

Abstract
Rationale
Sarcoidosis is a multisystemic inflammatory disease characterized by the formation of granulomas in response to persistent stimuli. The long pentraxin PTX3 (pentraxin 3) has emerged as a component of humoral innate immunity with essential functions in the resolution of inflammation, but its role during granuloma formation is unknown.
Objectives
To evaluate PTX3 as a modulator of pathogenic signals involved in granuloma formation and inflammation in sarcoidosis.
Methods
Peripheral blood mononuclear cells obtained from patients with sarcoidosis harboring loss-of-function genetic variants and gene-deleted mice were used to assess the role of PTX3 in experimental models of granuloma formation in vitro and in vivo. The identified mechanisms of granulomatous inflammation were further evaluated in tissue and BAL samples and correlated with the disease course.
Measurements and Main Results
We have identified a molecular link between PTX3 deficiency and the pathogenic amplification of complement activation to promote granuloma formation. Mechanistically, PTX3 deficiency licensed the complement component C5a-mediated activation of the metabolic checkpoint kinase mTORC1 (mammalian target of rapamycin complex 1) and the reprogramming of macrophages toward increased glycolysis to foster their proliferation and aggregation. This process sustained the further recruitment of granuloma-promoting immune cells and the associated proinflammatory microenvironment and influenced the clinical course of the disease.
Conclusions
Our results identify PTX3 as a pivotal molecule that regulates complement-mediated signaling cues in macrophages to restrain granulomatous inflammation and highlight the therapeutic potential of this signaling axis in targeting granuloma formation in sarcoidosis.
Keywords: sarcoidosis, humoral immunity, granuloma, macrophages, complement system proteins
At a Glance Commentary
Scientific Knowledge on the Subject
Sarcoidosis is a multisystemic inflammatory disease characterized by the formation of epithelioid granulomas in affected organs, predominantly the lungs. The granulomatous response involves a stepwise program in which a series of macrophage transformations recruits additional cells to produce structural modifications. The relevance of humoral innate immunity, particularly complement activation, in sarcoidosis has been recently suggested. However, the mechanistic bases linking the functional activity of humoral mediators and the pathogenesis of sarcoidosis remain unknown.
What This Study Adds to the Field
We report that the long pentraxin PTX3 (pentraxin 3), a fluid-phase pattern recognition molecule that exerts essential roles in innate immunity, is a major regulator of granulomatous inflammation. PTX3 acts as a physiological break of granuloma formation by restraining the pathogenic activation of complement and the downstream metabolic reprogramming of macrophages required to sustain their functional activity and proliferation, leading to further recruitment of granuloma-promoting immune cells. This study unveils the missing link in the connection between humoral innate immunity and the regulation of granulomatous inflammation while suggesting complement inhibition as a therapeutic strategy in sarcoidosis.
Sarcoidosis is a multisystemic granulomatous disease of unknown etiology that can lead to chronic debilitating disease (1, 2). The pathological hallmark of sarcoidosis is the formation of epithelioid, nonnecrotizing granulomas in involved organs, predominantly the lungs, characterized by activated monocytes/macrophages and highly polarized Th1 (T helper 1) and Th17 cells consistent with persistent antigen stimulation, and the production of immunoregulatory cytokines (3). Despite the clinical significance of granulomas, the molecular and cellular events promoting macrophage aggregation and transformation into epithelioid cells that initiate and maintain these structures remain elusive.
The humoral arm of innate immunity includes several molecules, such as complement components, collectins, ficolins, and pentraxins (4). The long pentraxin PTX3 (pentraxin 3) is a fluid-phase pattern recognition molecule with ancestral antibody-like properties that exerts essential roles in resistance to pathogens (5, 6), tissue remodeling (7), and regulation of inflammation (8, 9). In particular, PTX3 fine-tunes inflammatory responses by interacting with endothelial P-selectin to control leukocyte recruitment (9) and by regulating the activation of the complement cascade by interacting with C1q, the recognition unit of the classical pathway (10), and components of the lectin and alternative pathways (11, 12).
Proteome analysis of BAL-derived exosomes from patients with sarcoidosis revealed significant enrichment in complement-activating factors, including complement factors B, C1q, and C3 (13), and increased alveolar concentrations of activated C5a were detected in sarcoidosis, but not other interstitial lung diseases (14). Genetic variation in complement receptor 1 has also been proposed as a susceptibility factor for sarcoidosis (15), although these findings lack replication. Nevertheless, monocytes from patients display an increased expression of complement receptors and an enhanced phagocytic activity which could, at least partly, explain the persistent antigenemia in sarcoidosis (16).
Although the importance of humoral innate immunity in sarcoidosis has been suggested, no study to date has determined the mechanistic bases linking the functional activity of humoral mediators and disease pathogenesis. Herein, we report that PTX3 is an essential regulator of granulomatous inflammation that acts by inhibiting the complement-mediated metabolic reprogramming and proliferation of macrophages to restrain granuloma formation. These observations unveil the missing link in the connection between humoral innate immunity and the regulation of granulomatous inflammation in sarcoidosis.
Methods
The materials and methods used, including the mouse model of lung granulomatous inflammation and the in vitro human granuloma model, patient sample collection and analysis, immunohistochemistry and immunofluorescence, cytokine and metabolic measurements, flow cytometry, gene expression and single-cell RNA sequencing analysis, single-nucleotide polymorphism (SNP) genotyping, and statistical analysis, are reported in the online supplement.
Results
PTX3 Deficiency Enhances Lung Granulomatous Inflammation
We examined the susceptibility of Ptx3−/− mice to experimental granuloma formation using a peptide from the mycobacterial sodA protein (17) (Figure 1A). Histological analysis of lung sections revealed larger epithelioid granulomas localized to the broncho–arterial bundle in Ptx3−/− mice as early as Day 2 and until Day 4 after embolization (Figure 1B). On Day 4, the mean area of granulomas in Ptx3−/− and wild-type (WT) mice was 0.037 and 0.023 mm2, respectively (Figure 1C). No differences were found in the number of granulomas per area of lung section (Figure E1A in the online supplement), and no granulomas were detected in other organs (data not shown). The granuloma size also did not differ after challenge with peptide-naked beads (Figure E1B). Administration of rPTX3 (recombinant PTX3) using either a prophylactic or a therapeutic regimen rescued the phenotype of Ptx3−/− mice, with the mean granuloma area decreasing from 0.035 to 0.013 mm2 and from 0.033 to 0.019 mm2, respectively, whereas in both cases, it did not affect the lesion size in WT animals (Figures 1D and E1C).
Figure 1.
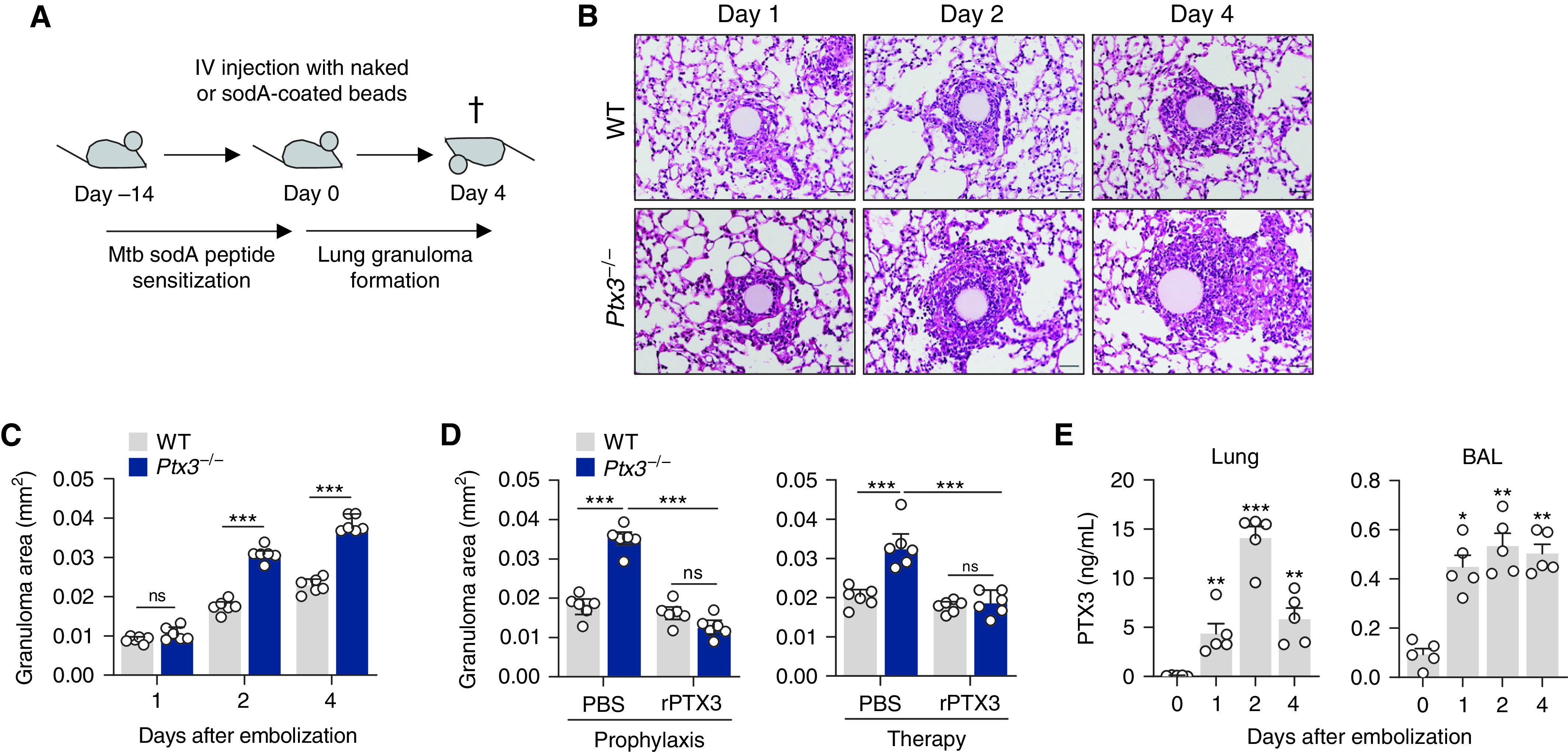
PTX3 (pentraxin 3) regulates lung granulomatous inflammation. (A) Experimental setup for lung granuloma formation in vivo using sodA peptide-coated Sepharose beads. (B) Hematoxylin and eosin-stained lung granulomas from wild-type (WT) and Ptx3−/− mice on Days 1, 2, and 4 after sodA bead embolization (representative of three independent experiments). Scale bars indicate 50 µm. (C) Area of sodA-elicited lung granulomas in WT and Ptx3−/− mice on Days 1, 2, and 4 after bead embolization (n = 6), and (D) on Day 4 after bead embolization and treatment with rPTX3 (recombinant PTX3) or phosphate-buffered saline (PBS) using a prophylactic (from Day −1) or therapeutic (from Day 1) scheme of administration (n = 6). (E) Concentrations of PTX3 in supernatants from lung cell suspensions and BAL from WT mice on Days 1, 2, and 4 after sodA bead embolization (n = 5). *P < 0.05, **P < 0.01, and ***P < 0.001. IV = intravenous; ns = not significant.
The expression of PTX3 was significantly and durably induced in the lungs and BAL from sodA-elicited WT animals (Figure 1E) relative to uncoated bead control subjects (Figure E1D). Analysis of PTX3 expression in the lung revealed its preferential localization to sites of sodA-elicited granulomas, with positive staining interspersed throughout the lesion and close to the bead nidus (Figure E1E).
PTX3 Regulates Immune Cell Dynamics During Granulomatous Inflammation
We next evaluated the influx of inflammatory cells in the BAL (Figure E2A). The number of total CD45+ leukocytes was higher in Ptx3−/− than in WT mice on Day 4 after embolization (Figure 2A). The increased leukocyte infiltration resulted primarily from an accumulation of alveolar macrophages, inflammatory monocytes, and CD4+ T cells, whereas the number of neutrophils and CD8+ T cells did not differ between backgrounds (Figure 2B). These findings were recapitulated in the lung, in which an expanded number of macrophages and CD4+ T cells, but also neutrophils, was detected (Figure E2B). Immunostaining of granulomas confirmed that macrophages were the predominant immune cell type at the innermost layers of granulomas from Ptx3−/− mice, whereas neutrophils and T cells were less abundant and preferentially localized to the outer regions (Figure 2C). Alveolar cells were located primarily outside of the granulomatous core and were present at similar frequencies in both genetic backgrounds (Figure 2D). Moreover, macrophages were confirmed as the relevant cell source of PTX3 since the adoptive transfer of WT, but not Ptx3−/−, macrophages decreased the areas of forming granulomas in Ptx3−/−-recipient mice to values similar to those in WT animals (Figure 2E). Phenotypic analysis of lung leukocytes revealed that alveolar macrophages from Ptx3−/− mice exhibited a higher rate of hypertrophy than WT cells (Figure 2F). Although interstitial macrophages from Ptx3−/− mice also displayed increased granularity and cell size, their relative percentage was lower than that of resident cells (Figure E2C). Treatment with rPTX3 reduced the numbers of alveolar macrophages in the BAL of Ptx3−/− mice, abolishing the differences between genotypes (Figure 2G). The expanded number of CD4+ T cells was also decreased after administration of rPTX3 (Figure E2D).
Figure 2.
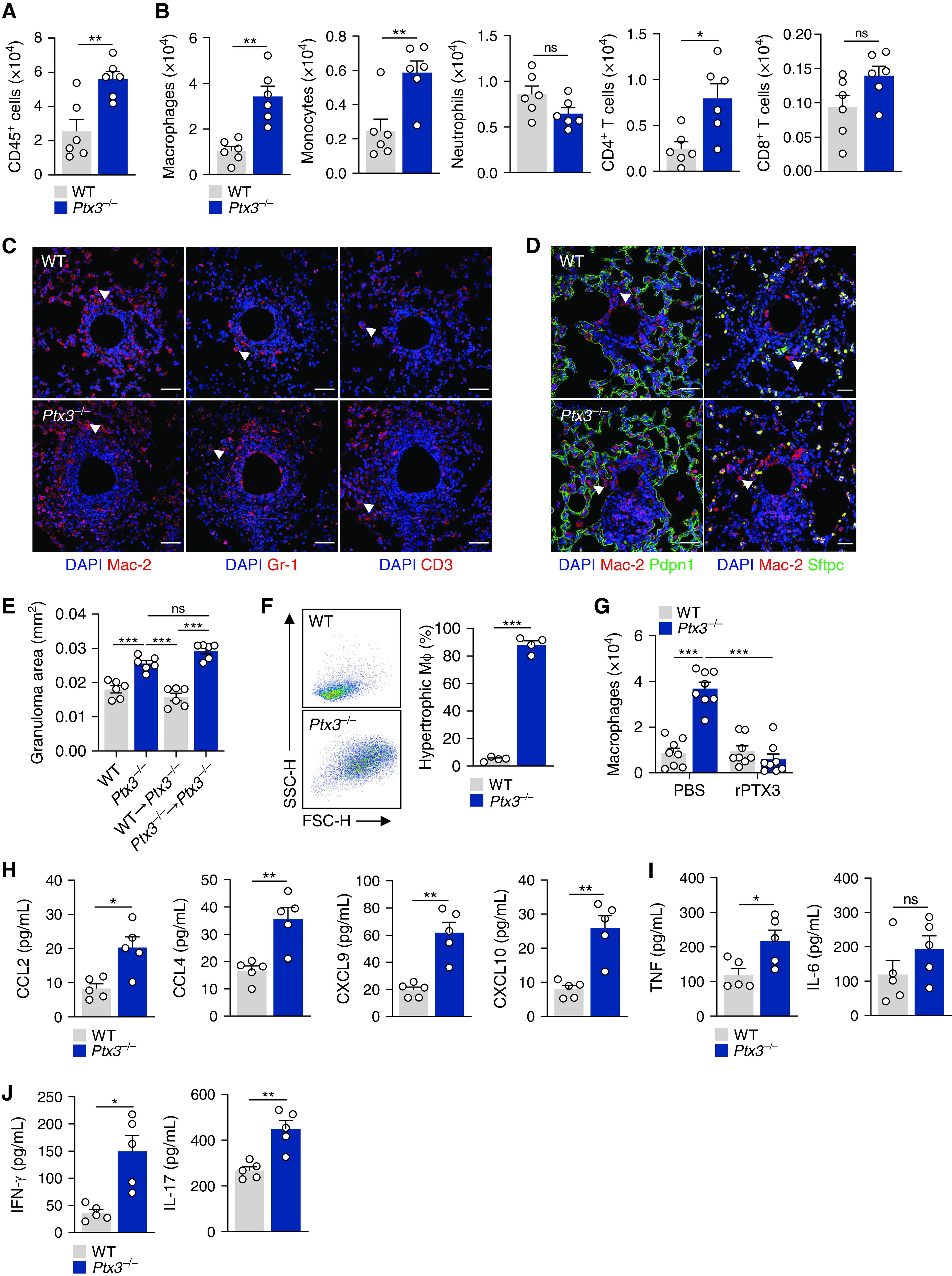
PTX3 (pentraxin 3) restrains leukocyte infiltration during granuloma formation. (A) The number of CD45+ leukocytes and (B) alveolar macrophages, inflammatory monocytes, neutrophils, CD4+ T cells, and CD8+ T cells in the BAL of wild-type (WT) and Ptx3−/− mice on Day 4 after sodA bead embolization (n = 6). (C) Immunofluorescence for macrophages (Mac-2), neutrophils (Gr-1), and T cells (CD3), and (D) type I (Pdpn1) and type II (Sftpc) alveolar cells in granulomas from WT and Ptx3−/− mice on Day 4 after sodA bead embolization (representative of three independent experiments). Nuclei were stained with DAPI. Scale bars indicate 50 µm. The arrows indicate examples of cells of interest. (E) Area of sodA-elicited granulomas in Ptx3−/−-recipient mice on Day 4 after bead embolization after the intratracheal adoptive transfer of WT or Ptx3−/− macrophages (n = 6). (F) Flow cytometry scatterplot and percentage of hypertrophic cells within total alveolar macrophages on Day 4 after sodA bead embolization (n = 4, representative of two independent experiments). (G) Number of alveolar macrophages in the BAL of WT and Ptx3−/− mice on Day 4 after sodA bead embolization and treatment with rPTX3 (recombinant PTX3) or PBS (n = 8). (H) Concentrations of CCL2, CCL4, CXCL9, and CXCL10 (n = 5), (I) TNF and IL-6 (n = 5), and (J) IFN-γ and IL-17 (n = 5) in lung supernatants from WT and Ptx3−/− mice on Day 4 after sodA bead embolization. *P < 0.05, **P < 0.01, and ***P < 0.001; FSC-H = forward scatter height; ns = not significant; PBS = phosphate-buffered saline; SSC-H = side scatter height.
The first steps toward granuloma formation are orchestrated by a complex network of chemokines and cytokines (18). Accordingly, we observed increased concentrations of the chemokines CCL2, CCL4, CXCL9, and CXCL10 (which are produced by and act on myeloid cells) in the lungs of Ptx3−/− mice on Day 4 after embolization (Figure 2H). The concentrations of cytokines, including TNF (Figure 2I), and T-cell–derived cytokines such as IFN-γ and IL-17, were also higher in Ptx3−/− mice (Figure 2J).
PTX3 Restrains Complement-mediated Inflammation During Granuloma Formation
PTX3 fine-tunes inflammatory responses by modulating P-selectin–dependent leukocyte recruitment and complement activation (8, 9). We first evaluated granuloma formation after treatment with recombinant anti–P-selectin (CD62P) or the corresponding isotype. In these conditions, PTX3 deficiency promoted the formation of larger granulomas regardless of P-selectin activity (Figures 3A and E3A), therefore discarding a major role for this adhesion molecule in this model.
Figure 3.

PTX3 (pentraxin 3) deficiency promotes complement activation during granuloma formation. (A) Area of sodA-elicited granulomas in wild-type (WT) and Ptx3−/− mice on Day 4 after bead embolization and treatment with anti-CD62P or IgG isotype control (n = 6). (B) Immunofluorescence for C3b/iC3b/C3c in granulomas from WT and Ptx3−/− mice on Day 4 after sodA bead embolization and treatment with rPTX3 (recombinant PTX3) or PBS (representative of three independent experiments). Nuclei were stained with DAPI. Scale bars indicate 50 µm. Fluorescence was quantified as MFI units. (C) Concentrations of C5a-desArg determined in lung supernatants from WT and Ptx3−/− mice on Day 4 after sodA bead embolization (n = 5). (D) Immunofluorescence for FH (factor H) and C4BP (C4-binding protein) in granulomas from WT and Ptx3−/− mice on Day 4 after sodA bead embolization (representative of three independent experiments). Nuclei were stained with DAPI. Scale bars indicate 50 µm. Fluorescence was quantified as MFI units. (E) Area of sodA-elicited granulomas in WT and Ptx3−/− mice on Day 4 after bead embolization and treatment with C5aR1 antagonist PMX53 or PBS (n = 6). (F) The number of alveolar macrophages in the lungs (n = 4) and (G) concentrations of CXCL9 and CXCL10 in lung supernatants (n = 5) from WT and Ptx3−/− mice on Day 4 after sodA bead embolization and treatment with PMX53 or PBS. *P < 0.05, **P < 0.01, and ***P < 0.001; MFI = mean fluorescence intensity; ns = not significant; PBS = phosphate-buffered saline.
We next evaluated complement activation during granuloma formation. Immunostaining of granulomas revealed enhanced deposition of C3 in granulomas from Ptx3−/− mice relative to WT control subjects (Figure 3B). Treatment with rPTX3 reduced the immunoreactivity for C3 in Ptx3−/− mice to values similar to those in WT animals (Figure 3B). The concentrations of activated C5a were also higher in lung supernatants from Ptx3−/− mice than in control subjects (Figure 3C), although the mRNA expression of C3a and C5a receptors was not affected (Figure E3B). Because PTX3 interacts with FH (factor H) and C4BP (C4-binding protein) to amplify their regulatory activity over complement activation (11, 19), we analyzed the immunoreactivity of these molecules. Immunostaining for FH was significantly lower in granulomas from Ptx3−/− mice, whereas the deposition of C4BP did not differ between genotypes (Figure 3D).
To further assess the pathogenic role of complement activation, we evaluated granuloma formation after treatment with the C5aR1 (C5a receptor 1) antagonist PMX53. In these conditions, the size of granulomas in Ptx3−/− mice decreased from 0.032 to 0.019 mm2 without affecting their formation in WT animals (Figures 3E and E3C). This effect was accompanied by a decrease in the number of alveolar macrophages in Ptx3−/− mice (Figure 3F). Consistent with a role for complement activation in promoting chemokine production (20), the concentrations of CXCL9 and CXCL10 (Figure 3G), but not CCL2 or CCL4 (Figure E3D), in Ptx3−/− mice were also decreased after PMX53 treatment. Likewise, the augmented production of TNF, IL-6, IFN-γ, and IL-17 was also rescued on the C5aR1 blockade (Figure E3E).
PTX3 Regulates the Metabolic Activity of Macrophages to Control Aggregation and Proliferation
To decipher the molecular mechanisms whereby PTX3 might regulate macrophage activity during granuloma formation, we assessed the functional phenotypes of macrophages. The analysis of lungs from Ptx3−/− mice revealed an increased expression of Arg1, Chil3, Lgals3, Mrc1, and Igf1, but reduced expression of Il12b, compared with WT control subjects (Figure 4A), indicating the polarization of macrophages toward an alternative (M2-like) phenotype.
Figure 4.
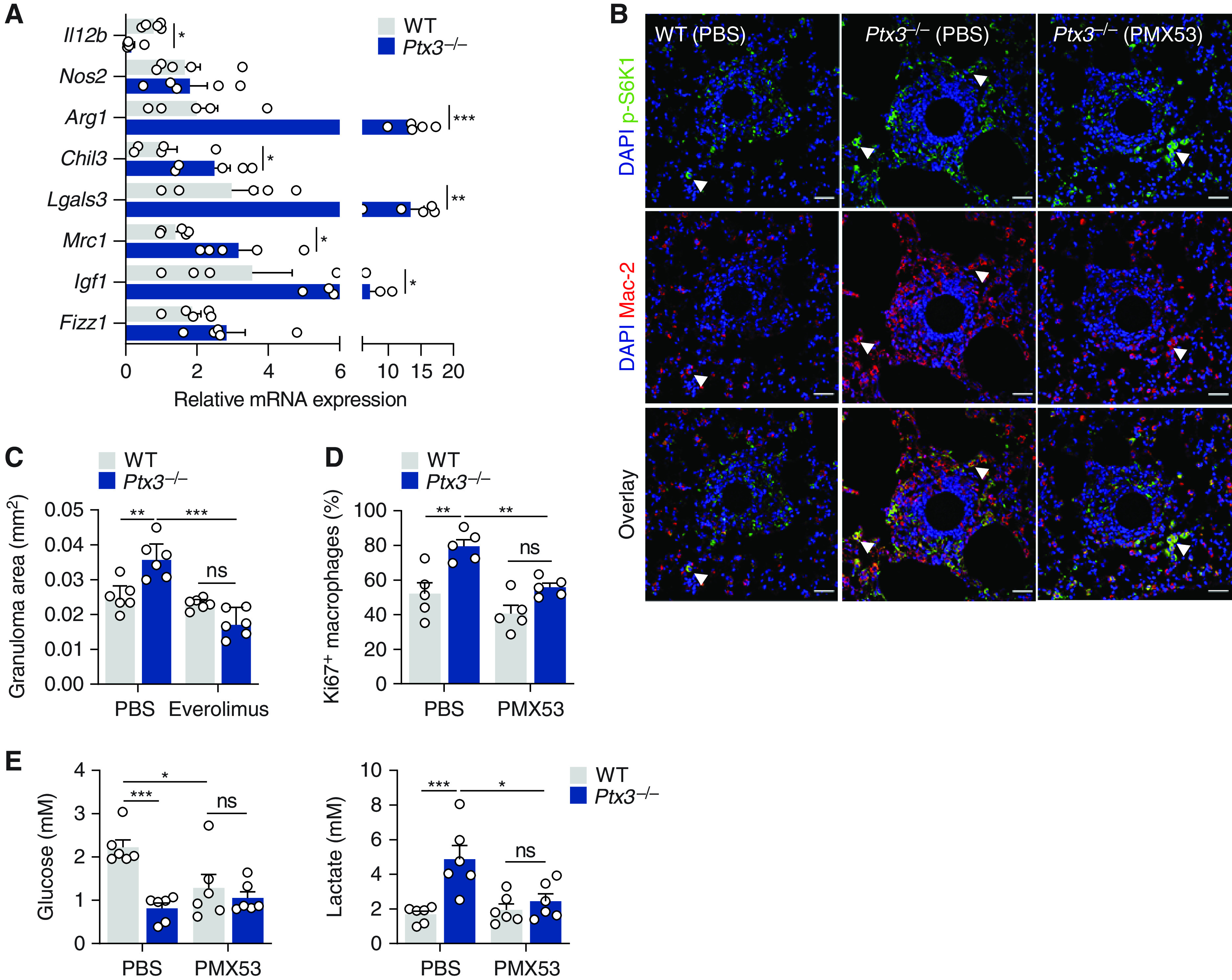
PTX3 (pentraxin 3) inhibits the aggregation and proliferation of macrophages. (A) mRNA expression of Il12b, Nos2, Arg1, Chil3, Lgals3, Mrc1, Igf1, and Fizz1 in lung tissue from wild-type (WT) and Ptx3−/− mice on Day 4 after sodA bead embolization (n = 5). (B) Immunofluorescence for p-S6K1 and Mac-2 in granulomas from WT and Ptx3−/− mice on Day 4 after sodA bead embolization and treatment with PMX53 or PBS (representative of three independent experiments). Nuclei were stained with DAPI. Scale bars indicate 50 µm. The arrows indicate macrophages with active S6K1 (cells that are positive for both Mac-2 and p-S6K1). (C) Area of sodA-elicited granulomas in WT and Ptx3−/− mice on Day 4 after bead embolization and treatment with everolimus or PBS (n = 6). (D) Proliferation (expressed as the percentage of Ki67+ cells; n = 5) of alveolar macrophages from WT and Ptx3−/− mice on Day 4 after sodA bead embolization and treatment with PMX53 or vehicle. (E) Concentrations of glucose and lactate in lung supernatants from WT and Ptx3−/− mice on Day 4 after sodA bead embolization and treatment with PMX53 or PBS (n = 6). *P < 0.05, **P < 0.01, and ***P < 0.001. ns = not significant.
Because the mTORC1-dependent proliferation of macrophages is required for granuloma formation (21), we assessed the phosphorylation of the p70S6 kinase (S6K1), a downstream target of mTORC1. The concentrations of p-S6K1 were higher in granulomas from Ptx3−/− mice than in WT control subjects and displayed robust colocalization with the macrophage Mac-2 marker (Figures 4B and E4A). Treatment with PMX53 restrained mTORC1 activation in Ptx3−/− mice and the number of macrophages infiltrating the granulomas. Inhibition of mTORC1 with everolimus abrogated the enhanced granuloma formation observed in Ptx3−/− mice (Figures 4C and E4B). Moreover, the percentage of proliferating macrophages in Ptx3−/− mice was also higher than in WT animals, and their number decreased after treatment with PMX53 (Figure 4D). In support of enhanced proliferation under PTX3 deficiency, there were no differences in the viability of lung macrophages between backgrounds (Figure E4C).
Metabolic adaptations support cellular proliferation during granuloma formation (21). Analysis of lung homogenates demonstrated that the expression of genes involved in glycolysis, such as the glucose transporter Slc2a1 (Glut1), Hk2 (hexokinase 2), and Pfkfb3 (6-phosphofructose-2-kinase/fructose-2,6-bisphosphatase 3) was higher in Ptx3−/− than WT mice (Figure E4D). The induction of glycolysis was confirmed by the lower concentrations of glucose and the higher concentrations of lactate detected in the lungs of Ptx3−/− mice (Figure 4E). In support of a relevant role for C5aR1 signaling in macrophage metabolism, treatment with PMX53 restored the concentrations of both lactate and glucose detected in Ptx3−/− mice to values similar to those in WT animals (Figure 4E).
Genetic Variation in Human PTX3 Regulates Granuloma Formation
SNPs in PTX3 were reported to impair PTX3 expression in infectious diseases (5, 6, 22) and inflammatory conditions (23, 24). We examined an in vitro model of granuloma formation using peripheral blood mononuclear cells (PBMCs) from patients with sarcoidosis (25). Patients were grouped according to the presence of homozygous genotypes for the major or minor alleles in rs2305619 and rs3816527 SNPs, known to underlie distinct profiles of PTX3 expression and hereafter referred to as h1/h1 or h2/h2, respectively (6). The frequency of the h1/h1, h1/h2, and h2/h2 haplotypes was 16%, 50%, and 34%, and 22%, 47%, and 31%, in patients with sarcoidosis and healthy subjects, respectively, and was not different between groups.
In response to sodA-coated polystyrene beads, h2/h2 PBMCs elicited the formation of larger granuloma-like structures as early as Day 4 and until Day 7 of culture, whereas only minimal cell aggregation was apparent in h1/h1 PBMCs (Figure 5A). No differences were observed in the number of aggregates formed (Figure E5A), and the size of the granulomas elicited by naked beads was negligible (Figure E5B). On Day 7 of culture, the median granuloma area elicited by h2/h2 and h1/h1 PBMCs was 10.0 (8.3–12.8) μm2 and 5.9 (4.7–6.4) μm2, respectively. The enhanced granuloma formation by h2/h2 PBMCs was associated with lower secretion of PTX3 (Figure 5B), and treatment with rPTX3 decreased the size of the forming structures from 10.1 (7.9–12.0) μm2 to 4.1 (2.4–6.7) μm2, whereas it did not affect h1/h1 control subjects (Figures 5C and E5C).
Figure 5.
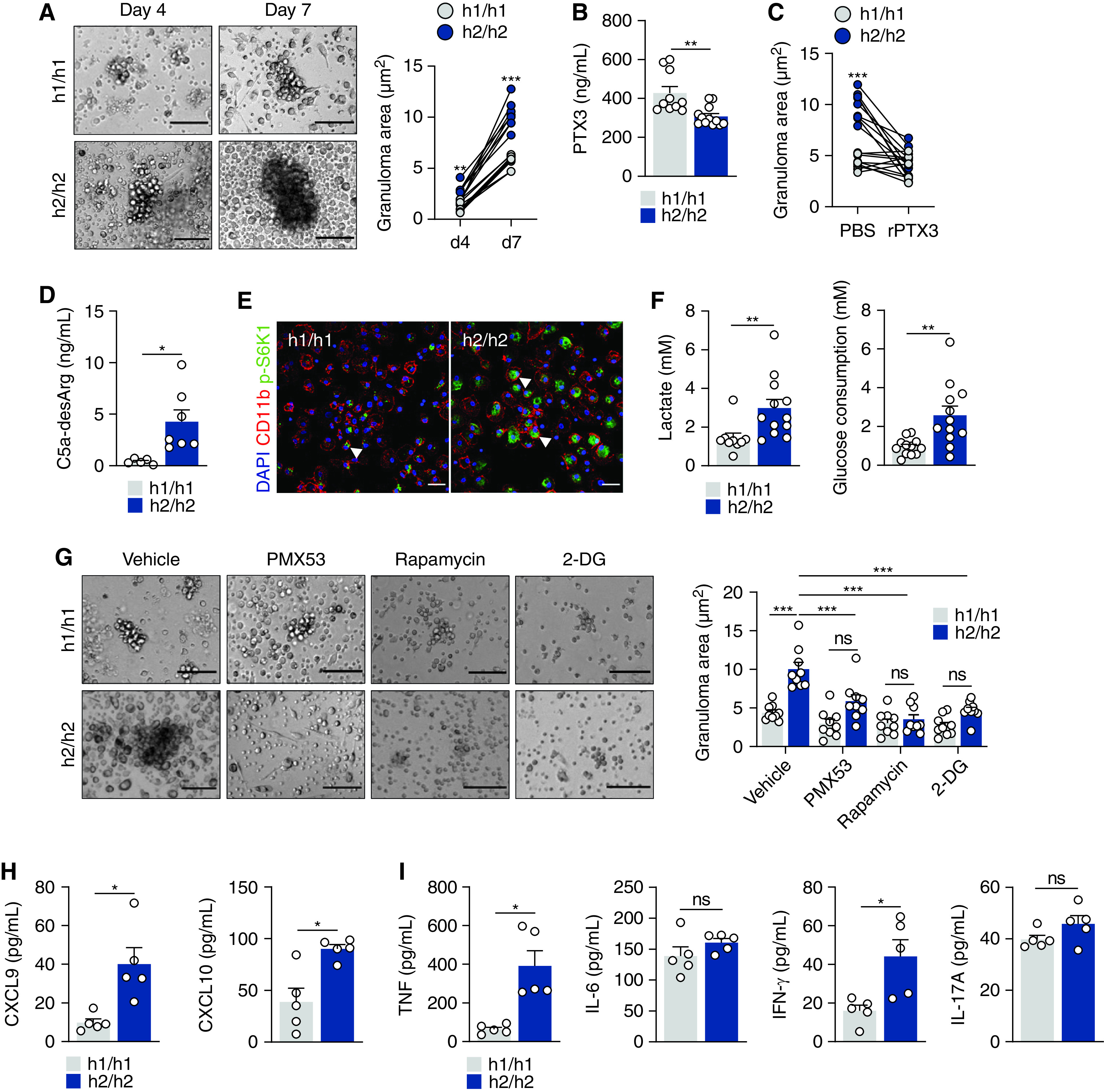
Genetic variants in PTX3 (pentraxin 3) influence human granuloma formation in vitro. (A) Granuloma formation by peripheral blood mononuclear cells (PBMCs) from h1/h1 and h2/h2 patients on Days 4 and 7 after culture with sodA beads (representative of three independent experiments). Dots represent the mean area of the granulomas per well in each patient (n = 9). Scale bars indicate 100 µm. (B) Concentrations of PTX3 in supernatants from h1/h1 (n = 10) and h2/h2 (n = 12) PBMCs on Day 1 after culture with sodA beads. (C) Area of sodA-elicited granulomas by h1/h1 and h2/h2 PBMCs on Day 7 after culture and treatment with rPTX3 (recombinant PTX3) or PBS (n = 9). (D) Concentrations of C5a-desArg in supernatants from h1/h1 and h2/h2 PBMCs on Day 7 after culture with sodA beads (n = 9). (E) Immunofluorescence for CD11b and p-S6K1 in sodA-elicited granulomas by h1/h1 and h2/h2 PBMCs on Day 7 after culture and treatment with PMX53 or PBS (representative of three independent experiments). Nuclei were stained with DAPI. Scale bars indicate 100 µm. The arrows indicate CD11b-positive cells with positive p-S6K1 staining. (F) Concentrations of lactate secretion and glucose consumption in supernatants from h1/h1 and h2/h2 PBMCs on Day 4 after culture with sodA beads (n = 12). (G) Granuloma formation by PBMCs from h1/h1 and h2/h2 patients on Days 4 and 7 after culture with sodA beads and treatment with PMX53, rapamycin, 2-DG, or vehicle (representative of three independent experiments). Dots represent the mean area of the granulomas per well in each patient (n = 9). Scale bars indicate 100 µm. (H) Concentrations of CXCL9 and CXCL10 and (I) TNF, IL-6, IFN-γ, and IL-17 in sodA-elicited granulomas by h1/h1 and h2/h2 PBMCs on Day 4 after culture (n = 5). *P < 0.05, **P < 0.01, and ***P < 0.001. ns = not significant; PBS = phosphate-buffered saline.
On Day 7, increased concentrations of C5a were detected in supernatants from h2/h2 PBMCs relative to h1/h1 control subjects (Figure 5D). The granulomas elicited by h2/h2 PBMCs displayed stronger S6K1 phosphorylation, particularly among CD11b-positive cells (Figure 5E), and the concentrations of secreted lactate and glucose uptake were also higher in h2/h2 PBMCs (Figure 5F). In support of the relevance of these functional features to granuloma formation, blocking complement activation (PMX53), mTOR signaling (rapamycin), or glycolysis (2-DG) restrained the larger size of the h2/h2 aggregates (Figure 5G). None of the treatments significantly affected the aggregation of h1/h1 PBMCs. In line with the complement-dependent production of chemokines, the concentrations of CXCL9 and CXCL10 were higher in granulomas elicited by h2/h2 PBMCs (Figure 5H). Likewise, the concentrations of TNF and IFN-γ, but not IL-6 and IL-17A (Figure 5I), were increased in supernatants from h2/h2 PBMCs.
PTX3 Influences the Clinical Features and Outcome of Sarcoidosis
PTX3 expression is increased across several pulmonary diseases, including aspergillosis, chronic obstructive pulmonary disease, and asthma (6, 24, 26). To assess the relevance of PTX3 to the phenotype and clinical course of sarcoidosis, we performed an in silico analysis of single-cell transcriptome data of BAL from patients with granulomatous diseases (27). The results revealed that PTX3 expression was restricted to macrophages (Figure 6A) and that cells from patients with sarcoidosis displayed higher concentrations of PTX3 relative to chronic beryllium disease (Figure 6B). These findings were confirmed in a peripheral blood cell dataset, in which PTX3 expression was limited to CD14+ and CD16+ monocytes and higher in patients with sarcoidosis relative to healthy control subjects (Figure 6C).
Figure 6.
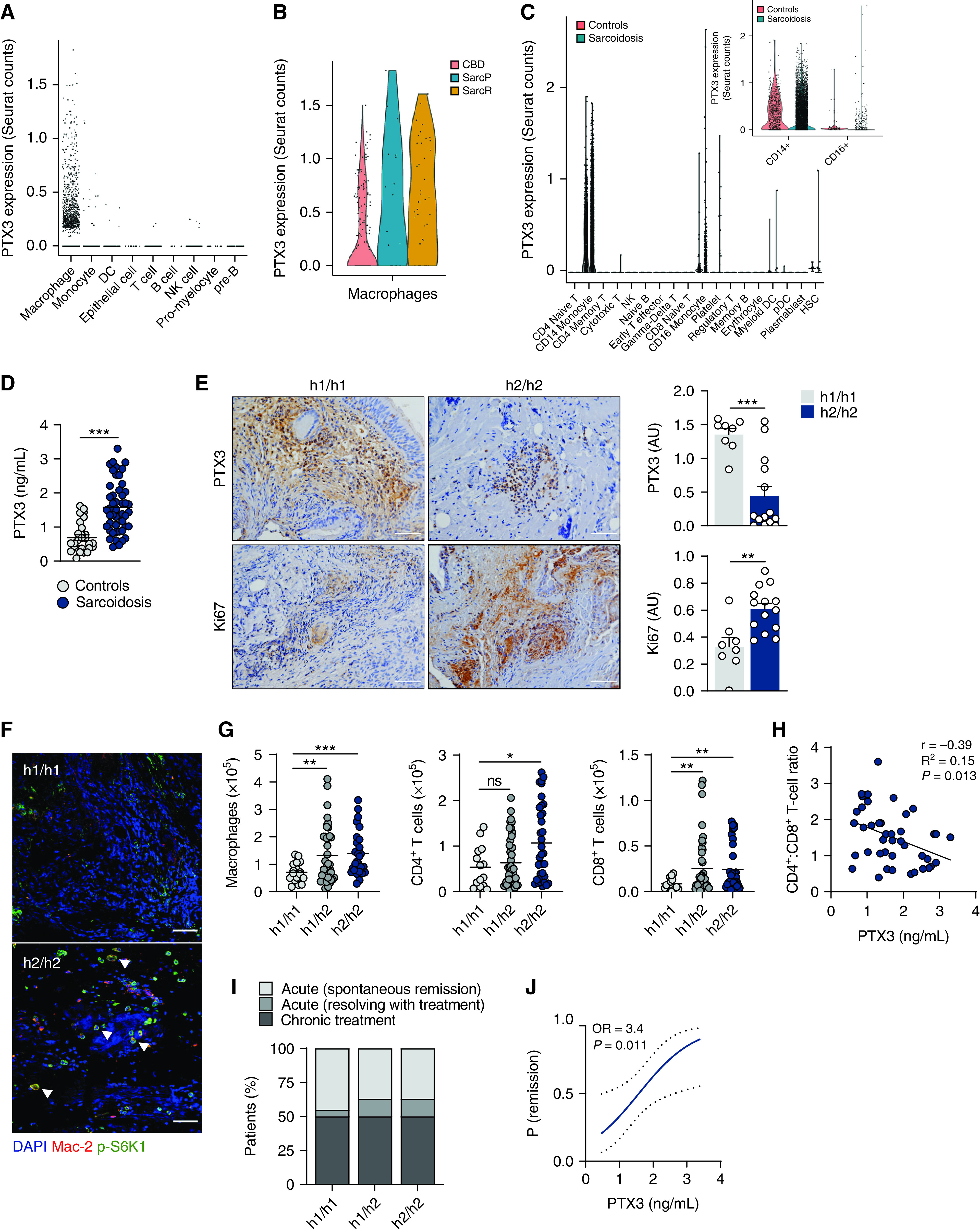
PTX3 (pentraxin 3) influences human granulomatous inflammation and the course of sarcoidosis. (A) PTX3 expression in single-cell RNA-seq performed on BAL samples from patients with granulomatous diseases, including sarcoidosis and chronic beryllium disease (CBD). Data were obtained from GEO under the identifier GSE184735. (B) Violin plot depicting the expression of PTX3 in the isolated cluster of PTX3-expressing macrophages in chronic beryllium disease and progressive sarcoidosis (SarcP) and remitting sarcoidosis (SarcR). (C) PTX3 expression in single-cell RNA-seq performed on peripheral blood mononuclear cells from patients with sarcoidosis and healthy control subjects. Data were obtained from GEO under the identifier GSE132338. The inset shows the results for CD14+ and CD16+ monocytes. (D) Concentrations of PTX3 in plasma from patients with active sarcoidosis (n = 48) and age- and sex-matched healthy control subjects (n = 28). This subgroup of patients was selected based on the availability of plasma samples at diagnosis. (E) Immunohistochemistry of PTX3 and Ki67 in lung granulomatous lesions from h1/h1 (n = 8) and h2/h2 (n = 14) patients (representative images are shown). Scale bars indicate 50 µm. Concentrations of PTX3 and Ki67 were estimated as arbitrary units. (F) Immunofluorescence for p-S6K1 and Mac-2 in granulomatous lesions from h1/h1 (n = 8) and h2/h2 (n = 14) patients (representative images are shown). The arrows indicate macrophages with positive p-S6K1 staining. Scale bars indicate 50 µm. (G) Number of macrophages and CD4+ T cells and CD8+ T cells in the BAL from h1/h1 (n = 17 and 14, respectively), h1/h2 (n = 40 and 44, respectively), and h2/h2 (n = 27 and 35, respectively) patients with sarcoidosis. Numbers were normalized against total cell counts in the BAL. (H) Correlation analysis of the CD4+:CD8+ T-cell ratio in the BAL and the concentrations of PTX3 in the plasma (n = 42). (I) Percentage of h1/h1, h1/h2, and h2/h2 patients diagnosed with spontaneous remission, remission after treatment, or chronic disease after a 2-year follow-up period. (J) Predictive modeling of remission according to the concentrations of PTX3 in the plasma at baseline (n = 39). The plot shows the probability of remitting the disease within 2 years after diagnosis. Dotted lines indicate the 95% confidence interval. *P < 0.05, **P < 0.01, and ***P < 0.001. GEO = gene expression omnibus; ns = not significant; OR = odds ratio; RNA-seq = RNA sequencing.
To confirm the transcriptional data, we examined the concentrations of PTX3 in plasma samples from patients with sarcoidosis (Table 1). In line with a response to detrimental inflammation, patients at the initial diagnosis of sarcoidosis exhibited higher concentrations of circulating PTX3 than healthy donors (1.59 ± 0.08 vs. 0.69 ± 0.11 ng/ml) (Figure 6D). Although systemic PTX3 was not influenced by genetic variation (Figure E6A), analysis of PTX3 expression in granulomatous lung lesions revealed that h2/h2 patients presented lower concentrations of PTX3 and, in contrast, higher expression of Ki67, indicating enhanced proliferation (Figure 6E). The analysis of each tissue sample revealed that PTX3 and Ki67 were inversely correlated (Pearson r = −0.47; P = 0.028) (Figure E6B). Moreover, the reduced expression of PTX3 within h2/h2 granulomas was associated with increased infiltration of Mac-2–expressing hypertrophic cells that displayed pronounced mTORC1 activation, as revealed by the enhanced S6K1 phosphorylation (Figure 6F).
Table 1.
Demographic and Clinical Features of Patients with Sarcoidosis
| Clinical Features | N = 188 |
|---|---|
| Age, mean ± SD | 35.3 ± 9.4 |
| Female, n (%) | 110 (58.5) |
| White, n (%) | 181 (96.3) |
| Smoking status, n (%)* | |
| Smoker | 24 (13.3) |
| Ex-smoker | 18 (9.9) |
| Nonsmoker | 139 (76.8) |
| Scadding stage† | |
| Stage 0 | 2 (1.1) |
| Stage 1 | 75 (40.8) |
| Stage 2 | 83 (45.1) |
| Stage 3 | 15 (8.2) |
| Stage 4 | 9 (4.9) |
| Multiorgan involvement‡ | 74 (41.3) |
| Pulmonary function tests§ | |
| FVC, % predicted | 95.5 ± 17.3 |
| FEV1, % predicted | 91.1 ± 18.8 |
| FEV1/FVC, % | 81.7 ± 8.3 |
| TLC, % predicted | 94.0 ± 16.5 |
| RV, % predicted | 97.0 ± 30.7 |
| DLCO, % predicted | 81.4 ± 17.3 |
| KCO, % predicted | 85.5 ± 17.3 |
| BAL cellular markers | |
| Total cell count, ×105/mlǁ | 2.29 ± 1.63 |
| Lymphocytes, % | 43.0 ± 17.2 |
| CD4+/CD8+ T-cell ratio | 5.92 ± 4.68 |
| Macrophages, % | 54.3 ± 16.9 |
| Neutrophils, % | 2.6 ± 3.5 |
| Eosinophils, % | 1.3 ± 2.3 |
| Elevated SACE (>70 IU/L), %¶ | 83 (51.9) |
| Outcome | |
| Acute (spontaneous remission) | 64 (39.5) |
| Acute (resolving with treatment) | 18 (11.1) |
| Chronic treatment | 80 (49.4) |
Definition of abbreviations: DLCO = diffusion capacity for carbon monoxide; KCO = carbon monoxide transfer coefficient; RV = residual volume; SACE = serum angiotensin-converting enzyme; TLC = total lung capacity.
Missing values: *7, †4, ‡9, §33, ǁ67, and ¶28.
We next evaluated whether genetic variation in PTX3 regulated the influx of inflammatory cells in the BAL. Our analysis revealed a general effect on leukocyte infiltration that was genotype-dependent, with h2/h2 patients displaying increased numbers of macrophages, and CD4+ and CD8+ T cells (Figure 6G), but also neutrophils, eosinophils, and mast cells (Figure E6C) compared with h1/h1 patients. The BAL CD4+:CD8+ lymphocyte ratio was not affected by PTX3 genotypes (Figure E6D), although it was inversely correlated with plasma PTX3 (Pearson r = −0.38; P = 0.013) (Figure 6H). The number of circulating CD4+ and CD8+ T cells was instead diminished in h1/h2 and h2/h2 compared with h1/h1 patients (Figure E6E). To substantiate these findings, we analyzed patients from the ACCESS (A Case Control Etiologic Study of Sarcoidosis) study with available immunophenotyping data at the time of diagnosis (28) and confirmed that h2/h2 patients displayed an increased percentage of BAL macrophages and CD3+ T cells compared with h1/h1 control subjects (Figure E6F).
To determine whether PTX3 expression at diagnosis could influence disease outcome, we performed predictive modeling analyses. Out of the patients included, 162 reached a 2-year follow-up at the time of analysis, with 82 remitting and 80 developing chronic disease (Table 1). Although the disease course was not influenced by genetic variation in PTX3 (Figure 6I), the probability of remitting disease 2 years after diagnosis, either spontaneously or after treatment, was instead 3.4-fold higher when the concentrations of circulating PTX3 at diagnosis were also elevated (Figure 6J).
Discussion
PTX3 exerts a critical role in pulmonary homeostasis by mediating immunity against respiratory infections, including fungal, and also allergy, and tissue damage. The finding that T cells are specifically primed by an airborne mold-derived peptide in acute sarcoidosis (29) suggests the involvement of PTX3 in sarcoidosis. We now show that PTX3 acts as a physiological break of granuloma formation by restraining the pathogenic activation of complement and the downstream metabolic reprogramming of macrophages required to sustain their functional activity and proliferation. Activated macrophages produce chemokines that further recruit granuloma-promoting immune cells to support the formation of sarcoid granulomas. Chemokines, notably CXCL9 and CXCL10, recruit both lymphocytes and cells of the monocytic lineage in sarcoidosis and have been associated with systemic organ involvement and the severity of pulmonary disease (30).
Besides PTX3, innate immunity components such as SAA (serum amyloid A) have also been implicated in granuloma formation (31). Because SAA orchestrates alveolar macrophage differentiation via CSF-1 (colony-stimulating factor 1) receptor-dependent signals (32) and regulates PTX3 secretion by endothelial cells (33), PTX3 and SAA likely act in concert to coordinate the functional activity of macrophages. Exposure of macrophages to SAA activated the NLRP3 (NLR family pyrin domain containing 3) inflammasome and promoted IL-1β production (34), and these pathways are upregulated in sarcoidosis and involved in granuloma formation (35). Therefore, PTX3 may represent the missing link between humoral innate immunity to an unknown signal and the activation of pathogenic signatures that converge to drive granuloma formation.
The macrophages in tuberculous granulomas undergo a mesenchymal–epithelial reprogramming and express canonical epithelial determinants, such as E-cadherin (36). Because PTX3 inhibits mesenchymal–epithelial transitions in melanoma cells (37), this finding suggests its protective role during granuloma formation. In addition, transitions between quiescent and activated states require the rewiring of cellular metabolism to promote granulomatous inflammation. The chronic mTORC1 activation was found to redirect metabolism toward glycolysis and sustain the differentiation and proliferation of hypertrophic macrophages and the formation of epithelioid granulomas (21). Whether mTORC1 activity directly induces the expression of E-cadherin or other genes involved in the epithelioid transformation of macrophages remains unknown. However, mutations in mTOR-related genes have been detected in familial sarcoidosis (38), and phagosome-regulated mTORC1 signaling is required for granuloma formation in vitro (39), even if the exacerbated signaling in granulomatous lesions failed to predict disease course (40). Our results position PTX3 as a critical upstream regulator that inhibits the detrimental activation of mTORC1 and the associated granuloma-promoting cues in macrophages.
Complement activation regulates key metabolic pathways and impacts fundamental cellular processes, such as proliferation and survival (41). Although the impact of C5aR1 in macrophages is unclear, its ablation was found to limit the alkalinization and activation of glucose metabolism in neutrophils (42) and alleviate the severity of graft-versus-host-disease in stem cell transplantation by impairing the glycolytic activity and Th1 differentiation of donor T cells (43). In contrast, C5aR1 stimulation promoted an aberrant NLRP3 activation and the secretion of IL-1β by T cells which, in turn, acted as an autocrine signal for the production of IFN-γ and Th1 differentiation (44).
Genetic variation plays a significant role in the etiology of sarcoidosis (45, 46), despite not all the contributing factors having been elucidated. We now implicate genetic variation in PTX3 in the regulation of macrophage activity and granuloma formation in human disease. Whether PTX3 variants are involved in the actual onset of sarcoidosis requires the investigation of larger and carefully controlled cohorts of patients. Nevertheless, these findings support the rational discovery of therapeutic approaches based on the functional properties of PTX3 but also highlight the integration of immunogenetic markers into clinical stratification processes of disease progression. Our data suggest, however, that PTX3 may display therapeutic benefits only in the context of functional defects that drive the pathogenic amplification of complement activation. Because this pathway is already highly regulated by PTX3 in competent individuals, it is likely that further inhibition is not achieved. In support of this, rPTX3 failed to improve disease phenotypes in WT mice subjected to other experimental models in which complement-related inflammation is also detrimental, most notably cancer (8). This is in line with the dominant phenotype of the heterozygous carriage of PTX3 variants because it is likely that the concentrations of PTX3 in these patients are not sufficient to restrain exacerbated complement activation.
A few case reports have suggested a clinical benefit for rapamycin in the treatment of sarcoidosis (47). Complement-inhibitory strategies might also be relevant in balancing physiological mechanisms required for the metabolic activity and proliferation of granuloma-promoting macrophages. Although complement inhibitors (e.g., eculizumab and pegcetacoplan) are available (48), clinical trials assessing their safety and efficacy in sarcoidosis are lacking. Likewise, no PTX3-based formulations are currently available, and its therapeutic potential has been evaluated only in preclinical models. A recombinant human pentraxin 2 (PRM-151) formulation displayed beneficial effects on lung function in idiopathic pulmonary fibrosis over a 76-week open-label extension study of the phase 2 PRM-151–202 trial (49). Notably, PRM-151 prevents the differentiation of profibrotic M2 macrophages while it fosters the development of regulatory macrophages that promote epithelial healing and resolution of inflammation (50). Therefore, immunotherapeutic approaches to target detrimental macrophage functions to granuloma formation might also have clinical value in sarcoidosis.
Ultimately, understanding how metabolic networks regulate immune cell function may lead to innovative host-directed therapeutic interventions based on the metabolic repurposing of host cells toward immune protection against granuloma formation.
Acknowledgments
Acknowledgment
The authors thank Sara Ferreira for excellent technical assistance in the in vivo studies and Marilia Beltrão for the analysis of BAL from patients.
Footnotes
Supported by the Fundação para a Ciência e a Tecnologia (FCT) (PTDC/MED-GEN/28778/2017 [A.C.], PTDC/MED-OUT/1112/2021 [A.C.], UIDB/50026/2020 and UIDP/50026/2020, SFRH/BD/141127/2018 [C.D.-O.], PD/BD/137680/2018 [D.A.], CEECIND/03070/2020 [E.T.], CEECIND/00185/2020 [R.S.], and CEECIND/04058/2018 [C.C.]); the “la Caixa” Foundation and FCT under the agreement LCF/PR/HR17/52190003; the European Union’s Horizon 2020 research and innovation programme (H2020 Societal Challenges) under grant agreement no. 847507; the Northern Portugal Regional Operational Programme (NORTE 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (ERDF) (NORTE-01-0145-FEDER-000039); the Associazione Italiana Ricerca sul Cancro (AIRC IG-21714 [C.G.]); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (206780/2014-1/PDE [S.D.S.]), the ICVS Scientific Microscopy Platform, member of the national infrastructure PPBI - Portuguese Platform of Bioimaging (PPBI-POCI-01-0145-FEDER-022122); and the Clinical Academic Center - Braga (2-CA Braga).
Author Contributions: R.A.G., H.N.B., C.C., and A.C. conceived the study and designed all experiments. R.A.G., C.D.-O., D.A., C.F.C, and S.D.S. developed, performed, and analyzed in vivo and in vitro models of lung granulomatous inflammation. H.N.B., O.S., M.J., R.R., C.S.M., L.D., and A. Morais recruited patients and provided clinical specimens and metadata. E.T. and R.S. performed flow cytometry analysis. R.A.G., C.D.-O., and S.C. performed immunofluorescence and immunochemistry analyses. A.D., S.N.M., and C.G. analyzed single-cell RNA-seq datasets. F.L.v.d.V., T.W., J.D.L., A. Mantovani, and C.G. critically revised the manuscript for important intellectual content. R.A.G., H.N.B., C.C., and A.C. drafted the manuscript. All experiments were supervised by A.C.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202112-2771OC on June 29, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Crouser ED, Maier LA, Wilson KC, Bonham CA, Morgenthau AS, Patterson KC, et al. Diagnosis and detection of sarcoidosis. An official American Thoracic Society clinical practice guideline. Am J Respir Crit Care Med . 2020;201:e26–e51. doi: 10.1164/rccm.202002-0251ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drent M, Crouser ED, Grunewald J. Challenges of sarcoidosis and its management. N Engl J Med . 2021;385:1018–1032. doi: 10.1056/NEJMra2101555. [DOI] [PubMed] [Google Scholar]
- 3. Cinetto F, Scarpa R, Dell’Edera A, Jones MG. Immunology of sarcoidosis: old companions, new relationships. Curr Opin Pulm Med . 2020;26:535–543. doi: 10.1097/MCP.0000000000000711. [DOI] [PubMed] [Google Scholar]
- 4. Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol . 2010;28:157–183. doi: 10.1146/annurev-immunol-030409-101305. [DOI] [PubMed] [Google Scholar]
- 5. Campos CF, Leite L, Pereira P, Vaz CP, Branca R, Campilho F, et al. PTX3 polymorphisms influence cytomegalovirus reactivation after stem-cell transplantation. Front Immunol . 2019;10:88. doi: 10.3389/fimmu.2019.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cunha C, Aversa F, Lacerda JF, Busca A, Kurzai O, Grube M, et al. Genetic PTX3 deficiency and aspergillosis in stem-cell transplantation. N Engl J Med . 2014;370:421–432. doi: 10.1056/NEJMoa1211161. [DOI] [PubMed] [Google Scholar]
- 7. Doni A, Musso T, Morone D, Bastone A, Zambelli V, Sironi M, et al. An acidic microenvironment sets the humoral pattern recognition molecule PTX3 in a tissue repair mode. J Exp Med . 2015;212:905–925. doi: 10.1084/jem.20141268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bonavita E, Gentile S, Rubino M, Maina V, Papait R, Kunderfranco P, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell . 2015;160:700–714. doi: 10.1016/j.cell.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 9. Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, et al. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol . 2010;11:328–334. doi: 10.1038/ni.1854. [DOI] [PubMed] [Google Scholar]
- 10. Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, et al. Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem . 1997;272:32817–32823. doi: 10.1074/jbc.272.52.32817. [DOI] [PubMed] [Google Scholar]
- 11. Deban L, Jarva H, Lehtinen MJ, Bottazzi B, Bastone A, Doni A, et al. Binding of the long pentraxin PTX3 to factor H: interacting domains and function in the regulation of complement activation. J Immunol . 2008;181:8433–8440. doi: 10.4049/jimmunol.181.12.8433. [DOI] [PubMed] [Google Scholar]
- 12. Ma YJ, Doni A, Skjoedt MO, Honoré C, Arendrup M, Mantovani A, et al. Heterocomplexes of mannose-binding lectin and the pentraxins PTX3 or serum amyloid P component trigger cross-activation of the complement system. J Biol Chem . 2011;286:3405–3417. doi: 10.1074/jbc.M110.190637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martinez-Bravo MJ, Wahlund CJ, Qazi KR, Moulder R, Lukic A, Rådmark O, et al. Pulmonary sarcoidosis is associated with exosomal vitamin D-binding protein and inflammatory molecules. J Allergy Clin Immunol . 2017;139:1186–1194. doi: 10.1016/j.jaci.2016.05.051. [DOI] [PubMed] [Google Scholar]
- 14. Vogt S, Trendelenburg M, Tamm M, Stolz D, Hostettler KE, Osthoff M. Local and systemic concentrations of pattern recognition receptors of the lectin pathway of complement in a cohort of patients with interstitial lung diseases. Front Immunol . 2020;11:562564. doi: 10.3389/fimmu.2020.562564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zorzetto M, Bombieri C, Ferrarotti I, Medaglia S, Agostini C, Tinelli C, et al. Complement receptor 1 gene polymorphisms in sarcoidosis. Am J Respir Cell Mol Biol . 2002;27:17–23. doi: 10.1165/ajrcmb.27.1.4805. [DOI] [PubMed] [Google Scholar]
- 16. Dubaniewicz A, Typiak M, Wybieralska M, Szadurska M, Nowakowski S, Staniewicz-Panasik A, et al. Changed phagocytic activity and pattern of Fcγ and complement receptors on blood monocytes in sarcoidosis. Hum Immunol . 2012;73:788–794. doi: 10.1016/j.humimm.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 17. Swaisgood CM, Oswald-Richter K, Moeller SD, Klemenc JM, Ruple LM, Farver CF, et al. Development of a sarcoidosis murine lung granuloma model using Mycobacterium superoxide dismutase A peptide. Am J Respir Cell Mol Biol . 2011;44:166–174. doi: 10.1165/rcmb.2009-0350OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pagán AJ, Ramakrishnan L. The formation and function of granulomas. Annu Rev Immunol . 2018;36:639–665. doi: 10.1146/annurev-immunol-032712-100022. [DOI] [PubMed] [Google Scholar]
- 19. Braunschweig A, Józsi M. Human pentraxin 3 binds to the complement regulator c4b-binding protein. PLoS One . 2011;6:e23991. doi: 10.1371/journal.pone.0023991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol . 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 21. Linke M, Pham HT, Katholnig K, Schnöller T, Miller A, Demel F, et al. Chronic signaling via the metabolic checkpoint kinase mTORC1 induces macrophage granuloma formation and marks sarcoidosis progression. Nat Immunol . 2017;18:293–302. doi: 10.1038/ni.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jaillon S, Moalli F, Ragnarsdottir B, Bonavita E, Puthia M, Riva F, et al. The humoral pattern recognition molecule PTX3 is a key component of innate immunity against urinary tract infection. Immunity . 2014;40:621–632. doi: 10.1016/j.immuni.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 23. Diamond JM, Meyer NJ, Feng R, Rushefski M, Lederer DJ, Kawut SM, et al. Lung Transplant Outcomes Group Variation in PTX3 is associated with primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med . 2012;186:546–552. doi: 10.1164/rccm.201204-0692OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y, Tedrow J, Nouraie M, Li X, Chandra D, Bon J, et al. Elevated plasma level of pentraxin 3 is associated with emphysema and mortality in smokers. Thorax . 2021;76:335–342. doi: 10.1136/thoraxjnl-2020-215356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crouser ED, White P, Caceres EG, Julian MW, Papp AC, Locke LW, et al. A novel in vitro human granuloma model of sarcoidosis and latent tuberculosis infection. Am J Respir Cell Mol Biol . 2017;57:487–498. doi: 10.1165/rcmb.2016-0321OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balhara J, Shan L, Zhang J, Muhuri A, Halayko AJ, Almiski MS, et al. Pentraxin 3 deletion aggravates allergic inflammation through a TH17-dominant phenotype and enhanced CD4 T-cell survival. J Allergy Clin Immunol . 2017;139:950–963.e9. doi: 10.1016/j.jaci.2016.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liao SY, Atif SM, Mould K, Konigsberg IR, Fu R, Davidson E, et al. Single-cell RNA sequencing identifies macrophage transcriptional heterogeneities in granulomatous diseases. Eur Respir J . 2021;57:2003794. doi: 10.1183/13993003.03794-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Semenzato G. ACCESS: a case control etiologic study of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis . 2005;22:83–86. [PubMed] [Google Scholar]
- 29. Greaves SA, Ravindran A, Santos RG, Chen L, Falta MT, Wang Y, et al. CD4+ T cells in the lungs of acute sarcoidosis patients recognize an Aspergillus nidulans epitope. J Exp Med . 2021;218:e20210785. doi: 10.1084/jem.20210785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arger NK, Ho ME, Allen IE, Benn BS, Woodruff PG, Koth LL. CXCL9 and CXCL10 are differentially associated with systemic organ involvement and pulmonary disease severity in sarcoidosis. Respir Med . 2020;161:105822. doi: 10.1016/j.rmed.2019.105822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen ES, Song Z, Willett MH, Heine S, Yung RC, Liu MC, et al. Serum amyloid A regulates granulomatous inflammation in sarcoidosis through Toll-like receptor-2. Am J Respir Crit Care Med . 2010;181:360–373. doi: 10.1164/rccm.200905-0696OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Anthony D, McQualter JL, Bishara M, Lim EX, Yatmaz S, Seow HJ, et al. SAA drives proinflammatory heterotypic macrophage differentiation in the lung via CSF-1R-dependent signaling. FASEB J . 2014;28:3867–3877. doi: 10.1096/fj.14-250332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong Z, An F, Wu T, Zhang C, Zhang M, Zhang Y, et al. PTX3, a key component of innate immunity, is induced by SAA via FPRL1-mediated signaling in HAECs. J Cell Biochem . 2011;112:2097–2105. doi: 10.1002/jcb.23128. [DOI] [PubMed] [Google Scholar]
- 34. Niemi K, Teirilä L, Lappalainen J, Rajamäki K, Baumann MH, Öörni K, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol . 2011;186:6119–6128. doi: 10.4049/jimmunol.1002843. [DOI] [PubMed] [Google Scholar]
- 35. Huppertz C, Jäger B, Wieczorek G, Engelhard P, Oliver SJ, Bauernfeind FG, et al. The NLRP3 inflammasome pathway is activated in sarcoidosis and involved in granuloma formation. Eur Respir J . 2020;55:1900119. doi: 10.1183/13993003.00119-2019. [DOI] [PubMed] [Google Scholar]
- 36. Cronan MR, Beerman RW, Rosenberg AF, Saelens JW, Johnson MG, Oehlers SH, et al. Macrophage epithelial reprogramming underlies mycobacterial granuloma formation and promotes infection. Immunity . 2016;45:861–876. doi: 10.1016/j.immuni.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ronca R, Di Salle E, Giacomini A, Leali D, Alessi P, Coltrini D, et al. Long pentraxin-3 inhibits epithelial-mesenchymal transition in melanoma cells. Mol Cancer Ther . 2013;12:2760–2771. doi: 10.1158/1535-7163.MCT-13-0487. [DOI] [PubMed] [Google Scholar]
- 38. Calender A, Lim CX, Weichhart T, Buisson A, Besnard V, Rollat-Farnier PA, et al. in the frame of GSF (Group Sarcoidosis France) Exome sequencing and pathogenicity-network analysis of five French families implicate mTOR signalling and autophagy in familial sarcoidosis. Eur Respir J . 2019;54:1900430. doi: 10.1183/13993003.00430-2019. [DOI] [PubMed] [Google Scholar]
- 39. Crouser ED, Locke LW, Julian MW, Bicer S, Sadee W, White P, et al. Phagosome-regulated mTOR signalling during sarcoidosis granuloma biogenesis. Eur Respir J . 2021;57:2002695. doi: 10.1183/13993003.02695-2020. [DOI] [PubMed] [Google Scholar]
- 40. Pizzini A, Bacher H, Aichner M, Franchi A, Watzinger K, Tancevski I, et al. High expression of mTOR signaling in granulomatous lesions is not predictive for the clinical course of sarcoidosis. Respir Med . 2021;177:106294. doi: 10.1016/j.rmed.2020.106294. [DOI] [PubMed] [Google Scholar]
- 41. Hess C, Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity . 2016;45:240–254. doi: 10.1016/j.immuni.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Denk S, Neher MD, Messerer DAC, Wiegner R, Nilsson B, Rittirsch D, et al. Complement C5a functions as a master switch for the pH balance in neutrophils exerting fundamental immunometabolic effects. J Immunol . 2017;198:4846–4854. doi: 10.4049/jimmunol.1700393. [DOI] [PubMed] [Google Scholar]
- 43. Nguyen H, Kuril S, Bastian D, Kim J, Zhang M, Vaena SG, et al. Complement C3a and C5a receptors promote GVHD by suppressing mitophagy in recipient dendritic cells. JCI Insight . 2018;3:e121697. doi: 10.1172/jci.insight.121697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science . 2016;352:aad1210. doi: 10.1126/science.aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fischer A, Ellinghaus D, Nutsua M, Hofmann S, Montgomery CG, Iannuzzi MC, et al. GenPhenReSa Consortium Identification of immune-relevant factors conferring sarcoidosis genetic risk. Am J Respir Crit Care Med . 2015;192:727–736. doi: 10.1164/rccm.201503-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rivera NV, Ronninger M, Shchetynsky K, Franke A, Nöthen MM, Müller-Quernheim J, et al. High-density genetic mapping identifies new susceptibility variants in sarcoidosis phenotypes and shows genomic-driven phenotypic differences. Am J Respir Crit Care Med . 2016;193:1008–1022. doi: 10.1164/rccm.201507-1372OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gupta N, Bleesing JH, McCormack FX. Successful response to treatment with sirolimus in pulmonary sarcoidosis. Am J Respir Crit Care Med . 2020;202:e119–e120. doi: 10.1164/rccm.202004-0914IM. [DOI] [PubMed] [Google Scholar]
- 48. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med . 2021;384:1028–1037. doi: 10.1056/NEJMoa2029073. [DOI] [PubMed] [Google Scholar]
- 49. Raghu G, van den Blink B, Hamblin MJ, Brown AW, Golden JA, Ho LA, et al. Long-term treatment with recombinant human pentraxin 2 protein in patients with idiopathic pulmonary fibrosis: an open-label extension study. Lancet Respir Med . 2019;7:657–664. doi: 10.1016/S2213-2600(19)30172-9. [DOI] [PubMed] [Google Scholar]
- 50. Murray LA, Rosada R, Moreira AP, Joshi A, Kramer MS, Hesson DP, et al. Serum amyloid P therapeutically attenuates murine bleomycin-induced pulmonary fibrosis via its effects on macrophages. PLoS One . 2010;5:e9683. doi: 10.1371/journal.pone.0009683. [DOI] [PMC free article] [PubMed] [Google Scholar]