

Abstract
Given the highly variable clinical phenotype of Coronavirus disease 2019 (COVID-19), a deeper analysis of the host genetic contribution to severe COVID-19 is important to improve our understanding of underlying disease mechanisms. Here, we describe an extended genome-wide association meta-analysis of a well-characterized cohort of 3255 COVID-19 patients with respiratory failure and 12 488 population controls from Italy, Spain, Norway and Germany/Austria, including stratified analyses based on age, sex and disease severity, as well as targeted analyses of chromosome Y haplotypes, the human leukocyte antigen region and the SARS-CoV-2 peptidome. By inversion imputation, we traced a reported association at 17q21.31 to a ~0.9-Mb inversion polymorphism that creates two highly differentiated haplotypes and characterized the potential effects of the inversion in detail. Our data, together with the 5th release of summary statistics from the COVID-19 Host Genetics Initiative including non-Caucasian individuals, also identified a new locus at 19q13.33, including NAPSA, a gene which is expressed primarily in alveolar cells responsible for gas exchange in the lung.
Introduction
In the past year, Coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has evolved into a global pandemic with more than 532 million confirmed cases and 6.3 million COVID-19-related deaths worldwide (frequencies reported by the World Health Organization, June 11, 2022). The clinical manifestations of COVID-19 are variable and range from complete absence of symptoms to severe respiratory failure and death. Severe COVID-19 requires intensive medical care with respiratory support and can result in long-term damages detrimental to the individual. The pathogenesis of severe COVID-19 is, however, still poorly understood. This condition has been associated with clinical risk factors such as old age, male sex and comorbidities including diabetes, active cancer, hypertension and coronary artery disease (CAD) as well as solid organ transplant or other conditions that promote an immunosuppressive state (1–4).
Different studies, including the first genome-wide association analysis (GWAS) by of our Severe COVID-19 GWAS study group, have shown that genetic predisposition plays a role in COVID-19 susceptibility and severity (5–7). In particular, we reported significant associations between genetic variants at loci 3p21.31 (around LZTF1) and 9q34.2 (ABO blood group locus) to severe respiratory failure and SARS-CoV-2 infection in 1610 severe COVID-19 patients and 2205 population controls of European ancestry. Since then, these loci have been replicated in subsequent studies and extended also to non-European cohorts (8–10). Eleven additional genome-wide significant loci, associated with SARS-CoV-2 infection or COVID-19 manifestations, have been reported by various studies, including the Genetics Of Mortality In Critical Care (GenOMICC) Initiative and, more recently, the largest genetic consortium for COVID-19, the COVID-19 Host Genetics initiative (HGI) (6,7). Six of these loci have been linked to critical illness by COVID-19 and include genes that were previously associated with pulmonary or autoimmune and inflammatory diseases (7).
Since our primary publication, the Severe COVID-19 GWAS study group dataset has been extended to 3255 severe COVID-19 patients with respiratory failure (5) and 12 488 controls with unknown COVID-19 status from 30 study centers across Italy, Spain, Norway and Germany/Austria (after quality control (QC); Supplementary Material, Table S1A and D). Compared with our previous study, we increased the number of patients and controls by approximate factors of 2 and 5.5, respectively (from 1610 and 2205). Furthermore, detailed information on age, sex and comorbidities as hypertension, diabetes and CAD is available in this dataset. The control cohort included predominantly population controls recruited at the respective centers as blood or bone marrow donors with unknown or negative COVID-19 status (Supplementary Material, Table S1C). With this unique resource at hand, we performed an extended genome-wide association study (GWAS) and meta-analysis of severe COVID-19 as well as a meta-analysis with the COVID-19 HGI release 5 GWAS summary statistics, followed by stratified analyses of age, sex and disease and a detailed analysis of ABO secretor status. Using Y-chromosomal genotype calls from the Global Screening Array (GSA), we additionally performed an imputation and disease association of chromosome Y haplotypes. Compared with our first study, we also performed an even more detailed analysis of the human leukocyte antigen (HLA) which includes classical fine-mapping of the HLA region based on local imputation of SNP, amino acid and classical allele information, as well as a broad range of other approaches, including a peptidome-wide association study (PepWAS) (11) computational prediction of SARS-CoV-2 peptide presentation, HLA class I supertype association analysis and tests for heterozygote advantage, divergent allele advantage and molecular mimicry. Finally, by inversion imputation, we present a functional analysis of the established 17q21.31 locus (7), which has been described by COVID-19 HGI but not characterized in depth.
Results
GWAS meta-analyses for severe COVID-19 with respiratory failure
Genotyping was carried out using Illumina’s GSA, followed by genotype QC analysis and TOPMed genotype imputation (Material and Methods, Supplementary Material, Figure S1, patient numbers before and after QC are shown in Supplementary Material, Table S1A and C). Respiratory failure was defined as respiratory support with supplemental oxygen [class 1] or non-invasive and invasive ventilation [classes 2 and 3, respectively], or by extracorporeal membrane oxygenation (ECMO) [class 4]).(5) Analogously to the COVID-19 HGI (7), we conducted two GWAS discovery meta-analyses for two main categories of COVID-19 disease state: First, ‘hospitalization with respiratory support’ (respiratory support classes 1–4 with a total of 3255 patients and 12 488 controls, main analysis), and second, a more stringent definition of severe COVID-19 ‘hospitalization with mechanical ventilation’ (classes 2–4 with 1911 critically ill individuals and 12 226 controls, subtype analysis). Details of per cohort patient numbers are shown in Supplementary Material, Table S1E. The characteristics of patients and controls included in our analyses are shown in Table 1 and Supplementary Material, Table S2. After imputation, we carried out a GWAS of 9 223 806 and 9175283 high-quality genetic variants [imputation R2 ≥ 0.6 and minor allele frequency (MAF) ≥1%] stratified by ancestry (Italy, Spain, Norway and Germany/Austria) using a logistic mixed model analysis as implemented in SAIGE (12), followed by a fixed-effect inverse variance-weighted meta-analysis using METAL (13) (low genomic inflation of 1.017; Supplementary Material, Figs S2 and S3). Genome-wide comparison of the case–control frequencies [conservatively adjusted for age, sex, age*age, sex*age and top 10 principal components (PCs) from principal component analysis (PCA) as employed by the COVID-19 HGI; Material and Methods]‚ revealed genome-wide significant associations (P < 5 × 10−8; Table 2 and Supplementary Material, Table S3, Figs S4 and S5) at three known loci in the main analysis: chr3:45848457:C:T (rs35731912) within LZTFL1 at the 3p21.31 locus, chr9:133261703:A:G (rs687289) within ABO at 9q34.2, chr19:10351837:C:T (rs11085725) within TYK2 at 19p13.2. In addition to the same TYK2 variant, chr19:4717660:A:G (rs12610495) within DPP9 at 19p13.3 was genome-wide significant in the subtype analysis. Additional suggestive loci (P < 10−6) from both analyses include previously reported associations at 17q21.31 (MAPT), 19p13.3 (DPP9) and 21q22.11 (IFNAR2) (Table 2 and Supplementary Material, Table S3) (7). Of note, the respective lead risk variants (this study versus COVID-19 HGI) at reported loci are linked with r2 or D’ > 0.9 across our study cohorts (Supplementary Material, Table S4). Although hierarchical mixture model analysis with MAMBA (14) (Materials and Methods) showed a high posterior probability of replication (PPR > 0.8) for suggestive associations at PCDH7 at 4p15.1, FREM1 at 9p22.3 (main analysis), OLMF4 at 13q21.1 and PTPRM at 18p11.23 (subtype analysis), they were not observed as significantly associated with COVID-19 in the COVID-19 HGI 5 release dataset, which is why we did not consider them further (forest plots including all data are shown in Supplementary Material, Fig. S6). Future analyses will be required to determine the significance of these associations to COVID-19.
Table 1.
Overview of patients included in the genome-wide discovery analysis
| Italy | Spain | Norway | Germany/Austria | |
|---|---|---|---|---|
| Total N severe COVID-19 patients | 1536 | 1421 | 262 | 241 |
| Respiratory support categories N (%) | ||||
| Supplemental oxygen only (1) | 276 (17.97) | 897 (63.53) | 80.65 | 109 (45.23) |
| Non-invasive ventilation (2) | 933 (60.74) | 122 (8.62) | 3.23 | 21 (8.71) |
| Ventilator (3) | 320 (20.83) | 381 (26.91) | 16.13 | 86 (35.68) |
| ECMO (4) | 7 (0.46) | 16 (1.13) | 0 | 25 (10.37) |
| Median age (IQR)—years (1) | 75 (21) | 69 (21) | 59 (20) | 63 (23) |
| Median age (IQR)—years (2) | 66 (23) | 72 (17) | 68.5 (10) | 68 (20) |
| Median age (IQR)—years (3) | 63 (14) | 65 (15) | 66 (19) | 64 (18) |
| Median age (IQR)—years (4) | 49 (12) | 57 (10) | / | 56 (12) |
| Main analysis | ||||
| N severe COVID-19 patients | 1536 | 1421 | 262 | 241 |
| Female sex—% | 32.81 | 35.88 | 33.87 | 26.97 |
| Median age (IQR)—year | 67 (22) | 68 (18) | 59.5 (21) | 63 (19) |
| Hypertension—% affected (% missing) | 40.55 (21.16) | 49.6 (1.91) | 41.3 (25.81) | 53.09 (32.78) |
| CAD—% affected (% missing) | 17.09 (21.16) | 10.16 (1.98) | 25.81 (0) | 15.23 (37.34) |
| Diabetes—% affected (% missing) | 14.86 (21.16) | 24.55 (1.91) | 14.52 (0) | 19.63 (32.37) |
| Subtype analysis | ||||
| N severe COVID-19 patients | 1260 | 519 | 132 | |
| Female sex—% | 29.68 | 27.55 | / | 22.73 |
| Hypertension % affected (% missing) | 38.26 (23.25) | 52.19 (3.28) | / | 62.96 (38.64) |
| CAD—% affected (% missing) | 16.03 (23.25) | 12.55 (3.28) | / | 18.75 (39.39) |
| Diabetes—% affected (% missing) | 14.58 (23.25) | 25.3 (3.28) | / | 19.75 (38.64) |
Overview of patients included in our first analysis (3260 patients) and second analysis (1911 patients). Individuals of the Italian, Spanish, Norwegian and German/Austrian cohorts were recruited at 5, 7, 8 and 10 different hospitals/centers, respectively. Shown are respiratory support status groups 1–4, age and median age across all individuals as well as within each respiratory support group, percentage of females within each cohort, as well as percentage of individuals affected by known comorbidities of COVID-19. Commonly reported comorbidities in COVID-19 are shown, hypertension, CAD and diabetes. Characteristics of control individuals are shown in Supplementary Material, Table S2.
Table 2.
Candidate variants from the main and subtype analysis
| snpid | rsid | A1 | A2 | FRQ | OR | OR_95L | OR_95U | PPR | P | gene | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Main analysis | chr3:45848457:C:T | rs35731912 | T | C | 0.09 | 1.784 | 1.723 | 1.847 | 1.000 | 2.32E−21 | LZTFL1 |
| chr19:10351837:C:T | rs11085725 | T | C | 0.273 | 1.269 | 1.254 | 1.283 | 1.000 | 1.02E−10 | TYK2 | |
| chr9:133261703:A:G | rs687289 | A | G | 0.358 | 1.240 | 1.219 | 1.262 | 1.000 | 4.49E−10 | ABO | |
| chr19:4717660:A:G | rs12610495 | G | A | 0.27 | 1.216 | 1.188 | 1.244 | 0.990 | 9.91E−08 | DPP9 | |
| chr9:14754866:A:G | rs7023573 | A | G | 0.285 | 1.207 | 1.185 | 1.231 | 0.954 | 5.21E−07 | FREM1 | |
| chr17:45933112:G:A | rs8065800 | G | A | 0.328 | 1.194 | 1.145 | 1.247 | 0.925 | 8.90E−07 | MAPT | |
| chr4:30946048:T:C | rs12512667 | C | T | 0.444 | 0.850 | 0.828 | 0.873 | 0.922 | 8.95E−07 | PCDH7 | |
| Subtype analysis | chr19:10351837:C:T | rs11085725 | T | C | 0.272 | 1.309 | 1.294 | 1.325 | 0.999 | 3.24E−09 | TYK2 |
| chr19:4717660:A:G | rs12610495 | G | A | 0.267 | 1.286 | 1.257 | 1.316 | 0.993 | 2.87E−08 | DPP9 | |
| chr13:55031860:T:C | rs111671068 | C | T | 0.012 | 2.802 | 2.794 | 2.810 | 0.806 | 1.43E−07 | OLFM4 | |
| chr9:133271182:T:C | rs550057 | T | C | 0.266 | 1.262 | 1.238 | 1.286 | 0.939 | 4.56E−07 | ABO | |
| chr18:7897309:A:C | rs17565758 | C | A | 0.078 | 1.510 | 1.470 | 1.550 | 0.938 | 4.84E−07 | PTPRM | |
| chr21:33247902:C:T | rs2834161 | C | T | 0.371 | 1.250 | 1.175 | 1.331 | 0.923 | 6.10E−07 | IFNAR2 |
We show only variants that have a PPR > 0.8 in the MAMBA analysis. A more detailed overview of all variants is shown in Supplementary Material, Table S3. snpid: id of the marker as chr:pos:alleles, genome build hg38; A1: minor allele from TopMed imputation; A2: major allele from TopMed imputation; OR/OR_95L/OR_95U: OR and 95% CI of A1; PPR: posterior probability of replication; P: P value of association.
Next, we performed a replication analysis of the 13 variants reported to be genome-wide significantly associated with COVID-19 by the COVID-19 HGI. Since data from the Severe COVID-19 group where shared with the COVID-19 HGI, release 5 of the HGI contains 3815 individuals from our previous analysis. We therefore included only individuals from the Severe COVID-19 GWAS group not contained in the COVID-19 HGI release 5 datasets (Material and Methods and Supplementary Methods) for this analysis. 8 of the 13 variants reported as genome-wide significant by the COVID-19 HGI (7) replicated at least a nominal P-value of 0.05 (located near LZTFL1, ABO, TYK2, DDP9, IFNAR2, MAPT/KANSL1, OAS1; Supplementary Material, Table S5). Subsequently, we performed a fixed-effect inverse variance-weighted meta-analysis using METAL (13) across the main (hospitalized with respiratory support) analysis and COVID-19 HGI B2 statistics as well as the subtype analysis (critically ill: hospitalized with mechanical ventilation) and COVID-19 A2 statistics, resulting in a total of 1 320 760 (14 467 and cases and 1 306 293 controls) and 725 601 (6526 cases and 719 075 controls) analysed individuals in the respective analyses as well as 9 163 456 and 9 309 373 high quality variants (imputation R2 ≥ 0.6 in our cohorts and overall MAF ≥ 1%; Supplementary Material, Figs S7 and S8). This analysis prioritized rs1819040 at the 17q21.31 locus within the KANSL1 gene as the most strongly associated variant in this region (P = 3.27 × 10−11 for rs1819040; OR = 0.88 for minor allele T; 95%CI = 0.84–0.92) over rs8065800 from the main analysis (Supplementary Material, Tables S3, S4 and S6). The meta-analysis of our subtype analysis with the COVID-19 HGI A2 summary statistics revealed an additional genome-wide significant locus not previously associated with severe COVID-19, chr19:50379362:T:C (rs1405655, P = 3.25 × 10−8; OR = 1.09; 95%CI = 1.06–1.13) located near the NAPSA gene at 19q13.33 (regional association and forest plots are shown in Supplementary Material, Fig. S9, Table S6). Bayesian fine-mapping analysis of this locus with FINEMAP (15) (Material and Methods) identified a total of 15 (log10(Bayes factor) = 5.95) variants that belong to the 95% most likely to be causal (Fig. 1, Supplementary Material, Table S7), indicating that a small number of SNPs could be considered causal in subsequent analyses.
Figure 1.

Forest plot of candidates from the in-depth stratified analysis. The plots show the variants chr3:45823240, chr3:45848457:C:T, chr17:46142465:T:A which were significantly associated with age (interaction P-value [P_FDR(I) < 0.05]) when comparing age groups ≤60 and >60. We additionally show variants chr19:4717660:A:G and chr21:33242905:T:C with insignificant, though strong trends for association with age. ORs and their respective 95% confidence intervals (CIs) are visualized for each of our four cohorts separately. The size of the dots indicates the size of the respective cohort (N). The OR value is displayed in addition as a numerical value. Only cohorts in which N > 50 in both cases and controls are shown. The headers are built as follows: gene variant id (chr:pos(hg38):allele)—rs-id—effect allele */**. * indicates a variant that was observed as associated from data in this study; **indicates a variant that was observed as associated in the release 5 of the COVID-19 HGI.
In-depth stratified analysis of lead variants
To estimate differences in genetic effects across different age groups and different biological sex categories, we performed an in-depth stratified analysis for selected variants. These included genome-wide and suggestively significant variants from this study (LZTFL1, ABO, TYK2, DPP9; MAPT, IFNAR2; 7 lead variants in total from the main and the subtype analysis). We additionally included all of the 13 variants from the COVID-19 HGI release 5 analysis (7) and the novel association, chr19:50379362:T:C, at the NAPSA locus. A detailed overview of these loci is shown in Supplementary Material, Table S8. We additionally investigated the association of these variants to known comorbidities such as hypertension, CAD and diabetes in cases only and performed sex-stratified analysis within age groups and with respect to disease severity groups. We performed interaction analysis of the SNP with sex and age (age group ≤ 60 and >60), respectively, to investigate sex-specific and age-specific effects, i.e. to determine whether differences in effects within these groups were statistically significant (Materials and Methods and Supplementary Methods). Results of these analyses are shown in Figure 1, Supplementary Material, Tables S8 and S9, Figures S10 and S11. We confirmed our previously described associations with age (Fig. 1) and with disease severity at 3p21.31 for chr3:45848457:C:T (severity: P = 9.73 × 10−7, OR = 1.65; 95%CI = 1.35–2.03), which is also discussed in detail in a study by Nakanishi et al. (16). Differential association with age was also observed for chr17:46142465:T:A (rs1819040, MAPT) (Fig. 1). Strong but non-significant trends for an association were also observed for chr19:4717660:A:G (rs12610495; DPP9) at the 19p13.3 locus, and chr21:33242905:T:C (rs13050728; IFNAR2) which was significantly associated with severe COVID-19 only in younger individuals before correction for multiple testing (Fig. 1). We analysed these 5 variants even more in-depth within the age groups of 41–50, 51–60 and 61–70 as well as 41–60 and 61–80 (Supplementary Material, Fig. S10C) and observed a tendency for stronger effects in the younger population at the four loci. Interestingly, the German population showed opposite effect sizes than the other cohorts in the broad age group analysis for chr17:46142465:T:A (see also below).
We did not observe any statistically significant associations with age, sex, severity or comorbidities for the remaining analysed lead variants. Additional age and sex stratified forest plots of this analysis are shown in Supplementary Material, Figure S11A and B.
As the genome-wide significant association near LTZF1, TYK2, DDP9 and ABO have been followed up in detail before, we next focused on the novel association at the 19q13.33 (NAPSA) locus from the meta-analysis with COVID-19 HGI release 5 summary statistics and performed an in-depth analysis of the 17q21.31 (MAPT, KANSL1) locus associated with age. The 17q21.31 locus has been previously reported to be linked to a common inversion polymorphism of two highly divergent haplotypes H1 and H2 (17). Indeed, Bayesian fine-mapping (15) at this locus showed that the 95% credible set includes 1530 variants among these chr17:45933112:G:A (rs8065800) from our initial discovery analysis and chr17:46142465:T:A (rs1819040) from the COVID-19 HGI analysis with certainty <0.3%, indicating, based on the overlap of LD boundaries of highly associated SNP variants with the inversion boundaries (Fig. 2A), that the individual SNP associations may be proxy variants for the associated inversion polymorphism.
Figure 2.

Association of the 17q21.31 locus with severe COVID-19 with respiratory failure. (A) Regional association plot showing the variant most strongly associated with severe COVID-19 (rs1819040, purple diamond) a ~ 0.9 Mb inversion polymorphism at 17q21.21 (56) (white line with blue rectangles representing the variable segmental duplication (SD) blocks at the breakpoints), and the large credible set obtained by statistical fine-mapping including 2178 SNPs in high LD (median [LD] = 0.97) with the inversion (Supplementary Material, Table S7). Pairwise LD values (r2) with lead variant rs1819040 were calculated from merged Italian, Spanish, German/Austrian and Norwegian GWAS main anlysis datasets. The dotted line indicates the genome-wide significance threshold (P = 5 × 10−8). Below, organization of the 17q21.31 inversion genomic region, with the extended haplotypes associated with each orientation (H1 and H2) shown as red and blue arrows, respectively, and breakpoint SDs as dark rectangles. Protein-coding genes for which the inversion is a lead eQTL in at least one GTEx tissue are shown as pointed rectangles indicating the direction of transcription. (B) Forest plot and extended meta-analysis of our first discovery analysis and the COVID-19 HGI release 5 analysis B2 dataset (Material and Methods) of the association between severe COVID-19 and the 17q21.31 inversion based on the presence relative to the absence of the inversion haplotype H2. We visualized the ORs and their 95% confidence invervals (CIs) across all analysed cohorts of the main analysis and the COVID-19 HGI release 5 analysis B2 data. In this analysis, the overlap between the main analysis cohort and the COVID-19 HGI data was excluded from the main analysis cohort. The OR value is displayed in addition as a numerical value. The size of the dots indicates the size of the respective cohort (N). (C) Phenome-wide association study (PheWAS) results for the 17q21.31 inversion allele H2 showing only potentially COVID-19-related phenotypes from the GWAS Catalog (P = 10−7) grouped by disease categories using different colors. The effect direction of known SNP-trait associations from the corresponding GWAS is shown using triangles pointing upward (increase) and downward (decrease), whereas dots represent unknown effect direction. Phenotypes shown were selected according to previously reported COVID-19 links with lung damage, blood cell alterations and exacerbated immune response, as well as some potential co-morbidities. The whole list of phenotypic associations is included in Supplementary Material, Table S11.
We therefore imputed the inversion haplotypes H1 and H2 for our cohorts by genotype imputation with IMPUTE2 (18), employing as reference 109 individuals from the 1000 Genomes Project for which 17q21.31 inversion genotypes were obtained experimentally by FISH and droplet digital polymerase-chain-reaction (PCR; Material and Methods). LD between the chr17:46142465:T:A (rs1819040) variant, which was prioritized over chr17:45933112:G:A (rs8065800) in the meta-analysis with the COVID-19 HGI summary statistics, and the inversion in our cohorts is near perfect (r2 = 0.98, D′ = 0.99). Genome-wide significant association with severe respiratory COVID-19 for the inversion was confirmed using logistic regression followed by meta-analysis across this study’s panels and summary statistics derived for the from the COVID-19 HGI data (Materials and Methods) (meta-analysis main analysis and COVID-19 HGI release 5 B2: P = 7.61 × 10−10, OR = 0.89; 95%CI = 0.84–0.92; meta-analysis subtype analysis and COVID-19 HGI release 5 A2: P = 1.5 × 10−4, OR = 0.90; 95%CI = 0.85–0.95; Fig. 2B, Supplementary Material, Table S10). Using stratified analysis, we also show an age effect for the inversion (Supplementary Material, Table S7, Fig. S10). The opposite effects direction observed for the German cohort for linking SNP chr17:46142465:T:A was shifted for individuals aged 40–61 when analysing the inversion. We did not observe any association of the inversion with disease severity.
Functional analysis of 17q21.31 and 19q13.33 using publicly available datasets
We next performed several follow-up analyses to better understand possible functional implications of the associations at the 17q21.31 inversion and at the novel 19q13.33 locus, including a phenome-wide association study (PheWas) and exploratory gene expression analysis.
We queried variants in high LD (r2 > 0.9) with chr19:50379362:T:C (rs1405655) or the 17q21.31 inversion, using a wide range of phenotypes from the NHGRI GWAS Catalog (Material and Methods) (19). While no known phenotypes were found to be linked to chr19:50379362:T:C (rs1405655) or its proxy variants, 161 GWAS associations were identified for variants in high LD (r2 > 0.9) with the 17q21.31 inversion, illustrating its multiple effects (Supplementary Material, Table S11). These associations included several traits potentially related to COVID-19 pathology, such as blood and immune cell composition or lung function (Fig. 2C).
Variants within the credible set from Bayesian fine mapping at 19q13.33 overlap with several genes, including for instance NAPSA, NR1H2 and KCNC3, while the 17q21.31 inversion spans multiple genes including MAPT, KANSL1, FMNL1 and CRHR1 (Supplementary Material, Figs S4, S5 and S9 and Fig. 2). To identify possible target genes relevant in COVID-19 disease pathology, we performed an exploratory gene expression analysis using several publicly available datasets to: (i) examine the direct effect of both loci on gene expression by analysing colocalization of lead SNPs with expression and splicing quantitative trait loci (eQTL and sQTL), (ii) identify in which tissues or cell types our candidate genes are expressed by analysing their RNA expression at bulk and single-cell levels and (iii) infer the possible contribution of these genes to COVID-19 pathology by looking at their expression patterns in (a) monocytes exposed to different viral and non-viral immune stimulators; (b) organoids infected with COVID-19 and (c) single-cell RNA-seq of lung and other tissues from patients who died after experiencing a SARS-CoV-2 severe disease (Material and Methods). Results of these analyses are displayed in Figure 3.
Figure 3.

Expression analysis of the most plausible candidate genes associated with the 17q.21.31 and 19q.13.33 loci in organ tissues and COVID-19 relevant cell types. (A) GTEx tissue-specific expression QTL (eQTL, upper panel) and splicing QTL (sQTL, middle panel) effects of the 17q21.31 and 19q.13.33 loci on selected candidate genes as well as expression of these genes in GTEx (20) tissues (lower panel). The direction of the normalized eQTL and sQTL effect size (NES) of the lead SNP rs1405655 and the inversion tagger rs62055540 in perfect LD with the inversion is represented by color intensities, and statistical significance by dot size. Black rectangles indicate genes for which the expression colocalizes (regional probability > 0.9) with GWAS loci in a given human tissue from the GTEx dataset. Heatmap displays gene-wise centered median by tissue expression values (represented by color intensities), showing in which tissues candidate genes are mostly enriched. (B) Expression levels of candidate genes in scRNA-seq datasets from healthy upper airways (nasal, bronchi) and lung (parenchyma) cells (47) and adult human brain cells from recently deceased, non-diseased donors (48). Figure displays log-normalized mean expression (represented by color) and fraction of cells expressing those genes (indicated by dot size). Processed and cell-type-annotated gene expression levels from studies were retrieved from COVID-19 Cell Atlas (49). (C) The figure shows differential expression of candidate genes in lung cells of COVID-19 patients compared with healthy controls. Log2 fold change (log2FC) values are presented as color gradient. Nominal P-values in −log10 scale are shown proportionally to dot size. Black-bordered circles indicate significantly differentially expressed genes after FDR correction. Results were obtained from pseudo-bulk differential expression analysis by Delorey et al. (21). More detailed figures are shown in Supplementary Material, Figures S11, S12 and S15.
The potential functional role of the 17q21.31 inversion is supported by the fact that the inversion locus (chr17:45883776 ± 75 kb) strongly colocalizes (regional probability > 0.9) with eQTLs and sQTLs (i.e. displaying the strongest association with the target), for 36 (eQTL) and 15 (sQTL) genes, respectively (Fig. 3A, Supplementary Material, Table S12, Fig. S12). Expression patterns of protein-coding genes with eQTLs associated to the inversion are shown in Supplementary Material, Figure S13, with the best candidates to play a role in the effects of the inversion on COVID-19, including MAPT, KANSL1, FMNL1 and CRHR1, being summarized in Figure 3. Many of these genes are highly expressed in neural tissues and testis (Supplementary Material, Fig. S13). However, several of them are also expressed in major immune cell types in COVID-19 relevant tissues (Fig. 3B, Supplementary Material, Fig. S13A/B), such as KANSL1, which is expressed in lung tissue-resident alveolar macrophages, or FMNL1 (Fig. 3B, Supplementary Material, Fig. S13B). These two genes especially show significantly higher expression in different lung cell types in acute COVID-19 patients who died with the disease as compared with healthy controls (20) (Fig. 3C, Supplementary Material, Fig. S13C). Moreover, RNA-seq data of monocytes under different bacterial and viral stimuli that activate toll-like receptor pathways, including bacterial lipopolysaccharide, Pam3CSK4, R848 and influenza A virus, show expression changes in several genes affected by the inversion (21) (Supplementary Material, Fig. S14, Table S13). For example, KANSL1, that could have anti-inflammatory effects, shows higher expression in H2 carrier monocytes stimulated with Influenza A virus infection-like conditions which appears to be related to a significant increase of coding and non-coding isoforms (Supplementary Material, Fig. S15). Similarly, in SARS-CoV-2 infected brain organoids, the expression of MAPT was significantly downregulated in premature and mature neuronal cells (Supplementary Material, Fig. S16, Table S14).
In the case of the 19q13.33 locus, expression of candidate genes shows high tissue specificity, with NAPSA mRNA being specific to lung and lung parenchyma and KCNC3 being highly expressed in brain and thyroid tissue, while NR1H2 is more broadly expressed among human tissues, including many immune cell types (Fig. 2B, Supplementary Material, Fig. S12). Of those candidates, expression of KCNC3 and especially NAPSA appears to be clearly affected by the rs1405655 lead SNP which is also displayed by strong colocalization [regional probability > 0.9] of the loci with eQTLs in multiple tissues (Fig. 3A). Single SNP Mendelian randomization analysis using cis-eQTL data and assessment of effect heterogeneity identified higher expression of NAPSA as a potentially causal protective factor for severe COVID-19 (Beta = −0.39; P = 1.26 × 10−7) (Supplementary Material, Table S15). Notably, NAPSA shows significantly increased expression in type 1 alveolar cells of COVID-19 patients as compared with healthy controls (20) (Fig. 3, Supplementary Material,Fig. S13). On the other hand, the NR1H2 gene is significantly down-regulated in some parenchymal (basal, ciliated, club) and endothelial (pericyte) lung cells and is up-regulated in monocytes of COVID-19 patients when compared with healthy controls (Fig. 3C, Supplementary Material, Fig. S13) (20).
Association analysis of specific candidate genomic regions: ABO locus, HLA locus and Y-chromosome haplogroups
The ABO locus has one of the most significant associations with COVID-19 infection, as highlighted in several publications (5,7). In our initial publication, we showed that the genetic association at the ABO locus could be explained by differences in the actual ABO blood type. ABO secretor status, i.e. secretion of ABO antigens into body fluids, is determined by genetic variation in the FUT2 gene. Recent studies suggest that A secretors have a higher risk of COVID-19 susceptibility (22). Here, we sought to replicate this finding (Material and Methods) 81% of all individuals were secretors irrespective of blood group on average across all control cohorts. In our study, though not statistically significant in an interaction analysis of blood group and secretion status, a trend was observed for increased risk of COVID-19 infection for A secretors when comparing COVID-19 cases to the general population (A secretors: OR = 1.32; 95%CI = 1.20–1.46; A non-secretors: OR = 1.08; 95%CI = 0.91–1.28) (Supplementary Material, Table S16). B secretion was not significantly associated with COVID-19 susceptibility and both A/B secretion were not significantly associated with disease severity.
The HLA fine-mapping approach yielded no association at the genome-wide or nominal (P < 10−5) significance threshold, neither in the overall meta-analysis across the four cohorts nor within the separate cohorts (Supplementary Material, Table S17, Fig. S17). Furthermore, we found no significant association for any HLA-presented viral peptide in a so-called PepWAS approach (Supplementary Material, Table S18, Figs S18 and S19), where associations between HLA-presented peptides and disease are unraveled by integrating similarities and differences in peptide binding among HLA alleles across patients nor robust statistical associations with any of the other tested HLA parameters (Supplementary Material, Table S19).
With male sex identified as a risk factor for severe COVID-19 and COVID-19-related death (3), we explored possible associations between genetic variants on the Y chromosome and the risk of developing severe COVID-19 and COVID-19 mortality in males (3). Variations on the Y chromosome describe so-called Y-chromosome haplogroups with letters A–Z (defined by the Y Chromosome Consortium) (23) and follow a pattern of ancestral population migrations in Europe and on a global scale.
Results of the Y-chromosome haplogroup analysis are shown in Supplementary Material, Table S20. We did not observe any statistically significant or consistent results for Y chromosomal haplotype association with COVID-19 across the Spanish, Italian and German populations in our main or subtype analysis after correction of P-values for multiple testing using Bonferroni. We found that cohort-specific suggestive (P < 0.05) associations within the Italian population for the R1b haplogroup were partially driven by single genotyping batches with diverging haplogroup frequencies (i.e. high coefficient of variation between control batches). COVID-19-related mortality showed a strong trend for association with haplogroup R1b1a2a1 (U106) (P = 9.49 × 10−3, OR = 2.8, 95%CI = 1.29–6.2) in both the Spanish and Italian populations. This association remained after adjusting for the comorbidities hypertension, CAD and diabetes. COVID-19-related death remains, however, a challenging endpoint influenced by many factors.
Discussion
We here present a large collaborative COVID-19 genetics study of different centers from Italy, Spain, Norway and Germany/Austria. With our clearly defined phenotype of severe respiratory COVID-19, a centralized genotyping and rigorous QC, we have generated a valuable resource for further COVID-19-related genetic studies. By conservatively including only severely affected COVID-19 patients in our analysis, we aimed to balance out a potential bias from selection of controls from the general population, which is however general practice in GWA analyses. Our study replicates known associations with COVID-19 from our previous and other studies and suggests associations at four novel loci, PCDH7 at 4p15.1; FREM1 at 9p22.3; OLMF4 at 13q21.1 and PTPRM at 18p11.23. These associations do not replicate in the COVID-19 HGI release 5 statistics despite their high level of replicability across all our different cohorts. Future studies in independent cohorts will be required to confirm their association to COVID-19 disease.
Genome-wide meta-analysis of COVID19 HGI release 5 summary statistics with our data revealed a so-far unreported association at the 19q13.33 locus, near the gene NAPSA. Our in-depth stratified analysis showed significant age-associations for LZTFL1 at the 3p21.31 locus and the inversion at 17q21.31. Effects of age in genetic variation are frequently observed in genetic studies; however, reasons for this are not clear. Possible models are discussed in detail in Jiang et al. (24).
Our functional analysis focused on two loci of interest associated with severe COVID-19, the previously known 17q21.31 inversion and the 19q13.33 locus, including the NAPSA gene. For the novel association at the 19q13.33 locus, additional analyses provided first hints for a functional involvement in COVID-19 through its regulation of the NAPSA gene. NAPSA encodes for a protease highly expressed in Type 1 (AT1) and Type 2 (AT2) alveolar cells, where two cell types required for the gas exchange at the lung surface and the secretion of surfactant proteins as well as immunomodulatory factors (AT2) (25). Complementary findings were observed in another recent study dissecting the lung transcriptome of COVID-19 infected patients in which NAPSA expression is increased in AT1 cells. This study also linked NAPSA to the marker gene expression signature of ‘damage-associated transient progenitors’, an intermediate cell state between AT1 and AT2 cells promoted by inflammation, characterized by a failure of AT2 cells to differentiate to AT1 cells (26). Thus, given that the NAPSA protein is involved in lung surfactant production, which is dysregulated in COVID-19 (27), the fact that the COVID-19 risk allele is associated with decreased NAPSA expression, while increased expression of NAPSA is a protective factor for severe COVID-19, suggests a potential role of NAPSA in susceptibility for severe COVID-19. These findings are in line with a recent report in which NAPSA has been identified as a candidate gene for COVID-19 severity through a cross methylome omnibus test combined with S-PrediXcan analyses in blood tissue and integrative multiomics (28), as well as with another recent report in which summary-based Mendelian randomization analysis showed an inverse correlation between the expression of NAPSA in blood and COVID-19 severity (29).
Though we cannot prove causality of the 17q21.31 inversion polymorphism in COVID-19 disease, it is linked to many traits potentially relevant to COVID-19 outcome. For instance, the inverted haplotype H2 was previously associated with higher number of red blood cells and hemoglobin levels, whereas each haplotype correlates with different proportions of lymphocytes and granulocytes, which could potentially modulate the immune response during SARS-CoV-2 infection (30). In addition, the H2 protective allele is also associated with decreased lung function and increased risk of chronic obstructive pulmonary disease but protects against the development of pulmonary fibrosis (31), and is associated with higher ventilatory response to corticosteroids in individuals with asthma (32), showing potential trade-offs and shared pathways that may be important in lung health. This variant has been proposed to be under positive selection in Europeans through its effect on fertility (17). Our results point to a role of this polymorphism in immunity and virus infection defence as well. Interestingly, inversion effects were found to be stronger in the younger age group in both severity classes, which could explain the weaker association in the HGI more severe A2 phenotype due likely to a larger proportion of older individuals (7). The inversion probably affects the COVID-19 disease course through its large effects on gene expression shown by us and others (7). Although the function of many of the affected genes is not well known, the inversion acts as an eQTL and sQTL of several interesting candidate genes for severe COVID-19. In particular, there are several genes potentially associated with immune function and immune response. For example, KANSL1, involved in histone acetylation, is broadly expressed in many types of immune cells in upper airways and lung tissue (Fig. 3) and has been proposed to play a role in the macrophage transition to an anti-inflammatory phenotype in mice (33). Here, we have found that the expression decrease in infection-like stimulated monocytes is partially compensated in homozygotes for the H2 inversion haplotype. CHRH1, which is associated to higher expression in the H2 haplotype in several tissues (Fig. 3), encodes a receptor that binds to corticotropin-releasing hormones, which are major regulators of the hypothalamic–pituitary–adrenal axis, and regulates immune and inflammatory responses (34). Finally, FMNL1, which is also located in the inversion locus, shows high expression levels in macrophages, dendritic cells and B and T lymphocytes in different COVID-19-related tissues (Fig. 3) and it is involved in cell motility and T cell trafficking (35). In addition, many of the genes in the 17q21.31 inversion regions show a predominant expression in brain tissues, which could also play an important role in COVID-19. The clearest example is MAPT, which is downregulated in SARS-CoV-2 infected neuronal cells (Supplementary Material, Fig. S16) and lung-related cells and tissues. The H2 haplotype is linked to increased expression of MAPT in the lung and its MAPT exon 3 in brain tissues (36), which could compensate for the downregulation during viral infection and have a protective effect against COVID-19. However, despite the potential implication of these and other genes, it is not possible to single out just one as the most likely candidate. In fact, inversions are well-known for keeping together a combination of alleles from different genes that generate complex phenotypic traits in different organisms (37,38).
Our hypothesis-driven analysis of associations in the HLA, as well as COVID-19-specific PepWAS analyses, yielded no significant results, indicating no major role for HLA variability in mediating the severity of COVID-19 in our cohorts. These results are in line with a recent, and so-far the largest, HLA analysis from Shachar et al. (39). Within our analysis of Y-chromosome haplogroups, none of the results remain significant after correction for multiple testing, though single haplogroups showed trends for association (Supplementary Text) and larger study samples are necessary to obtain reliable conclusions. For individual haplogroups, we observed different frequencies between batches that may also arise from different versions of the same genotyping platform. In general, the analysis of Y-chromosomal disease-relevant SNPs is mainly neglected in GWAS, hence the curation of Y-chromosomal SNPs on genotyping arrays is potentially also a confounding factor. Therefore, to gain more knowledge regarding the potential role of the Y-chromosome haplogroups in disease in general, this should be investigated in more depth.
In summary, our findings add to the number of genome-wide significant hits for COVID-19—totalling now 14 independent loci—and provide new insights to the molecular basis of COVID-19 severity that could potentially trigger subsequent and more targeted experiments to develop therapies for severe COVID-19.
Materials and Methods
Study participants and recruitment
We recruited 5228 patients with mild to severe COVID-19, which was defined as hospitalization only (mild) or with respiratory failure (severe) with a confirmed SARS-CoV-2 viral RNA PCR test from nasopharyngeal swabs or other relevant biologic fluids, cross sectionally, from intensive care units and general wards at different hospitals from Italy (4 centers, N = 1857), Spain (6 centers, N = 2795), Norway (7 centers, N = 127) and Germany/Austria (8 German, 1 Austrian center, N = 449). For comparison, we included 13 705 control participants from Italy (4 centers, N = 5247), Spain (3 centers, N = 4552), Norway (1 center, N = 288) and Germany (1 center, N = 3582). Details on the centers and origin of the control panels are shown in Supplementary Material, Table S1a and b. Though all patient samples that were sent to our study center were processed, only the severe COVID-19 individuals were analysed in this study (Supplementary Material, Table S1e). Respiratory failure was defined in the simplest possible manner to ensure feasibility: the use of oxygen supplementation or mechanical ventilation, with severity graded according to the maximum respiratory support received at any point during hospitalization (1: supplemental oxygen therapy only, 2: noninvasive ventilatory support, 3: invasive ventilatory support or 4: extracorporeal membrane oxygenation) (5).
Recruiting centers and ethics committee approval IDs
The project protocol involved the rapid recruitment of patient-participants and no additional project-related procedures (we primarily used material from clinically indicated venipunctures) and afforded anonymity, owing to the minimal dataset collected. Differences in recruitment and consent procedures among the centers arose because some centers integrated the project into larger COVID-19 biobanking efforts, whereas other centers did not, and because there were differences in how local ethics committees provided guidance on the handling of anonymization or deidentification of data as well as consent procedures. Written informed consent was obtained, sometimes in a delayed fashion, from the study patients at each center when possible. In some instances, informed consent was provided verbally or by the next of kin, depending on local ethics committee regulations and special policies issued for COVID-19 research. For some severely ill patients, an exemption from informed consent was obtained from a local ethics committee or according to local regulations to allow the use of completely anonymized surplus material from diagnostic venipuncture. Centers from which samples were obtained are listed together with their ethics approval reference numbers from each ethic committee in Supplementary Material, Table S1b.
Sample processing and genotype calling
DNA extraction was either performed at the Institute of Clinical Molecular Biology (IKMB, Christian-Albrechts-University of Kiel, Germany) or the respective study centers (Supplementary Material, Table S1a, Supplementary Methods) from whole-blood or buffy coat samples and for a very small subset also saliva. Genotype calling was performed at the IKMB for all samples with the Illumina GenomeStudio Version 2.0 software using cluster definition files GSAMD24v2-0_20024620_A1-762Samples-LifeBrain (GSA Version 2.0; 712 189 SNPs), GSAMD-24v3–0-EA_200034606_A1 (GSA Version 3.0; 730 059 SNPs) and GSAMD-24v1-0-A_4349HNR_Samples (GSA Version 1.0; 700 078 SNPs). After genotyping, a total of 4067 German/Austrian (449 cases/3618 controls), 7347 Spanish (2795 cases/4552 controls), 415 Norwegian (127 cases/288 controls) and 7114 Italian (1857 cases/5247 controls) samples were available with non-missing core-phenotype (COVID-19 case–control status) information. For individuals with missing sex information, sex was inferred from the genotypic sex if possible.
SNP and sample QC and PCA
Based on initial genotype data, we removed samples with <90% call rate using PLINK (40,41). We additionally removed individuals with non-matching genotypic and phenotypic sex. After genotype calling, a QC procedure was carried out for the Spanish, Italian, Norwegian and German/Austrian case–controls GWAS datasets, respectively. Variants that had >2% missing data, an MAF < 0.1% in disease sets or in controls, different missing genotype rates in affected and unaffected individuals (PFisher < 10−5) or deviated from Hardy–Weinberg equilibrium (with a false discovery rate (FDR) threshold of 10−5 in controls) (i) across the entire collection with at most one batch being removed or (ii) falling below in two single batches, were excluded. Samples that had overall increased/decreased heterozygosity rates (i.e. ±5 SD away from the sample mean) were removed. For robust duplicate/relatedness testing (IBS/IBD estimation) and population structure analysis, we used a linkage disequilibrium (LD)-pruned subset of SNPs on the basis of a set of independent (MAF ≥ 5%) SNPs excluding X- and Y-chromosomes, SNPs in LD (leaving no pairs with r2 > 0.2) and 11 high-LD regions as described by Price et al. (42). Pairwise percentage IBD values were computed using PLINK. By definition, Z0: P(IBD = 0), Z1: P(IBD = 1), Z2: P(IBD = 2), Z0 + Z1 + Z2 = 1 and PI_HAT: P(IBD = 2) + 0.5 * P(IBD = 1) (proportion IBD). One individual (the one showing greater missingness) from each pair with PI_HAT > 0.1875 was removed. A value of 0.1875 (proportion IBD) corresponds to a theoretical relationship of halfway between the expected IBD for third- and second-degree relatives. To identify ancestry outliers, i.e. subjects of non-European ancestry, we performed PCA for the remaining QCed cases and controls including reference samples from the 1000 Genomes Phase 3 reference panel (43). We used the PCA method, as implemented in FlashPCA (44) on an LD-pruned subset of SNPs (see text above). Ancestry outliers not matching European populations were removed (Supplementary Material, Fig. S1A–F). After QC, PCA revealed no non-European ancestry outliers (Supplementary Material, Fig. S1E–H) when performing PCA.
SNP genotype imputation
The QCed Italian, Spanish, Norwegian and German GWAS datasets comprised 1563 Italian COVID-19 cases, 4759 Italian controls, 2174 Spanish COVID-19 cases, 4406 Spanish controls, 81 Norwegian cases, 283 Norwegian controls, 336 German COVID-19 cases and 3303 German controls, and contained 567 131 (Italy), 564 856 (Spain), 525 836 (Norway) and 476 562 (Germany/Austria) variants after QC and filtering of SNPs with alleles AT or CG (the latter often leading to strand issues during imputation). Genotype imputation was conducted for chromosomes 1–22 and X data using the novel TOPMed Freeze 5 on genome build GRCh38 and the Michigan Imputation Server (45). We provided the input data in ‘vcf.gz’ format as GRCh38 build. We used the offered population panel ‘ALL’ and applied the server-side option to filter by an imputation R2 with threshold 0.1. The final imputed results contained 80 794 511 variants in the Italian dataset, 75 346 562 variants in the Spanish dataset, 20 195 513 variants in the Norwegian dataset and 53 164 135 variants in the German/Austrian dataset after TOPMed imputation. For the imputation of the X chromosome, we coded males as diploid in the non-pseudoautosomal (non-PAR) region. After QC and imputation using TOPMed, in total, 8 910 172 variants were included for the Italian panel, 9 089 877 variants for the Spanish panel, 8 841 609 variants for the Norwegian panel and 9 019 898 variants for the German/Austrian panel with post-imputation R2 ≥ 0.6 and MAF ≥ 1%.
Statistical analysis
Previous to the statistical analysis, we excluded qced individuals with a mild disease phenotype (did not receive respiratory support) or missing age information. This left 1536 Italian COVID-19 cases, 4734 Italian controls, 1416 Spanish COVID-19 cases, 4382 Spanish controls, 62 Norwegian cases, 262 Norwegian controls, 241 German COVID-19 cases and 3110 German controls for analysis (Supplementary Material, Table S1e).
GWAS and meta-analysis of our main and subtype cohorts
Using genome-wide SNP information, we tested for phenotypic associations with allele dosage data separately for the Italian, Spanish, German/Austrian and Norwegian case–control data. Including age, biological sex, age*age, biological sex*age and the first 10 PCs from PCA as covariates, we performed a logistic association analysis assuming additive effects with SAIGE for chromosomes 1–22 an X (12). For chromosome X association analysis, haplotypic allele calls outside the pseudo-autosomal regions (PARs) in males are converted to homozygous calls by doubling the haplotypic allele (assuming inactivation of large parts of 1 of the 2 female X chromosomes (46) and sex is used as a covariate for association testing of the non-PAR regions on chromosome X. For each of the analyses, sample numbers are given in Supplementary Material, Table S1e.
An inverse-variance weighted fixed-effects meta-analysis was conducted with the meta-analysis tool METAL (13) on the cohort of the main analysis including 3255 cases and 12 488 population controls of unknown COVID-19 status from Italy, Spain, Norway and Germany/Austria and the cohort from the subtype analysis including 1911 critically ill cases and 12 226 population controls (Supplementary Material, Table S1e). Only variants that were common to at least 2 datasets with respective post-imputation R2 ≥ 0.6 and that had an overall MAF of ≥1% were considered in the analysis. For each variant, we computed across-cohort heterogeneity P-values and I2 values using METAL. We considered a P-value of 10−3 to indicate significant heterogeneity and used a significance threshold of P < 10−6 for joint P-values to determine suggestive statistical significance and the common threshold of P < 5 × 10−8 to determine genome-wide significance.
Replication analysis of 13 release 5 COVID-19 HGI associations
To replicate 13 genome-wide significant associations reported by the COVID-19 HGI (7), we excluded 3837 individuals overlapping between our current study and COVID-19 HGI release 5. We subsequently carried our logistic regression and meta-analysis as described above in 1579 cases and 10 372 population controls for the main analysis and 944 cases and 10 065 population controls for the subtype analysis (Supplementary Material, Table S1e). A threshold of 0.05 was used to define replicability of the results. Meta-analysis with the COVID-19 HGI release 5 summary statistics included up to 12 888 hospitalized cases (including 5582 critically ill cases; COVID19_HGI_B2_ALL_leave_23andme_20210107.txt.gz; COVID19_HGI_A2_ALL_leave_23andme_20210107.txt.gz) and 1 295 966 population controls of the COVID-19 HGI. We performed meta-analysis with METAL as described above. To unify case definitions by our study and the COVID-19 HGI, we performed meta-analysis of summary statistics from our main analysis with COVID-19 release 5 A2 summary statistics and meta-analysis of our subtype analysis and COVID-19 release 5 B2 summary statistics. We used the common threshold of P < 5 × 10−8 to determine genome-wide significance.
Bayesian fine-mapping analysis
Statistical fine-mapping analysis was conducted using FINEMAP (15) Version 1.4 for each of the loci of interest to calculate the posterior inclusion probability for each lead SNP and every other SNP within 250 kb flanking regions. In the case where the association signal extended over a larger region, as in the case of 17q21.21, the region was expanded to include this. FINEMAP determined the 95% credible set of SNPs assuming a single causal variant using shotgun stochastic search (—n-causal-snps 1—sss), i.e. the minimum set of variants containing the causal variant with ≥95% certainty. The union of the genotypes of the Italian, Spanish, Norwegian and German/Austrian cohorts was used as the LD reference population.
Meta-analysis model-based assessment of replicability
We tested the replicability of our candidate variants from the meta-analysis using the Meta-Analysis Model-based Assessment of replicability (14) (MAMBA) method, as single outlier studies can drive a false positive meta-analysis association. MAMBA takes the genetic effects and standard deviations from participating studies to test each SNP for a true non-zero effect. This is achieved by fitting a two-level mixture model to genome-wide LD-pruned SNPs that reduces to a fixed effect meta-analysis in absence of outliers, in which case it has similar power. Since this is a likelihood model, the result is a posterior probability of replicability (PPR) that the SNP has a non-zero replicable effect. If the PPR is low, MAMBA tests for excess in variation of effects sizes and outputs the probability that each study is an outlier. For each of the first and second analyses, we used the lead variants and a genome-wide set of LD-pruned SNPs (PLINK v1.90b6.16 64-bit, —indep-pairwise 500 kb 1 0.1) (40,41) from each participating study for the algorithm to estimate the null distribution. Suggestive variants and LD-pruned background SNPs are entered as a single table and the algorithm has no prior knowledge of which SNPs are considered significant. MAMBA was run with default settings and always terminated before 10 000 iterations. We considered a PPR value >0.8 to indicate a high probability of replication.
Association analysis of candidate SNPs
We performed age- and sex-stratified on 7 candidate SNPs from our main and subtype analysis and 13 candidate SNPs from the COVID-19 HGI analysis, as well as the novel variant identified in this study (7). We additionally analysed disease severity and comorbidities hypertension, CAD and diabetes in cases only. We carried out a logistic regression analysis including age, biological sex, age*age, biological sex*age and the first 10 PCs from PCA as covariates dependent on the stratification level (i.e. for sex-stratified analyses, only age and PCs were included; for age-stratified analysis, only the biological sex and PCs were included; detailed formulas are shown in the Supplementary Methods). All analysis was done in R version 3.6.2. An inverse-variance weighted fixed-effects meta-analysis was conducted using the R-package metafor (47) to combine cohort-specific summary statistics, including only statistics from cohorts with NCase and NControl > 50. Age was analysed in groups ≤60 to >60, which were chosen in reference to the analyses performed by the COVID-19 HGI. We additionally performed an interaction analysis of SNP and sex as well as SNP and binarized age (≤60 versus >60), to determine statistical significance of differences between the groups, respectively (Supplementary Methods). For variants that showed a significant age effect, we additionally analysed age groups 40–51, 51–60 and 61–70 for the Italian and Spanish cohorts and performed a meta-analysis on the summary statistics from these two cohorts only (Supplementary Material, Fig. S10). We were interested in observing more detailed age effects in these age intervals with sufficient data for analysis. For the meta-analyses, P-values were adjusted for multiple testing using the P-value correction of Benjamini–Hochberg.
Imputation of the ~0.9-Mb inversion polymorphism at 17q21.31
In 2005, Stefansson and colleagues discovered a 900 kb inversion polymorphism at 17q21.31, a region that contains several genes, including those encoding corticotropin releasing hormone receptor 1 (CRHR1) and microtubule-associated protein tau (MAPT) (17). Chromosomes with the inverted segment in different orientations represent two distinct lineages, H1 and H2, that have diverged for up to 3 million years and show no evidence of recombination (17). For the Italian, Spanish, Norwegian and German/Austrian GWAS discovery cohorts, we inferred the 17q21.31 inversion status (H1 or H2) with IMPUTE v2.3.2 (18) using genotype information and an imputation reference panel consisting of 109 individuals (from different continents [EUR, EAS, AFR]) from the 1000 Genomes Project Phase 3 (43), for which 17q21.31 inversion genotypes, obtained experimentally by FISH (48,49) and droplet digital PCR (50), as well as SNP genotype data are available for the region of the inversion. Imputed inversion genotypes were determined according to the highest posterior probability and were further confirmed by examining consensus genotypes of known inversion tag SNPs in perfect LD (r2 = 1) with the inversion in the imputation reference panel. H1 and H2 alleles were coded according to an additive model as 0,1 and 2.
We additionally imputed the 17q21.31 inversion for the COVID-19 HGI release 5 A2 and B2 analyses from the files COVID19_HGI_A2_ALL_leave_23andme_20210107.txt.gz and COVID19_HGI_B2_ALL_leave_23andme_20210107.txt.gz. Here, the inversion P-value was imputed from summary statistics using Fast and accurate P-value Imputation for genome-wide association study (FAPI) (51). The odds ratio (OR) and 95% confidence intervals (CIs) were estimated from the SNP rs62061809, which is in perfect LD with the inversion (r2 = 1). The inversion was coded as 0 for the major allele H1 and 1 for the minor allele H2. All association analyses were carried out as described above on the minor allele H2.
Functional analysis
Phenome-wide Association study (PheWAS)
Phenotype associations with the different variants were obtained from the NHGRI-EBI GWAS Catalog curated collection of established genome-wide significant disease/trait associations (http://www.ebi.ac.uk/gwas/; release 2020-12-02) (19). We queried GWAS hits in high LD (r2 > 0.9) with the inversion as well as hits in high LD (r2 > 0.9) with rs1405655 (19q13.33) and P-value < 10−7. Since each GWAS study is focused on populations from different origins, the LD patterns employed to evaluate the association between the inversion and GWAS signals were based on individuals with the corresponding ancestry or the closest one available from our inversion imputation panel (of 109 experimentally genotyped individuals), whereas the global LD was selected if populations from underrepresented ancestries were studied .27 proxy variants were used for r1405655 and 2904 proxy variants were used for the inversion.
Colocalization of GWAS and tissue-specific expression and splicing quantitative trait loci (eQTL, sQTL)
Colocalization of GWAS and QTL (eQTL and sQTL) data from 49 different human tissues of the GTEx Project (GTEx Analysis Release V8) (19) was performed using the HyPrColoc method (52) implemented in the ezQTL tool (53). The analysis was conducted including all cis-QTL variants falling within ±75 kb from coordinates (GRCh38) of the two variants of interest: chr17:46142465:T:A—lead SNP for the 17q21.31 inversion—and chr19:50379362 (rs1405655—lead SNP of the 19q13.33 locus). LD values used in the colocalization analysis were calculated based on the 1000 Genomes population European (2) population only, to account for the ethnic bias of GTEx donors. The loci of tested traits were considered to be colocalized when a regional probability of colocalization was greater than 0.9. Additionally, to assess the actual effect (and its direction) of the variants on neighboring gene expression, normalized effect score (NES) and P-values of the two variants (inversion tagged by rs62061809) were retrieved from the GTEx portal.
Selection and definition of candidate genes at 17q21.31 and 19q13.33
To identify candidate genes most likely to play a causative role at 17q21.31 and 19q13.33, all protein-coding genes that are located within locus boundaries or that are candidates based on GWAS colocalization with eQTL (eGenes) or sQTL (sGenes)-variants were chosen. More precisely, the boundaries for the 19q13.33 locus were defined by Bayesian fine mapping (GRCh38: chr19:50344768–50379362), while extended boundaries (GRCh38: chr17:45495836–46707123) (54) were used for the 17q21.21.31 locus, since it lies within a ~0.9 Mb inversion region of high LD that affects expression and splicing of numerous genes in GTEx (55). To retrieve candidate genes that overlap the boundaries, GENCODE v36 (56) annotations for genome build GRCh38 were used. A complete list of candidate protein-coding genes from both loci is provided in Supplementary Material, Table S21.
Gene expression analysis for candidate genes from genome-wide significant susceptibility loci
Publicly available bulk tissue and immune cell type RNA-seq data for all available candidate genes were retrieved from GTEx v8 (55) and from the Expression Atlas (57) (BLUEPRINT consortium data [accession E-MTAB-3827] (58)) portals, respectively. Gene-level expression values (transcripts per million, TPM) by tissue or by cell type were obtained as median-summarized in the case of the GTEx data and as mean-summarized in the case of the BLUEPRINT data. The summarized TPM values were centered gene-wise and z-score scaled for visualization using the ggplot2 R-package. The single-cell RNA-seq (scRNA-seq) data in COVID-19 relevant tissues from non-diseased individuals, such as lung and upper airways (59) or brain (60), were obtained from the COVID-19 Cell Atlas (61). The pre-processed and cell type annotated scRNA-seq datasets were retrieved as AnnData objects in.h5ad format files. Log-normalized average expression values of available candidate genes by cell type were visualized using the scanpy v1.4.6 package (62). For gene expression analysis of candidate genes in SARS-CoV-2 infected brain organoids, pre-processed and cell-annotated scRNA-seq data were obtained upon request from Song et al. (63). Differential gene expression analysis of scRNA-seq data was performed using the R package MAST (64). More precisely, hurdle models were used to evaluate differentially expressed genes in each brain organoid cell type (neural progenitors, interneurons, neurons and cortical neurons) comparing SARS-CoV-2 infected and mock (non-infected) cells. The models were fitted using the condition, sample identification, number of detected genes (centered) and total counts of SARS-CoV-2 transcripts (centered) as covariates, thus adjusting for the cellular detection rate, batch effects and viral load. Genes with PFDR < 0.01 and absolute value of log2 fold change >0.1 were considered as significantly differentially expressed. Finally, the status, log2 fold change and P-values of candidate gene differential expression in COVID-19-infected lung cells were obtained from pseudo-bulk differential expression analysis performed by Delorey et al. (20)
Analysis and fine mapping of the HLA
Detailed descriptions of this analysis can be found in the Supplementary Methods. In brief, quality-controlled genotypes at the HLA region (chr6:29-34 Mb) were extracted. HLA allele, amino acid and SNP imputation were performed using the random-forest-based HLA genotype imputation with attribute bagging (HIBAG) and applying specially tailored as well as publicly available reference panels (5,65,66). The resulting data were used as a basis for several subsequent analyses, including: (i) basic association analysis (fine mapping) as described in the section Statistical Analysis and the Supplementary Methods, (ii) a peptidome-wide association study (11) (pepWAS), to screen for disease-relevant peptides from SARS-CoV-2, that may present a possible functional link between severe COVID-19 and variation at classical HLA loci, (iii) quantitative HLA analyses directed at the number of peptides bound by an HLA allele as well as (iv) an analysis of HLA-presentation of shared peptides (‘molecular mimicry’).
Analysis of the Y-chromosome haplotypes
First, we produced high-quality Y-chromosome genotypes by manually calling and visually inspecting Y-chromosome SNPs in the male individuals from all cohorts only. Next, we used 22 Y-chromosome SNPs to distinguish known Y-chromosome haplogroups as described in the Supplementary Methods at different haplogroup resolutions. We here focused on haplogroup R, the most prevalent Y-chromosome haplogroup across Europe. Association analysis was carried out as described in the Section Statistical Analysis—Association Analysis of Candidate SNPs on Y-chromosome haplogroups coded as absent (0) or present (1). We additionally analysed the end-point mortality (Supplementary Methods). The coefficient of variation (CV) across frequencies was calculated for Y-chromosome haplogroups between individual batches (i.e. contributing center/hospital or different versions of the GSA). Here, only frequencies calculated from batches including more than 100 individuals were considered.
Analysis of the ABO secretor status
ABO blood group typing was performed as described by Ellinghaus et al. (5). Briefly, genotypes of the SNPs rs8176747 (c.803C > T, ABO*B), rs41302905 (c.802G > A, ABO*O2) and rs8176719 (c.261delG, ABO*O1) were extracted from the imputed data (R2 = 1 for all SNPs and cohorts) and used to infer the A, B and O blood types. The ABO-‘secretor’ status was inferred from the genotypes of the rs601338 SNP (c.428G > A, FUT2*01 N.02) at the FUT2 gene, located at 19q13.33 and extracted from the imputed data (R2 = 0.98–0.99 for all cohorts). Individuals carrying genotypes GA or GG were assigned secretor status and individuals carrying genotype AA were assigned non-secretor status based on the genotype dosages, ranging from 0 to 2, retrieved from the imputed data. Individuals with allelic dosages 1.3–1.7 were called as ‘no call’, individuals with dosages 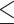 1.3 were called ‘secretors’ and individuals with dosages
1.3 were called ‘secretors’ and individuals with dosages 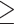 1.7 were called ‘non-secretors’. Association analysis was carried out as described in the Section Statistical Analysis—Association Analysis of Candidate SNPs on blood type or blood type secretor status coded as absent (0) or present (1).
1.7 were called ‘non-secretors’. Association analysis was carried out as described in the Section Statistical Analysis—Association Analysis of Candidate SNPs on blood type or blood type secretor status coded as absent (0) or present (1).
Availability of summary statistics
The GWAS summary statistics are available through GWAS Catalog (https://www.ebi.ac.uk/gwas/), study accession number GCST90129579.
Supplementary Material
Acknowledgements
We would like to thank the COVID-19 Human Genetics Initiative (HGI) for collecting and openly sharing extensive summary statistics with the community.
For the cohort from Milan Fondazione Ca’ Granda, we would like to acknowledge specifically Rossana Carpani. A full list of the investigators who contributed to the generation of the GCAT data is available from www.genomesforlife.com/. We thank Dr Luis Puig and Vanessa Pleguezuelos on behalf of the Blood and Tissue Bank from Catalonia (BST) who collaborated in the GCAT/COVICAT recruitment, and all the GCAT volunteers that participated in the study. We would also like to acknowledge the Task Force Humanitas and the Task Force Humanitas Gavazzeni Castelli (Supplementary Note). We would like to acknowledge all the participants from the ‘Grupo de Trabajo en Medicina Personalizada contra el COVID-19 de Andalucia’. We also thank ‘Consejeria de Salud y Familias’ and ‘Junta de Andalucía’ for their support and funding that is currently supporting the COVID-19-GWAS and the COVID-PREMED initiatives. We are indebted to the HCB-IDIBAPS Biobank for the biological human samples and data procurement and to the Fundació Glòria Soler for its support to the COVIDBANK collection. We acknowledge the work of the Norwegian SARS-CoV-2 Study group (Supplementary Note). We also acknowledge Benedicte A. Lie at the Department of Immunology, Oslo University Hospital and the Norwegian Bone Marrow Donor Registry are acknowledged for providing healthy controls. We thank the support of the German COVID-19 multiomics initiative (decoi.eu). We thank the members of the COVID-19 use and access committee (Prof. Reinhold Förster, Prof. Christine Falk, Prof. Thomas Fühner, Prof. Christoph Höner zu Siederdissen, Prof. Marius Höper, Prof. Thomas Friedrich Schulz, Prof. Markus Cornberg, Prof. Thomas Illig and Prof. Michael Manns), as well as the study nurses, medical doctors and biobank staff members involved in the project. For funding, we thank the Ministry of Science and Culture (MWK) of Lower-Saxony. We thank everybody involved in the COVID-19 registry at the University Hospital Regensburg (COVUR). Genotyping of the German control dataset was performed at the Regeneron Genetics Center. Genotyping of the COMRI study was performed by the Genotyping laboratory of Institute for Molecular Medicine Finland FIMM Technology Centre, University of Helsinki. We are very grateful to Prof. Akiko Iwasaki and Eric Song for generously sharing pre-processed single-cell RNA-seq data generated from SARS-CoV-2-infected brain organoids.
Conflict of Interest statement. P.K. has received lecture honoraria from or is advisor to Akademie für Infektionsmedizin e.V., Astellas Pharma, European Confederation of Medical Mycology, Gilead Sciences, GPR Academy Ruesselsheim, MSD Sharp & Dohme GmbH, Noxxon N.V. and University Hospital, LMU Munich outside the submitted work. He also received nonfinancial scientific grants from Miltenyi Biotec GmbH.
O.A.C. reports grants or contracts from Amplyx, Basilea, BMBF, Cidara, DZIF, EU-DG RTD (101037867), F2G, Gilead, Matinas, MedPace, MSD, Mundipharma, Octapharma, Pfizer, Scynexis; Consulting fees from Amplyx, Biocon, Biosys, Cidara, Da Volterra, Gilead, Matinas, MedPace, Menarini, Molecular Partners, MSG-ERC, Noxxon, Octapharma, PSI, Scynexis, Seres; Honoraria for lectures from Abbott, Al-Jazeera Pharmaceuticals, Astellas, Grupo Biotoscana/United Medical/Knight, Hikma, MedScape, MedUpdate, Merck/MSD, Mylan, Pfizer; Payment for expert testimony from Cidara; Participation on a Data Safety Monitoring Board or Advisory Board from Actelion, Allecra, Cidara, Entasis, IQVIA, Jannsen, MedPace, Paratek, PSI, Shionogi; a pending patent currently reviewed at the German Patent and Trade Mark Office; other interests from DGHO, DGI, ECMM, ISHAM, MSG-ERC, Wiley. S.D. received funding from the Banca Intesa San Paolo. A.M. has received research funding from or reports personal fees from Dolce&Gabbana Fashion Firm, Ventana, Pierre Fabre, Verily, AbbVie, Astra Zeneca, Verseau Therapeutics, Compugen, Myeloid Therapeutics, Third Rock Venture, Imcheck Therapeutics, Ellipses, Novartis, Roche, Macrophage Pharma, BiovelocIta, Merck, Principia, Cedarlane Laboratories Ltd, HyCult Biotechnology, eBioscience, Biolegend, ABCAM Plc, Novus Biologicals, Enzo Life (ex Alexis Corp.), Affymetrix, BMS, Johnson&Johnson; in addition, he has a patent WO2019057780 ‘Anti-human migration stimulating factor (MSF) and uses thereof’ issued, a patent WO2019081591 ‘NK or T cells and uses thereof’ issued, a patent WO2020127471 ‘Use of SAP for the treatment of Euromycetes fungi infections’ issued and a patent EP20182181.6 ‘PTX3 as prognostic marker in Covid-19’ pending. A.L. has consulted by Intercept Pharma, Takeda, AlfaSigma, Abbvie, Gilead, and Merck Sharp, and Dohme. M.R.G. has served as a speaker for AbbVie, Bristol-Myers Squibb, GENFIT, Gilead Sciences, Intercept, MSD and Roche; an advisory board member for GENFIT, Gilead Sciences, Intercept, Janssen-Cilag, Kaleido, NovoNordisk, Medimmune and Prosceinto; and has received research grants from Abbvie, Gilead Sciences and Intercept. J.C.H. received a philanthropic donation from Vivaldi Invest A/S owned by Jon Stephenson von Tetzchner during the conduct of this study. C.D.S. reports grants, personal fees from AstraZeneca, personal fees and non-financial support from BBraun Melsungen, personal fees from BioNtech, grants, personal fees and non-financial support from Gilead Sciences, grants and personal fees from Janssen-Cilag, personal fees from Eli Lilly, personal fees from Formycon, personal fees from Roche, other from Apeiron, grants and personal fees from MSD, grants from Cepheid, personal fees from GSK, personal fees from Molecular partners, other from Eli Lilly, personal fees from SOBI during the conduct of the study; personal fees from AbbVie, personal fees from Synairgen, grants and personal fees from ViiV Healthcare, outside the submitted work. F.B. reports grants and personal fees from Astrazeneca, grants and personal fees from Chiesi, grants and personal fees from GSK, personal fees from Grifols, personal fees from Guidotti, personal fees from Insmed, grants and personal fees from Menarini, personal fees from Novartis, grants and personal fees from Pfizer, personal fees from Zambon, personal fees from Vertex and personal fees from Viatris outside the submitted work. C.L. reports personal fees from Chiesi, Gilead, Janssen, Novartis, Oxfordimmunotec and Insmed outside the submitted work. J.H. reports personal fees from Chiesi, Gilead and Janssen outside the submitted work. T.H.K. has received consulting fees from Engitix and Intercept and speaker fees from Gilead and Alfasigma.
Contributor Information
Frauke Degenhardt, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
David Ellinghaus, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Novo Nordisk Foundation Center for Protein Research, Disease Systems Biology, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Simonas Juzenas, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Life Science Centre, Institute of Biotechnology, Vilnius University, Vilnius, Lithuania.
Jon Lerga-Jaso, Institut de Biotecnologia i de Biomedicina, Universitat Autònoma de Barcelona, Barcelona, Spain.
Mareike Wendorff, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Douglas Maya-Miles, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain.
Florian Uellendahl-Werth, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Hesham ElAbd, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Malte C Rühlemann, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Institute for Medical Microbiology and Hospital Epidemiology, Hannover Medical School, Hannover, Germany.
Jatin Arora, Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Division of Rheumatology, Inflammation and Immunity, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA; Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA; Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA; Center for Data Sciences, Brigham and Women’s Hospital, Boston, MA, USA.
Onur Özer, Research Group for Evolutionary Immunogenomics, Max Planck Institute for Evolutionary Biology, Plön, Germany; Research Unit for Evolutionary Immunogenomics, Department of Biology, University of Hamburg, Hamburg, Germany.
Ole Bernt Lenning, Research Department, Stavanger University Hospital, Stavanger, Norway; Randaberg Municipality, Stavanger, Norway.
Ronny Myhre, Division of Health Data and Digitalization, Department of Genetics and Bioinformatics (HDGB), Norwegian Institute of Public Health, Oslo, Norway.
May Sissel Vadla, Randaberg Municipality, Stavanger, Norway; Department of Quality and Health Technology, Faculty of Health Sciences, University of Stavanger, Stavanger, Norway.
Eike M Wacker, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Lars Wienbrandt, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Aaron Blandino Ortiz, Department of Intensive Care, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Adolfo de Salazar, Ibs.Granada Instituto de Investigación Biosanitaria, Granada, Spain; Microbiology Unit. Hospital Universitario Clinico San Cecilio, Granada, Spain.
Adolfo Garrido Chercoles, Clinical Biochemistry Department, Osakidetza Basque Health Service, Donostialdea Integrated Health Organisation, San Sebastian, Spain.
Adriana Palom, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Barcelona, Spain; Liver Unit, Department of Internal Medicine, Hospital Universitari Vall d’Hebron, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain.
Agustín Ruiz, Research Center and Memory Clinic, Ace Alzheimer Center Barcelona, Universitat Internacional de Catalunya, Barcelona, Spain; CIBERNED, Network Center for Biomedical Research in Neurodegenerative Diseases, National Institute of Health Carlos III, Madrid, Spain.
Alba-Estela Garcia-Fernandez, Department of Biochemistry, University Hospital Vall d’Hebron, Barcelona, Spain.
Albert Blanco-Grau, Department of Biochemistry, University Hospital Vall d’Hebron, Barcelona, Spain.
Alberto Mantovani, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Alberto Zanella, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Aleksander Rygh Holten, Department of Acute Medicine, Oslo University Hospital, Oslo, Norway; Institute of Clinical Medicine, University of Oslo, Oslo, Norway.
Alena Mayer, Charite Universitätsmedizin Berlin, Berlin, Germany.
Alessandra Bandera, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Alessandro Cherubini, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Alessandro Protti, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Alessio Aghemo, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Alessio Gerussi, European Reference Network on Hepatological Diseases (ERN RARE-LIVER), San Gerardo Hospital, Monza, Italy; Division of Gastroenterology, School of Medicine and Surgery, Center for Autoimmune Liver Diseases, University of Milano-Bicocca, Milan, Italy.
Alfredo Ramirez, Division of Neurogenetics and Molecular Psychiatry, Department of Psychiatry and Psychotherapy, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Department of Neurodegenerative Diseases and Geriatric Psychiatry, University Hospital Bonn, Medical Faculty, Bonn, Germany; Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; German Center for Neurodegenerative Diseases (DZNE), Bonn, Germany; Department of Psychiatry, Glenn Biggs Institute for Alzheimer’s and Neurodegenerative Diseases, San Antonio, TX, USA.
Alice Braun, Charite Universitätsmedizin Berlin, Berlin, Germany.
Almut Nebel, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Ana Barreira, Liver Unit, Department of Internal Medicine, Hospital Universitari Vall d’Hebron, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain.
Ana Lleo, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Ana Teles, Research Group for Evolutionary Immunogenomics, Max Planck Institute for Evolutionary Biology, Plön, Germany; Research Unit for Evolutionary Immunogenomics, Department of Biology, University of Hamburg, Hamburg, Germany.
Anders Benjamin Kildal, Department of Anesthesiology and Intensive Care, University Hospital of North Norway, Tromsø, Norway.
Andrea Biondi, Pediatric Departement, Centro Tettamanti-European Reference Network (ERN) PaedCan, EuroBloodNet, MetabERN-University of Milano-Bicocca-Fondazione MBBM/Ospedale, San Gerardo, Italy.
Andrea Caballero-Garralda, Biochemistry Department, Echevarne Laboratory, Barcelona, Spain.
Andrea Ganna, Institute for Molecular Medicine Finland, University of Helsinki, Helsinki, Finland.
Andrea Gori, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy; Centre for Multidisciplinary Research in Health Science (MACH), University of Milan, Milan, Italy.
Andreas Glück, Klinik für Innere Medizin I, Universitätsklinikum Schleswig-Holstein, Kiel, Germany.
Andreas Lind, Department of Microbiology, Oslo University Hospital, Oslo, Norway.
Anja Tanck, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Anke Hinney, Department of Child and Adolescent Psychiatry, University Hospital Essen, University of Duisburg-Essen, Essen, Germany.
Anna Carreras Nolla, Genomes for Life-GCAT Lab, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Anna Ludovica Fracanzani, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Anna Peschuck, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Annalisa Cavallero, Laboratory of Microbiology, San Gerardo Hospital, Monza, Italy.
Anne Ma Dyrhol-Riise, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Department of Infectious diseases, Oslo University Hospital, Oslo, Norway.
Antonella Ruello, Humanitas Gavazzeni-Castelli, Bergamo, Italy.
Antonio Julià, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Barcelona, Spain.
Antonio Muscatello, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Antonio Pesenti, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Antonio Voza, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Ariadna Rando-Segura, Microbiology Department, Hospital Universitari Vall d'Hebron, Barcelona, Spain; Universitat Autònoma de Barcelona, Bellatera, Spain.
Aurora Solier, Department of Respiratory Diseases, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain; Centro de Investigación Biomédica en Red en Enfermedades Respiratorias (CIBERES), University of Alcalá, Madrid, Spain.
Axel Schmidt, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Beatriz Cortes, Genomes for Life-GCAT Lab, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Beatriz Mateos, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Gastroenterology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Beatriz Nafria-Jimenez, Clinical Biochemistry Department, Osakidetza Basque Health Service, Donostialdea Integrated Health Organisation, San Sebastian, Spain.
Benedikt Schaefer, Department of Medicine I, Gastroenterology, Hepatology and Endocrinology, Medical University of Innsbruck, Innsbruck, Austria; Christian Doppler Laboratory of Iron and Phosphate Biology at the Department of Medicine I, Medical University of Innsbruck, Innsbruck, Austria.
Björn Jensen, Department of Gastroenterology, Hepatology and Infectious Diseases, University Hospital Duesseldorf, Medical Faculty Heinrich Heine University, Duesseldorf, Germany.
Carla Bellinghausen, Department of Respiratory Medicine and Allergology, University Hospital, Goethe University, Frankfurt am Main, Germany.
Carlo Maj, Institute of Genomic Statistics and Bioinformatics, University Hospital Bonn, Medical Faculty University of Bonn, Bonn, Germany.
Carlos Ferrando, Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Madrid, Spain; Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Carmen de la Horra, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Consejo Superior de Investigaciones Científicas, Sevilla, Spain.
Carmen Quereda, Department of Infectious Diseases, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Carsten Skurk, Charite Universitätsmedizin Berlin, Berlin, Germany.
Charlotte Thibeault, Charite Universitätsmedizin Berlin, Berlin, Germany.
Chiara Scollo, Department of Transfusion Medicine and Haematology Laboratory, San Gerardo Hospital, Monza, Italy.
Christian Herr, Department of Internal Medicine V—Pneumology, Allergology and Intensive Care Medicine, University Hospital Saarland, Homburg, Germany.
Christoph D Spinner, Department of Internal Medicine II, School of Medicine, University Hospital rechts der Isar, Technical University of Munich, Munich, Germany.
Christoph Gassner, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Institute of Translational Medicine, Private University in the Principality of Liechtenstein (UFL), Triesen, Liechtenstein.
Christoph Lange, Respiratory Medicine and International Health, University of Lübeck, Lübeck, Germany; Division of Clinical Infectious Diseases, Research Center Borstel, Borstel, Germany; Clinical Tuberculosis Unit, German Center for Infection Research (DZIF), Borstel, Germany.
Cinzia Hu, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Cinzia Paccapelo, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Clara Lehmann, Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany; German Center for Infection Research (DZIF), Cologne, Germany.
Claudio Angelini, Humanitas Clinical and Research Center, IRCCS, Milan, Italy.
Claudio Cappadona, Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Clinton Azuure, Research Group for Evolutionary Immunogenomics, Max Planck Institute for Evolutionary Biology, Plön, Germany; Research Unit for Evolutionary Immunogenomics, Department of Biology, University of Hamburg, Hamburg, Germany.
Cristiana Bianco, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Cristina Cea, Department of Biochemistry, University Hospital Vall d’Hebron, Barcelona, Spain.
Cristina Sancho, Osakidetza Basque Health Service, Basurto University Hospital, Respiratory Service, Bilbao, Spain.
Dag Arne Lihaug Hoff, Department of Clinical and Molecular Medicine, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology, Trondheim, Norway; Department of Medicine, Møre and Romsdal Hospital Trust, Ålesund, Norway.
Daniela Galimberti, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Daniele Prati, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
David Haschka, Department of Internal Medicine II, Medical University of Innsbruck, Innsbruck, Austria.
David Jiménez, Department of Respiratory Diseases, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain; Centro de Investigación Biomédica en Red en Enfermedades Respiratorias (CIBERES), University of Alcalá, Madrid, Spain.
David Pestaña, Department of Anesthesiology and Critical Care, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
David Toapanta, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Eduardo Muñiz-Diaz, Immunohematology Department, Banc de Sang i Teixits, Autonomous University of Barcelona, Barcelona, Spain.
Elena Azzolini, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Elena Sandoval, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Eleonora Binatti, European Reference Network on Hepatological Diseases (ERN RARE-LIVER), San Gerardo Hospital, Monza, Italy; Division of Gastroenterology, School of Medicine and Surgery, Center for Autoimmune Liver Diseases, University of Milano-Bicocca, Milan, Italy.
Elio Scarpini, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Elisa T Helbig, Charite Universitätsmedizin Berlin, Berlin, Germany.
Elisabetta Casalone, Department of Medical Sciences, Università degli Studi di Torino, Turin, Italy.
Eloisa Urrechaga, Osakidetza Basque Health Service, Galdakao Hospital, Respiratory Service, Galdakao, Spain; Biocruces Bizkaia Health Research Institute, Barakaldo, Spain.
Elvezia Maria Paraboschi, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Emanuele Pontali, Department of Infectious Diseases, E.O. Ospedali Galliera, Genova, Italy.
Enric Reverter, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Enrique J Calderón, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Consejo Superior de Investigaciones Científicas, Sevilla, Spain.
Enrique Navas, Department of Infectious Diseases, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Erik Solligård, Geminicenter for Sepsis Research, Institute of Circulation and Medical Imaging (ISB), NTNU, Trondheim, Norway; Clinic of Anesthesia and Intensive Care, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway.
Ernesto Contro, Accident and Emergency and Emergency Medicine Unit, San Gerardo Hospital, Monza, Italy.
Eunate Arana-Arri, Biocruces Bizkaia Health Research Institute, Barakaldo, Spain.
Fátima Aziz, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Federico Garcia, Ibs.Granada Instituto de Investigación Biosanitaria, Granada, Spain; Microbiology Unit. Hospital Universitario Clinico San Cecilio, Granada, Spain; Centro de Investigación Biomédica en Red en Enfermedades Infecciosas (CIBERINFEC), Instituto de Salud Carlos III (ISCIII), Madrid, Spain.
Félix García Sánchez, Histocompatibilidad y Biologia Molecular, Centro de Transfusion de Madrid, Madrid, Spain.
Ferruccio Ceriotti, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Filippo Martinelli-Boneschi, Department of Pathophysiology and Transplantation, Dino Ferrari Center, University of Milan, Milan, Italy; Neurology Unit, IRCCS Fondazione Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Flora Peyvandi, Department of Pathophysiology and Transplantation, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Angelo Bianchi Bonomi Hemophilia and Thrombosis Center, Milan, Italy.
Florian Kurth, Charite Universitätsmedizin Berlin, Berlin, Germany; Department of Tropical Medicine, Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany; Department of Medicine I, University Medical Centre Hamburg-Eppendorf, Hamburg, Germany.
Francesco Blasi, Respiratory Unit, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy; Department of Pathophysiology and Transplantation, Università degli Studi di Milano, Milan, Italy.
Francesco Malvestiti, University of Milan, Milan, Italy.
Francisco J Medrano, Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Consejo Superior de Investigaciones Científicas, Sevilla, Spain; Internal Medicine Department, Virgen del Rocio University Hospital, Sevilla, Spain.
Francisco Mesonero, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Gastroenterology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Francisco Rodriguez-Frias, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Barcelona, Spain; Department of Biochemistry, University Hospital Vall d’Hebron, Barcelona, Spain; Microbiology Department, Hospital Universitari Vall d'Hebron, Barcelona, Spain; Universitat Autònoma de Barcelona, Bellatera, Spain; Biochemistry Department, University Hospital Vall d'Hebron, Barcelona, Spain.
Frank Hanses, Emergency Department, University Hospital Regensburg, Regensburg, Germany; Department for Infectious Diseases and Infection Control, University Hospital Regensburg, Regensburg, Germany.
Fredrik Müller, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Department of Microbiology, Oslo University Hospital, Oslo, Norway.
Georg Hemmrich-Stanisak, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Giacomo Bellani, Department Emergency, Anesthesia and Intensive Care, San Gerardo Hospital, Monza, Italy; School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy.
Giacomo Grasselli, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Gianni Pezzoli, Fondazione Grigioni per il Morbo di Parkinson and Parkinson Institute, ASST Gaetano Pini-CTO, Milan, Italy.
Giorgio Costantino, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Giovanni Albano, Humanitas Gavazzeni-Castelli, Bergamo, Italy.
Giulia Cardamone, Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Giuseppe Bellelli, School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy; Acute Geriatric Unit, San Gerardo Hospital, Monza, Italy.
Giuseppe Citerio, School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy; Neurointensive Care Unit, San Gerardo Hospital, Monza, Italy.
Giuseppe Foti, Department Emergency, Anesthesia and Intensive Care, San Gerardo Hospital, Monza, Italy; School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy.
Giuseppe Lamorte, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Giuseppe Matullo, Department of Medical Sciences, Università degli Studi di Torino, Turin, Italy.
Guido Baselli, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Hayato Kurihara, Humanitas Clinical and Research Center, IRCCS, Milan, Italy.
Holger Neb, Department of Anesthesiology, Intensive Care Medicine and Pain Therapy, University Hospital Frankfurt, Frankfurt am Main, Germany.
Ilaria My, IRCCS Humanitas Research Hospital, Milan, Italy.
Ingo Kurth, Institute of Human Genetics, Medical Faculty, RWTH Aachen University, Aachen, Germany.
Isabel Hernández, Research Center and Memory Clinic, Ace Alzheimer Center Barcelona, Universitat Internacional de Catalunya, Barcelona, Spain; CIBERNED, Network Center for Biomedical Research in Neurodegenerative Diseases, National Institute of Health Carlos III, Madrid, Spain.
Isabell Pink, Department of Pneumology, Hannover Medical School, Hannover, Germany.
Itziar de Rojas, Research Center and Memory Clinic, Ace Alzheimer Center Barcelona, Universitat Internacional de Catalunya, Barcelona, Spain; CIBERNED, Network Center for Biomedical Research in Neurodegenerative Diseases, National Institute of Health Carlos III, Madrid, Spain.
Iván Galván-Femenia, Genomes for Life-GCAT Lab, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Jan Cato Holter, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Department of Microbiology, Oslo University Hospital, Oslo, Norway.
Jan Egil Afset, Osakidetza Basque Health Service, Basurto University Hospital, Respiratory Service, Bilbao, Spain; Department of Medical Microbiology, Clinic of Laboratory Medicine, St. Olavs hospital, Trondheim, Norway.
Jan Heyckendorf, Respiratory Medicine and International Health, University of Lübeck, Lübeck, Germany; Division of Clinical Infectious Diseases, Research Center Borstel, Borstel, Germany; Clinical Tuberculosis Unit, German Center for Infection Research (DZIF), Borstel, Germany.
Jan Kässens, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Jan Kristian Damås, Department of Infectious Diseases, St. Olavs Hospital, Trondheim University Hospital, Trondheim, Norway; Department of Clinical and Molecular Medicine, NTNU, Trondheim, Norway.
Jan Rybniker, Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany; German Centre for Infection Research (DZIF), Cologne, Germany.
Janine Altmüller, Cologne Center for Genomics (CCG), University of Cologne, Cologne, Germany.
Javier Ampuero, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Digestive Diseases Unit, Virgen del Rocio University Hospital, Institute of Biomedicine of Seville, University of Seville, Seville, Spain.
Javier Martín, Institute of Parasitology and Biomedicine Lopez-Neyra, CSIC, Granada, Spain.
Jeanette Erdmann, Institute for Cardiogenetics, University of Lübeck, Lübeck, Germany; German Research Center for Cardiovascular Research, Lübeck, Germany; University Heart Center Lübeck, Lübeck, Germany.
Jesus M Banales, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Liver and Gastrointestinal Diseases, Biodonostia Health Research Institute—Donostia University Hospital, University of the Basque Country (UPV/EHU), CIBERehd, Ikerbasque, San Sebastian, Spain; Ikerbasque, Basque Foundation for Science, Bilbao, Spain.
Joan Ramon Badia, Respiratory ICU, Institut Clínic Respiratory, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Joaquin Dopazo, Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; Bioinformatics Area, Fundación Progreso y Salud, and Institute of Biomedicine of Sevilla (IBIS), Sevilla, Spain.
Jochen Schneider, Department of Internal Medicine V—Pneumology, Allergology and Intensive Care Medicine, University Hospital Saarland, Homburg, Germany.
Jonas Bergan, Department of Research, Ostfold Hospital Trust, Gralum, Norway.
Jordi Barretina, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Jörn Walter, Department of Genetics & Epigenetics, Saarland University, Saarbrücken, Germany.
Jose Hernández Quero, Ibs.Granada Instituto de Investigación Biosanitaria, Granada, Spain; Department of Infectious Diseases, Hospital Universitario Clinico San Cecilio, Granada, Spain.
Josune Goikoetxea, Infectious Diseases Service, Osakidetza, Biocruces Bizkaia Health Research Institute, Barakaldo, Spain.
Juan Delgado, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Consejo Superior de Investigaciones Científicas, Sevilla, Spain.
Juan M Guerrero, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain.
Julia Fazaal, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Julia Kraft, Charite Universitätsmedizin Berlin, Berlin, Germany.
Julia Schröder, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Kari Risnes, Department of Clinical and Molecular Medicine, NTNU, Trondheim, Norway; Department of Research, St. Olav Hospital, Trondheim University Hospital, Trondheim, Norway.
Karina Banasik, Novo Nordisk Foundation Center for Protein Research, Disease Systems Biology, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Karl Erik Müller, Medical Department, Drammen Hospital, Vestre Viken Hospital Trust, Drammen, Norway.
Karoline I Gaede, Research Center Borstel, BioMaterialBank Nord, Borstel, Germany; German Center for Lung Research (DZL), Airway Research Center North (ARCN), Borstel, Germany; Popgen 2.0 Network (P2N), Kiel, Germany.
Koldo Garcia-Etxebarria, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Liver and Gastrointestinal Diseases, Biodonostia Health Research Institute—Donostia University Hospital, University of the Basque Country (UPV/EHU), CIBERehd, Ikerbasque, San Sebastian, Spain.
Kristian Tonby, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Department of Infectious diseases, Oslo University Hospital, Oslo, Norway.
Lars Heggelund, Medical Department, Drammen Hospital, Vestre Viken Hospital Trust, Drammen, Norway; Department of Clinical Science, University of Bergen, Bergen, Norway.
Laura Izquierdo-Sanchez, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Liver and Gastrointestinal Diseases, Biodonostia Health Research Institute—Donostia University Hospital, University of the Basque Country (UPV/EHU), CIBERehd, Ikerbasque, San Sebastian, Spain; Biodonostia Health Research Institute, Donostia University Hospital, San Sebastian, Spain.
Laura Rachele Bettini, Pediatric Departement, Centro Tettamanti-European Reference Network (ERN) PaedCan, EuroBloodNet, MetabERN-University of Milano-Bicocca-Fondazione MBBM/Ospedale, San Gerardo, Italy.
Lauro Sumoy, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Leif Erik Sander, Charite Universitätsmedizin Berlin, Berlin, Germany.
Lena J Lippert, Charite Universitätsmedizin Berlin, Berlin, Germany.
Leonardo Terranova, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Lindokuhle Nkambule, Analytic and Translational Genetics Unit, Massachusetts General Hospital, Boston, MA, USA; Stanley Center for Psychiatric Research & Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA.
Lisa Knopp, Department of Gastroenterology, Hepatology and Infectious Diseases, University Hospital Duesseldorf, Medical Faculty Heinrich Heine University, Duesseldorf, Germany.
Lise Tuset Gustad, Geminicenter for Sepsis Research, Institute of Circulation and Medical Imaging (ISB), NTNU, Trondheim, Norway; Clinic of Medicine and Rehabilitation, Levanger Hospital, Nord-Trondelag Hospital Trust, Levanger, Norway.
Lucia Garbarino, HLA Laboratory, E.O. Ospedali Galliera, Genova, Italy.
Luigi Santoro, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Luis Téllez, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Gastroenterology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Luisa Roade, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Liver Unit, Department of Internal Medicine, Hospital Universitari Vall d’Hebron, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain; Department of Respiratory Diseases, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Mahnoosh Ostadreza, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Maider Intxausti, Osakidetza Basque Health Service, Basurto University Hospital, Respiratory Service, Bilbao, Spain.
Manolis Kogevinas, Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; ISGlobal, Barcelona, Spain; Universitat Pompeu Fabra (UPF), Barcelona, Spain; IMIM (Hospital del Mar Medical Research Institute), Barcelona, Spain.
Mar Riveiro-Barciela, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Liver Unit, Department of Internal Medicine, Hospital Universitari Vall d’Hebron, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain; Universitat Autònoma de Barcelona, Bellatera, Spain.
Marc M Berger, Department of Anesthesiology and Intensive Care Medicine, University Hospital Essen, University Duisburg-Essen, Essen, Germany.
Marco Schaefer, Stefan-Morsch-Stiftung, Birkenfeld, Germany.
Mari E K Niemi, Institute for Molecular Medicine Finland, University of Helsinki, Helsinki, Finland.
María A Gutiérrez-Stampa, Osakidetza, OSI Donostialdea, Altza Primary Care, Biodonostia Health Research Institute, San Sebastián, Spain.
Maria Carrabba, Internal Medicine Department, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Maria E Figuera Basso, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Maria Grazia Valsecchi, Center of Bioinformatics, Biostatistics and Bioimaging, School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy.
María Hernandez-Tejero, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Maria J G T Vehreschild, Department of Internal Medicine, Infectious Diseases, University Hospital Frankfurt & Goethe University Frankfurt, Frankfurt am Main, Germany.
Maria Manunta, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Marialbert Acosta-Herrera, Institute of Parasitology and Biomedicine Lopez-Neyra, CSIC, Granada, Spain.
Mariella D'Angiò, Pediatric Departement, Centro Tettamanti-European Reference Network (ERN) PaedCan, EuroBloodNet, MetabERN-University of Milano-Bicocca-Fondazione MBBM/Ospedale, San Gerardo, Italy.
Marina Baldini, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Marina Cazzaniga, Phase 1 Research Centre, ASST Monza, School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy.
Marit M Grimsrud, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Research Institute for Internal Medicine, Oslo University Hospital Rikshospitalet and University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Department of Transplantation Medicine, Norwegian PSC Research Center, Oslo University Hospital Rikshospitalet, Oslo, Norway.
Markus Cornberg, Department of Gastroenterology, Hepatology and Endocrinology, Hannover Medical School, Hannover, Germany.
Markus M Nöthen, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Marta Marquié, Research Center and Memory Clinic, Ace Alzheimer Center Barcelona, Universitat Internacional de Catalunya, Barcelona, Spain; CIBERNED, Network Center for Biomedical Research in Neurodegenerative Diseases, National Institute of Health Carlos III, Madrid, Spain.
Massimo Castoldi, Humanitas Gavazzeni-Castelli, Bergamo, Italy.
Mattia Cordioli, Institute for Molecular Medicine Finland, University of Helsinki, Helsinki, Finland.
Maurizio Cecconi, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Mauro D'Amato, Ikerbasque, Basque Foundation for Science, Bilbao, Spain; Gastrointestinal Genetics Lab, CIC bioGUNE—BRTA, Derio, Spain; Department of Medicine and Surgery, LUM University, Casamassima, Italy.
Max Augustin, Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany; German Center for Infection Research (DZIF), Cologne, Germany.
Melissa Tomasi, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Mercè Boada, Research Center and Memory Clinic, Ace Alzheimer Center Barcelona, Universitat Internacional de Catalunya, Barcelona, Spain; CIBERNED, Network Center for Biomedical Research in Neurodegenerative Diseases, National Institute of Health Carlos III, Madrid, Spain.
Michael Dreher, Department of Pneumology and Intensive Care Medicine, University Hospital Aachen, Aachen, Germany.
Michael J Seilmaier, Munich Clinic Schwabing, Academic Teaching Hospital, Ludwig-Maximilians-University (LMU), Munich, Germany.
Michael Joannidis, Division of Intensive Care and Emergency Medicine, Department of Internal Medicine, Medical University Innsbruck, Innsbruck, Austria.
Michael Wittig, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Michela Mazzocco, HLA Laboratory, E.O. Ospedali Galliera, Genova, Italy.
Michele Ciccarelli, Humanitas Clinical and Research Center, IRCCS, Milan, Italy.
Miguel Rodríguez-Gandía, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Gastroenterology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Monica Bocciolone, Humanitas Clinical and Research Center, IRCCS, Milan, Italy.
Monica Miozzo, University of Milan, Milan, Italy; Internal Medicine Department, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Natale Imaz Ayo, Biocruces Bizkaia Health Research Institute, Barakaldo, Spain.
Natalia Blay, Genomes for Life-GCAT Lab, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Natalia Chueca, Microbiology Unit. Hospital Universitario Clinico San Cecilio, Granada, Spain.
Nicola Montano, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Nicole Braun, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; University Hospital Schleswig-Holstein, Kiel, Germany.
Nicole Ludwig, Department of Human Genetics, Center of Human and Molecular Biology, University Hospital Saarland, Homburg/Saar, Germany.
Nikolaus Marx, Department of Internal Medicine I, RWTH Aachen University Hospital, Aachen, Germany.
Nilda Martínez, Department of Anesthesiology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), Madrid, Spain.
Oliver A Cornely, Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; German Center for Infection Research (DZIF), Cologne, Germany; Faculty of Medicine and University Hospital Cologne, Clinical Trials Centre Cologne (ZKS Köln), University of Cologne, Cologne, Germany.
Oliver Witzke, Department of Infectious Diseases, University Hospital Essen, University Duisburg-Essen, Essen, Germany.
Orazio Palmieri, Gastroenterology Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy.
Paola Faverio, School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy; Pulmonary Unit, San Gerardo Hospital, Monza, Italy.
Paoletta Preatoni, Humanitas Clinical and Research Center, IRCCS, Milan, Italy.
Paolo Bonfanti, School of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy; Infectious Diseases Unit, San Gerardo Hospital, Monza, Italy.
Paolo Omodei, Humanitas Clinical and Research Center, IRCCS, Milan, Italy.
Paolo Tentorio, IRCCS Humanitas Research Hospital, Milan, Italy.
Pedro Castro, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain.
Pedro M Rodrigues, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Liver and Gastrointestinal Diseases, Biodonostia Health Research Institute—Donostia University Hospital, University of the Basque Country (UPV/EHU), CIBERehd, Ikerbasque, San Sebastian, Spain; Ikerbasque, Basque Foundation for Science, Bilbao, Spain; Biodonostia Health Research Institute, Donostia University Hospital, San Sebastian, Spain.
Pedro Pablo España, Osakidetza Basque Health Service, Galdakao Hospital, Respiratory Service, Galdakao, Spain.
Per Hoffmann, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Philip Rosenstiel, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Philipp Schommers, Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany; German Center for Infection Research (DZIF), Cologne, Germany.
Phillip Suwalski, Charite Universitätsmedizin Berlin, Berlin, Germany.
Raúl de Pablo, Department of Intensive Care, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Ricard Ferrer, Intensive Care Department, Vall d'Hebron University Hospital, SODIR-VHIR research group, Barcelona, Spain.
Robert Bals, Department of Internal Medicine V—Pneumology, Allergology and Intensive Care Medicine, University Hospital Saarland, Homburg, Germany.
Roberta Gualtierotti, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Rocío Gallego-Durán, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain.
Rosa Nieto, Department of Respiratory Diseases, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain; Centro de Investigación Biomédica en Red en Enfermedades Respiratorias (CIBERES), University of Alcalá, Madrid, Spain.
Rossana Carpani, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Rubén Morilla, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Consejo Superior de Investigaciones Científicas, Sevilla, Spain.
Salvatore Badalamenti, IRCCS Humanitas Research Hospital, Milan, Italy.
Sammra Haider, Department of Medicine, Møre & Romsdal Hospital Trust, Molde, Norway.
Sandra Ciesek, Institute of Medical Virology, University Hospital Frankfurt, Goethe University, Frankfurt am Main, Germany; German Centre for Infection Research (DZIF), Frankfurt am Main, Germany.
Sandra May, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Sara Bombace, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Sara Marsal, Vall d’Hebron Institut de Recerca (VHIR), Vall d’Hebron Hospital Universitari, Barcelona, Spain.
Sara Pigazzini, Institute for Molecular Medicine Finland, University of Helsinki, Helsinki, Finland.
Sebastian Klein, Division of Intensive Care and Emergency Medicine, Department of Internal Medicine, Medical University Innsbruck, Innsbruck, Austria.
Serena Pelusi, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Sibylle Wilfling, Department for Infectious Diseases and Infection Control, University Hospital Regensburg, Regensburg, Germany; Zentrum für Humangenetik Regensburg, Regensburg, Germany; Department of Neurology, Bezirksklinikum Regensburg, University of Regensburg, Regensburg, Germany.
Silvano Bosari, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Sonja Volland, Hannover Unified Biobank, Hannover Medical School, Hannover, Germany.
Søren Brunak, Novo Nordisk Foundation Center for Protein Research, Disease Systems Biology, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Soumya Raychaudhuri, Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, USA; Division of Rheumatology, Inflammation and Immunity, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA, USA; Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA; Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA; Center for Data Sciences, Brigham and Women’s Hospital, Boston, MA, USA; Centre for Genetics and Genomics Versus Arthritis, Centre for Musculoskeletal Research, Manchester Academic Health Science Centre, The University of Manchester, Manchester, UK.
Stefan Schreiber, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Klinik für Innere Medizin I, Universitätsklinikum Schleswig-Holstein, Kiel, Germany.
Stefanie Heilmann-Heimbach, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Stefano Aliberti, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Stephan Ripke, Charite Universitätsmedizin Berlin, Berlin, Germany.
Susanne Dudman, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Department of Microbiology, Oslo University Hospital, Oslo, Norway.
Tanja Wesse, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Tenghao Zheng, School of Biological Sciences, Monash University, Clayton, VIC, Australia.
Thomas Bahmer, Klinik für Innere Medizin I, Universitätsklinikum Schleswig-Holstein, Kiel, Germany.
Thomas Eggermann, Institute of Human Genetics, Medical Faculty, RWTH Aachen University, Aachen, Germany.
Thomas Illig, Hannover Unified Biobank, Hannover Medical School, Hannover, Germany.
Thorsten Brenner, Department of Anesthesiology and Intensive Care Medicine, University Hospital Essen, University Duisburg-Essen, Essen, Germany.
Tomas Pumarola, Department of Microbiology, University Hospital Vall d’Hebron, Barcelona, Spain; Autonoma University of Barcelona, Barcelona, Spain.
Torsten Feldt, Department of Gastroenterology, Hepatology and Infectious Diseases, University Hospital Duesseldorf, Medical Faculty Heinrich Heine University, Duesseldorf, Germany.
Trine Folseraas, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Research Institute for Internal Medicine, Oslo University Hospital Rikshospitalet and University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Department of Transplantation Medicine, Norwegian PSC Research Center, Oslo University Hospital Rikshospitalet, Oslo, Norway; Division for Cancer Medicine, Surgery and Transplantation, Section for Gastroenterology, Department of Transplantation Medicine, Oslo University Hospital Rikshospitalet, Oslo, Norway.
Trinidad Gonzalez Cejudo, Biochemistry Unit, Hospital Universitario Clinico San Cecilio, Granada, Spain.
Ulf Landmesser, Berlin Institute of Health, Charite Universitätsmedizin Berlin, Berlin Germany.
Ulrike Protzer, Institute of Virology, Technical University Munich/Helmholtz Zentrum München, Munich, Germany; German Center for Infection Research (DZIF), Munich, Germany.
Ute Hehr, Zentrum für Humangenetik Regensburg, Regensburg, Germany.
Valeria Rimoldi, Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Valter Monzani, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Vegard Skogen, Department of Infectious Diseases, University Hospital of North Norway, Tromsø, Norway; Faculty of Health Sciences, UIT The Arctic University of Norway, Norway.
Verena Keitel, Department of Gastroenterology, Hepatology and Infectious Diseases, University Hospital Duesseldorf, Medical Faculty Heinrich Heine University, Duesseldorf, Germany.
Verena Kopfnagel, Hannover Unified Biobank, Hannover Medical School, Hannover, Germany.
Vicente Friaza, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Consejo Superior de Investigaciones Científicas, Sevilla, Spain.
Victor Andrade, Division of Neurogenetics and Molecular Psychiatry, Department of Psychiatry and Psychotherapy, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Department of Neurodegenerative Diseases and Geriatric Psychiatry, University Hospital Bonn, Medical Faculty, Bonn, Germany.
Victor Moreno, Centro de Investigación Biomédica en Red de Epidemiología y Salud Pública (CIBERESP), Madrid, Spain; Catalan Institute of Oncology (ICO), Barcelona, Spain; Bellvitge Biomedical Research Institute (IDIBELL), Barcelona, Spain; Universitat de Barcelona (UB), Barcelona, Spain.
Wolfgang Albrecht, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Wolfgang Peter, Stefan-Morsch-Stiftung, Birkenfeld, Germany; Faculty of Medicine and University Hospital Cologne, Institute for Transfusion Medicine, University of Cologne, Cologne, Germany.
Wolfgang Poller, Charite Universitätsmedizin Berlin, Berlin, Germany.
Xavier Farre, Genomes for Life-GCAT Lab, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Xiaoli Yi, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany.
Xiaomin Wang, Charite Universitätsmedizin Berlin, Berlin, Germany.
Yascha Khodamoradi, Department of Internal Medicine, Infectious Diseases, University Hospital Frankfurt & Goethe University Frankfurt, Frankfurt am Main, Germany.
Zehra Karadeniz, Charite Universitätsmedizin Berlin, Berlin, Germany.
Anna Latiano, Gastroenterology Unit, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy.
Siegfried Goerg, Institute of Transfusionsmedicine, University Hospital Schleswig-Holstein (UKSH), Kiel, Germany.
Petra Bacher, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Institute of Immunology, Christian-Albrechts-University of Kiel and UKSH Schleswig-Holstein, Kiel, Germany.
Philipp Koehler, Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Department I of Internal Medicine, Faculty of Medicine and University Hospital Cologne, University of Cologne, Cologne, Germany; Center for Molecular Medicine Cologne (CMMC), University of Cologne, Cologne, Germany.
Florian Tran, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; Klinik für Innere Medizin I, Universitätsklinikum Schleswig-Holstein, Kiel, Germany.
Heinz Zoller, Department of Medicine I, Gastroenterology, Hepatology and Endocrinology, Medical University of Innsbruck, Innsbruck, Austria; Christian Doppler Laboratory of Iron and Phosphate Biology at the Department of Medicine I, Medical University of Innsbruck, Innsbruck, Austria.
Eva C Schulte, Institute of Virology, Technical University Munich/Helmholtz Zentrum München, Munich, Germany; Institute of Psychiatric Phenomics and Genomics, University Medical Center, University of Munich, Munich, Germany; Department of Psychiatry, University Medical Center, University of Munich, Munich, Germany.
Bettina Heidecker, Charite Universitätsmedizin Berlin, Berlin, Germany.
Kerstin U Ludwig, Institute of Human Genetics, University of Bonn School of Medicine & University Hospital Bonn, Bonn, Germany.
Javier Fernández, Hospital Clinic, University of Barcelona, and IDIBAPS, Barcelona, Spain; European Foundation for the Study of Chronic Liver Failure (EF-CLIF), Barcelona, Spain.
Manuel Romero-Gómez, Hospital Universitario Virgen del Rocío de Sevilla, Sevilla, Spain; Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Instituto de Biomedicina de Sevilla (IBIS), Sevilla, Spain; University of Sevilla, Sevilla, Spain; Digestive Diseases Unit, Virgen del Rocio University Hospital, Institute of Biomedicine of Seville, University of Seville, Seville, Spain.
Agustín Albillos, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Gastroenterology, Hospital Universitario Ramón y Cajal, Instituto Ramón y Cajal de Investigación Sanitaria (IRYCIS), University of Alcalá, Madrid, Spain.
Pietro Invernizzi, European Reference Network on Hepatological Diseases (ERN RARE-LIVER), San Gerardo Hospital, Monza, Italy; Division of Gastroenterology, School of Medicine and Surgery, Center for Autoimmune Liver Diseases, University of Milano-Bicocca, Milan, Italy.
Maria Buti, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Liver Unit, Department of Internal Medicine, Hospital Universitari Vall d’Hebron, Vall d’Hebron Barcelona Hospital Campus, Barcelona, Spain; Universitat Autònoma de Barcelona, Bellatera, Spain.
Stefano Duga, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Luis Bujanda, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), Madrid, Spain; Department of Liver and Gastrointestinal Diseases, Biodonostia Health Research Institute—Donostia University Hospital, University of the Basque Country (UPV/EHU), CIBERehd, Ikerbasque, San Sebastian, Spain.
Johannes R Hov, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Research Institute for Internal Medicine, Oslo University Hospital Rikshospitalet and University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Department of Transplantation Medicine, Norwegian PSC Research Center, Oslo University Hospital Rikshospitalet, Oslo, Norway; Division for Cancer Medicine, Surgery and Transplantation, Section for Gastroenterology, Department of Transplantation Medicine, Oslo University Hospital Rikshospitalet, Oslo, Norway.
Tobias L Lenz, Research Group for Evolutionary Immunogenomics, Max Planck Institute for Evolutionary Biology, Plön, Germany; Research Unit for Evolutionary Immunogenomics, Department of Biology, University of Hamburg, Hamburg, Germany.
Rosanna Asselta, IRCCS Humanitas Research Hospital, Milan, Italy; Department of Biomedical Sciences, Humanitas University, Milan, Italy.
Rafael de Cid, Genomes for Life-GCAT Lab, Germans Trias i Pujol Research Institute (IGTP), Badalona, Spain.
Luca Valenti, University of Milan, Milan, Italy; Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy.
Tom H Karlsen, Institute of Clinical Medicine, University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Research Institute for Internal Medicine, Oslo University Hospital Rikshospitalet and University of Oslo, Oslo, Norway; Division of Surgery, Inflammatory Diseases and Transplantation, Department of Transplantation Medicine, Norwegian PSC Research Center, Oslo University Hospital Rikshospitalet, Oslo, Norway; Division for Cancer Medicine, Surgery and Transplantation, Section for Gastroenterology, Department of Transplantation Medicine, Oslo University Hospital Rikshospitalet, Oslo, Norway.
Mario Cáceres, Institut de Biotecnologia i de Biomedicina, Universitat Autònoma de Barcelona, Barcelona, Spain; ICREA, Barcelona, Spain.
Andre Franke, Institute of Clinical Molecular Biology, Christian-Albrechts-University, Kiel, Germany; University Hospital Schleswig-Holstein (UKSH), Kiel, Germany.
Funding
S.E.H. and C.A.S. partially supported genotyping through a philanthropic donation. A.F. and D.E. were supported by a grant from the German Federal Ministry of Education and COVID-19 grant Research (BMBF; ID:01KI20197); A.F., D.E. and F.D. were supported by the Deutsche Forschungsgemeinschaft Cluster of Excellence ‘Precision Medicine in Chronic Inflammation’ (EXC2167). D.E. was supported by the German Federal Ministry of Education and Research (BMBF) within the framework of the Computational Life Sciences funding concept (CompLS grant 031L0165). D.E., K.B. and S.B. acknowledge the Novo Nordisk Foundation (NNF14CC0001 and NNF17OC0027594). T.L.L., A.T. and O.Ö. were funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project numbers 279645989; 433116033; 437857095. M.W. and H.E. are supported by the German Research Foundation (DFG) through the Research Training Group 1743, ‘Genes, Environment and Inflammation’. L.V. received funding from: Ricerca Finalizzata Ministero della Salute (RF-2016-02364358), Italian Ministry of Health ‘CV PREVITAL’—strategie di prevenzione primaria cardiovascolare primaria nella popolazione italiana; The European Union (EU) Programme Horizon 2020 (under grant agreement No. 777377) for the project LITMUS- and for the project ‘REVEAL’; Fondazione IRCCS Ca’ Granda ‘Ricerca corrente’, Fondazione Sviluppo Ca’ Granda ‘Liver-BIBLE’ (PR-0391), Fondazione IRCCS Ca’ Granda ‘5permille’ ‘COVID-19 Biobank’ (RC100017A). A.B. was supported by a grant from Fondazione Cariplo to Fondazione Tettamanti: ‘Bio-banking of Covid-19 patient samples to support national and international research (Covid-Bank). This research was partly funded by an MIUR grant to the Department of Medical Sciences, under the program ‘Dipartimenti di Eccellenza 2018–2022’. This study makes use of data generated by the GCAT-Genomes for Life. Cohort study of the Genomes of Catalonia, Fundació IGTP (The Institute for Health Science Research Germans Trias i Pujol) IGTP is part of the CERCA Program/Generalitat de Catalunya. GCAT is supported by Acción de Dinamización del ISCIII-MINECO and the Ministry of Health of the Generalitat of Catalunya (ADE 10/00026); the Agència de Gestió d’Ajuts Universitaris i de Recerca (AGAUR) (2017-SGR 529). M.M. received research funding from grant PI19/00335 Acción Estratégica en Salud, integrated in the Spanish National RDI Plan and financed by ISCIII-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (European Regional Development Fund (FEDER)-Una manera de hacer Europa’). B.C. is supported by national grants PI18/01512. X.F. is supported by the VEIS project (001-P-001647) (co-funded by the European Regional Development Fund (ERDF), ‘A way to build Europe’). Additional data included in this study were obtained in part by the COVICAT Study Group (Cohort Covid de Catalunya) supported by IsGlobal and IGTP, European Institute of Innovation & Technology (EIT), a body of the European Union, COVID-19 Rapid Response activity 73A and SR20-01024 La Caixa Foundation. A.J. and S.M. were supported by the Spanish Ministry of Economy and Competitiveness (grant numbers: PSE-010000-2006-6 and IPT-010000-2010-36). A.J. was also supported by national grant PI17/00019 from the Acción Estratégica en Salud (ISCIII) and the European Regional Development Fund (FEDER). The Basque Biobank, a hospital-related platform that also involves all Osakidetza health centres, the Basque government’s Department of Health and Onkologikoa, is operated by the Basque Foundation for Health Innovation and Research-BIOEF. M.C. received Grants BFU2016-77244-R and PID2019-107836RB-I00 funded by the Agencia Estatal de Investigación (AEI, Spain) and the European Regional Development Fund (FEDER, EU). M.R.G., J.A.H., R.G.D. and D.M.M. are supported by the ‘Spanish Ministry of Economy, Innovation and Competition, the Instituto de Salud Carlos III’ (PI19/01404, PI16/01842, PI19/00589, PI17/00535 and GLD19/00100) and by the Andalussian government (Proyectos Estratégicos-Fondos Feder PE-0451-2018, COVID-Premed, COVID GWAs). The position held by Itziar de Rojas Salarich is funded by grant FI20/00215, PFIS Contratos Predoctorales de Formación en Investigación en Salud. Enrique Calderón’s team is supported by CIBER of Epidemiology and Public Health (CIBERESP), ‘Instituto de Salud Carlos III’. J.C.H. reports grants from Research Council of Norway grant no 312780 during the conduct of the study. E.S. reports grants from Research Council of Norway grant no. 312769. The BioMaterialBank Nord is supported by the German Center for Lung Research (DZL), Airway Research Center North (ARCN). The BioMaterialBank Nord is member of popgen 2.0 network (P2N). P.K. Bergisch Gladbach, Germany and the Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Cologne, Germany. He is supported by the German Federal Ministry of Education and Research (BMBF). O.A.C. is supported by the German Federal Ministry of Research and Education and is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—CECAD, EXC 2030–390661388. The COMRI cohort is funded by Technical University of Munich, Munich, Germany. This work was supported by grants of the Rolf M. Schwiete Stiftung, the Saarland University, BMBF and The States of Saarland and Lower Saxony. K.U.L. is supported by the German Research Foundation (DFG, LU-1944/3-1). Genotyping for the BoSCO study is funded by the Institute of Human Genetics, University Hospital Bonn. F.H. was supported by the Bavarian State Ministry for Science and Arts. Part of the genotyping was supported by a grant to A.R. from the German Federal Ministry of Education and Research (BMBF, grant: 01ED1619A, European Alzheimer DNA BioBank, EADB) within the context of the EU Joint Programme—Neurodegenerative Disease Research (JPND). Additional funding was derived from the German Research Foundation (DFG) grant: RA 1971/6-1 to A.R. P.R. is supported by the DFG (CCGA Sequencing Centre and DFG ExC2167 PMI and by SH state funds for COVID19 research). F.T. is supported by the Clinician Scientist Program of the Deutsche Forschungsgemeinschaft Cluster of Excellence ‘Precision Medicine in Chronic Inflammation’ (EXC2167). C.L. and J.H. are supported by the German Center for Infection Research (DZIF). T.B., M.M.B., O.W. und A.H. are supported by the Stiftung Universitätsmedizin Essen. M.A.-H. was supported by Juan de la Cierva Incorporacion program, grant IJC2018-035131-I funded by MCIN/AEI/10.13039/501100011033. E.C.S. is supported by the Deutsche Forschungsgemeinschaft (DFG; SCHU 2419/2-1).
Authors’ contributions
Contributions are shown in the Supplementary Note.
References
- 1. Booth, A., Reed, A.B., Ponzo, S., Yassaee, A., Aral, M., Plans, D., Labrique, A. and Mohan, D. (2021) Population risk factors for severe disease and mortality in COVID-19: a global systematic review and meta-analysis. PLoS One, 16, e0247461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong, C.K.H., Wong, J.Y.H., Tang, E.H.M., Au, C.H. and Wai, A.K.C. (2020) Clinical presentations, laboratory and radiological findings, and treatments for 11,028 COVID-19 patients: a systematic review and meta-analysis. Sci. Rep., 10, 19765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peckham, H., de Gruijter, N.M., Raine, C., Radziszewska, A., Ciurtin, C., Wedderburn, L.R., Rosser, E.C., Webb, K. and Deakin, C.T. (2020) Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun., 11, 6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu, Y., Sun, J., Dai, Z., Deng, H., Li, X., Huang, Q., Wu, Y., Sun, L. and Xu, Y. (2020) Prevalence and severity of corona virus disease 2019 (COVID-19): a systematic review and meta-analysis. J. Clin. Virol., 127, 104371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Severe Covid-19 GWAS Group (2020) Genomewide association study of severe Covid-19 with respiratory failure. N. Engl. J. Med., 383, 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pairo-Castineira, E., Clohisey, S., Klaric, L., Bretherick, A.D., Rawlik, K., Pasko, D., Walker, S., Parkinson, N., Fourman, M.H., Russell, C.D. et al. (2021) Genetic mechanisms of critical illness in COVID-19. Nature, 591, 92–98. [DOI] [PubMed] [Google Scholar]
- 7. COVID-19 Host Genetics Initiative, C (2021) Mapping the human genetic architecture of COVID-19. Nature, 600, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Velavan, T.P., Pallerla, S.R., Rüter, J., Augustin, Y., Kremsner, P.G., Krishna, S. and Meyer, C.G. (2021) Host genetic factors determining COVID-19 susceptibility and severity. EBioMedicine, 72, 103629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Namkoong, H., Edahiro, R., Fukunaga, K., Shirai, Y., Sonehara, K., Tanaka, H., Lee, H., Hasegawa, T., Kanai, M., Naito, T. et al. (2021) Japan COVID-19 task force: a nation-wide consortium to elucidate host genetics of COVID-19 pandemic in Japan. medRxiv. 2021.05.17.21256513; 10.1101/2021.05.17.2125651. [DOI] [Google Scholar]
- 10. Li, Y., Ke, Y., Xia, X., Wang, Y., Cheng, F., Liu, X., Jin, X., Li, B., Xie, C., Liu, S. et al. (2021) Genome-wide association study of COVID-19 severity among the Chinese population. Cell Discov., 7, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arora, J., McLaren, P.J., Chaturvedi, N., Carrington, M., Fellay, J. and Lenz, T.L. (2019) HIV peptidome-wide association study reveals patient-specific epitope repertoires associated with HIV control. Proc. Natl. Acad. Sci. U. S. A., 116, 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou, W., Nielsen, J.B., Fritsche, L.G., Dey, R., Gabrielsen, M.E., Wolford, B.N., LeFaive, J., VandeHaar, P., Gagliano, S.A., Gifford, A. et al. (2018) Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet., 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Willer, C.J., Li, Y. and Abecasis, G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McGuire, D., Jiang, Y., Liu, M., Weissenkampen, J.D., Eckert, S., Yang, L., Chen, F., Liu, M., Jiang, Y., Wedow, R. et al. (2021) Model-based assessment of replicability for genome-wide association meta-analysis. Nat. Commun., 12, 1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Benner, C., Spencer, C.C.A., Havulinna, A.S., Salomaa, V., Ripatti, S. and Pirinen, M. (2016) FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics, 32, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakanishi, T., Pigazzini, S., Degenhardt, F., Cordioli, M., Butler-Laporte, G., Maya-Miles, D., Nafría-Jiménez, B., Bouysran, Y., Niemi, M., Palom, A. et al. (2021) Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J. Clin. Invest., 131, e152386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stefansson, H., Helgason, A., Thorleifsson, G., Steinthorsdottir, V., Masson, G., Barnard, J., Baker, A., Jonasdottir, A., Ingason, A., Gudnadottir, V.G. et al. (2005) A common inversion under selection in Europeans. Nat. Genet., 37, 129–137. [DOI] [PubMed] [Google Scholar]
- 18. Howie, B., Marchini, J. and Stephens, M. (2011) Genotype imputation with thousands of genomes. G3, 1, 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. MacArthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., Junkins, H., McMahon, A., Milano, A., Morales, J. et al. (2017) The new NHGRI-EBI catalog of published genome-wide association studies (GWAS catalog). Nucleic Acids Res., 45, D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Delorey, T.M., Ziegler, C.G.K., Heimberg, G., Normand, R., Yang, Y., Segerstolpe, Å., Abbondanza, D., Fleming, S.J., Subramanian, A., Montoro, D.T. et al. (2021) COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature, 595, 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quach, H., Rotival, M., Pothlichet, J., Loh, Y.H.E., Dannemann, M., Zidane, N., Laval, G., Patin, E., Harmant, C., Lopez, M. et al. (2016) Genetic adaptation and Neandertal admixture shaped the immune system of human populations. Cell, 167, 643–656.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mankelow, T.J., Singleton, B.K., Moura, P.L., Stevens-Hernandez, C.J., Cogan, N.M., Gyorffy, G., Kupzig, S., Nichols, L., Asby, C., Pooley, J. et al. (2021) Blood group type a secretors are associated with a higher risk of COVID-19 cardiovascular disease complications. eJHaem, 2, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hammer, M. (2002) A nomenclature system for the tree of human Y-chromosomal binary haplogroups. Genome Res., 12, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang, X., Holmes, C. and McVean, G. (2021) The impact of age on genetic risk for common diseases. PLoS Genet., 17, e1009723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ward, H.E. and Nicholas, T.E. (1984) Alveolar type I and type II cells. Aust. NZ J. Med., 14, 731–734. [PubMed] [Google Scholar]
- 26. Melms, J.C., Biermann, J., Huang, H., Wang, Y., Nair, A., Tagore, S., Katsyv, I., Rendeiro, A.F., Amin, A.D., Schapiro, D. et al. (2021) A molecular single-cell lung atlas of lethal COVID-19. Nature, 595, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Islam, A.B.M.M.K. and Khan, M.A.A.K. (2020) Lung transcriptome of a COVID-19 patient and systems biology predictions suggest impaired surfactant production which may be druggable by surfactant therapy. Sci. Rep., 10, 19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu, L., Zhu, J., Liu, D., Sun, Y. and Wu, C. (2021) An integrative multiomics analysis identifies putative causal genes for COVID-19 severity. Genet. Med., 23, 2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.American Journal of Respiratory and Critical Care Medicine; 203, 2021. This seems to be from an online abstract submission. 203.1_MeetingAbstracts.A3765. [Google Scholar]
- 30. Astle, W.J., Elding, H., Jiang, T., Allen, D., Ruklisa, D., Mann, A.L., Mead, D., Bouman, H., Riveros-Mckay, F., Kostadima, M.A. et al. (2016) The allelic landscape of human blood cell trait variation and links to common complex disease. Cell, 167, 1415–1429.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sakornsakolpat, P., Prokopenko, D., Lamontagne, M., Reeve, N.F., Guyatt, A.L., Jackson, V.E., Shrine, N., Qiao, D., Bartz, T.M., Kim, D.K. et al. (2019) Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet., 51, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tantisira, K.G., Lazarus, R., Litonjua, A.A., Klanderman, B. and Weiss, S.T. (2008) Chromosome 17: association of a large inversion polymorphism with corticosteroid response in asthma. Pharmacogenet. Genomics, 18, 733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. den Dekker, A.D., Davis, F.M., Joshi, A.D., Wolf, S.J., Allen, R., Lipinski, J., Nguyen, B., Kirma, J., Nycz, D., Bermick, J. et al. (2020) TNF-α regulates diabetic macrophage function through the histone acetyltransferase MOF. JCI Insight, 5, e132306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cursano, S., Battaglia, C.R., Urrutia-Ruiz, C., Grabrucker, S., Schön, M., Bockmann, J., Braumüller, S., Radermacher, P., Roselli, F., Huber-Lang, M. et al. (2020) A CRHR1 antagonist prevents synaptic loss and memory deficits in a trauma-induced delirium-like syndrome. Mol. Psychiatry., 26, 3778–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thompson, S.B., Sandor, A.M., Lui, V., Chung, J.W., Waldman, M.M., Long, R.A., Estin, M.L., Matsuda, J.L., Friedman, R.S. and Jacobelli, J. (2020) Formin-like 1 mediates effector t cell trafficking to inflammatory sites to enable t cell-mediated autoimmunity. elife, 9. 10.7554/eLife.58046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pickett, B.E., Greer, D.S., Zhang, Y., Stewart, L., Zhou, L., Sun, G., Gu, Z., Kumar, S., Zaremba, S., Larsen, C.N. et al. (2012) Virus pathogen database and analysis resource (ViPR): a comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses, 4, 3209–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kirkpatrick, M. (2010) How and why chromosome inversions evolve. PLoS Biol., 8, e1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wellenreuther, M. and Bernatchez, L. (2018) Eco-evolutionary genomics of chromosomal inversions. Trends Ecol. Evol., 33, 427–440. [DOI] [PubMed] [Google Scholar]
- 39. Ben Shachar, S., Barda, N., Manor, S., Israeli, S., Dagan, N., Carmi, S., Balicer, R., Zisser, B. and Louzoun, Y. (2021) MHC Haplotyping of SARS-CoV-2 patients: HLA subtypes are not associated with the presence and severity of COVID-19 in the Israeli population. J. Clin. Immunol., 41, 1154–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Purcell, S. PLINK 1.9. PLINK 1.9. http://pngu.mgh.harvard.edu/purcell/plink/.
- 41. Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M.A., Bender, D., Maller, J., Sklar, P., de Bakker, P.I., Daly, M.J. and Sham, P.C. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet., 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Price, A.L., Weale, M.E., Patterson, N., Myers, S.R., Need, A.C., Shianna, K.V., Ge, D., Rotter, J.I., Torres, E., Taylor, K.D.D. et al. (2008) Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet., 83, 132–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A. and Abecasis, G.R. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abraham, G., Qiu, Y. and Inouye, M. (2017) FlashPCA2: principal component analysis of biobank-scale genotype datasets. Bioinformatics, 33, 2776–2778. [DOI] [PubMed] [Google Scholar]
- 45. Taliun, D., Harris, D.N., Kessler, M.D., Carlson, J., Szpiech, Z.A., Torres, R., Taliun, S.A.G., Corvelo, A., Gogarten, S.M., Kang, H.M. et al. (2021) Sequencing of 53,831 diverse genomes from the NHLBI TOPMed program. Nature, 590, 290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. König, I.R., Loley, C., Erdmann, J. and Ziegler, A. (2014) How to include chromosome X in your genome-wide association study. Genet. Epidemiol., 38, 97–103. [DOI] [PubMed] [Google Scholar]
- 47. Viechtbauer, W. (2010) Conducting meta-analyses in R with the metafor. J. Stat. Softw., 36, 1–48. [Google Scholar]
- 48. Antonacci, F., Kidd, J.M., Marques-Bonet, T., Ventura, M., Siswara, P., Jiang, Z. and Eichler, E.E. (2009) Characterization of six human disease-associated inversion polymorphisms. Hum. Mol. Genet., 18, 2555–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Steinberg, K.M., Antonacci, F., Sudmant, P.H., Kidd, J.M., Campbell, C.D., Vives, L., Malig, M., Scheinfeldt, L., Beggs, W., Ibrahim, M. et al. (2012) Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat. Genet., 44, 872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Puig, M., Lerga-Jaso, J., Giner-Delgado, C., Pacheco, S., Izquierdo, D., Delprat, A., Gayà-Vidal, M., Regan, J.F., Karlin-Neumann, G. and Cáceres, M. (2020) Determining the impact of uncharacterized inversions in the human genome by droplet digital PCR. Genome Res., 30, 724–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kwan, J.S.H., Li, M.X., Deng, J.E. and Sham, P.C. (2016) FAPI: fast and accurate P-value imputation for genome-wide association study. Eur. J. Hum. Genet., 24, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Foley, C.N., Staley, J.R., Breen, P.G., Sun, B.B., Kirk, P.D.W., Burgess, S. and Howson, J.M.M. (2021) A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun., 12, 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang, T., Klein, A., Sang, J., Choi, J. and Brown, K.M. (2022) ezQTL: a web platform for interactive visualization and colocalization of quantitative trait loci and GWAS. Genomics Proteomics Bioinformatics. 25:S1672-0229(22)00069-9. doi: 10.1016/j.gpb.2022.05.004. Epub ahead of print. PMID: 35643189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Martínez-Fundichely, A., Casillas, S., Egea, R., Ràmia, M., Barbadilla, A., Pantano, L., Puig, M. and Cáceres, M. (2014) InvFEST, a database integrating information of polymorphic inversions in the human genome. Nucleic Acids Res., 42, D1027–D1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aguet, F., Barbeira, A.N., Bonazzola, R., Brown, A., Castel, S.E., Jo, B., Kasela, S., Kim-Hellmuth, S., Liang, Y., Oliva, M. et al. (2020) The GTEx consortium atlas of genetic regulatory effects across human tissues. Science (80-.), 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Frankish, A., Diekhans, M., Ferreira, A.M., Johnson, R., Jungreis, I., Loveland, J., Mudge, J.M., Sisu, C., Wright, J., Armstrong, J. et al. (2019) GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res., 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Papatheodorou, I., Moreno, P., Manning, J., Fuentes, A.M.P., George, N., Fexova, S., Fonseca, N.A., Füllgrabe, A., Green, M., Huang, N. et al. (2020) Expression atlas update: from tissues to single cells. Nucleic Acids Res., 48, D77–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stunnenberg, H.G., Abrignani, S., Adams, D., de Almeida, M., Altucci, L., Amin, V., Amit, I., Antonarakis, S.E., Aparicio, S., Arima, T. et al. (2016) The international human epigenome consortium: a Blueprint for scientific collaboration and discovery. Cell, 167, 1145–1149. [DOI] [PubMed] [Google Scholar]
- 59. Vieira Braga, F.A., Kar, G., Berg, M., Carpaij, O.A., Polanski, K., Simon, L.M., Brouwer, S., Gomes, T., Hesse, L., Jiang, J. et al. (2019) A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med., 25, 1153–1163. [DOI] [PubMed] [Google Scholar]
- 60. Habib, N., Avraham-Davidi, I., Basu, A., Burks, T., Shekhar, K., Hofree, M., Choudhury, S.R., Aguet, F., Gelfand, E., Ardlie, K. et al. (2017) Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods, 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sungnak, W., Huang, N., Bécavin, C., Berg, M., Queen, R., Litvinukova, M., Talavera-López, C., Maatz, H., Reichart, D., Sampaziotis, F. et al. (2020) SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med., 26, 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wolf, F.A., Angerer, P. and Theis, F.J. (2018) SCANPY: large-scale single-cell gene expression data analysis. Genome Biol., 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Song, E., Zhang, C., Israelow, B., Lu-Culligan, A., Prado, A.V., Skriabine, S., Lu, P., El Weizman, O., Liu, F., Dai, Y. et al. (2021) Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med., 218, e20202135. doi: 10.1084/jem.20202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Finak, G., McDavid, A., Yajima, M., Deng, J., Gersuk, V., Shalek, A.K., Slichter, C.K., Miller, H.W., McElrath, M.J., Prlic, M., Linsley, P.S. and Gottardo, R. (2015) MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol., 16, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Degenhardt, F., Wendorff, M., Wittig, M., Ellinghaus, E., Datta, L.W., Schembri, J., Ng, S.C., Rosati, E., Hübenthal, M., Ellinghaus, D. et al. (2019) Construction and benchmarking of a multi-ethnic reference panel for the imputation of HLA class I and II alleles. Hum. Mol. Genet., 28, 2078–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Degenhardt, F., Mayr, G., Wendorff, M., Boucher, G., Ellinghaus, E., Ellinghaus, D., ElAbd, H., Rosati, E., Hübenthal, M., Juzenas, S. et al. (2021) Trans-ethnic analysis of the human leukocyte antigen region for ulcerative colitis reveals common disease signatures. Hum. Mol. Genet., 30, 356–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.