10.3390/metabo11010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehnder D, Bland R, Williams MC, McNinch RW, Howie AJ, Stewart PM, & Hewison M (2001). Extrarenal expression of 25-hydroxyvitamin D3-1α-hydroxylase. The Journal of Clinical Endocrinology and Metabolism, 86(2), 888–894. 10.1210/jcem.86.2.7220 [DOI] [PubMed] [Google Scholar]
- Zhang P, Schatz A, Adeyemi B, Kozminski D, Welsh J, Tenniswood M, & Wang WW (2019). Vitamin D and testosterone co-ordinately modulate intracellular zinc levels and energy metabolism in prostate cancer cells. The Journal of Steroid Biochemistry and Molecular Biology, 189, 248–258. 10.1016/j.jsbmb.2019.01.006 [DOI] [PubMed] [Google Scholar]
- Zhang X, Li P, Bao J, Nicosia SV, Wang H, Enkemann SA, & Bai W (2005). Suppression of death receptor-mediated apoptosis by 1,25-dihydroxyvitamin D3 revealed by microarray analysis. The Journal of Biological Chemistry, 280(42), 35458–35468. 10.1074/jbc.M506648200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Guo Q, Zhang Z, Bai N, Liu Z, Xiong M, Wei Y, Xiang R, & Tan X (2014). VDR status arbitrates the prometastatic effects of tumor-associated macrophages. Molecular Cancer Research, 12(8), 1181–1191. 10.1158/1541-7786.MCR-14-0036 [DOI] [PubMed] [Google Scholar]
- Zheng W, Tayyari F, Gowda GA, Raftery D, McLamore ES, Shi J, Porterfield DM, Donkin SS, Bequette B, & Teegarden D (2013). 1,25-dihydroxyvitamin D regulation of glucose metabolism in Harvey-ras transformed MCF10A human breast epithelial cells. The Journal of Steroid Biochemistry and Molecular Biology, 138, 81–89. 10.1016/j.jsbmb.2013.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Zheng W, Nagana Gowda GA, Raftery D, Donkin SS, Bequette B, & Teegarden D (2016). 1,25-Dihydroxyvitamin D inhibits glutamine metabolism in Harvey-ras transformed MCF10A human breast epithelial cell. The Journal of Steroid Biochemistry and Molecular Biology, 163, 147–156. 10.1016/j.jsbmb.2016.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmijewski MA, & Carlberg C (2020). Vitamin D receptor(s): In the nucleus but also at membranes? Experimental Dermatology, 29(9), 876–884. 10.1111/exd.14147 [DOI] [PubMed] [Google Scholar]
- Zuo S, Wu L, Wang Y, & Yuan X (2020). Long non-coding RNA. Frontiers in Oncology, 10, 274. 10.3389/fonc.2020.00274 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article because no new data were created or analysed in this study.