

Abstract
There is abundant evidence that infectious sepsis both in humans and mice with polymicrobial sepsis results in robust activation of complement. Major complement activation products involved in sepsis include C5a anaphylatoxin and its receptors (C5aR1 and C5aR2) and, perhaps, the terminal complement activation product, C5b-9. These products (and others) also cause dysfunction of the innate immune system, with exaggerated early proinflammatory responses, followed by decline of the innate immune system, leading to immunosuppression and multiorgan dysfunction. Generation of C5a during sepsis also leads to activation of neutrophils and macrophages and ultimate appearance of extracellular histones, which have powerful proinflammatory and prothrombotic activities. The distal complement activation product, C5b-9, triggers intracellular Ca2+ fluxes in epithelial and endothelial cells. Histones activate the NLRP3 inflammasome, products of which can damage cells. C5a also activates MAPKs and Akt signaling pathways in cardiomyocytes, causing buildup of [Ca2+]i, defective action potentials and substantial cell dysfunction, resulting in cardiac and other organ dysfunction. Cardiac dysfunction can be quantitated by ECHO-Doppler parameters. In vivo interventions that block these complement-dependent products responsible for organ dysfunction in sepsis reduce the intensity of sepsis. The obvious targets in sepsis are C5a and its receptors, histones, and perhaps the MAPK pathways. Blockade of C5 has been considered in sepsis, but the FDA-approved antibody (eculizumab) is known to compromise defenses against neisseria and pneumonococcal bacteria, and requires immunization before the mAb to C5 can be used clinically. Small molecular blocking agents for C5aRs are currently in development and may be therapeutically effective for treatment of sepsis.
Keywords: C5 anaphylatoxin, histones, NLRP3 inflammasome, ROS, NETs, METs, C5b-9
Introduction
It is well established that intense activation of complement develops during sepsis, associated with activation of the classical, alternative and lectin pathways of complement. In mice, polymicrobial sepsis is induced by cecal ligation and puncture (CLP), which mimics sepsis in humans and was established by Irshad Chaudry in rodents (1979 in rats and 1983 in mice) (1, 2). Both in septic mice and in humans with sepsis, the innate immune system is first activated, featuring a flood of proinflammatory cytokines and chemokines, followed by evidence of decline of the innate immune system, resulting in immunosuppression (3–5). These events often lead to appearance of substantial amounts in plasma of extracellular histones (e.g. 25 μg/ml) (6), proinflammatory cytokines and chemokines in plasma, together with reactive oxygen species (ROS) (7), all of which cause multiorgan dysfunction involving liver, kidneys, lungs, heart, and brain as well as other organs (5, 8–12). Proof that proinflammatory cytokines and chemokines and their receptors play roles in multiorgan dysfunction of sepsis is very difficult because of the tremendous overlap between the biological activities of these peptides and their receptors. Resolution of this problem would require mice with multiple knockouts of these peptides or knockout of multiple receptors or use of multiple biochemical strategies for inhibition of the peptides or receptors. As emphasized in this review, many of the adverse events of sepsis are linked to effects of complement activation products (and relevant receptors) developing during infectious sepsis and after trauma. These events lead to increased morbidity, mortality and multiorgan failure (13–17). Progression of sepsis ultimately causes impaired responses of innate immune cells (neutrophils [PMNs], macrophages and monocytes) which early in sepsis release abundant amounts of ROS as well as proinflammatory cytokines and chemokines, resulting in dysfunction of immune cells (7, 18–20). We present a scheme that has a focus on how generation of harmful complement activation products may be reduced during polymicrobial sepsis in mice and, perhaps, in septic humans.
Overview of Literature and Summary of Our Findings
Effects of Complement Activation Products on Reponses of Phagocytes during Polymicrobial Sepsis (Figures 1–4)
Figure 1. Role of Complement in Polymicrobial Sepsis.

Infectious sepsis robustly activates the classical and alternative and lectin complement pathways, generating C5a which reacts with its receptors (C5aR1, C5aR2) on neutrophils and macrophages. This leads to formation of neutrophil extracellular traps (NETs) and macrophage extracellular traps (METs)that in release of histones.
Figure 4. Synopsis for Role of Complement in Damaging Events of Sepsis.
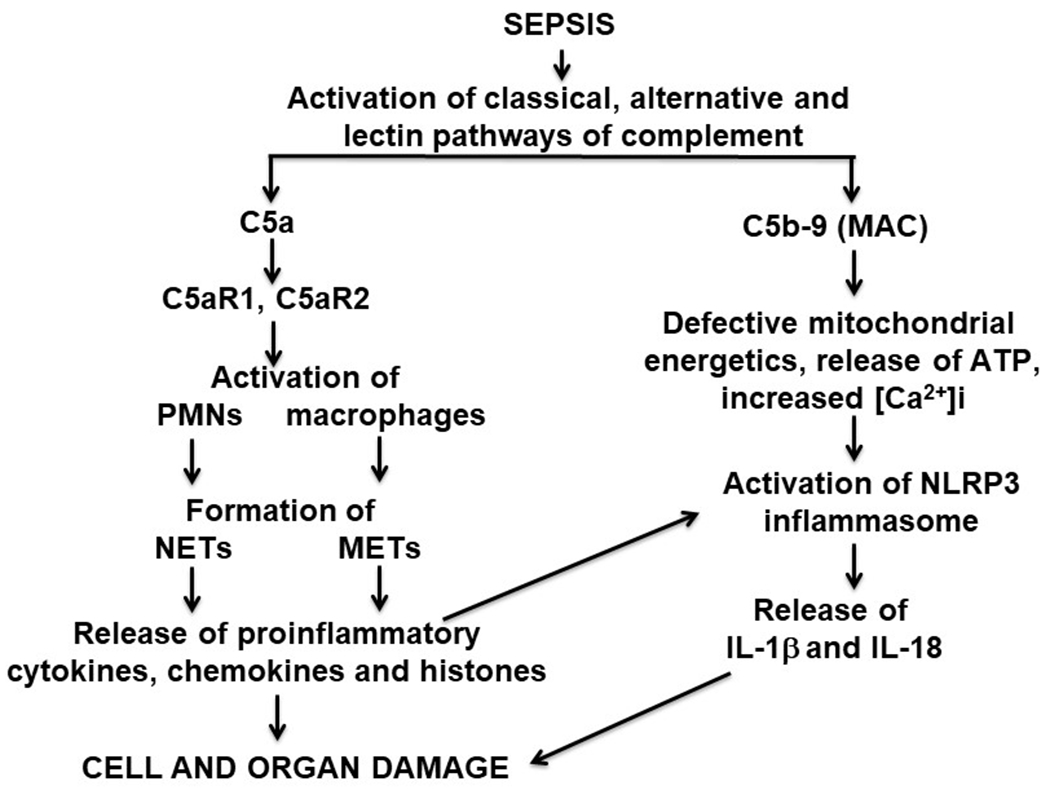
Composite of complement activation events during infectious sepsis. The most important complement activation products are C5a and C5b-9 (membrane attack complex, MAC) which result in cell and organ dysfunction.
Details of the relevant complement activation pathways during sepsis are described in Figures 1–4. Generation of complement anaphylatoxins (C3a, C5a) is linked to several adverse outcomes during sepsis (16, 21–23) as emphasized in these figures. In Figure 1, the emphasis is on generation of neutrophil extracellular traps (NETs) and macrophage extracellular traps (METs) following cell activation by C5a. Figure 2 is a scheme which describes the generation of strands (NETs) of DNA from activated neutrophils and macrophages after complement activation during sepsis, resulting in local clearance of bacteria in blood and in lymph fluids. In addition, extracellular histones appear, which are intensely prothrombotic and proinflammatory causing organ dysfunction (e.g. heart failure, as shown in Figure 2). Figure 3 has a focus on the generation of C5b-9, also known as the membrane attack complex (MAC), binding to the surfaces of target cells and causing cytolysis or cell dysfunction (24, 25). This causes damage to the mitochondrial metabolic chain along with activation of the NLRP3 inflammasome in target cells (26–29), resulting in widespread cell and organ damage. Generation of the anaphylatoxins is known to cause increased vascular permeability, edema formation, accumulation of PMNs, macrophages and monocytes in tissues (Figure 4). As emphasized in these figures, C5a anaphylatoxin is generated very early during sepsis. Figure 4 emphasizes that C5a interacts with its receptors (C5aR1, C5aR2) on phagocytes, resulting in activation of both PMNs and macrophages. Activation of these phagocytes leads to production of NETs and METs, which trap and kill bacteria (30–38). However, NETs and METs also contain products of neutrophils, such as myeloperoxidase, proteases, metalloproteases along with extracellular histones, which have strong proinflammatory, prothrombotic and cell damaging effects (including apoptosis) (30, 32, 39). Another important complement activation product appearing during sepsis is the terminal complement activation product, C5b-9. C5b-9 is generated during sepsis (Figure 4). It is cell-damaging, resulting in cell dysfunction and cell lysis. The cause for some of these outcomes is that sublytic levels of C5b-9 cause disruption of mitochondrial electron transport, resulting in functional defects in cardiomyocytes (CMs), in part due to buildup of [Ca2+]i in CMs during diastole and reductions in ATP. This results in defective action potentials, causing significant cardiac dysfunction. Histones can also activate the NLRP3 inflammasome, ultimately causing release of IL-1β and IL-18 along with activation of caspase 1 which may trigger the caspase cascade, leading to apoptosis (Figures 3, 4). Details of biological effects of C5b-9 during sepsis are provided below. Figure 4 also provides an overview of complement activation events developing during polymicrobial sepsis and indicates how effects of C5a and C5b-9 appear to be linked together in the setting of sepsis.
Figure 2. Formation of NETs and METs in Infectious Sepsis.
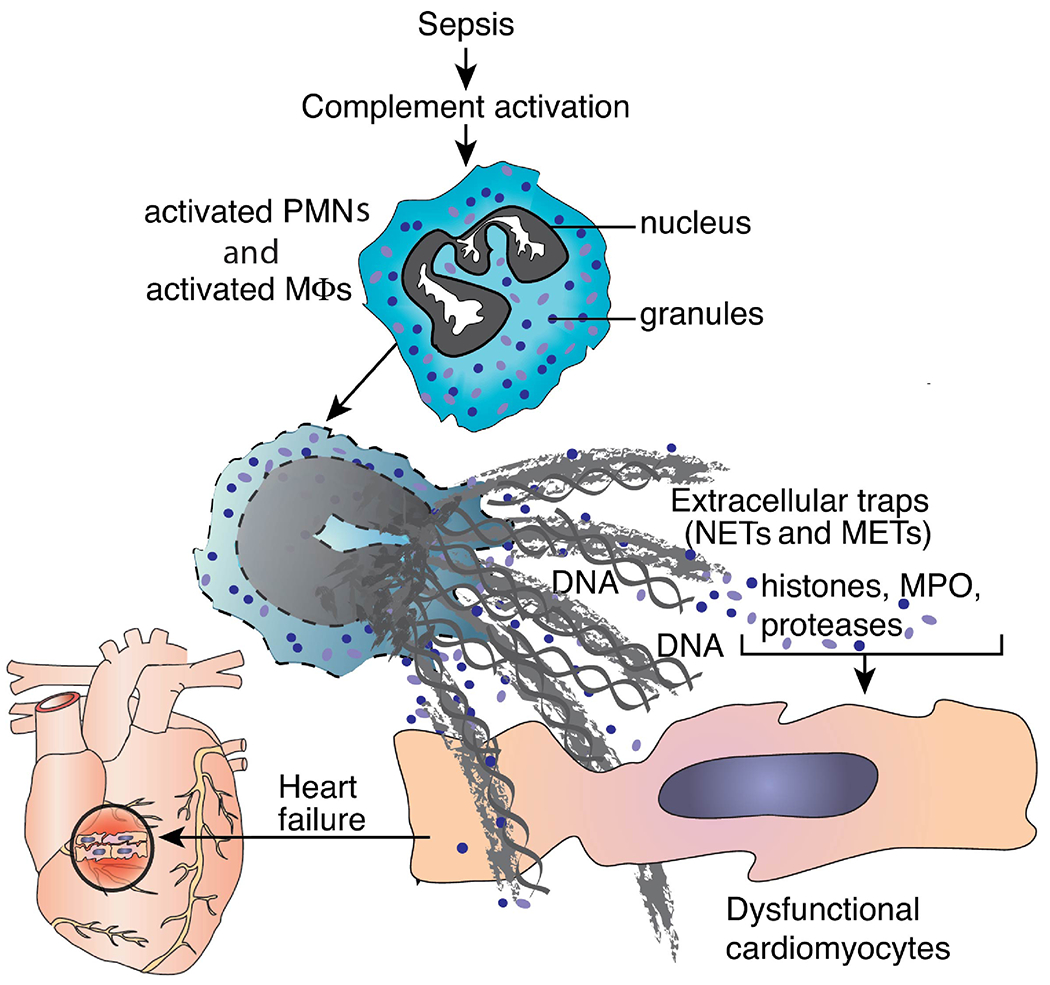
Events in sepsis following complement activation and phagocyte activation by C5a results in formation of NETs and METs and release of histones, leading to cell and heart dysfunction.
Figure 3. Role of Distal Complement Pathway in Polymicrobial Sepsis.

C5b-9 and histones activate the NLRP3 inflammasome resulting in mitochondrial damage, release of ATP and disturbed energetic pathways in mitochondria. In the process, there is intracellular buildup of ROS, increased [Ca2+]i, and release of IL-1β and IL-18, which are strong proinflammatory peptides.
It should be noted that complement activation products such as C5a and C3a may also play important roles in the activation of T cells. Liszewski et al (40) have described that human T cells contain protease cathepsin L, which causes release of C3a from C3, enhancing the function of T cells. There is developing evidence that C5 and C5a may also engage in similar activation events involving human T cells (41). Heeger and colleagues have described how innate immune cells generate C3a and C5a which costimulate human T cells in the setting of human organ allotransplants (42). Pio et al have recently described how anaphylatoxins regulate the anti-tumor activities of T cells (43). Recent data provide evidence that, like the story with phagocytes, complement anaphylatoxins may modify T cell functions in a variety of ways. Accordingly, complement anaphylatoxins may promote or block responses of T cells in the setting of allotransplants and in the presence of cancers. The lymphopenia developing during the first three days of infectious sepsis in mice has a requirement for C5a receptors (based on studies of C5aR1 and C5aR2 knockout mice), since no lymphopenia developed in septic mice lacking these C5aRs over a three-day period following onset of polymicrobial sepsis (44). These data suggest the C3a and C5a anaphylatoxins, together with their receptors to have diverse effects on a variety of cells related to the innate immune system.
In humans and mice, sepsis shows early evidence for strong activation of the clotting pathways, followed by intense fibrinolytic responses, which result in enzymatic breakdown of fibrin deposits in capillaries. We previously showed that thrombin, which is activated early in sepsis, also had the ability to cleave C5, generating both C5a and C5b (45). These results explained why C3 knockout (KO) mice were able to fully develop acute lung injury following IgG immune complex deposits in lung. M. Huber-Lang and his colleagues have also shown that several activated proteins involved in the intrinsic clotting cascade are able to cleave both C3 and C5, generating proinflammatory anaphylatoxins (46, 47).
Activation of Akt and mitogen-activated protein kinases (MAPKs) during Polymicrobial Sepsis or after In Vitro Exposure of CMs to C5a (Figure 5)
Figure 5. Activation of Akt and MAPKs during CLP or after Exposure of CMs to C5a.

Upper frames: Following CLP, Akt, p38, ERK1/2 and JNK1/2 were all activated (phosphorylated) in rat CMs 16 hr after onset of CLP, as determined by flow cytometry. Lower frames: Rat LV CMs were exposed in vitro to rrC5a (500 ng/ml) over a 4-hr time point. End points were determined by flow cytometry, revealing activation of Akt and all three MAPKs as a function of time of CM exposure to C5a.
There is evidence in the literature showing involvement of Akt as well as MAPKs activation pathway during sepsis in human (48–51) and in mice (52–57). In line, we have also shown in our recent study, a role for Akt and MAPKs activation during polymicrobial sepsis (53) (some data shown in Figure 5). In the upper frames of the Figure 5, Wt rats were subjected to polymicrobial sepsis. In the first 48 hr of sepsis, left ventricular (LV) samples (frozen sections) were assessed by immunofluorescence, indicating presence of phospho-Akt and phospho-p38, phospho-ERK1/2, and phospho-JNK1/2. After CLP, all of these proteins were activated, with phosphoproteins peaking 16 hr after CLP. In the lower frames, LV CMs from normal Wt rats were obtained and incubated with buffer or with rrC5a (500 ng/ml) for the times indicated (10-240 min) at 37°C. CMs were then washed with buffer and evaluated for activation (phosphorylation) of Akt, p38, ERK1/2 and JNK1/2 using flow cytometry. Analysis was done by flow cytometry, as described in one of our recent publication (53). In all cases, CMs that had been incubated with C5a showed evidence of activation of Akt and MAPKs. In most cases activation of CMs peaked 60-120 min after start of exposure of CMs to C5a. There was one exception involving JNK1/2 in which activation only occurred after 120 and 240 min of incubation with C5a. Together, these data indicate that both polymicrobial sepsis and direct contact of CMs with C5a leads to activation of Akt and all MAPKs.
Activation of ERK1/2 and p38 in LV CMs 16 hr after Polymicrobial Sepsis, Based on Immunofluorescence (Figure 6)
Figure 6. Complement-dependent Activation of ERK1/2 and p38 in CMs During Sepsis in Mice.

Frozen sections of LV CMs from mice 16 hr after CLP were obtained and IF was used to evaluate cells containing troponin T (TnT), ERK1/2 and p38, as indicated. The upper frames indicate the CM straining indicating phosphorylation of ERK1/2 was blocked by absence of C5aR1 or C5aR2. The lower frames show that green staining for activated p38 was in CMs as revealed by the staining for TnT (red).
LV frozen heart sections from mice were obtained 16 hr after CLP and evaluated by immunofluorescence (IF). CM staining indicated activation (phosphorylation) of Akt, p38, ERK1/2 and JNK1/2. The data in Figure 6 confirm in LV frozen sections that 16 hr after polymicrobial sepsis, both phospho-ERK1/2 and phospho-p38 could be visualized by green immunofluorescence staining. In the case of ERK1/2, staining in a normal heart showed very little evidence of green staining (left upper frame of Figure 6), while the septic heart (at 16 hr) showed diffuse green staining of CMs. The staining for phospho-ERK was sharply reduced in CMs from mice lacking either C5aR1 or C5aR2. In the lower frames in Figure 6, red staining indicated presence of troponin T (TnT) staining in CMs, together with a similar green pattern indicating phospho-p38 presence. These data indicate that LV CMs from septic mice were activated in the copresence of C5aR1 or C5aR2 as well as when CMs were exposed in vitro to C5a (Figure 5).
Pathways Leading to Complement-Dependent Cardiac Dysfunction after CLP (Figure 7)
Figure 7. Signaling Pathways Leading to Complement-Dependent Cardiac Dysfunction after Polymicrobial Sepsis.

Polymicrobial sepsis was induced in Wt C57BL/6 mice resulting in complement activation, appearance of C5a, binding to C5aRs on neutrophils and macrophages. This resulted in release of NETs and METs from phagocytes. These responses also induced activation of NETs and METs, causing extracellular appearance of plasma proinflammatory cytokines, chemokines and histones. The results of these responses led to reduced levels of three ATPases in CMs, causing defective action potentials in heart and elevated levels in CMs of [Ca2+]i and defective cardiac function.
Figure 7 summarizes the events in the complement signaling cascades that ultimately lead to cardiac dysfunction after the onset of polymicrobial sepsis in mice. As described in Figures 1–6, there are several complement-dependent signaling events that are triggered by sepsis. The onset of sepsis caused substantial amounts of C5a to appear in the circulation which initially caused production of NETs and METs (Figures 1, 2) along with the appearance in plasma of circulating histones. Soon thereafter, there was activation of signaling pathways leading to appearance of proinflammatory cytokines and chemokines in the circulation. The activation of MAPKs and Akt during sepsis resulted in the activation of MAPKs and Akt in CMs. At this time, LV CMs began to show defective action potentials (related to defective Na+/K+-ATPase) as well as buildup in the cytosol of Ca2+ associated with markedly reduced levels of two vital Ca2+-regulatory ATPases (SERCA2, NCX) that normally function to prevent the buildup of post diastolic [Ca2+]i in CMs. Sepsis caused substantial decreases in protein and mRNA amounts of SERCA2 and the Na+/Ca2 exchanger, the mechanisms of which are not understood. The markedly reduced protein levels of these ATPases early in sepsis (8-18 hr) caused defective action potentials that impaired CM function as well as a buildup of [Ca2+]i in CMs (58). These combined events greatly impaired cardiac function. In our recent studies, we showed that preventing blood buildup of C5a or absence of C5a receptors or blockade of MAPK (p38) prevented the series of outcomes leading to cardiac dysfunction which often is fatal (53, 58).
Role of NLRP3 Inflammasome in Polymicrobial Sepsis and in Acute Lung Injury
The NLRP3 inflammasome has been shown to play an important role in polymicrobial sepsis and also in LPS-induced acute lung injury (ALI) in mice (59, 60). IL-1β is the main product of the NLRP3 inflammasome activation. There is also abundant literature indicating that NLRP3 and its products (IL-1β and IL-18) are involved in sepsis. KO or inhibition of NLRP3 inflammasome greatly reduced multiorgan dysfunction in septic mice (60–71). Our own studies revealed that KO of NLRP3 greatly reduced multiorgan dysfunction in mice with polymicrobial sepsis (7, 72). Gene profile studies of inflammasome on patients with sepsis admitted to ICU showed increased levels of NLRP3 together with IL-1β and IL-18 expression in the septic patients compared to the control individuals, with higher levels observed in non-survivor patients (73), in line with earlier studies on human reporting persistent increase in levels of IL-1β in non-survivor septic patients (74). We also have shown that the absence of mRNA for the NLRP3 inflammasome (in mice genetically lacking NLRP3), resulting in reduced levels of proinflammatory mediators (cytokines, chemokines) in mice, and improved survival after CLP, and reduced apoptosis in ALI (7, 59, 60, 63). In addition, we have also shown the role for IL-18, another product of NLRP3 inflammasome activation (75) in development of ALI in mice. In our 2001 study, we assessed immune complex-induced ALI in mice and also used blocking monoclonal antibodies (mAb) to mouse IL-18, resulting in marked suppression of ALI (75).
As summary of our studies on sepsis, the absence of NLRP3 mRNA reduced the intensity of septic cardiomyopathy (7). Septic NLRP3 KO mice had greatly reduced plasma levels of IL-1β (as a key product of the NLRP3 inflammasome), IL-6 proinflammatory cytokine (7) and extracellular histones (6). In addition, mice with KO of C5aR1 or C5aR2 were protected from sepsis and the appearance in plasma of proinflammatory peptides and histones which were reduced (6). The absence of either C5aR1 or C5aR2 resulted in reduced phosphorylation of Akt and MAPKs and these mice were protected from sepsis (53). These data indicated that the NLRP3 inflammasome plays an important role in harmful events developing during polymicrobial sepsis and that these events are dependent on availability of C5aR1 and C5aR2 (7).
The lack of the intact NLRP3 inflammasome (due to KO of NLRP3 or caspase 1), both after CLP (6) and during ALI-induced by airway instillation of LPS (59), was associated with significantly reduced inflammatory injury. Development of ALI was dependent on the inflammasome as well as its products (IL-1β and IL-18). Mice that had KO of NLRP3 had suppressed ALI (59). In the case of CLP-induced sepsis in mice lacking NLRP3, the early acute inflammatory responses were suppressed (7), and there were reduced amounts of lipoxin B4 (LXB4) in vivo (in NLRP3 KO mice) and in vitro using the inflammasome protocol, as well as improved survival in sepsis (60). Sepsis can be functionally affected (either intensified or suppressed) by various lipid-related mediators that often appear during inflammatory responses. The data described above suggest that multiple proinflammatory responses involving lipid mediators were suppressed in mice that had KO of NLRP3. The inflammasome is a multiprotein complex which, when activated, generates and releases IL-1β and IL-18, which are strong proinflammatory cytokines. Morgan and colleagues were the first to demonstrate that in sheets of alveolar epithelial cells exposed to the C5b-9 complex, the development of mitochondrial dysfunction was associated with increased mitochondrial [Ca2+]i which was a key indicator of mitochondrial dysfunction (26).
The NLRP3 inflammasome has been found to be present in a variety of cells, including phagocytes (neutrophils, macrophages, CMs and many other cell types). It was also noted a few years ago that the NLRP3 inflammasome could also be activated by histones, but the details have not been defined (59, 76). We have recently reported that the rank order for purified histones to activate the NLRP3 inflammasome was: H1=H2A=H2B>H3=H4, based on release of IL-1β (76). Activation of the NLRP3 inflammasome by either C5b-9 or histones is summarized in Figure 4. For the inflammasome protocol, 1x106 cells/ml were incubated in a two-step procedure. First, cells were incubated with LPS (100 ng/ml) for 4 hr at 37°C. Very little IL-1β or IL-18 was released. This is referred to as the “priming step”. The next step is the “activation step” in which cells are incubated with ATP (1 mM), histones (50 μg/ml), or a variety of other “DAMPs” for 45 min at 37°C. This results in release of high concentrations of IL-1β, IL-18 and activation of caspase 1. As indicated above, the NLRP3 inflammasome was also activated by C5b-9 (26). For inflammasome activation, all terminal complement proteins are required (C5b, C6, C7, C8, and C9). As this process proceeds, the final complement component, C9, binds to C5b-8 and expresses a C9 “neoepitope”, which is a marker for MAC formation and can be used as an indicator of terminal complement activation. Presence of antibody to the epitope prevents the buildup of relatively large amounts of activated human C9, which is necessary to cause pore formation in cell membranes. However, currently there is no reliable neutralizing antibody for activated C9 in rodents or other mammals.
Mechanisms of Histone-induced Cell and Organ Damage in Polymicrobial Sepsis (Figure 8)
Figure 8. Mechanisms of Sepsis and Histone-induced Cell and Tissue Damage after Polymicrobial Sepsis.

Infectious sepsis triggers complement activation and appearance of extracellular histones. Histones generally bind to TLR2 and TLR4 on a variety of cell types, resulting in activation of platelets, PMNs and macrophages. Histones can also directly cause cell dysfunction and apoptosis. Collectively, extracellular histones have strong prothrombotic and proinflammatory activities.
It is well known that infectious sepsis and noninfectious sepsis are not associated with specific plasma markers that would allow differentiation between two types of sepsis, or, in general, allow diagnosis of “sepsis” (77). “Noninfectious sepsis” occurs after trauma, hemorrhagic shock, and drug-induced (e.g. acetaminophen) acute hepatic injury, traumatic head injury, to name just a few examples (78–82). As stated above, aside from blood cultures that yield bacteria, viruses, fungi or protozoa, clinical and laboratory markers are nonspecific. Figure 8 describes events in sepsis that may lead to severe multiorgan injury and/or death. As emphasized above, sepsis (infectious and noninfectious) leads to complement activation and appearance of C5a-dependent extracellular histones. If the clinical condition has been triggered by the presence of Gram negative bacteria, plasma lipopolysaccharide (LPS) may appear in the plasma. LPS interacts with TLR2 and TLR4, resulting in intracellular translocation, activation of platelet and prothrombotic pathways. LPS via TLR2 and TLR4 is quickly internalized into phagocytes, causing cell activation, followed by release of ROS, and a series of proinflammatory responses (activation on endothelial cells that promote leukocyte adhesion and transmigration) resulting in cell dysfunction and apoptosis. Numerous reports showed the involvement of TLR2 and TLR4 during sepsis, which were overexpressed (83–87). There is also new evidence suggesting that TLR2 and TLR4 are receptors for extracellular histones (88–91). Xu et al have shown that fatal liver injury caused by exposure to acetaminophen was related to the availability of TLR2 and TLR4. Absence of either of these TLRs protected mice from histone-mediated liver injury (90). Similar outcomes were also found using infusion of concanavalin A (90). Finally, the presence of histones can exacerbate cell damage and apoptosis (92). It is also clear that TLR3 or TLR9 may also play important roles in processing of LPS within cells, although the reasons for protection against cardiac dysfunction after polymicrobial sepsis are unknown (93–95). Clearly, our understanding of the roles of TLRs in sepsis is quite inadequate.
It should be pointed out that in the last decade, two large and independent clinical trials in septic humans have shown that treatment of sepsis patients with either of two compounds (eritoran, TAK 242) (which block the ability of LPS to bind to TLRs) were not protective in the setting of human sepsis (96, 97). Clinical efficacy was so minimal in septic humans that the trials were discontinued early (96). This experience suggests that in the majority of humans with sepsis seen in emergency rooms, do not have symptoms that can be related to LPS.
Discussion
Emerging Strategies to Block Effects of Complement Activation Products in Infectious Sepsis (Table 1)
Table 1.
Interventions that Block Complement-related Adverse Events in Polymicrobial Sepsis
| Target | Intervention | Concerns |
|---|---|---|
| 1. C5a blockade | Neutralizing antibody (safe in humans, short term) | Small molecular weight (mw) inhibitors to C5a are being developed |
| 2. C5aR blockade | Neutralizing antibody or small mw inhibitors | Question of which C5aR to block? |
| 3. Histones blockade | Neutralizing antibody | Incomplete knowledge of histones as targets |
| 4. C5b-9 blockade | Blockade of C9 neoepitope | Theoretically, should block action of C5b-9 |
| 5. NLRP3 inflammasome blockade | Blockade with antibody | Efficacy and safety in humans is not established |
| 6. MAPK blockade | Water-soluble inhibitor of p38 | Efficacy and safety in humans is not clear |
| 7. C5 blockade | Using mAb to C5, which prevents its activation and buildup of C5a and C5b-9 | Compromises defenses to pneumococcal and neisserial infections |
This report emphasizes that certain activation products of complement and their receptors play important roles in the sequence of harmful events during polymicrobial sepsis (72, 98–101). These events include morbidity, mortality, cell and organ injury or apoptosis as well as long term sequelae (reduced life span after “recovery” from acute sepsis, cognition defects, increased hospital readmission beyond the first year of sepsis, reduced quality of life beyond the first year of sepsis) (102, 103). There have been a limited number of studies in humans who have “recovered” from sepsis. In typical early cases of sepsis in humans, blockade of either the classical, alternative or lectin pathways of complement activation would be debatable because of lack of specificity of the blockade and the potential for interfering with important innate immune pathways that are protective in the face of infectious conditions. Blockade of C5a anaphylatoxin is attractive because of our earlier studies which clearly implicated C5a as playing a major role in adverse outcomes of sepsis in rodents (17, 104, 105), including activation of PMNs and macrophages and C5a interactions with C5aR1 and C5aR2 resulting in histone release (6, 59, 106, 107). As has been emphasized, C5a-induced activation of PMNs and macrophages resulted in production of NETs and METs and appearance of extracellular histones. A humanized mAb neutralized human C5a and was developed by InflaRx, a small biotech company in Jena, Germany. Phase I clinical trials with this mAb in healthy humans indicated no harm, and escalating doses of the antibody were safe, over a period of one week, with no developing infectious problems. Studies using the mAb in phase II clinical trials in humans with sepsis have not been completed. Attempts to develop small molecular weight inhibitors to block C5a are underway.
Blockade of either C5aR1 or C5aR2 has not yet been studied systematically in humans, so the issue of whether either blockade would have undesirable effects is problematic, and whether or not this intervention would be effective in sepsis is currently unknown. Assuming that low molecular weight compounds that block either C5aR1 or C5aR2 would be effective, the question becomes: which C5aR would be the preferred target? Originally, when both C5aR1 and C5aR2 were cloned, C5aR1 seemed to be predominant and preferred target. C5aR2 (formerly known as C5L2) was originally described as a “scavenger receptor” which would bind C5a and C5a des arg but not trigger effector responses (108). Such data have suggested that C5aR1 might be the preferred target for blockade in sepsis. However, the controversy related to the function of C5aR2 remains unresolved. This impedes progress to resolve this dilemma.
Experimental studies have shown that our neutralizing antibody to C5a was highly protective in mice with polymicrobial sepsis (17, 104, 105). The monoclonal antibody to histones H2A/H4 (clone BWA3) (109) was purified from human ascites fluids by protein A/G chromatography. This antibody was highly effective for treatment of mice with polymicrobial sepsis, reducing the numerous harmful events, improving survival, reducing the intensity of defective, innate immune responses, reducing multiorgan dysfunction, etc. (6, 59, 110). There are data (described above) showing that the neutralizing antibodies to histones were highly protective against cardiac dysfunction in the setting of polymicrobial sepsis (6). It is known that in both human sepsis and mouse sepsis, extracellular histones are present in the plasma and play a key role in adverse events in sepsis (6, 37, 111–115). However, the amount of histones appearing in plasma and to what extent modifications in histones (acetylation, methylation, phosphorylation, ubiquination) affect histone biological functions is not clear. Based on all of these features of histones, much more basic information related to structure-functions of histones is needed before clinical trials would likely receive FDA approval for treatment of septic humans.
Another complement target would be C5b-9, which plays an important role in activation of the NLRP3 inflammasome (26–29), resulting in release of IL-1β and IL-18 which have strong proinflammatory functions (116, 117). The ideal strategy would likely be to use a blocking mAb to the C9 neoepitope which should prevent the ability of C5b-9 to cause lysis of cells (26). Blockade of the C9 neoepitope would seem to be an effective way to block the cell damaging effects of C5b-9 but a neutralizing antibody to the activation epitope of mouse C9 is not commercially available. Related to item 7 in Table 1, C5 blockade with mAb is highly effective in treatments of humans with paroxysmal nocturnal hemoglobinemia or with hemolytic-uremic syndrome. This antibody has received FDA approval, but its use comes with the caveat that patients must be pretreated with vaccines to prevent the development of pneumococcal or neisserial infections. Obviously, such pretreatment in humans with sepsis would not be possible.
For many years it was widely thought that endotoxemia was probably an important cause in the development of sepsis, because infusion of lipopolysaccharide (LPS) caused shock in mammals and led to many symptoms developing that are seen in humans with sepsis. However, in two large international clinical trials, eritoran (118) or TAK242 (97) were used. These drugs prevent LPS from activating TLR4 which usually promotes cell responses to LPS. The trials were terminated after 6 months because there was no evidence of clinical benefit. It is likely that the use of these inhibitors in proven Gram-negative sepsis might be clinically useful, but not in situations described in the two large clinical trials.
Acknowledgments
These studies were supported in part by the National Institutes of Health grants NIGMS R01 GM-29507 and R01 GM-61656, NIA R56 AG061207, and by the Stobbe Endowment, Department of Pathology, University of Michigan Medical School.
Funding:
These studies were supported in part by National Institutes of Health grants NIGMS R01-GM29507 and R01-GM61656, NIA R56-AG061207, and by the Stobbe Endowment, Department of Pathology, University of Michigan Medical School.
Abbreviations
- Akt
protein kinase B (PKB)
- ALI
acute lung injury
- BWA3
designation for mAb that blocks histones H2A/H4
- C5a
complement C5 anaphylatoxin
- C5aRs
C5a receptors (C5aR1, C5aR2)
- CLP
cecal ligation and puncture
- CMs
cardiomyocytes
- KO
knockout
- LPS
lipopolysaccharide
- mAb
monoclonal antibody
- MAC
membrane attack complex (C5b-9)
- MAPKs
mitogen-activated protein kinases
- METs
macrophage extracellular traps
- MPO
myeloperoxidase
- NCX
sodium-calcium exchanger ATPase
- NETs
neutrophil extracellular traps
- NLRP3
NACHT, LRR, and PYD domains-containing protein
- PMNs
polymorphonuclear cells (neutrophils)
- ROS
reactive oxygen species
- SERCA
sarco/endoplasmic reticulum Ca2+-regulatory ATPase
- TLRs
toll-like receptors
- Wt
wild type
Footnotes
Financial Disclosure: The authors declare no relevant conflicts of interest.
References
- 1.Wichterman KA, Baue AE, Chaudry IH: Sepsis and septic shock--a review of laboratory models and a proposal. J Surg Res 29:189–201, 1980. [DOI] [PubMed] [Google Scholar]
- 2.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, 3rd, Bland KI, Chaudry IH: Cecal ligation and puncture. Shock 24 Suppl 1:52–7, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Fattahi F, Ward PA: Understanding Immunosuppression after Sepsis. Immunity 47:3–5, 2017. [DOI] [PubMed] [Google Scholar]
- 4.Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, Nagarkatti M: Role of cytokines as a double-edged sword in sepsis. In Vivo 27:669–84, 2013. [PMC free article] [PubMed] [Google Scholar]
- 5.Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD 2nd, Kreisel D, Krupnick AS, et al. : Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA 306:2594–605, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalbitz M, Grailer JJ, Fattahi F, Jajou L, Herron TJ, Campbell KF, Zetoune FS, Bosmann M, Sarma JV, Huber-Lang M, et al. : Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J 29:2185–93, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalbitz M, Fattahi F, Grailer JJ, Jajou L, Malan EA, Zetoune FS, Huber-Lang M, Russell MW, Ward PA: Complement-induced activation of the cardiac NLRP3 inflammasome in sepsis. FASEB J 30:3997–4006, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bozza FA, Salluh JI, Japiassu AM, Soares M, Assis EF, Gomes RN, Bozza MT, Castro-Faria-Neto HC, Bozza PT: Cytokine profiles as markers of disease severity in sepsis: a multiplex analysis. Crit Care 11:R49, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koivikko P, Arola O, Inkinen O, Tallgren M: One-Year Survival After In-Hospital Cardiac Arrest- does Pre-Arrest Sepsis Matter? Shock, 2017. [DOI] [PubMed] [Google Scholar]
- 10.Power C, Fanning N, Redmond HP: Cellular apoptosis and organ injury in sepsis: a review. Shock 18:197–211, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Seeley EJ, Matthay MA, Wolters PJ: Inflection points in sepsis biology: from local defense to systemic organ injury. Am J Physiol Lung Cell Mol Physiol 303:L355–63, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Libert C, Ayala A, Bauer M, Cavaillon JM, Deutschman C, Frostell C, Knapp S, Kozlov AV, Wang P, Osuchowski MF, et al. : Part II: Minimum Quality Threshold in Preclinical Sepsis Studies (MQTiPSS) for Types of Infections and Organ Dysfunction Endpoints. Shock 51:23–32, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo RF, Riedemann NC, Ward PA: Role of C5a-C5aR interaction in sepsis. Shock 21:1–7, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Gressner OA, Koch A, Sanson E, Trautwein C, Tacke F: High C5a levels are associated with increased mortality in sepsis patients--no enhancing effect by actin-free Gc-globulin. Clin Biochem 41:974–80, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Hoesel LM, Niederbichler AD, Ward PA: Complement-related molecular events in sepsis leading to heart failure. Mol Immunol 44:95–102, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Huber-Lang M, Sarma VJ, Lu KT, McGuire SR, Padgaonkar VA, Guo RF, Younkin EM, Kunkel RG, Ding J, Erickson R, et al. : Role of C5a in multiorgan failure during sepsis. J Immunol 166:1193–9, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Czermak BJ, Sarma V, Pierson CL, Warner RL, Huber-Lang M, Bless NM, Schmal H, Friedl HP, Ward PA: Protective effects of C5a blockade in sepsis. Nat Med 5:788–92, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Salomao R, Brunialti MK, Rapozo MM, Baggio-Zappia GL, Galanos C, Freudenberg M: Bacterial sensing, cell signaling, and modulation of the immune response during sepsis. Shock 38:227–42, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Czermak BJ, Friedl HP, Ward PA: Complement, cytokines, and adhesion molecule expression in inflammatory reactions. Proc Assoc Am Physicians 110:306–12, 1998. [PubMed] [Google Scholar]
- 20.Hoesel LM, Neff TA, Neff SB, Younger JG, Olle EW, Gao H, Pianko MJ, Bernacki KD, Sarma JV, Ward PA: Harmful and protective roles of neutrophils in sepsis. Shock 24:40–7, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Sarma VJ, Huber-Lang M, Ward PA: Complement in lung disease. Autoimmunity 39:387–94, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Ward PA: Sepsis, apoptosis and complement. Biochem Pharmacol 76:1383–8, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albrecht EA, Ward PA: Complement-induced impairment of the innate immune system during sepsis. Curr Allergy Asthma Rep 4:359–64, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Bhakdi S, Tranum-Jensen J: Complement lysis: a hole is a hole. Immunol Today 12:318–20; discussion 321, 1991. [DOI] [PubMed] [Google Scholar]
- 25.Hammer CH, Nicholson A, Mayer MM: On the mechanism of cytolysis by complement: evidence on insertion of C5b and C7 subunits of the C5b,6,7 complex into phospholipid bilayers of erythrocyte membranes. Proc Natl Acad Sci U S A 72:5076–80, 1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Triantafilou K, Hughes TR, Triantafilou M, Morgan BP: The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci 126:2903–13, 2013. [DOI] [PubMed] [Google Scholar]
- 27.Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, Morgan BP, Sivasankar B, Mortellaro A: Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1beta release. J Immunol 191:1006–10, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Towner LD, Wheat RA, Hughes TR, Morgan BP: Complement Membrane Attack and Tumorigenesis: A SYSTEMS BIOLOGY APPROACH. J Biol Chem 291:14927–38, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ichinohe T, Pang IK, Iwasaki A: Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 11:404–10, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A: Neutrophil extracellular traps kill bacteria. Science 303:1532–5, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M: Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol 8:81, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doster RS, Rogers LM, Gaddy JA, Aronoff DM: Macrophage Extracellular Traps: A Scoping Review. J Innate Immun 10:3–13, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma R, O’Sullivan KM, Holdsworth SR, Bardin PG, King PT: Visualizing Macrophage Extracellular Traps Using Confocal Microscopy. J Vis Exp, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A: Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–41, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knopf J, Leppkes M, Schett G, Herrmann M, Munoz LE: Aggregated NETs Sequester and Detoxify Extracellular Histones. Front Immunol 10:2176, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boe DM, Curtis BJ, Chen MM, Ippolito JA, Kovacs EJ: Extracellular traps and macrophages: new roles for the versatile phagocyte. J Leukoc Biol 97:1023–35, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margraf S, Logters T, Reipen J, Altrichter J, Scholz M, Windolf J: Neutrophil-derived circulating free DNA (cf-DNA/NETs): a potential prognostic marker for posttraumatic development of inflammatory second hit and sepsis. Shock 30:352–8, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Araujo CV, Campbell C, Goncalves-de-Albuquerque CF, Molinaro R, Cody MJ, Yost CC, Bozza PT, Zimmerman GA, Weyrich AS, Castro-Faria-Neto HC, et al. : A PPARgamma AGONIST ENHANCES BACTERIAL CLEARANCE THROUGH NEUTROPHIL EXTRACELLULAR TRAP FORMATION AND IMPROVES SURVIVAL IN SEPSIS. Shock 45:393–403, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT: Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One 7:e32366, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, Subias M, Pickering MC, Drouet C, Meri S, et al. : Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39:1143–57, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, Dutow P, Woodruff TM, Yu ZX, O’Neill LA, et al. : T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 352:aad1210, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cravedi P, Leventhal J, Lakhani P, Ward SC, Donovan MJ, Heeger PS: Immune cell-derived C3a and C5a costimulate human T cell alloimmunity. Am J Transplant 13:2530–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pio R, Ajona D, Ortiz-Espinosa S, Mantovani A, Lambris JD: Complementing the Cancer-Immunity Cycle. Front Immunol 10:774, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grailer JJ, Fattahi F, Dick RS, Zetoune FS, Ward PA: Cutting edge: critical role for C5aRs in the development of septic lymphopenia in mice. J Immunol 194:868–72, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, et al. : Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med 12:682–7, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, Lambris JD, Huber-Lang M: Interaction between the coagulation and complement system. Adv Exp Med Biol 632:71–9, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Bruckner UB, Nilsson B, Gebhard F, Lambris JD, et al. : Molecular intercommunication between the complement and coagulation systems. J Immunol 185:5628–36, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pathan N, Franklin JL, Eleftherohorinou H, Wright VJ, Hemingway CA, Waddell SJ, Griffiths M, Dennis JL, Relman DA, Harding SE, et al. : Myocardial depressant effects of interleukin 6 in meningococcal sepsis are regulated by p38 mitogen-activated protein kinase. Crit Care Med 39:1692–711, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Liang Y, Li X, Zhang X, Li Z, Wang L, Sun Y, Liu Z, Ma X: Elevated levels of plasma TNF-alpha are associated with microvascular endothelial dysfunction in patients with sepsis through activating the NF-kappaB and p38 mitogen-activated protein kinase in endothelial cells. Shock 41:275–81, 2014. [DOI] [PubMed] [Google Scholar]
- 50.Abraham E: Alterations in cell signaling in sepsis. Clin Infect Dis 41 Suppl 7:S459–64, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Wang YY, Myhre AE, Pettersen SJ, Dahle MK, Foster SJ, Thiemermann C, Bjornland K, Aasen AO, Wang JE: Peptidoglycan of Staphylococcus aureus induces enhanced levels of matrix metalloproteinase-9 in human blood originating from neutrophils. Shock 24:214–8, 2005. [DOI] [PubMed] [Google Scholar]
- 52.Gao M, Ha T, Zhang X, Wang X, Liu L, Kalbfleisch J, Singh K, Williams D, Li C: The Toll-like receptor 9 ligand, CpG oligodeoxynucleotide, attenuates cardiac dysfunction in polymicrobial sepsis, involving activation of both phosphoinositide 3 kinase/Akt and extracellular-signal-related kinase signaling. J Infect Dis 207:1471–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fattahi F, Kalbitz M, Malan EA, Abe E, Jajou L, Huber-Lang MS, Bosmann M, Russell MW, Zetoune FS, Ward PA: Complement-induced activation of MAPKs and Akt during sepsis: role in cardiac dysfunction. FASEB J 31:4129–4139, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bosmann M, Patel VR, Russkamp NF, Pache F, Zetoune FS, Sarma JV, Ward PA: MyD88-dependent production of IL-17F is modulated by the anaphylatoxin C5a via the Akt signaling pathway. FASEB J 25:4222–32, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Singleton KD, Beckey VE, Wischmeyer PE: GLUTAMINE PREVENTS ACTIVATION OF NF-kappaB AND STRESS KINASE PATHWAYS, ATTENUATES INFLAMMATORY CYTOKINE RELEASE, AND PREVENTS ACUTE RESPIRATORY DISTRESS SYNDROME (ARDS) FOLLOWING SEPSIS. Shock 24:583–9, 2005. [DOI] [PubMed] [Google Scholar]
- 56.Maitra SR, Bhaduri S, Chen E, Shapiro MJ: Role of chemically modified tetracycline on TNF-alpha and mitogen-activated protein kinases in sepsis. Shock 22:478–81, 2004. [DOI] [PubMed] [Google Scholar]
- 57.Song GY, Chung CS, Jarrar D, Chaudry IH, Ayala A: Evolution of an immune suppressive macrophage phenotype as a product of P38 MAPK activation in polymicrobial sepsis. Shock 15:42–8, 2001. [DOI] [PubMed] [Google Scholar]
- 58.Kalbitz M, Fattahi F, Herron TJ, Grailer JJ, Jajou L, Lu H, Huber-Lang M, Zetoune FS, Sarma JV, Day SM, et al. : Complement Destabilizes Cardiomyocyte Function In Vivo after Polymicrobial Sepsis and In Vitro. J Immunol 197:2353–61, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, Zetoune FS, Ward PA: Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol 192:5974–83, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee S, Nakahira K, Dalli J, Siempos II, Norris PC, Colas RA, Moon JS, Shinohara M, Hisata S, Howrylak JA, et al. : NLRP3 Inflammasome Deficiency Protects against Microbial Sepsis via Increased Lipoxin B4 Synthesis. Am J Respir Crit Care Med 196:713–726, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang C, Xia W, Liu X, Lin J, Wu A: Role of TXNIP/NLRP3 in sepsis-induced myocardial dysfunction. Int J Mol Med 44:417–426, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Tao A, Lan T, Cepinskas G, Kao R, Martin CM, Rui T: Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res Cardiol 112:16, 2017. [DOI] [PubMed] [Google Scholar]
- 63.Jin L, Batra S, Jeyaseelan S: Deletion of Nlrp3 Augments Survival during Polymicrobial Sepsis by Decreasing Autophagy and Enhancing Phagocytosis. J Immunol 198:1253–1262, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Long H, Xu B, Luo Y, Luo K: Artemisinin protects mice against burn sepsis through inhibiting NLRP3 inflammasome activation. Am J Emerg Med 34:772–7, 2016. [DOI] [PubMed] [Google Scholar]
- 65.Sui DM, Xie Q, Yi WJ, Gupta S, Yu XY, Li JB, Wang J, Wang JF, Deng XM: Resveratrol Protects against Sepsis-Associated Encephalopathy and Inhibits the NLRP3/IL-1beta Axis in Microglia. Mediators Inflamm 2016:1045657, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gong Z, Zhou J, Li H, Gao Y, Xu C, Zhao S, Chen Y, Cai W, Wu J: Curcumin suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Mol Nutr Food Res 59:2132–42, 2015. [DOI] [PubMed] [Google Scholar]
- 67.Zhang B, Liu Y, Sui YB, Cai HQ, Liu WX, Zhu M, Yin XH: Cortistatin Inhibits NLRP3 Inflammasome Activation of Cardiac Fibroblasts During Sepsis. J Card Fail 21:426–433, 2015. [DOI] [PubMed] [Google Scholar]
- 68.Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, Yun M, Kim CK, Howrylak J, Ryter SW, et al. : UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 125:665–80, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Wang P, Huang J, Li Y, Chang R, Wu H, Lin J, Huang Z: Exogenous Carbon Monoxide Decreases Sepsis-Induced Acute Kidney Injury and Inhibits NLRP3 Inflammasome Activation in Rats. Int J Mol Sci 16:20595–608, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang W, Xu X, Kao R, Mele T, Kvietys P, Martin CM, Rui T: Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: the role of NLRP3 inflammasome activation. PLoS One 9:e107639, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo YP, Jiang L, Kang K, Fei DS, Meng XL, Nan CC, Pan SH, Zhao MR, Zhao MY: Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol 20:24–32, 2014. [DOI] [PubMed] [Google Scholar]
- 72.Fattahi F, Frydrych LM, Bian G, Kalbitz M, Herron TJ, Malan EA, Delano MJ, Ward PA: Role of complement C5a and histones in septic cardiomyopathy. Mol Immunol 102:32–41, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Esquerdo KF, Sharma NK, Brunialti MKC, Baggio-Zappia GL, Assuncao M, Azevedo LCP, Bafi AT, Salomao R: Inflammasome gene profile is modulated in septic patients, with a greater magnitude in non-survivors. Clin Exp Immunol 189:232–240, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mera S, Tatulescu D, Cismaru C, Bondor C, Slavcovici A, Zanc V, Carstina D, Oltean M: Multiplex cytokine profiling in patients with sepsis. APMIS 119:155–63, 2011. [DOI] [PubMed] [Google Scholar]
- 75.Jordan JA, Guo RF, Yun EC, Sarma V, Warner RL, Crouch LD, Senaldi G, Ulich TR, Ward PA: Role of IL-18 in acute lung inflammation. J Immunol 167:7060–8, 2001. [DOI] [PubMed] [Google Scholar]
- 76.Fattahi F, Grailer JJ, Lu H, Dick RS, Parlett M, Zetoune FS, Nunez G, Ward PA: Selective Biological Responses of Phagocytes and Lungs to Purified Histones. J Innate Immun 9:300–317, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huber-Lang M, Lambris JD, Ward PA: Innate immune responses to trauma. Nat Immunol 19:327–341, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gill R, Ruan X, Menzel CL, Namkoong S, Loughran P, Hackam DJ, Billiar TR: Systemic inflammation and liver injury following hemorrhagic shock and peripheral tissue trauma involve functional TLR9 signaling on bone marrow-derived cells and parenchymal cells. Shock 35:164–70, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lissauer ME, Johnson SB, Siuzdak G, Bochicchio G, Whiteford C, Nussbaumer B, Moore R, Scalea TM: Coagulation and complement protein differences between septic and uninfected systemic inflammatory response syndrome patients. J Trauma 62:1082–92; discussion 1092–4, 2007. [DOI] [PubMed] [Google Scholar]
- 80.Donnelly MC, Hayes PC, Simpson KJ: Role of inflammation and infection in the pathogenesis of human acute liver failure: Clinical implications for monitoring and therapy. World J Gastroenterol 22:5958–70, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tremoleda JL, Watts SA, Reynolds PS, Thiemermann C, Brohi K: Modeling Acute Traumatic Hemorrhagic Shock Injury: Challenges and Guidelines for Preclinical Studies. Shock 48:610–623, 2017. [DOI] [PubMed] [Google Scholar]
- 82.Meireles CZ, Pasarin M, Lozano JJ, Garcia-Caldero H, Gracia-Sancho J, Garcia-Pagan JC, Bosch J, Abraldes JG: Simvastatin Attenuates Liver Injury in Rodents with Biliary Cirrhosis Submitted to Hemorrhage/Resuscitation. Shock 47:370–377, 2017. [DOI] [PubMed] [Google Scholar]
- 83.Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ: Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A 106:4018–23, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zou L, Feng Y, Chen YJ, Si R, Shen S, Zhou Q, Ichinose F, Scherrer-Crosbie M, Chao W: Toll-like receptor 2 plays a critical role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med 38:1335–42, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alves-Filho JC, de Freitas A, Russo M, Cunha FQ: Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit Care Med 34:461–70, 2006. [DOI] [PubMed] [Google Scholar]
- 86.Roger T, Froidevaux C, Le Roy D, Reymond MK, Chanson AL, Mauri D, Burns K, Riederer BM, Akira S, Calandra T: Protection from lethal gram-negative bacterial sepsis by targeting Toll-like receptor 4. Proc Natl Acad Sci U S A 106:2348–52, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H: Role of Toll-like receptors in the development of sepsis. Shock 29:315–21, 2008. [DOI] [PubMed] [Google Scholar]
- 88.Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C, et al. : Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23:1375–88, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT: Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118:1952–61, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT: Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 187:2626–31, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang X, Li L, Liu J, Lv B, Chen F: Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-kappaB and AP-1. Thromb Res 137:211–218, 2016. [DOI] [PubMed] [Google Scholar]
- 92.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT: Extracellular histones are major mediators of death in sepsis. Nat Med 15:1318–21, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fattahi F, Russell MW, Malan EA, Parlett M, Abe E, Zetoune FS, Ward PA: Harmful Roles of TLR3 and TLR9 in Cardiac Dysfunction Developing during Polymicrobial Sepsis. Biomed Res Int 2018:4302726, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gao M, Ha T, Zhang X, Liu L, Wang X, Kelley J, Singh K, Kao R, Gao X, Williams D, et al. : Toll-like receptor 3 plays a central role in cardiac dysfunction during polymicrobial sepsis. Crit Care Med 40:2390–9, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lohner R, Schwederski M, Narath C, Klein J, Duerr GD, Torno A, Knuefermann P, Hoeft A, Baumgarten G, Meyer R, et al. : Toll-like receptor 9 promotes cardiac inflammation and heart failure during polymicrobial sepsis. Mediators Inflamm 2013:261049, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, et al. : Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309:1154–62, 2013. [DOI] [PubMed] [Google Scholar]
- 97.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, et al. : A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med 38:1685–94, 2010. [DOI] [PubMed] [Google Scholar]
- 98.Albrecht EA, Ward PA: Complement-induced Impairment of the Innate Immune System During Sepsis. Curr Infect Dis Rep 7:349–54, 2005. [DOI] [PubMed] [Google Scholar]
- 99.Flierl MA, Schreiber H, Huber-Lang MS: The role of complement, C5a and its receptors in sepsis and multiorgan dysfunction syndrome. J Invest Surg 19:255–65, 2006. [DOI] [PubMed] [Google Scholar]
- 100.Fattahi F, Ward PA: Complement and sepsis-induced heart dysfunction. Mol Immunol 84:57–64, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu G, Feng Y, Li D, Zhou Q, Chao W, Zou L: Importance of the Complement Alternative Pathway in Serum Chemotactic Activity During Sepsis. Shock 50:435–441, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Delano MJ, Ward PA: The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev 274:330–353, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Delano MJ, Ward PA: Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest 126:23–31, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huber-Lang MS, Sarma JV, McGuire SR, Lu KT, Guo RF, Padgaonkar VA, Younkin EM, Laudes IJ, Riedemann NC, Younger JG, et al. : Protective effects of anti-C5a peptide antibodies in experimental sepsis. FASEB J 15:568–70, 2001. [DOI] [PubMed] [Google Scholar]
- 105.Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, Zetoune FS, Gerard NP, Cianflone K, Kohl J, et al. : Functional roles for C5a receptors in sepsis. Nat Med 14:551–7, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ward PA, Fattahi F: New strategies for treatment of infectious sepsis. J Leukoc Biol 106:187–192, 2019. [DOI] [PubMed] [Google Scholar]
- 107.Fattahi F, Grailer JJ, Jajou L, Zetoune FS, Andjelkovic AV, Ward PA: Organ distribution of histones after intravenous infusion of FITC histones or after sepsis. Immunol Res 61:177–86, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li XX, Lee JD, Kemper C, Woodruff TM: The Complement Receptor C5aR2: A Powerful Modulator of Innate and Adaptive Immunity. J Immunol 202:3339–3348, 2019. [DOI] [PubMed] [Google Scholar]
- 109.Monestier M, Fasy TM, Losman MJ, Novick KE, Muller S: Structure and binding properties of monoclonal antibodies to core histones from autoimmune mice. Mol Immunol 30:1069–75, 1993. [DOI] [PubMed] [Google Scholar]
- 110.Bosmann M, Grailer JJ, Ruemmler R, Russkamp NF, Zetoune FS, Sarma JV, Standiford TJ, Ward PA: Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J 27:5010–21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Garcia-Gimenez JL, Roma-Mateo C, Carbonell N, Palacios L, Peiro-Chova L, Garcia-Lopez E, Garcia-Simon M, Lahuerta R, Gimenez-Garzo C, Berenguer-Pascual E, et al. : A new mass spectrometry-based method for the quantification of histones in plasma from septic shock patients. Sci Rep 7:10643, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee KH, Cavanaugh L, Leung H, Yan F, Ahmadi Z, Chong BH, Passam F: Quantification of NETs-associated markers by flow cytometry and serum assays in patients with thrombosis and sepsis. Int J Lab Hematol 40:392–399, 2018. [DOI] [PubMed] [Google Scholar]
- 113.Wildhagen KC, Wiewel MA, Schultz MJ, Horn J, Schrijver R, Reutelingsperger CP, van der Poll T, Nicolaes GA: Extracellular histone H3 levels are inversely correlated with antithrombin levels and platelet counts and are associated with mortality in sepsis patients. Thromb Res 136:542–7, 2015. [DOI] [PubMed] [Google Scholar]
- 114.Alhamdi Y, Abrams ST, Cheng Z, Jing S, Su D, Liu Z, Lane S, Welters I, Wang G, Toh CH: Circulating Histones Are Major Mediators of Cardiac Injury in Patients With Sepsis. Crit Care Med 43:2094–103, 2015. [DOI] [PubMed] [Google Scholar]
- 115.Gould TJ, Lysov Z, Swystun LL, Dwivedi DJ, Zarychanski R, Fox-Robichaud AE, Liaw PC, Canadian Critical Care Translational Biology G: Extracellular Histones Increase Tissue Factor Activity and Enhance Thrombin Generation by Human Blood Monocytes. Shock 46:655–662, 2016. [DOI] [PubMed] [Google Scholar]
- 116.van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA: Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol 32:110–6, 2011. [DOI] [PubMed] [Google Scholar]
- 117.Barker BR, Taxman DJ, Ting JP: Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Curr Opin Immunol 23:591–7, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, Wheeler J, Gogate J, Opal SM, Eritoran Sepsis Study G: Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit Care Med 38:72–83, 2010. [DOI] [PubMed] [Google Scholar]