

Abstract
Aging is characterized by an increased vulnerability to infection and the development of inflammatory diseases like atherosclerosis, frailty, cancer, and neurodegeneration. Here, we find that aging is associated with the loss of diurnally rhythmic innate immune responses, including monocyte trafficking from bone marrow to blood, phagocytosis, and resistance to bacterial infection. This decline in homeostatic immune responses was associated with a striking disappearance of circadian gene transcription in aged as compared to young tissue macrophages. Chromatin accessibility was significantly greater in young versus aged macrophages, however this difference did not explain the loss of rhythmic gene transcription in aged macrophages. Rather, diurnal expression of Kruppel-like Factor 4 (Klf4), a transcription factor well established in regulating cell differentiation and reprogramming, was selectively diminished in aged macrophages. Moreover, Klf4 binding to chromatin was lost in aging macrophages at a binding motif distinct from that involved in cellular reprogramming, suggesting a distinct role of Klf4 in maintenance of critical youthful rhythmic immune responses. Examination in human subjects with genetic variants of Klf4 revealed an association with age-dependent susceptibility to death caused by bacterial infection. Our results indicate that loss of rhythmic Klf4 expression in aged macrophages is associated with disruption of circadian homeostasis, and this mechanism may underlie age-associated loss of protective immune responses.
Keywords: Circadian, Aging, Immunology, Phagocytosis, Macrophages, Klf4
Circadian rhythms are endogenous, free-running cycles with periodicities approximately, but not exactly, 24 hours. Circadian rhythms are entrained to 24 hours by environmental cues, and they enable organisms to alter their physiology and behavior to anticipate changes in their environment1. In mammals, these rhythms are coordinated by the light-entrained master pacemaker in the suprachiasmatic nucleus (SCN) in the hypothalamus. Within SCN neurons, transcription-translation feedback loops involving the activator core clock protein heterodimer BMAL1/CLOCK and the repressor PER/CRY generate a circadian molecular oscillator. These transcription factors translocate to the nucleus, where they bind to consensus enhancer-box (E-box) DNA motifs of circadian-regulated genes.
Peripheral cells similarly possess endogenous circadian clocks that are governed by the same transcription factor feedback loop2. Comparisons of circadian-regulated genes across multiple organ systems have demonstrated that almost half of all protein coding genes are regulated in a diurnal fashion across different tissues3,4. This tissue-specific circadian gene expression regulates biological processes essential for the maintenance and dynamic changes of individual organ functions over the circadian cycle.
In the immune system, the diurnal rhythmicity of immune responses ranges from leukocyte trafficking and maintenance of immunosurveillance to recognition of pathogens and engagement of defensive responses5–8. Within the innate immune system, major components of myeloid responses, including phagocytic capacity, release of cytokines and chemokines, cell trafficking, and expression of innate immune Toll like receptors, are tightly regulated and follow distinct circadian phases8–16. The strict temporal gating of innate immune cell functions ensures the maintenance of homeostasis through an organized sequence of immune defensive responses to pathogens. Indeed, experimental disruption of immune circadian rhythmicity amplifies disease-causing inflammation and aggravates pathology in models of infection or high-fat diet8,17,18.
Aging is associated with an increased incidence of cancer, cardiovascular disease, metabolic syndrome, and neurodegeneration, and the onset and progression of these common diseases are strongly linked to age-associated changes in immune function. Aging is also characterized by marked dysregulation of immune function, including the development of a bias towards myeloid over lymphoid cell differentiation19, disrupted mitochondrial respiration and energy metabolism20, increased vulnerability to viral and bacterial infections, and persistent low-grade inflammation. We hypothesized that immune circadian rhythmicity might be disrupted with aging and lead to maladaptive responses that contribute to changes seen in the aging immune system.
We examined whether the diurnal features of the innate immune system were preserved in aged mice. We first investigated diurnal trafficking of Ly6Chi inflammatory monocytes from the bone-marrow to the blood, a homeostatic myeloid process critical to anti-microbial immune defense 8. Blood monocytes and bone marrow and spleen macrophages were isolated from young (2–3 months old) and aged (18–20 months old) male mice every 4 h over a 24 h period, and dynamic changes in cell abundance in these three myeloid compartments were quantified by flow-cytometry (FACS) (Fig. 1a–f; Supplementary Fig. 1). Consistent with previously published data8, young Ly6Chi monocytes demonstrated circadian trafficking from the bone marrow to the blood, manifested as phase-shifted changes in abundance (p=0.00026 and 0.046 for bone marrow and blood, respectively; rhythmicity assessed using JTK_CYCLE). However, the circadian rhythmicity of monocytes/macrophage numbers in blood and bone marrow was completely lost in aged mice (p=1 for each compartment).
Figure 1. Aging disrupts the diurnal rhythmicity of innate immune functions.
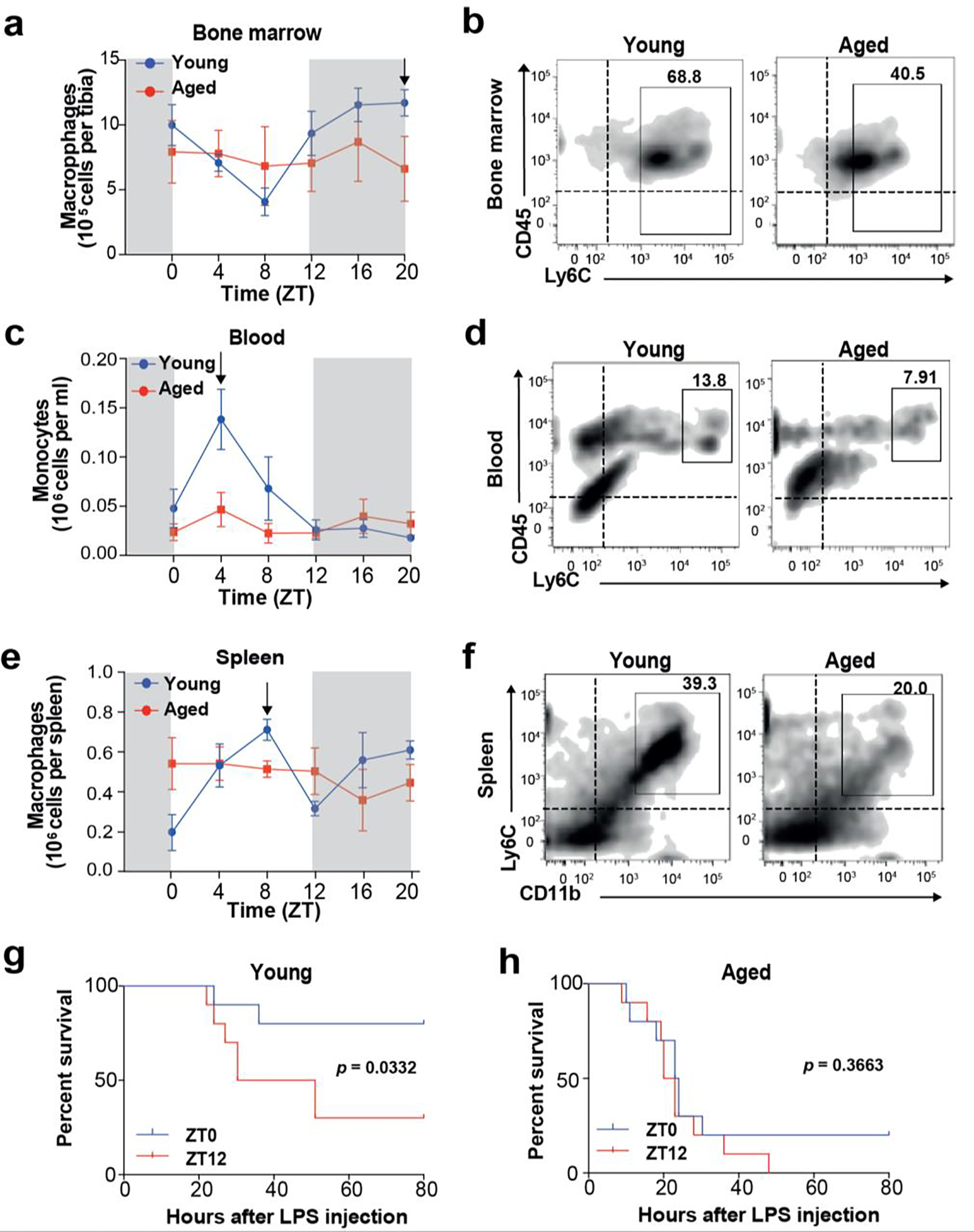
a-f. Young (2 mo) and aged (20–22 mo) C57B6/J male mice were examined for monocyte trafficking from the bone marrow to blood and spleen (n=3 per time point per age). Samples were analyzed by flow cytometry at ZT 4, 8, 12, 16, 20, and 24 hours. Vertical black arrows in a, c, and e denote maximum number of cells in the young age group featured in the representative plots b, d, and f.
a, b. CD45+CD11b+Ly6C+ bone marrow macrophages in young vs aged mice (p=0.00026 and p=1 for young and aged, respectively, by JTK_CYCLE).
c, d. CD45+CD11b+Ly6C+ blood monocytes in young vs aged mice (p=0.046 and p=1 for young and aged, respectively, by JTK_CYCLE)
e, f. CD45+CD11b+Ly6C+ spleen macrophages in young vs aged mice (p=1 for young and aged by JTK_CYCLE)
g-h, Young (g) and aged (h) mice were administered 25 mg/kg LPS either at ZT0 (blue line, “lights on”) or ZT12 (red line, “lights off”) and monitored for 7 days; n=10 mice in each group; *p<0.05, log-rank test.
Given the importance of diurnal migration of inflammatory monocytes in protecting against bacterial infection8,21, we tested whether disrupted circadian myeloid cell trafficking in aged mice was associated with increased vulnerability to sepsis. Susceptibility to sepsis has been shown to also be diurnally regulated, with increased mortality after lipopolysaccharide (LPS) challenge at night (ZT12)8,12,21,22. Indeed, we found that young mice injected with LPS at ZT0 showed significantly better survival (p=0.033) compared to young mice injected at ZT12 (Fig. 1g). However, aged mice did not show any difference in survival when LPS challenge occurred at ZT0 or at ZT12 (Fig. 1h), supporting the importance of diurnal myeloid trafficking and its disruption in aging.
To understand the molecular basis underlying loss of myeloid circadian rhythmicity with age, we carried out unbiased transcriptomics and compared the diurnal rhythms of peritoneal macrophages derived from young (2 months old) and aged (20–22 months old) C57BL/6J male mice collected at 4 h time intervals over 24 h (Fig. 2a, Supplementary Fig. 2a). The percentage of live macrophages was approximately 15% lower in aged as compared to young mice; however, there were no differences in macrophage enrichment at ZT0 (“lights on”), compared to ZT12 (“lights off”), for either age group (Supplementary Fig. 2b). RNA-seq expression of macrophage-specific markers was significantly higher than that of non-macrophage cell types (Supplementary Fig. 2c), indicating that samples were highly enriched for macrophages across time points and age groups. Of note, cell counts, RNA amounts, RIN scores and the percent of mapped genes were similar between the two age groups (Supplementary Fig. 2d–k). We next investigated diurnal rhythms in gene expression in young and aged macrophages over the 24 h time period. JTK_CYCLE analysis23 identified 680 genes undergoing rhythmic transcription in macrophages from young mice but only 53 genes in aged macrophages (Fig. 2b). Circadian transcription was dramatically disrupted in aged macrophages (Fig. 2c), with the majority of transcripts losing rhythmicity (Fig. 2b–e). Principal component analysis (PCA) of transcripts that were rhythmically expressed in young macrophages showed a significant clustering by age (Fig. 2f). Moreover, within the young macrophages, we observed sub-clustering by time of day over the 4-hour intervals, with ZT0 and ZT24 clustering together; this sub-clustering was not evident in aged macrophages (Figure 2f).
Figure 2. Aging abolishes rhythmic gene expression in macrophages.
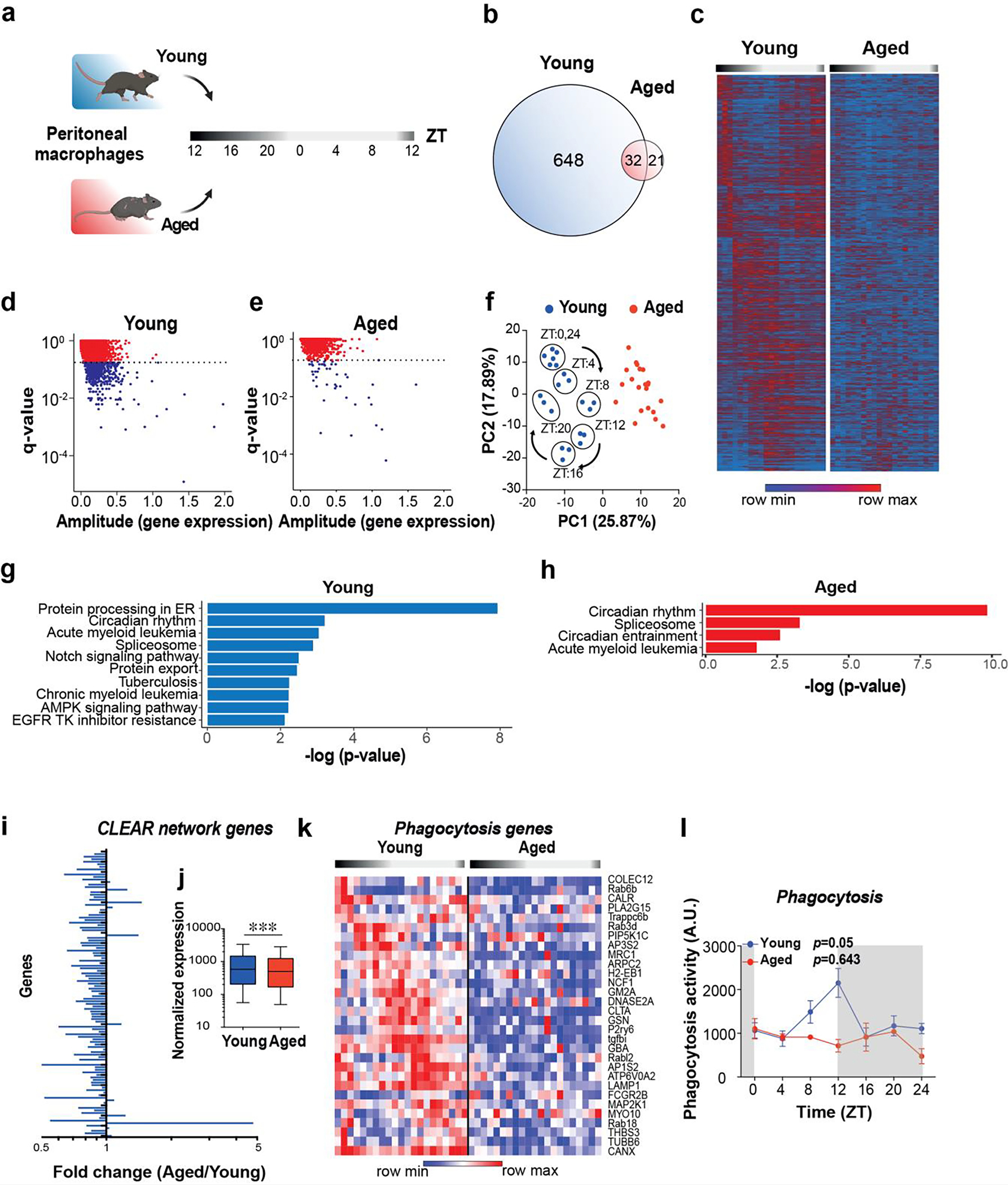
a. Schematic of experimental design. Peritoneal macrophages were collected from young (2 mo) and aged (20–22 mo) male mice at 4-hour intervals over a 24-hour period for RNA-seq; n=21 mice in each age group and n=3 in each time interval.
b. Venn diagram of unique and shared rhythmically expressed transcripts in young vs. aged macrophages.
c-e. Heatmap (c) and scatterplots of JTK_CYCLE results (d-e) of diurnally oscillating transcripts in young and aged peritoneal macrophages. The dotted lines in d and e represent q-value cutoffs of 0.2.
f. Principal component analysis (PCA) of rhythmic transcripts from young and aged macrophages. Note circadian clustering observed in young mice that is absent in the aged group.
g-h. KEGG pathway analyses of transcripts that show diurnal oscillations in young (g) and aged (h) macrophages. Genes with p<0.05 and q<0.1 by JTK_CYCLE are shown.
i. Fold change of Coordinated Lysosomal Expression and Regulation (CLEAR) network gene expression levels in aged vs young peritoneal macrophages over all time points.
j. Pooled normalized expression levels of CLEAR network transcripts in young and aged peritoneal macrophages; p < 0.001, 2-sided Mann Whitney U test.
k. A heatmap showing genes belonging to the KEGG pathway of phagocytosis over a 24 h time period in young and aged peritoneal macrophages. Each column represents one mouse. (n=3 mice per age per time interval). q<0.2, JTK_CYCLE.
l. Young and aged peritoneal macrophages were assayed for phagocytosis of fluorescent E coli particles over a 24 hour period, p=0.05 and p=0.643 for young and aged, respectively, by JTK_CYCLE (n=3 mice per time point per age).
Rhythmic transcripts in young peritoneal macrophages showed significant enrichment for many biological pathways, most prominently protein processing in the endoplasmic reticulum, a pathway that encompasses a broad range of homeostatic cellular processes including protein folding, modification, degradation, and antigen presentation (Fig. 2g). Rhythmic expression of this homeostatic pathway was completely lost in aged macrophages, suggesting a profound disruption of homeostatic immune responses that depend on intact ER function (Fig. 2h). Importantly, the expression of core clock genes was not altered between ages (Supplementary Fig. 3) and reflected by KEGG pathway enrichment of ‘circadian rhythms’ as the most significantly enriched pathway in aged macrophages (Fig. 2h).
We then focused on the Coordinated Lysosomal Expression and Regulation (CLEAR) network, a class of genes central to regulation of fundamental immune functions involving lysosomal biogenesis and function, autophagy, exo- and endocytosis, and phagocytosis24,25. We observed a striking reduction in global expression levels of CLEAR network genes in aged macrophages (Fig. 2i–j; Supplementary Fig. 4a). Within the CLEAR network, phagocytosis-related genes showed a significant loss of rhythmicity in aged macrophages (Figure 2k and Supplementary Fig. 4b). We compared the ability of young and aged primary peritoneal macrophages to engulf fluorescent E. coli particles every 4 h over the course of 24 h. Young macrophages showed rhythmic phagocytic activity over the course of 24 h (p=0.05 by JTK_CYCLE), with peak phagocytic activity at the beginning of the dark phase (Figure 2l). This diurnal variation was completely lost in aged macrophages (Figure 2l, p=0.643). These results indicate that the diurnal rhythmicity of macrophage-mediated phagocytosis of bacteria is lost with aging.
We next tested whether the loss of oscillatory gene expression in aged macrophages was mediated by age-associated epigenetic alterations. To this end, we conducted transposase-accessible chromatin sequencing (ATAC-seq) on peritoneal macrophages harvested from young and aged mice every 4 h over the course of 24 h. We integrated peak sizes of chromatin accessibility across 500 bp regions and assigned peaks to genomic loci. A peak-calling algorithm was used to identify 67,992 peaks, or 500-bp regions of accessible chromatin, tested using DESeq226. Of the 7,098 differentially accessible peaks (q < 0.05), 4,828 were more accessible in young macrophages, while only 2,270 were more accessible in old macrophages (Fig. 3a–b). Both PCA and hierarchical clustering of the peaks segregated the two age groups (Fig. 3c–d), indicating that age is the main factor driving the variability in chromatin openness among the samples27,28
Figure 3. Chromatin accessibility is globally decreased in aged macrophages but does not account for loss of diurnal transcription.
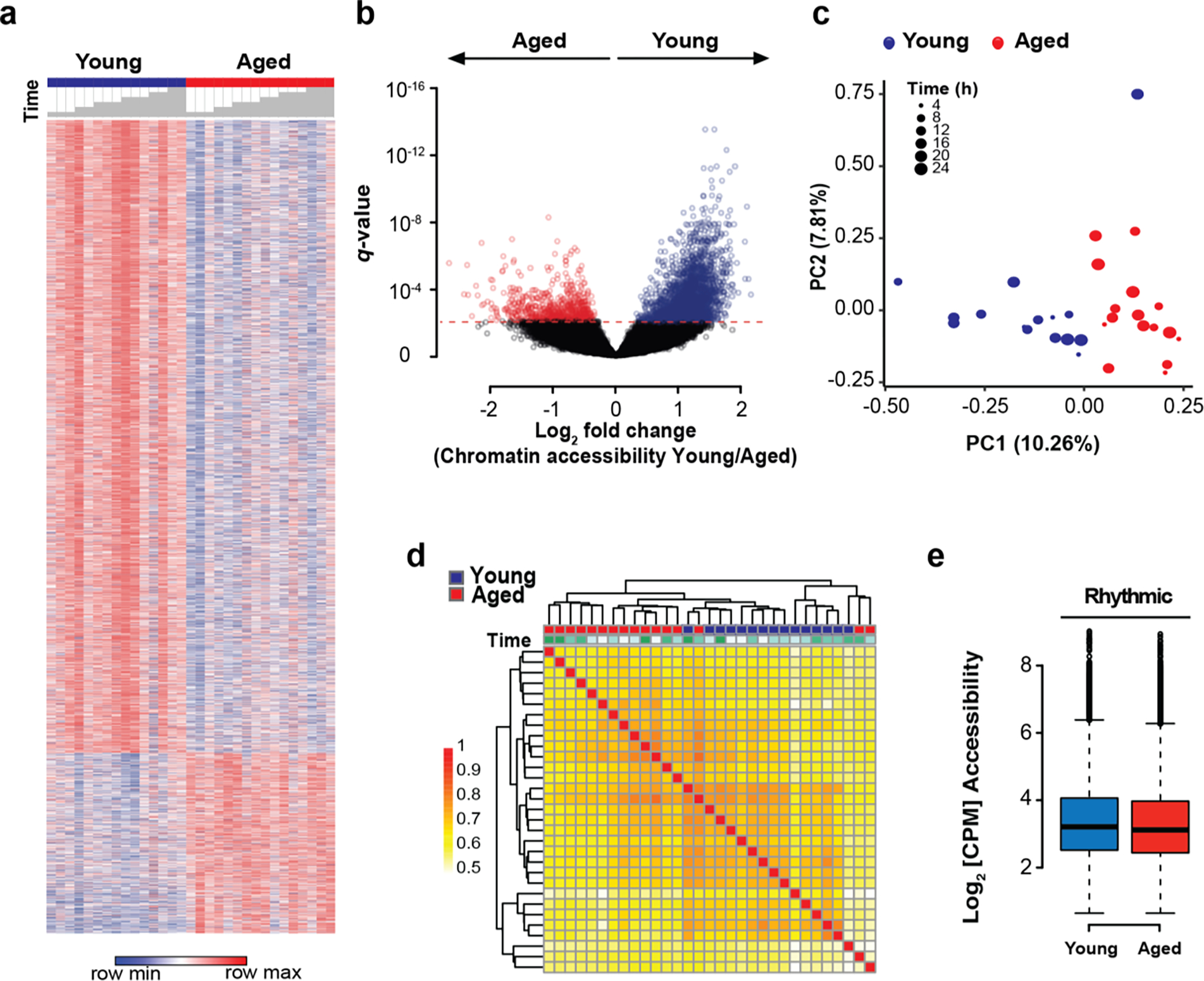
a. Heatmap of differential chromatin accessibility peaks in young vs. aged peritoneal macrophages over a 24-hour cycle at 4h intervals; n=15–16 mice in each age group and n=2–3 mice per 4 h time interval. Time is indicated with grey bars above the heatmap starting at ZT12. Differential accessibility was determined using DESeq2.
b. Volcano plot of differentially accessible peaks shows 4,828 vs 2,270 open chromatin peaks in young vs. aged macrophages.
c. PCA plot of accessible chromatin peaks shows separation by age between groups.
d. Correlation matrix of Spearman correlation coefficients of chromatin accessibility between young and aged macrophages. Time is indicated by color code above the matrix, with ZT12 being white, and 4-hour intervals being represented by increasingly stronger tones of green.
e. Chromatin accessibility (as normalized and log2-transformed values) at gene loci with rhythmic expression in young mice is not altered in aged macrophages.
To determine whether these chromatin alterations accounted for the loss of oscillatory gene expression in aged macrophages, we used three orthogonal approaches to link loci of differentially rhythmic genes to chromatin accessibility (Supplementary Fig. 5a). First, we determined whether differentially rhythmic genes were characterized by differentially accessible chromatin in young and old macrophages. To this end, we considered all 67,992 genomic regions and assigned gene loci to peaks using GREAT29. However, chromatin accessibility at differentially rhythmic loci was not different between young and old macrophages (Fig. 3e). We then tested whether differences in circadian gene expression might be explained by differentially accessible transcription start sites (TSSs), rather than by global ATAC-seq peaks. We therefore focused on promoter areas and TSSs of differentially rhythmic genes. While rhythmic genes in both groups showed generally higher accessibility than average genes, their accessibility was not different between young and old macrophages (Supplementary Fig. 5b). In fact, only 33 out of 648 differentially rhythmic genes also showed differential chromatin accessibility at the promoter (Supplementary Fig. 5c).
Given that overall chromatin accessibility was unlikely to account for differences in oscillatory gene expression between young and old macrophages, we then tested the possibility that the differential rhythmicity in gene expression between age groups might be mediated by rhythmic chromatin accessibility over the course of a day. We therefore explored the degree of rhythmicity in chromatin accessibility in both groups. However, JTK_CYCLE applied to ATAC-seq peaks in both young and aged macrophages did not reveal circadian rhythmicity (Supplementary Fig. 5d), even under multiple independent filtering conditions prior to p-value adjustment (data not shown). A comparison of the unadjusted p-value distributions for chromatin peaks (Supplementary Fig. 5e) and transcripts (Supplementary Fig. 5f) revealed that there is no enrichment of significant p-values relative to the background uniform distribution of non-significant p-values. These analyses led us to conclude that chromatin oscillations cannot be detected when applying the same methodological rigor as used for transcripts. These results suggest that differential 24 h oscillations in chromatin accessibility cannot explain the loss of rhythmic gene expression in aged macrophages.
Given that neither alterations in the core clock machinery nor differentially rhythmic chromatin accessibility provided mechanistic explanations for the circadian transcriptional reprogramming in aged macrophages, we examined whether differential binding of transcription factor/s (TFs) may drive the observed age-dependent loss of transcript oscillations. We identified candidate trans-acting factors using three conditions (Fig. 4a): (1) differential chromatin access between young and old macrophages, (2) differential binding to genes that show distinct oscillatory patterns between both age groups, and (3) rhythmic expression of the transcription factor that is lost in aged macrophages. Using chromVAR analysis30 on all differentially accessible peaks, we identified TFs that showed significantly higher activity in young macrophages (Fig. 4b). Next, we assessed whether this differential TF activity could account for the alterations in rhythmic transcription between both age groups. We therefore focused on genes with differentially rhythmic expression and assessed TF binding activity in corresponding ATAC-seq peaks using chromVAR (Fig. 4c) and transcripts using oPossum (Fig. 4d).
Figure 4. Age-dependent loss of Klf4 circadian activity leads to reduced macrophage functionality.

a. Selection criteria for candidate regulatory elements include (1) differential chromatin access between young and old macrophages, (2) differential binding to genes that show distinct oscillatory patterns between both age groups, and (3) TF rhythmic expression that is lost in aged macrophages
b. Heatmap of differentially accessible TF motifs in chromatin data from young vs aged macrophages. Analysis performed using chromVAR30. Arrow denotes KLF4 binding to MA0039.1 motif and range for q value < 0.05 is highlighted by vertical black line on right side of heatmap.
c. Heatmap of differentially accessible TF motifs in chromatin data from loci with differential rhythmic expression in young vs aged macrophages. Analysis performed using chromVAR30. Arrow denotes KLF4 binding to MA0039.1 motif and range for q value < 0.05 is highlighted by vertical black line on right side of heatmap.
d. Enrichment of TF motifs in rhythmic genes in young vs aged macrophages. Analysis performed using oPossum.
e. Klf4 mRNA rhythmicity in young and aged macrophages; p=0.01 and p=1 for young and aged, respectively, JTK_CYCLE (n=3 mice per age per time interval).
f. chromVAR30 deviations within all 500 bp peaks indicating Klf4 binding by estimating accessibility within peaks sharing the MA0039.1 motif or annotation. p=4.55×10−6, two-sided Mann-Whitney U test.
g. chromVAR30 deviations within peaks associated with differentially rhythmic genes indicating Klf4 binding by estimating accessibility within peaks sharing the MA0039.1 motif or annotation. p=1.25×10−3, two-sided Mann-Whitney U test.
h. Phagocytosis of fluorescent E coli particles by young and aged peritoneal macrophages transfected with Klf4 shRNA or scrambled vector as control. n=5 mice in each group. The experiment was repeated twice. Data are mean ± s.e.m. *p<0.05, **p<0.005, two-sided Mann-Whitney U test.
i. Crystal structure of the zinc-finger domain of KLF4 in complex with DNA and rs2236599 synonymous mutation site predicted by SWISS-MODEL48.
j. Percentages of E. coli infections of non-carrier vs T/T Klf4 variant carriers using UK BioBank data analysis. n=329,757 non-carrier and 15,537 T/T carrier individuals, p=0.003, two-sided Mann-Whitney U test.
k. UK BioBank data analysis of 12-year survival of non-carrier vs T/T variant who succumbed to microbial infection; n=19,791 non-carrier and 949 T/T carrier deceased individuals. p=0.047, log-rank test.
l. Overall survival of participants is not different between non-carriers and T/T variant. n=329,757 non-carrier and 15,537 T/T carrier individuals, p>0.05, log-rank test.
m. Odds ratio to develop an E coli infection of participants older than 65 years who carry the rs2236599 KLF4 variant (T/T) vs. non-carrier controls shows that increased susceptibility to infection with age is less pronounced in individuals carrying the Klf4 variant. n=266,771 non-carriers younger than 65 and 62,986 older than 65. n=12,593 T/T carrier individuals younger than 65 and 2,944 older than 65.
The only TF that fulfilled all three criteria was the zinc finger-containing TF KLF4. KLF4 is a member of the Kruppel-like factor (KLF) family of transcription factors and has been extensively investigated for its role in regulation of cell differentiation and stem cell reprogramming31–35. We found that KLF4 was not only associated with differential chromatin access (Fig. 4b) and differential binding to rhythmic genes (Fig. 4c, d), but also showed oscillatory expression in young macrophages that was lost in aged cells (Fig. 4e). The properties of differential chromatin access, differential binding to rhythmic genes, and rhythmic expression were unique to KLF4 among TFs and within the KLF family (Supplementary Fig. 6a, b). A de novo search of KLF4 binding motifs using ENCODE revealed two alternative motifs (MA0039.1 and MA0039.2), consistent with the motifs deposited in the JASPAR database (Supplementary Fig. 6c, d). Importantly, KLF4 binding to the MA0039.1 was significantly higher in young macrophages across all chromatin accessible regions and peaks associated with differentially rhythmic genes (Figure 4f, g), while the MA0039.2 motif was not different between both groups (Supplementary Fig. 6e, f). In young macrophages, diurnal increases of Klf4 gene expression occurred at ZT12 (Fig. 4e), similar to the diurnal peak in phagocytosis (Fig. 2l). Knockdown of Klf4 in young and aged macrophages significantly disrupted bacterial phagocytosis (Fig. 4h), indicating a central role for this transcription factor in regulating this circadian process.
Given the loss of Klf4 rhythmicity in aged macrophages, we investigated whether variants of Klf4 might be linked to age-associated deficits in anti-microbial immunity in human subjects. To this end, we investigated phenotypes associated with the rs2236599 genetic variant of Klf436–38 in the UK BioBank (Supplementary Table 1). This synonymous variant leads to an adenine-to-guanine transition at glycine39 (Fig. 4i). We found that carriers of this variant had a significantly elevated risk of developing E coli infection in the entire study population (Fig. 4j). In addition, carriers of the Klf4 variant show selectively enhanced infection-related mortality in the setting of normal overall survival (Fig. 4k, l). Interestingly, while non-carriers over the age of 65 years showed an elevated odds ratio to contract an E coli infection compared to younger subjects, this age-associated risk was less pronounced with the Klf4 variant (Fig. 4m). Thus, the rs2236599 genetic variant of Klf4 reveals a significant association with age-dependent susceptibility to death from bacterial infection.
The transcription factor KLF4 is traditionally studied in the context of somatic cell reprogramming and cell fate determination 34,35 where it binds the MA0039.2 motif 31,39; recent studies further indicate a role for KLF4 in regulation of enhancer networks in cell fate transition40. KLF4 also functions in terminally differentiated cells in the intestinal epithelium41, in monocyte cell differentiation42 and in establishment of M2 macrophage polarization states43,44. In regulating circadian gene transcription in young macrophages, KLF4 bound to the motif MA0039.1, which is distinct from that involved in stem cell reprogramming31,39. We found the MA0039.1 motif significantly enriched in young macrophages across all open chromatin regions and peaks associated with differentially rhythmic genes, while no such enrichment was found for the MA0039.2 motif involved in cell differentiation and reprogramming. This difference may be due to different chromatin states in different cell types, pluripotency potential, and age, and points toward a novel context-dependent function of KLF4.
Our work builds on the observation that the immune system changes significantly with aging and the mechanisms responsible are just beginning to be explored. The aging immune system is not only skewed towards the myeloid cell lineage 19,45,46, but macrophages lose their homeostatic polarization states and phagocytic capacities, phenotypes that are driven in part by an age-associated decline in cellular energy metabolism 47. The loss of oscillatory gene expression in aged macrophages is likely to further compound this functional decline. We found that aging was characterized by loss of diurnal phagocytosis and migration of Ly6Chi monocytes from bone marrow to blood; this phenotype would be expected to impair coordination of immune defenses against microbial pathogens, and in a sepsis model, diurnally-controlled survival differences were abolished with aging. Mechanistically, the core clock genes remained rhythmic in aged macrophages, and while dramatic differences were found in chromatin architecture of aged as compared to young macrophages, there were no diurnal fluctuations in chromatin accessibility that could explain the loss of transcriptional rhythmicity. Rather, we identified the upstream transcription factor KLF4, binding through a second motif distinct from that involved in cellular reprogramming, as responsible for oscillatory gene expression and phagocytic activity. Examination of a human genetic variant of Klf4 revealed a significant association with age-dependent susceptibility to death from bacterial infection.
In summary, we demonstrate a marked disruption of circadian rhythmicity of the macrophage transcriptome and function with aging. It is possible that cells of the immune system, and macrophages in particular, are subject to more varied and sustained cell-extrinsic cues with aging that then could disrupt coordinated immune gene expression, leading to age-associated dysregulation of diurnal and homeostatic immune functions. We also identify a novel role for KLF4 as a cell-intrinsic regulator of homeostatic circadian regulation in aging macrophages.
Supplementary Material
Acknowledgments
This work was supported by RO1AG048232 (KIA), RF1AG058047 (KIA), American Heart Association 19PABH134580007 (KIA), 1P50 AG047366 (KIA), T32 Neuroscience Institute (CTP), NSF GRFP (CTP). DP2AG067492 (CAT), the Edward Mallinckrodt, Jr. Foundation (CAT), UPenn Institute for Immunology (CAT), UPenn Diabetes Research Center P30-DK-019525 (CAT), and the UPenn Institute on Aging (CAT), Marie Skłodowska-Curie Grant 888494 (EB), Stanford School of Medicine Dean’s Postdoctoral Fellowship (EB), Medical Scientist Training Program T32 GM07170 (LL). The authors would like to thank Luis de Lecea for support in housing mice, the Stanford Shared FACS facility for flow cytometry analysis on LSR instruments (S10RR027431-01), and SCGPM for sequencing on HiSeq2000. The ATAC-seq sequencing data was generated on an Illumina HiSeq 4000 that was purchased with funds from NIH S10OD018220 for the Stanford Functional Genomics Facility.
References
- 1.Bhadra U, Thakkar N, Das P & Pal Bhadra M Evolution of circadian rhythms: from bacteria to human. Sleep Medicine 35, 49–61, doi: 10.1016/j.sleep.2017.04.008 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Herzog ED & Tosini G The mammalian circadian clock shop. Semin Cell Dev Biol 12, 295–303, doi: 10.1006/scdb.2001.0257 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Storch KF et al. Extensive and divergent circadian gene expression in liver and heart. Nature 417, 78–83, doi: 10.1038/nature744 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Zhang R, Lahens NF, Ballance HI, Hughes ME & Hogenesch JB A circadian gene expression atlas in mammals: Implications for biology and medicine. Proceedings of the National Academy of Sciences of the United States of America 111, 16219–16224, doi: 10.1073/pnas.1408886111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fonken LK et al. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain, Behavior, and Immunity 45, 171–179, doi: 10.1016/j.bbi.2014.11.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He W et al. Circadian Expression of Migratory Factors Establishes Lineage-Specific Signatures that Guide the Homing of Leukocyte Subsets to Tissues. Immunity 49, 1175–1190.e1177, doi: 10.1016/j.immuni.2018.10.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller M et al. A circadian clock in macrophages controls inflammatory immune responses. Proceedings of the National Academy of Sciences of the United States of America 106, 21407–21412, doi: 10.1073/pnas.0906361106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen KD et al. Circadian gene Bmal1 regulates diurnal oscillations of Ly6Chi inflammatory monocytes. Science 341, 1483–1488, doi: 10.1126/science.1240636 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geiger SS, Curtis AM, O’Neill LAJ & Siegel RM Daily variation in macrophage phagocytosis is clock-independent and dispensable for cytokine production. Immunology 157, 122–136, doi: 10.1111/imm.13053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitchen GB et al. The clock gene Bmal1 inhibits macrophage motility, phagocytosis, and impairs defense against pneumonia. Proceedings of the National Academy of Sciences of the United States of America 117, 1543–1551, doi: 10.1073/pnas.1915932117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliva-Ramírez J, Moreno-Altamirano MMB, Pineda-Olvera B, Cauich-Sánchez P & Javier Sánchez-García F Crosstalk between circadian rhythmicity, mitochondrial dynamics and macrophage bactericidal activity. Immunology 143, 490–497, doi: 10.1111/imm.12329 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curtis AM et al. Circadian control of innate immunity in macrophages by miR-155 targeting Bmal1. Proceedings of the National Academy of Sciences of the United States of America 11, 7231–7236, doi: 10.1073/pnas.1501327112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi M, Shimba S & Tezuka M Characterization of the molecular clock in mouse peritoneal macrophages. Biological and Pharmaceutical Bulletin 30, 621–626, doi: 10.1248/bpb.30.621 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Knyszynski A & Fischer H Circadian fluctuations in the activity of phagocytic cells in blood, spleen, and peritoneal cavity of mice as measured by zymosan-induced chemiluminescence. Journal of immunology (Baltimore, Md. : 1950) 127, 2508–2511 (1981). [PubMed] [Google Scholar]
- 15.Leone MJ, Marpegan L, Duhart JM & Golombek DA Role of proinflammatory cytokines on lipopolysaccharide-induced phase shifts in locomotor activity circadian rhythm. Chronobiology International 29, 715–723, doi: 10.3109/07420528.2012.682681 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Rahman SA et al. Endogenous circadian regulation of pro-inflammatory cytokines and chemokines in the presence of bacterial lipopolysaccharide in humans. Brain, Behavior, and Immunity 47, 4–13, doi: 10.1016/j.bbi.2014.11.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huo M et al. Myeloid Bmal1 deletion increases monocyte recruitment and worsens atherosclerosis. FASEB Journal 31, 1097–1106, doi: 10.1096/fj.201601030R (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheiermann C, Kunisaki Y & Frenette PS Circadian control of the immune system. Nature Reviews Immunology 13, 190–198, doi: 10.1038/nri3386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossi DJ, Jamieson CH & Weissman IL Stems cells and the pathways to aging and cancer. Cell 132, 681–696, doi: 10.1016/j.cell.2008.01.036 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Liu Q et al. Peripheral TREM1 responses to brain and intestinal immunogens amplify stroke severity. Nature Immunology 20, 1023–1034, doi: 10.1038/s41590-019-0421-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheiermann C et al. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 37, 290–301, doi: 10.1016/j.immuni.2012.05.021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng W et al. The Circadian Clock Controls Immune Checkpoint Pathway in Sepsis. Cell Reports 24, 366–378, doi: 10.1016/j.celrep.2018.06.026 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes ME, Hogenesch JB & Kornacker K JTK-CYCLE: An efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. Journal of Biological Rhythms 25, 372–380, doi: 10.1177/0748730410379711 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmieri M et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Human Molecular Genetics 20, 3852–3866, doi: 10.1093/hmg/ddr306 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Sardiello M et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477, doi: 10.1126/science.1174447 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550–550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moskowitz DM et al. Epigenomics of human CD8 T cell differentiation and aging. Science Immunology 2, eaag0192–eaag0192, doi: 10.1126/sciimmunol.aag0192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ucar D et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. Journal of Experimental Medicine 214, 3123–3144, doi: 10.1084/jem.20170416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501, doi: 10.1038/nbt.1630 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schep AN, Wu B, Buenrostro JD & Greenleaf WJ ChromVAR: Inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nature Methods 14, 975–978, doi: 10.1038/nmeth.4401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X et al. Integration of External Signaling Pathways with the Core Transcriptional Network in Embryonic Stem Cells. Cell 133, 1106–1117, doi: 10.1016/j.cell.2008.04.043 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Maherali N et al. Directly Reprogrammed Fibroblasts Show Global Epigenetic Remodeling and Widespread Tissue Contribution. Cell Stem Cell 1, 55–70, doi: 10.1016/j.stem.2007.05.014 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Okita K, Ichisaka T & Yamanaka S Generation of germline-competent induced pluripotent stem cells. Nature 448, 313–317, doi: 10.1038/nature05934 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Takahashi K & Yamanaka S Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126, 663–676, doi: 10.1016/j.cell.2006.07.024 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Wernig M et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448, 318–324, doi: 10.1038/nature05944 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Liu H et al. Irf6 directly regulates Klf17 in zebrafish periderm and Klf4 in murine oral epithelium, and dominant-negative KLF4 variants are present in patients with cleft lip and palate. Human Molecular Genetics 25, 766–776, doi: 10.1093/hmg/ddv614 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stratopoulos A et al. Genomic variants in members of the Krüppel-like factor gene family are associated with disease severity and hydroxyurea treatment efficacy in β-hemoglobinopathies patients. Pharmacogenomics 20, 791–801, doi: 10.2217/pgs-2019-0063 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Stremitzer S et al. Genetic variants associated with colorectal brain metastases susceptibility and survival. Pharmacogenomics Journal 17, 29–35, doi: 10.1038/tpj.2015.86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soufi A et al. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568, doi: 10.1016/j.cell.2015.03.017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Giammartino DC et al. KLF4 is involved in the organization and regulation of pluripotency-associated three-dimensional enhancer networks. Nat Cell Biol 21, 1179–+, doi: 10.1038/s41556-019-0390-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McConnell BB, Ghaleb AM, Nandan MO & Yang VW The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. BioEssays 29, 549–557, doi: 10.1002/bies.20581 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alder JK et al. Kruppel-Like Factor 4 Is Essential for Inflammatory Monocyte Differentiation In Vivo. The Journal of Immunology 180, 5645–5652, doi: 10.4049/jimmunol.180.8.5645 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapoor N et al. Transcription Factors STAT6 and KLF4 Implement Macrophage Polarization via the Dual Catalytic Powers of MCPIP. J Immunol 194, 6011–6023, doi: 10.4049/jimmunol.1402797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao X et al. Krüppel-like factor 4 regulates macrophage polarization. Journal of Clinical Investigation 121, 2736–2749, doi: 10.1172/JCI45444 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dykstra B & de Haan G Hematopoietic stem cell aging and self-renewal. Cell and tissue research 331, 91–101, doi: 10.1007/s00441-007-0529-9 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Dykstra B, Olthof S, Schreuder J, Ritsema M & de Haan G Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med 208, 2691–2703, doi: 10.1084/jem.20111490 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minhas PS et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat Immunol 20, 50–63, doi: 10.1038/s41590-018-0255-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waterhouse A et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, doi: 10.1093/nar/gky427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.