

ABSTRACT
The bacteria-derived short-chain fatty acids (SCFAs) butyrate and propionate play important (distinct) roles in health and disease, and understanding the ecology of respective bacteria on a community-wide level is a top priority in microbiome research. Applying sequence data (metagenomics and 16S rRNA gene) to predict SCFAs production in vitro and in vivo, a clear split between butyrate- and propionate-forming bacteria was detected with only very few taxa exhibiting pathways for the production of both SCFAs. After in vitro growth of fecal communities from distinct donors (n = 8) on different substrates (n = 7), abundances of bacteria exhibiting pathways correlated with respective SCFA concentrations, in particular in the case of butyrate. For propionate, correlations were weaker, indicating that its production is less imprinted into the core metabolism compared with butyrate-forming bacteria. Longitudinal measurements in vivo (n = 5 time-points from 20 subjects) also revealed a correlation between abundances of pathway-carrying bacteria and concentrations of the two SCFAs. Additionally, lower bacterial cell concentrations, together with higher stool moisture, promoted overall bacterial activity (measured by flow cytometry and coverage patterns of metagenome-assembled genomes) that led to elevated SCFA concentrations with over-proportional levels of butyrate. Predictions on pathway abundances based on 16S rRNA gene data using our in-house database worked well, yielding similar results as metagenomic-based analyses. Our study indicates that stimulating growth of butyrate- and propionate-producing bacteria directly leads to more production of those compounds, which is governed by two functionally distinct bacterial groups facilitating the development of precision intervention strategies targeting either metabolite.
KEYWORDS: Gut microbiota, SCFA, butyrate, propionate, function, quantitative biology, communities, metagenomics, modeling, anaerobic cultivation
Introduction
Short-chain fatty acids (SCFAs), mainly acetate, butyrate and propionate, are major products of bacterial fermentation in the human large intestine and have increasingly become the focus of research due to their importance in host metabolism and health. They are known to reduce local and systemic inflammation processes by immunomodulatory properties and maintenance of gut epithelial integrity.1–3 Scarcities of SCFAs are associated with emerging noncommunicable metabolic disorders, such as cardiovascular disease, obesity and type II diabetes,4 and an impairment of colonization resistance against enteric pathogens.5,6 Despite common actions of SCFAs, they also markedly differ in their effects on the human body. For instance, the main target site of butyrate are colonocytes that use this compound for energy generation, whereas the bulk of propionate reaches the liver and promotes gluconeogenesis.7 Circulating SCFAs bind to G-protein-coupled receptors that are expressed throughout the body; however, SCFA affinities to individual receptor types differ,1 promoting distinct levels of response.8 Furthermore, only butyrate shows major epigenetic properties that play a role in diverse diseases.9,10
Direct measurements of fecal SCFAs represent the gold standard for assessing a given community’s capability to produce those compounds. However, SCFAs are volatile molecules demanding immediate preparation of samples for exact measurements, which is often difficult in practice. Furthermore, it is estimated that 90–95% of SCFAs produced are absorbed by the colonic epithelium and inferences on production of individual SCFAs from measured concentrations are, hence, doubtful.11,12
While acetate is produced by most members of gut microbiota, only specific, phylogenetically diverse bacterial groups form butyrate and propionate.13 Butyrate formation from carbohydrates is performed via the Acetyl-CoA pathway,14 whereas propionate is largely produced via the succinate (Suc) and propanediol (Pdiol) pathways that are both fed by carbohydrates as well.15 The former two pathways are anchored in the core metabolism of bacteria, making them essential biochemical routes for growth of respective bacteria. Less is known of the propanediol pathway, but it is expected that it also plays an important role in respective bacteria to occupy ecological niches in vivo.15 SCFA-production and bacteria involved have been in focus for many decades; however, a detailed community-wide understanding on a system level is still in its infancy. A comprehensive screening of (meta)genomes for exhibiting butyrate synthesis pathways in gut microbiota has been performed, demonstrating that primarily members of the Lachnospiraceae and Ruminococcaceae of the Firmicutes serve as butyrate producers. In case of propionate, the Suc pathway is suspected to be predominantly encoded on gut bacteria of the phylum Bacteroidetes, including the abundant Bacteroides, and a few Negativicutes of the Firmicutes, whereas propanediol pathway carriers almost exclusively belong to the Lachnospiraceae, mainly to the genera Ruminococcus and Blautia.15 However, a systematic screening for those pathways in genomes derived from the gut environment is lacking.
A major goal in gut microbiota research is to get (quantitative) insights into bacterial functions affecting host physiology. In this context, deciphering contributions of individual bacteria of a given community to the total SCFA pool is a top priority.16 While metagenomic data allow for exact determination of SCFA pathway distributions in a given sample,17 analyses are often tedious and inferring functionality from low-cost, high-throughput data, such as 16S rRNA gene results, is desirable. An additional aspect that has increasingly become recognized is bacterial load, as it was demonstrated that in healthy individuals, cell numbers per gram stool differ by an order of magnitude, which probably has profound influences on overall functioning as well as actual metabolite concentrations of a given sample.18
The aim of this study was to reveal butyrate- and propionate-forming communities of gut microbiota in quantitative terms and to assess the ability to predict the production of those two SCFAs based on sequence data. To this end, we comprehensively screened reference organisms of gut bacteria for exhibiting respective pathways and performed a series of in vitro incubations together with a longitudinal in vivo experiment including human subjects, where a multitude of parameters considered important for SCFA production were analyzed.
Results
Establishing a database of gut bacteria harboring major pathways for the production of butyrate and propionate
We screened in total 3,754 genomes, involving 3,207 species representative genomes, originating from the Unified Human Gastrointestinal Genome (UHGG) collection19 for exhibiting butyrate- and propionate-forming pathways. In total, 18.0% (n = 675) of genomes were classified as butyrate producers harboring the acetyl-CoA (ACoA) pathway, while 14.9% (n = 558) and 9.3% (n = 350) were exhibiting the Suc and Pdiol pathways for propionate synthesis, respectively (Figure 1). For the former pathway, 50.9% carried butyryl-CoA:acetate CoA transferase (but) as the terminal enzyme. Butyrate kinase (buk) was detected in 20.9% of genomes and 1.2% (all members of the genus Coprococcus) exhibited both enzymes, whereas in 27% of cases, neither gene was detected. While pathways were present on a wide range of distinct taxa, the distribution of both the ACoA and the Suc pathway was largely consistent on the genus level. For instance, almost all members of the key butyrate-producing genera Faecalibacterium and Agathobacter exhibited the ACoA pathway and most members of the Bacteroides and Phocaeicola displayed the Suc pathway (Figure S1). Only a few metagenome-assembled genomes (MAGs) within those genera were predicted lacking those pathways. The Pdiol pathway on the other hand clustered less homogeneously; however, members of several abundant genera of gut microbiota, such as Blautia_A, consistently exhibited this pathway. Overall, results suggest that main pathways are largely split between bacterial groups, where genomes either contained genes for the formation of butyrate or propionate. Of the 675 genomes harboring the ACoA pathway, only 8.6% and 10.8% also exhibited the Suc and Pdiol pathway, respectively. This functional division into butyrate- and propionate-forming communities was even more pronounced in in vitro and in vivo communities (results below).
Figure 1.
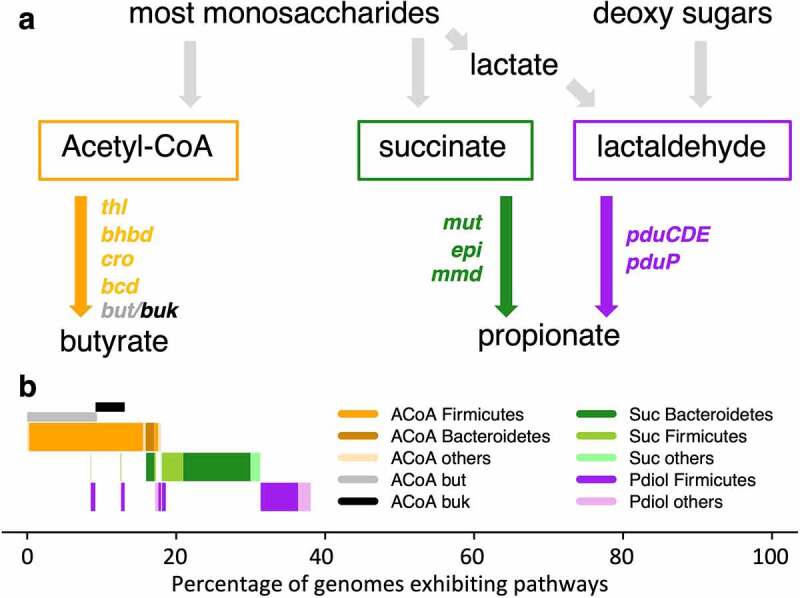
Overview of pathways and results from genome screenings. Panel a shows a simplification of main pathways involved in the formation of butyrate and propionate including gene names encoding enzymes catalyzing individual steps. For detailed description of pathways, refer to the study by Louis and Flint.13 Genomes of individual species of the Unified Human Gastrointestinal Genome (UHGG) collection were screened for exhibiting those pathways and taxonomic affiliations on the phylum level are indicated (panel b). ACoA: main butyrate-forming pathway including acetyl-CoA acetyltransferase (thl) β-hydroxybutyryl-CoA dehydrogenase (bhbd); crotonase (cro); butyryl-CoA dehydrogenase (bcd) as well as genes encoding the terminal enzymes butyryl-CoA:acetate CoA transferase (but) and butyrate kinase (buk). Suc: main propionate-forming pathway from carbohydrates including methylmalonyl-CoA mutase (mut), methylmalonyl-CoA epimerase (epi) and methylmalonyl-CoA decarboxylase (mmd). Pdiol: additional propionate-forming pathway with the key enzymes propanediol dehydratase (pduCDE) and propionaldehyde dehydrogenase (pduP).
Inferring pathways from genomes based on annotations derived from the Kyoto Encyclopedia of Genes and Genomes (KEGG) showed several discrepancies compared with our in-house database. Most obvious was the prediction of the ACoA pathway on genomes of many members of the Proteobacteria, such as Acinetobacter spp., Aeromonas spp., Citrobacter spp. and Yersinia spp. and several Bacilli, which have not been described as butyrate producers (Figure S1). Also for propionate-forming bacteria, inconsistencies with KEGG were detected. For instance, KEGG suggested that a specific clade of the Verrucomicrobiota including Akkermansia spp., which are known propionate producer, lacks the Suc pathway. Furthermore, based on KEGG, only a few members of the genus Blautia_A exhibit the Pdiol pathway, whereas our data indicate that this pathway was highly prevalent in this genus (Figure S1).
Of the total 3,754 genomes analyzed, 41.4% (n = 1,556) exhibited high-quality 16S rRNA gene sequences that were used as references for predicting SCFA pathways based on the picrust algorithm (Figure S2). In particular, many MAGs were devoid of adequate sequences and could, hence, not be included. Overall, predictions were largely following reference data, especially for the ACoA and Suc pathways and their presence/absence was wrongly predicted for only a few genomes. The Pdiol pathway was predicted correctly for most genomes as well; however, for a few taxa that disparately exhibit this pathway, such as Enterocloster and Escherichia, predictions deviated from references. Predictions based on input sequences trimmed to the variable regions V3V4 were largely mirroring full-length gene results (Figure S2).
Incubations of gut communities in vitro
To investigate the predictability of sequence data for the production of butyrate and propionate, we conducted a series of in vitro experiments, where freshly provided stool samples, derived from eight individuals, were incubated with six different growth substrates, namely, resistant starches type 2 and type 3, pectin from apple, mucin, inulin and protein. After 24 h, bacterial growth, i.e., cell numbers grown (measured by flow-cytometric (FCM) analysis), relative pathway abundances based on metagenomic analyses and SCFA concentrations (acetate, butyrate and propionate) were determined. Overall, bacterial composition after in vitro growth comprised common gut bacteria (Figure 2) and was similar to in vivo communities (see following section).
Figure 2.
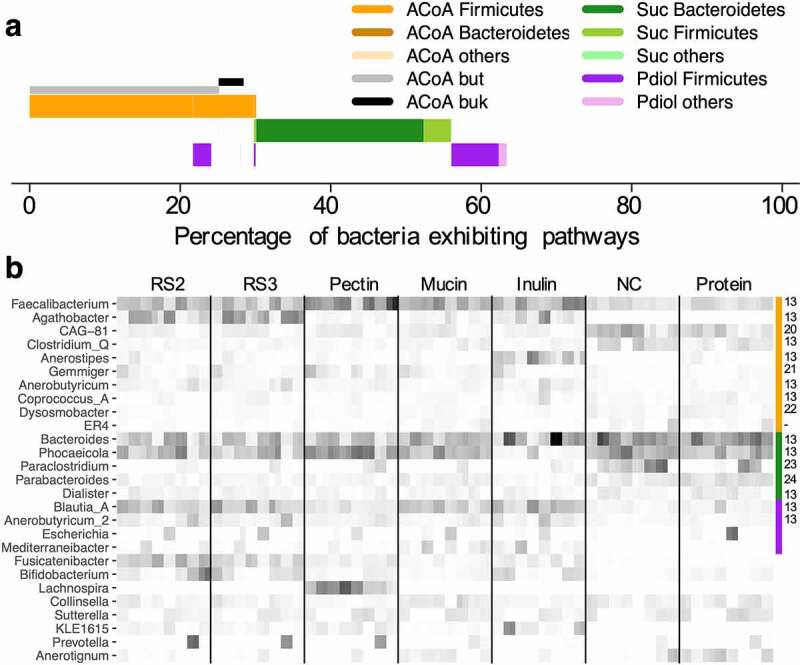
Overview of pathway abundances and associated taxonomic composition after in vitro growth of fecal communities derived from eight subjects grown on different substrates (n = 7). Panel a gives average abundances of pathways including taxonomic affiliations on the phylum level, whereas abundances of major genera comprising individual pathway communities (indicated by the color bar on the right) are given in panel b. Abundances are relative to housekeeping genes of the total bacterial community. Literature references supporting pathway presence based on biochemical testing in members of individual taxa shown are given on the right of the heatmap (-: MAGs, no isolate available).13,20–24 Incubations were performed in duplicate samples. For abbreviations of pathways in panel a, see Figure 1. RS2/3: resistant starch type 2/3; NC: basal medium.
On average, we detected growth of 4.07 × 108 ± 2.72 × 108 mL−1 butyrate producers, i.e., bacteria exhibiting the ACoA pathway, comprising 32.6% ± 6.9% of the total community, while 2.95 × 108 ± 1.62 × 108 (mL−1) Suc and 1.44 × 108 ± 1.00 × 108 (mL−1) Pdiol pathway carrying bacteria were detected representing 27.8% ± 12.8% and 10.9% ± 5.0% of the overall community, respectively. A clear split between butyrate- and propionate-producers was observed and only 2.77% of bacteria (mainly Anaerobutyricum) harbored pathways for both butyrate and propionate synthesis (Figure 2). The butyrate-forming community was primarily composed of Firmicutes, with several abundant genera including Faecalibacterium (27.7%) and Agathobacter (10.2%), whereas members of the Bacteroidetes, primarily Bacteroides (37.7%) and Phocaeicola (33.6%), comprised the Suc pathway; Blautia_A (44.6%) and Anaerobuytricum (18.3%) of the Firmicutes were the main members carrying the Pdiol pathway (Figure 2). Results of major taxa predicted to exhibit respective pathways were largely supported by literature reports.
Global community structures on the species level clustered strongly according to their donors and communities growing on proteins and only the baseline medium showed unique patterns forming a separate group (Figure S3a). Composition of functional communities, i.e., individual pathway carriers, showed strong subject-specific signatures as well (Figure S3b, c).
The average concentration of butyrate formed in all incubations was 5.27 mM ± 2.63 mM (Figure 3a) and we found a strong correlation (R2 = 0.63; p ≪ 0.01) with final growth of bacteria exhibiting the ACoA pathway. Relative butyrate concentrations (percentage of total SCFAs) was related with abundances of respective bacteria as well (R2 = 0.30; p ≪ 0.01; Figure 3b). Overall, average (relative) butyrate production and yields, i.e. butyrate produced per cell harboring the ACoA pathway, were in a similar range for communities derived from the different subjects; samples inoculated with bacteria from subjects e and h showed lower relative production and their yields were increased (Figure 3c-e). Growth on inulin and the resistant starches resulted in higher butyrate concentrations compared with results from mucin and pectin, whereas values for growth on proteins and the basal medium were the lowest (Figure 3f-h). Some growth was detected on the basal medium as it contained yeast extract and casitone (both at 1 g L−1) and we, hence, included those results into our analyses.
Figure 3.
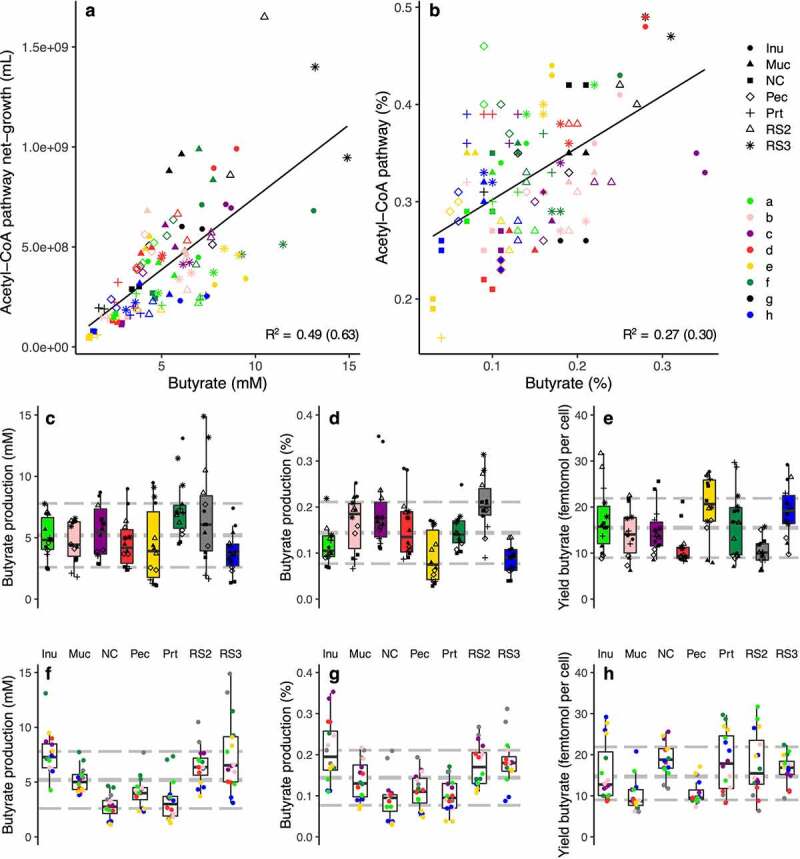
Correlation between ACoA pathway abundances and butyrate concentrations of in vitro experiments. Panel a shows the correlation between net-grown bacteria exhibiting the pathway and concentrations of butyrate formed, whereas associations between relative abundances of those bacteria with proportions of butyrate from total SCFAs are given in panel b. Values from communities derived from different donors and substrates are indicated. The Pearson correlation coefficient is given (values in brackets are based on log-transformed data). Panels c and f display concentrations of formed butyrate grouped into individual donors and substrates, respectively. Panels d and g give corresponding results for relative butyrate concentrations (from total SCFAs), whereas panels e and h show respective yields, i.e., butyrate formed per grown bacterium harboring the ACoA pathway. Gray lines depict average values along with standard deviations. Inu: inulin, Muc: mucin, NC: basal medium, Pec: pectin, Prt: protein, RS2/3: resistant starch type 2/3.
For propionate, a positive correlation between grown bacteria that contain the Suc and Pdiol pathways and propionate concentrations was observed (R2 = 0.24; p ≪ 0.01; Figure 4a). The fraction of propionate of total SCFAs was, however, not associated with abundances of those bacteria (Figure 4b). Average (relative) production was similar between communities derived from different subjects (Figure 4c-e) and concentrations of 3.97 mM ± 1.35 mM were lower than those of butyrate. Most propionate was formed during growth with mucin compared with other substrates, whereas the yield was highest on the basal medium (Figure 4f-h). The yield for propionate was lower, namely 10.9 ± 5.4 fmol propionate per propionate producer, compared with that of butyrate-producing bacteria (15.5 ± 6.4 fmol butyrate per butyrate producer). Total SCFA concentrations did only slightly correlate with pH (R2 = 0.11; p < .01) and were not associated with total bacterial growth (Figure S4a, b). The fraction of acetate showed strong negative correlations with both butyrate (R2 = 0.81; p ≪ 0.01) and propionate (R2 = 0.46; p ≪ 0.01) (Figure S4c), whereas relative concentrations of the latter two SCFAs were not associated (data not shown).
Figure 4.

Correlation between propionate pathway abundances and propionate concentrations of in vitro experiments. Cumulative abundances of the Suc and Pdiol pathways are shown. Panel a shows the correlation between net-grown bacteria exhibiting those pathways and concentrations of propionate formed, whereas associations between relative abundances of those bacteria with proportions of propionate from total SCFAs are given in panel b. Values from communities derived from different donors and substrates are indicated. The Pearson correlation coefficient is given (values in brackets are based on log-transformed data). Panels c and f display concentrations of formed propionate grouped into individual donors and substrates, respectively. Panels d and g give corresponding results for relative propionate concentrations (from total SCFAs), whereas panels e and h show respective yields, i.e., propionate formed per grown bacterium harboring the Suc/Pdiol pathways. Gray lines depict average values along with standard deviations. Inu: inulin, Muc: mucin, NC: basal medium, Pec: pectin, Prt: protein, RS2/3: resistant starch type 2/3.
Predicted pathway abundances from 16S rRNA gene data correlated well with results derived from metagenomes displaying R2s of 0.60, 0.70 and 0.59 (all p ≪ 0.01) for the ACoA, Suc and Pdiol pathways, respectively (Figure S5a). Average abundances of pathways and composition of associated bacteria were also similar to metagenome-derived data (Figure S5b, c). As for metagenome-based results, overall as well as pathway-specific communities clustered according to subjects with communities grown on proteins and the basal medium forming a separate group; donor communities (inocula) clustered with samples of respective subjects (Figure S6a-c).
SCFA-producing communities in vivo
In vitro experiments above indicated that it is possible to predict SCFA production from sequence data to a certain extent, in particular in the case of butyrate. As a next step, we investigated how those results relate to in vivo conditions by monitoring pathway abundances, bacterial concentrations and SCFA concentrations in 20 individuals, who provided five fresh stool samples over a period of 3 months.
On average, 28.1% ± 5.5% of bacteria carried the ACoA pathway, 14.4% ± 7.5% the Suc and 8.7% ± 3.7% the Pdiol pathway, respectively (Figure 5a); only 2.38% of bacteria overlapped and carried the ACoA together with a propionate-forming pathway. Community composition of bacteria harboring the ACoA pathway was in accordance with previously published data17 and that of in vitro results, where the bulk was classified as Firmicutes (95.9%), with Faecalibacterium (19.3%), Agathobacter (16.9%) and Gemmiger (9.9%) as the main taxa, and only a tiny fraction of Bacteroidetes (0.4%) (Figure 5b). The Suc pathway exhibiting community was primarily composed of Bacteroidetes (68.7%), with members of the genus Bacteroides (23.5%) and Phocaeicola (20.1%) representing the majority (Figure 5b), and of Dialister from the Firmicutes (20.0%) (Figure 5b). Pdiol pathway carriers were almost exclusively of the Firmicutes (94.8%), mainly of the genera Blautia_A (56.5%) and Anaerobutyricum (21.7%); the latter additionally exhibit the ACoA pathway representing the only noteworthy overlap between butyrate- and propionate-producers. Total as well as individual pathway community compositions showed strong subject-specific patterns (Figure S7).
Figure 5.
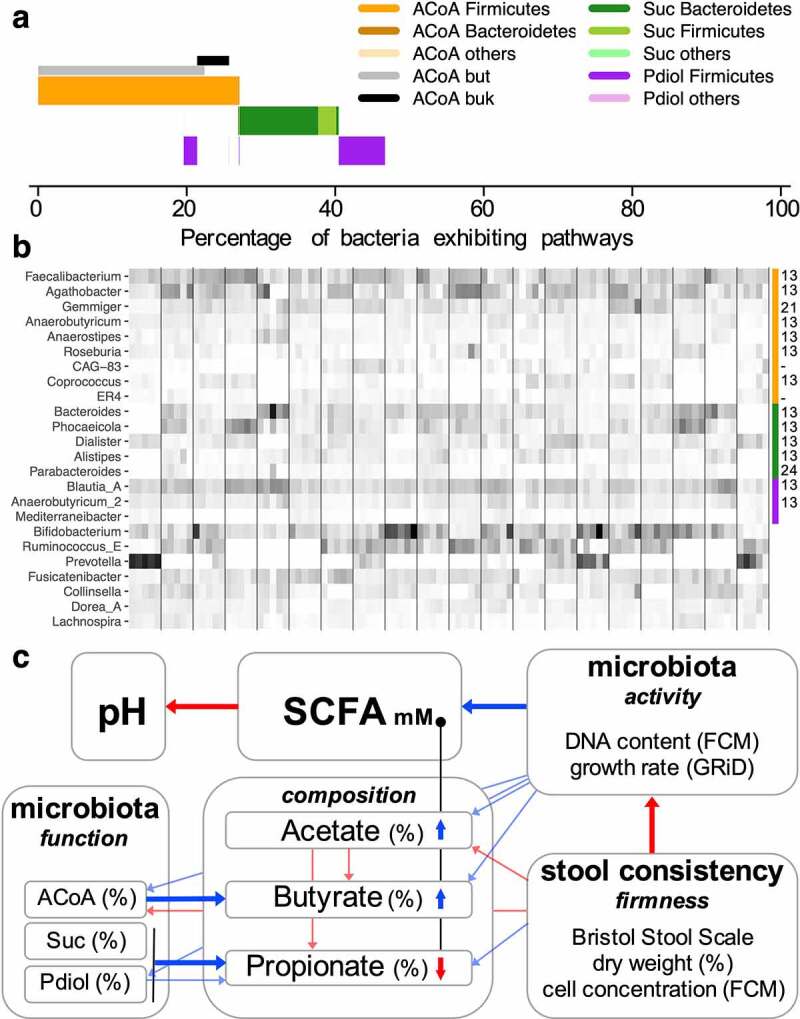
Overview of pathway abundances and taxonomic composition of the in vivo experiment (20 subjects were sampled at five time-points over the period of 3 months). Panel a gives average abundances of pathways including taxonomic affiliations on the phylum level, whereas abundances of major genera comprising individual pathway communities (indicated by the color bar on the right) are given in panel b; abundances are relative to housekeeping genes of the total bacterial community. Literature references supporting pathway presence based on biochemical testing in members of individual taxa shown are given on the right (-: MAGs, no isolate available).13,21,24 In panel c, a mechanistic model of in vivo fecal SCFA concentrations based on individual parameters measured is given. Blue refers to positive correlations based on linear mixed-effect models that included subject as a random effect, whereas red depicts negative associations. Correlations that are considered most important are highlighted as thick arrows. For explanations, see text.
Additional measured parameters from stool displayed strong variations between samples (Figure S8). SCFA concentrations varied by an order of magnitude for acetate (17.9–164.1 mM; average: 62.4 mM) and propionate (4.3–49.8 mM; average: 21.0 mM), while butyrate concentration varied by a factor of 40 (1.6–70.1 mM; average: 18.8 mM). Bacterial concentrations ranged from 4.94 × 1010 to 5.98 × 1011 (average: 2.50 × 1011) cells per gram wet stool and from 2.77 × 1011 to 1.39 × 1012 (average: 8.84 × 1011) cells per gram dry fecal matter, respectively; fecal moisture content displayed wide variations (51.8–85.4%; average: 72.2%). Values of the Bristol Stool Scale stretched over six of the seven categories (BSS 1–6).
Correlation analyses between all parameters allowed us to formulate a mechanistic model on factors governing fecal SCFA concentrations that is shown in Figure 5c. All stool parameters displayed high subject-specific patterns (Figure S8) and we, hence, included subject as a random effect in our correlation analyses (individual results from generalized linear models are given in Table S1). Contrary to our expectations, no association between fecal SCFA concentrations and the total amount of bacteria per gram stool was detected. However, bacterial activity, in particular green fluorescence signal intensities based on FCM analyses, which are a proxy of nucleic acid content, correlated positively with levels of fecal SCFAs (p < .01; Figure 5c). However, this parameter was negatively correlated with stool firmness (p ≪ 0.01 – p < .01), which was measured as percent dry weight, stool texture (according to the BSS) and fecal cell concentrations that all correlated highly with each other (p ≪ 0.01 – p < .01) (Table S1). The other parameter used to describe activity, namely, the growth rate index (GRiD) based on coverage ratios between ori and ter of constructed MAGs, correlated with FCM results (p < .05) and was trending (p = .12) with total SCFA concentrations. Stool firmness parameters did not correlate with total SCFA concentrations. (Relative) butyrate concentrations were associated with both activity parameters (p < .01 – p = .073) and with total SCFA concentrations (p ≪ 0.01), but not with stool firmness. However, firmness parameters correlated negatively with relative acetate concentrations (p < .01 – p < .05) and displayed positive associations with relative propionate concentrations (p < .05 – p = .069; Figure 5c). The proportion of both butyrate and acetate was increased at higher total SCFA concentrations (p ≪ 0.01 and p < .01), while that of propionate was reduced (p < .05). Relative acetate concentrations were negatively correlated with those of butyrate and propionate (p ≪ 0.01; Figure 5c). Abundances of ACoA, Pdiol and Suc pathways correlated with relative concentrations of respective SCFAs (p < .01 and p < .05; Figure 5c). Firmness was negatively correlated with ACoA pathway carrying bacteria (p < .01 – p < .05). A strong correlation between total SCFA concentrations and fecal pH was recorded (p ≪ 0.01); however, pH was not associated with abundance of any pathway (Table S1).
Predicted pathway abundances from 16S rRNA gene data were similar as those based on metagenomic analyses displaying R2s of 0.67, 0.80 and 0.52 (all p ≪ 0.01) for the ACoA, Suc and Pdiol pathways, respectively (Figure S9a); also the overall composition was comparable between the two techniques (Figure S9b). Detected average abundances of Suc were similar between the two methods, whereas concentrations of the ACoA and Pdiol pathways were higher compared with metagenomic results (Figure S9c).
Temporal stability of pathway abundances and SCFA concentrations
The longitudinal character of our study enabled insights into temporal dynamics of SCFA concentrations and bacteria harboring pathways for their formation. We observed a high volatility of butyrate concentrations displaying 41.2% ± 22.8% average difference between the first time-point and all other time-points (Figure 6a). Fluctuations in its relative concentration were much less (20.7% ± 15.7%) (Figure 6b), which was in accordance with relative abundances of ACoA pathway carriers that were rather constant varying on average by only 15.1% ± 11.6% (Figure 6c). Propionate pathway carriers showed higher dynamics for both the Suc (22.3% ± 16.6%) and the Pdiol (23.5% ± 17.7%) pathway (Figure 6c). Measured concentrations of propionate were, however, less volatile (31.2% ± 19.3%) as those of butyrate, as were relative propionate concentrations (17.7% ± 13.4%) (Figure 6a, b). Acetate showed highest temporal stability for both absolute (29.5% ± 19.5%) and relative concentrations (8.0% ± 7.4%).
Figure 6.
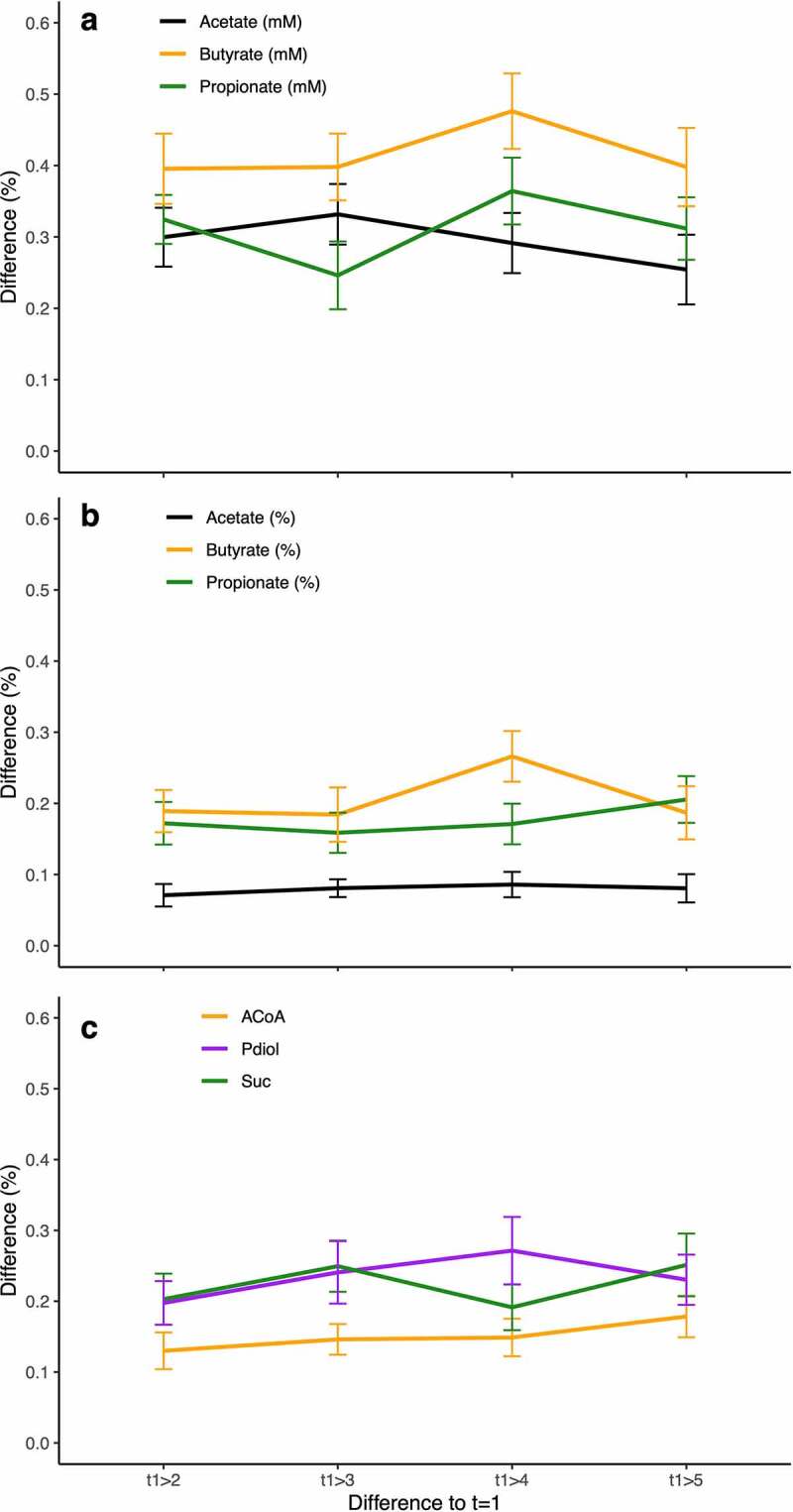
Temporal stability of SCFA concentrations and pathway abundances in vivo. Subjects (n = 20) were sampled (n = 5) over a period of 3 months and results relative to the first time-point are shown. Panel a displays relative concentration changes of the SCFAs acetate, butyrate and propionate, whereas variations based on proportions (relative to total SCFA concentrations) and of abundances of bacteria exhibiting individual pathways are given in panels b and c, respectively.
Discussion
The aim of this study was to gain quantitative insights into functional communities that form butyrate and propionate, and to predict the production of those SCFAs based on sequence data derived from both metagenomes and the 16S rRNA gene. Several criteria have to be fulfilled in order to achieve those goals: 1) accurate databases of gut bacteria carrying SCFA synthesis pathways are required, 2) pathways for production of the two SCFAs should not co-occur on genomes, 3) for predictions based on 16S rRNA gene data, pathways have to be distributed following phylogenetic patterns, 4) yields, i.e., amount of SCFAs produced per cell harboring respective pathways, should be equal between taxa and 5) should not be governed by environmental factors (e.g. type of growth substrate).
We comprehensively screened for pathways in isolates and MAGs that were specifically derived from the gut environment provided by the UHGG. A manually curated database for butyrate-producing bacteria was already established previously,17 but no systematic genome screening for propionate-forming pathways has been performed so far. On a genome level, our results do largely agree with KEGG, representing one of the most widely used public database; however, several crucial discrepancies were revealed. Most importantly, many Enterobacteriaceae were wrongly predicted to have the ACoA pathway by KEGG. Furthermore, Akkermansia and several Veillonella, which are known propionate-producers,15,25 were suggested to lack this function by KEGG. It should be noted that no information on terminal enzymes of the ACoA pathway, namely butyryl-CoA:acetate CoA transferase (but) and butyrate kinase (buk), is available within KEGG. Specifically for the ACoA pathway, it is important to consider pathway completeness, because a multitude of mis-annotations based on gene-homology alone are present.14,26 Those points highlight the use of manually curated databases for specific functions of interest.
The largely coherent distribution of the ACoA and Suc pathway on the genus level suggests strong selection for those features and verifies the view that both pathways serve as core fermentative routes in respective bacteria.15,17 We have previously demonstrated that taxonomy-based approaches (on the genus level) for predicting ACoA pathway abundances are valuable.17 Nevertheless, our analyses here suggested that within a few genera, such as Blautia, this metabolic route is not homogenously present. However, it still follows phylogenetic clustering making phylogenetic-based predictions superior over plain taxonomic-based analyses, as suggested earlier.27,28 This is also true in the case of propionate, where the Pdiol pathway displayed phylogenetic clustering within certain genera, where not all members exhibited the pathway (e.g. Mediterraneibacter, Eisenbergiella).
Our data revealed a profound separation between bacteria forming either butyrate or propionate. While this verifies observations based on metabolic profiles of major gut bacterial taxa, comprehensive community-wide analyses based on metagenomic data incorporating detailed, manually curated pathway annotations have not been performed so far on this topic. In vivo results demonstrated that a tiny part (2.38%) of bacteria cannot be unequivocally assigned either group. Bacteria harboring the genetic make-up for both functions can switch their metabolism according to the prevailing environmental conditions, also known as metabolic flexibility, hindering predictions on SCFA profiles on the DNA level. For instance, R. inulinivorans forms propionate during growth on fucose due to substrate-induced gene expression instead of butyrate, which is the primary SCFA formed when growing on glucose.29 Protein-fed butyrate production routes have not been considered here, as they are not vital for growth of individual bacterial carriers and abundances of those pathways on the DNA level are, hence, not directly coupled to activity, i.e. production of butyrate.17 Furthermore, proteins are believed to play only minor roles as growth substrates in the large bowel and their conversion to butyrate can also be catalyzed by the ACoA pathway, making this pathway the primary route for butyrate formation from proteins in the colonic environment.13 In summary, the first three criteria mentioned above are principally fulfilled and form the basis for predicting butyrate- and propionate-forming communities along with metabolite concentrations from sequencing data.
An additional crucial aspect for predicting SCFA production is functional redundancy, where pathway activity has to be constant between taxa and environmental conditions (e.g. growth substrates). Our in vitro experiments illustrated that yields were in a similar range, regardless of the type of substrate supplied and independent of individual’s community composition. In other words, the amounts of butyrate and propionate produced per grown bacterium harboring a pathway were similar. Those results are in line with previous observations.30 However, exact values on yields of whole functional communities have not been reported so far, as their determination requires adequate methodologies for enumerating pathway-carrying bacteria that was achieved here by coupling enumeration of bacterial cells by flow cytometry with metagenomics analyses on pathway abundances. It should be noted that the term yield does not refer to absolute final cell growth. We did observe substantial differences between substrates in terms of final SCFA concentrations/compositions and abundances of functional communities. For instance, the resistant starches were confirmed to promote formation of butyrate (and bacteria containing the ACoA pathway),31,32 which was also the case for inulin.33 On the other hand, mucin rather promoted formation of propionate, which is in accordance with major mucin-degrading taxa, such as specific Bacteroides, exhibiting the Suc pathway.34 For gaining such insights, the experimental set-up has to be designed in order to assure growth, i.e., multiplication of bacteria on the supplied substrates. In our experiments, we specifically diluted starting communities to provide growth over two orders of magnitudes (from ~107 mL−1 to ~109 mL−1). Often, communities are incubated at high cell concentrations with relatively little amount of substrates, which works well for assessing production capabilities for individual SCFAs,35 but hampers accurate calculations of yields and determination of bacterial taxa involved.
Largely complying with all five requirements introduced above, our fecal incubation experiments have demonstrated that in vitro it is indeed possible to calculate the absolute production of butyrate, and its proportion of the total SCFA pool, based on enumerating bacteria that exhibit the ACoA pathway. For propionate, predictions on absolute concentrations were possible, however, with less accuracy, and the fraction of propionate from total SCFAs was not explainable based on pathway abundances in vitro (in vivo this was, however, possible). We do attribute this observation rather to physiology of those bacteria than to inaccurate pathway callings. For instance, the Pdiol pathway is considered to be a major route of metabolic cross-feeding taking lactate produced by other bacteria as input,15 which is not essential for growth of those bacteria uncoupling its abundance from metabolite concentration. For instance, B. obeum can grow on sugars without producing any propionate,36 and specific induction of genes from this pathway was shown to depend on the growth substrate, as discussed above.29 Nevertheless, calculating propionate concentrations from the Suc pathway abundance alone was less effective and we, hence, used cumulative pathway abundances in our analyses. For bacteria exhibiting the Suc pathway, physiological adaptations were reported as well, where rather Suc and acetate than propionate are produced under certain conditions.36 The former compound is an intermediate and does usually not accumulate in the SCFA pool of gut communities.13,30
In vivo, relative abundances of the ACoA pathway and of the two propionate-forming routes did correlate with relative concentrations (proportions) of the two SCFAs, demonstrating that functional communities are reflected in SCFA composition. Against our expectations, total SCFA concentration did not correlate with absolute abundances of bacteria, nor did we observe associations between absolute abundances of pathways and corresponding SCFA concentrations. Results suggest that bacterial concentration is merely governed by stool firmness that is directly connected to retention time.37 The longer the colonic transit time of fecal matter, the more moisture is absorbed, resulting in a higher dry weight and higher bacterial cell concentrations per gram of stool. Water, ions and SCFAs are absorbed alike decoupling bacterial concentrations from SCFA concentrations.38 Furthermore, it can be assumed that conditions comprising less water content caused by slower transit provide challenging environments for bacterial growth, which is mirrored by the negative correlation observed between microbial activity and stool firmness, further decoupling SCFA concentrations from bacterial cell numbers. Community structure39 and functionality40 was previously associated with stool consistency, where longer transit (firmness) was negatively associated with ACoA pathway abundance and an enrichment for propionate production. According to our calculations based on in vitro yields, the amounts of SCFAs absorbed by the host are 97.7% ± 2.2% and 94.5% ± 5.4% for butyrate and propionate, respectively, which is even higher than previously suspected11,12 and stresses the distinction between SCFA production (defined as the total amount of SCFAs formed in a defined period of time) and SCFA concentrations. The latter can be regarded as a snapshot parameter, which is highly influenced by a series of factors irrespective of the source, i.e., bacterial concentrations, which was also reflected in high temporal variabilities of SCFA concentrations. On the other hand, proportions of individual SCFAs were rather constant, as were corresponding pathway abundances, and variances of neither parameters correlated with time intervals between sampling points, indicating that individual’s SCFA pattern, and corresponding functional communities, respectively, are fairly stable over time.
In conclusion, we give detailed insights into butyrate- and propionate-producing bacteria on a community-wide level demonstrating that they form two taxonomically distinct groups in gut microbiota, whose abundances determine SCFA composition in vivo. Overall, it was possible to predict relative metabolite concentrations from bacteria carrying respective pathways to a certain extent, demonstrating that altering the community structure is a valuable strategy to promote production of those specific SCFAs. The successful use of the 16S rRNA gene for function prediction provides a high-throughput, low-cost screening alternative over more tedious metagenomic analyses, which facilitates investigations on SCFA-forming communities in broad-scale applications.
Materials and methods
In vitro incubation experiments
Stool from eight healthy subjects (5 females/3 males), who were also participating in the in vivo studywas collected at the institute and immediately transferred to a vinyl anaerobic chamber (Coy Laboratory Products, Grass Lake, MI, USA; fed by N2 and an anaerobic gas-mixture consisting of 10% CO2, 10% H2 and 80% N2) for experiments. Samples were diluted (1:100) in pre-reduced 1× PBS, subjected to 30 µm filtration (Miltenyi Biotec, Bergisch Gladbach, Germany) and added to anaerobic basal medium (described in the study by Reichardt et al.30 with modifications (see supplemental information), ph = 6.8) to achieve a starting concentration of ~1−3 × 107 cells mL−1 (an aliquot for enumerating cell concentrations by flow cytometry was diluted fivefold in 1× PBS, snap frozen in liquid nitrogen and stored at −20°C). Suspensions were aliquoted (10 ml) into Hungate tubes and 1 ml of individual growth substrates was added (final concentration of 2 g L−1). The following growth substrates were used: resistant starch types 2 and 3 (Hylon VII (PCR) and Novelose330; both from Ingredion, Manchester, UK), pectin from apple (Sigma Aldrich, St. Louis, MO, USA), mucin (Sigma Aldrich, St. Louis, MO, USA), inulin (Orafti HP; from Beneo-Orafti, Oreye, Belgium) and protein (Bacto Casitone, BD, Franklin Lakes, NJ, USA); a negative control (stool in basal medium) was included as well. Substrates were boiled for 5 min in a microwave and pre-reduced overnight under the anaerobic chamber before being used in the experiments. Incubations were carried out in duplicate samples at 37°C for 24 h (200 rpm). The pH was determined using a pH-Meter (Knick International, Berlin, Germany) with an Inlab semi-micro electrode (Mettler Toledo, Columbus, OH, USA). Two milliliters of cultures was centrifuged (15,400 g, 4°C), diluted in NaOH (5 mM) and stored at −80°C before determination of SCFA concentrations; the pellet was used for DNA extraction. Bacterial concentrations were determined by flow cytometry (see below).
Monitoring of gut microbiota in vivo
Twenty volunteers (11 females/9 males) provided five fresh stool samples over a period of 3 months; three samples were collected in November/December 2019 (2 weeks interval), whereas another two samples (2 weeks interval) were collected in January 2020. Approximately 2 g stool was collected into feces collection tubes (Sarstedt, Nümbrecht, Germany) at the institute, put at 4°C and processed within 30 min; samples of three subjects were collected at home and immediately transported (cooled) to the institute within 15 min. Samples were diluted fivefold in 1× PBS; undiluted aliquots (~200 mg) for DNA extraction were snap frozen in liquid nitrogen and stored at −80°C until further analysis. For flow cytometric analyses, an aliquot of 100 µl from the dilution was snap frozen, whereas for the determination of SCFA concentrations, 20 μl was added to 980 μl NaOH (5 mM), centrifuged (5 min, 4,500 g, 4°C) and 100 μl of the supernatant was collected in gas chromatography (GC) glass vials (Macherey-Nagel, Düren, Germany); both were stored at −80°C. Fecal pH was directly measured in stool suspensions (fivefold dilution in distilled water). The Bristol Stool Scale (BSS) was recorded by individual donors themselves and determinations of dry weight was performed by weighing aliquots of approximately 200 mg stool before and after drying via SpeedVac RVC 2-18 CD plus (Martin Christ Gefriertrocknungsanlagen, Osterode am Harz, Germany), at 37°C (1,300 rpm for 4 h).
Flow-cytometric analyses and determination of SCFA concentrations
For flow-cytometric measurements (FCM), the fivefold dilutions of stool samples (in vivo experiments) were thawed at room temperature and diluted 100× with 1× PBS, including a 30 µm filtration step (Miltenyi Biotec, Bergisch Gladbach, Germany). For in vitro samples, 1:500 dilutions (1× PBS) were directly prepared from growth cultures; samples taken at the beginning of the experiment were thawed. All suspensions were stained with 10 µl EDTA and 10 µl SYBR Green (Thermo Fisher Scientific, USA) according to Hammes et al.41 and incubated for 15 min at 37°C in the dark. Before measurements, stained samples were diluted 10-fold in 1× PBS and cell concentrations as well as green fluorescence intensities were recorded on a MACSQuant Analyzer 10 (Miltenyi Biotec, Germany).
Concentrations of acetate, butyrate and propionate of fecal samples (in vivo experiments) and in vitro incubations were quantified at the RCU Metabolomics of Hannover Medical School based on a GC-MS method including a derivatization step and addition of a labeled standard (see Supplemental Methods).
DNA extraction, library preparation and sequencing
DNA was extracted (DNeasy PowerSoil Pro Kit, Qiagen, Germany; including a beat-beating step (2× 20 sec on Fastprep System (MP Biomedicals, Santa Ana, CA, USA) at speed 5.5)) and libraries for shotgun-sequencing were prepared (Illumina DNA Prep, Illumina, USA) that were subsequently sequenced on Illumina NovaSeq 6000 (at Helmholtz Center for Infection Research (HZI)) in paired-end mode (2 × 150 bp). For in vivo experiments, 2 × 107 reads per sample were sequenced (for the first and last samples 5 × 107 (2 × 250 bp) were obtained), whereas shallower sequencing (5 × 106 reads) were performed for in vitro samples. Libraries for 16S rRNA gene sequencing were prepared according to Rath et al.,42 but targeting the V3V4 region using primers from43 with an annealing temperature of 55°C. Obtained amplicons were pooled and sequenced on Illumina MiSeq (2 × 300 bp).
Constructing the catalog of SCFA pathway genes
All representative genomes from the UHGG collection that displayed decent quality (completeness >80 and contamination <10) were included into analysis (n = 3,207). To increase diversity, high-quality isolates (completeness >95%, contamination <2% and a 16S rRNA gene length >70%) that showed an average nucleotide identity (ANI, determined via fastANI (v1.32)44) below 98% to the representative genome (and to each other) were included as well (n = 522). A few were manually selected (n = 25). Genomes were downloaded and gene sequences were extracted with GffRead.45 UBCG (v3.0) was used to construct a phylogenetic tree based on 92 housekeeping genes (HKGs)46 and genomes were screened for SCFA pathways. For butyrate, the same approach as described previously was used17 consisting of a multi-level approach involving Hidden Markov Models (HMM) for all genes of the ACoA pathway and analyses on gene-synteny and pathway completeness. For propionate, a similar multilevel-screening approach for the Suc and Pdiol pathways based on key genes defined by Reichardt et al.15 was developed. Details are given in the Supplemental Methods. All results of pathway screenings were manually checked and a few genomes, which were filtered-out due to fragmented pathway genes, were included. For annotations based on KEGG, all genomes were subjected to GhostKOALA47 and subsequent filtering based on key genes of individual pathways (for ACoA, only bhbd, cro and bcd were considered) was performed including manual inspections (e.g. lacking of single genes). For 16S rRNA gene analyses, 1,623 genomes that had decent length genes (>900 bp) were included. The longest gene from each genome was aligned in Clustal Omega (web server), and a phylogenetic tree was constructed via FastTree 2 (v2.1.10).48 Manual inspections led to removal of 24 sequences as they clustered with the wrong phylum. Duplicates were removed, and finally 1,556 16S rRNA gene sequences were used for follow-up analyses.
Metagenomic analyses
The metaWRAP pipeline (v1.3.0) was used for genome-resolved metagenomic analyses.49 Raw sequences of in vivo samples were quality filtered, assembled via MEGAHIT and binned via a combination of MaxBin 2, MetaBAT 2 and CONCOCT using the BIN_REFINEMENT module; for assembly, all samples of a person were merged, whereas they were treated separately during the binning process. Finally, bins were reassembled (REASSEMBLE_BINs module) yielding MAGs. Taxonomic annotations of MAGs were done via the GTDB-Tk (1.7.0),50 HKGs were extracted via UBCG and SCFA-forming pathways were detected as described above. GRiD (v1.3.0) was used to infer growth rates by calculating coverage ratios between ori and ter;51 and a cumulative value for each sample was calculated (average of all MAGs normalized for their relative abundances).
For determining SCFA pathway abundances, HKGs and pathway genes from UHGG references and from constructed MAGs were used as a catalog for mapping reads via BBmap2 (non-target pathway genes showing sequence similarity, but displayed HMM scores below the set cutoffs, were included as well, as described previously17). Resulting counts were gene-length corrected and normalized to HKGs (mean abundance of all HKGs) yielding relative abundances of genes from respective pathways; mean results of pathway genes were used in follow-up analyses as done previously.17 Taxonomic affiliations of individual pathways were determined on the species level, where a taxon was recorded if at least three genes of a pathway were detected (two genes for the Pdiol pathway), which included 95.5% ± 2.7% (in vivo) and 91.3% ± 8.3% (in vitro) of all reads mapped to pathway genes. Results of taxonomies were subsequently merged for insights at higher orders. For overall taxonomic compositions, HKGs were used, where a species was considered present if at least 20 HKGs were detected (96.4% ± 1.0% (in vivo) and 92.5% ± 2.5% (in vitro) of all reads that mapped to HKGs were included).
16S rRNA gene analyses
Sequences were processed via the DADA2 pipeline (v1.20) in default mode and annotated based on RDP’s taxonomy.52 Chimera were removed and only sequences displaying a length >300bp, counts >10 that were annotated at the phylum level were included (sequences derived from Chloroplasts were excluded). Samples were rarefied to equal depths of 4,247 and 22,777 counts for in vitro and in vivo experiments, respectively. SCFA pathway predictions were done via the picrust2 algorithm (v2.3.0b)53 by placing sequences into our reference tree (place_seqs.py) followed by hidden-state predictions (hsp.py).
Statistics and generation of plots
Growth of SCFA pathway exhibiting bacteria was calculated from final cell concentrations determined by FCM and metagenomic results that provided relative abundances of bacteria carrying individual pathways. All plots were constructed in R via ggplot2 (v3.3.5) and ggtree (v1.14.6). Correlations for in vitro results were calculated (function lm) from original and log-transformed data (log(data+1)). Non-metric multidimensional scaling (NMDS) analyses were done in phyloseq (v1.36.0) on relative abundance data of taxa on the species level. Correlations of in vivo parameters from all subjects at all time points were determined via linear mixed-effects models using the function lmer from the lme4 package (v1.1–27.1) on log-transformed data (log(data+1)) including subject as a random effect.
Supplementary Material
Acknowledgments
We greatly thank all our participants contributing to the in vivo experiment. Furthermore, thanks to Maren Scharfe and Michael Jarek from the HZI for their technical assistance and to Colin Davenport and Jannes Gless for maintaining the HPCSeq at MHH.
Funding Statement
This work was funded by the DFG (project #456214861) and intramural funds (HiLF II of MHH).
Disclosure statement
The authors report no conflict of interest.
Data availability statement
All raw data are available at the European Nucleotide Archive (PRJEB51501). Reference pathway genes from the UHGG along with files needed for predicting pathways based on 16S rRNA gene data are available at https://github.com/ag-vital/predict_SCFA_producers
Consent
The study was approved by local ethic authorities (#8566_BO_K_2019) and all subjects have given informed consent.
Authors’ contributions
M.V. and B.K. conceived the study, B.K., S.W., F.G., H.B., R.G. performed experiments, M.V. and B.K. analyzed data, B.K. and M.V. drafted the manuscript, all authors commented on the text.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19490976.2022.2149019
References
- 1.Blacher E, Levy M, Tatirovsky E, Elinav E.. Microbiome-modulated metabolites at the interface of host immunity. J Immunol. 2017;198:572–17. doi: 10.4049/jimmunol.1601247. [DOI] [PubMed] [Google Scholar]
- 2.Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN, Hermoso MA. Short chain fatty acids (SCFAs)-Mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota–gut–brain communication. Nat Rev Gastroenterol Hepatol. 2019;16:461–478. [DOI] [PubMed] [Google Scholar]
- 4.Cani PD. Human gut microbiome: hopes, threats and promises. Gut. 2018;67:1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorbara MT, Pamer EG. Interbacterial mechanisms of colonization resistance and the strategies pathogens use to overcome them. Mucosal Immunol. 2019;12:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Osbelt L, Thiemann S, Smit N, Lesker TR, Schröter M, Gálvez EJC, Schmidt-Hohagen K, Pils MC, Mühlen S, Dersch P, et al. Variations in microbiota composition of laboratory mice influence citrobacter rodentium infection via variable short-chain fatty acid production. PLoS Pathog. 2020;16:e1008448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.den Besten G, Lange K, Havinga R, van Dijk TH, Gerding A, van Eunen K, Müller M, Groen AK, Hooiveld GJ, Bakker BM, et al. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. Am J Physiol Gastrointest Liver Physiol. 2013;305:G900–10. [DOI] [PubMed] [Google Scholar]
- 8.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic treg cell homeostasis. Science. 2013;341:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Theiler A, Bärnthaler T, Platzer W, Richtig G, Peinhaupt M, Rittchen S, Kargl J, Ulven T, Marsh LM, Marsche G, et al. Butyrate ameliorates allergic airway inflammation by limiting eosinophil trafficking and survival. J Allergy Clin Immunol. 2019;144:764–776. [DOI] [PubMed] [Google Scholar]
- 10.Yu L, Zhong X, He Y, Shi Y. Butyrate, but not propionate, reverses maternal diet-induced neurocognitive deficits in offspring. Pharmacol Res. 2020;160:105082. doi: 10.1016/j.phrs.2020.105082. [DOI] [PubMed] [Google Scholar]
- 11.Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81:1031–1064. doi: 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- 12.McNeil NI, Cummings JH, James WPT. Short chain fatty acid absorption by the human large intestine. Gut. 1978;19:819–822. doi: 10.1136/gut.19.9.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41. doi: 10.1111/1462-2920.13589. [DOI] [PubMed] [Google Scholar]
- 14.Vital M, Howe A, Tiedje J. Revealing the bacterial butyrate synthesis pathways by analyzing (meta) genomic data. MBio. 2014;5:e00889–14. doi: 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deehan EC, Yang C, Perez-Muñoz ME, Nguyen NK, Cheng CC, Triador L, Zhang Z, Bakal JA, Walter J. Precision microbiome modulation with discrete dietary fiber structures directs short-chain fatty acid production. Cell Host Microbe. 2020;27:389–404.e6. [DOI] [PubMed] [Google Scholar]
- 17.Vital M, Karch A, Pieper DH. Colonic butyrate-producing communities in humans: an overview using omics data. mSystems. 2017;2:e00130–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandeputte D, De Commer L, Tito RY, Kathagen G, Sabino J, Vermeire S, Faust K, Raes J. Temporal variability in quantitative human gut microbiome profiles and implications for clinical research. Nat Commun. 2021;12:6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, Pollard KS, Sakharova E, Parks DH, Hugenholtz P, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. 2021;39:105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Louis P, Duncan SH, McCrae SI, Millar J, Jackson MS, Flint HJ. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J Bacteriol. 2004;186:2099–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gossling J, Moore WEC. Gemmiger formicilis, n.gen., n.sp., an anaerobic budding bacterium from intestines. Int J Syst Evol Microbiol. 1975;25:202–207. [Google Scholar]
- 22.Le Roy T, Van Der Smissen P, Paquot A, Delzenne N, Muccioli GG, Collet J, Cani PD. Dysosmobacter welbionis gen. nov., sp. nov., isolated from human faeces and emended description of the genus Oscillibacter. Int J Syst Evol Microbiol. 2020;70:4851–4858. [DOI] [PubMed] [Google Scholar]
- 23.Popoff M, Guillou J-P, Carlier J-P. Taxonomic position of lecithinase-negative strains of Clostridium sordellii. J Gen Microbiol. 1989;131:1697–1703. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Xu X, Zhou N, Sun Y, Liu C, Liu S, You X. Parabacteroides acidifaciens sp . nov ., isolated from human faeces. Int J Syst Evol Microbiol. 2019;69:761–766. [DOI] [PubMed] [Google Scholar]
- 25.Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia municiphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–1476. [DOI] [PubMed] [Google Scholar]
- 26.Anand S, Kuntal BK, Mohapatra A, Bhatt V, Mande SS. FunGeCo: a web-based tool for estimation of functional potential of bacterial genomes and microbiomes using gene context information. Bioinformatics. 2020;36:2575–2577. [DOI] [PubMed] [Google Scholar]
- 27.Louis P, Flint HJ. Development of a semiquantitative degenerate real-time PCR-based assay for estimation of numbers of butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Appl Environ Microbiol. 2007;73:2009–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vital M, Penton CR, Wang Q, Young VB, Antonopoulos DA, Sogin ML, Morrison HG, Raffals L, Chang EB, Huffnagle GB, et al. A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome. 2013;1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott KP, Martin JC, Campbell G, Mayer C-D, Flint HJ. Whole-genome transcription profiling reveals genes up-regulated by growth on fucose in the human gut bacterium “Roseburia inulinivorans”. J Bacteriol. 2006;188:4340–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reichardt N, Vollmer M, Holtrop G, Farquharson FM, Wefers D, Bunzel M, Duncan SH, Drew JE, Williams LM, Milligan G, et al. Specific substrate-driven changes in human faecal microbiota composition contrast with functional redundancy in short-chain fatty acid production. ISME J. 2018;12:610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venkataraman A, Sieber JR, Schmidt AW, Waldron C, Theis KR, Schmidt TM. Variable responses of human microbiomes to dietary supplementation with resistant starch. Microbiome. 2016;4:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scott KP, Martin JC, Duncan SH, Flint HJ. Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria, in vitro. FEMS Microbiol Ecol. 2014;87:30–40. [DOI] [PubMed] [Google Scholar]
- 34.Tailford LE, Crost EH, Kavanaugh D, Juge N. Mucin glycan foraging in the human gut microbiome. Front Genet. 2015;6:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin M, Kalainy S, Baskota N, Chiang D, Deehan EC, McDougall C, Tandon P, Martínez I, Cervera C, Walter J, et al. Faecal microbiota from patients with cirrhosis has a low capacity to ferment non-digestible carbohydrates into short-chain fatty acids. Liver Int. 2019;39:1437–1447. [DOI] [PubMed] [Google Scholar]
- 36.Lawson PA, Finegold SM. Reclassification of ruminococcus obeum as blautia obeum comb. nov. Int J Syst Evol Microbiol. 2015;65:789–793. [DOI] [PubMed] [Google Scholar]
- 37.Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924. [DOI] [PubMed] [Google Scholar]
- 38.Lewis SJ, Heaton KW. Increasing butyrate concentration in the distal colon by accelerating intestinal transit. Gut. 1997;41:245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roager HM, Hansen LBS, Bahl MI, Frandsen HL, Carvalho V, Gøbel RJ, Dalgaard MD, Plichta DR, Sparholt MH, Vestergaard H, et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat Microbiol. 2016;1:16093. [DOI] [PubMed] [Google Scholar]
- 41.Hammes F, Berney M, Wang Y, Vital M, Köster O, Egli T. Flow-cytometric total bacterial cell counts as a descriptive microbiological parameter for drinking water treatment processes. Water Res. 2008;42:269–277. [DOI] [PubMed] [Google Scholar]
- 42.Rath S, Heidrich B, Pieper DH, Vital M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome. 2017;5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. Development of a prokaryotic universal primer for simultaneous analysis of bacteria and archaea using next-generation sequencing. PLoS One. 2014;9:e105592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pertea G, Pertea M. GFF utilities: gffRead and GffCompare. F1000Research. 2020;9:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Na SI, Kim YO, Yoon SH, Ha S, Baek I, Chun J. UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J Microbiol. 2018;56:281–285. [DOI] [PubMed] [Google Scholar]
- 47.Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428:726–731. [DOI] [PubMed] [Google Scholar]
- 48.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2019;36:1925–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emiola A, Oh J. High throughput in situ metagenomic measurement of bacterial replication at ultra-low sequencing coverage. Nat Commun. 2018;9:4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MGI. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available at the European Nucleotide Archive (PRJEB51501). Reference pathway genes from the UHGG along with files needed for predicting pathways based on 16S rRNA gene data are available at https://github.com/ag-vital/predict_SCFA_producers