

Abstract
NADK2 encodes the mitochondrial form of nicotinamide adenine dinucleotide (NAD) kinase, which phosphorylates NAD. Rare recessive mutations in human NADK2 are associated with a syndromic neurological mitochondrial disease that includes metabolic changes, such as hyperlysinemia and 2,4 dienoyl CoA reductase (DECR) deficiency. However, the full pathophysiology resulting from NADK2 deficiency is not known. Here, we describe two chemically induced mouse mutations in Nadk2—S326L and S330P—which cause severe neuromuscular disease and shorten lifespan. The S330P allele was characterized in detail and shown to have marked denervation of neuromuscular junctions by 5 weeks of age and muscle atrophy by 11 weeks of age. Cerebellar Purkinje cells also showed progressive degeneration in this model. Transcriptome profiling on brain and muscle was performed at early and late disease stages. In addition, metabolomic profiling was performed on the brain, muscle, liver and spinal cord at the same ages and on plasma at 5 weeks. Combined transcriptomic and metabolomic analyses identified hyperlysinemia, DECR deficiency and generalized metabolic dysfunction in Nadk2 mutant mice, indicating relevance to the human disease. We compared findings from the Nadk model to equivalent RNA sequencing and metabolomic datasets from a mouse model of infantile neuroaxonal dystrophy, caused by recessive mutations in Pla2g6. This enabled us to identify disrupted biological processes that are common between these mouse models of neurological disease, as well as those processes that are gene-specific. These findings improve our understanding of the pathophysiology of neuromuscular diseases and describe mouse models that will be useful for future preclinical studies.
Introduction
The pyridine nucleotide nicotinamide adenine dinucleotide (NAD) and its phosphorylated form, NADP, are found in all organisms and are involved in a myriad of biological functions, including serving as substrates and cofactors for numerous enzymes and metabolic processes. NAD is converted to NADP by NAD kinases (NADKs). Since NAD and NADP are membrane impermeant, these kinases are compartmentalized within the cell (1). The cytosolic form of human NADK was identified in 2001 (2), but it was not until 2012 that the mitochondrial form of NADK was discovered in yeast and in 2013 in mammals (NADK2, also called MNADK for mitochondrial NAD kinase, and C5ORF33, prior to the functional characterization of the human gene product) (3,4). The initial characterization of NADK2 showed that this kinase phosphorylates NAD using ATP or inorganic polyphosphate as a substrate, and localizes to mitochondria due to an amino-terminal mitochondrial targeting sequence (3).
In human genetic studies, three patients have been described with recessive NADK2 mutations, as summarized later. The first had hyperlysinemia and 2,4 dienoyl CoA reductase (DECR) deficiency, indicated by elevated levels of C10:2 carnitine (5). At 8 weeks of age, the patient had failed to thrive and exhibited microcephaly, central hypotonia and dysmorphic features. With age, neurological features increased with encephalopathy, dystonia, spastic quadriplegia and epilepsy. The patient died at 5 years of age. Whole-exome sequencing revealed a homozygous R340X truncating mutation in NADK2 in this patient. One previous patient with similar presentation, but even earlier mortality, was described in 1990, but no genetic results were reported (6).
In the second patient described, the general metabolic and neurological presentations were very similar to those described earlier (7). However, the patient was placed on a lysine-restricted diet at 1 month of age, and treatment with pyridoxal phosphate at 3 years of age improved electroclinical symptoms. At age 10, the patient had ataxia, poor coordination, cerebellar atrophy and oromotordysphasia but understood fluid speech and was social. She was diagnosed as having a similar homozygous truncating mutation, W319Cfs*21.
The third patient has much milder symptoms and later-onset (8). She was ascertained at 9 years of age with normal intelligence but decreased visual acuity, optic atrophy, nystagmus and peripheral neuropathy. Hyperlysinemia was present, as well as hyperprolinemia, but C10:2 carnitine levels were normal. Sequencing through the Undiagnosed Disease Network identified a homozygous Met1Val change in NADK2. NADK2 has multiple transcripts, though the initiation of translation at the next downstream ATG is not expected to produce a protein with a mitochondrial targeting sequence. Consistent with this, patient’s fibroblasts revealed residual protein expression, though primarily in the cytosolic fraction. This and the milder phenotype suggest that this is a hypomorphic allele of NADK2, producing protein through an unknown mechanism.
Hyperlysinemia is caused by mutations in AASS, encoding α-aminoadipic semialdehyde synthase, which degrades lysine (9,10). Similarly, defects in DECR cause elevated C10:2 carnitine due to changes in beta-oxidation of polyunsaturated fatty acids (11). Both AASS and DECR enzymatic activities depend on NADP as a co-substrate. Furthermore, in the absence of mitochondrial NADP due to NADK2 mutations, these enzymes are either inactive (DECR1, the mitochondrial DECR enzyme, DECR2 localizes to peroxisomes), or reduced in their levels (AASS), suggesting a molecular chaperone role for NADP (5). Thus, the molecular basis for hyperlysinemia and C10:2 carnitine elevation in NADK2-deficient patients may be understood. However, AASS deficiency is considered a benign metabolic variant (9,12), and mice lacking Decr1 are viable and have sensitivity to fasting and cold stress, but no reported neurological phenotype or shortened lifespan (13,14). Therefore, these pathways alone are unlikely to account for the severe disease observed in patients with mutations in NADK2, and a deeper analysis of the pathophysiology of this rare disorder is needed.
Model organisms have the potential to help elucidate the pathophysiology of NADK2 deficiency and to provide a platform for preclinical studies. Two independent null alleles of mouse Nadk2, tm1b (EUCOMM)Wtsi (MGI:5637034) and em1(IMPC)J (MGI:5689888), produced and analyzed through the Knockout Mouse Program (KOMP), are early lethal with complete penetrance. The em1J homozygotes die at 12.5 days of gestation (E12.5), indicating null alleles of Nadk2 are embryonic lethal in mice. Analysis has also been performed on the tm1a(EUCOMM)Wtsi allele (MGI:4842252), which includes a LacZ reporter and gene-trapping insertion, but does not include the deletion of a critical, frameshifting exon as in the tm1b allele, and may therefore result in a hypomorph (https://www.infrafrontier.eu/sites/infrafrontier.eu/files/upload/public/pdf/Resources%20and%20Services/eucomm-alleles-overview_infrafrontier-2016.pdf). The homozygous Nadk2tm1a(EUCOMM)Wtsi mice survive and have relevant elevated lysine and C10:2 carnitine levels (15). They were analyzed primarily for liver metabolic phenotypes and develop nonalcoholic fatty liver when fed a high-fat diet. No neurological symptoms or shortened lifespan were reported.
Here, we describe two chemically induced point mutations in Nadk2 that present with clear neuromuscular phenotypes, neurodegeneration, shortened lifespan and metabolic deficits. To better understand the pathophysiology underlying these phenotypes, we performed extensive metabolomic and transcriptomic analyses in these mice and compared them to mice carrying a mutation in Pla2g6 (16), a model of infantile neuroaxonal dystrophy (INAD), (17) to separate changes associated with general neuromuscular dysfunction from gene- and disease-specific effects.
Results
Identification of Nadk2 mouse mutations
We identified two independent strains of mice with mutations in Nadk2. The mice were first analyzed because of their overt neuromuscular phenotypes, including hind limb wasting, gait abnormalities and shakiness. In addition, although a formal lifespan study was not performed affected mice begin dying at 3–4 months of age, indicating early mortality. The mutations were produced in chemical mutagenesis programs performed at The Jackson Laboratory and were recessive (unaffected parents produced ~25% affected offspring). They were mapped using standard positional cloning strategies (see ‘Materials and Methods’), and both were located on proximal Chromosome 15 (Fig. 1A). The first mutation was identified in the Nadk2 gene, located at 9.1 mb on Chromosome 15 (designated Nadk2m1Jcs, MGI:6825802) by sequencing polymerase chain reaction (PCR)-amplified cDNA of candidate genes in the interval, which revealed a homozygous Nadk2 mutation, changing Serine 326 to Leucine (S326L) (Fig. 1B). Given the overlapping map positions and similar phenotypes of the two mutations, we sequenced the Nadk2 gene in the second strain (Nadk2nmf421, MGI:6825801), and also identified a homozygous Nadk2 mutation changing Serine 330 to Proline (S330P) (Fig. 1B). Both point mutations are predicted to be ‘probably damaging’ by PolyPhen with both having the maximum score of 1.00 (18). To confirm these mutations were causal, we performed a complementation test, breeding mice heterozygous for each mutation to produce compound heterozygotes (S326L/S330P). This produced affected offspring with a similar phenotype, including two mice that were small and shaky and died by ~3 months of age and a third with a similar overt phenotype. These mice were confirmed to be compound heterozygotes by genotyping. Thus, we have identified two independent mutations in the Nadk2 gene in mice that cause an overt phenotype and shortened lifespan. Given the similar phenotype of the two strains, only the nmf421 S330P allele was used in the experiments described later and homozygotes are referred to as Nadk2−/− for simplicity. The m1Jcs S326L allele is cryopreserved.
Figure 1.
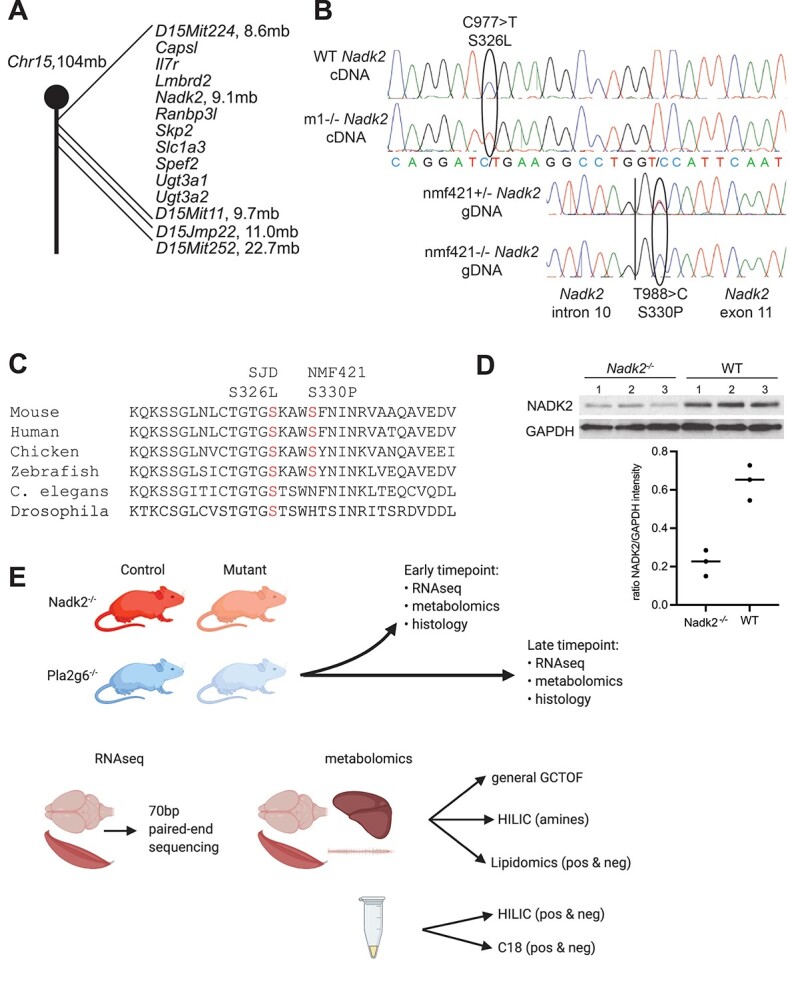
Nadk2 mouse mutations and study design. (A) The ENU-induced mutations mapped to proximal mouse Chromosome 15, with the m1Jcs allele mapping between D15Mit224 at 8.6 mb and D15Mit11 at 9.7 mb. This interval contains 10 protein coding genes. The nmf421 allele mapped proximally to D15Jmp22 at 11.0 mb. (B) Sequencing chromatograms showing the C977 > T mutation in cDNA prepared from wild type and m1Jcs homozygous mice (m1), and the T988 > C mutation in PCR-amplified genomic DNA from heterozygous and homozygous nmf421 mice. The vertical line in genomic DNA sequence is the intron 10/exon 11 boundary. (C) Evolutionary conservation of S326 and S330 in NADK2. (D) Western blot analysis of NADK2 versus a GAPDH loading control revealed the mutant protein were significantly reduced compared to littermate controls. (E) Overview of RNA-Seq and metabolomics studies. Samples from Nadk2 and Pla2g6 mutant mice, as well as wild-type littermates for each, were collected at both early and late time points as described in the ‘Materials and Methods’. At each time point, samples were collected for RNA-Seq and metabolomics, as well as for histology. Samples for RNA-Seq derived from whole brain and muscle, and were sequenced using 100-bp paired-end sequencing. Samples for metabolomics included whole brain, muscle, spinal cord, and liver, as well as plasma, which was previously omitted. These samples were analyzed by general GCTOF mass spectrometry, HILIC for biogenic amines, and lipidomics (positive and negative ionization modes).
The mouse NADK2 protein is made of 452 amino acids, encoded by the 14 exon Nadk2 gene. The mutations identified are located at the 3′ end of exon 10 and the 5′ end of exon 11 (transcript Nadk2-205). Alternatively spliced transcripts lacking either exon 5 or exon 8 are reported in the Ensembl genome browser (transcripts Nadk2-204 and − 206), but exons 10 and 11 are included in all mRNAs producing full length protein. The two mutated serine residues at positions 326 and 330 are conserved through vertebrate evolution, and S326 is also conserved in Caenorhabditis elegans and Drosophila melanogaster (Fig. 1C). The serine residues fall at the C-terminal end of the NAD kinase domain. The mouse NADK2 protein is made of 452 amino acids, whereas the human protein is made of 442 amino acids, but the domain structure and the mutated serines are conserved. Western blotting liver homogenates from 3 Nadk2−/− and 3 littermate control mice revealed a 65.6 ± 10.2% decrease in NADK2 protein abundance in mice with the S330P mutation (Fig. 1D). An analysis of mitochondrial content in muscle and liver of 5 week-old mutant mice and littermate controls using quantitative PCR (qPCR) to assess mitochondrial genomes versus nuclear genomes revealed no significant changes, although muscle samples showed a trend toward reduced mitochondrial abundance.
To better understand the pathogenic mechanisms that lead to the Nadk2 phenotype, we performed both metabolomic and transcriptomic analyses (Fig. 1E), after confirming that the Nadk2−/− mice do indeed have neuromuscular and neurodegenerative phenotypes by histology. Furthermore, we compared the Nadk2−/− mice to a mouse mutation in Pla2g6 (Pla2g6m1J, MGI:4412026, homozygotes hereafter referred to as Pla2g6−/− for simplicity), a model of infantile neuroaxonal dystrophy (16). The Pla2g6−/− mice also have a neuromuscular phenotype that is outwardly similar to the Nadk2−/− mice, but through an independent gene and possibly unrelated pathogenic mechanism. We compared these genotypes to gain a better understanding of both Nadk2 and Pla2g6 mutation pathogenicity, to identify metabolomic and transcriptomic signatures that may be shared as general neuromuscular disease perturbations, to identify genomic and metabolomic changes that are genotype-specific, and to validate these mice as useful models for future preclinical studies.
Neuromuscular and neurodegenerative phenotypes in Nadk2 −/− mice
The Nadk2−/− mice have overt neuromuscular phenotypes, including muscle wasting, tremor, abnormal gait, and hind limb dysfunction. To better understand the basis for this, we performed histopathology on 3 males and 3 females at 5 weeks and 11 weeks of age, compared to wild-type littermates. These ages represent an early time point when overt phenotypes are just discernable, and a late-stage time point, when mice are clearly affected and may soon die. Consistent with the overt phenotype, cross-sections of the gastrocnemius muscle showed striking progressive abnormalities (Fig. 2A–C). At 5 weeks of age, occasional central nuclei were seen, indicating regenerated muscle fibers, and some atrophied fibers were present, but defects were subtle (Fig. 2B). By 11 weeks of age, the muscle was severely atrophied, and most fibers were smaller than wild type (Fig. 2C). This atrophy may be largely neurogenic, secondary to denervation, but myogenic defects may also be involved, as indicated by our omics analyses later. Neuromuscular junctions (NMJs) in the hind limb showed marked denervation, with many postsynaptic sites being completely devoid of overlying motor nerve terminals (Fig. 2D–F). In 107 NMJs examined in the plantaris muscle of the lower leg from two Nadk2−/− mice at 5 weeks of age, 35 were fully innervated, 32 were partially innervated, where the nerve terminal did not completely overlie the postsynaptic acetylcholine receptor field and 40 were completely denervated. In contrast, in littermate controls, all 119 NMJs examined were fully innervated. The generally well-preserved integrity of the denervated postsynaptic sites at 5 weeks of age is consistent with recent denervation (Fig. 2E) before the postsynaptic apparatus has disintegrated and before the muscle fiber has atrophied. In the motor branch of the femoral nerve, which innervates the quadriceps muscle in the hind limb, myelinated axons were not reduced in number, but were reduced in size by 11 weeks of age (Fig. 2G–L). The mutant axons were not smaller at 5 weeks of age, and while the wild-type axons significantly increased their size between 5 and 11 weeks, the mutant axons did not.
Figure 2.
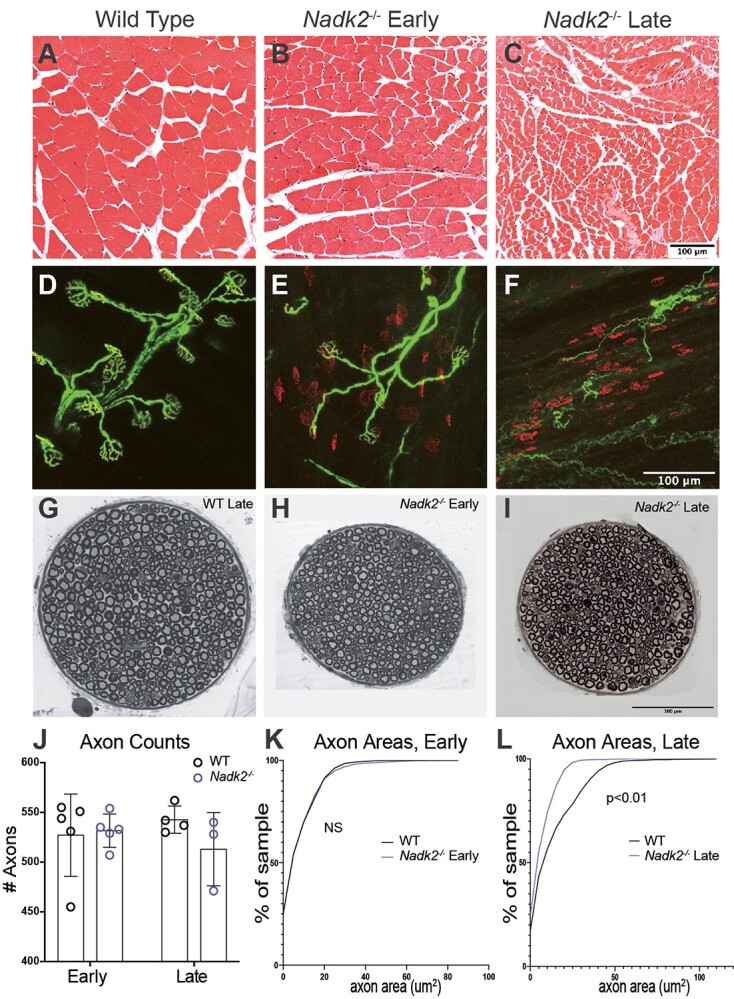
Nadk2 −/− muscle and nerve histology. (A–C) Cross-sections of the gastrocnemius muscle stained with H&E in wild type (A), 5-week-old Nadk2−/− (B), and 11-week-old Nadk2−/− mice (C). Severe muscle atrophy was present at 11 weeks. (D–F) Neuromuscular junctions in the plantaris muscle in wild type (D), 5-week-old Nadk2−/− (E), and 9-week-old Nadk2−/− mice (F). Neurofilament and SV2 label the nerve (green) and a-Bungarotoxin labels the postsynaptic acetylcholine receptors (red). Many denervated NMJs are observed at both 5 and 11 weeks of age. (G–I) Cross-sections of the motor branch of the femoral nerve in 11-week-old wild-type (G), 5-week-old Nadk2−/− (H), and 11-week-old Nadk2−/− mice (I). (J) The number of myelinated axons in the femoral motor nerve was not reduced at either age in Nadk2−/− mice. (K–L) The distribution of axon cross-sectional diameters was not different than control at 5 weeks of age (K), but axon size did not increase with age and axons in Nadk2−/− mice were smaller than wild type at 11 weeks of age. Scale bar in (C) is 100 μm for (A–C), in (F) is 100 μm for (D–F), and in (I) is 500 μm for (G–I). Significance tested by the Student t-test (J), or Kologmorov–Smirnoff and Mann–Whitney U tests (K, L).
In addition to the denervation and muscle atrophy described earlier, we also observed neurodegeneration in other regions of the central nervous system in Nadk2−/− mice. Since optic neuropathy was described in at least one NADK2 patient, we examined the retina in Nadk2−/− mice. There was no evidence of optic neuropathy, with no cupping at the optic nerve head (Fig. 3A–C), and retinal ganglion cell density was not reduced (14.2 ± 1.6 cells per 100 μm in control versus 15.2 ± 0.8 in 11-week Nadk2−/− mice). The outer nuclear layer of photoreceptors appeared less dense in mutant samples, but quantification revealed no changes in outer nuclear layer thickness or photoreceptor cell density (Fig. 3D). In the cerebellum, there was progressive Purkinje cell loss, with a significant reduction in Purkinje cell density at 5 weeks of age, which was even more pronounced at 11 weeks of age (Fig. 3E–H). An examination of sagittal sections of the brain did not reveal evidence of widespread neurodegeneration (pyknosis, chromatolysis, karyorrhexis) or astrocyte gliosis in regions, such as the hippocampus or cortex (not shown). However, while our data discussed earlier indicate that the Nadk2 mutant mice have aspects of neurodegeneration, we acknowledge that this is not an exhaustive analysis and other cell populations may undergo neurodegeneration that we did not note with this high-level analysis.
Figure 3.

Nadk2 −/− retina and cerebellum histology. (A–C) Cross-sections of the retina and the optic nerve head for wild type (A), 5-week-old (B), and 11-week-old (C) Nadk2−/− mice. No optic nerve cupping indicative of optic neuropathy was observed. (D) Quantification of photoreceptor number did not reveal a decrease in the Nadk2−/− mice at either early or late time points. (E-G) Sagittal sections of the cerebellum in wild type (E), 5-week-old (F), and 11-week old (G) Nadk2−/− mice. (H) Quantification of Purkinje cell number revealed a decrease in 5-week-old Nadk2−/− mice that was even more pronounced at 11 weeks of age. * P < 0.05, **** P < 0.0001. Pairwise comparisons tested using Student t-test. Scale bar in (C) is 100 μm for (A–C), and in F is 100 μm for (D–F).
RNA sequencing
To better understand the pathological mechanisms of NADK2 deficiency, we performed RNA sequencing (RNA-Seq) of a sagittal half brain and skeletal muscle (triceps surae) from Nadk2−/− and Pla2g6−/− mice, as well as littermate controls for each (Fig. 1E, Table 1). Two time points were used, 5 weeks and 11 weeks for Nadk2 mutants, and 6 weeks and 13 weeks for Pla2g6 mutants. These ages were selected to be either early or late in disease progression and to match those used for the histological analyses described earlier. To identify the tissues and time points that exhibit transcriptomic and metabolomic disease signatures that differentiate between mutant samples and wild-type controls, principal component analysis (PCA) was used (Fig. 4A and B). These plots indicate that clear separation of mutant samples from controls was only obtained from muscle at the late time point in Nadk2−/− mice, suggesting that muscle may be more strongly affected than the brain in these animals. The lack of separation in other tissues and time points could not readily be explained but may arise from subtle effects of the mutation or other sources of experimental variation. To examine the magnitude of the changes to the transcriptome and the similarity between data sets, we first generated lists of differentially expressed genes (DEGs) by comparing mutant to control within each time point and tissue type with cutoffs for false discovery rate (FDR) of <0.05 and fold change |Log2FC| > 1.5). These lists were then compared as shown in the Venn diagrams (Fig. 4C and D). These diagrams suggest that the disease signature seen in muscle from late Nadk2−/− mice is already at least partly present in the early time point, given the overlap in the gene lists. In contrast, the number of DEGs is smaller and there is little overlap between muscle samples from early and late Pla2g6−/− mice, suggesting that this mutation has a milder effect on the transcriptome of muscle than does Nadk2−/−. When DEGs are compared between the skeletal muscle of Nadk2−/− and Pla2g6−/− mice at the later time point, although there are many fewer DEGs in Pla2g6−/−, more than 50% of these DEGs are also seen in Nadk2−/−. This shared subset potentially includes genes that are generally misregulated in neuromuscular disease, while the majority of DEGs in Nadk2−/− are private and may represent pathways specific to that disease process. Using our stringent thresholds, many fewer DEGs were found in the brain, and while early and late time points for each mutation overlapped, no DEGs were shared between Nadk2−/− and Pla2g6−/− samples (Fig. 4D). DEGs for each mutation versus littermate controls, each tissue and each time point are provided (Supplementary Material, Table S1). We determined the number of DEGs from each mutant, tissue and time point that had annotations in each of the databases we used for subsequent enrichment analyses (Supplementary Material, Table S2).
Table 1.
RNA-Seq samples
| Genotypes | Ages | N | Tissues |
|---|---|---|---|
| Nadk2−/− | 5 weeks | 3 F, 3 M | Brain, muscle |
| 11 weeks | 3 F, 3 M | ||
| Pla2g6−/− | 6 weeks | 2 F, 3 M | |
| 13 weeks | 2 F, 2 M | ||
| Wild type | 5–6 weeks | 3 F, 3 M | |
| 11–13 weeks | 3 F, 3 M |
Genotypes, ages, sex and tissues used in RNA-Seq studies.
Figure 4.

PCA plots and differential expression analysis of the transcriptome of Nadk2−/− and Pla2g6−/− mice. (A) PCA plots of DEGs from brain and muscle of Nadk2−/− mice (M, blue) compared to wild-type littermate controls (C, red) at early and late time points. Clear separation of the genotypes is seen only at the late time point in muscle. (B) PCA plots of DEGs from brain and muscle of Pla2g6−/− mice compared to wild-type littermate controls at early and late time points. Plots indicate little separation of the transcriptomes from the two groups. (C, D) Venn diagrams comparing the number of DEGs and their overlap between the indicated genotypes and time points for muscle (C), and brain (D).
Metabolomics
To complement the RNA-Seq data, samples were obtained from sagittal hemi-brains and muscle at early and late time points, as well as from spinal cord and liver, from both Nadk2−/− and Pla2g6−/− mice and littermate controls for metabolomics studies (Fig. 1E, Table 2). All samples were analyzed by gas chromatography (GC) coupled to time-of-flight (GC-TOF) mass spectrometry to screen for primary metabolites (Supplementary Material, Tables S3–S6), and brain and muscle whole-tissue samples were additionally analyzed for biogenic amines by hydrophilic interaction liquid chromatography (HILIC) (Supplementary Material, Table S7) and for lipidomics by CSH-QTOF-MS/MS (Supplementary Material, Table S8). Several approaches were taken to analyze the extent to which mutant samples differed from matched wild-type controls. First, random forest analysis was performed on each group, and the out of bag (OOB) errors were determined (Fig. 5A). As lower OOB scores indicate better separation of mutant from control, Nadk2−/− overall showed a greater degree of difference from wild type than did Pla2g6−/−. Specifically, samples from the late time point showed a greater difference from wild type than did samples from the early time point, and samples from muscle showed the greatest degree of difference from wild type. Samples from late Nadk2−/− muscle yielded OOB error scores of 0 on all three metabolomics platforms utilized, suggesting a large magnitude of change within the metabolome of muscle by the late stages of the disease. The degree of separation between select Nadk2−/− and wild-type samples was also visualized by plotting scores from PCA (Fig. 5B and C). In agreement with the random forest OOB scores, clear separation of mutant and control samples was only seen in Nadk2−/− muscle at the late time point and this separation was seen using data from any of the three metabolomics platforms. To better compare to patient data, we also performed a 4-mode exploratory metabolomics analysis on plasma from 5-week-old Nadk2−/− mice using mass spectrometry with C18 and HILIC chromatography columns in both positive and negative ionization modes (Table 3, two technical replicates, MS data in Supplementary Material, Tables S9–12). Principle component analyses of these data separated mutants from controls (Fig. 5D). PCA was not performed on Pla2g6−/− samples, given the comparatively small number of differentially detected metabolites. Thus, our metabolomics results agree with the major conclusion of the RNA-Seq described earlier that the strongest differences are seen in Nadk2−/− muscle at the late time point.
Table 2.
GC-TOF, HILIC, lipidomics WCMC metabolomics samples
| Genotypes | Ages | N | Tissues |
|---|---|---|---|
| Nadk2−/− | 5 weeks | 4 F, 6 M | Brain, muscle, spinal cord, liver, |
| 11 weeks | 5 F, 5 M | ||
| Nadk2+/+ | 5 weeks | 2 F, 5 M | |
| 11 weeks | 3 F, 6 M | ||
| Pla2g6−/− | 6 weeks | 5 F, 4 M | |
| 13 weeks | 4 F, 6 M | ||
| Pla2g6+/+ | 6 weeks | 2 F, 7 M | |
| 13 weeks | 4 F, 5 M |
Genotypes, ages, sex and tissues used in GC-TOF, HILIC and lipidomics studies.
Figure 5.

Random forest OOB error rates and PCA scores plots for metabolomics analysis of Nadk2 and Pla2g6 mutant mice versus littermate controls. (A) OOB error rates from random forest analysis of all metabolomics data from mutant mice compared to littermate controls. Scores are color-coded to indicate the magnitude of the error score, with scores of zero shaded dark orange, those between zero and 0.3 shaded medium orange, and those between 0.3 and 0.5 shaded pale orange. The lowest error rates are seen in Nadk2−/− mice at the late time point, especially in muscle samples. (B) PCA score plots of general GCTOF metabolomics data of Nadk2−/− mice (M, blue) versus wild-type littermate controls (C, red) from brain and muscle samples at early and late time points. Separation of the genotype groups is only observed at the late time point in muscle. (C) PCA score plots of HILIC and lipidomics analyses from muscle collected from Nadk2−/− mice at the late time point also display separation from matched wild-type samples. (D) PCA score plots of C18 and HILIC metabolomics in both positive and negative ionization modes for plasma from Nadk2−/− mice and controls at the early time point.
Table 3.
Plasma metabolomics samples
| Genotypes | Ages | N | Tissues |
|---|---|---|---|
| Nadk2−/− | 5 weeks | 2 M, 1 F | Plasma |
| Nadk2+/+ | 2 M, 1 F |
Genotypes, ages, sex and tissues used in plasma metabolomics studies.
Analysis of transcriptomic and metabolomic data from Nadk2-deficient mice
We next performed enrichment and pathway analyses using brain and muscle RNA-Seq and primary metabolite levels from the GC-TOF platform for Nadk2−/− mice at 5- and 11 weeks of age. Only data from the GC-TOF metabolomics were used in these analyses because of challenges in analyte identification and annotation in the HILIC and CSH-QTOF-MS/MS results. We find that lysine is the only metabolite that is significantly elevated relative to controls across all tissues at both early and late time points (Fig. 6A–C, Supplementary Material, Fig. S1A). A variety of other metabolites also exhibited differences in abundance between Nadk2−/− and controls (Supplementary Material, Fig. S1B). In particular, 3-hydroxybutyric acid is elevated in the liver, spinal cord and muscle at the late time point. To improve the comparability between existing patient data and our Nadk2−/− mice, we performed a metabolomics analysis of plasma, which also revealed an elevation of lysine (Fig. 6D and E). Importantly, at the early time point, the metabolite list from muscle tissue is strongly associated with a variety of metabolic disorders, including hyperlysinemia and 2,4-Dienoyl-COA reductase deficiency (Fig. 6F).
Figure 6.
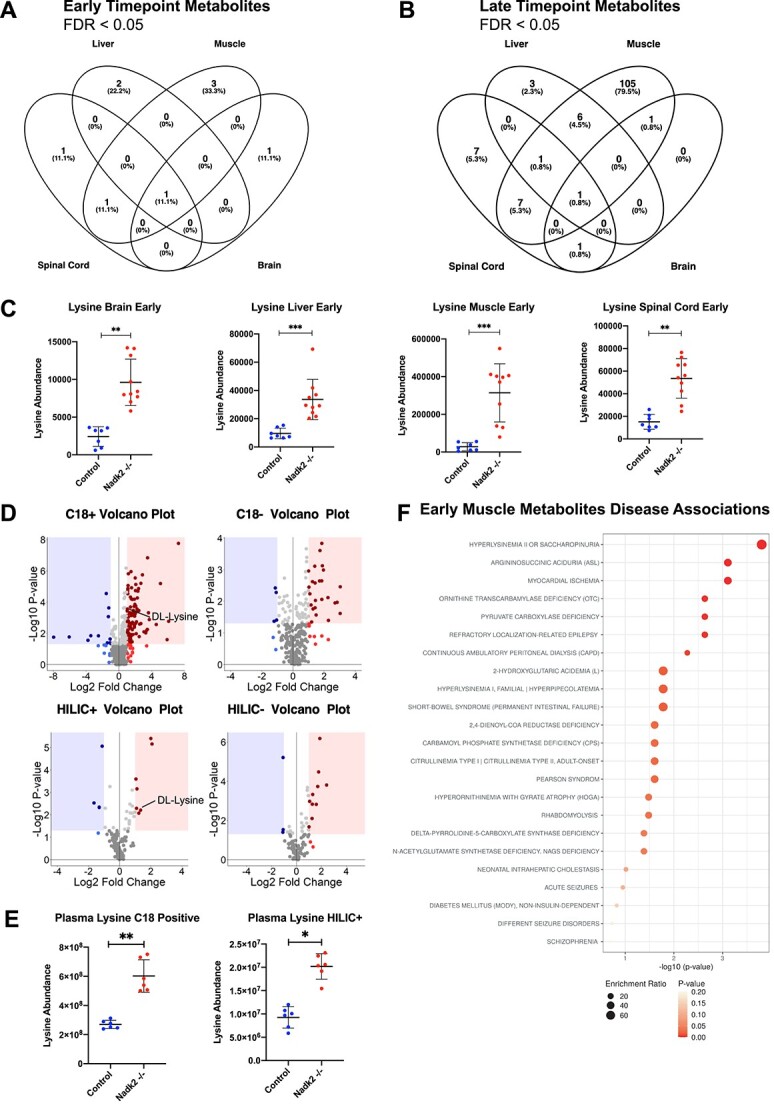
Metabolomic analyses of Nadk2−/− Muscle. (A) Lists of GC-TOF metabolites with significantly different abundance between Nadk2−/− mice and controls are compared between all tissues at the early time point. (B) Comparison of significant GC-TOF metabolites between Nadk2−/− mice and controls between all tissues at the late time point. Significance is determined by MetaboAnalyst5.0 t-test and Adj. P < 0.05. (C) The abundance of lysine in brain, liver, spinal cord, and skeletal muscle at the early time point. Significance is determined by the MetaboAnalyst 5.0 t-test. (D) Volcano plots for C18 and HILIC from plasma metabolomics in Nadk2−/− mice and controls at 5 weeks of age. (E) Abundance of lysine in plasma for C18 positive and HILIC positive ionization modes. (F) MetaboAnalyst5.0 disease association dot plot for Nadk2−/− early time point metabolite lists. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s.: not significant.
There are 932 DEGs in Nadk2−/− muscle relative to wild-type controls at the late time point, which is more than any other tissue or time point for Nadk2−/− mice: 557 DEGs in the early muscle, 5 DEGs in the early brain and 1 DEG in the late brain (Figs 4C and 7A). Given the number of DEGs and clear separation between mutants and controls by PCA we chose to focus our analyses on muscle at the late time point for Nadk2−/− mice. DEGs in this set with a positive Log2FC are associated with Reactome Pathway terms involving acetylcholine signaling, likely reflecting a response to denervation (Fig. 2), whereas DEGs with a negative Log2FC are associated with metabolic processes (Fig. 7B). Gene set enrichment analysis (GSEA) of trimmed mean of M-values (TMM)-normalized counts data revealed enrichment of three metabolic pathways: glycogen metabolism, glycolysis and glucose metabolism (Fig. 7C).
Figure 7.

Transcriptomic and metabolomic analyses of Nadk2−/− muscle at the late time point. (A) Volcano plot of DEGs in skeletal muscle samples of Nadk2 −/− collected at 11 weeks relative to age-matched controls. Genes were filtered using |Log2FC| > 1.5 and FDR < 0.05 cutoffs. (B) Reactome pathway enrichment for filtered genes with negative Log2FC in blue and genes with positive Log2FC in red. Ranked by −log10 of the enrichment P-value. (C) GSEA diagrams for the three top Nadk2 deficiency pathways by FDR value in control relative to Nadk2−/− muscle at the late time point. (D) MetaboAnalyst5.0 joint-pathway analysis plot (gene expression and metabolomics data) depicting significance and impact of pathway disruption in skeletal muscle samples Nadk2−/− mice relative to controls at the late time point. Key pathways are labeled. (E) KEGG pathway diagram of glycolysis and gluconeogenesis color-coded by MetaboAnalyst5.0 to show involvement of differential genes/metabolites from Nadk2−/− mice. (F) Top five dysfunctional enzymes predicted to produce metabolomic signatures similar to that from Nadk2−/− mice using MetaboAnalyst5.0 enrichment analysis functionality. Predicted enzymes are ranked by -log10 of the prediction P-value.
We also performed a targeted search for NADK2 deficiency-associated pathways, ‘lysine degradation’ and ‘beta oxidation’, in the KEGG pathway database and used the mapper tool to project DEGs onto these pathways (Supplementary Material, Fig. S2A and B). In both cases, numerous DEGs from our lists are involved in the target pathways (beta oxidation: M00087: 40/55, lysine degradation: M00032: 4/10). Given the mitochondrial compartmentalization of NADK2, we compared all DEGs from Nadk2−/− late muscle to genes in the MitoCarta3.0 database of genes with mitochondrial function, revealing a significant overlap by Jaccard index (Supplementary Material, Fig. S2C). Multiple mitochondria-specific pathways were implicated by the list of overlapping genes, including detoxification, carbohydrate and lipid metabolism (Supplementary Material, Fig. S2D).
Next, gene expression and metabolomic data were integrated using the MetaboAnalyst5.0 DEGs were uploaded alongside metabolite lists in the MetaboAnalyst5.0 Joint-Pathway module to identify those pathways most impacted by the gene expression and metabolite abundance changes from both datasets, which include purine metabolism, glycolysis and gluconeogenesis and fatty acid degradation (Fig. 7D). The top predicted pathway by impact score with FDR < 0.05 is glycolysis and gluconeogenesis (Fig. 7E). Metabolite enrichment analysis in MetaboAnalyst5.0 enabled prediction of enzymes whose deficiency would produce metabolomic changes consistent with our uploaded lists, and the top predicted enzyme by significance is pyruvate carboxylase (Fig. 7F).
Enrichment analysis of metabolite lists identified by metabolomics in plasma revealed enrichment of linolenic acid pathways and several pathways related to fatty acid metabolism (arachidonic acid, beta-oxidation), as well as pyrimidine metabolism and pyruvate metabolism (Supplementary Material, Fig. S3A). Top disease associations, compared to 344 human blood metabolite sets in MetaboAnalyst5.0 include hyperlysinemia, Hartnup disease, glycogenosis and lactic acidosis. Several other mitochondrial disorders were identified (Supplementary Material, Fig. S3B). Predicted dysfunctional enzyme pathways identified carnitine O-palmitoyltransferase and beta-oxidation of fatty acid. Other top sets involve carnitine metabolism, glutathione metabolism and linolenic acid metabolism (Supplementary Material, Fig. S3C). These results, taken together, indicate that lysine is strongly implicated in all Nadk2−/− metabolomic datasets and that disruption of metabolic pathways is the most prominent ‘omics’ feature of Nadk2−/− mice, with beta-oxidation dysfunction identified across multiple tissues.
Transcriptomic analysis of Pla2g6−/− mice
Because no GC-TOF metabolites from Pla2g6−/− mice had significant differences in abundance after correction for multiple testing (Adj. P < 0.05), we focused our analysis on DEGs. There are 14 DEGs in Pla2g6−/− brain at the early time point, and 10 at the late time point. There are 349 DEGs in Pla2g6−/− muscle at the early time point, and 52 DEGs at the later time point. Only three DEGs overlap between both tissues and both time points, and one of these is Pla2g6 (Supplementary Material, Fig. S4A and B). The other two DEGs encode ribosomal proteins (Supplementary Material, Fig. S4B). Though no pathways or disease-associated terms reached significance after correction for multiple testing in Pla2g6−/− brain tissue gene lists, ‘neurodegeneration with brain iron accumulation’ produced a nominal P-value of <0.05 at both early and late time points (Supplementary Material, Fig. S4C). It should be noted that these highly specific associations arose with a relatively small number of annotated genes (Supplementary Material, Table S2). Given the paucity of DEGs in the Pla2g6−/− brain, we focused our analysis on the muscle (Supplementary Material, Fig. S4D and F). DEGs with a positive Log2FC from early muscle tissue are associated with the Reactome pathways ‘formation of the cornified envelope’ and ‘keratinization’, whereas DEGs with a negative Log2FC are associated with numerous pathways related to the nervous system (Supplementary Material, Fig. S4E). Upregulated DEGs at the late time point are not significantly associated with any Reactome pathways while downregulated DEGs are associated with muscle contraction (Supplementary Material, Fig. S4G). These results suggest that neurological dysfunction in Pla2g6−/− mice produces a decreased expression of a relatively low number of key genes, a small effect that may explain the lack of separation by PCA, and does not produce detectable metabolomic signatures of disease.
Comparative analysis of Nadk2 and Pla2g6 mutant mice
To better understand the transcriptomic changes private to Nadk2 or Pla2g6 mutants, and those changes that overlap between neuromuscular disorders, we examined the shared DEGs of each tissue, genotype and time point. Only DEGs from muscle overlapped between mutants, and this overlap was greatest at the early time point (Fig. 8A). However, the overlap between DEGs in muscle at both early and late time points is significantly greater than would be expected due to chance, as indicated by GeneWeaver Jaccard analysis (Fig. 8A and B). There are 251 genes that overlap between Nadk2−/− and Pla2g6−/− mice at the early time point and 32 genes that overlap between Nadk2−/− and Pla2g6−/− mice at the late time point. Reactome pathways associated with the 251 DEGs shared between Nadk2−/− and Pla2g6−/− muscle at the early time point involve the nervous system and include numerous calcium-dependent processes and neurotransmitter release processes by over-representation analysis (Fig. 8C). GSEA identified a wider array of overlapping Reactome pathways, with the pathways exhibiting the highest normalized enrichment score pertaining to translation (Fig. 8D).
Figure 8.
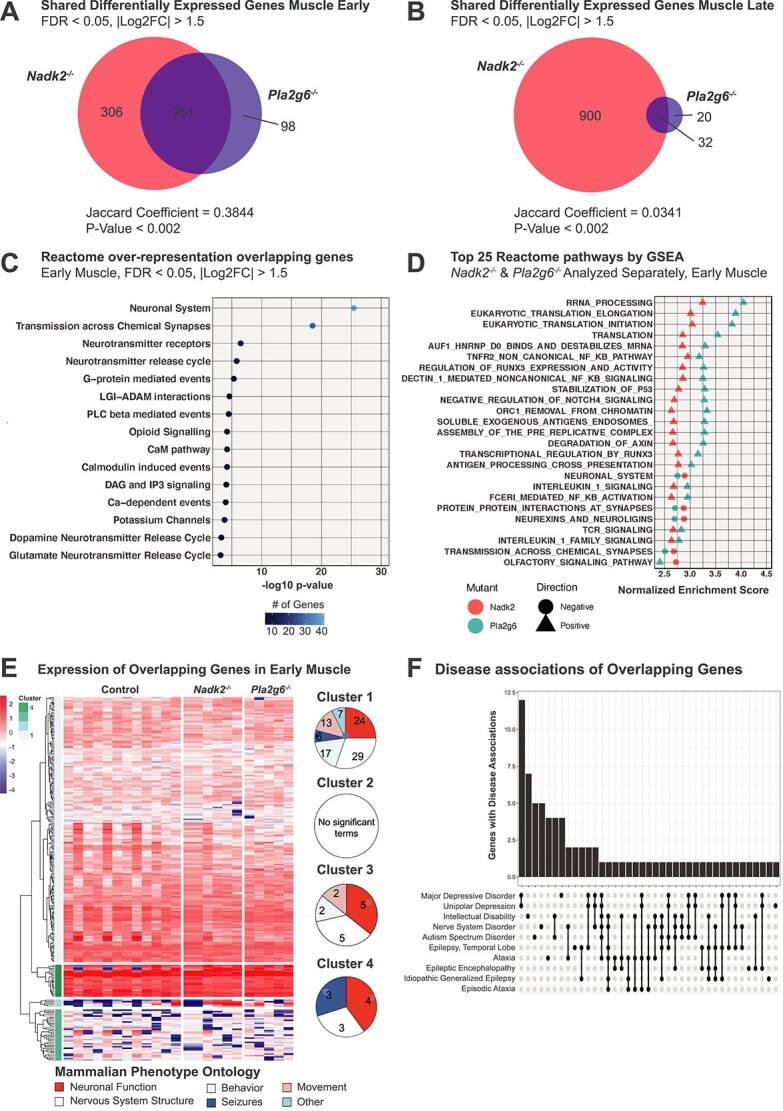
Comparative transcriptomic analysis of Nadk2 and Pla2g6 mutants. (A) Venn diagrams depicting overlapping DEGs in Nadk2−/− and Pla2g6−/− muscle at the early time point. (B) Venn diagram depicting overlapping DEGs in Nadk2−/− and Pla2g6−/− at the late time point. |Log2FC| > 1.5 and FDR < 0.05 cutoffs applied. Jaccard coefficients and significance of overlap from GeneWeaver. (C) Reactome pathway terms associated with DEGs shared between Nadk2−/− and Pla2g6−/− muscle at the early time point. Color indicates the number of genes associated with each ontology term. (D) Top 25 Reactome pathways by absolute normalized enrichment score with FWER < 0.05 implicated by gene set enrichment analysis shared between Nadk2−/− and Pla2g6−/− muscle at the early time point. Color indicates genotype and shape differentiates positive and negative normalized enrichment scores. (E) Heatmap and dendrogram depicting expression levels of DEGs shared between Nadk2−/− and Pla2g6−/− mice in muscle tissue at the early time point. Genes are clustered into four groups based on the similarity of gene expression patterns across samples. Mammalian phenotype ontology terms for each cluster are grouped into major categories, and the representation of major categories in each cluster is presented as circular plots. (F) Upset plot of gene–disease associations from human orthologs of DEGs that overlap between Pla2g6−/− and Nadk2−/− early muscle tissue.
To determine if the direction of differential expression was consistent between mutants, we created a heatmap using TMM-normalized counts. We applied a supervised clustering approach based on dendrogram height to segment these genes into four clusters based on expression level: genes with moderate expression across all samples (cluster 1), genes with highly variable expression (very high or very low outliers) that vary widely between samples (cluster 2), genes with moderate expression that vary widely across samples (cluster 3), and genes with high expression across all samples (cluster 4). We examined mammalian phenotype ontology terms separately for each cluster and determined that the cluster 4 genes were disproportionately associated with neuronal functions and seizures, whereas the cluster 1 genes have diverse associations with movement- and behavior-related terms (Fig. 8E). We examined the gene–disease associations of human orthologs of DEGs shared between Nadk2−/− and Pla2g6−/− mutant muscle at the early time point. Top disease associations were limited to disorders of the central nervous system, including ataxia (Fig. 8F).
Discussion
Here, we describe two mutations in mouse Nadk2 that were identified because of their overt neuromuscular symptoms. Both mutations alter serine residues near the C-terminal end of the catalytic domain of NADK2 and are likely partial loss-of-function alleles. Histological examination of Nadk2−/− mice indicates there are indeed neuromuscular and neurodegeneration phenotypes, with muscle atrophy and denervation, as well as loss of cerebellar Purkinje cells. Analysis of both differential metabolites and DEGs in several tissues demonstrates that changes in muscle best distinguish mutant mice from controls. The pathways perturbed in Nadk2−/− mice were largely separate from those affected in Pla2g6−/− mice, despite the similar overt presentation of the mutant animals. An integrated analysis of altered genes and metabolites demonstrates that the Nadk2−/− mice recapitulate major hallmarks of the patients, including hyperlysinemia and deficiencies in beta-oxidation of fatty acids, mediated partially by DECR1.
Although a limited number of NADK2 patients have been described, they consistently present with neurological and metabolic conditions, including hyperlysinemia and DECR deficiency. The identification of similar symptoms in the Nadk2−/− mice suggests that the S330P mutation creates a valid model of the human disease. This mutation is likely a hypomorph, as only a single amino acid is changed and the phenotype is milder than null mice produced through the KOMP program, which die embryonically. However, the mice have a more severe phenotype than described for the Nadk2tm1a(EUCOMM)Wtsi allele, which may be an even weaker hypomorph with reduced levels of wild-type Nadk2 produced. The relevance of our findings in this mouse model to human disease is discussed later.
Our genetic analysis also indicates that serines 326 and 330 are both important for NADK2 function, as mutations at either of these residues produce a strong partial-loss-of-function phenotype. We have admittedly not explored the impact of these mutations on NADK2 enzymatic activity in this study. These mutations could have multiple (and not mutually exclusive) possible impacts on NADK2, including protein instability, supported by reduced protein abundance (Fig. 1D), and enzymatic activity (kinetics, substrate specificity and so forth) or regulation through modifications, such as phosphorylation or other post-translational modifications. The serine at 326 is predicted to be a possible protein kinase C target using online tools, but this has not been experimentally verified. These studies would best be done by investigators with strong expertise in kinase biochemistry.
The hyperlysinemia and C10:2 carnitine elevation may be understood through the requirement for NADP as a substrate for AASS and DECR1 in lysine degradation and beta-oxidation of fatty acids, respectively. However, the severe disease phenotype and neurological symptoms are likely not wholly attributable to these pathways. To characterize the broader cellular dysfunction underlying neuromuscular deficits in our Nadk2−/− mice we performed transcriptomic and metabolomic analyses at the level of individual genes and metabolites, ontology terms associated with gene and metabolite lists, and pathway analysis that integrated both DEGs and metabolites. Our Nadk2−/− mice exhibited significant increases in lysine abundance in plasma at 5 weeks of age, and in all other tissues at both early and late time points, recapitulating the lysine elevation of patients. At the early time point, the metabolite list from muscle showed very high enrichment ratios against Metaboanalyst5.0 hyperlysinemia and DECR deficiency metabolites sets, with hyperlysinemia annotations also appearing in plasma metabolomics. These terms are directly relevant to NADK2 deficiency in patients, though many additional sets were identified in our analyses. The identification of many enriched metabolic processes is not surprising given the important role of NADP in myriad biological reactions and pathways. At the later time point, metabolite sets ranked by enrichment ratio pertained to dysfunction of metabolic enzymes, such as pyruvate carboxylase, an NADP-dependent enzyme, and elevation of 3-hydroxybutyric acid is observed across all tissues except the brain. Plasma metabolomics additionally revealed changes in carnitine metabolism and beta-oxidation of fatty acids. It should be noted that while C10:2-carnitine was not specifically identified in our plasma metabolomics datasets, that is likely due to the exploratory methods employed and the absence of a high confidence structure in the metabolomics database, although dysfunctional carnitine metabolism was identified at the level of the pathway. Further, mapping DEGs onto NADK2 deficiency KEGG pathways ‘lysine degradation’ and ‘beta-oxidation of fatty acids’ revealed numerous overlaps at the later time point (8). Together, these findings suggest that early onset deficiencies exist in key pathways and are later masked by a progressive generalized metabolic dysfunction in ‘omics’ datasets. Reactome terms from upregulated DEGs in late muscle tissue pertain to acetylcholine signaling, a finding that parallels the denervation of NMJs we observe, whereas downregulated genes are generally related to metabolism. We conclude that our Nadk2 mutant mice broadly recapitulate biochemical signatures of NADK2-deficient patients.
We also analyzed a Pla2g6 mouse model of INAD using similar methods. A major difference between Nadk2 and Pla2g6 mutant mice is the absence of metabolites that passed Adj. P < 0.05 filtering in any tissue or time point of Pla2g6−/−. Analysis of DEGs in early time point muscle identified numerous annotations involving the nervous system. These terms are replaced by annotations associated with muscle contraction at a later time point. Very few ontology terms reached significance in the brain tissue from Pla2g6 mutants, and for this reason, we considered terms that fell short of Holm–Bonferroni multiple testing correction, using a nominal P-value as an exploratory method. Interestingly, this approach revealed a small number of highly specific disease associations, such as ‘neurodegeneration with brain iron accumulation’, likely driven by the reduced expression of Pla2g6 itself. Muscle pathology has been documented in PLA2G6 neuroaxonal dystrophy patients, and generally brain iron accumulation is considered a defining feature of this disorder (19,20). These results assured us that our analysis was sensitive enough to detect disease-relevant molecular signatures in both Nadk2 and Pla2g6 mutant mice.
However, we were interested in determining if our identification of disease-relevant biochemical signatures had high specificity for each mutant. Given the absence of metabolites that passed Adj. P filtering for Pla2g6−/−, we compared lists of DEGs. No DEGs identified from the brain overlap between the two mutants, and so our comparison is limited to the muscle. At a later time point, a small number of mammalian phenotype ontology terms related to skeletal muscle were significant. We then focused on the early time point where 251 DEGs are shared. Over-representation analysis suggested these shared genes are involved in nervous system function and chemical synaptic transmission, perhaps driven by postsynaptic response to denervation. GSEA generally agreed with our over-representation analysis but also identified shared positive enrichment of translation pathways between mutants. Our cluster analysis indicated that neurological terms are consistently implicated by DEGs regardless of expression level, with a few minor deviations noted earlier. Gene–disease associations implicate a nonspecific set of nervous system disorders. Our failure to identify annotation enrichment for Nadk2-related processes, such as metabolism and lysine degradation or Pla2g6-related processes, such as brain iron accumulation in the shared set suggests our analysis reliably discriminated pathways that are specific to each condition. However, our transcriptomic and metabolomic analyses probably do not fully reflect the underlying pathophysiology of each mutant with complete fidelity, given the tendency for annotation bias to skew results in favor of well-studied processes (21). Nonetheless, the gene expression and metabolomics datasets reported here will aid in the study of human NADK2 mutations and infantile neuroaxonal dystrophies. The S330P Nadk2 mutant mice may be better suited for in vivo studies than previous mouse alleles and may serve as valid models for eventual preclinical studies to treatments for this rare recessively inherited condition.
Materials and Methods
Mapping and identification of Nadk2 mutations
We identified two strains of mice with an overt neuromuscular phenotype (hind limb wasting, kyphosis, poor gait and motor ability) from two independent screens using N-ethyl-N-nitrosourea (ENU) to mutagenize mice. The screens have been previously described (22,23). Both mutations were assumed to be single-gene recessive events based on pedigrees.
The first allele, referred to as m1Jcs, was ENU induced in a C57BL/6 J background. The mutation was mapped by transferring ovaries from an affected (presumed homozygous) mouse into NOD.CB17-Prkdcscid/J recipient female. This mouse was subsequently bred to CAST/Ei, producing F1 mice that were presumed obligate heterozygotes. F1 mice were intercrossed to produce F2 mapping animals. A genome scan and subsequent fine-mapping showed complete linkage to proximal Chromosome 15 between D15Mit224 and D15Mit11. This region includes 10 protein-coding genes (Capsl, Il7r, Lmbrd2, Nadk2, Ranbp3l, Skp2, Slc1a3, Spef2, Ugt3a1 and Ugt3a2). The coding portion of the candidate genes in the interval was evaluated by Sanger sequencing of PCR amplified whole-brain cDNA produced from mutant and heterozygote mapping animals.
The second allele, designated nmf421, was induced in a C57BL/6 J background and was mapped by breeding an early-stage affected mouse (presumed homozygote) to BALB/cByJ to produce F1 mice (presumed obligate heterozygotes). The F1 offspring were then intercrossed to produce F2 mice used in the mapping. A genome scan using simple sequence length polymorphisms (SSLPs, Mit markers) was performed and a significant association with markers D15Jmp22 (11.0 mb) and D15Mit252 (22.7 mb) on proximal Chromosome 15 was identified. Of 13 affected mice screened, 12 were homozygous C57BL/6 J at both markers, one was heterozygous (chi-square test; 2.3×10−8), and more distal markers on Chromosome 15 were less well associated, suggesting the mutation was proximal to D15Jmp22. Given the identification of the m1Jcs mutation in Nadk2, exons of Nadk2 were sequenced in nmf421, identifying the Nadk2nmf421 S330P allele.
Tissue collection
For all studies, Nadk2 and Pla2g6 homozygous mutant mice were produced in heterozygous matings, and littermates were used as controls (in rare cases, strain and age-matched wild-type mice were used to fill out a cohort). For metabolomics, submandibular cheek bleeds were performed before mice were euthanized by cervical dislocation and organs were collected and snap-frozen in liquid nitrogen as quickly as possible. Tissue was then powdered while frozen and separated into three subsamples for extraction and mass spectrometry at the West Coast Metabolomics Center (see ‘Supplementary Methods’). Sample numbers, genotypes, ages and sexes used for GC-TOF, HILIC and lipidomics are given in Table 2. Blood samples were centrifuged at 2000g for 10 min at 4°C in Microvette CB 300 K2 EDTA collection tubes to separate plasma for extraction and mass spectrometry at the Mass Spectrometry and Protein Chemistry Service at The Jackson Laboratory (see ‘Supplementary Methods’). Sample numbers, genotypes, ages and sexes used for plasma metabolomics are given in Table 3. For RNA-Seq, a sagittal hemi-brain or triceps surae muscle was snap-frozen in liquid nitrogen for subsequent RNA isolation. The other half-brain and muscles from the other leg were processed for histology as described later. Tissue was collected in batches (litter by litter), and banked at −80°C. All samples were processed and analyzed (metabolomics or RNA-Seq) in single batches. Sample numbers, genotypes, ages and sexes for RNA-Seq are given in Table 1.
Histology
Tissue from the same cohort of mice used for RNA-Seq was also processed for histological analysis. Samples were dissected free and fixed by immersion in Bouin’s fixative overnight. The tissue was then processed by standard protocols for paraffin embedding and microtome sectioning at 4–6 microns of sections. After sectioning, the tissue was mounted on slides and stained using a Leica ST5010 automated slide stainer with hematoxylin and eosin (H&E) by standard protocols.
Purkinje cell density was quantified by defining a line of known length along the granule cell/molecular layer boundary in H&E stained sections and counting the number of Purkinje cell bodies per unit length. Photoreceptor cell density was quantified by defining a region of interest in the outer nuclear layer and counting the number of photoreceptor nuclei per square micron. The same analysis provided outer nuclear layer thickness, as areas were defined from the outer plexiform layer to the outer boundary of the outer nuclear layer. Retinal ganglion cell number was determined by counting the number of cells in the ganglion cell layer across a known distance in the central retina near the nerve head.
Femoral nerves were fixed by immersion in 2% paraformaldehyde and 2% glutaraldehyde in a 0.1 M cacodylate buffer. Sections were then dehydrated and plastic embedded as described, and 0.5-micron sections were stained by toluidine blue (24). Images were captured with a 40X objective lens and myelinated axons were counted.
Neuromuscular junction staining
The plantaris muscle was fixed in freshly prepared 4% paraformaldehyde in phosphate-buffered saline (PBS) for ~ 4 h at 4°C and rinsed in PBS. The tissue was then teased and stained as a whole mount for neurofilament light chain (2H3, DHSB) and SV2 (DHSB) and Alexa594 conjugated alpha-bungarotoxin as described (25).
Analysis of mtDNA/nDNA ratio
The protocol is adapted from Quiros et al. (26). DNA isolation was performed from frozen medial gastrocnemius and liver tissue from NADK2 or wild-type mice using the Qiagen DNEasy Blood and Tissue Kit (ID 69504) according to the kit’s protocol. DNA concentration was measured using NanoDrop, and the samples were diluted using double-distilled water to produce a final concentration of 10 ng DNA/microliter to be used for qPCR. The mitochondrial genes analyzed are ND1 and 16sRNA, and were compared to nuclear gene HK2. The following primer pairs were each made into a 100 μM stock and working stocks of 10 μM (ND1 F: 5′-CTAGCAGAAACAAACCGGGC-3′ R: 5′-CCGGCTGCGTATTCTACGTT-3′, 16sRNA F: 5′-CCGCAAGGGAAAGATGAAAGAC-3′R: 5′-TCGTTTGGTTTCGGGGTTTC-3′, and HK2 F: 5′-GCCAGCCTCTCCTGATTTTAGTGT-3′R: 5′-GGGAACACAAAAGACCTCTTCTGG-3′). All qPCR reactions were carried out in quadruplicate in a 384-well plate with a reaction consisting of 2 μl DNA, 2.5 μl Syber Green Mix and 0.5 μl of 10 μM primer pair. The plate was then covered with a plate seal, spun down at a top speed of plate centrifuge for 10 s and then placed in a quantitative thermocycler. The conditions for the qPCR are (preamplification step) 95°C for 5 min, (amplification step) 45 cycles of 95°C for 10 s, 60°C for 10 s and 72°C for 20 s. Finally, a melting curve is calculated to confirm the presence of a single PCR product following these steps: 95°C for 5 s, 66°C for 1 min and gradual increase in temperature up to 97°C. Analysis was then carried out in two different ways to ensure confidence in the results. The ΔΔCt method is used to calculate the expression of each of the samples. ΔCt is calculated using the following equation: ΔCt = Ct(mtDNA gene)—Ct(nDNA gene). ΔΔCt was then calculated using the mean of the wild-type cohort as the control sample ΔΔCt = ΔCt(sample of interest)—ΔCt(control sample). Finally, calculate the expression of each sample as 2-ddCt. Alternatively, the number of copies of mtDNA was also calculated using the following formulas, first calculating ΔCt with ΔCt = Ct(nDNA gene)—Ct(mtDNA gene), then calculated copies of mtDNA using mtDNA = 2 × 2dCt. The relative mtDNA content is then calculated with relative mtDNA content = mtDNA experimental/mtDNA control. The data were imported into GraphPad Prism for statistical analysis.
Western blot
Snap-frozen liver from three 5-week-old Nadk2−/− and three littermate control mice were homogenized in RIPA buffer containing protease and phosphatase inhibitors (AG Scientific T-2495) and centrifuged at 15 k ×g for 15 min at 4°C to clear. Laemmli buffer was added and 15 μg of protein per sample was run on a 4–20% SDS-PAGE gel (Bio-Rad #4561094), then transferred to PVDF using standard techniques. Blots were sequentially probed with anti-NADK2 (rabbit, 1:1000, Abnova PAB23271) and anti-GAPDH(rabbit, 1:2000, Cell Signaling Technology #2118) antibodies; 0.02% sodium azide was included in the anti-GAPDH primary solution to quench the first HRP reaction. Blots were visualized by incubation with HRP-conjugated secondary antibody (1:50 000, Jackson ImmunoResearch #711-035-152, anti-rabbit) followed by HRP substrate (Millipore #WBLUR0100). Quantification of protein levels was performed by measuring NADK2 band intensity in FIJI and normalizing it to GAPDH band intensity. P-values were determined in Prism using an unpaired t-test.
RNA-Seq
RNA-Seq was performed on the following samples: brain and muscle from Nadk2 and Pla2g6 mutants, along with wild-type littermates, at early (5 weeks for Nadk2−/− and 6 weeks for Pla2g6−/−) and late (11 weeks for Nadk2−/− and 13 weeks for Pla2g6−/−) time points (Table 1). Tissue was collected, snap-frozen in liquid nitrogen and stored at −80°C until it was processed for RNA extraction and library preparation. RNA isolation: RNA was isolated from tissue using the MagMAX mirVana Total RNA Isolation Kit (ThermoFisher) and the KingFisher Flex purification system (ThermoFisher). Tissues were lysed and homogenized in TRIzol Reagent (ThermoFisher). After the addition of chloroform, the RNA-containing aqueous layer was removed for RNA isolation according to the manufacturer’s protocol, beginning with the RNA bead-binding step. RNA concentration and quality were assessed using the Nanodrop 2000 spectrophotometer (Thermo Scientific) and the RNA Total RNA Nano assay (Agilent Technologies). Library preparation: Libraries were prepared using the KAPA mRNA HyperPrep Kit (KAPA Biosystems), according to the manufacturer’s instructions. Briefly, the protocol entails isolation of polyA containing mRNA using oligo-dT magnetic beads, RNA fragmentation, first- and second-strand cDNA synthesis, and ligation of Illumina-specific adapters containing a unique barcode sequence for each library, and PCR amplification. Libraries were checked for quality and concentration using the D5000 ScreenTape assay (Agilent Technologies) and qPCR (KAPA Biosystems), according to the manufacturers’ instructions. Sequencing: Libraries were pooled and sequenced 70 bp single-end on the HiSeq 4000 (Illumina) using HiSeq 3000/4000 SBS Kit reagents (Illumina), targeting 30 million reads per sample.
A standard RNA-Seq pipeline comprising tools to perform read quality assessment, alignment and variant calling was developed at The Jackson Laboratory. The pipeline takes sequence reads for each sample as raw fastq files and outputs read counts. Sequence base qualities ≥30 over 70% of reading length criteria were used in downstream analyses. Sequence reads that passed the quality were aligned to a mouse reference (GRCm38.74) using Bowtie v2.2.0 (27,28). To prevent genes with low read counts from biasing results, all genes with fewer than one aligned read per million mapped reads were removed before differential expression analysis. Gene expression estimates were determined using RSEM v1.2.19 (rsem-calculate-expression) with default parameters (29). Expression estimates were further normalized by using upper quantile normalization of nonzero expected counts and scaling to 1000. R package edgeR was used to perform the normalization and the test for differential expression (30). Normalization factors were calculated using the TMM method (31). Constrained Regression Recalibration (ConReg-R) was used to recalibrate the empirical P-values by modeling their distribution to improve the FDR estimates (32). Gene lists were filtered using cutoffs for significance and fold change (FDR < 0.05 and |Log2FC| > 1.5). Raw data were deposited into GEO, accession number 188592, and differential expression data are included in Supplementary Material, Table S1.
Metabolomics
Metabolomics analysis was performed at the West Coast Metabolomics Center and the Mass Spectrometry and Protein Chemistry Service at The Jackson Laboratory. Three platforms were used for the hemi-brain, muscle, liver and spinal cord, which were examined for primary metabolism by GC-TOF-MS. Brain and muscle were also analyzed for lipidomics using CSH-QTOF-MS/MS (positive and negative electrospray ionization) and for biogenic amines by HILIC-QTOF-MS/MS. Exploratory metabolomics of plasma was performed using a Thermo Q-Exactive Orbitrap mass spectrometer coupled to a dual-channel Vanquish Ultra-Performance Liquid Chromatography system with a hydrophobic C18 column and hydrophilic HILIC column under both positive and negative polarity. Detailed methods provided by the WCMC and by the Mass Spectrometry and Protein Chemistry Service at The Jackson Laboratory are provided in ‘Supplementary Methods’. The ages, genotypes, sex and numbers of the mice analyzed are provided in Tables 2 and 3. Concentration data were uploaded as comma-separated values (.csv) files to MetaboAnalyst. Data integrity checks, missing value imputations and data filtering were performed before normalization. Statistical analysis was performed with the null hypothesis of no difference between the mutant and control samples at each time point. Multiple exploratory, univariate and multivariate and machine learning methods were used in the data analysis (33,34). Samples with 50% or more values missing in either mutant or control or both samples were discarded from further analysis. Missing values were then imputed using half the detection limit and filtered using the interquartile range method (35,36). The data was then log2 transformed and auto-scaled (37,38). Student t-test, fold change and PCA were performed as exploratory data analysis methods. Machine learning models were developed using a support vector machine algorithm (39), with a linear kernel method and random forest analysis (40) to classify the mutant and control samples and rank the importance of metabolites in discriminating the two types of samples (see ‘Supplementary Methods’ for full details of data processing and analysis).
Data filtering
DEGs and TMM-normalized reads were output from a standard differential expression analysis script based on edgeR (30) implemented by the Computational Sciences Core at The Jackson Laboratory. Lists of differentially expressed genes from edgeR were filtered to remove those with an FDR > 0.05. Only DEGs with an absolute log2 fold-change Log2FC > 1.5 were considered in downstream analysis. Unless otherwise stated ‘metabolites’ refer to those metabolites identified by the GC-TOF platform from powdered tissue (indicated by PT) samples. Metabolites output from MetaboAnalyst5.0 comparisons were filtered to remove those with Adj. P > 0.05, and all changes in abundance were considered regardless of the fold change value. DEGs were compared between tissues and time points using Venny2.0 or Deep Venn interactive Venn Diagram web applications and GeneWeaver to test for significance of overlap by Jaccard index using binary score type and Jaccard Similarity module parameters: homology excluded, pairwise deletion disabled and P-value 0.05 (41–43).
Over-representation analysis
Lists of filtered DEGs were uploaded to MouseMine ‘list’ query (44). Gene ontology terms (biological process, cellular compartment, molecular function) (45–47), Reactome pathway ontologies (48), and mammalian phenotype ontology (49) terms were ranked by Holm–Bonferroni adjusted P-value and exported as comma-separated value (.csv) files. The number of genes annotated in each database at the time of analysis (December 1, 2021) is provided in Supplementary Material, Table S2. Significance values were −log10 transformed for visualization via bar plots and dot plots in R studio.
Gene set enrichment analysis
TMM-normalized counts were input to GSEA software (50). We performed comparisons against numerous gene sets from the online database, and Reactome pathways were included in the final report (48). As many as 1500 phenotype permutations with No Collapse were entered as required parameters. Default basic fields were used with a weighted enrichment statistic and Signal2Noise was used for ranking genes. We performed a leading-edge analysis in GSEA to determine which subsets of genes contribute most to the enrichment of a given process, and to find the most significant core processes by adjusted P-values, which we cross-referenced with differential expression data to improve our biological interpretation.
NetworkAnalyst3.0
Lists of filtered DEGs from tissues where no metabolites passed Adj. P filtering were uploaded to NetworkAnalyst3.0 (51). Generic protein–protein interaction networks, transcription factor–gene interactions, and interactive cord diagram functionalities were used to understand gene set interaction, regulation and similarity.
MetaboAnalyst5.0
Metabolites that passed Adj. P filtering were uploaded to the enrichment module of MetaboAnalyst5.0. The uploaded list was cross-referenced against the Metaboanalyst5.0 internally curated ‘Disease signatures’ metabolite sets for blood and CSF. Filtered DEGs and metabolites lists were uploaded to the MetaboAnalyst5.0 Joint-Pathway module using the ‘Metabolic pathways’ setting (52,53). MetaboAnalyst5.0 parameters are as follows: enrichment analysis was performed with Fisher’s exact test using betweenness centrality as a topology measure and combining P-values (overall) as the integration method. Pathways were ranked by impact score. Lists of metabolites and DEGs implicated in top pathways were then projected onto KEGG pathway maps using the MetaboAnalyst5.0 function.
Data visualization in GraphPad prism and R studio
Ranked ontologies from NetworkAnalyst3.0, MetaboAnalyst5.0 and MouseMine were visualized as bar plots using GraphPad Prism 8 or as dot plots made with GGPlot2 for R statistical software in R Studio (54,55). TMM normalized counts were visualized using the Pheatmap package for R studio (56). Clusters were identified in a supervised manner using dendrogram height (h = 15, k = 4) to separate genes into four categories based on expression level and consistency of expression between samples. Ontologies for each cluster were queried in MouseMine as earlier, and grouped into major categories: neuronal function, nervous system structure, behavior, seizures, movement and others. Category membership is visualized using circular plots. Gene–disease associations are visualized using the enrichplotR package (57). NCBI GeneIDs were mapped to Ensembl IDs (GRCm38.78) from differential expression analysis pipeline using Ensembl Biomart.
Targeted pathway mapping
Lists of DEGs identified by Ensembl IDs (GRCm38.78) were filtered to remove those with FDR > 0.05, and the NCBI GeneIDs were assigned to each using the KEGG Mapper—Convert ID function. The NCBI GeneID lists were then entered into the KEGG Mapper—Search Pathway module using the organism-specific search mode (set to Mus musculus) (58). Mappings of our gene lists to lysine degradation and fatty acid degradation pathways were exported. We have received permission from Kanehisa Laboratories to reprint KEGG pathway images. Lists of DEGs were compared to the MitoCarta3.0 database to assess the mitochondria-specific functions of gene products (59).
Supplementary Material
Acknowledgements
The authors would like to thank the Scientific Services at The Jackson Laboratory for their technical assistance in this project, including the fine mapping service, the histology and electron microscopy service, mass spectrometry and protein chemistry within protein sciences, the genome technologies service, and the computational sciences service. The services are a Shared Resource of the JAX Cancer Center (P30 CA034196).
Conflict of Interest statement. None declared.
Contributor Information
G C Murray, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; The Graduate School of Biomedical Science and Engineering, University of Maine, Orono, ME 04469, USA.
P Bais, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
C L Hatton, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
A L D Tadenev, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
B R Hoffmann, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
T J Stodola, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
K H Morelli, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; The Graduate School of Biomedical Science and Engineering, University of Maine, Orono, ME 04469, USA.
S L Pratt, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; Neuroscience Program, Graduate School of Biomedical Sciences, Tufts University, Boston, MA 02111, USA.
D Schroeder, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
R Doty, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA.
O Fiehn, West Coast Metabolomics Center, University of California Davis, 451 Health Science Dr., Davis, CA 95618, USA.
S W M John, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; Department of Ophthalmology, Columbia University Irving Medical Center, New York, NY 10032, USA; Zuckerman Mind Brain Behavior Institute, Columbia University, New York, NY 10032, USA.
C J Bult, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; The Graduate School of Biomedical Science and Engineering, University of Maine, Orono, ME 04469, USA.
G A Cox, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; The Graduate School of Biomedical Science and Engineering, University of Maine, Orono, ME 04469, USA; Neuroscience Program, Graduate School of Biomedical Sciences, Tufts University, Boston, MA 02111, USA.
R W Burgess, The Jackson Laboratory, 600 Main St., Bar Harbor, ME 04609, USA; The Graduate School of Biomedical Science and Engineering, University of Maine, Orono, ME 04469, USA; Neuroscience Program, Graduate School of Biomedical Sciences, Tufts University, Boston, MA 02111, USA.
Funding
National Institutes of Health (R21 NS082666 and R37 NS054154 to R.W.B.; R01 EY011721 and R01 EY032507 to S.W.M.J; U2 ES030158 to The West Coast Metabolomics Center O.F.; and T32 GM132006 to G.C.M.); RBP and the Precision Medicine Initiative at Columbia University to S.W.M.J.
References
- 1. Zhang, R. (2015) MNADK, a long-awaited human mitochondrion-localized NAD kinase. J. Cell. Physiol., 230, 1697–1701. [DOI] [PubMed] [Google Scholar]
- 2. Lerner, F., Niere, M., Ludwig, A. and Ziegler, M. (2001) Structural and functional characterization of human NAD kinase. Biochem. Biophys. Res. Commun., 288, 69–74. [DOI] [PubMed] [Google Scholar]
- 3. Ohashi, K., Kawai, S. and Murata, K. (2012) Identification and characterization of a human mitochondrial NAD kinase. Nat. Commun., 3, 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang, R. (2013) MNADK, a novel liver-enriched mitochondrion-localized NAD kinase. Biol. Open, 2, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Houten, S.M., Denis, S., Te Brinke, H., Jongejan, A., van Kampen, A.H., Bradley, E.J., Baas, F., Hennekam, R.C., Millington, D.S., Young, S.P. et al. (2014) Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia. Hum. Mol. Genet., 23, 5009–5016. [DOI] [PubMed] [Google Scholar]
- 6. Roe, C.R., Millington, D.S., Norwood, D.L., Kodo, N., Sprecher, H., Mohammed, B.S., Nada, M., Schulz, H. and McVie, R. (1990) 2,4-Dienoyl-coenzyme a reductase deficiency: a possible new disorder of fatty acid oxidation. J. Clin. Invest., 85, 1703–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tort, F., Ugarteburu, O., Torres, M.A., Garcia-Villoria, J., Giros, M., Ruiz, A. and Ribes, A. (2016) Lysine restriction and pyridoxal phosphate administration in a NADK2 patient. Pediatrics, 138, e20154534. [DOI] [PubMed] [Google Scholar]
- 8. Pomerantz, D.J., Ferdinandusse, S., Cogan, J., Cooper, D.N., Reimschisel, T., Robertson, A., Bican, A., McGregor, T., Gauthier, J., Millington, D.S. et al. (2018) Clinical heterogeneity of mitochondrial NAD kinase deficiency caused by a NADK2 start loss variant. Am. J. Med. Genet. A, 176, 692–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Houten, S.M., Te Brinke, H., Denis, S., Ruiter, J.P., Knegt, A.C., de Klerk, J.B., Augoustides-Savvopoulou, P., Haberle, J., Baumgartner, M.R., Coskun, T. et al. (2013) Genetic basis of hyperlysinemia. Orphanet J. Rare Dis., 8, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sacksteder, K.A., Biery, B.J., Morrell, J.C., Goodman, B.K., Geisbrecht, B.V., Cox, R.P., Gould, S.J. and Geraghty, M.T. (2000) Identification of the alpha-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia. Am. J. Hum. Genet., 66, 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gurvitz, A., Wabnegger, L., Yagi, A.I., Binder, M., Hartig, A., Ruis, H., Hamilton, B., Dawes, I.W., Hiltunen, J.K. and Rottensteiner, H. (1999) Function of human mitochondrial 2,4-dienoyl-CoA reductase and rat monofunctional Delta3-Delta2-enoyl-CoA isomerase in beta-oxidation of unsaturated fatty acids. Biochem. J., 344, 903–914. [PMC free article] [PubMed] [Google Scholar]
- 12. Dancis, J., Hutzler, J., Ampola, M.G., Shih, V.E., van Gelderen, H.H., Kirby, L.T. and Woody, N.C. (1983) The prognosis of hyperlysinemia: an interim report. Am. J. Hum. Genet., 35, 438–442. [PMC free article] [PubMed] [Google Scholar]
- 13. Miinalainen, I.J., Schmitz, W., Huotari, A., Autio, K.J., Soininen, R., Loren, V., van Themaat, E., Baes, M., Herzig, K.H., Conzelmann, E. and Hiltunen, J.K. (2009) Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. PLoS Genet., 5, e1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Makela, A.M., Hohtola, E., Miinalainen, I.J., Autio, J.A., Schmitz, W., Niemi, K.J., Hiltunen, J.K. and Autio, K.J. (2019) Mitochondrial 2,4-dienoyl-CoA reductase (Decr) deficiency and impairment of thermogenesis in mouse brown adipose tissue. Sci. Rep., 9, 12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang, K., Kim, H., Fu, Z., Qiu, Y., Yang, Z., Wang, J., Zhang, D., Tong, X., Yin, L., Li, J. et al. (2018) Deficiency of the mitochondrial NAD kinase causes stress-induced hepatic steatosis in mice. Gastroenterology, 154, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Strokin, M., Seburn, K.L., Cox, G.A., Martens, K.A. and Reiser, G. (2012) Severe disturbance in the Ca2+ signaling in astrocytes from mouse models of human infantile neuroaxonal dystrophy with mutated Pla2g6. Hum. Mol. Genet., 21, 2807–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morgan, N.V., Westaway, S.K., Morton, J.E., Gregory, A., Gissen, P., Sonek, S., Cangul, H., Coryell, J., Canham, N., Nardocci, N. et al. (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet., 38, 752–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rosenberg, A.S., Puig, M., Nagaraju, K., Hoffman, E.P., Villalta, S.A., Rao, V.A., Wakefield, L.M. and Woodcock, J. (2015) Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med., 7, 299rv294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kurian, M.A., Morgan, N.V., MacPherson, L., Foster, K., Peake, D., Gupta, R., Philip, S.G., Hendriksz, C., Morton, J.E., Kingston, H.M. et al. (2008) Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology, 70, 1623–1629. [DOI] [PubMed] [Google Scholar]
- 21. Haynes, W.A., Tomczak, A. and Khatri, P. (2018) Gene annotation bias impedes biomedical research. Sci. Rep., 8, 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldowitz, D., Frankel, W.N., Takahashi, J.S., Holtz-Vitaterna, M., Bult, C., Kibbe, W.A., Snoddy, J., Li, Y., Pretel, S., Yates, J. and Swanson, D.J. (2004) Large-scale mutagenesis of the mouse to understand the genetic bases of nervous system structure and function. Brain Res. Mol. Brain Res., 132, 105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munroe, R.J., Bergstrom, R.A., Zheng, Q.Y., Libby, B., Smith, R., John, S.W., Schimenti, K.J., Browning, V.L. and Schimenti, J.C. (2000) Mouse mutants from chemically mutagenized embryonic stem cells. Nat. Genet., 24, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgess, R.W., Cox, G.A. and Seburn, K.L. (2010) Neuromuscular disease models and analysis. Methods Mol. Biol., 602, 347–393. [DOI] [PubMed] [Google Scholar]
- 25. Morelli, K.H., Griffin, L.B., Pyne, N.K., Wallace, L.M., Fowler, A.M., Oprescu, S.N., Takase, R., Wei, N., Meyer-Schuman, R., Mellacheruvu, D. et al. (2019) Allele-specific RNA interference prevents neuropathy in Charcot-Marie-tooth disease type 2D mouse models. J. Clin. Invest., 129, 5568–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Quiros, P.M., Goyal, A., Jha, P. and Auwerx, J. (2017) Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol, 7, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langmead, B. and Salzberg, S.L. (2012) Fast gapped-read alignment with bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead, B., Trapnell, C., Pop, M. and Salzberg, S.L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li, B. and Dewey, C.N. (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Robinson, M.D., McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson, M.D. and Oshlack, A. (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol., 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li, J., Paramita, P., Choi, K.P. and Karuturi, R.K. (2011) ConReg-R: extrapolative recalibration of the empirical distribution of p-values to improve false discovery rate estimates. Biol. Direct, 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xia, J., Psychogios, N., Young, N. and Wishart, D.S. (2009) MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res., 37, W652–W660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xia, J., Mandal, R., Sinelnikov, I.V., Broadhurst, D. and Wishart, D.S. (2012) MetaboAnalyst 2.0--a comprehensive server for metabolomic data analysis. Nucleic Acids Res., 40, W127–W133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hackstadt, A.J. and Hess, A.M. (2009) Filtering for increased power for microarray data analysis. BMC Bioinformatics, 10, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stacklies, W., Redestig, H., Scholz, M., Walther, D. and Selbig, J. (2007) pcaMethods--a bioconductor package providing PCA methods for incomplete data. Bioinformatics, 23, 1164–1167. [DOI] [PubMed] [Google Scholar]
- 37. Dieterle, F., Ross, A., Schlotterbeck, G. and Senn, H. (2006) Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem., 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- 38. van den Berg, R.A., Hoefsloot, H.C., Westerhuis, J.A., Smilde, A.K. and van der Werf, M.J. (2006) Centering, scaling, and transformations: improving the biological information content of metabolomics data. BMC Genomics, 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vapnik, V.N. (1995) The nature of statistical learning theory. Springer, New York. [Google Scholar]
- 40. Liaw, A. and Wiener, M. (2002) Classification and regression by random forest. R News, 2, 18–22. [Google Scholar]
- 41. Oliveros, J.C. (2007) VENNY. In An Interactive Tool for Comparing Lists with Venn Diagrams. BioinfoGP, Spanish National Biotechnology Centre.
- 42. Hulsen, T., de Vlieg, J. and Alkema, W. (2008) BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics, 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bubier, J.A., Langston, M.A., Baker, E.J. and Chesler, E.J. (2017) Integrative functional genomics for systems genetics in GeneWeaver.org. Method. Mol. Biol., 1488, 131–152. [DOI] [PubMed] [Google Scholar]
- 44. Motenko, H., Neuhauser, S.B., O'Keefe, M. and Richardson, J.E. (2015) MouseMine: a new data warehouse for MGI. Mamm. Genome, 26, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology consortium. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harris, M.A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., Eilbeck, K., Lewis, S., Marshall, B., Mungall, C. et al. (2004) The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res., 32, D258–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gene Ontology, C. (2021) The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res., 49, D325–D334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jassal, B., Matthews, L., Viteri, G., Gong, C., Lorente, P., Fabregat, A., Sidiropoulos, K., Cook, J., Gillespie, M., Haw, R. et al. (2020) The reactome pathway knowledgebase. Nucleic Acids Res., 48, D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith, C.L. and Eppig, J.T. (2012) The mammalian phenotype Ontology as a unifying standard for experimental and high-throughput phenotyping data. Mamm. Genome, 23, 653–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Subramanian, A., Tamayo, P., Mootha, V.K., Mukherjee, S., Ebert, B.L., Gillette, M.A., Paulovich, A., Pomeroy, S.L., Golub, T.R., Lander, E.S. and Mesirov, J.P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A., 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhou, G., Soufan, O., Ewald, J., Hancock, R.E.W., Basu, N. and Xia, J. (2019) NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res., 47, W234–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chong, J., Soufan, O., Li, C., Caraus, I., Li, S., Bourque, G., Wishart, D.S. and Xia, J. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res., 46, W486–W494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pang, Z., Chong, J., Zhou, G., de Lima Morais, D.A., Chang, L., Barrette, M., Gauthier, C., Jacques, P.E., Li, S. and Xia, J. (2021) MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res., 49, W388–W396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wickham, H. (2016) ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag, New York. [Google Scholar]
- 55. (2014), R. C. T . (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/. [Google Scholar]
- 56. Kolde, R. (2012) Pheatmap: pretty heatmaps. R package version, 1, 726. [Google Scholar]
- 57. Yu, G. (2021) Enrichplot: visualization of functional enrichment result. R package, version 1.12.3. [Google Scholar]
- 58. Kanehisa, M. and Goto, S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rath, S., Sharma, R., Gupta, R., Ast, T., Chan, C., Durham, T.J., Goodman, R.P., Grabarek, Z., Haas, M.E., Hung, W.H.W. et al. (2021) MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res., 49, D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.