

Abstract
Background
The Creutzfeldt-Jakob disease (CJD) is a spongiform encephalopathy that manifests as a rapidly progressive dementia syndrome. Currently, CJD has no cure, and many patients die within the first year, but some drugs are being studied as options for managing this condition.
Objective
To evaluate the effectiveness of pharmacological treatments offered to patients with CJD as a means to increase survival and reduce cognitive deterioration.
Methods
A systematic review of the literature was performed using 4 independent reviewers and 1 extra reviewer to resolve possible divergences in the search and analysis of papers indexed in MedLINE (PubMed), SciELO and Lilacs databases. The Medical Subject Heading (MeSH) terms used were: prion diseases, Creutzfeldt-Jakob disease, pharmacologic therapy, therapeutics, quinacrine, doxycycline, flupirtine, and pentosan polysulfate, with the Boolean operators AND and OR. This search included controlled clinical trials, uncontrolled clinical trials, and case series published from the year 2000 onwards, in the English language.
Results
A total of 85 papers were found using the descriptors used. At the end of the selection analyses, 9 articles remained, which were analyzed fully and individually.
Conclusions
None of the drugs evaluated proved significantly effective in increasing survival in patients with CJD. Flupirtine appears to have a beneficial effect in reducing cognitive deterioration in patients with CJD. However, additional studies are needed to establish better evidence and therapeutic options for the management of patients with CJD.
Keywords: Creutzfeldt-Jakob Syndrome, Prion Diseases, Therapeutics
Resumo
Antecedentes
A doença de Creutzfeldt-Jakob (DCJ) é uma encefalopatia espongiforme que se manifesta como síndrome demencial rapidamente progressiva. Atualmente, a DCJ não possui cura e muitos pacientes morrem no primeiro ano de doença, mas alguns medicamentos vêm sendo estudados como opções no manejo desta condição.
Objetivo
Avaliar a eficácia dos tratamentos farmacológicos oferecidos aos pacientes com DCJ no aumento de sobrevida e na redução da deterioração cognitiva.
Métodos
Foi realizada uma revisão sistemática da literatura utilizando 4 revisores independentes e 1 extra para resolver divergências eventuais na busca e na análise de trabalhos indexados nas bases de dados MedLINE (via PubMed), SciELO e Lilacs. Os termos Medical Subjects Heading (MeSH) utilizados foram: prion diseases, creutzfeldt jakob disease, pharmacologic therapy, therapeutics, quinacrine, doxycycline, flupirtine e pentosan polysulfate, com os operadores booleanos AND e OR. Essa pesquisa incluiu ensaios clínicos controlados, não controlados e séries de casos, publicados a partir do ano 2000 no idioma inglês.
Resultados
Ao todo, foram encontrados 85 trabalhos através dos descritores utilizados. Ao final das análises de seleção, restaram 9 artigos, que foram analisados na íntegra individualmente.
Conclusões
Nenhuma das drogas avaliadas se mostrou significativamente eficaz no aumento da sobrevida dos pacientes com DCJ. A flupirtina parece ter um efeito benéfico na redução da deterioração cognitiva dos pacientes com DCJ. Entretanto, estudos adicionais são necessários para o estabelecimento de melhores evidências e opções terapêuticas para o manejo dos pacientes com DCJ.
Palavras-chave: Síndrome de Creutzfeldt-Jakob, Doenças Priônicas, Terapêutica
INTRODUCTION
Prion diseases make up a large group of neurodegenerative conditions known as spongiform encephalopathies, affecting both animals and humans. 1,2 These conditions are fatal, with no current effective form of treatment. 1 However, increased understanding of their pathogenesis has recently led to the promise of effective therapeutic interventions, particularly for the Creutzfeldt-Jakob disease (CJD).
The CJD has a long incubation period, but once its clinical manifestations appear, a very rapid progression to a fatal outcome can be expected. 3,4 It can have a myriad of causes, namely sporadic, genetic, iatrogenic, or idiopathic. 5 The current accepted hypothesis for CJD's etiological agent is mainly an abnormal conformation of a host-encoded glycoprotein called prion protein. 6,7
The global incidence of CJD is estimated to be approximately 1 to 2 cases per million per year. 8 Sporadic CJD has a rapidly progressive clinical course, invariably progressing to death within 6 to 14 months, with an average of 8 months, 8 occurring worldwide without significant differences between ethnic groups or genders. The mean age of onset of sporadic CJD is 62 years, and among individuals aged 60 to 74 years the incidence is 5 cases per million population. 9 Although it is very uncommon, it can affect individuals under the age of 55.
Clinically, CJD is characterized by rapidly progressive dementia associated with myoclonic, pyramidal, extrapyramidal, and cerebellar symptoms, with abnormalities in the electroencephalogram. In some cases, patients may have symptoms of peripheral neuropathy preceding the central involvement. In addition to these elements, elevated cerebrospinal fluid 14-3-3 protein and magnetic resonance imaging (MRI) scans with findings of cortical ribbons help in determining a suspected case of CJD. 3,4,10 Because it is a diagnosis of exclusion, patients with suspected CJD undergo extensive complementary evaluations to rule out other possible causes that make a differential diagnosis.
Several potential therapeutic interventions are under development over the last years, 10,11,12 but no treatment has been proven effective for CJD. In this perspective, some chemotherapeutic, immunological, or pharmacological neuroprotection stimulation approaches have been studied in patients with CJD to extend the supportive therapeutic possibilities already employed. 13-15 Thus, it is important to know in detail the advances in the pharmacological management of CJD in the international medical literature to improve patient care and enlightenment.
METHODS
The present study consists of a systematic review of the literature, using controlled and uncontrolled clinical trials and case series, on the efficacy of pharmacological treatments offered to prolong survival time and reduce cognitive deterioration in patients with CJD. The studies included in this paper focus on the effects of drugs studied and prescribed in the management of CJD, and include pharmacological intervention groups whose effect are compared with placebo, other drugs, or simply compared with previous literature findings.
The inclusion criteria were patients with probable or possible cases of CJD regardless of gender, age, comorbidities, or concomitant or prior use of any type of pharmacological management. The probable case of CJD is characterized by the neuropsychiatric disorder plus positive real-time quaking-induced conversion (RT-QuIC) in the cerebrospinal fluid or other tissues, or rapidly progressive dementia syndrome with at least two of the listed findings: myoclonus, visual changes, cerebellar syndrome, pyramidal or extrapyramidal symptoms, akinetic mutism with typical findings on electroencephalogram (EEG), or a positive 14-3-3 CSF assay in patients with a disease duration of less than 2 years, or typical findings on MRI and without routine investigations indicating an alternative diagnosis. 16 The probable case of CJD requires progressive dementia syndrome, in addition to duration of illness of less than 2 years, absence of a positive result for any of the 4 tests listed above that would classify a case as “probable”, and without routine investigations indicating an alternative diagnosis, and, finally, at least two out of the following four clinical features: myoclonus, visual changes, cerebellar syndrome, pyramidal or extrapyramidal symptoms, or akinetic mutism. 16
The efficacy of pharmacological treatments in CJD was evaluated by assessing the number of patients per group that demonstrated increased survival time in months or reduction in cognitive deterioration, using specific scales. The studies that were selected for this review were identified through an electronic search conducted in July 2021 in the MedLINE (PubMed), SciELO, and Lilacs databases. The search was conducted in the English language, including articles from the year 2000 onwards, using the following descriptors: prion diseases, Creutzfeldt-Jakob disease, pharmacologic therapy, therapeutics, quinacrine, doxycycline, flupirtine, pentosan polysulfate, associated with the Boolean operators AND and OR. In addition to the directly searched works, the bibliographic references of relevant studies, metanalyses, and review articles found on the subject were also analyzed, with the aim of expanding the number of eligible works that were eventually not located in the previous steps.
Using the aforementioned methodology, four independent reviewers evaluated the titles, keywords, and abstracts identified as of interest in the initial steps. The potentially relevant studies were analyzed in full. Following this, the same reviewers independently selected the articles that met the inclusion criteria, and all reviewers completed a data extraction form for each paper. Any discrepancies were resolved by a fifth independent reviewer.
This is a systematic review built upon secondary data made available through selected articles, with no form of personal interest whatsoever, this research did not present any risk of violating normative ethical rights and, therefore, did not need to be submitted to the Ethics Committee on Research with Human Beings.
RESULTS
In the initial stages of article collection, a total of 85 papers were identified. After analyzing the titles, keywords, and abstracts, 20 studies were considered potentially relevant in the methodological terms described. Of these, only 9 articles met the inclusion criteria. Divergences were resolved by the fifth reviewer (►Figure 1 ). Of the total number of studies included in this systematic review (►Table 1 ), 4 evaluated the effects of quinacrine, 17-20 2 of pentosan polysulfate (PPS), 21,22 and 2 of doxycycline 23,24 on increasing survival times (months) of CJD patients, while 1 paper analyzed the effects of flupirtine on cognitive deterioration in this population. 25 Among the articles elected for this review, 4 studies conducted double-blind randomized clinical trials 18,23-25 (►Table 2 ).
Figure 1. Abbreviations: NRCT, nonrandomized clinical trial; RCT, randomized clinical trial. Methodological flowchart of the systematic review steps, PRISMA 2020 statement. 42 *The study by Varges et al. 24 was composed of 2 groups, one as RCT and the other as NRCT.
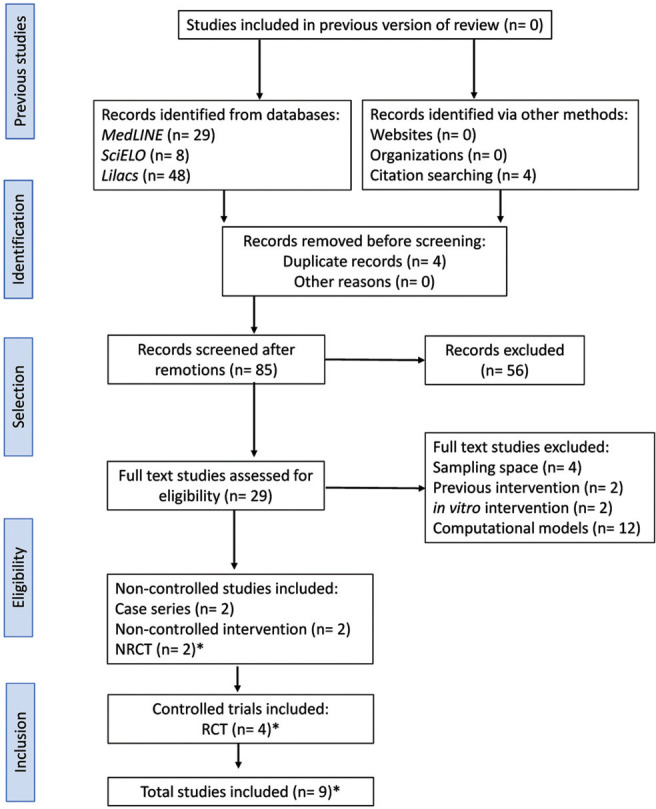
Table 1. Selected studies at the end of the analyses.
| Authors | Study design | Sampling | Intervention | Control | Primary outcome |
|---|---|---|---|---|---|
| Bone et al. (2008) 21 | Case series | n = 7 | PPS | − | Increased survival |
| Collinge et al. (2009) 17 | NRCT | n = 107 | Quinacrine | − | Increased survival |
| Geschwind et al. (2013) 18 | RCT | n = 51 (I= 23, C= 28) | Quinacrine | Placebo | Increased survival |
| Haïk et al. (2004) 19 | Case series | n = 32 | Quinacrine | − | Increased survival |
| Haïk et al. (2014) 23 | RCT | n = 121 (I= 62, C= 59) | Doxycycline | Placebo | Increased survival |
| Nakajima et al. (2004) 20 | NCI | n = 4 | Quinacrine | − | Increased survival |
| Otto et al. (2004) 25 | RCT | n = 28 (I= 13, C= 15) | Flupirtine | Placebo | Cognitive decline reduction |
| Tsuboi et al. (2009) 22 | NCI | n = 11 | PPS | − | Increased survival |
| Varges et al. (2017) 24 | RCT* | n = 13 (I= 7, C= 6) | Doxycycline | Placebo | Increased survival |
| Varges et al. (2017) 24 | NRCT* | n = 88 (I= 55, C= 33) | Doxycycline | − | Increased survival |
Abbreviations: C, control group; I, intervention group; NCI, non-controlled intervention; NRCT, non-randomized clinical trial; PPS, pentosan polysulfate; RCT, randomized clinical trial.
Notes: *The study by Varges et al. 24 was composed of 2 groups, one as RCT and the other as NRCT.
Table 2. Double-blind randomized clinical trials.
| Study | Geschwind et al. (2013) 18 | Haïk et al. (2014) 23 | Otto et al. (2004) 25 | Varges et al. (2017) 24 |
|---|---|---|---|---|
| Design | Crossover | Crossover | Crossover | Crossover |
| Intervention (I) | Quinacrine | Doxycycline | Flupirtine | Doxycycline |
| Dosage | 300 mg/day | 100 mg/day | 300 mg/day | 100 mg/day |
| Median follow-up time (months) | 2 | 5.5 | 1.3 | 7 |
| Control (C) | Placebo | Placebo | Placebo | Placebo |
| Sampling (I:C) | 51 (23:28) | 121 (62:59) | 28 (13:15) | 13 (7:6) |
| Median age (±DP) | 63 (±9.4) | 63 (±10.1) | 59 (±9.9) | 65 (±10.0) |
| Gender (M–F) | 31–20 | 51–70 | 13–15 | 7–5 |
| Previous intervention | No | No | No | No |
| Primary outcome | Increased survival | Increased survival | Cognitive decline reduction* | Increased survival |
| Results | I = C | I = C | I > C | I = C |
| Measure of association | HR: 95% CI = 1.43 (0.58–3.53); p= 0.43 |
HR: 95% CI= 1.10 (0.8–1.7); p= 0.50 |
One-tailed t-test; p= 0.02 |
HR: 95% CI = 0.84 (0.24–2.97); p ≤ 0.50** |
Abbreviations: CI, confidence interval.
Notes: *Assessed by ADAS-Cog: cognitive subscale of the Alzheimer disease assessment scale (ADAS); ** the study did not specify the p-value associated with this HR on the double-blind randomized clinical trial group.
In a case series written by Haïk et al., 19 the quinacrine efficacy was evaluated in 32 patients, 30 of whom were carriers of sporadic CJD (sCJD) and 2 of variant Creutzfeldt-Jakob disease (vCJD). Patients were reevaluated every 30 days until death. The experimental group did not demonstrate a significant increase in survival compared with untreated patients. It should be noted that this same study demonstrated a slight increase in survival time among treated patients, but that was attributed to the excess presence of methionine/valine and valine/valine genotypes when compared with methionine/methionine.
In a study of 11 patients by Tsuboi et al. 22 the increased survival with PPS infusion was investigated. During the research, 7 of the patients died with infectious complications and the remaining 4 continued receiving the treatment. According to the results of the study, all patients had increased survival times when compared with similar cases, at a mean time of 24.2 months. All treated cases reported deterioration of cognitive functions to varying degrees, in addition to subdural fluid collection. No other side effects were observed in the patients of the group, although side effects such as thrombocytopenia and coagulation disturbances are common in the administration of this drug.
This study has shown that the infusion of the drug does not directly affect the reversal or preservation of the patients' pathology, although it does result in an improvement in life expectancy. It has not been established whether higher doses of the drug would result in a better outcome. Postmortem analysis of the patients showed that administration of the drug reduced the prion deposition in the brain. 22
In a double-blind, randomized clinical trial with 28 patients conducted by Otto et al., 25 the use of flupirtine maleate (n= 13) and placebo (n= 15) were compared regarding cognitive function improvement in patients with CJD. For inclusion and continuation in the study, patients were required to achieve at least 50% on 2 of the 12 subscales of the dementia tests used. A battery of standardized questionnaires was employed to monitor disease progression. The main outcome variable was the cognitive portion of the Alzheimer disease assessment scale (ADAS-Cog). The difference between baseline and the best score under treatment was defined as the primary efficacy variable for hypothesis testing.
Patients treated with flupirtine were found to have significantly less deterioration in dementia tests for memory and orientation items, when compared with patients treated with placebo, while there was no improvement in communicative functions of this population. This outcome was restricted to the patients' cognitive issues, and was not observed positively in patients' survival time. This shows that flupirtine has beneficial effects in partially reducing the deterioration of cognitive functions in patients with CJD, but does not alter the survival time of these patients. 25
In a study using a daily dose of 100 mg doxycycline, Varges et al. 24 randomized 13 patients with sCJD into group 1, which was double-blinded; 7 patients were placed in the intervention group and 6 in the placebo control group. There was no significant difference between the groups in the increase in survival time, which was the primary endpoint; the hazard ratio (HR), measured with 95% confidence interval (CI), was 0.84 (0.24–2.97). There was also no significant difference in the quality of life of this population, as measured by the mini mental status examination and the Barthel index.
In this same study, another 88 patients were followed in a non-randomized fashion, of which 55 were enrolled in the doxycycline protocol, and 33 were not, making up group 2 of this study. Patients with sCJD started the study with a longer time to diagnosis and received the treatment for a longer time, when compared with group 1. In the stratified analysis, there was increased survival time in the intervention group (HR: 95% CI = 0.61, 0.37–0.99), and the association with the genetic profile was also a significant factor in the primary outcome analysis, with the methionine/valine genotype having the greatest increase in survival, followed by the valine/valine genotype. The authors merged the 2 groups in a meta-analysis, with heterogeneity estimated at 0 and observed superiority of doxycycline use (HR: 95% CI = 0.63, 0.40–0.99; p= 0.049) when compared with the control group. 24
In another study using doxycycline, Haïk et al. 23 conducted a double-blind, randomized clinical trial, in which all selected patients were over 18-years-old, with probable or definitive diagnosis of CJD through Italian and French reference centers, with age and gender in similar proportions. The outcome analyzed was the increase in survival times after the intervention, compared with the placebo group. For this, 121 patients were included. The intervention group had a total of 62 patients, who received a dosage of 100 mg of doxycycline per day, while the control group, containing 59 patients, received placebo.
In this study, it was observed that using a dosage of 100 mg/day of doxycycline compared with placebo brought no increase in survival times (HR: 95% CI = 1.1, 0.8–1.7). There were no significant side effects related to the therapy implementation. The authors stated that even with the failure of the study, the benefits of using this drug in higher dosages, such as 200 mg/day, are not ruled out, and can be researched further. Additionally, it was suggested that further research involving the use of doxycycline should select patients with early stages of the disease, or even presymptomatic patients, where benefit may be more possible, as demonstrated in animal models. 23
In a study evaluating the quinacrine efficacy in the treatment of prion diseases, Nakajima et al. 20 included 4 patients, 3 with probable sCJD and 1 with iatrogenic sCJD. As a result, quinacrine was well tolerated by all 4, who had improved arousal levels after quinacrine treatment. Other changes in global brain function included decreased reflex frequencies or action myoclonus, startle response, and mitigation of the hyperkinetic state. No other medications were administered during the quinacrine administration period.
In this study, the four patients received 300 mg/day of quinacrine via nasogastric tube for 3 months. Transaminase values were elevated in 3 of the 4 patients, but never reached 5 times the upper normal limits. Patients 1 and 2 had quinacrine temporarily suspended because of aspiration pneumonia, urinary tract infection, and diarrhea, but completed the 3-month course of treatment. 20
In an observational study evaluating the efficacy of continuous PPS infusion in the treatment of prion diseases, Bone et al. 21 evaluated 7 patients, 3 with vCJD. In this study, there was no standardization of the dose administered and the results indicated that the drug was well tolerated over a wide therapeutic range (11–110 µg/kg/day). According to the study results, the time for disease onset of all patients in this study exceeded the average reported in the literature.
Another study, conducted by Collinge et al., 17 had a total of 107 patients with prion diseases who were recruited through the UK national referral system and had the option of choosing between quinacrine (300 mg/day), no quinacrine, or randomization to immediate quinacrine or deferred quinacrine in an open trial of patient preference. The primary endpoints were death and serious adverse events related to the experimental drug.
In this study, among the 107 patients included, 45 were sporadic, two iatrogenic, 18 variant, and 42 hereditary. From those, 23 patients were allocated to a pilot study, and 84 to the main study. Only two patients chose randomization; 40 took quinacrine during follow-up; and 37 of these chose it at enrollment. Treatment choice was associated with disease severity, with the least and most severely affected being more likely to choose not to receive quinacrine. Of the 78 (73%) patients who died, 1 was randomly assigned to deferred treatment, 26 (68%) chose immediate quinacrine, and 51 (75%) did not choose quinacrine. There was no difference in mortality between the groups after adjustment. 17
A double-blind, placebo-controlled, stratified randomization, clinical trial was conducted by Geschwind et al. 18 to evaluate the effect of 300 mg quinacrine on survival in patients with sCJD. Patients were randomized 1:1 between quinacrine or placebo, with hospitalizations for patient evaluation planned at months 2, 6, and 12 of treatment. Patients who returned for the 2-month evaluation were offered an open trial of quinacrine. Of the 425 patients referred, only 51 participated in the trial and survival analysis, with 28 in the placebo group and 23 in the intervention group.
The survival for the randomized portion of this trial over the first 2 months showed no significant difference between both groups (HR: 95% CI = 1.43, 0.58–3.53). Patients who took quinacrine treatment, however, showed a smaller decline in 2 of 3 functionality assessment scales (the modified Rankin and the clinical dementia rating) when compared with the placebo group in the first 2 months. Starting at 2 months, subjects who returned for evaluation were able to choose whether or not to start quinacrine, eliminating randomization. Because of this, the secondary survival analysis was performed using a Cox proportional hazards model with a time-dependent treatment group variable. The survival of patients who chose quinacrine did not differ significantly from those who did not (HR: 95% CI = 0.86, 0.44–1.70; p= 0.67). 18
DISCUSSION
The planning and execution of clinical trials in patients with CJD is hampered by several difficulties inherent to the context of this condition. Moreover, as expected for systematic reviews and metanalyses, the quality of the aggregated results is directly proportional to, among other things, the individual robustness and homogeneity of the selected primary studies. Thus, the adoption of quality assessment criteria and in-depth critical analysis of the available evidence allows for a greater possibility of extrapolation of the results to this population of interest, since it reduces the influence of systematic and random errors on the final product of the analysis.
Due to the low incidence of CJD, there is a difficulty in patients' clinical management, both due to the assisting professionals' lack of knowledge and the difficulty of having well-defined flowcharts. 3,15 It is also important to highlight that the diagnostic criteria are not fully elucidated or consolidated, which contributes to the delay in diagnosis, classification for studies, as well as the underreporting of cases. As it is known, CJD progresses to death in a few months, 4 making the quality of life of these patients of paramount importance in this short period. Good communication with the family, along with a multidisciplinary care is fundamental for the patient to have the best assistance possible, since most patients with CJD will be indicated for palliation, to reduce their suffering and improve their quality of life.
Studies have shown that the malaria treatment drug quinacrine and the antipsychotic chlorpromazine are effective in clearing prions in non-human models. 26-28 However, chlorpromazine probably has a higher risk of toxicity at its estimated therapeutic dose when compared with quinacrine. 28 Although the antiprionic mechanism of quinacrine in ScN2a cells is unknown, some investigators suggest that quinacrine works through binding to the cellular prion protein (PrPC). 14,29,30 Favorable results were obtained in inhibiting the accumulation of protease-resistant prion protein (PrPres) in N2a58/22L and ScN2a cells, but this drug did not show any defibrillogenic effects, nor did it alter prion resistance to Protease K. 31 This suggests that quinacrine is not effective in breaking down preformed PrPres aggregates, and, consequently, its effects on the course of the disease are limited, since these neuronal-damaging aggregates are very slowly degraded in the brain. Quinacrine has also been observed to reduce microglia-mediated cell death in ScN2a cells, possibly due to an inhibition of PrPres formation and consequent reversal of cell membrane changes that are recognized by mycroglia. 28,31 However, in mouse studies it has been shown that quinacrine has a balancing role, so that the drug interferes with PrPres synthesis and catabolism at different levels depending on the cell type, proving it is not an efficient treatment. 31
Flupirtine is a triaminopyridine derivative that acts as a non-opioid analgesic. It is a well-tolerated drug that acts on the CNS, and has demonstrated a high cytoprotective effect in vitro and in vivo on neurons induced to apoptosis. 14,32,33 The antiapoptotic effect has a potential for good results in patients with dementia, although more studies are needed. Its pharmacodynamic effects are mainly attributed to its function of selectively opening neuronal potassium channels and its NMDA antagonist property. 34 It has also been hypothesized that patients treated with flupirtine have a neuroprotective effect, due to positive regulation of the bcl 2 proto-oncogene, in addition to normalization of glutathione levels. 35 In vitro and in vivo tests suggest that flupirtine antagonizes neurotoxicity caused by the abnormal prion protein (PrPSc). 14,29,35 The study conducted by Otto et al. 25 demonstrated partial beneficial effects in reducing cognitive deterioration in patients with CJD.
Doxycycline is an antibiotic from the tetracycline group, whose effect is directly aimed at inhibiting protein synthesis, which prevents the nutrition of bacteria. Unlike quinacrine, evidence suggests that this drug has a defibrillogenic role and reverses proteinase K resistance in CJD patients. 36,37 Tetracyclines have an amphiphilic structure, which allows strong interaction with the lipophilic domains of PrPSc. 35,36 Additionally, tetracyclines interact with oligomeric structures and inhibit the protein folding failure that is associated with PrPSc formation. 14,29,36-38 It was clear from the studies reviewed that the drug is able to cross the blood-brain barrier and remain there for days, possibly due to its ability to remain bound to prion aggregates.
Finally, PPS is a drug with anticoagulant and fibrinolytic effects. 14,29 Studies conducted have hypothesized that infusion of the drug could inhibit PrPSc formation. 14,29,39 Its mechanism of action has not yet been identified, but it is suggested that PPS competitively interferes with the binding of PrPC and PrPSc with endogenous glycosaminoglycans, which are essential for the formation or stabilization of PrPSc. 39,40 However, PPS does not penetrate the blood-brain barrier when administered orally or parenterally. To address this, Doh-ura et al. 41 administered it intraventricularly via an infusion pump device in an animal model. This work evaluated a wide therapeutic range and demonstrated that PPS infusion decreased not only PrPSc deposition, but also neurodegenerative and infectious changes. These changes were observed within the cerebral hemisphere that received the intraventricular infusion, but not in the contralateral hemisphere. 41 In contrast, human studies have been plagued by small sample sizes and many of the expected complications for this drug. 21,22
The difficulty of grouping a significant number of patients is evident in all published studies. The study with the largest population was performed by Haïk et al., in 2014, with 121 patients, which added strength to their findings. 23 With less robustness, the same can be said of Geschwind et al. (n= 51). 18 Another well-structured factor of the selected articles was the division of patients into groups according to presentation, with majority use of sporadic CJD cases. Studies such as Geschwind et al. used only sCJD patients, 18 whereas Haïk et al. used both sCJD and genetic CJD patients. 23 Additionally, articles such as Tsuboi et al. presented cases of sCJD, genetic CJD, and iatrogenic CJD. 22 Thus, a wide distribution among the subtypes of CJD studied was observed, which has made it possible to compare therapeutic action within a wide spectrum of the disease, and helped to give strength to the resulting outcomes.
As a rapidly progressive dementia with high mortality, added to the presence of a vacuum in viable treatment alternatives, a range of studies have been mounted in the hopes of achieving minimal progression in the treatment of CJD. However, the fact that many of the applied drugs have never reported significant reduction in prion accumulation in vitro or shown positive development in animal models raises doubts in the initial validity of clinical trials, as is the case, for instance, with preclinical data from studies of doxycycline. 23
It is important to highlight that the analyzed studies have as common characteristics association measures anchored in p-values very close to the significance limit, a fact present in 3 of the randomized clinical trials, 18,23,24 except for the study by Otto et al., 25 which observed a p-value far from the significance margin. Although the analyzed randomized clinical trials had exposed their treatment protocol's follow-up flowcharts, some deficiencies were observed. The simplicity of the variables exposed in this flowchart, the intermediate number of patients submitted to randomization, and unclear treatment periods, 25 borderline confidence intervals, with a mixture of observational design and unfavorable clinical trial, and the small sample size 22 are some of the weaknesses of these studies.
Regarding the non-controlled studies, they are basically divided into non-randomized clinical trials and case series. Case series have major limitations for analyzing outcomes, mainly because there is no control group, and therefore they are restricted to analyses within the limits of the sample studied, without much power of extrapolation. The article conducted by Bone et al., 21 besides the limitations inherent to the design itself, also used a very small number of patients (n= 7), of which only 4 were followed throughout the study, thus compromising the reach of the results obtained. The study by Haïk et al., 19 although with a larger sample (n= 32), is also limited to affirm some advantage in the use of quinacrine in patients with CJD, since it analyzes outcomes from observation, with p-values at levels below those required. The study conducted by Varges et al., 24 despite having a slightly larger sample (n= 88) and a form of patient allocation that aimed to include only patients in the early stage of the disease, did not randomly distribute its patients, with a large numerical predominance in the intervention group over the control group.
These non-randomized clinical trials also presented important limitations in the sense of extrapolation of results, since the randomization of patients avoids several biases that can be introduced when allocating patients. The study by Collinge et al. 17 had the largest sample size of all these studies. In it, patients had the option to choose whether or not to be treated with quinacrine from the start, or even during the course of the study. This type of design meant that the vast majority of patients did not opt for randomization. Moreover, the severity profile of the patients within the study was diverse, preventing a proper conclusion of the inefficiency of the drug by the advanced stage of the pathology in some, or by the lack of response to the drug itself.
Given the above, this review has demonstrated that the vast majority of the papers analyzed here lack strong methodologies and, consequently, possibilities for extrapolation relevant to clinical practice on the management of CJD patients. To date, there are no robust studies that provide support for pharmacological approaches with doxycycline, PPS, or quinacrine in increasing survival of CJD patients. However, one clinical trial has suggested success of flupirtine in partially reducing cognitive deterioration in this population, when compared with the control group. Therefore, this review supports the need for further studies to evaluate the efficacy of different pharmacological options used in patients with CJD, to establish more solid treatment alternatives for these patients in the future.
References
- 1.Geschwind MD. Prion Diseases. Continuum (Minneap Minn) 2015;21(6 Neuroinfectious Disease):1612–1638. doi: 10.1212/CON.0000000000000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson RT. Prion diseases. Lancet Neurol. 2005;4(10):635–642. doi: 10.1016/S1474-4422(05)70192-7. [DOI] [PubMed] [Google Scholar]
- 3.Groveman BR, Foliaki ST, Orru CD, et al. Sporadic Creutzfeldt-Jakob disease prion infection of human cerebral organoids. Acta Neuropathol Commun. 2019;7(01):90. doi: 10.1186/s40478-019-0742-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma S, Mukherjee M, Kedage V, Muttigi MS, Rao A, Rao S. Sporadic Creutzfeldt-Jakob disease-a review. Int J Neurosci. 2009;119(11):1981–1994. doi: 10.1080/00207450903139762. [DOI] [PubMed] [Google Scholar]
- 5.Ministério da Saúde Protocolo de notificação e investigação da doença de Creutzfeldt-Jakob com foco na identificação da nova variante. Brasília, DF; 2018 [Google Scholar]
- 6.Sigurdson CJ, Bartz JC, Glatzel M. Cellular and Molecular Mechanisms of Prion Disease. Annu Rev Pathol. 2019;14(01):497–516. doi: 10.1146/annurev-pathmechdis-012418-013109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terry C, Wadsworth JDF. Recent Advances in Understanding Mammalian Prion Structure: A Mini Review. Front Mol Neurosci. 2019;12:169. doi: 10.3389/fnmol.2019.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maddox RA, Person MK, Blevins JE, et al. Prion disease incidence in the United States: 2003-2015. Neurology. 2020;94(02):e153–e157. doi: 10.1212/WNL.0000000000008680. [DOI] [PubMed] [Google Scholar]
- 9.Uttley L, Carroll C, Wong R, Hilton DA, Stevenson M. Creutzfeldt-Jakob disease: a systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect Dis. 2020;20(01):e2–e10. doi: 10.1016/S1473-3099(19)30615-2. [DOI] [PubMed] [Google Scholar]
- 10.Al-Ansari A, Robertson NP. Creutzfeldt-Jacob disease: new directions in diagnosis and therapeutics. J Neurol. 2017;264(05):1029–1031. doi: 10.1007/s00415-017-8473-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korth C, Peters PJ. Emerging pharmacotherapies for Creutzfeldt-Jakob disease. Arch Neurol. 2006;63(04):497–501. doi: 10.1001/archneur.63.4.497. [DOI] [PubMed] [Google Scholar]
- 12.Vetrugno V, Puopolo M, Cardone F, Capozzoli F, Ladogana A, Pocchiari M. The future for treating Creutzfeldt-Jakob disease. Expert Opin Orphan Drugs. 2015;3(01):57–74. [Google Scholar]
- 13.Appleby BS, Yobs DR. In: Handbook of Clinical Neurology. Pocchiari M, Manson J, editors. Cambridge: Elsevier;; 2008. Symptomatic treatment, care, and support of CJD patients; pp. 399–408. [DOI] [PubMed] [Google Scholar]
- 14.Chen C, Dong X. Therapeutic implications of prion diseases. Biosafety Health. 2021;3(02):92–100. [Google Scholar]
- 15.Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(05):E2. doi: 10.3171/2015.8.FOCUS15328. [DOI] [PubMed] [Google Scholar]
- 16.CDC’s Diagnostic Criteria for Creutzfeldt-Jakob Disease (CJD) Adapted from: a) Global surveillance, diagnosis, and therapy of human transmissible spongiform encephalopathies: Report of a WHO consultation, February 9-11, 1998, Geneva, Switzerland; b) Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009, 132; 2659-2668; and c) National CJD Research & Surveillance Unit. 2018. [Accessed 18 May 2022]. < https://www.cdc.-gov/prions/cjd/diagnostic-criteria.html>.
- 17.Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patientpreference trial. Lancet Neurol. 2009;8(04):334–344. doi: 10.1016/S1474-4422(09)70049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geschwind MD, Kuo AL, Wong KS, et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology. 2013;81(23):2015–2023. doi: 10.1212/WNL.0b013e3182a9f3b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haïk S, Brandel JP, Salomon D, et al. Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology. 2004;63(12):2413–2415. doi: 10.1212/01.wnl.0000148596.15681.4d. [DOI] [PubMed] [Google Scholar]
- 20.Nakajima M, Yamada T, Kusuhara T, et al. Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord. 2004;17(03):158–163. doi: 10.1159/000076350. [DOI] [PubMed] [Google Scholar]
- 21.Bone I, Belton L, Walker AS, Darbyshire J. Intraventricular pentosan polysulphate in human prion diseases: an observational study in the UK. Eur J Neurol. 2008;15(05):458–464. doi: 10.1111/j.1468-1331.2008.02108.x. [DOI] [PubMed] [Google Scholar]
- 22.Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology. 2009;29(05):632–636. doi: 10.1111/j.1440-1789.2009.01058.x. [DOI] [PubMed] [Google Scholar]
- 23.Haïk S, Marcon G, Mallet A, et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(02):150–158. doi: 10.1016/S1474-4422(13)70307-7. [DOI] [PubMed] [Google Scholar]
- 24.Varges D, Manthey H, Heinemann U, et al. Doxycycline in early CJD: a double-blinded randomised phase II and observational study. J Neurol Neurosurg Psychiatry. 2017;88(02):119–125. doi: 10.1136/jnnp-2016-313541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology. 2004;62(05):714–718. doi: 10.1212/01.wnl.0000113764.35026.ef. [DOI] [PubMed] [Google Scholar]
- 26.Korth C, May BC, Cohen FE, Prusiner SB. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A. 2001;98(17):9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74(10):4894–4897. doi: 10.1128/jvi.74.10.4894-4897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witt A, Campeau J, Sim V. Combination Therapy with Doxycycline, Chlorpromazine or Quinacrine Inhibits Prion Protein Replication in a N2A Model (P5.204) Neurology. 2016;86(16) [Google Scholar]
- 29.Teruya K, Doh-Ura K. Insights from Therapeutic Studies for PrP Prion Disease. Cold Spring Harb Perspect Med. 2017;7(03):24–43. doi: 10.1101/cshperspect.a024430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vogtherr M, Grimme S, Elshorst B, et al. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J Med Chem. 2003;46(17):3563–3564. doi: 10.1021/jm034093h. [DOI] [PubMed] [Google Scholar]
- 31.Barret A, Tagliavini F, Forloni G, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003;77(15):8462–8469. doi: 10.1128/JVI.77.15.8462-8469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dhar S, Bitting RL, Rylova SN, et al. Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3- and CLN2-deficient neurons. Ann Neurol. 2002;51(04):448–466. doi: 10.1002/ana.10143. [DOI] [PubMed] [Google Scholar]
- 33.Müller WE, Romero FJ, Perovic S, Pergande G, Pialoglou P. Protection of flupirtine on beta-amyloid-induced apoptosis in neuronal cells in vitro: prevention of amyloid-induced glutathione depletion. J Neurochem. 1997;68(06):2371–2377. doi: 10.1046/j.1471-4159.1997.68062371.x. [DOI] [PubMed] [Google Scholar]
- 34.Kornhuber J, Bleich S, Wiltfang J, Maler M, Parsons CG. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2þ block via activation of voltage independent potassium channels. Rapid communication. J Neural Transm (Vienna) 1999;106(9-10):857–867. doi: 10.1007/s007020050206. [DOI] [PubMed] [Google Scholar]
- 35.Perovic S, Schröder HC, Pergande G, Ushijima H, Müller WE. Effect of flupirtine on Bcl-2 and glutathione level in neuronal cells treated in vitro with the prion protein fragment (PrP106-126) Exp Neurol. 1997;147(02):518–524. doi: 10.1006/exnr.1997.6559. [DOI] [PubMed] [Google Scholar]
- 36.Tagliavini F, Forloni G, Colombo L, et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrPSc in vitro11Edited by J. Karn. Journal of Molecular Biology. 2000;300(05):1309–1322. doi: 10.1006/jmbi.2000.3840. [DOI] [PubMed] [Google Scholar]
- 37.Forloni G, Salmona M, Marcon G, Tagliavini F. Tetracyclines and prion infectivity. Infect Disord Drug Targets. 2009;9(01):23–30. doi: 10.2174/1871526510909010023. [DOI] [PubMed] [Google Scholar]
- 38.Di Fede G, Giaccone G, Salmona M, Tagliavini F. Translational Research in Alzheimer’s and Prion Diseases. J Alzheimers Dis. 2018;62(03):1247–1259. doi: 10.3233/JAD-170770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petrosyan R, Patra S, Rezajooei N, Garen CR, Woodside MT. Unfolded and intermediate states of PrP play a key role in the mechanism of action of an antiprion chaperone. Proc Natl Acad Sci U S A. 2021;118(09):e2010213118. doi: 10.1073/pnas.2010213118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caughey B, Raymond GJ. Sulfated polyanion inhibition of scrapieassociated PrP accumulation in cultured cells. J Virol. 1993;67(02):643–650. doi: 10.1128/jvi.67.2.643-650.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doh-ura K, Ishikawa K, Murakami-Kubo I, et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78(10):4999–5006. doi: 10.1128/JVI.78.10.4999-5006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi: 10.1136/bmj.n71. [DOI] [PMC free article] [PubMed] [Google Scholar]